

Summary
Mitogenomics has improved our understanding of medusozoan phylogeny. However, sequenced medusozoan mitogenomes remain scarce, and Medusozoa phylogeny studies often analyze mitogenomic sequences without incorporating mitogenome rearrangements. To better understand medusozoan evolution, we analyzed Medusozoa mitogenome phylogeny by sequencing and assembling eight mitogenomes from three classes (Cubozoa, Hydrozoa, and Scyphozoa). We reconstructed the mitogenome phylogeny using these mitogenomes and 84 other existing cnidarian mitogenomes to study mitochondrial gene rearrangements. All reconstructed mitogenomes had 13 mitochondrial protein-coding genes and two ribosomal genes typical for Medusozoa. Non-cubozoan mitogenomes were all linear and had typical gene orders, while arrangement of genes in the fragmented Cubozoa (Morbakka sp.) mitogenome differed from other Cubozoa mitogenomes. Gene order comparisons and ancestral state reconstruction suggest minimal rearrangements within medusozoan classes except for Hydrozoa. Our findings support a staurozoan ancestral medusozoan gene order, expand the pool of available medusozoan mitogenomes, and enhance our understanding of medusozoan phylogenetic relationships.
Subject areas: genomics, phylogeny
Graphical abstract
Highlights
-
•
Mitogenomes of two Hydrozoa, five Scyphozoa, and one Cubozoa sequenced
-
•
Hydrozoa, Staurozoa, and Cubozoa are monophyletic; Scyphozoa is paraphyletic
-
•
Medusozoan gene orders highly conserved, except in Hydrozoa
-
•
Staurozoa gene order inferred as ancestral to rest of Medusozoa
Introduction
Cnidaria is an ancient and diverse group of organisms dating back to the Precambrian.1,2 It comprises three major clades3,4,5,6—subphyla Anthozoa (classes Octocorallia and Hexacorallia), Endocnidozoa (classes Myxozoa and Polypodiozoa), and Medusozoa (classes Cubozoa, Hydrozoa, Scyphozoa, and Staurozoa). The large morphological diversity and phenotypic plasticity within Medusozoa have hampered efforts to resolve evolutionary relationships between medusozoans based solely on morphology.7,8 Consequently, molecular techniques have been employed both alongside morphology9 and independently3,7,10 to disentangle the evolutionary history of medusozoans.
Among the different molecular techniques used, including transcriptomics11,12 and nuclear phylogenomics,3 mitogenomic analysis has played an important role in improving our understanding of cnidarian, and in particular medusozoan, phylogenetic relationships. An early hypothesis based on molecular analyses by Bridge et al.13 placed Anthozoa as sister to Medusozoa on the basis that the former has circular mitogenomes while the latter has linear mitogenomes. Since then, numerous cnidarian studies have found further support for circular mitogenomes in Anthozoa14,15 and linear mitogenomes in Medusozoa.16,17,18,19 However, recent findings suggested that some anthozoans (e.g., Ceriantharia) may also have linear mitogenomes20 (but see Smith21).
Mitogenomics has also been used to investigate the relationships between the medusozoan classes. Studies on mitochondrial protein-coding genes (PCGs) by Zou et al.22 recovered the monophyletic classes Cubozoa, Hydrozoa, and Staurozoa, as well as a paraphyletic Scyphozoa, with Staurozoa being inferred to be sister to the other medusozoan classes (Figure 1A). Similarly, Kayal et al.18 also recovered a paraphyletic Scyphozoa alongside monophyletic classes Cubozoa, Hydrozoa, and Staurozoa using mitochondrial PCGs, although they recovered Staurozoa and Cubozoa as sister clades instead (Figure 1B). Notably, while mitogenomics in the aforementioned studies recovered a paraphyletic Anthozoa and placed Discomedusae as the sister group to Hydrozoa, a phylotranscriptomic reconstruction by Pratlong et al.12 suggested that the paraphyly of Anthozoa in mitogenomic studies likely resulted from saturation bias. More recently, analyses by Feng et al.23 on the PCGs of 266 complete cnidarian mitogenomes retrieved a monophyletic Anthozoa and paraphyletic Medusozoa (Figure 1C). Within Medusozoa, classes Staurozoa, Hydrozoa, and Scyphozoa were each inferred to be monophyletic as well. The study grouped Hydrozoa and Scyphozoa as sister taxa but placed Staurozoa as the sister group to Anthozoa instead (Figure 1C). However, Feng et al.23 omitted Cubozoa from their study due to insufficient publicly available complete cubozoan mitogenomes and did not include any Coronamedusae (Scyphozoa) mitogenomes. Mitogenomics has also narrowed the phylogenetic position of Myxozoa. Reclassified from Protista to Cnidaria in the late 20th century,8,9,24,25 early studies have inferred Myxozoa to be either part of Medusozoa or sister to the subphyla.8 Since then, additional phylogenomic studies3,11 and further reconstructions of non-linear Myxozoa mitogenomes25,26,27 have provided greater support for its phylogenetic position as sister to Medusozoa. These various competing hypotheses proposed based on mitogenomic studies throughout the years highlight the complexity involved in resolving the phylogenetic relationships within Medusozoa.
Figure 1.

Hypotheses of cnidarian phylogenetic relationships based on previous mitogenomic analyses
(A–C) (A) Zou et al.22; (B) Kayal et al.18; and (C) Feng et al.23
More recently, advancements in genomic sequencing technologies have enabled large volumes of genomic data to be produced and analyzed, leading to a push toward phylogenomic reconstructions across the tree of life.28,29,30 Consequently, proposed mitogenomic phylogeny hypotheses have also been more recently corroborated by nuclear phylogenomics. For instance, both nuclear31 and mitogenomic32,33 analyses show that Hydrozoa orders Anthoathecata and Leptothecata are paraphyletic groups and support the monophyly of subphylum Medusozoa.
However, there have also been disagreements between mitochondrial and nuclear phylogeny reconstructions. While the paraphyly of anthozoans has been proposed by mitogenomics22,34 (but see Feng et al.23), nuclear phylogenomic reconstructions have consistently established Anthozoa as a clade sister to all remaining cnidarians.11,24 The phylogenetic position of Meduosozoa classes has also been disputed. Almost two decades ago, scyphozoan paraphyly, with Cubozoa as a sister group to the [Discomedusae + Hydrozoa] clade (Figure S1A) was inferred by Dawson7 based on 5.8S and partial 28S ribosomal DNA (rDNA) sequences. Since then, other nuclear phylogenomic studies have found different phylogenetic relationships among the medusozoan classes (Figures S1B and S1C) based on nuclear protein genes.3,11 These discrepancies arise as mitochondrial DNA (mtDNA) is known to have higher substitution saturation compared to nuclear DNA (nDNA) and suggest that mitogenomics may not be as suitable as nDNA for inferring deep Medusozoa relationships.12 Together, these discrepancies highlight the challenges of resolving the medusozoan phylogeny using mitogenomics alone.
Nevertheless, despite the rise of nuclear phylogenomics, the study of mitochondrial evolution remains valuable for elucidating medusozoan phylogeny. Mitochondrial sequences generally evolve at a faster rate than nDNA.35,36 While the rate of mtDNA evolution in Anthozoa is much lower compared to other taxa37,38 (but see Stampar et al.39), mtDNA evolution rates in Medusozoa remain comparable to other metazoans,39,40 so mtDNA remains useful in providing informative sites for studying Medusozoa phylogeny. Additionally, mitogenomic sequences potentially provide greater phylogenetic resolution between organisms at lower taxonomic levels.41,42 The low recombination rates of mtDNA43 also contribute to its utility in studying closely related taxa.44 Moreover, the use of mtDNA, which has a much smaller genomic size, and genes of a common ancestry, as well as a relatively conserved genome structure,18 provides additional characters to base phylogenetic inference, such as mitochondrial gene order18,23,45 and mitochondrial sequence evolution.44 Indeed, the smaller size of mitogenomes allows mtDNA to serve as a cost-effective marker for conducting preliminary investigations on phylogenies compared to sequencing whole genomes.29,46 Consequently, investigating mitochondrial evolution remains important even with recent developments in nuclear phylogenomics.
In this study, we sequenced and assembled eight medusozoan mitochondrial genomes and reconstructed a mitogenome phylogeny with 84 other existing partial and complete cnidarian mitogenomes to study gene rearrangements in medusozoan mitogenomes. Our study expands upon the pool of medusozoan mitochondrial genomes available and enhances our understanding of medusozoan phylogenetic relationships.
Results
Newly assembled mitogenomes
A total of five new complete and three new partial mitogenomes were assembled (GenBank ID: OR400200 – OR400214) comprising five Scyphozoa (one partial, four complete), one complete Cubozoa, and two partial hydrozoan mitogenomes. The assembled scyphozoan and hydrozoan mitogenomes were assembled as single linear molecules, whereas the cubozoan mtDNA recovered eight linear mitochondrial chromosomes. All mitogenomes had 19 mitochondrial genes, with the exception of the two hydrozoan mitogenomes (missing orf314 and dnaB) and Morbakka sp. (missing trnW). A list of the mitochondrial genes present in each mitogenome can be found in Table S1 while DNA mapping figures of assembled mitogenomes can be found in Figure S2.
Phylogenetic analyses
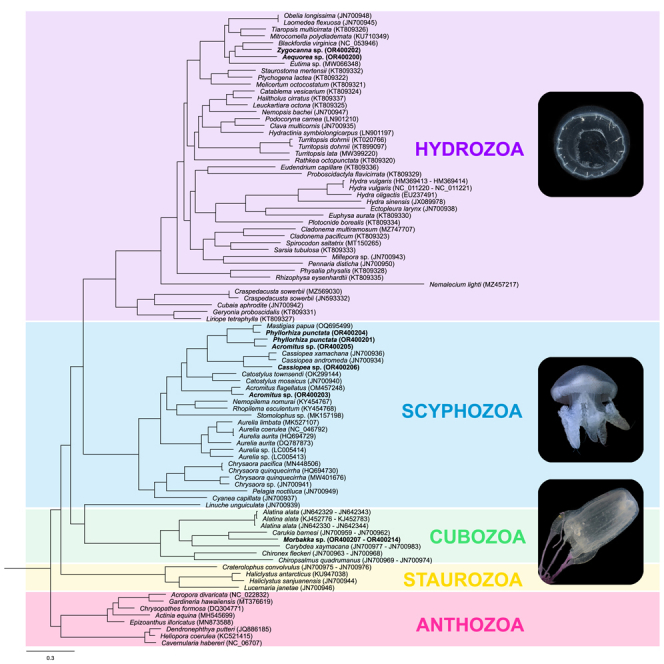
A monophyletic Medusozoa was recovered in both the maximum likelihood (ML) and Bayesian (BI) phylogenetic trees. As the topology of both analyses was largely conserved (save for Hydrozoa), only the ML tree is presented in Figure 2 (the complete ML and BI trees are provided in Figures S3 and S4, respectively). Of the four medusozoan classes, all except Scyphozoa were monophyletic with maximum bootstrap support and posterior probability (Figure 2). For Scyphozoa, the subclass Discomedusae formed a clade (bootstrap support [BS] = 100, posterior probability [pp] = 1) while subclass Coronamedusae (represented by Linuche unguiculata, GenBank: JN700939) was inferred to be sister to Cubozoa (BS = 51, pp = 0.98). The phylogenetic relationships between medusozoan classes were conserved in both analyses: Hydrozoa and Discomedusae were recovered as sister clades (BS = 97, pp = 1), the [Cubozoa + Coronamedusae] clade was designated as the sister group to the [Hydrozoa + Discomedusae] clade (BS = 50, pp = 0.99), and Staurozoa was placed as the outgroup to the rest of the medusozoans, with maximum bootstrap support and posterior probability.
Figure 2.

Phylogenetic relationships among Cnidaria mitogenomes based on maximum likelihood (ML) and Bayesian (BI) analyses
(A and B) (A) Ulmaridae; (B) [Mastigiidae + Acromitus] clade. Support values correspond to the bootstrap values from the ML phylogeny (BS ≥ 50%) and posterior probabilities from the BI analysis (pp ≥ 0.8). For nodes with differences in topology, only the bootstrap values (BS ≥ 50%) were provided. Posterior probabilities for these nodes were denoted with a dash (−). GenBank ascension numbers of mitogenomes are provided in parentheses. Bolded specimens indicate newly assembled mitogenomes from the current study. The degree (°) symbol denotes available mitogenome recorded as circular on GenBank.
Intra-class relationships were identical for all medusozoan classes less Hydrozoa between ML and BI analyses with strong bootstrap support and high posterior probability. For Staurozoa, a monophyletic suborder Myostaurida was inferred from both analyses (BS = 77, pp = 0.97). The sole Amyostaurida analyzed in the current study (Craterolophus convolvulus, GenBank: JN700975 – JN700976) was designated as the sister taxa to the Myostaurida. Within Cubozoa, Carybdeida (BS = 99, pp = 0.98) and Chirodropida (BS = 100, pp = 1) were recovered as monophyletic orders. For Discomedusae scyphozoans, the order Rhizostomeae was reconstructed as monophyletic (BS = 63, pp = 99) while Semaeostomeae was inferred to be paraphyletic, with the family Ulmaridae forming a sister clade to the Rhizostomeae (Figure 2) and the remainder of Semaeostomeae being recovered as the sister group to the [Rhizostomeae + Ulmaridae] clade (BS = 100, pp = 1).
For Hydrozoa (Figure 2; Figure S4), both ML and BI analyses retrieved the subclass Trachylinae as monophyletic with maximum bootstrap support and posterior probability. Additionally, most clades within Hydroidolina remained consistent across both analyses as well. Both ML and BI analyses recovered order Siphonophorae as monophyletic (BS = 100, pp = 1) and both Leptothecata and Anthothecata as paraphyletic. In Leptothecata, all taxa less Nemalecium lighti (GenBank: MZ457217) formed a single clade with high bootstrap support and posterior probability (BS = 100, pp = 0.99). Meanwhile, within Anthoathecata, both ML and BI analyses inferred monophyletic suborders Aplanulata (BS = 98, pp = 0.99) and Capitata (BS = 99, pp = 1), alongside a paraphyletic Filifera.
Intra-suborder phylogenetic relationships within Hydroidolina were more uncertain (Figure 2; Figure S4). In the ML phylogeny reconstruction, Filifera IV members were recovered as paraphyletic, forming a clade with Filifera III, and together, they were sister to Leptothecata (less Nemalecium lighti) with moderate bootstrap support (BS = 63). The remainder of Filifera (Filifera I + II) was inferred to be sister to Aplanulata, and the [Aplanulata + Filifera I and II] clade was sister to Capitata, although both phylogenetic relationships have low bootstrap support (BS < 50). The siphonophores were designated as the sister group to the [Leptothecata + Anthothecata] group (less Nemalecium lighti; BS = 51), while Nemalecium lighti was inferred to be sister to the rest of Hydroidolina, with maximum bootstrap support. In comparison, BI analysis recovered Filifera III and Filifera IV as clades (pp = 1 and 0.93, respectively). Of the remaining Anthoathecata, Aplanulata was inferred to be sister to the Leptothecata Nemalecium lighti (pp = 0.99). In turn, the [Aplanulata + Nemalecium lighti] clade was inferred to be sister to Capitata (pp = 0.84). Unlike the ML analysis, Siphonophorae was deeply nested within Hydroidolina (pp = 1) while Leptothecata (less Nemalecium lighti) was inferred to be the first diverging group among Hydroidolina (pp = 1).
The taxonomic positions of the newly assembled mitogenomes largely corresponded to their expected taxonomic groupings (Figure 2). However, within Rhizostomeae, both ML and BI analysis reconstructed Phyllorhiza punctata (GenBank: OR400201) and Acromitus sp. (GenBank: OR400205) as sister taxa (BS = 100, pp = 1), with the other Phyllorhiza punctata (GenBank: OR400204) being designated as the sister taxa to Mastigias papua (GenBank: OQ695499). The remaining Acromitus sp. (GenBank: OR400203) was inferred to be sister to Acromitus flagellates (GenBank: OM457248) (BS = 100).
Medusozoan mitochondrial gene rearrangement
A total of 92 complete and partial mitogenomes (eight from the current study and 84 existing genomes from GenBank) representing 82 unique taxa were analyzed for medusozoan mitogenome PCG rearrangements. Overall, the mitochondrial gene orders in the current study largely corroborate those previously analyzed by Kayal et al.18
Inter-class gene order for medusozoan PCGs remained highly conserved between most of the medusozoans, less Hydrozoa, while minor rearrangements were found within classes. As most of the gene order rearrangements were consistent between the ML and BI analyses (less that of Nemalecium lighti), only the gene order rearrangements relative to the ML phylogeny are presented in Figure 3 (gene order rearrangements for the BI analysis are provided in Figure S5). Gene order for all but two Scyphozoa was conserved and identical to that of the four Staurozoa examined in the current study. Of the remaining two scyphozoans, Linuche unguilata (GenBank: JN700939), the sole coronate, lacks trnW, rnl, polB, and orf314 (although the latter three genes may still be present in Coronomedusae given that the Linuche unguilata mitogenome is incomplete; see Kayal et al.18) while Stomolophus sp. (GenBank: MK157198) lacks both polB and orf314 and has an inverted rnl translocated downstream of cob.
Figure 3.

Comparison of gene order of Medusozoa mitochondrial genes with reference to the Maximum Likelihood (ML) phylogeny, using complete cnidarian mitogenomes
For mitogenomes with multiple chromosomes, vertical lines denote breaks between each chromosomal fragment. Complete mitochondrial genes are indicated in color, while pseudogenes (as indicated on GenBank) are colored but outlined with dotted lines. Missing genes are represented by uncolored boxes outlined with dotted lines.
(A–I) Mitochondrial rearrangements indicated using black arrows: (A) Ancestral medusozoan gene order (AMGO); (B) fragmentation of mitochondrial genome, loss of trnW; (C) loss of trnW, rnl, polB, and orf314, (D) inverse transposition of rnl and loss of polB and orf314; (E) loss of gene pair polB-orf314, and duplication, transposition, and inversion of cox1; (F) inversion of rnl and transposition of trnW; (G) transposition of trnM; (H) fragmentation and duplication of cox1; (I) inversion and transposition of rnl, as well as transposition of rns, cox1, atp6, and nad1. Asterisks (∗) denote the phylogenetic positions of specimens assembled in current study: Scyphozoa – Acromitus sp., Cassiopea sp., Phyllorhiza punctata; Hydrozoa – Zygocanna sp., Aequorea sp.; Cubozoa – Morbakka sp.
Likewise, gene order was conserved in six out of eight of the chromosomes across all Cubozoa mitogenomes, including the Morbakka sp. reconstructed in the current study. Similar to Kayal et al.,18 we recovered single-gene chromosomes containing cob, cox1, and rnl, chromosomes with gene pairs nad2-nad5 and orf314-polB, and a chromosome with the gene cluster cox2-atp8-atp6-cox3. However, the remaining two chromosomes recovered in the Morbakka sp. mitogenome comprised a single-gene chromosome containing rns and a chromosome with gene cluster nad6-nad3-nad4L-nad1-nad4, rather than a single-gene chromosome containing nad4 and a chromosome with gene cluster rns-nad6-nad3-nad4L-nad1-nad4 found in all other Cubozoa mitogenome. Additionally, while trnM was recovered downstream of cox3 in the Morbakka sp. chromosome containing gene cluster cox2-atp8-atp6-cox3, we failed to identify additional copies of trnM downstream of cox1 and cob as previously identified in all three Alatina alata mitogenomes. With the inclusion of the Morbakka sp. mitogenome, the mitochondrial gene order of cubozoans across all eight chromosomes remained highly similar to that of Staurozoa and Scyphozoa save the absence of trnW, supporting previous findings from Kayal et al.18
Mitochondrial gene orders among Hydrozoa exhibited greater variation (Figure 3). While the gene orders for Trachylinae were identical to Staurozoa, the gene arrangement in Hydroidolina differed from the rest of the medusozoans: within Siphonophorae, Anthoathecata, and Leptothecata, there was a transposition, inversion, and duplication of cox1, along with a loss of the gene pair polB-orf314 with respect to the staurozoan gene order. This gene order was conserved across Hydroidolina, except in Aplanulata and Nemalecium lighti (GenBank: MZ457217). Within Aplanulata, a transposition of trnW upstream of cox2 and inversion of rnl relative to the Hydroidolina gene order were observed, while a second transposition of trnM upstream of rnl was also found specifically within Hydridae. Additionally, as noted by Kayal et al.,18 the mitogenome of Hydra vulgaris (GenBank: HM369413 – HM369414) was derived from the fragmentation of the Hydridae gene order into two similarly sized linear chromosomes. In comparison, Leptothecata Nemalecium lighti has a highly unique gene order, undergoing an inversion of rnl as well as transposition of trnW, rnl, rns, and cox1. The inversion of rnl was inferred to have occurred independently of that in Aplanulata in the ML analysis (Figure 3) but deemed to be synapomorphic in the BI analysis (Figure S5). While Kayal et al.18 noted the presence of two identical copies of cox1, one located at each end of the mitochondrial genome in non-trachyline hydrozoan species sequenced in their study, we were unable to find the cox1 psuedogene in both Hydroidolina (Aequorea sp. and Zygocanna sp.) mitogenomes assembled here.
Ancestral state of Medusozoa gene order
Of the 92 mitogenomes used in the phylogenetic analyses, only 82 mitogenomes had all 13 PCGs and both rRNA rnl and rns. These mitogenomes were examined in the ancestral state reconstructions of the medusozoan mitochondria gene order. Differences in the ML and BI topology did not involve taxa that had differing relative gene orders except Nemalecium lighti; thus, only the ancestral state reconstructions carried out using the RAxML phylogeny are presented here (ancestral state reconstructions using the BI phylogeny can be found in Figure S6). Results showed that the ancestral medusozoan gene order (AMGO) following linearization corresponded to that of the staurozoan gene order (Figure 4), which remained conserved among all stuarozoans analyzed in the current study. Minor genome rearrangements occurred within most of the medusozoan classes. For Scyphozoa, an inverse transposition of rnl in Stomolophus sp. (GenBank: MK157198) was inferred to have occurred relative to the AMGO. The AMGO was conserved in all other scyphozoans.
Figure 4.

Results from the TreeRex analysis of the RAxML medusozoa phylogeny
Where applicable, inferred mitochondrial genome rearrangements are listed at the corresponding nodes. Gene order rearrangements are as follows: I – inversion; T - transposition; iT – inverse transposition; TDRL – tandem duplication random loss. Genes undergoing rearrangements are indicated in parentheses. Labels at nodes denote the inferred gene order for the common ancestor as listed in the ML TreeRex analysis output (Data S1).
In Hydrozoa, the AMGO was conserved among all Trachylinae while an inverse transposition of cox1 downstream of cob in the AMGO gave rise to the Hydroidolina AMGO. A further inversion of rnl in the Hydroidolina gene order gave rise to the Aplanulata gene order. This inversion of rnl was determined to have occurred independently in Aplanulata and Nemalecium lighti in the ML phylogeny but inferred to be a shared occurrence in the BI analysis (Figure S6). Besides the inversion of rnl, further transpositions of cox1, atp6, and nad1 and a tandem duplication random loss (TDRL) event relative to the Hydroidolina gene order were inferred in the gene order for Nemalecium lighti. Gene order rearrangements for Cubozoa could not be determined due to the fragmented nature of cubozoan mitogenomes. Additionally, genome duplication events, such as those common in Hydrozoa, and loss of genes (e.g., trnW in Cubozoa) were not represented in the ancestral state reconstruction. TreeRex outputs for the RAxML and BI phylogenies can be found in Datas S1 and S2, respectively.
Discussion
The linear structure of medusozoan mitogenomes renders them difficult to sequence and assemble.18 As a result, the pool of available medusozoan mitogenomes is smaller compared to anthozoans, which have circular mitogenomes.23 This scarcity of complete medusozoan mitogenomes contributes to the challenge of ensuring sufficient taxon representation when studying medusozoan phylogeny.18,23 Furthermore, most medusozoan mitochondrial phylogenetic studies focus on studying mitochondrial sequences, with only the study by Kayal et al.18 attempting to integrate both mitochondrial gene order and existing medusozoan phylogeny into a comprehensive hypothesis on medusozoan mitochondrial evolution. Our study aimed to improve our understanding of medusozoan mitogenome evolution by presenting five new complete and three new partial medusozoan mitogenomes and revisiting medusozoan mitochondrial gene order rearrangements in the context of their phylogeny.
Reconstructed medusozoan topology in context of existing phylogenetic hypotheses
The current study reconstructed a monophyletic Medusozoa topology similar to Zou et al.22 (Figure 1B). Within Scyphozoa, a monophyletic Rhizostomeae and a paraphyletic Semaeostomeae were recovered following the addition of five new Rhizostomeae mitogenomes (two Phyllorhiza punctata, two Acromitus sp., and one Cassiopea sp.). Ulmaridae remained more closely related to order Rhizostomeae than the rest of Semaeostomeae (Figure 2), with maximum bootstrap support and posterior probability. In Hydrozoa, orders Limnomedusae and Siphonophorae were reconstructed as monophyletic clades, while Lepthothecata and Anthoathecata were reconstructed as paraphyletic clades (Figure 2) following addition of two newly reconstructed Leptothecata mitogenomes (Zygocanna sp. and Aequorea sp.). Filifera was also consistently paraphyletic across ML and BI analyses, forming two clades [Filifera I and II] and [Filifera III and IV]. However, our analyses failed to resolve phylogenetic relationships within Hydroidolina. While ML analysis found support for Siphonophorae as the first diverging clade within Hydroidolina (Figure 2) and recovered the clades [Leptothecata-Filifera III-IV] and [Aplanulata-Filifera I-II-Capitata], as per Kayal et al.,32 bootstrap supports for both clades were low (BS = 63 and 45, respectively). In comparison, Leptothecata was inferred to be the first diverging Hydroidolina clade in the BI analysis, with maximum posterior probability. Additionally, while the [Aplanulata-Filifera I-II-Capitata] clade was still recovered in the BI analysis, the clade now includes Nemalecium lighti and is sister to Siphonophorae (Figure S4).
As Hydroidolina showed higher rates of gene order rearrangements relative to the rest of Medusozoa (Figure 3), discrepancies in the hydroidolinan topologies, particularly the phylogenetic position of Nemalecium lighti, may have arisen due to long-branch attraction (LBA).33 Since LBA hinders accurate phylogenetic tree reconstructions by grouping rapidly evolving taxa together based on homoplastic traits rather than evolutionary history,47 we employed the following methods to minimize the impacts of LBA. Firstly, we used a large number of Hydroidolina mitogenomes (40 mitogenomes) to break up potentially long branches.47,48 Next, we cleaned all mitochondrial gene alignments using Gblocks49 (v0.91b) to remove poorly mapped regions that may affect accurate phylogenetic reconstructions.49,50 Lastly, we also used PhyloBayes (CAT-Poisson model) for our BI analyses, since it may be better able to account for LBA artifacts.51
Discrepancies between morphological identities and molecular phylogeny
From the reconstructed medusozoan phylogeny, one Acromitus sp. (GenBank: OR400205) was recovered alongside Mastigiidae (Figure 2) instead of being grouped with the remaining Acromitus spp. (GenBank: OR400203 and OM457248). This discrepancy may have arisen due to the large morphological diversity and phenotypic plasticity exhibited within and between Medusozoa species.52 Furthermore, many Meduosozoa species exhibit ontogenetic morphological changes that contribute to the absence or presence of key morphological characters during development, including several traits used for morphological identification (e.g., number of tentacles and statocysts).53 As two of the specimens analyzed in the current study were juveniles (Acromitus sp. [GenBank: OR400205] and Phyllorhiza punctata [GenBank: OR400204]), ontogenetic morphological variation may contribute to difficulties in identifying specimens via morphological characters. We caution that sample OR400205 could indeed be a Phyllorhiza species, or comprise a hitherto unknown taxon, and that taxonomic revisions to Rhizostomeae are sorely needed. Indeed, previous molecular evidence has resolved the systematics of the Rhizostomeae from earlier, more subjective morphology-based taxonomic approaches.54
Revisiting mitochondrial genome evolution in Medusozoa
The addition of eight newly reconstructed mitogenomes spanning three classes (Cubozoa, Scyphozoa, and Hydrozoa) to the list of available medusozoan mitogenomes on GenBank allowed us to revisit medusozoan phylogenetic relationships and mitogenome evolution previously examined by Kayal et al.18 Analyses of medusozoan gene rearrangements (Figure 3) and ancestral state reconstructions (Figure 4) using 13 PCGs and two rRNA genes both suggested that gene order within Medusozoa was highly conserved,18,23 with minimal inter-class gene order rearrangements within Staurozoa, Cubozoa, and Scyphozoa. Furthermore, reconstructed medusozoan topology (Figure 2), Medusozoa gene order rearrangements (Figure 3), and ancestral state reconstructions (Figure 4) all supported a staurozoan mitochondrial gene order ancestral to the rest of Medusozoa. This finding corroborates the staurozoan AMGO put forth by Kayal et al.18 and contrasts against the hydrozoan AMGO inferred by Feng et al.23
At the class level, gene orders in Staurozoa, Scyphozoa, and Cubozoa were largely conserved. Gene orders of the four Staurozoa mitogenomes analyzed in the current study were mostly identical. While polB, orf314, trnM, and nad2 were missing from some of these mitogenomes, the absence of these genes was likely due to incomplete assemblies instead of evolutionary differences within staurozoan mitogenomic sequences. Within Scyphozoa, the gene orders of the five new scyphozoan mitogenomes assembled in the current study followed the staurozoan AMGO and were identical to all other existing scyphozoan mitogenomes with the exception of Stomolophus sp. Similarly, despite cubozoans having multiple chromosomes due to fragmentation (Figure 3), gene order among Cubozoa mitogenomes, including the newly reconstructed Morbakka sp., was largely identical to that of the AMGO save for missing trnW. All complete Cubozoa mitogenomes thus far have also recovered eight mitochondrial chromosomes.
Nevertheless, some variations within Cubozoa were observed with regards to the gene arrangements among cubozoan mitochondrial chromosomes. While gene orders were conserved for six out of eight cubozoan mitochondrial chromosomes, the grouping of mitochondrial genes in the remaining two chromosomes differed between the newly assembled Morbakka sp. (rns and nad6-nad3-nad4L-nad1-nad4) and the rest of Cubozoa (rns-nad6-nad3-nad4L-nad1-nad4 and nad4) (Figure 3). Additionally, while the Alatina alata mitogenomes all contained three identical copies of trnM, only a single copy of trnM, located downstream of cox3, was reconstructed in the Morbakka sp. Partial mitogenomes of remaining cubozoans analyzed in the current study were all missing both tRNA genes. While these differences could have arisen due to errors in assembly, it is unlikely given the lack of spuriously mapped regions in the DNA mapping figure of the Morbakka sp. mitogenome (Figure S2). Alternatively, these discrepancies could imply the presence of further gene rearrangements among cubozoan mitochondrial chromosomes, although further work on Cubozoa mitogenomes is needed to verify this hypothesis.
In comparison, Hydrozoa displayed the greatest variation in gene order among all the medusozoan classes, with at least six different gene orders among the hydrozoans investigated in the current study (Figure 3). Trachylinae gene orders were identical to the staurozoan AMGO,18,23 while most Hydroidolina (save Aplanulata and Nemalecium lighti) had a conserved gene order featuring the transposition, replication, and inversion of cox1 and the loss of the polB-orf314 gene pair relative to the AMGO (Figure 3). Most of the gene rearrangements in Hydrozoa were found within Aplanulata, with non-Hydridae Aplanulata, Hydridae, and Hydra vulgaris all having different gene orders from the rest of the subclass.18 The Hydra vulgaris mitogenome is also fragmented into two chromosomes, a feature that is suggested to have arisen independently from mtDNA fragmentation in Cubozoa.18 Among Leptothecata, Nemalecium lighti is the only taxon positioned separately from the rest of Leptothecata. Its unique gene order among Hydrozoa and large number of gene order rearrangements (Figure 3) relative to the rest of Leptothecata appear to validate the concerns raised by Macher et al.33 regarding its phylogenetic position within Leptothecata. While the gene order of the remaining Leptothecata comprises two cox1,18 the absence of a second copy of cox1 in both Leptothecata mitogenomes reconstructed in the current study (Zygocanna sp. and Aequorea sp.) is likely an artifact of incomplete assembly rather than evolutionary differences, given that copies of cox1 pseudogenes were recovered from other hydrozoans (see Kayal et al.32).
Ancestral state analyses in our study largely support the mitogenome rearrangements described earlier, although our ML and BI phylogenies differed in whether the inversion of rnl was synapomorhic to both Aplanulata and Nemalecium lighti. However, as TreeRex is unable to handle mitogenomic datasets without a fixed gene set,55 these analyses are not able to infer duplication, deletion, and fragmentation events within Medusozoa mitogenomes. Since the deletion of gene pair polB-orf314 and cox1 duplication are common in Hydroidolina (Figure 4), while mtDNA fragmentation is ubiquitous in Cubozoa (and several Hydra species),18 understanding the evolution of mitogenomic arrangements among the hydrozoans and cubozoans remains a challenge. Finally, the high variability in the rates of gene rearrangements within the various medusozoan groups, particularly between non-hydroidolinan medusozoans and hydroidolinans (Figures 3 and 4), also highlights the limitations of using only gene order for phylogenetic reconstructions.
While mitochondrial sequences and gene orders have been used to infer phylogenetic relationships,18,23 the evolutionary implications of mitochondrial gene order rearrangements remain understudied.56 Nevertheless, recent studies on metazoan mitochondrial gene order by Shtolz and Mishmar57 reported that differences in the mitochondrial gene orders of taxonomic groups that shared mtDNA gene content (e.g., arthropods and chordates) resulted in changes in mtDNA transcription rates between different organisms. Such changes arise as mtDNA rearrangements reshuffle regulatory elements (e.g., transcription binding sites) within the mitogenome that regulate mtDNA transcription,58,59 contributing to changes in interactions between transcription factors and mtDNA regulatory sites that affect mitochondrial gene expression.57 Furthermore, since the mitochondrion regulates respiration, differences in mtDNA transcription rates arising from mtDNA rearrangements57 may in turn introduce physiological or ecological trade-offs among different taxa (see Ballard and Pichaud56), highlighting the potential implications of mitochondrial gene order rearrangements on evolutionary fitness. However, research in this area, generally for Metozoa56,57 and specific to Medusozoa, remains lacking. Further work is required to elucidate the impacts of mitochondrial gene order on Medusozoa ecology.
Limitations of mitochondrial studies in deciphering deep Medusozoa phylogeny
As mtDNA has higher substitution saturation compared to nDNA, similarities in Medusozoa mitogenomic sequences may not accurately reflect their phylogenetic relationships, leading to a significant loss of phylogenetic information when studying deep phylogenetic relationships.12 Considering that inter-class relationships within Cnidaria have largely been resolved (see Kayal et al.3), and that current studies seek to decipher the topologies of higher taxonomic clades within medusozoan classes (e.g., Hydroidolina) (see Bentlage and Collins60), nuclear genes, which have lower rates of substitution than mtDNA, will be required especially when dealing with relationships at taxonomic levels higher than subclass.44
Nevertheless, mitogenomics can play a supplementary role alongside nuclear phylogenomics in unraveling medusozoan phylogeny. While low rates of mtDNA in sister subphyla Anthozoa may hamper the use of mitogenomic sequences for inferring anthozoan phylogeny at lower taxonomic levels,38,61 evolution rates of Medusozoa mitochondrial sequences remain comparable to those of other Metazoa.40 Consequently, mitogenomic sequences can still provide greater phylogenetic resolution at the species and genus levels,41 complementing the use of nuclear phylogenetics for higher-level Medusozoa relationships.
Additionally, the scarcity of complete medusozoan mitogenomes, owing to difficulties in assembling linear mitogenomes, has curtailed taxon representation for reconstructing the medusozoan phylogeny.18,23 As Zou et al.22 highlighted, there is currently a lack of complete mitogenomes of Coronamedusae available on GenBank; as of the study, most available coronate mitochondrial sequences on GenBank are cox1 sequences, with only a handful of rnl and cox3 sequences and one partial mitochondrial genome (Linuche unguiculata) published by Kayal et al.18 Beyond the coronates, several other clades of interest within Hydroidolina (Filifera I, Filifera II, Siphonophorae, and leptothecatan family Haleciidae), as well as Staurozoa and Cubozoa in general, are also underrepresented. The reconstruction and incorporation of additional mitogenomes from these groups in future studies will be required to better understand phylogenetic relationships within the various Medusozoa classes.
Critically, both the structure and chromosome numbers of Myxozoa mitogenomes remain poorly characterized. Work by Yahalomi et al.26 on Enteromyxum leei indicated a Myxozoa mitogenome with eight circular chromosomes of roughly 23 kb each, while other findings by Takeuchi et al.25 returned circular fragmented mitogenomes of between 15 kb and 19 kb. As both studies also highlighted the variable number of PCGs within Myxozoa, they were excluded from the current phylogenetic analysis. Once the characteristics of Myxozoa mitogenomes are better characterized, future studies can incorporate additional Myxozoa mitogenomes into Medusozoa phylogenetic analyses to investigate if the current hypotheses for the phylogenetic position of Myxozoa in Cnidaria (Figures S1B and S1C) are supported by both mitogenomic sequences and mitochondrial gene orders.
Conclusion
Despite developments in nuclear phylogeneomics, the unique characteristics of mitochondrial genomes, including the availability of mitochondrial gene orders18,23,45 and mitochondrial sequence evolution44 as additional characters for phylogenetic inferences, provide a compelling case for the continued study of mitochondrial phylogenomics. As current medusozoan mitochondrial phylogenomic studies are limited by a shortage of taxonomic representation,18,23 the present study contributes to the existing database of medusozoan mitogenomes by reconstructing and annotating an additional five complete and three partial mitochondrial genomes spanning three Medusozoa classes (Cubozoa, Hydrozoa, and Scyphozoa).
Findings here have enabled us to revisit both medusozoan phylogeny and mitogenome evolution. Analysis of the newly reconstructed mitogenomes alongside 84 existing medusozoan mitogenomes corroborates previous medusozoan phylogenies hypothesized by Zou et al.22 Furthermore, mitogenome organizations of the newly reconstructed mitogenomes are largely consistent with gene orders of existing medusozoans, and patterns of mitochondrial gene rearrangements in the context of the phylogeny support a hypothesis of medusozoan mitogenome evolution largely consistent with that of Kayal et al.18 As our present findings are limited by the current lack of complete mitogenomes from some key taxa (e.g., Coronamedusae, Staurozoa, Cubozoa, Hydroidolina, and Myxozoa), future studies ought to examine mitogenomes from these groups to better improve our understanding of medusozoan mitogenome evolution.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Aequorea sp. | This study | ZRC.CNI.1440 |
| Phyllorhiza punctata | This study | ZRC.CNI.1437 |
| Zygocanna sp. | This study | ZRC.CNI.1433 |
| Acromitus sp. | This study | ZRC.CNI.1396 |
| Phyllorhiza punctata | This study | ZRC.CNI.1439 |
| Acromitus sp. | This study | ZRC.CNI.1436 |
| Cassiopea sp. | This study | ZRC.CNI.3022 – ZRC.CNI.3023 |
| Morbakka sp. | This study | ZRC.CNI.1418 |
| Critical commercial assays | ||
| E.Z.N.A.® Mollusc DNA Kit | Omega Bio-Tek | D3373 |
| Genomic DNA Clean & Concentrator – 25 | Zymo Research | D4065 |
| KAPA HyperPrep Kit | KAPA Biosystems | KK8502 |
| Deposited data | ||
| Raw reads of Medusozoa samples | This study | NCBI SRA BioProject PRJNA934731 |
| Aequorea sp. mitogenome | This study | GenBank: OR400200 |
| Phyllorhiza punctata mitogenome | This study | GenBank: OR400201 |
| Zygocanna sp. mitogenome | This study | GenBank: OR400202 |
| Acromitus sp. mitogenome | This study | GenBank: OR400203 |
| Phyllorhiza punctata mitogenome | This study | GenBank: OR400204 |
| Acromitus sp. mitogenome | This study | GenBank: OR400205 |
| Cassiopea sp. mitogenome | This study | GenBank: OR400206 |
| Morbakka sp. mitogenome | This study | GenBank: OR400207 – OR400214 |
| 84 publicly available Cnidaria mitogenome sequences | NCBI GenBank | Refer to Tables S2 and S3 |
| Software and algorithms | ||
| fastp v0.23.2 | Chen et al., 201870 | https://github.com/OpenGene/fastp |
| SPAdes v3.15.4 | Bankevich et al., 201271 | https://cab.spbu.ru/software/spades/ |
| BLAST v2.90 | Camacho et al., 200972 | https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ |
| NOVOplasty v4.3.1 | Dierckxsens et al., 201773 | https://github.com/ndierckx/NOVOPlasty |
| minimap2 v2.17-r941 | Li, 201874 | https://github.com/lh3/minimap2 |
| Samtools v1.13 | Danecek et al., 202175 | https://www.htslib.org/ |
| Geneious Prime v2022.0.1 | Kearse et al., 201276 | https://www.geneious.com/ |
| Inverted Repeats Finder v3.08 | Warburton et al., 200477 | https://tandem.bu.edu/irf/home |
| IUPACpal | Alamro et al., 202178 | https://sourceforge.net/projects/iupacpal/ |
| MITObim v1.9 | Hahn et al., 201379 | https://github.com/chrishah/MITObim |
| MITOS2 | Bernt et al., 201380 | http://mitos2.bioinf.uni-leipzig.de/index.py |
| Infernal cmscan v1.1.2 | Nawrocki & Eddy, 201381 | http://eddylab.org/infernal/ |
| tRNAscan-SE v2.0 | Chan et al., 202182 | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| MAFFT v7.453 | Katoh & Standley, 201384 | https://mafft.cbrc.jp/alignment/server/index.html |
| TranslatorX | Abascal et al., 201086 | http://translatorx.co.uk/ |
| Gblocks v0.91b | Talavera & Castresana, 200749 | http://phylogeny.lirmm.fr/phylo_cgi/one_task.cgi?task_type=gblocks |
| ModelTest-NG v0.2.0 | Darriba et al., 201987 | https://github.com/ddarriba/modeltest |
| RAxML-NG v0.8.1 | Kozlov et al., 201988 | https://github.com/amkozlov/raxml-ng |
| PhyloBayes v3.3e | Lartillot et al., 200951 | https://github.com/bayesiancook/phylobayes |
| Tracer v1.7.1 | Rambaut et al., 201889 | http://beast.community/tracer |
| TreeRex v1.85 | Bernt et al., 200890 | https://siks.informatik.uni-leipzig.de/185-0-TreeREx.html |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Min Kang Ling (minkangling@u.nus.edu).
Materials availability
This study did not generate new unique reagents. Specimens used in this study were deposited in the Lee Kong Chian Natural History Museum. Catalog numbers of the specimens are provided in the Supplementary Materials (Table S2).
Method details
Specimen collection and identification
Eight specimens comprising Cubozoa, Hydrozoa and Scyphozoa were collected at seven sites in the coastal waters of Singapore. Tissue samples subsampled from the specimens were cryo-preserved in 100% ethanol at -175°C in the Lee Kong Chian Natural History Museum prior to DNA extraction and sequencing, while whole specimens were stored in formalin. A list of the sampling localities and storage details for specimens used in the current study can be found in Table S2.
Taxonomic identities of the specimens were determined based on morphology. Formalin-preserved specimens were examined while suspended in freshwater for the duration of observation. Medusozoans were identified following the taxonomic descriptions by Mayer,62 Kramp,63 Cornelius,64 Bouillon et al.,65 Calder,66 as well as Bentlage and Lewis.67
DNA sequencing and mitogenome assembly
DNA extraction and library preparation were conducted following Quek et al.68 Briefly, genomic DNA (gDNA) was extracted using E.Z.N.A. Mollusc DNA Kit (Omega Bio-tek) with a modified elution step. The first elute of 50 μL was discarded to remove smaller fragments. The remaining high quality gDNA was retained and cleaned using Zymo Genomic DNA Clean and Concentrator. Purified gDNA was sonicated to a mode fragment length of 200 bp using Bioruptor Pico (Diagenode), and dual-indexed libraries were prepared using KAPA HyperPrep Kit (KK8502; KAPA Biosystems) according to manufacturer’s recommendations. A final double-size selection was conducted to narrow fragment size distribution prior to library amplification (six cycles). All cnidarian libraries were then pooled with six other samples of sponges69 in equimolar amounts and sequenced on a single HiSeq 4000 lane (150 × 150 bp).
Raw reads for Scyphozoa and Hydrozoa specimens were trimmed using fastp70 (v0.23.2) under default settings to remove adapters and low-quality reads. Trimmed reads were then de novo assembled using different k-mer values (21, 33, 55, 77, 99 and 127) by SPAdes71 (v3.15.4). To identify mitochondrial sequences from our assemblies, assembled contigs were compared to reference mitochondrial sequences from GenBank (Table S3) using BLASTn72 (v2.90) (e-value = 10−6). Subsequently, each SPAdes-assembled contig was directly extended with untrimmed reads using NOVOplasty73 (v4.3.1) (k-mer = 127). To validate the mitogenome assemblies, we mapped the untrimmed paired reads for each sample against the assembled mitochondrial chromosomes using minimap274 (v2.17-r941) at default settings. Thereafter, Samtools75 (v1.13) was used to process mapped files (-b -F 4 -q 30) and filter out low quality reads (MAPQ < q30). Mapped assemblies were visualised in Geneious Prime76 (v2022.0.1) to identify potential misassemblies (Figure S2). Additionally, the Inverted Repeats Finder77 (v3.08) webserver (default settings) and IUPACpal78 (-m = 20, -M = 1000, -g = [contig length – 40]) were used to identify and annotate inverted tandem repeats (ITRs) present in reconstructed mitogenomes. Mappings were visually inspected and spuriously extended regions were manually removed. After trimming, all reconstructed mitochondrial chromosomes were inspected for circularisation using circules.py from MITObim79 (v1.9).
Initial annotation of the assembled mitochondrial chromosomes was carried out using the MITOS280 webserver (RefSeq89 Metazoa, translation code 4). Thereafter, Infernal cmscan81 (v1.1.2) was used to validate MITOS2 ribosomal RNA (rRNA) annotations and recover rRNA genes not annotated by MITOS2. Similarly, the tRNAscan-SE82 (v2.0) webserver was used to validate the MITOS2 transfer RNA (tRNA) annotations, and recover tRNA genes not annotated by MITOS2. As the annotation of mitochondrial genes may erroneously be extended or truncated,83 we aligned the above annotations against existing medusozoan mitochondrial gene sequences obtained from GenBank (Table S3) using MAFFT84 (v7.453) (--auto) and visually inspected the alignments on Geneious Prime76 (v2022.0.1) to verify the accuracy of the annotations. Where necessary, the mitochondrial genes were then manually reannotated.
Separately, as the Cubozoa mitochondrial genome (Morbakka sp.; GenBank: OR400207 – OR400214) consisted of multiple chromosomes, we took a different approach to identify sequences of mitochondrial origin. Raw reads were trimmed using fastp70 (v0.23.2) and de novo assembled by SPAdes71 (v3.15.4) as described previously. As ribosomal and mitochondrial genes are typically enriched85 and are expected to have high coverage, we removed assembled contigs with an average k-mer coverage <10 from the assembly. To identify the Morbakka sp. mitochondrial chromosomes, we translated the assembled contigs and carried out a BLASTx72 (v2.9.0) search of the assembled contigs against translated sequences of existing cubozoan mitochondrial PCGs from GenBank under default settings. Once the mitochondrial contigs were identified, the corresponding nucleotide sequences were extended with NOVOplasty73 (v4.3.1) and annotated for ITRs as described above for non-Cubozoa mitogenomes. As the initial BLASTx search only recovered six out of the eight expected Morbakka sp. chromosomes, we carried out BLASTn72 (v2.9.0) searches using the Morbakka sp. ITRs to identify the remaining two chromosomes from the assembled contigs. These two chromosomes were extended using NOVOplasty73 (v4.3.1) and annotated for ITRs as described above. Subsequently, the eight Morbakka sp. chromosomes were mapped and annotated as described previously for non-cubozoan mitogenomes.
Phylogenetic analyses
Phylogeny reconstruction was performed with Anthozoa as the outgroup. Nucleotide sequences of 84 previously published Cnidaria mitochondrial genomes (partial and complete) were first downloaded from GenBank (Table S4) and the following mitochondrial gene sequences extracted: ATP synthase F0 subunit 6 and 8 (atp6 and atp8), cytochrome b (cob), cytochrome c oxidase subunit I – III (cox1 – 3), NADH dehydrogenase subunit 1–6 and 4L (nad1 – 6 and nad4L), 16S ribosomal RNA (rnl), 12S ribosomal RNA (rns), methionine tRNA (trnM), tryptophan tRNA (trnW), DNA polymerase beta (polB) and open reading frame 314 (orf314). Coding sequences of the 13 PCGs, polB and orf314 were aligned in the TranslatorX86 webserver using genetic code translation table 4, with protein alignment and trimming performed using MAFFT84 (v7.453) and Gblocks49 (v0.91b) (default settings) respectively to obtain a nucleotide alignment. For rnl, rns, trnM and trnW, nucleotide sequence alignment was carried out using MAFFT84 (v7.453) (L-INS-i) and cleaned with the Gblocks49 (v0.91b) webserver (settings as per the TranslatorX webserver defaults). To determine the appropriate evolutionary models for analyses, we ran ModelTest-NG87 (v0.2.0) for each of the mitochondrial gene alignments (Table S5). The nucleotide alignments were then concatenated into a single matrix for phylogeny reconstructions, consisting of 92 nucleotide sequences spanning across 82 distinct taxa.
A maximum likelihood (ML) phylogeny was inferred using RAxML-NG88 (v0.8.1), partitioned by gene models as determined above, with 50 random and 50 maximum parsimony starting trees, and 1000 bootstrap pseudoreplicates to determine node support. Bayesian analysis (BI) was carried out in PhyloBayes51 (v3.3e) using default settings across two independent runs. Each run comprised one MCMC chain with 39800 cycles (10,731,594 generations) per chain, with sampling carried out once every 100 generations. Convergence between runs were determined using the bpcomp (maxdiff <0.1) and tracecomp commands (maximum discrepancy <0.1, minimum effective size >100), and visually inspected with Tracer89 (v1.7.1). A majority rule consensus tree was obtained using the bpcomp command after discarding one-fifth of the total length of each chain (7960 cycles) as burn-in.
Ancestral state reconstruction
Ancestral state reconstruction of the medusozoan mitogenomes was carried out using TreeRex90 (v1.85) as per Tyagi et al.55 As several Medusozoa clades have missing genes (e.g., polB and orf314 for most Hydrozoa, and trnW for Cubozoa), ancestral state reconstruction was conducted using the relative order of the 13 mitochondrial PCGs (atp6, atp8, cob, cox1-3, nad1-6 and nad4L) and the genes for both ribosomal subunits (rnl and rns). Mitogenomes missing any of the above genes were omitted from the TreeRex analyses (Table S4). Thereafter, the mitochondrial gene orders of each of the remaining 82 mitogenomes were compiled into a single dataset (Data S3). Where applicable, cox1 pseudogenes present in the mitogenomes of several taxa were identified using their GenBank records and removed from the dataset. For taxa with mitogenomes comprising multiple chromosomes (Cubozoa and Hydra vulgaris), chromosomes were arranged such that the gene order corresponded to that of the most closely related taxa based on the phylogenetic analysis. Subsequently, TreeREx analysis was conducted using the default settings (-s -w -W -o -m = 0). Replicate analyses were carried out using the trees inferred from the RAxML and BIphylogenies as reference (Datas S4 and S5). The pipeline for mitogenome assembly, phylogenetic analyses and ancestral reconstruction is provided in File S1.
Acknowledgments
Funding for this study was awarded under the IntraCreate Seed Grant 08, a partnership between the National University of Singapore with the Singapore-Hebrew University of Jerusalem Alliance for Research and Enterprise (SHARE), supported by the National Research Foundation (NRF), Prime Minister's Office, Singapore, under its Campus for Research Excellence and Technological Enterprise (CREATE) program. This research was also supported by the Lee Kong Chian Natural History Museum and Singapore Ministry of Education Academic Research Fund Tier 1 (A-0008517-00-00). Parts of this research were conducted on the St John’s Island National Marine Laboratory (SJINML), a National Research Infrastructure under the NRF, which provided the facilities necessary for conducting this study. Collection for the animals used in this study was permitted through the National Parks Board Singapore (NParks Permit: NP/RP22-025). We would like to thank the two anonymous reviewers for their invaluable comments and suggestions, which greatly improved the manuscript.
Author contributions
Conceptualization, M.K.L., D.H., and Z.B.R.Q.; Methodology, M.K.L., I.B.I., Z.T.Y., and Z.B.Q.R.; Software, M.K.L. and Z.B.Q.R.; Formal Analysis, M.K.L. and Z.B.Q.R.; Investigation, M.K.L., I.B.I., Z.T.Y., and Z.B.Q.R.; Resources, N.W.L.Y., I.B.I., Z.T.Y., D.H., and Z.B.Q.R.; Data Curation, M.K.L. and Z.B.Q.R.; Writing – Original Draft, M.K.L., I.B.I., Z.T.Y., and Z.B.R.Q.; Writing – Review & Editing, all authors have read, edited, and approved the manuscript; Visualization, M.K.L.; Supervision, D.H. and Z.B.Q.R.; Funding Acquisition, N.W.L.Y. and D.H.
Declaration of interests
The authors declare no competing interests.
Published: October 18, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108252.
Contributor Information
Min Kang Ling, Email: minkangling@u.nus.edu.
Zheng Bin Randolph Quek, Email: randolphquek@u.nus.edu.
Supplemental information
Data and code availability
Raw reads used for mitogenome assembly have been deposited at NCBI SRA under BioProject PRJNA934731. The mitogenomes assembled in this study have been deposited into GenBank under accession numbers OR400200 – OR400214. Additionally, this paper also analyses existing, publicly available mitogenomes provided on GenBank (refer to key resources table for accession numbers). Gene orders for TreeRex analyses have been provided under Data S3. ML and BItrees used for TreeRex analyses have been provided under Datas S4 and S5 respectively, while TreeRex outputs for ML and BIanalyses have been provided under Datas S1 and S2 respectively.
All original code used for this study has been provided as a pipeline under File S1.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Park E., Hwang D.-S., Lee J.-S., Song J.-I., Seo T.-K., Won Y.-J. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol. Phylogenet. Evol. 2012;62:329–345. doi: 10.1016/j.ympev.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Quattrini A.M., Rodríguez E., Faircloth B.C., Cowman P.F., Brugler M.R., Farfan G.A., Hellberg M.E., Kitahara M.V., Morrison C.L., Paz-García D.A., et al. Palaeoclimate ocean conditions shaped the evolution of corals and their skeletons through deep time. Nat. Ecol. Evol. 2020;4:1531–1538. doi: 10.1038/s41559-020-01291-1. [DOI] [PubMed] [Google Scholar]
- 3.Kayal E., Bentlage B., Sabrina Pankey M., Ohdera A.H., Medina M., Plachetzki D.C., Collins A.G., Ryan J.F. Phylogenomics provides a robust topology of the major cnidarian lineages and insights on the origins of key organismal traits. BMC Evol. Biol. 2018;18:68. [Google Scholar]
- 4.McFadden C.S., Ofwegen L.P., Quattrini A.M. Revisionary systematics of Octocorallia (Cnidaria: Anthozoa) guided by phylogenomics. BSSB. 2022;1:1–79. [Google Scholar]
- 5.Novosolov M., Yahalomi D., Chang E.S., Fiala I., Cartwright P., Huchon D. The phylogenetic position of the enigmatic, Polypodium hydriforme (Cnidaria, Polypodiozoa): Insights from mitochondrial genomes. Genome Biol. Evol. 2022;14 doi: 10.1093/gbe/evac112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao B., Guo Q., Zhai Y., Gu Z. Transcriptomic insights into the diversity and evolution of Myxozoa (Cnidaria, Endocnidozoa) toxin-like proteins. Mar. Drugs. 2022;20:291. doi: 10.3390/md20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson M.N. Some implications of molecular phylogenetics for understanding biodiversity in jellyfishes, with emphasis on Scyphozoa. Hydrobiologia. 2004;530–531:249–260. [Google Scholar]
- 8.Foox J., Siddall M.E. The road to Cnidaria: History of phylogeny of the Myxozoa. J. Parasitol. 2015;101:269–274. doi: 10.1645/14-671.1. [DOI] [PubMed] [Google Scholar]
- 9.Siddall M.E., Martin D.S., Bridge D., Desser S.S., Cone D.K. The demise of a phylum of protists: Phylogeny of Myxozoa and other parasitic Cnidaria. J. Parasitol. 1995;81:961–967. [PubMed] [Google Scholar]
- 10.Collins A.G. Phylogeny of Medusozoa and the evolution of cnidarian life cycles. J. Evol. Biol. 2002;15:418–432. [Google Scholar]
- 11.Chang E.S., Neuhof M., Rubinstein N.D., Diamant A., Philippe H., Huchon D., Cartwright P. Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Evolution. 2015;112:14912–14917. doi: 10.1073/pnas.1511468112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pratlong M., Rancurel C., Pontarotti P., Aurelle D. Monophyly of Anthozoa (Cnidaria): why do nuclear and mitochondrial phylogenies disagree? Zool. Scr. 2017;46:363–371. [Google Scholar]
- 13.Bridge D., Cunningham C.W., Schierwater B., DeSalle R., Buss L.W. Class-level relationships in the phylum Cnidaria: evidence from mitochondrial genome structure. Proc. Natl. Acad. Sci. USA. 1992;89:8750–8753. doi: 10.1073/pnas.89.18.8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beaton M.J., Roger A.J., Cavalier-Smith T. Sequence analysis of the mitochondrial genome of Sarcophyton glaucum: conserved gene order among octocorals. J. Mol. Evol. 1998;47:697–708. doi: 10.1007/pl00006429. [DOI] [PubMed] [Google Scholar]
- 15.van Oppen M.J.H., Catmull J., McDonald B.J., Hislop N.R., Hagerman P.J., Miller D.J. The mitochondrial genome of Acropora tenuis (Cnidaria; Scleractinia) contains a large group I intron and a candidate control region. J. Mol. Evol. 2002;55:1–13. doi: 10.1007/s00239-001-0075-0. [DOI] [PubMed] [Google Scholar]
- 16.Shao Z., Graf S., Chaga O.Y., Lavrov D.V. Mitochondrial genome of the moon jelly Aurelia aurita (Cnidaria, Scyphozoa): A linear DNA molecule encoding a putative DNA-dependent DNA polymerase. Gene. 2006;381:92–101. doi: 10.1016/j.gene.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 17.Kayal E., Lavrov D.V. The mitochondrial genome of Hydra oligactis (Cnidaria, Hydrozoa) sheds new light on animal mtDNA evolution and cnidarian phylogeny. Gene. 2008;410:177–186. doi: 10.1016/j.gene.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Kayal E., Bentlage B., Collins A.G., Kayal M., Pirro S., Lavrov D.V. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol. Evol. 2012;4:1–12. doi: 10.1093/gbe/evr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith D.R., Kayal E., Yanagihara A.A., Collins A.G., Pirro S., Keeling P.J. First complete mitochondrial genome sequence from a box jellyfish reveals a highly fragmented linear architecture and insights into telomere evolution. Genome Biol. Evol. 2012;4:52–58. doi: 10.1093/gbe/evr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stampar S.N., Broe M.B., Macrander J., Reitzel A.M., Brugler M.R., Daly M. Linear mitochondrial genome in Anthozoa (Cnidaria): A case study in Ceriantharia. Sci. Rep. 2019;9:6094. doi: 10.1038/s41598-019-42621-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith D.R. Revisiting Ceriantharian (Anthozoa) mitochondrial genomes: Casting doubts about their structure and size. Genome Biol. Evol. 2020;12:1440–1443. doi: 10.1093/gbe/evaa130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zou H., Zhang J., Li W., Wu S., Wang G. Mitochondrial genome of the freshwater jellyfish Craspedacusta Sowerbyi and phylogenetics of Medusozoa. PLoS One. 2012;7 doi: 10.1371/journal.pone.0051465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng H., Lv S., Li R., Shi J., Wang J., Cao P. Mitochondrial genome comparison reveals the evolution of Cnidarians. Ecol. Evol. 2023;13 doi: 10.1002/ece3.10157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins A.G. Recent insights into cnidarian phylogeny. Smithson. Contrib. Mar. Sci. 2009;38:139–149. [Google Scholar]
- 25.Takeuchi F., Sekizuka T., Ogasawara Y., Yokoyama H., Kamikawa R., Inagaki Y., Nozaki T., Sugita-Konishi Y., Ohnishi T., Kuroda M. The mitochondrial genomes of a Myxozoan genus Kudoa are extremely divergent in Metazoa. PLoS One. 2015;10 doi: 10.1371/journal.pone.0132030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yahalomi D., Haddas-Sasson M., Rubinstein N.D., Feldstein T., Diamant A., Huchon D. The multipartite mitochondrial genome of Enteromyxum leei (Myxozoa): Eight fast-evolving megacircles. Mol. Biol. Evol. 2017;34:1551–1556. doi: 10.1093/molbev/msx072. [DOI] [PubMed] [Google Scholar]
- 27.Yahalomi D., Atkinson S.D., Neuhof M., Chang E.S., Philippe H., Cartwright P., Bartholomew J.L., Huchon D. A cnidarian parasite of salmon (Myxozoa: Henneguya) lacks a mitochondrial genome. Proc. Natl. Acad. Sci. USA. 2020;117:5358–5363. doi: 10.1073/pnas.1909907117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quek R.Z.B., Jain S.S., Neo M.L., Rouse G.W., Huang D. Transcriptome-based target-enrichment baits for stony corals (Cnidaria: Anthozoa: Scleractinia) Mol. Ecol. Resour. 2020;20:807–818. doi: 10.1111/1755-0998.13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quek Z.B.R., Huang D. Application of phylogenomic tools to unravel anthozoan evolution. Coral Reefs. 2022;41:475–495. [Google Scholar]
- 30.Santander M.D., Maronna M.M., Ryan J.F., Andrade S.C.S. The state of Medusozoa genomics: current evidence and future challenges. GigaScience. 2022;11 doi: 10.1093/gigascience/giac036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maggioni D., Puce S., Galli P., Seveso D., Montano S. Description of Turritopsoides marhei sp. nov. (Hydrozoa, Anthoathecata) from the Maldives and its phylogenetic position. Mar. Biol. Res. 2017;13:983–992. [Google Scholar]
- 32.Kayal E., Bentlage B., Cartwright P., Yanagihara A.A., Lindsay D.J., Hopcroft R.R., Collins A.G. Phylogenetic analysis of higher-level relationships within Hydroidolina (Cnidaria: Hydrozoa) using mitochondrial genome data and insight into their mitochondrial transcription. PeerJ. 2015;3 doi: 10.7717/peerj.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macher J.-N., Kayal E., Duijm E., van der Hoorn B., Montano S., Speksnijder A. The mitochondrial genome of Nemalecium lighti (Hydrozoa, Leptothecata) Mitochondrial DNA. B Resour. 2021;6:3196–3198. doi: 10.1080/23802359.2021.1989335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayal E., Roure B., Philippe H., Collins A.G., Lavrov D.V. Cnidarian phylogenetic relationships as revealed by mitogenomics. BMC Evol. Biol. 2013;13:5. doi: 10.1186/1471-2148-13-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown W.M., George M., Jr., Wilson A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA. 1979;76:1967–1971. doi: 10.1073/pnas.76.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boore J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shearer T.L., van Oppen M.J.H., Romano S.L., Wörheide G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria) Mol. Ecol. 2002;11:2475–2487. doi: 10.1046/j.1365-294x.2002.01652.x. [DOI] [PubMed] [Google Scholar]
- 38.Bilewitch J.P., Degnan S.M. A unique horizontal gene transfer event has provided the octocoral mitochondrial genome with an active mismatch repair gene that has potential for an unusual self-contained function. BMC Evol. Biol. 2011;11:228. doi: 10.1186/1471-2148-11-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stampar S.N., Maronna M.M., Kitahara M.V., Reimer J.D., Morandini A.C. Fast-evolving mitochondrial DNA in Ceriantharia: a reflection of Hexacorallia paraphyly? PLoS One. 2014;9 doi: 10.1371/journal.pone.0086612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang D., Meier R., Todd P.A., Chou L.M. Slow mitochondrial COI sequence evolution at the base of the metazoan tree and its implications for DNA barcoding. J. Mol. Evol. 2008;66:167–174. doi: 10.1007/s00239-008-9069-5. [DOI] [PubMed] [Google Scholar]
- 41.Rubinoff D., Holland B.S. Between two extremes: Mitochondrial DNA is neither the panacea nor the nemesis of phylogenetic and taxonomic inference. Syst. Biol. 2005;54:952–961. doi: 10.1080/10635150500234674. [DOI] [PubMed] [Google Scholar]
- 42.Chan A.H.E., Chaisiri K., Saralamba S., Morand S., Thaenkham U. Assessing the suitability of mitochondrial and nuclear DNA genetic markers for molecular systematics and species identification of helminths. Parasit. Vectors. 2021;14:233. doi: 10.1186/s13071-021-04737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piganeau G., Gardner M., Eyre-Walker A. A broad survey of recombination in animal mitochondria. Mol. Biol. Evol. 2004;21:2319–2325. doi: 10.1093/molbev/msh244. [DOI] [PubMed] [Google Scholar]
- 44.Figueroa D.F., Baco A.R. Octocoral mitochondrial genomes provide insights into the phylogenetic history of gene order rearrangements, order reversals, and cnidarian phylogenetics. Genome Biol. Evol. 2014;7:391–409. doi: 10.1093/gbe/evu286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavrov D.V. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr. Comp. Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- 46.Chen R., Aldred M.A., Xu W., Zein J., Bazeley P., Comhair S.A.A., Meyers D.A., Bleecker E.R., Liu C., Erzurum S.C., et al. Comparison of whole genome sequencing and targeted sequencing for mitochondrial DNA. Mitochondrion. 2021;58:303–310. doi: 10.1016/j.mito.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lartillot N., Brinkmann H., Philippe H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007;7:S4. doi: 10.1186/1471-2148-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hendy M.D., Penny D. A framework for the quantitative study of evolutionary trees. Syst. Zool. 1989;38:297–309. [Google Scholar]
- 49.Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 50.Qu X.J., Jin J.J., Chaw S.M., Li D.-Z., Yi T.-S. Multiple measures could alleviate long-branch attraction in phylogenomic reconstruction of Cupressoideae (Cupressaceae) Sci. Rep. 2017;7 doi: 10.1038/srep41005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lartillot N., Lepage T., Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- 52.Anthony C.J., Heagy M., Bentlage B. Phenotypic plasticity in Cassiopea ornata (Cnidaria: Scyphozoa: Rhizostomeae) suggests environmentally driven morphology. Zoomorphology. 2022;141:115–131. [Google Scholar]
- 53.Cunha A.F., Maronna M.M., Marques A.C. Variability on microevolutionary and macroevolutionary scales: a review on patterns of morphological variation in Cnidaria Medusozoa. Org. Divers. Evol. 2016;16:431–442. [Google Scholar]
- 54.Dawson M.N. Morphological variation and systematics in the Scyphozoa: Mastigias (Rhizostomeae, Mastigiidae) - a golden unstandard? Hydrobiologia. 2005;537:185–206. [Google Scholar]
- 55.Tyagi K., Chakraborty R., Cameron S.L., Sweet A.D., Chandra K., Kumar V. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta) Sci. Rep. 2020;10:695. doi: 10.1038/s41598-020-57705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ballard J.W.O., Pichaud N. Mitochondrial DNA: more than an evolutionary bystander. Funct. Ecol. 2014;28:218–231. [Google Scholar]
- 57.Shtolz N., Mishmar D. The metazoan landscape of mitochondrial DNA gene order and content is shaped by selection and affects mitochondrial transcription. Commun. Biol. 2023;6:93. doi: 10.1038/s42003-023-04471-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blumberg A., Sri Sailaja B., Kundaje A., Levin L., Dadon S., Shmorak S., Shaulian E., Meshorer E., Mishmar D. Transcription factors bind negatively selected sites within human mtDNA genes. Genome Biol. Evol. 2014;6:2634–2646. doi: 10.1093/gbe/evu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barshad G., Marom S., Cohen T., Mishmar D. Mitochondrial DNA transcription and its regulation: an evolutionary perspective. Trends Genet. 2018;34:682–692. doi: 10.1016/j.tig.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 60.Bentlage B., Collins A.G. Tackling the phylogenetic conundrum of Hydroidolina (Cnidaria: Medusozoa: Hydrozoa) by assessing competing tree topologies with targeted high-throughput sequencing. PeerJ. 2021;9 doi: 10.7717/peerj.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McFadden C.S., Benayahu Y., Pante E., Thoma J.N., Nevarez P.A., France S.C. Limitations of mitochondrial gene barcoding in Octocorallia. Mol. Ecol. Resour. 2011;11:19–31. doi: 10.1111/j.1755-0998.2010.02875.x. [DOI] [PubMed] [Google Scholar]
- 62.Mayer A.G. Medusae of the World, 3. The Scyphomedusae. Carnegie Institution of Washington; 1910. [Google Scholar]
- 63.Kramp P.L. Synopsis of the medusae of the world. J. Mar. Biol. Assoc. U. K. 1961;40:7–382. [Google Scholar]
- 64.Cornelius P.F. In: The Proceedings of the 6th International Conference on Coelenterate Biology. The Leeuwenhorst, Noordwijkerhout, The Netherlands, 16-21 July 1995. den Hartog J.C, editor. Nationaal Natuurhistorisch Museum; 1997. Keys to the genera of Cubomedusae and Scyphomedusae (Cnidaria) pp. 109–122. [Google Scholar]
- 65.Bouillon J., Gravili C., Pagès F., Gili J.-M., Boero F. An Introduction to Hydrozoa. Muséum National d’Histoire Naturelle; 2006. [Google Scholar]
- 66.Calder D.R. Cubozoan and scyphozoan Jellyfishes of the Carolinian Biogeographic Province, southeastern USA. R. Ont. Mus. Contrib. Sci. 2009;3:1–58. [Google Scholar]
- 67.Bentlage B., Lewis C. An illustrated key and synopsis of the families and genera of carybdeid box jellyfishes (Cnidaria: Cubozoa: Carybdeida), with emphasis on the “Irukandji family” (Carukiidae) J. Nat. Hist. 2012;46:2595–2620. [Google Scholar]
- 68.Quek Z.B.R., Chang J.J.M., Ip Y.C.A., Huang D. Complete mitochondrial genome of the sea star Archaster typicus (Asteroidea: Archasteridae) Mitochondrial DNA Part B. 2019;4:3130–3132. doi: 10.1080/23802359.2019.1666676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quek Z.B.R., Ng J.Y., Jain S.S., Long J.X.S., Lim S.C., Tun K., Huang D. Low genetic diversity and predation threaten a rediscovered marine sponge. Sci. Rep. 2022;12 doi: 10.1038/s41598-022-26970-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinf. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dierckxsens N., Mardulyn P., Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45:e18. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warburton P.E., Giordano J., Cheung F., Gelfand Y., Benson G. Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 2004;14:1861–1869. doi: 10.1101/gr.2542904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alamro H., Alzamel M., Iliopoulos C.S., Pissis S.P., Watts S. IUPACpal: efficient identification of inverted repeats in IUPAC-encoded DNA sequences. BMC Bioinf. 2021;22:51. doi: 10.1186/s12859-021-03983-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hahn C., Bachmann L., Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129. doi: 10.1093/nar/gkt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bernt M., Donath A., Jühling F., Externbrink F., Florentz C., Fritzsch G., Pütz J., Middendorf M., Stadler P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 81.Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan P.P., Lin B.Y., Mak A.J., Lowe T.M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49:9077–9096. doi: 10.1093/nar/gkab688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quek Z.B.R., Chang J.J.M., Ip Y.C.A., Chan Y.K.S., Huang D. Mitogenomes reveal alternative initiation codons and lineage-specific gene order conservation in Echinoderms. Mol. Biol. Evol. 2021;38:981–985. doi: 10.1093/molbev/msaa262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katoh K., Standley D.M. MAFFT multiple sequence alignment software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Diroma M.A., Modi A., Lari M., Sineo L., Caramelli D., Vai S. New insights into mitochondrial DNA reconstruction and variant detection in ancient samples. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.619950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abascal F., Zardoya R., Telford M.J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq291. W7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Darriba D., Posada D., Kozlov A.M., Stamatakis A., Morel B., Flouri T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020;37:291–294. doi: 10.1093/molbev/msz189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kozlov A.M., Darriba D., Flouri T., Morel B., Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bernt M., Merkle D., Middendorf M. An algorithm for inferring mitogenome rearrangements in a phylogenetic tree. RECOMB-CG 2008. Lect. Notes Comput. Sci. 2008;5267:143–157. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads used for mitogenome assembly have been deposited at NCBI SRA under BioProject PRJNA934731. The mitogenomes assembled in this study have been deposited into GenBank under accession numbers OR400200 – OR400214. Additionally, this paper also analyses existing, publicly available mitogenomes provided on GenBank (refer to key resources table for accession numbers). Gene orders for TreeRex analyses have been provided under Data S3. ML and BItrees used for TreeRex analyses have been provided under Datas S4 and S5 respectively, while TreeRex outputs for ML and BIanalyses have been provided under Datas S1 and S2 respectively.
All original code used for this study has been provided as a pipeline under File S1.
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.