


Keywords: chronic kidney disease, fibrosis, hemodynamics, homeostasis, ureteral obstruction
Abstract
The kidneys play a key role in maintaining total body homeostasis. The complexity of this task is reflected in the unique architecture of the organ. Ureteral obstruction greatly affects renal physiology by altering hemodynamics, changing glomerular filtration and renal metabolism, and inducing architectural malformations of the kidney parenchyma, most importantly renal fibrosis. Persisting pathological changes lead to chronic kidney disease, which currently affects ∼10% of the global population and is one of the major causes of death worldwide. Studies on the consequences of ureteral obstruction date back to the 1800s. Even today, experimental unilateral ureteral obstruction (UUO) remains the standard model for tubulointerstitial fibrosis. However, the model has certain limitations when it comes to studying tubular injury and repair, as well as a limited potential for human translation. Nevertheless, ureteral obstruction has provided the scientific community with a wealth of knowledge on renal (patho)physiology. With the introduction of advanced omics techniques, the classical UUO model has remained relevant to this day and has been instrumental in understanding renal fibrosis at the molecular, genomic, and cellular levels. This review details key concepts and recent advances in the understanding of obstructive nephropathy, highlighting the pathophysiological hallmarks responsible for the functional and architectural changes induced by ureteral obstruction, with a special emphasis on renal fibrosis.
CLINICAL HIGHLIGHTS.
Chronic kidney disease (CKD) affects 10% of the world population and is a leading cause of death. CKD, characterized by inflammation and renal fibrosis, can progress to end-stage renal disease (ESRD), which is fatal if left untreated.
Obstructive nephropathy, the renal disease caused by impaired urine flow, can become manifest at all ages, and the causes, either benign or malignant, are manifold.
Kidney function and architecture are severely impacted by ureteral obstruction; however, the extent of the alterations, and thus the pathological consequences, depend greatly on the duration, degree, and site of the obstruction.
Experimental ureteral obstruction has been used as a model to study renal pathophysiology since the 1800s and is currently mainly used to elucidate the molecular and cellular mechanisms underlying renal fibrosis as well as for identifying therapeutic targets. At the time of writing, almost 18,500 publications on experimental ureteral obstruction are indexed in PubMed, testifying to the widespread use of this model. However, the often-used complete and irreversible model of ureteral obstruction has limitations when it comes to studying tubular injury and repair.
Several cell types and signaling pathways are involved in renal fibrosis, which greatly hampers the development of efficacious therapeutics. Combining models of obstructive nephropathy with next-generation analysis techniques will greatly advance our understanding of renal fibrosis and support drug development.
1. INTRODUCTION
The global incidence of chronic kidney disease (CKD) is steadily increasing, presenting a significant clinical challenge because CKD may lead to end-stage renal disease (ESRD), which requires interventions such as dialysis and kidney transplantation to prevent death. CKD affects more than 0.5 billion people and has a global prevalence rate approaching 10% (1, 2). A prominent feature of CKD is inflammation of the tubulointerstitial compartment, which leads to renal fibrosis, a complex process involving the loss of capillary networks, decline in renal functions, and progressive accumulation of fibrillary collagens, activated myofibroblasts, and inflammatory cells (3, 4). As fibrosis progresses, kidney functions gradually deteriorate, characterized by reduced renal blood flow (RBF) and tissue perfusion, decreased glomerular filtration rate (GFR), impaired tubular handling of water and electrolytes, and elevated urinary protein excretion.
There are numerous diseases causing CKD, including hypertension, diabetes, and cardiovascular disease, and the speed and severity of CKD development vary among these different diseases. Another important condition causing CKD is obstructive nephropathy, which is also characterized by renal dysfunction and renal interstitial fibrosis. Obstructive nephropathy is caused by chronic urinary tract obstruction that is the consequence of numerous urinary tract diseases, including congenital anatomic abnormalities, renal calculi, prostatic hyperplasia, and bladder tumors, and it is characterized by a complex array of pathophysiological processes leading to ESRD (FIGURE 1). The understanding of these functional changes has been significantly enhanced through advances in cell and molecular biology, and much of this knowledge was accumulated through animal studies.
Figure 1.

Hallmarks of obstructive nephropathy showing key characteristics of functional and cellular consequences of ureteral obstruction as a time lapse from hours to months. ESRD, end-stage renal disease; GFR, glomerular filtration rate; RBF, renal blood flow; RVR, renal vascular resistance.
Unilateral ureteral obstruction (UUO), also a model for experimental hydronephrosis, is among the most important animal models for identifying novel mechanisms underlying renal fibrosis. The use of different species with UUO as an experimental model to examine renal (patho)physiology dates back centuries. In 1919, Dr. Hinman wrote in the Journal of Urology “The literature relative to experimental hydronephrosis is enormous. Original contributions personally reviewed number more than three hundred which is only a partial list” (5). In the same paper, the earliest work referred to is from 1859. The literature concerning ureteral obstruction (UO) has expanded dramatically ever since, with almost 18,500 publications indexed in PubMed at the time of writing this review. The fact that the UUO model has been in use for such a long time underscores its value in studying the molecular mechanisms of renal fibrosis, but it is important to acknowledge that the model has limitations when it comes to investigating tubular injury and repair.
This review provides an overview of the hallmarks of obstructive nephropathy, highlighting the key molecular mechanisms currently known to contribute to the decline in kidney function and the progression of renal interstitial fibrosis (FIGURE 2).
Figure 2.

Schematic overview of the functional, cellular, and molecular consequences of unilateral ureteral obstruction (UUO) leading to acute kidney injury (AKI) or chronic kidney disease (CKD).
2. CLINICAL OBSTRUCTIVE NEPHROPATHY
Obstructive uropathy refers to the presence of structural or functional changes in the urinary tract that interrupt the normal flow of urine (6), whereas obstructive nephropathy is the renal disease caused by impaired urine flow. Obstructive nephropathy can manifest at all ages, stretching from the antenatal period to adulthood, and is caused by chronic urinary tract obstruction, which may be associated with hydronephrosis (i.e., dilation of the urinary tract) (6). Obstructive nephropathy has many causes, both benign and malignant. The most common causes of intrinsic obstruction in children are congenital anatomic abnormalities, whereas renal calculi (6), affecting ∼10% of adults worldwide (7), are the most common cause in young adults. In older adults, benign prostatic hyperplasia, prostate cancer, and bladder tumors are common causes of intrinsic obstruction (6). On the other hand, extrinsic obstruction is caused by processes outside the urinary tract, such as retroperitoneal fibrosis and Crohn’s disease (8–10), as well as pregnancy (11). Moreover, obstructive uropathy can be classified according to the underlying cause (congenital or acquired), duration (acute or chronic), degree (partial or complete), and site (unilateral or bilateral). Clinical signs and symptoms may be absent or vary from flank pain, urinary retention, overflow incontinence, and decreased urine flow to complicated urinary tract infections and acute renal failure. The overall incidence and prevalence of obstructive nephropathy are difficult to assess accurately. A review based on 59,064 autopsies, ranging from neonates to elderly, identified hydronephrosis in 3.1% (3.3% in males and 2.9% in females) (12). Provided that hydronephrosis can be used as a surrogate marker of obstruction, the study showed that until the age of 20 yr there was no substantial sex difference in the frequency of abnormalities. Between the ages of 20 and 60 yr urinary tract obstruction was more frequent among women than among men, and above the age of 60 yr prostatic disease raised the frequency of urinary tract obstruction among men above that observed among women. Because a high proportion of these autopsy-detected cases of obstruction likely went undetected during life, the overall prevalence of urinary tract obstruction is most likely far greater than reports suggest. This conclusion is reinforced by the fact that obstruction can be asymptomatic and transient, for instance, when the obstruction is caused by pregnancy or renal calculi. To date, surgery remains the primary intervention for urinary tract obstruction; however, for many patients with moderate or severe loss of renal function, improving ureteric drainage does not necessarily lead to an improvement in renal function (13). Thus, there remains a clinical need for improved treatment modalities. One potential drug candidate that holds promise for renal function recovery following relief of obstruction is losartan, an angiotensin II (ANG II) receptor blocker with proven efficacy in both experimental UUO (14–16) (further details regarding the model can be found below) and patients with UO (17). Patients with UO due to benign prostatic hyperplasia often receive α-blocker therapy to improve urinary flow. Interestingly, recent findings have revealed that pharmacological blockade of α1-adrenergic receptors can reduce UUO-induced renal fibrosis in mice (18). Furthermore, patients experiencing both UO and renal colic, frequently resulting from kidney stones, are commonly prescribed analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) (19). NSAIDs function by modulating the prostaglandin system. However, it is crucial to acknowledge that these drugs can also potentially induce acute kidney injury (AKI) (20–22). Therefore, it is imperative to exercise caution when using this class of drugs. The duality of the prostaglandin system in renal disease is elaborated upon below in this review.
3. EXPERIMENTAL MODELS OF URETERAL OBSTRUCTION
3.1. Animal Models of Ureteral Obstruction
Even though the clinical features of UO have been recognized for ages, our understanding of the pathophysiology of renal dysfunction has been markedly enhanced by detailed studies of experimental animal models of UO. The effects of UO on renal anatomy and function are significantly impacted both during and after release of obstruction, and the degree of change is greatly influenced by whether the obstruction is unilateral or bilateral, acute or chronic, and partial or complete. Most mechanistic studies on UO-related renal dysfunction use experimental models of acute complete unilateral (UUO) or bilateral (BUO) ureteral obstruction, usually for 24 h. In these “classical” animal models, the extent of UO is clear and reproducible, and the obtained results are not confounded by extensive inflammation- or fibrosis-induced changes in the renal parenchyma; the BUO model especially is often used to explore the pathophysiological regulation of transport proteins (23–25). Complete obstruction (UUO/BUO) for a short duration strikingly alters renal blood flow (RBF), glomerular filtration, and tubular functions but produces minimal anatomical changes in blood vessels, glomeruli, and tubules (6). The responses to prolonged obstruction following complete UUO include interstitial inflammation (peak at day 2–3), tubular dilation and atrophy, as well as tubulointerstitial fibrosis (TIF), which is mostly observable from day 7 onward (26). However, the direct translation of results from these models has limitations and leaves a gap of knowledge compared with the chronic disease development of CKD as a consequence of obstruction in humans. Because most cases of clinical UO involve partial rather than complete obstruction, animal models of partial UUO have been established in both neonatal and adult mice and rats (27, 28). These models can be used to study progressive renal injury and TIF (29); however, the partial UUO model is technically very challenging, with frequent adhesions leading to complete obstruction. As detailed in this review, paramount knowledge of the pathophysiology has been obtained from these experimental models of urinary tract obstruction. However it is important to underline that all these animal models of obstructive nephropathy have limitations for direct comparison of the pathophysiological changes in humans with chronic obstruction of the urinary tract.
3.2. The UUO Model—Strengths and Limitations
UUO is the standard experimental model for TIF because it is not complicated by hypertension, proteinuria, hyperlipidemia, or toxicity-driven renal injury (30–32). UUO has emerged as a central model to provide information on myofibroblast activation and extracellular matrix (ECM) accumulation in the context of renal TIF (26, 33), and the model recapitulates fundamental pathophysiological processes that typify human CKD in an accelerated time span (4, 34), although the model misses important aspects of tubular repair.
The major advantages of the UUO model are that it is easy to perform and fast (generally, experiments last for 3–14 days) and it has a high degree of reproducibility, even in mice. Moreover, the model can be modulated with respect to timing, severity, and duration of the obstruction. However, even though UUO is one of the most popular models of CKD, it has limitations to take into consideration. One major disadvantage is the inability to precisely monitor changes in renal function. The contralateral nonobstructed kidney compensates for the loss of function in the obstructed kidney, so the levels of endogenous filtration markers, e.g., plasma creatinine and blood urea nitrogen (BUN), which are often used to assess renal function, hardly change. Moreover, in the UUO model there is no tubular recovery, so it is not an ideal model to study tubular injury and repair. Also, the precise cytopathological mechanisms driving UUO-induced renal injury are poorly understood (35). In addition, fibrosis develops very rapidly in the model, which limits the experimental window for testing potential ameliorating interventions unless they are introduced before the UUO injury or are expected to have profound effects (36, 37). Nevertheless, numerous putative antifibrotic compounds have shown therapeutic potential in this model (32), including dapagliflozin and empagliflozin (38, 39), which are both approved by the FDA for the treatment of CKD, as well as pirfenidone and nintedanib (40, 41), which are both approved by the FDA for the treatment of idiopathic pulmonary fibrosis. Another caveat of the UUO model, which is rarely discussed, is suture placement. This is an important variable because localization of the suture will affect the time course of renal injury and variation in placement can lead to problems of reproducibility.
Whether the UUO model can be used to study the structural and functional recovery of the kidneys after relief of the obstruction is controversial. Some studies have demonstrated that the obstructed kidney is able to regenerate after reversal of the obstruction (42–45), whereas others observed that renal damage persists after relief of obstruction (46–49). However, on the basis of the above-mentioned studies, it appears that there are three important aspects for achieving a successful reversible UUO model: 1) optimal timing of the relief to allow for recovery; 2) ability to achieve complete relief of the obstruction; and 3) removing the contralateral kidney to avoid compensatory functional changes in the nonobstructed kidney (50). Overall, the reversible UUO model, although technically challenging, is considered to be an appropriate model to explore the process of tissue remodeling in the ipsilateral kidney. Moreover, the model can be of clinical relevance, as it reflects the pathophysiology of patients with UO who received decompression but are still at risk for ESRD due to persistent impairment of renal functions (51, 52).
3.3. Translation to Human CKD
For decades, the UUO model has been instrumental in preclinical research, and it is widely used to characterize the pathophysiology of CKD and test potential interventions. However, therapeutic strategies with proven efficacy in the UUO model cannot always be successfully translated into clinical practice because of several limitations: First, there are significant differences in physiology and pathophysiology between animals and humans, in particular because humans rarely present a picture with complete obstruction. Second, animal models do not reflect the variability of the patient population, since it is very common to choose a single inbred strain of animal of a single sex and with a specific age to minimize variance in animal experiments. Third, there is often a mismatch between the measured outcomes in preclinical research compared with clinical trials. To illustrate this, in UUO animals biochemical and histological parameters are mainly evaluated, whereas in humans more restricted end points such as GFR, proteinuria, or mortality are often employed. Fourth, and last, the UUO model does not reflect established disease, which is mostly seen in clinical practice. Therefore, it is paramount to validate findings in other CKD models, especially with regard to fibrosis progression. Moreover, there is a need for better animal models that closely resemble AKI-CKD transition (53). Despite these limitations, careful use of the UUO model can provide clinically relevant insight into the different pathophysiological mechanisms underlying CKD development and can unveil possible therapeutic targets.
4. FUNCTIONAL CHANGES IN OBSTRUCTIVE NEPHROPATHY
4.1. Hemodynamic Changes Associated with Obstructive Nephropathy
Obstructive nephropathy is characterized by severe vasoconstriction and profoundly changes all components of glomerular function. The extent to which glomerular function is affected depends on the duration and severity of the obstruction, whether it is unilateral or bilateral, and the extent to which the obstruction is persistent or has been relieved (54, 55). The hemodynamic changes can be divided into an early vasodilation phase (1–2 h of unilateral obstruction) and a late vasoconstrictive phase (>3 h of unilateral obstruction), as illustrated in FIGURE 3.
Figure 3.
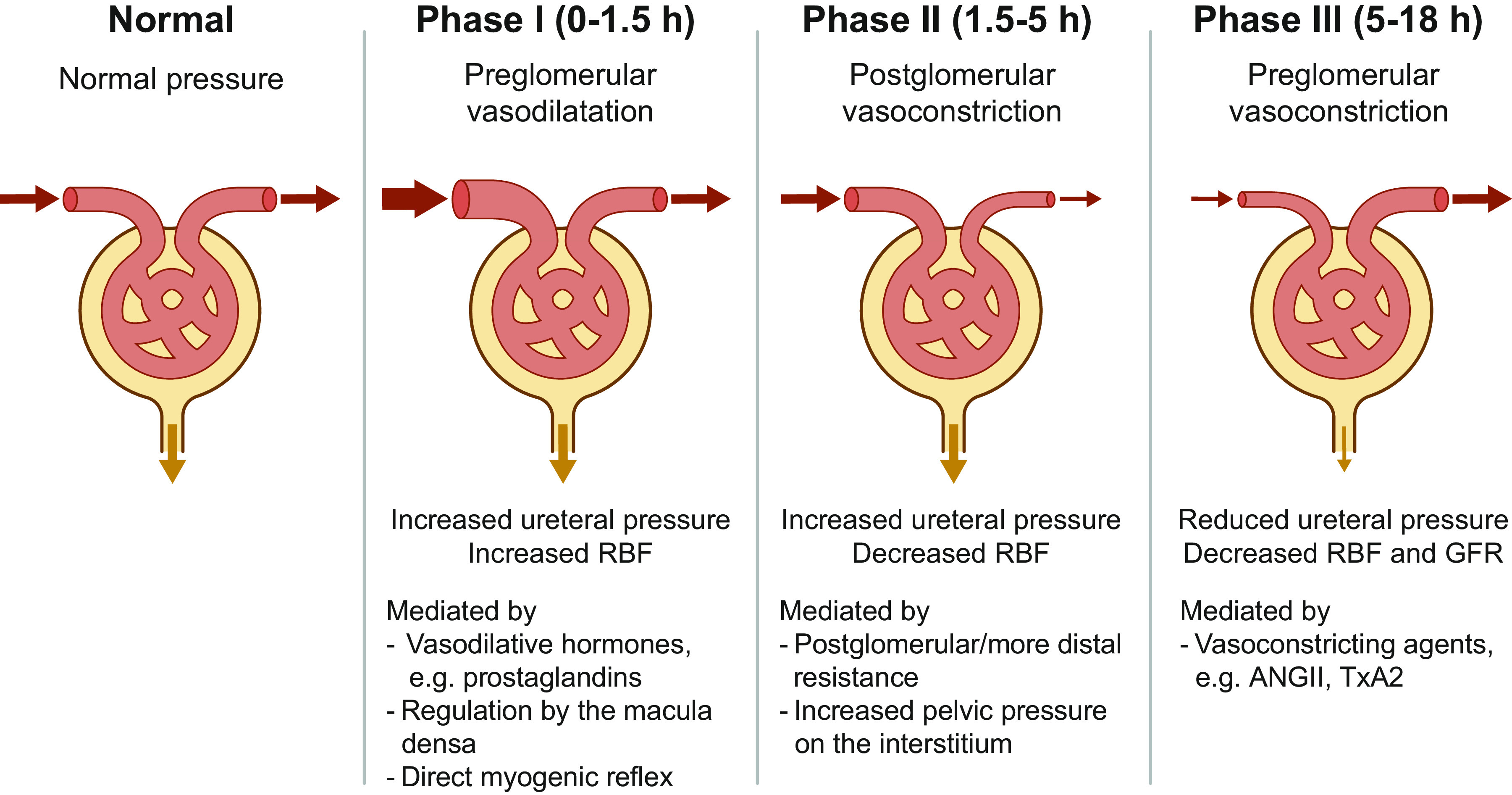
Characteristic phases of hemodynamic changes associated with obstructive nephropathy. ANG II, angiotensin II; GFR, glomerular filtration rate; RBF, renal blood flow; TxA2, thromboxane A2.
4.1.1. The early vasodilation phase of obstruction.
Moody et al. (56) demonstrated a direct relationship between the hydrodynamic and hemodynamic events in the ipsilateral kidney after acute UUO for 18 h in awake dogs. Three distinct phases follow after the onset of UO: phase I (0–1.5 h), characterized by an increase in both ureteral pressure and RBF, possibly due to a predominant preglomerular vasodilation; phase II (1.5–5 h), characterized by an increase in ureteral pressure and a decrease in RBF, explained by postglomerular vasoconstriction; and phase III (5–18 h), characterized by a reduction in both ureteral pressure and RBF, explained by a predominant preglomerular vasoconstriction. The initial transient increase in RBF in phase I has been demonstrated in many studies in dogs (56–58), rabbits (59), rats (60), and pigs (61). This afferent vasodilation is mediated by several mechanisms, including increases in vasodilator hormones, such as prostaglandins (see below), reduced NaCl concentration at the macula densa, and a direct myogenic reflex (62). The hyperemic response is not attenuated by renal nerve stimulation or infusion of catecholamines (63, 64) and may be linked to changes in interstitial pressure (65, 66).
The increase in RBF after the onset of UO is not a consistent finding in all models. RBF has been reported to remain constant or decline after the onset of UUO in awake dogs (67), rats (68), lambs (69), and pigs (70, 71), suggesting a complex balance between vasodilatory and vasoconstrictor mechanisms at this early time point after obstruction. Upon the onset of UO, a rapid increase in pelvic (ureter) pressure could increase the peritubular capillary pressure. A constant RBF in the face of an increasing peritubular capillary pressure is consistent with some degree of arteriolar vasodilation (72). Blood flow measurements revealed that the initial vasodilation is expected both in the cortex and in the medulla (73). The mechanisms underlying the changes in the pre- and postglomerular vascular resistance, after the onset of UO, are still not completely understood. However, infusion of indomethacin, a nonselective inhibitor of the cyclooxygenase enzymes (COX-1 and COX-2), dramatically reduced RBF during UUO in pigs (74), suggesting that vasodilatory prostaglandins (e.g., PGE2) might be involved in the increase or the maintenance of RBF during UUO.
4.1.2. The late vasoconstrictive phase of obstruction.
After the brief vasodilatory phase I, ipsilateral renal blood flow progressively decreases during UUO [phases II and III (56)]. In phase II, although pelvic (ureteral) pressure does not reach the maximum levels, the reduction in RBF is already ongoing, suggesting that primarily the postglomerular vasculature is vasoconstricted. Thus, the underlying mechanisms in phase II could be a selective increase in the postglomerular or more distal efferent arteriolar resistance or a direct result of the increasing pelvic pressure on the interstitium increasing the peritubular capillary pressure (56, 70, 71).
In phase III, RBF continues to decline and decreases by ∼30% of the preobstructive levels, with high plasma angiotensin II (ANG II) levels after 15 h of UUO (70), consistent with other studies demonstrating continued active vasoconstriction (56, 57). The mechanism underlying phase III is supposed to be the continuous vasoconstriction, localized predominantly at the preglomerular vasculature (67, 75). This finding is supported by micropuncture studies of individual surface glomeruli in the rat kidneys after blockade of tubules (76, 77). Consistent with this, UUO in rats for 24 h markedly increased the afferent arteriolar resistance and decreased afferent arteriolar blood flow (77). These studies highlighted the critical role of afferent arteriolar vasoconstriction in attenuating single-nephron GFR (SNGFR) during the established phase of obstruction and that of intrarenal mechanisms responsible for the late preglomerular vasoconstrictive response to UUO. Simultaneous with the progressive reduction in RBF, GFR, and renal tissue perfusion, oxygen delivery to the renal cortex and particularly to the renal medulla is reduced, leading to a gradual loss of the capillary network in the kidney, increased apoptosis, and stimulation of cell infiltration in the renal parenchyma (6). Numerous intrarenal vasoactive compounds, such as ANG II and thromboxane A2 (TxA2), are potentially implicated in the active vasoconstriction and subsequent stimulation of cellular and molecular processes, which involve inflammation, deposition of matrix proteins, and development of progressive interstitial fibrosis. Multiple studies revealed that several vasoactive mediators play significant roles in the reduction in RBF and GFR because prostaglandin E2 blockade, COX-2 inhibition (74), angiotensin II-converting enzyme (ACE) inhibitors (78–80), angiotensin II receptor blockade (81), inhibition of TxA2 synthesis (82), kallikrein inhibitor (83), and blockade of endothelin ETA receptor (84) changed the RBF and GFR to some extent upon release of the obstruction. The role of these mediators has been highlighted in detail in a previous review (4).
4.1.3. The renin-angiotensin-aldosterone system in the regulation of renal hemodynamics after ureteral obstruction.
The reduction in RBF in UUO is primarily the consequence of active vasoconstriction (57) and can be attenuated by specific pharmacological agonists or antagonists (85). Several such mediators are vasoconstricting agents, such as ANG II and TxA2; the changes in RBF in UUO in response to the blockade of ANG II or vasoactive eicosanoid prostaglandin system have been examined in several studies (74, 78, 81). Hammad et al. (86) demonstrated that ANG II infusion decreased RBF, GFR, and urine volume in rats. Vander and Miller (87) reported an increase in renin concentration in the renal vein when ureteral pressure was elevated. These findings evoked interest in the renin-angiotensin system (RAS) to understand the underlying pathophysiology of obstructive nephropathy as well as to introduce therapeutic interventions. Consistently, several studies demonstrated that administration of angiotensin-converting enzyme (ACE) inhibitor, captopril or enalapril, attenuated renal vasoconstriction during UO and improved postobstructive renal dysfunction in rat and guinea pig (79, 80, 88). In contrast, no significant improvement in postobstructive renal dysfunction was observed in UUO rats infused with an ANG II antagonist, saralasin, which might be partly attributable to vasoconstrictor properties of saralasin (78). Other investigators, however, demonstrated that ACE inhibitors did not decrease renal vasoconstriction in pigs (89). Infusion of an ACE inhibitor, captopril, in UUO pigs resulted in an increase in contralateral RBF, suggesting that a compensatory increase in RBF may in part be inhibited by ANG II-mediated vasoconstriction in the contralateral kidney during obstruction (89). These results are contrary to the notion that vasoconstriction in UUO is exclusively dependent on ANG II and suggest the role of other mediators in the vasoconstriction observed in obstructive nephropathy.
4.1.4. The prostaglandin system in the regulation of renal hemodynamics after ureteral obstruction.
Administration of indomethacin during UUO decreased RBF in both the ipsilateral and contralateral kidneys. Simultaneously, a decrease in the urinary excretion of PGE2 from the contralateral kidney was observed after UUO, demonstrating inhibition of PGE2 production in the kidney after indomethacin treatment (74, 90). The reduction in RBF is immediate after indomethacin administration, and the initial increase in RBF following the onset of UUO subsided (91). Thus, the vasodilatory prostaglandins (e.g., PGE2) likely play an important role in the maintenance of RBF during UUO (92, 93). Administration of nonselective COX inhibitors blocks both vasodilatory (PGE2) and vasoconstrictor arachidonic acid (TxA2) products. The maintenance of RBF during UUO could not be achieved upon suppression of vasodilatory prostaglandins. In contrast, inhibition of the thromboxane system with a COX inhibitor resulted in the preservation of renal function (79). Administration of aprotinin, a kallikrein inhibitor, also improved postobstructive function by decreasing kinin-stimulated production of TxA2 in rats subjected to 24-h UUO (83). These findings indicate multiple sites of action in the vasculature of obstructed kidneys by vasoactive prostanoids.
4.2. Changes in Urinary Concentration in Response to Obstructive Nephropathy
4.2.1. Urinary concentration defect in ureteral obstruction.
Urinary tract obstruction is associated with changes in renal water handling. Studies using isolated perfused tubules from animal models of BUO and UUO, including proximal straight tubule, medullary thick ascending limb (TAL), and cortical collecting tubule, revealed marked impairment of fluid and chloride reabsorption capacity (94, 95). Consistently, altered expression of water channel proteins and sodium transporters in the kidney has been demonstrated in obstructive nephropathy (FIGURE 4). The expression of aquaporin-1 (AQP1) in the proximal tubule and descending thin limb and of AQP2, -3, and -4 in the collecting duct was significantly decreased in experimental BUO for 24 h (23, 24). Moreover, downregulation of sodium transporters [type 3 Na/H exchanger (NHE3), type II Na-Pi cotransporters (NaPi-2), Na-K-2Cl cotransporter (NKCC2), Na-Cl cotransporter (NCC), epithelial sodium channel (ENaC)], Na-K-ATPase and urea transporters (UT-A1, UT-A3, and UT-B) in BUO kidneys contributes to the impaired urinary concentration capacity (25, 96, 97). Postobstructive polyuria and decreased urine concentration follow the release of 24-h BUO. Li et al. (24) demonstrated that AQP2 and AQP3 levels in the collecting duct remained decreased for ∼2 wk after the release of 24-h BUO and were normalized 30 days after the release. AQP1 levels were also significantly reduced and remained downregulated for 30 days after the release (24), indicating that prolonged downregulation of AQPs in the renal tubules in obstructive nephropathy contributes to changes in renal water handling, that is, postobstructive polyuria and decreased urine concentration.
Figure 4.

A: expression of renal water channel proteins (aquaporins), sodium transporters, and urea transporters (UTs) that contribute to tubular reabsorption of water, sodium, and urea in each segment of the nephron. B: changes in the expression of the aquaporins and sodium and urea transporters in different models of ureteral obstruction compared with the healthy situation (Sham, 100%), based on Refs. 24, 25, 96–99. 24hBUO, 24 h of bilateral ureteral obstruction; 24hBOU-3R, 24 h of bilateral ureteral obstruction followed by the release of obstruction for 3 h; 24hUUO, 24 h of unilateral ureteral obstruction; AQP, aquaporin; ENaC, epithelial sodium channel; NaPi-2, type II Na-Pi cotransporters; NCC, Na-Cl cotransporter; NHE3, type 3 Na/H exchanger.
In contrast to BUO, UUO does not significantly change the total urinary excretion of water and sodium because the nonobstructed contralateral kidney compensates for the dysfunction of the obstructed kidney to excrete water and solutes (98, 100). Importantly, these studies demonstrated long-term parallel reduction in AQP2 expression and development of polyuria, decreased urine osmolality, and increased free water clearance, indicating a functional association between AQP2 abundance and the capacity to excrete free water (24, 98, 100). The levels of AQP1, -2, -3, and -4 and phosphorylated AQP2 at serine 256 as well as major sodium transporters (NHE3, NaPi-2, NKCC2, NCC, and ENaC) and urea transporters (UT-A1, UT-A3, and UT-B) were significantly decreased in the obstructed kidney in 24-h UUO (96–100) (FIGURE 4). The downregulation of AQP2 in obstructed kidneys was associated with reduced expression of AQP2 mRNA, suggesting that AQP2 was regulated at the transcriptional level (100). No systematic comparison between the UUO model and human CKD has been performed related to the regulation of different transport proteins. However, there are reports suggesting a similar reduction in protein expression of AQP2 and -3 in biopsies from human kidneys with substantial interstitial fibrosis and nephron loss (101).
4.2.2. Underlying mechanisms responsible for the regulation of channels and transporters in response to ureteral obstruction.
BUO is associated with the induction of COX-2 and cellular infiltration in the renal medulla (102–105), which may contribute to the downregulation of AQP2. Consistently, local accumulation of COX-2-produced prostanoids (PGE2, PGF2α, 6-keto-PGF1α, PGD2, and thromboxane B2) was observed in BUO kidneys (104). UUO for 7 days was also associated with increased COX-1/2 and microsomal prostaglandin E synthase 1 levels in the kidney as well as PGE2 secretion in urine collected from the obstructed ureter (106). The infiltration of leukocytes following UO was associated with an increase in thromboxane B2 excretion by the kidney and coincided with a reduction in GFR (107). In contrast, treatment with parecoxib, a COX-2 inhibitor, attenuated the increase in the concentrations of PGE2, PGF2α, 6-keto-PGF1α, and thromboxane B2 in the inner medulla of 24-h BUO kidneys (104). Importantly, COX-2 inhibition in rats with 24-h BUO significantly attenuated the downregulation of AQP2 in the inner medulla and of sodium transporters (NHE3 and NKCC2) in the proximal tubule and medullary TAL (103). These findings indicate that COX-2 and associated prostanoids regulate, at least in part, the expression of water channel proteins and sodium transporters, which play a role in impaired renal water and sodium handling in response to BUO (103, 108, 109).
Renin mRNA levels and ANG II content in the kidneys increase in obstructive nephropathy in rats, coinciding with high systolic blood pressure and plasma ANG II levels (110). Notably, treatment of BUO rats with candesartan, an ANG II type 1 (AT1) receptor blocker, significantly attenuated the downregulation of NaPi-2 in the proximal tubule, NKCC2 in the medullary TAL, and AQP2 in the collecting duct at 2 days after the release of 24-h BUO (111). Moreover, AT1 receptor blockade attenuated the increased expression of COX-2 in the inner medulla, suggesting that COX-2 induction after BUO and related changes in the expression of sodium transporters and AQP2 could be regulated, at least in part, by ANG II (111). These observations were different from the findings in normal rats because ANG II per se potentiates the effect of vasopressin on the plasma membrane targeting of AQP2 (112) and regulates the expression of AQPs and sodium transporters in the renal tubule (113–115).
Consistent with the COX-2 induction observed in the kidney inner medulla in BUO, COX-2 mRNA and protein levels are significantly increased in the inner medulla in response to UUO (116, 117). The role of reactive oxygen species (ROS) in the induction of COX-2 was demonstrated in rats with 3-day UUO: antioxidants (NADPH oxidase inhibitor diphenyleneiodonium and the complex I inhibitor rotenone) decreased the UUO-mediated induction of COX-2 in the kidney inner medulla (118). Treatment with superoxide dismutase 2 (SOD2)-mimic manganese(III)tetrakis[(4-benzoic acid) porphyrin chloride (MnTBAP)] also significantly attenuated AQP2 downregulation in 7-day UUO and suppressed COX-2 induction in the kidney (106). This finding suggested that mitochondrial oxidative stress partly mediates AQP2 downregulation in obstructive nephropathy through the COX-2 pathway. However, the underlying mechanisms and signaling pathways responsible for the downregulation of channels and transporters in the renal tubule in response to UO are complex. Other possible factors include high hydrostatic pressure on tubular epithelial cells, increased interstitial pressure, changes in blood circulation in the kidney, absence of urine flow, production of natriuretic substances, and altered expression of genes associated with cell transformation/transition. Indeed, the effects of changes in the microenvironment, such as osmolality, urine flow, and pH, on the expression of AQP2 were demonstrated (119–121).
5. METABOLIC CHANGES IN OBSTRUCTIVE NEPHROPATHY
UO is associated with significant changes in metabolism that coincide with tubular cell injury and macrophage infiltration in the interstitium (26). Metabolites are endogenous and exogenous molecules that play a role in regulatory systems in the cells, and metabolic profiling has been performed to understand the metabolic responses of kidney cells under physiological and pathophysiological conditions (122–127).
Mass spectrometry imaging of the rat kidneys after 1 and 3 wk of UUO revealed altered renal metabolism, such as glycolysis (decreased levels of glucose and increased levels of pyruvic acid), tricarboxylic acid cycle (increased levels of citric acid, succinate, and glutamine and decreased level of aspartate), ATP metabolism (reduced ADP and AMP levels), fatty acid metabolism (reduced levels of linoleic acid, oleic acid, stearic acid, and arachidonic acid), antioxidants (reduced levels of taurine and GSH), and electrolytes (reduced levels of Na+ and K+) (128) (FIGURE 5). Moreover, a marked increase in triglyceride content and a decrease in total phospholipid content were observed in kidneys of rats subjected to 24 h of UUO (129). These changes in renal metabolism in UUO could contribute to the development of renal fibrosis. Hyperpolarized 13C magnetic resonance imaging revealed an increase in the lactate-to-pyruvate ratio in the kidneys of mice after partial UUO (130). Notably, these changes were associated with hydronephrosis, fibrosis, and macrophage infiltration in the obstructed kidney (130). In animal models, a metabolic shift from mitochondrial oxidative phosphorylation to aerobic glycolysis was observed in renal fibrosis (128, 131, 132).
Figure 5.

Overview of the renal metabolic changes in response to 1 and 3 wk of unilateral ureteral obstruction (UUO). Because of the myriad of tasks performed by the kidney to maintain total body homeostasis, it is one of the most energy-demanding organs in the body. In the healthy kidney, energy, in the form of ATP, is generated via glycolysis, in the citric acid cycle, and via β-oxidation of fatty acids. In obstructive nephropathy, ATP production is severely impaired because of 1) perturbations in glycolysis, resulting in decreased levels of glucose and increased levels of pyruvate; 2) interruption of the citric acid cycle, causing accumulation of citric acid, succinate, and glutamine; 3) changes in ATP metabolism; and 4) impairment of fatty acid metabolism, leading to a reduction in the levels of linoleic acid, oleic acid, stearic acid, and arachidonic acid. Moreover, there is an overall reduction in renal antioxidant systems. These metabolic changes are believed to promote fibrogenesis following UUO.
In several studies, kidney tissue and fibroblasts exhibited increased expression of mRNA and protein levels of glycolytic enzymes [hexokinase (HK2), pyruvate kinase M2 (PKM2), lactate dehydrogenase A (LDHA)] and lactic acid production in the process of fibrosis (132–134). Importantly, shikonin (an inhibitor of PKM2) and dichloroacetate [an inhibitor of pyruvate dehydrogenase kinase-1 (PDK1)] significantly inhibited the expression of fibronectin and type I collagen, tubular apoptosis, and macrophage infiltration in the obstructed kidney in a mouse model of 7-day UUO (135). In line with this, it was found that shikonin and 2-deoxyglucose, another aerobic glycolysis inhibitor, attenuated myofibroblast activation in UUO-induced fibrosis. Moreover, it was shown that shikonin inhibits aerobic glycolysis by reducing phosphorylation of PKM2, the rate-limiting glycolytic enzyme associated with cell reliance on aerobic glycolysis (136). However, another study of the 7-day UUO model revealed that only very few regulators of glucose utilization were differentially expressed in the obstructed kidneys (137). Clearly, more studies are needed to examine the changes in the protein levels and activity of each enzyme involved in the whole glycolytic pathway in the obstructed kidney. Mitochondrial fatty acid β-oxidation of long-chain fatty acids, in which carnitine palmitoyltransferase 1 (CPT1) is the critical regulatory enzyme (138), provides energy to renal tubular epithelial cells (139), and decreased fatty acid oxidation is associated with renal fibrosis (140, 141). Indeed, in carnitine palmitoyl-transferase 1A (Cpt1a)-knockin mice overexpression of Cpt1a decreased renal fibrosis, blunted proinflammatory responses, inhibited epithelial cell damage and macrophage infiltration, and prevented mitochondrial dysfunction (137). Thus, the metabolic alterations in renal fibrosis, including changes in glucose and lipid metabolism, are important pathophysiological features that potentially could become important therapeutic targets (142–145).
6. TRANSCRIPTIONAL CHANGES IN OBSTRUCTIVE NEPHROPATHY
Obstructive nephropathy is associated with tubular apoptosis, interstitial inflammation, and interstitial fibrosis, and there are complex changes in gene expression and protein abundance. The observed changes could serve as potential biomarkers of disease progression and therapeutic targets. These include signaling molecules involved in macrophage recruitment and proliferation, tubular cell activation/differentiation, cell adhesion, apoptosis, immune/inflammatory responses, cell cycle regulation, proteolysis, metabolic processes, transport functions, and epithelial-mesenchymal transition (EMT) (146–148). Microarray analysis of obstructed kidneys of rats with neonatal UUO revealed that mRNA expression of multiple immune modulators, including Krox24, interferon-γ regulating factor-1 (IRF-1), monocyte chemoattractant protein-1 (MCP-1), interleukin-1β (IL-1β), CCAAT/enhancer binding protein (C/EBP), p21, c-fos, c-jun, and pJunB, was significantly upregulated compared with sham-operated kidneys on day 12 after obstruction (149). Moreover, microarray analysis of mouse kidneys subjected to UUO revealed that >1,800 transcripts were changed during days 1 through 9 after obstruction (147). These changes include many transcripts, such as Fos protooncogene, AP-1 transcription factor subunit (FOS), CD44, clusterin (CLU), secreted phosphoprotein 1 (SPP1), and epidermal growth factor (EGF). Pathway analysis showed that upregulated transcripts were enriched in cell cycle/chromosome segregation, organization, immune/inflammatory responses, cell adhesion, cell activation/differentiation, and development terms. In contrast, downregulated transcripts were mainly involved in metabolic processes, transport functions, and oxidation-reduction (147). Network analysis using the Ingenuity Pathway Analysis (IPA) software showed that CCAAT/enhancer binding protein-β (CEBPB) and hepatocyte nuclear factor 4-α (HNF4A) played a central role in the transcriptional regulation of genes implicated in UO or renal fibrosis (147). Higgins et al. (150) identified differentially expressed genes in obstructed kidneys of C57BL/6 mice on days 4 and 10 after UUO, using Affymetrix microarrays. Several profibrogenic genes, whose mRNA levels were upregulated in obstructed kidneys after UUO, were fibroblast-inducible secreted protein, murine homolog of connective tissue growth factor, collagen XVIII alpha1, src-suppressed C-kinase substrate, and secreted protein acidic and rich in cysteine (SPARC) (150). Consistent with this, Gerarduzzi et al. (151) demonstrated that silencing secreted modular calcium-binding protein 2 (SMOC2), which belongs to the SPARC family, attenuated renal fibrosis by inhibiting transforming growth factor-β (TGFβ)-mediated fibroblast-to-myofibroblast transition.
RNA-seq technology has recently provided novel insights into gene expression and regulatory networks. In a mice model of UUO (2 days or 8 days after ligation), RNA-seq analysis revealed change in the expression of thousands of genes (148). Many novel protein-coding genes were identified, including many upregulated genes, such as Fblim1, Epha2, Wisp1, Parvg, Madcam1, Mybpc2, Cdh3, Gpr56, Troap, Pstpip1, Pcdh8, Nlgn2, and Mfap4, potentially associated with renal fibrosis. In addition, many critical transcription factors that could potentially be implicated in renal fibrosis, such as Sox9, Runx1, Uhrf1, and Ezh2, were upregulated. Several other critical transcription factors, including Fosl1, Fosl2, Fos, Jun, JunD, Egr2, Creb5, FoxJ1, Ikzf4, Atf5, Sox4, and Sox11, were also upregulated. p53 (Tp53) is also an important transcriptional coregulator of several TGFβ fibrotic-response genes (31). The contribution of all these transcription factors is still not completely understood with regard to their selective role in UUO-induced fibrosis. Moreover, long noncoding RNAs (lncRNAs) are differentially expressed in the mouse model of UUO, which is likely to affect the expression of fibrosis-related proteins and the cellular phenotype (148). In particular, lncRNAs TCONS_00088786 and TCONS_01496394 were regulated by TGFβ stimulation, and they can affect the expression of fibrosis-related genes through a feedback loop (152). Pavkovic et al. (153) used a comprehensive and combined multiomics data set (mRNAs, proteins, and microRNAs) in UUO mice to understand the molecular pathogenesis of kidney fibrosis. α-Smooth muscle actin (Acta2), collagen (Col1a1), and fibronectin (Fn1), which are fibrotic markers, were constantly increased over time (days 3, 7, and 14 after ligation) in the irreversible UUO model. Kidney injury markers, namely clusterin (Clu), kidney injury molecule 1 (Kim-1), and lipocalin-2 (Ngal), were increased early on without further significant increases over time in the obstructed kidneys. miR-192 was decreased, whereas miR-21 was increased, in obstructed kidneys in the UUO model. Further studies are warranted to elucidate the role of up- or downregulated transcripts in obstructed kidneys, particularly for new therapeutic approaches to renal fibrosis.
7. CELLULAR CHANGES IN OBSTRUCTIVE NEPHROPATHY
Surgical blockade of the urinary flow results in increased hydrostatic pressure, which initially affects the collecting ducts and then rapidly expands to the distal and proximal tubules (32). Long-term UUO causes outer medullary ablation and atrophy of the tubules. Within 14 days of UUO, the proximal tubule mass can decrease up to 65% and epithelial apoptosis/necrosis, infiltration of inflammatory cells, interstitial expansion with increased cell proliferation, as well as subsequent fibrosis are prominent in the cortical compartment of the obstructed kidney (30, 154). In addition, >80% of glomeruli were distinctly transformed after 14 days of UUO and displayed atubular glomeruli, indicating that the glomerulotubular junction is particularly vulnerable to UUO-induced damage (154).
7.1. Role of Glomeruli and Proximal Tubules in Obstructive Nephropathy
During the middle of the twentieth century, the glomerular and tubular epithelium received increased attention as primary targets of progressive renal damage (53, 154, 155). Progressive development of atubular glomeruli apparently plays an important role in obstructive nephropathy. An atubular glomerulus is one that is not connected to its proximal tubule, and this glomerulotubular disconnection is observed in many tubulointerstitial disorders. The presence of atubular glomeruli in diseased kidneys was originally described by Jean Oliver back in the 1930s (156). On the basis of sophisticated microdissection studies, Oliver demonstrated the presence of glomerulotubular disconnection in kidneys from patients with chronic Bright’s disease, which is now described as chronic glomerulonephritis. Subsequently, Marcussen published a series of articles (157–159) in the 1990s highlighting the importance of atubular glomeruli in a variety of patients with CKD due to different forms of renal diseases. He used serial sections rather than microdissection to demonstrate the discontinuity between glomeruli and tubules and reported that the percentage of atubular glomeruli in advanced cases of CKD may reach >35% (158).
The first description of atubular glomeruli in relation to obstructive nephropathy was in dogs subjected to UUO in 1965 in which the glomeruli were perfused but were not able to filter, indicating a glomerulotubular disconnection (160). Moreover, Konda et al. (161) reported an increase in renin-containing cells in the juxtaglomerular apparatus of atubular glomeruli in children with obstructive nephropathy. The presence of atubular glomeruli in fibrotic areas indicated that these areas may contribute to increased production of renin as well as of ANG II, which may promote progressive renal scarring. Forbes et al. (154) used histomorphometry to investigate the formation of atubular glomeruli in adult mice subjected to complete UUO. They showed that destruction of the glomerulotubular junction and formation of atubular glomeruli developed in 80% of nephrons after only 14 days of UUO. The glomerulotubular junction became atrophic because of autophagy and apoptosis along with concurrent remodeling of Bowman’s capsule to form atubular glomeruli. In this deterioration, epithelial cells in the urinary pole underwent transition to a mesenchymal phenotype and expanded to cover the capsule. At the same time, perfusion of the atubular glomeruli was maintained, although renin-positive cells were significantly increased along the afferent arterioles (154).
In line with this, Chevalier (53) highlights the proximal tubules as a primary sensor and effector in the progression of CKD as well as AKI. Because of its high rates of oxygen consumption and relative lack of endogenous antioxidant defenses, the proximal tubule is particularly vulnerable to injury. Interestingly, clinical studies of kidney injury reveal that targeting of the proximal tubule is adequate to induce AKI that can progress to CKD when the injury is repeated, indicating the proximal tubules as the central initiator of AKI-CKD progression. Another factor promoting renal injury in both AKI and CKD is the loss of tubular secretion (162), which can result in the accumulation of endogenous and exogenous waste products, including uremic toxins (163, 164). In CKD patients, elevated serum levels of uremic toxins are associated with an increased risk of adverse events, including cardiovascular mortality (165–167). It is also reported that uremic toxins accumulate and worsen the prognosis in AKI patients (168, 169). Interestingly, it was recently demonstrated that the plasma levels of uremic toxins were increased in 14-day UUO rats and positively correlated with TIF (170). The precise molecular mechanisms underlying uremic toxicity remain to be elucidated; however, it is clear that uremic toxins can act directly on various cell types, including proximal tubule cells (171, 172).
Taken together, these events indicate that both degenerative and regenerative processes are activated in response to the formation of atubular glomeruli following UUO and that the glomerular and tubular epithelium play an important role in progressive renal injury.
7.2. Tubulointerstitial Fibrosis
As mentioned above, a series of events is triggered after UUO, resulting in altered hemodynamics, changes in glomerular filtration, and architectural malformations (e.g., hydronephrosis); therefore, it is difficult to identify the earliest molecular or cellular response that provides the initial trigger for the ensuing fibrosis. Presumably, the increase in tubular pressure and mechanical stress results in tubular epithelial injury and interstitial macrophage infiltration, which in turn causes activation of resident fibroblasts and cellular differentiation into myofibroblasts, which are the main producers of ECM.
Fibrosis is characterized by the excessive deposition of ECM proteins, such as collagens and fibronectin, mainly by activated myofibroblasts. Irrespective of etiology, the fibrosis ultimately results in the loss of organ architecture and function, necessitating renal replacement therapy or transplantation. Most fibrosing renal diseases originate in the tubules, resulting in TIF.
7.3. Tubulointerstitial Fibrosis and the Origin of Myofibroblasts
This section describes the different origins of myofibroblasts, including interstitial resident fibroblasts, bone marrow-derived cells, tubular epithelial cells, endothelial cells, pericytes, and macrophages (FIGURE 6).
Figure 6.

To date, resident fibroblasts and hematopoietic cells migrating into the renal tissue are considered the most important progenitors of collagen-producing myofibroblasts, whereas epithelial and endothelial cells contribute to a lesser extent to the myofibroblast pool. EMT, epithelial-to-mesenchymal transition; EndoMT, endothelial-to-mesenchymal transition; MMT, macrophage-to-myofibroblast transition.
7.3.1. Role of interstitial fibroblasts in TIF.
Until the 1990s, it was generally accepted that myofibroblasts arose from local resident kidney fibroblasts (173, 174). Interstitial resident fibroblasts can be detected by electron and fluorescence microscopy based on strong expression of ecto-5′-nucleotidase (CD73) in their plasma membrane (175–177). Picard and colleagues (178) investigated the alterations in resident fibroblasts upon conversion into myofibroblasts during the first 4 days of UUO. Using double immunofluorescence, they observed the expression of the myofibroblast marker α-smooth muscle actin (αSMA) in ecto-5′-nucleotidase-positive cells already on the first day after UUO, and this coexpression became progressively more frequent up to day 4. Thus, it appears that during the first days of UUO the resident fibroblasts can acquire the phenotype of myofibroblasts, indicating that this phenotypic alteration of resident fibroblasts can occur at a very early stage after UUO. Certain subpopulations of resident fibroblasts can produce erythropoietin (EPO) (179). Asada and coworkers (180) demonstrated that EPO-producing resident fibroblasts from kidneys in 14-day UUO mice can transdifferentiate into αSMA-positive myofibroblasts, in the same way as other resident fibroblasts in renal tissue, at the cost of EPO production. Reversal of the UUO-induced injury restored the production of EPO and the physiological phenotype of EPO-producing cells, indicating that myofibroblasts maintain reversibility in response to improvement of the microenvironment (181). Moreover, LeBleu and colleagues (182) performed a comprehensive study in 2013 using >20 different genetically engineered mouse lines to address the question of myofibroblast linage in the UUO fibrosis model at days 2, 5, and 10 after UUO. They demonstrated that accumulation of myofibroblasts occurred predominantly from two primary sources: ∼50% were derived by local proliferation of resident interstitial fibroblasts, and ∼35% were derived from bone marrow cells without any evidence of proliferation in the kidney tissue (FIGURE 6).
7.3.2. Role of bone marrow-derived cells and macrophages in TIF.
Bone marrow-derived macrophages and monocytes play an important role in the pathophysiological processes following UUO. When describing these processes, the macrophage population is generally divided into two distinct groups with different functions, namely M1 and M2 macrophages. The classically activated M1 macrophages are regarded as proinflammatory, and alternatively activated M2 macrophages are considered profibrotic. Although this is a gross oversimplification, as delineated below, we use the same classification here. After obstruction, circulating classical monocytes (CD14++CD16− in humans and Ly6ChiCCR2hiCX3CR1lo in mice) (183) are recruited to the injured kidney, where they proliferate and acquire a proinflammatory M1 phenotype. Various chemokines, including C-C motif ligand (CCL)-2 (184), CCL5 (184), spleen tyrosine kinase (185), and osteopontin (186), are involved in the recruitment phase. Rovin and colleagues (187) demonstrated that macrophage infiltration peaks between 4 and 12 h after obstruction. M1 macrophages are predominant in the kidney up to day 3 after UUO, whereafter M2 macrophages become the dominant type (188). M1 polarization is induced by cytokines and danger signals, such as interferon (IFN)-γ, tumor necrosis factor (TNF)α, and granulocyte macrophage-colony stimulating factor (GM-CSF), present in the microenvironment of injured renal tissue (189). These M1 macrophages are procoagulant and produce cytokines (e.g., IL-1β, TNFα, and IL-6), oxygen radicals, and proteolytic enzymes [e.g., matrix metalloproteinase (MMP)-12] that not only enhance the inflammatory response but can also cause renal injury (190). The subsequent transition to an M2 phenotype is partly driven by tissue-type plasminogen activators (191). M2 macrophages (F4/80+CD206+) contribute to fibrosis by secreting profibrotic factors such as TGFβ (192). In addition, some older studies in which macrophage subpopulations were not differentiated showed that macrophages can promote fibrosis by expressing other factors, such as platelet-derived growth factor (PDGF)-C (193), galectin-3 (194), and MMP-9 (186).
As stated above, in vivo the macrophage population that is involved in UUO-induced renal injury is extremely heterogeneous. Using single-cell RNA sequencing (scRNAseq), Conway and colleagues (43) could identify 12 clusters of myeloid cells in a release of 7-day UUO (R-UUO) model, which were further divided into monocytes, macrophages, or dendritic cells (DCs). Monocytes could be classified as patrolling monocytes, proinflammatory Ly6C+ monocytes, and profibrotic Arg1+ monocytes. Interestingly, the latter were exclusively present on day 2 after UUO. Macrophages can be further classified as quiescent resident macrophages, Mrc1+ cells, which are closely aligned to embryonic macrophages; Ccr2+ macrophages, which are most likely derived from infiltrating Ly6C+ monocytes; IFN-induced macrophages; and Mmp12+ macrophages. Interestingly, the Mmp12+ macrophages represent a reparative phenotype, and these macrophages were solely present in kidneys that had undergone R-UUO. DCs could be categorized as type 1 and type 2 conventional DCs and lymph node DCs. Thus, it is clear that the population of immune cells involved in UUO comprises an extremely (functionally) diverse group of cells.
Moreover, bone marrow-derived circulating myofibroblast progenitors, either fibrocytes or M2 macrophages, can also contribute to renal fibrogenesis, but it is still unclear as to how much each of these cells contributes to the fibrosis. Fibrocyte-derived cells within the pool of collagen-producing cells have been estimated to range from nearly 0% to 50%, depending on the method used to identify these cells (182, 195–200). Additionally, it is unclear how many of the profibrotic M2 macrophages actually differentiate into collagen-producing myofibroblasts via a process termed “macrophage-to-myofibroblast transition” (MMT). Meng and colleagues (201) reported that in UUO mice as much as 65% of total αSMA+ myofibroblasts arose from MMT. Conversely, more recent studies have shown that only ∼10% of cells are F4/80+/αSMA+ (202–204), suggesting a modest contribution to the process of fibrosis. Importantly, in human fibrotic kidneys, only 0.01% of immune cells expressed both PDGFRβ and COL1α1 (205).
7.3.3. Role of epithelial cells in TIF.
For decades, it was widely believed that EMT plays a key role in renal fibrogenesis. EMT is a process via which polarized epithelial cells acquire a mesenchymal phenotype. This phenotypical switch is characterized by the downregulation of epithelial markers, such as E-cadherin and zonula occludens-1, and upregulation of mesenchymal markers, such as αSMA and vimentin (FIGURE 6). These changes result in the cells exhibiting an enhanced capacity to migrate, resist apoptosis, and produce ECM components (206, 207). In 1995, Strutz et al. (208) postulated that during fibrogenesis the parenchymal epithelium might, at least in part, be converted into fibroblasts at the site of injury. This observation greatly stimulated research into the role of EMT in renal fibrosis; however, because of the absence of proper cellular markers, EMT remained a controversial topic (209). To overcome this hurdle, several research teams performed genetic fate-mapping studies to identify the progenitor cells of myofibroblasts in rodents subjected to UUO, and they demonstrated that only a minor fraction of collagen-producing myofibroblasts arose through EMT (182, 199, 210, 211). These findings were recently corroborated by Kuppe and coworkers. Using single-cell RNA sequencing (scRNAseq), they demonstrated that in human fibrotic kidneys only 0.28% of epithelial cells express both PDGFRβ and COL1α1, indicating that dedifferentiated proximal tubule cells hardly contribute to ECM production (205).
Although the majority of epithelial cells do not become ECM-producing fibroblasts, they do dedifferentiate upon injury and contribute to the fibrotic process. Recently, Wu and colleagues reported that after 14 days of UUO two distinct epithelial cell populations could be distinguished by scRNAseq: one was characterized by a strong proliferative gene signature next to increased expression of injury markers, such as Kim-1 (212), and the other population, consisting of dedifferentiated proximal tubule cells, was characterized by a strong cell movement genotype. Notably, these cells seem to be a source of proinflammatory cytokines, including CCL2 and IL-34 (212). In line with this observation, injured proximal tubule cells were reported to represent one of the main cellular sources of TGFβ signaling ligands to pericytes, fibroblasts, and myofibroblasts (205). These nonproliferative dedifferentiated epithelial cells also undergo metabolic reprogramming, as illustrated by the downregulation of genes related to solute transport, fatty acid metabolism, and β-oxidation (213). In particular, the loss of fatty acid oxidation appears to play a central role in renal fibrogenesis (214).
7.3.4. Role of endothelial cells in TIF.
Endothelial-to-mesenchymal transition (EndoMT) is a process similar to EMT, wherein endothelial cells can undergo mesenchymal transition to form myofibroblasts (215–217) (FIGURE 6). In 2008, Zeisberg et al. (218) reported the contribution of EndoMT to renal fibrosis in mice subjected to 7-day UUO. They found that a considerable percentage (around 25–35%) of (myo)fibroblasts coexpress the endothelial marker CD31 and markers of fibroblast and myofibroblasts, such as FSP-1 and αSMA. Using an endothelial lineage-traceable transgenic mouse line (Tier2-Cre; R26R-stop-EYFP), they showed that endothelial cells labeled with yellow fluorescent protein (YFP) coexpressed FSP1 and αSMA, suggesting an endothelial origin of these fibroblasts. LeBleu et al. (182) demonstrated that ∼10% of myofibroblasts coexpressed the markers of endothelial cells and activated fibroblasts in lineage-tagged transgenic mice after 10-day UUO. Thus, these studies indicate that EndoMT may account for a substantial portion of myofibroblasts in UUO mice. However, in human fibrotic kidneys, only 0.16% of endothelial cells were shown to express both PDGFRβ and COL1α1 (205).
Recently, it was suggested that the soluble proteoglycan endothelial cell-specific molecule 1 (ESM1) may serve as an EndoMT marker of progression of renal fibrosis at day 7 and day 14 after UUO (219). In addition, Zhao and colleagues (220) showed that MMP-9, a major proteolytic enzyme, contributed to UUO-induced fibrosis via EndoMT. Deficiency of MMP-9 contributed to a 30% reduction in total αSMA-positive myofibroblasts arising from EndoMT of peritubular endothelial cells. However, these results need confirmation using lineage-tracing techniques. Although these studies provide some evidence of EndoMT in vivo, the essential role of EndoMT in renal fibrogenesis remains highly controversial.
7.3.5. Role of pericytes in TIF.
Pericytes, which are ECM-producing cells localized in the subendothelial region, have been suggested to be another source of myofibroblasts (FIGURE 6). Lin et al. (217) and Humphreys et al. (210) identified pericytes and perivascular fibroblasts as myofibroblast precursors in the UUO model of kidney fibrosis. Kramann et al. (221) identified a group of Gli1-positive perivascular mesenchymal stem cell-like pericytes as myofibroblast progenitors using lineage tracing and cell ablation. Furthermore, using transgenic Gli-CreERt2;tdTomato 10-day UUO mice, they showed that Gli1-positive cells proliferate within the interstitial area and express αSMA, indicating perivascular Gli1+ mesenchymal-like cells to be a major cellular origin of renal fibrosis. In contrast, LeBleu et al. (182) questioned the specific contribution of pericytes to the myofibroblast population in the UUO model. Using genetic pericyte tagging approaches, they observed that most pericytes did not express αSMA. Moreover, they showed that specific deletion of pericytes neither changed the recruitment of myofibroblasts nor improved UUO-induced fibrosis. Based on these studies, LeBleu and coworkers concluded that pericytes have a negligible role as a source of myofibroblasts. Thus, additional studies are essential before the controversial role of pericytes as myofibroblast precursors is resolved.
In conclusion, the current literature strongly supports the notion that myofibroblasts arise from a variety of cellular sources. Based on the latest research, ∼50% of myofibroblasts are supposed to be derived from resident fibroblasts, 35% from bone marrow-derived cells and macrophages, 5% from epithelial cells, and 10% from endothelial cells. However, these numbers need to be interpreted with caution. To date, resident fibroblasts and hematopoietic cells migrating into the renal tissue are considered the most important progenitors of myofibroblasts, and it is clear that the cellular composition of the kidney markedly changes during renal fibrosis (FIGURE 7). More insights can be obtained with the UUO model, and the implementation of advanced genetic analysis techniques, such as spatial transcriptomics, will hopefully provide an even better understanding of the origins of myofibroblasts in renal fibrosis.
Figure 7.

Time lapse of the cellular composition of the kidney during unilateral ureteral obstruction (UUO)-induced fibrogenesis. Before injury, the kidney mainly consists of fibroblasts, epithelial cells, and endothelial cells. Directly after UUO, the number of healthy cells starts to decline and there is a marked influx of proinflammatory M1 macrophages. Upon persistent injury, the resulting fibrotic lesions mainly consist of collagen-producing myofibroblasts and profibrotic M2 macrophages.
7.4. The Distal Nephron—a Lesser-Known Player in TIF
The UUO model has primarily focused on the role of the proximal tubule in TIF and on late time points when the interstitial and fibrotic responses predominate (31, 32). However, the distal nephron, including the collecting duct, does significantly contribute to and modulate the pathophysiology and progression of renal injury following UUO. Hiatt and coworkers (222) investigated time points ranging from 1 to 14 days of UUO and demonstrated that obstruction of the urinary tract caused a two- to threefold increase in tubular dilation as well as a sixfold increase in accumulation of αSMA-positive myofibroblasts in the vicinity of the distal nephron. Myofibroblast accumulation was reported to occur rapidly in both the outer medullary region and, to a smaller extent, the cortical region. They also observed that the structural and cellular composition of the collecting duct was changed in response to obstruction. The number of AQP2-expressing principal cells and V-type proton ATPase (vATPase)-positive intercalated cells decreased by 65% and 75%, respectively, along with the disruption of E-cadherin localization. Notably, these features are also found in the distal and connecting tubules, indicating that the distal nephron is a major target of UUO-induced injury.
Several studies have demonstrated that collecting duct principal cells play a pivotal role in the development of tubulointerstitial fibrosis (223–225). Using a fetal monkey model in which UUO was performed at 70 days of gestation, Butt and coworkers (223) demonstrated that an abundance of collecting duct (CD) cells coexpress the intercalated cell marker carbonic anhydrase II and αSMA and these cells migrate through the basement membrane, suggesting a CD EMT, and thereby contribute to the development of progressive tubulointerstitial fibrosis. In other studies, tubulointerstitial inflammation and fibrosis were induced by collecting duct-specific disruption of β1-integrin (226) as well as of the E3 ubiquitin ligase Mib1, which is required for Notch signaling (227). In a recent study, specific inactivation of histone H3 K79 methyltransferase Dot1 in connecting tubules and collecting ducts facilitated the development of kidney fibrosis by increasing endothelin-1 levels in 14-day UUO mice (228). Moreover, it was revealed that kidney fibrosis after UUO is epigenetically regulated by Dot1 action in the connecting tubule and collecting ducts.
Taken together, the distal nephron contributes markedly to UUO-induced injury, indicating that the pathophysiology of obstructive nephropathy is complex and likely involves the entire nephron. This suggests that nephron segment-specific fibrotic factors might need to be taken into consideration in the preparation of targeted therapies.
8. MAJOR FACTORS DRIVING UUO-INDUCED DAMAGE AND POTENTIAL THERAPEUTICS
The molecular processes driving kidney fibrosis are complex and have far-reaching implications for the impairment of kidney function. In this section, we provide an overview of the most important signaling pathways and interactions related to UUO-induced fibrosis (summarized in FIGURE 8). Moreover, in FIGURE 9 we showcase potential therapeutic targets for preventing CKD progression supported by knowledge obtained from experimental UUO studies and delineate, based on these targets, which drugs are currently in the drug development pipeline or are already approved for the treatment of CKD (FIGURE 11). Details of the clinical trials are provided in TABLE 1.
Figure 8.

Schematic overview of the main signaling pathways involved in unilateral ureteral obstruction (UUO)-induced fibrogenesis. Binding of the various factors to their specific receptors mostly activates profibrotic signaling pathways, resulting in the formation and deposition of extracellular matrix (ECM) proteins; however, bone morphogenetic protein 7 (BMP7) is regarded as a protein that counteracts the profibrotic effects of transforming growth factor beta (TGFβ). In addition, prostaglandins (PGs) can be either pro- or antifibrotic depending on which receptor is activated. ANG II, angiotensin II; ATR1/2, angiotensin II receptor type 1 and type 2; BMPRII, bone morphogenetic protein receptor type II; CCN2, cellular communication network factor 2; DAMPs, damage-associated molecular patterns; IL-1R, interleukin-1 receptor; MAPK, mitogen-activated protein kinase; miR, microRNA; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOX, nicotinamide adenine dinucleotide phosphate oxidases; PDGF, platelet-derived growth factor; PG-R, prostaglandin receptors; RHO/ROCK, Rho/Rho-associated protein kinase; ROS, reactive oxygen species; RTKs, receptor tyrosine kinases; TNFR, tumor necrosis factor alpha receptor; TNFα, tumor necrosis factor alpha; TβRII, transforming growth factor beta receptor II.
Figure 9.

Overview of potential therapeutic targets for chronic kidney disease based on experimental unilateral ureteral obstruction (UUO) studies. In general, anti-inflammatory therapies are targeted against key cytokines such as interleukin 1 (IL-1) and tumor necrosis factor (TNF)α. Vascular targets are mainly associated with reducing hypertension-induced renal injury. Renal fibrosis can be mitigated by inhibition of the transforming growth factor beta (TGFβ) pathway, hampering integrin signaling, or reducing the number of collagen-producing myofibroblasts. ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blockers; BMP7, bone morphogenetic protein 7; CAR T cells, chimeric antigen receptor T cells; CCN2, cellular communication network factor 2; DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; ETA, endothelin receptor type A; GLP-1, glucagon-like peptide 1; RAAS, renin-angiotensin-aldosterone system; SGLT2, sodium-glucose cotransporter 2.
Figure 11.

Overview of the current drug development pipeline for chronic kidney disease containing drugs that target pathways that have been shown to prevent renal injury in experimental models of unilateral ureteral obstruction (UUO). ACE, angiotensin-converting enzyme; AT1, angiotensin II receptor type 1; CCN2, cellular communication network factor 2; GLP-1, glucagon-like peptide 1; IL-1, interleukin 1; SGLT2, sodium-glucose cotransporter 2; TGFβ, transforming growth factor beta; TNFα, tumor necrosis factor alpha.
Table 1.
Characteristics of clinical trials pertaining to selected chronic kidney disease targets
| Target | Drug | Phase | Condition | Clinical Trial | Status | Outcome | Ref. |
|---|---|---|---|---|---|---|---|
| TGF-β | LY2382770 | II | DKD | NCT01113801 | Terminated | No change in serum creatinine from baseline to end of treatment | (229) |
| Fresolimumab | II | FSG | NCT01665391 | Completed | The study was underpowered and did not meet the primary or secondary end points: remission in urinary protein-to- creatinine ratio, reduction in proteinuria, change in GFR. | (230) | |
| Pirfenidone | II | CKD | NCT04258397 | Recruiting (estimated study completion date: December 2024) | |||
| αv Integrins | STX-100 | II | CAD | NCT00878761 | Withdrawn | Withdrawn | |
| GCS-100 | II | CKD | NCT01843790 | Completed | Pending | ||
| CCN2 | FG-3019 | II | DKD | NCT00913393 | Terminated | Terminated early for business purposes | |
| IL-1 | Anakinra | II/III | CKD* | NCT02578394 | Completed | Pending | (231) |
| Rilonacept | II/IV | CKD | NCT00897715; NCT01663103 | Completed | Treatment reduced systemic inflammation. | (232) | |
| TNFα | Adalimumab | II | FSG | NCT00814255 | Completed | The study was underpowered and did not meet the primary end point: reduction in proteinuria and stable GFR. | (233) |
| Etanercept | II | ESRD | NCT00293202 | Terminated | Unable to recruit sufficient patients | (234) | |
| Prostacyclin | TRK-100STP | II/III | CKD | NCT01090037 | Completed | No significant effect on the renal composite end point: doubling of serum creatinine or occurrence of ESRD | (235) |
| Endothelin | Atrasentan | III | DKD | NCT01858532 | Terminated | Increased risk of heart failure hospitalization | (236,237) |
| Avosentan | III | DKD | NCT00120328 | Terminated | Induced significant fluid overload and congestive heart failure | (238) | |
| GLP-1 | Semaglutide | III | CKD | NCT04865770 | Recruiting (estimated study completion date: July 2024) | ||
| Dulaglutide | IV | DKD | NCT05218915 | Recruiting (estimated study completion date: January 2025) | |||
| Exenatide | IV | DKD | NCT02690883 | Completed | Significant reduction of albuminuria | (239) | |
| SGLT2 | Canagliflozin | IV | CKD | NCT05309785 | Recruiting (estimated study completion date: February 2025) | ||
| Dapagliflozin | III | CKD | NCT03036150 | Completed (stopped early because of efficacy) | Significant effect on the composite end point: decline in GFR, reaching ESRD or death from renal or cardiovascular causes | (240) | |
| Empagliflozin | III | CKD | NCT03594110 | Completed (stopped early because of efficacy) | Significant effect on the primary end point: first occurrence of progression of kidney disease or death from cardiovascular causes | (241) |
CAD, chronic allograft dysfunction; CCN2, cellular communication network factor 2; CKD, chronic kidney disease; DKD, diabetic kidney disease; ESRD, end-stage renal disease; FSG, focal segmental glomerulosclerosis; GFR, glomerular filtration rate; GLP-1, glucagon-like peptide 1; IL-1, interleukin-1; SGLT2, sodium-glucose cotransporter 2; TGFβ, transforming growth factor beta; TNFα, tumor necrosis factor alpha; *As treatment for acute gout attacks.
8.1. Roles of TGFβ and BMP-7 in Obstructive Nephropathy
The TGFβ cascade plays a major role in tubulointerstitial inflammatory and fibrotic responses to experimental UUO (31, 242). The expression of Tgfb1 mRNA increases in several nephron segments after 1 day of UUO, followed by upregulation of the protein (243–247). TGFβ transcripts are more abundant in the injured tubular epithelium and, to a lesser degree, in the fraction of resident and infiltrative macrophages (245, 248). Moreover, urinary TGFβ levels are increased in patients with ureteropelvic junction obstruction (249–251), and changes in these levels over time are associated with similar changes in the grade of hydronephrosis (252, 253), indicating that urine TGFβ can be a useful noninvasive tool for diagnosis of upper urinary tract obstruction in children.
TGFβ is a potent stimulator of the synthesis of ECM proteins and plays a central role in transdifferentiation of myofibroblasts (254–256). Blocking TGFβ with inhibitors, neutralizing antibodies, antisense oligonucleotides, or synthetic oligodeoxynucleotides (41, 257–261) can attenuate renal fibrosis in UUO models. The TGFβ signaling cascade involves phosphorylation of Smad2 and Smad3, which serve as downstream effectors, primarily by modulating target gene expression (256, 262) (FIGURE 8). Smad2 has a probable renoprotective role (263), whereas Smad3 is verified to be pathogenic because its targeted deletion (264) and deacetylation (265), or specific inhibition of TGFβ/Smad3 signaling (266), prevents UUO-induced fibrosis, suggesting that Smad3 may be a therapeutic target for renal damage related to obstructive nephropathy.
Bone morphogenetic protein-7 (BMP-7), formerly called osteogenic protein-1/OP-1, is a member of the TGFβ superfamily and is regarded as a protein that counteracts the profibrotic effects of TGFβ (262, 267). Moreover, BMP-7 can increase ECM degradation as well as reduce ECM formation by inactivating matrix-producing cells (267). Under disease conditions, BMP-7 expression is significantly downregulated, as detected in obstructive nephropathy (268, 269), and introduction of exogenous recombinant BMP-7 or reactivation of endogenous BMP-7 signaling pathways protects against UUO-induced fibrosis (47, 268, 270–273).
The TGFβ superfamily involves multiple signaling cascades and can interact with numerous other cell pathways, the details of which are too complex to review here and have been reviewed in detail previously (31, 256, 262). Taken together, the regulation of TGFβ-induced renal fibrosis is complex.
8.1.1. TGFβ as therapeutic target in CKD.
Substantial preclinical as well as clinical evidence indicates that TGFβ plays a crucial role in promoting renal fibrosis, suggesting TGFβ to be a major therapeutic target in CKD patients (FIGURE 9). However, because TGFβ regulates many biological functions in normal human physiology as well as in pathological conditions, developing successful therapies that interrupt the TGFβ pathway has been very challenging. Global blockade of TGFβ can have deleterious effects, especially in CKD, where long-term treatment is required (274). Nevertheless, several clinical trials have been conducted to evaluate the effect of pharmacological blockade of TGFβ in fibrotic kidney disease with variable success. A multicenter phase II clinical trial using a humanized neutralizing antibody against TGFβ (LY2382770) in diabetic kidney disease showed no clinical benefits (229). A subsequent phase II multicenter, double-blind, placebo-controlled randomized study of another neutralizing antibody (fresolimumab) against all three TGFβ isoforms was evaluated in patients with primary focal segmental glomerulosclerosis (FSGS). Although fresolimumab was well tolerated, no statistical difference in estimated GFR (eGFR) was observed between the groups (230). A single-center pilot study showed that oral administration of pirfenidone, a small-molecule compound inhibiting TGFβ synthesis, significantly slowed the decline in renal function in patients with FSGS (275). Moreover, another phase I/II randomized placebo-controlled trial in patients with established diabetic nephropathy showed promising results for pirfenidone in improving kidney function, but without improving proteinuria (276). Interestingly, another phase II randomized, double-blind, placebo-controlled study on pirfenidone is planned for patients with CKD to evaluate whether pirfenidone can prevent CKD progression. This study will be completed in December 2024 (ClinicalTrials.gov: NCT04258397). However, despite promising results in preclinical studies, therapeutic interventions targeting TGFβ in humans have been disappointing. Further research is needed on the pathways that trigger TGFβ-induced fibrosis to identify alternative therapeutic strategies for mitigating kidney fibrosis in CKD.
8.2. Role of Integrins in Obstructive Nephropathy
Integrins are a large family of transmembrane cell adhesion and signaling receptors, consisting of α- and β-subunits that are able to connect the cytoskeleton with the ECM (FIGURE 8). In mammals, 18 α- and 8 β-subunits have been identified, forming a total of 24 different integrin heterodimers (277). Several studies have demonstrated that integrins, specifically αv (alpha-vee)-containing integrins, play an important role in controlling the activation of TGFβ. Myofibroblasts express the αv-containing integrins (αvβ1, αvβ3, αvβ5, αvβ6, αvβ8), which can recognize the amino acid sequence Arg-Gly-Asp (RGD), allowing them to bind to and activate latent TGFβ (reviewed in Refs. 278, 279). In line with this, Ma and coworkers (280) showed that disruption of αvβ6-mediated TGFβ activation could protect against tubulointerstitial fibrosis in mice after 14-day UUO. Likewise, inhibition of integrin αvβ1 or integrin β3 ameliorated UUO-induced fibrosis (281, 282). Moreover, in a recent study, an integrin αvβ3-based drug delivery system that targets myofibroblasts was constructed and it was shown that specific targeting of αvβ3-positive myofibroblasts alleviated UUO-induced fibrosis (283).
Integrin-linked kinase (ILK) also plays a role in the development of kidney fibrosis. ILK is an intracellular serine/threonine protein kinase that interacts with the cytoplasmic domains of the β-integrin subunit, resulting in integrin-mediated signaling (284). ILK expression is increased in response to UUO in a time-dependent manner (285), and its overexpression aggravates renal fibrosis (286). Li and coworkers (287) demonstrated that inhibition of ILK activity attenuates ECM production and myofibroblast activation in UUO-induced fibrosis.
Overall, these studies indicate that the integrin signaling pathways play a critical role in the development and progression of kidney fibrosis and might represent novel targets for antifibrotic therapies (FIGURE 9). However, the complexity of integrin biology, with multiple integrin subtypes involved in kidney fibrosis, suggests that a small-molecule integrin modulator targeting several integrin subtypes might be an ideal antifibrotic therapy. Interestingly, Zhou and coworkers (288) demonstrated that MK-0429, an equipotent pan-inhibitor of multiple αv integrins, reduces proteinuria, kidney fibrosis, and collagen accumulation in a rat model of diabetic nephropathy. This pan-αv inhibitor has recently been patented by Merck for CKD treatment (289).
8.3. Role of Growth Factors in Obstructive Nephropathy
8.3.1. Platelet-derived growth factor.
The platelet-derived growth factor (PDGF) family consists of five dimeric glycoproteins (PDGF-AA, -AB, -BB, -CC, and -DD), which signal via either receptor PDGFRα or PDGFRβ (FIGURE 8). These two receptors are expressed by mesenchymal cells in all organs, and their activation drives proliferation, migration, and production of ECM, reflecting a key role of PDGF in numerous (patho)physiological processes, including fibrosis of the lung, kidney, liver, and heart (290–292). With regard to UUO-induced renal fibrosis, Sommer and colleagues (293) used Northern blot analysis to demonstrate that Pdgfb mRNA levels slowly increased after obstruction and reached the highest levels on days 10 and 15. In contrast, expression of Pdgfrb remained elevated for 25 days (293). Taneda and coworkers (294) used immunohistochemistry to evaluate the expression profile of PDGF ligands and receptors in UUO kidneys. They observed in mice that PDGF-A was expressed in vascular smooth muscle cells and some infiltrating cells in UUO kidneys but not in cortical interstitial cells. On day 4 after UUO, PDGF-B was observed in proximal and distal tubules as well as in interstitial cells; in the latter, PDGF-B was still present on day 14. In healthy kidneys, PDGF-C was observed in various endothelial cells, and its expression profile did not change after UUO; however, in another study PDGF-C was highly expressed in infiltrating macrophages in UUO kidneys (193). PDGF-D was present in interstitial cells on day 4 after UUO, especially around injured tubules, and expression levels were even higher on day 14. In another study on fibrotic kidneys, dilated tubules coexpressed PDGF-D and AQP1, indicating that proximal tubules and/or descending limbs of Henle’s loop can express PDGF-D de novo upon injury (295). Furthermore, PDGF-D is dispensable for normal renal development and physiological function and may represent an attractive therapeutic target in CKD.
In UUO mice, PDGF receptors PDGFRα and PDGFRβ are mainly expressed in interstitial cells from day 4 (294). Chen and colleagues (296) reported a time-dependent increase in PDGF isoforms and its receptors after UUO as well as in the phosphorylation of PDGFRα and PDGFRβ. Moreover, using collagen 1-green fluorescent protein reporter mice, they demonstrated that PDGFRα and PDGFRβ were mainly expressed in pericytes and interstitial myofibroblasts in both healthy and UUO kidneys (296). Despite the clear role of PDGF signaling in fibrogenesis, it has been difficult to utilize this pathway as a therapeutic target, especially in clinical settings. Therefore, direct targeting of the PDGF pathway might not be the therapeutic way forward; however, the distinct, cell-specific expression profile of PDGF receptors opens other possibilities as described by Poosti and coworkers (297), who used PDGFRβ as a docking receptor for myofibroblast-specific delivery of IFNγ to mitigate UUO-induced fibrosis.
8.3.2. Connective tissue growth factor/cellular communication network factor 2.
In 2018, the HUGO Gene Nomenclature Committee decided that the official name for connective tissue growth factor (CTGF) would become cellular communication network factor 2 (CCN2), partly based on the fact that the old name was misleading considering that the gene does not encode a growth factor (298). Historically, CCN2 was regarded as a growth factor and a proinflammatory cytokine; however, it is now clear that CCN2 is a matricellular protein involved in various cellular functions, including matrix remodeling (299) (FIGURE 8). The multiplicity of CCN2 functions owes to its complex protein structure. CCN2 consists of four functional domains, viz. module I, homologous to insulin-like growth factor binding protein; module II, a von Willebrand factor type-C repeat; module III, a thrombospondin type-1 repeat; and module IV, a cysteine knot-containing domain (CT), with a “hinge” region between the second and third domains (300). Based on its pericellular localization and complex structure, it is believed that CCN2 can regulate cell functions through interactions with diverse receptors and macromolecules in various extracellular and intracellular compartments. Moreover, the complete protein can be cleaved by proteases, such as MMP-2, resulting in the release of various biologically active fragments (301, 302). CCN2 expression is associated with a variety of fibrotic diseases, including renal fibrosis (303–305). CCN2 can stimulate fibrogenesis by promoting the recruitment of inflammatory cells, stimulating fibroblast proliferation and activation, and enhancing ECM production. These profibrotic effects are mediated by interaction with, among others, TGFβ, BMPs, MMPs, integrins (e.g., αvβ3, αvβ5), Wnt, and fibronectin (304, 305). In 1998, Ito and coworkers (306) reported that CCN2 was highly expressed in fibrotic lesions and at sites of chronic tubulointerstitial damage in patients with CKD of various origins. In 2004, the role of CCN2 in UUO-induced renal fibrosis was proven by Yokoi and colleagues (307). Using a CCN2 antisense oligonucleotide, they demonstrated that CCN2 blockade mitigated UUO-induced fibronectin and collagen 1 expression and deposition without impacting the proliferation of tubular and interstitial cells (307). In 2010, a phase 1 trial showed that treatment of patients with microalbuminuric diabetic kidney disease with a monoclonal antibody to CCN2 reduced albuminuria (308). Since then, a variety of therapeutics targeting CCN2 have been developed (300); however, unfortunately, no clinical trials are ongoing for CCN2 and CKD.
8.4. Role of Cytokines in Obstructive Nephropathy
8.4.1. Damage-associated molecular patterns.
UUO causes sterile inflammation, which is an inflammatory response to physical, chemical, or metabolic noxious stimuli, in the absence of pathogens, characterized by the release of damage-associated molecular patterns (DAMPs) (309). Several DAMPs, including high-mobility group box 1, IL-1α, IL-33, soluble uric acid, extracellular ATP (eATP), and S100A8/A9, are involved in UUO-induced fibrosis (310–314). These DAMPs interact with various receptors, such as toll-like receptors, the IL-1 receptor family, and P2 purinergic receptors (314–316), and receptor binding results in inflammasome assembly, caspase activation, and maturation of IL-1β and IL-18 and promotes fibrogenesis (314, 317, 318) (FIGURE 8 AND FIGURE 10). Considering the central role of the IL-1 family of cytokines in renal fibrosis, they are potentially interesting therapeutic targets (FIGURE 9). Several clinical trials have been conducted in CKD patients using anakinra, a nonglycosylated form of the naturally occurring human IL-1 receptor antagonist (IL-1Ra). At the time of writing, one trial (NCT04844814) was about to start, one was withdrawn because of failure in enrolling patients (NCT03062176), and four others were completed [NCT03141983, NCT02578394 (231), NCT02278562, and NCT00420290]. The completed trials showed that administration of anakinra to maintenance hemodialysis patients for 4 wk successfully controlled the inflammatory response, as evidenced by a significant reduction in C-reactive protein and IL-6 levels (319). In another two-site, double-blind trial (NCT01663103 and NCT00897715), the effect of an IL-1 trap, rilonacept, was tested in CKD patients, and it was found to reduce systemic inflammation (232). Recently, canakinumab, a human monoclonal antibody against IL-1β, was shown to mitigate EMT of HK-2 cells (320). However, the efficacy of other (in)direct cytokine inhibitors, such as emricasan, serelaxin, and salidroside, in the treatment of CKD needs to be further evaluated (317). Of note, another cytokine that might be a promising therapeutic target is IL-11 (321). With regard to eATP-mediated purinergic signaling, it has been demonstrated that the P2X7 receptor is involved in UUO-induced inflammation and fibrosis (322), and treatment of UUO rats with brilliant blue G, a P2X7 receptor antagonist, reduced renal inflammation, collagen synthesis, and renal cell apoptosis (323). Noteworthy, several P2X7 receptor inhibitors are already being tested in clinical trials for the treatment of inflammatory diseases (324); however, whether these compounds can be used for the treatment of CKD still needs to be assessed.
Figure 10.

Simplified representation of a signaling network driving renal fibrosis. Factors related to the damage-associated molecular pattern (DAMP), prostanoid, purinergic, and transforming growth factor beta (TGFβ) signaling pathway can stimulate key cellular players in both an autocrine (green arrows) and a paracrine (blue arrows) fashion, thereby creating an autoinductive feedforward loop resulting in perpetual activation of the profibrotic machinery. For instance, autocrine extracellular ATP (eATP) signaling promotes inflammasome activation in macrophages, resulting in the production of interleukin-1 beta (IL-1β), which, in a paracrine manner, activates fibroblasts. As a consequence, fibroblasts increase the expression of integrins that are capable of activating latent, extracellular matrix (ECM)-bound TGFβ. Subsequently, autocrine TGFβ signaling maintains fibroblast activation, and paracrine TGFβ signaling promotes the release of cytokines by epithelial cells resulting in macrophage recruitment. Ado, adenosine; PGs, prostaglandins; TNFα, tumor necrosis factor alpha.
8.4.2. Adenosine.
Extracellular adenosine is an endogenous signaling molecule that regulates inflammatory and fibrotic processes in various organs (325–327). Under normal, normoxic conditions, adenosine is present in low concentrations in the extracellular space of the kidney. However, after hypoxia or renal injury, the levels of extracellular adenosine rise significantly, in part due to cell death-mediated release of ATP into the pericellular space (328). Extracellular adenosine can signal through four different G protein-coupled P1 receptors (A1, A2A, A2B, and A3) that either stimulate (A2A and A2B) or inhibit (A1 and A3) adenylyl cyclase activity and cAMP production in the cell (328, 329). The various receptor subtypes are differentially expressed depending on the cell type: A2A is the predominant receptor subtype on immune cells (330), whereas A2B is the predominant adenosine receptor expressed on isolated renal fibroblasts (327). As a consequence, the receptors also contribute differently to renal fibrogenesis. Using the UUO model, several studies demonstrated that deletion of the A2A receptor increases renal fibrosis (331, 332) whereas activation of the receptor slows down the fibrotic process; however, treatment did not fully protect against UUO-induced injury (331, 333). Conversely, it has been demonstrated that deletion of the A2B receptor attenuates UUO-induced fibrosis (334). Taken together, these studies reveal that the adenosine receptor subtypes should be modulated differently in order to achieve therapeutic efficacy. Along these lines, Pak et al. (335) recently demonstrated that LJ-4459, a newly synthesized dual-acting ligand that acts as an A2A agonist and an A3 antagonist, reduced fibrosis in 6-day UUO mice.
8.4.3. NLRP3 inflammasome.
Inflammasomes, first described in 2002 by Martinon and colleagues (336), are intracellular multiprotein complexes, mainly consisting of a pattern recognition receptor, an apoptosis-associated speck-like protein, and pro-caspase-1, that are known to play an important role in organ fibrosis (337). Classical activation of the inflammasomes, for instance by DAMPs, results in caspase-1-mediated maturation of IL-1β, IL-18, and IL-33, thereby promoting inflammation and fibrogenesis (337). The NLRP3 inflammasome is currently the most studied and characterized inflammasome and well known to contribute to CKD (337–339). In 2010, Vilaysane et al. (340) reported that NLRP3 gene and protein expression increased progressively over a 14-day time course in UUO mice. During that time, they also observed increased levels of activated caspase-1 and mature IL-1β and IL-18 (340). Moreover, they demonstrated that NLRP3 deletion attenuated tubular injury, tubulointerstitial inflammation, and fibrosis following UUO (340). These findings were corroborated by Guo and coworkers (341), who demonstrated that NLRP3 deficiency ameliorates mitochondrial dysfunction and alleviates renal fibrosis in 14-day UUO mice. Conversely, Pulskens and colleagues (342) reported in mice that after 1-day UUO NLRP3 preserved renal integrity and provided protection against early tubular injury and interstitial edema. Despite these conflicting results, the inflammasome–IL-1/IL-18 axis appears to a promising therapeutic target for CKD. Recently, it was demonstrated that gemigliptin, a dipeptidyl peptidase-4 inhibitor, mitigated UUO-induced tubular atrophy and renal fibrosis in mice by downregulating NLRP3 activity (343). As described previously, several drugs that target cytokines downstream of NLRP3 activation, e.g., anakinra and rilonacept, are currently being evaluated for the treatment of CKD; however, selective NLRP3 inhibitors, such as CY-09 and OLT1177 (dapansutrile) (344, 345), are still only undergoing preclinical testing (346, 347).
8.4.4. Tumor necrosis factor-α.
Tumor necrosis factor-α (TNFα) is produced by various cells including macrophages, glomerular mesangial cells, and tubular epithelial cells (348). It acts via two receptors, TNF receptor (TNFR)1 and 2, the activation of which results in the expression of a variety of transcription factors, cytokines (e.g., IL-1β and CCL2), growth factors (e.g., TGFβ), receptors, cell adhesion molecules (e.g., ICAM-1 and VCAM-1), mediators of inflammatory processes, acute phase proteins, and major histocompatibility complex proteins (291, 348) (FIGURE 8). Notably, TNFRs can be shed, and soluble TNFR can mitigate inflammation and fibrosis by neutralizing free TNFα (291). More than 25 years ago, Kaneto et al. (349) demonstrated significant increase in the expression of Tnf mRNA in obstructed kidneys at 1 (×2), 2 (×2.7), 4 (×3.6), 24 (×2.7), 72 (×1.8), and 120 (×2.8) h after UUO compared with that in the contralateral kidney of the same rats. In 1999, Guo and colleagues (350) demonstrated that TNFα and its two receptors are involved in UUO-induced fibrosis. Using knockout (KO) mice, they demonstrated that deletion of TNFR1 markedly reduced the interstitial volume of the 5-day UUO kidney from 33% [wild-type (WT) mice] to 19.4%, whereas that of TNFR2 KO mice was significantly decreased to 25.4% (350). Moreover, they observed a reduction in collagen IV and αSMA matrix scores and reduced nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) activation (350). They postulated that TNFα and ANG II together accounted for ∼70–80% of the pathophysiological changes in obstructive nephropathy (351). Inhibition of TNFα with infliximab, etanercept, or PEG-sTNFR1, a pegylated form of soluble TNFR1, decreased albuminuria and slowed down CKD progression in animal models (352–354); however, no clinical studies have been conducted. The FONT phase II clinical trial (NCT00814255) was designed to assess the efficacy of adalimumab in patients with resistant focal segmental glomerulosclerosis. Unfortunately, recruitment fell short of enrollment goals (233). In a pilot study to evaluate the efficacy of etanercept in chronic hemodialysis patients (NCT00293202), no changes in inflammation markers were observed (234).
8.5. Role of Vasoactive Peptides in Obstructive Nephropathy
8.5.1. Angiotensin II.
As described above, RAS and ANG II have been known for decades to play an important role in the pathogenesis of UO (78, 79). In UUO kidneys, activation of RAS is related to mechanical stretching, leading to inflammation, oxidative stress, and fibrosis (355, 356). ANG II, via AT(1) and AT(2) receptors, activates the NFκB signaling pathways, contributing to increased production of TNFα, IL-6, and MCP-1, which promotes inflammation, apoptosis, and oxidative stress (FIGURE 8). Consequently, blockade of the ANG II system, for example, with ACE inhibitors, AT1 receptor antagonists, and thymoquinone, reduces the expression of NFκB-related proinflammatory genes in UUO kidneys, thereby improving inflammation and oxidative damage (357–360). The interaction between ANG II and TNFα was studied in further details by using a genetic-pharmacological approach with a combination of AT1- and TNFα receptor- (TNFR1 and TNFR2) KO mice and pharmacological manipulation (350, 351). Guo and coworkers subjected double TNFα receptor KO mice to 5-day UUO and then treated them with the ACE inhibitor enalapril to assess the contribution of both systems to the development of renal fibrosis (351). Treatment with enalapril further decreased fibrosis progression in the double TNFα receptor KO mice after 5-day UUO, suggesting an interaction between ANG II and TNFα systems. Moreover, ANG II activates TGFβ signaling, leading to increased production of ECM as well as fibrosis; specific ANG II receptor antagonists or ACE inhibition was reported to ameliorate the UUO-induced fibrosis (361–363). In line with these studies, it has been shown that KO or transgenic mice for the AT1 receptor or angiotensinogen gene develop less TIF than WT mice during UUO (364, 365). In contrast, Nishida and coworkers (366) demonstrated that stimulation of the AT1 receptor can also be beneficial in 14-day UUO. They performed bone marrow transplantation from AT1 receptor KO mice to WT mice and showed that kidneys from mice with AT1-deficient bone marrow display more fibrotic changes and fewer infiltrating macrophages than those from nontransplanted WT mice after 14 days of UUO. Because infiltrating macrophages are assumed to play a critical role in the evolution of renal fibrosis, this study suggests that the AT1 receptors on bone marrow-derived macrophages might promote preservation of the renal parenchymal architecture during development of renal fibrosis in response to UUO.
Activation of RAS and the increase in ANG II levels leads to overproduction of reactive oxygen species (ROS) in UUO kidneys (355). The mitochondria play a central role in controlling ROS production, and in UUO mitochondrial dysfunction is associated with increased generation of ROS leading to oxidative stress. In addition to mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) and endoplasmic reticulum (ER) are important sources of ROS that can activate different signaling pathways, thereby contributing to inflammation and fibrosis (367). ANG II is able to stimulate NOX-derived ROS production via binding to AT1 receptors (368), and in UUO models NOX2 and NOX4 are upregulated after 7 days of UUO (369, 370). Treatment with an ANG II receptor blocker, fimasartan, attenuated the UUO-induced expression of NOX2 and NOX4 but enhanced a nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated antioxidant defense response, which is critical for neutralizing ROS and reducing oxidative stress as well as inflammation and fibrosis (369). Furthermore, in 14-day UUO mice, ANG II upregulation of NOX2 could be attenuated by β-aminoisobutyric acid (BAIBA), a natural thymine catabolite, leading to decreased oxidative stress and fibrosis (371). In line with this, Cheng and coworkers (372) showed that treatment with apocynin, a NOX2/NOX4 inhibitor, reduced oxidative stress and myofibroblast accumulation in 7-day UUO rats. Likewise, ANG II activation of the AT1 receptor increased the levels of NOX subunits, including p22/p47/p67, in 7-day UUO mice, resulting in ROS overproduction, fibrosis, and loss of kidney function (373). Thus, these studies indicate that increased production of ANG II by RAS activation is associated with oxidative stress, promoting renal damage and inflammation, which leads to kidney fibrosis.
It has been proposed that the intrarenal RAS is as important as systemic RAS in terms of renal pathophysiology, including in UO (374–376). Recently, Figueroa and colleagues (374) demonstrated that the intrarenal RAS is activated in the cortex of UUO kidneys as reflected by increased renin expression. Moreover, it has been shown that suppression of intrarenal RAS by angiotensinogen (AGT) antisense RNA prevents UUO-induced TGFβ, fibronectin, and collagen type I expression (377). Together, this suggests that targeting the intrarenal RAS may have significant clinical implications for the treatment of renal fibrosis.
8.5.2. Renin-angiotensin system as a therapeutic target in CKD.
Current therapeutic strategies to delay CKD progression focus primarily on treating the underlying conditions, such as high blood pressure and glucose levels. Clinically, ACE inhibitors and ANG II receptor blockers are therefore cornerstone therapeutics to reduce hypertension, proteinuria, progression of CKD, and cardiovascular risk (378, 379) (FIGURE 9 AND FIGURE 11). Several clinical studies initiated in the late 1980s have shown that blockers of RAS activity can improve kidney function in patients with progressive CKD over the long term (379). In most of these studies, the primary end points were defined as the occurrence of ESRD, doubling of serum creatinine levels, or a combination of both. Moreover, a recent double-blind randomized placebo-controlled trial demonstrated that losartan administration contributes to renal function recovery, as shown by renographic GFR improvement, in anuric and oliguric patients with a solitary obstructed kidney (17). However, the promising preclinical effect demonstrating that RAS blockade can reduce renal inflammation and fibrosis remains to be demonstrated clinically.
8.5.3. The prostaglandin system.
The prostaglandin system is activated in response to obstructive nephropathy. Numerous studies have shown that obstruction of the urinary tract is associated with a marked induction of COX-2, as well as an increase in prostaglandin and thromboxane synthesis (78, 103, 104, 108, 116–118, 380, 381). COX-2 distributes differentially in various regions of the kidneys upon UUO. Compared with the contralateral nonobstructed kidney, cortical COX-2 expression is reduced in the obstructed kidney but is markedly induced in both the outer and inner medulla, indicating that COX-2 may contribute to distinct regional hemodynamic alterations in response to UUO. Conversely, COX-1 expression is identical in the obstructed and contralateral nonobstructed kidneys, highlighting COX-2 as the main player in obstructive nephropathy (116).
There is some controversy regarding the role of COX-2 in renal injury secondary to UO. Inhibition of COX-2 prevented UUO-induced apoptosis and fibrosis (382, 383), indicating that COX-2 has detrimental effects. In line with these findings, we inhibited COX-2 using target-specific gene silencing via RNA interference (RNAi). We designed chitosan/COX-2 siRNA nanoparticles that silenced COX-2 specifically in the macrophages. These nanoparticles were administered via intraperitoneal injection, and the character of these chitosan nanoparticles and the macrophage-rich milieu in the peritoneum aided their proficient uptake by the macrophages (384). These macrophages were then subsequently recruited to the obstructed kidney; in 3-day UUO mice with macrophage-specific knockdown of COX-2, renal inflammation, apoptosis, and fibrosis were diminished (385, 386). Thus, COX-2 inhibition using RNAi may represent a potential therapeutic approach.
Contrary to these findings, Kamata et al. (387) showed that meloxicam, a selective COX-2 inhibitor, aggravated 5-day UUO-induced fibrosis, indicating a protective effect of COX-2-derived prostaglandins against UUO-induced renal damage. Consistent with these findings, we demonstrated higher levels of apoptosis as well as tubular damage in COX-2 KO mice subjected to 3 and 7 days of UUO (388), supporting the protective role of COX-2. The microsomal PGE2 synthase, which is responsible for the production of PGE2, has likewise been shown to exert a protective effect against inflammation and renal fibrosis in 7-day UUO mice (389). This was further supported by studies focusing on the PGE2, EP2, and EP4 receptors (390). PGE2 can bind and activate four different EP receptors, EP1–4. We and others have demonstrated increased expression of EP2 and EP4 receptors in response to 7-day UUO (388, 389, 391). Moreover, we recently demonstrated that stimulation of the EP2 receptor mitigates renal fibrogenesis in 7-day UUO mice and human precision-cut kidney slices (PCKS) (392). PCKS is a unique translation ex vivo model of renal fibrosis that is ideal for studying multicellular processes because cellular diversity and organ architecture are maintained in the slices. The slices are prepared from functional and macroscopically healthy cortical tissue obtained from tumor nephrectomies (393). In addition, Nakagawa and coworkers (394) showed aggravated renal injury in mice lacking the EP4 receptor in response to 7-day UUO. Taken together, these studies confirm that the activation of EP2 and EP4 receptors might also protect against UUO-induced damage, indicating that these receptors may be involved in the cytoprotective effect of COX-2 activation during UUO (FIGURE 8). In contrast, we demonstrated that inhibition of the EP1 receptor diminished fibrosis in 7-day UUO mice and mitigated early- and late-stage renal fibrosis in human PCKS prepared from patients with established renal fibrosis (395), indicating that the EP1 receptor is also a promising target for preventing renal fibrosis.
Prostaglandin I2 (PGI2) or prostacyclin also plays an important role in UUO-induced damage. PGI2 is primarily synthesized in endothelial cells and has a strong vasodilation function. In this regard, it was recently demonstrated that a prostacyclin analog, beraprost sodium, could attenuate renal fibrosis by improving renal microcirculation and suppressing EndoMT progression in mice subjected to 7-day UUO (396). Moreover, ONO-1301, a PGI2 receptor (IP) agonist, mitigated TIF in 7-day UUO mice (397).
Prostaglandin D2 (PGD2) has likewise been demonstrated to affect the development of renal fibrosis. It can interact with two receptors, viz., the DP1 receptor and the DP2/CRTH2 receptor. In a study on renal fibrosis, the PGD2-CRTH2 pathway was found to be involved in the progression of inflammation and TIF (398). Ito and coworkers (399) blocked the activation of CRTH2, using an antagonist or genetic KO, and showed that UUO-induced TIF was ameliorated in CRTH2 KO mice or in mice treated with the CRTH2 antagonist, suggesting that the PGD2-CRTH2 pathway promotes renal fibrosis.
Taken together, these studies reveal a counterbalance between the intrarenal handling of COX-2, that is, the detrimental versus beneficial effects of COX-2 may depend on the cellular expression and localization of COX-2 in the obstructed kidney. This suggests that protective COX-2-mediated endogenous prostaglandin production may take place in resident cells in response to UUO. Hence, the COX-2/prostaglandin signaling pathway is complex and can be regulated at multiple steps. Among these steps, the COXs and prostaglandin synthases are of importance in the production of prostaglandins, which can then bind and activate specific cytoprotective receptors and subsequently reduce renal injury and fibrosis.
8.5.4. The COX/prostaglandin system as a therapeutic target in CKD.
Nonsteroidal anti-inflammatory drugs (NSAIDs), which are nonspecific inhibitors of both COX-1 and COX-2 as well as selective COX-2 inhibitors, are among the most commonly used drugs for treatment of pain, fever, and inflammation. However, the use of NSAIDs is associated with an increased risk of renal and cardiovascular complications, such as compromised GFR, elevated blood pressure, and peripheral edema (400, 401). Gooch and coworkers (402) performed a prospective community-based study of >10,000 elderly patients to determine the association between NSAID use and CKD progression. They showed a linear relationship between a high cumulative NSAID exposure and an increased risk for CKD progression in an elderly community-based cohort. Moreover, Whelton and coworkers (403) reviewed >50 clinical studies involving >13,000 subjects to study the renal safety profile of the selective COX-2 inhibitor celecoxib. They reported that the overall incidence of renal adverse events was greater for celecoxib treatment compared with that for placebo but was similar to that for NSAIDs.
Considering the high adverse effects of NSAIDs on the kidney, an increasing number of preclinical studies are now focusing on evaluating the effect of prostaglandin analogs or receptor agonist/antagonists in CKD animal models. In the future, relevant clinical studies will be crucial to verify the effect of prostaglandin analogs or receptor agonist/antagonist in the treatment of renal fibrosis in CKD patients. A few clinical trials have been conducted. The CASSIOPEIR trial studied the effect of prostacyclin analog beraprost sodium (TRK-100STP) in patients with CKD (either primary glomerular disease or nephrosclerosis). This randomized, double-blind placebo-controlled study, including 892 CKD patients, has been conducted at 160 sites in seven Asia-Pacific countries and regions. Unfortunately, no beneficial effects of TRK-100STP in preventing CKD progression were observed over placebo (235).
8.5.5. Endothelin.
Endothelins (ETs) are a family of three potent vasoactive peptides (ET-1, ET-2, and ET-3). ET-1 is the main isoform in the kidney and plays an important role in the regulation of renal function (404). ET-1 can bind two different receptors, namely ETA and ETB (FIGURE 8). Both receptor subtypes are present in the kidney but have different localization and functions. The ETA receptor is mainly expressed in the renal vessels but has also been observed in the distal tubules and cortical collecting ducts, whereas the ETB receptor is primarily found in vasa recta and inner medullary collecting duct (IMCD) but has also been found in proximal tubules and glomerular cells (405). This differential distribution of the ET receptors in the vascular and tubule system might suggest that the receptors can have differential effects.
ET-1 can promote cell proliferation, inflammation, and fibrosis. Increased ET-1 mRNA levels and plasma levels have been observed in 24-h UUO mice (406, 407). In addition, several studies have reported increased expression of Ednra mRNA in response to 5-day UUO, whereas the level of the Ednrb mRNA expression was not affected by UUO (408–410), suggesting that ET-1 interaction with the ETA receptors might play a role in the progression of interstitial fibrosis in response to UUO. Moreover, treatment with an ETA and ETB receptor antagonist, bosentan, attenuated UUO-induced fibrosis (407) as well as renal dysfunction associated with UUO (86, 406). Cotreatment with bosentan and the AT1 antagonist valsartan had additive protective effects against UUO-induced fibrosis (407). Taken together, these data suggest that inhibition of ET-1 signaling has favorable effects on renal hemodynamics as well as anti-inflammatory and antifibrotic effects in UUO models (FIGURE 9). Interestingly, in a recent study, Neder and colleagues (409) showed increased expression of Edn1 mRNA in the 5-day UUO kidney in endothelial and tubular cells. They also showed that the increased expression of Ednra in the UUO mice mainly occurred in the stromal cell compartment, including renal interstitial fibroblasts. Nevertheless, in a genetic mouse model with constitutive deletion of both endothelin receptors from the stromal cell compartment, there was no change in the UUO-induced expression of proinflammatory and profibrotic markers (409), suggesting that endothelin signaling in stromal cells has a minor effect on renal fibrosis.
8.5.6. Endothelin system as therapeutic target in CKD.
Previous studies have demonstrated increased ET-1 plasma levels in CKD patients, which were associated with kidney function and albuminuria (404). Several clinical trials have tested the therapeutic potential of ETA antagonists in CKD, and early-phase clinical trials suggest that ETA antagonists might be beneficial as nephroprotective and antiproteinuric drugs for diabetes, hypertensive nephropathy, and CKD (411–414). A phase 3 study (ASCEND) assessed the effect of the ETA antagonist avosentan on renal disease progression or death in 1,392 patients with stage 3–4 CKD. At the dosages used, avosentan induced a marked decrease in proteinuria after a median follow-up of 4 mo, but the trial was prematurely terminated because of adverse cardiovascular events in the avosentan groups (238). It was suggested that the use of doses of 25 and 50 mg avosentan may activate the ETB receptor to promote additional fluid retention (415). Taken together, it has been shown that inhibition of ETA receptors can mitigate renal injury and fibrosis at multiple levels. However, the major drawback of endothelin receptor antagonists is the adverse effects of fluid retention, which can potentially be minimized by careful patient selection, diuretic administration, and drug dosing.
8.6. Signaling Networks
To add to the complexity, all of the above-described signaling pathways interact, and work in concert, in both an autocrine and a paracrine fashion. In the healthy kidney, this can convey renoprotection (416); however, in the setting of renal fibrosis, this interaction creates an autoinductive feedforward loop resulting in perpetual activation of the profibrotic machinery (326, 417). In FIGURE 10 we provide a simplified overview of how the DAMP, prostanoid, purinergic, and TGFβ signaling pathways can interact and influence the key cellular players in fibrosis. Much more research is needed to elucidate the complex signaling networks, but doing so will hopefully unveil means to attenuate the self-sustaining character of renal fibrosis.
8.7. MicroRNA in Obstructive Nephropathy
MicroRNAs (miRNAs) are short noncoding RNAs that interact with 3′-untranslated regions of mRNAs (418). miRNAs modulate gene expression by regulating mRNA translation as a posttranscriptional regulator and provide novel insights into the regulation of protein expression and function (419, 420). For example, miR-32 and miR-137 target AQP2, providing a novel mechanism for the regulation of AQP2 abundance (420, 421). Moreover, aldosterone decreased miR-34c-5p levels in mouse cortical collecting duct cells, and a consequent increase in calcium/calmodulin-dependent protein kinase (CaMK)-IIβ expression could play a role in aldosterone-induced fibrosis (419). miRNAs and miRNA-expressing exosomes produced by cells are involved in the development of renal fibrosis (422–425). For instance, several miRNAs, such as miR-34a (426), miR-21 (424), miR-150-5p (427), miR-216a (428), miR-153-3p (429), miR-215, miR-199a-5p, and miR-199a-3p, have been demonstrated to be upregulated and/or be involved in the development of renal fibrosis in the UUO model (430) (FIGURE 8). miR-21-expressing exosomes promote renal fibrosis after 7-day UUO and target phosphatase and tensin homolog (PTEN) (424). Exosomal miR-150-5p and miR-216a derived from the tubular cells are also involved in renal fibrosis (427, 428). The results indicate that cell-cell communication via exosomes derived from tubular cells could play a role in the pathogenesis of renal fibrosis after UUO, and miRNAs expressed in the exosome are novel therapeutic targets against the progression to CKD.
In contrast, miR-29, which has antifibrotic activity through direct inhibition of YY1 and TGFβ3, partly suppressed renal fibrosis in mice with 14-day UUO when injected in exosome-encapsulated form (431, 432). RNA-seq study in mice subjected to UUO for 5 and 10 days revealed that miR-9-5p mitigated renal fibrosis by preventing downregulation of genes associated with critical metabolic pathways in obstructive nephropathy, including mitochondrial function, oxidative phosphorylation, fatty acid oxidation, and glycolysis (433). Overexpression of the miR-200b/c family attenuated renal fibrosis by controlling EMT via targeting fascin-1 and CD44 (422). Moreover, overexpression of miR-27b-3p in UUO inhibited renal fibrosis via downregulation of signal transducers and activators of transcription 1 (STAT1) expression (434). Thus, miRNAs could be novel therapeutic targets against CKD progression.
8.8. Mesenchymal Stem Cell-Derived Extracellular Vesicles in Obstructive Nephropathy
Intrarenal delivery of autologous mesenchymal stem cell (MSC)-derived extracellular vesicles (EVs) has been demonstrated in several studies to ameliorate structural and functional decline and to attenuate renal inflammation, manifested as decreased levels of proinflammatory cytokines, including TNFα, IL-6, and IL-1β (435). Intravenous administration of MSC-derived EVs mitigated tubular injury and fibrosis after 14-day UUO (436). Importantly, miRNAs transferred through EVs are capable of modulating fibrosis and EMT. Consistent with these findings, in vitro experiments using tubular cells treated with TGFβ showed that coincubation with kidney-resident MSC-derived EVs reversed EMT and TGFβ-induced morphological changes. This mechanism was also confirmed in another study on TGFβ-treated endothelial cells, in which MSC-derived EVs ameliorated EMT and improved cell proliferation 7 days after UUO (437). In light of these few studies, MSC-derived EVs may have a role as antifibrotic and renoprotective treatment. Moreover, genome-engineered mesenchymal stem cells secreting a specific protein expressed in the EV could be exploited for effective renoprotective treatment against kidney injury (438). In a recent review, it was concluded that the concept of EV-based treatment for CKD remains promising, but more research is needed regarding standardization of EV protocols, improving study quality, determining the optimal delivery and dosage of EVs, and, most importantly, understanding the global biological mechanisms of the observed protective effects (439).
9. CONCLUSIONS
Ureteral obstruction, and its impact on the kidney, has fascinated scientists for decades. To this day, experimental UO remains an important tool in nephrology research. As comprehensively described above, the models of UO have led to the elucidation of numerous aspects of renal (patho)physiology; however, there are many outstanding questions.
One of the challenges to comprehensively understanding the pathophysiological effects of UO has been the lack of studies on molecular changes and changes in renal functions in the same individual. Recently, Wagner and colleagues (440) developed a novel approach to quantify the effects of chronic 5-wk UUO on glomerular processes using two-photon microscopy. This technological improvement may represent a paradigm shift in the possibility of investigating physiological and pathophysiological processes, because it allows in vivo quantification of glomerular capillary blood flow, vasoconstriction, and vasodilation responses to drugs, permeability, and inflammation in the renal tissue.
The pathological processes following UO involve a wide variety of cells; however, proximal tubule cells, (myo)fibroblasts, and macrophages have garnered the most attention from the scientific community, especially in relation to renal fibrosis. In several studies, it has been clearly shown that cells from the distal nephron also contribute to the development of fibrosis. We expect that combining the UO models with advanced genomic analysis techniques, such as scRNA-seq, snATAC-seq, or spatial transcriptomics, will greatly improve our understanding of the pathogenesis of renal fibrosis and will aid in the identification of novel therapeutic targets. Unfortunately, all these genomic techniques share a caveat, that is, a lack of consensus regarding cell type markers, a problem plaguing lineage-tracing studies as well. This is a major issue that the scientific community needs to tackle. Fortunately, reference atlases are continuously growing and annotation protocols are improving.
Besides changes in the cellular composition of the kidney following UO (FIGURE 7), there are also clear changes in the activity of metabolic pathways, such as reduced operation of gluconeogenesis and fatty acid oxidation (FIGURE 5). This aspect is relatively unexplored, especially with regard to distinct metabolic changes at the cellular level as well as their functional consequences. Moving forward, single-cell proteomic and metabolomic analyses should provide essential spatially resolved biochemical information. Together with an improved understanding of key cellular players, we believe that these mass spectrometry studies will increase the catalog of clinically relevant targets and biomarkers.
Another area that needs more attention is the identification and validation of proper biomarkers of CKD-associated renal fibrosis, especially since the extent of fibrosis correlates with, and predicts, renal function loss (441, 442). To date, renal biopsy remains the most widely used method to assess the extent of fibrosis, yet needle-based biopsies are associated with several limitations, one of the most important being that they are not suitable for long-term monitoring of disease progression or therapy responses. This has a big impact on the clinical management of CKD as well as on drug development. In this light, models of UO might aid in the search for novel fibrosis biomarkers, for instance, related to ECM turnover (443, 444). Moreover, we believe that the addition of the “omics” techniques will expedite the identification of novel liquid biomarkers; however, we expect that noninvasive imaging methods, such as elastin imaging (445), may be an important part of the solution and therefore deserve more attention.
Finally, it is of paramount importance to develop new therapies for the treatment of CKD. The most commonly used therapies target conventional causes of CKD, such as hypertension and diabetes, but do not act directly on the kidney. This partly changed with the introduction of SGLT2 inhibitors as CKD therapeutics (FIGURE 11 and TABLE 1). This class of drugs includes dapagliflozin, which was recently approved by the FDA to reduce the risk of decline in kidney function, kidney failure, death caused by cardiovascular diseases, and hospitalization for heart failure in adults with CKD (446). Despite these exciting advances, there remains an urgent need for therapies that can attenuate renal fibrosis, which plays a key role in CKD progression.
One of the main hurdles in developing efficacious antifibrotic therapies is that the majority of profibrotic factors, such as TGFβ, also play key roles in normal physiological processes, thereby markedly increasing the risk for adverse effects following systemic drug therapy. This issue could be mitigated by developing therapeutics that solely act on the diseased organ, for instance, by conjugating drugs to delivery systems that are directed to transporters exclusively expressed in the kidney. Advances have been made in this field (447), but, unfortunately, targeted therapeutics are still not available in clinical practice. With regard to obstructive nephropathy, an aspect that requires attention is the increased risk of CKD and ESRD in some patients after obstruction release. Clinical management of the obstruction and underlying cause might not be sufficient to prevent the deterioration of renal function, and new treatment modalities should incorporate means to reduce the risk of renal failure. Since all experimental models have limitations, future research initiatives with new models should aim to parallel clinical disorders more closely.
To achieve these goals, the various preclinical models of UO will remain an indispensable and relevant tool, with a proven track record dating back to the 1800s.
GRANTS
This work was supported by grants from the Novo Nordisk foundation (NNF19OC0054481) to R.N., Lundbeck foundation (R368-2021-726) to H.A.M.M., Danish Council for Independent Research (DFF-7016-00305B) to J.F., and the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MIST) (2021R1A5A2021614) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Korea (HI15C0001) to T.-H.K.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.N., H.A.M.M., J.F. and T.-H.K. prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
ACKNOWLEDGMENTS
We thank medical illustrator Ken Peter Kragsfeldt for preparing the figures.
REFERENCES
- 1. Cockwell P, Fisher LA. The global burden of chronic kidney disease. Lancet 395: 662–664, 2020. doi: 10.1016/S0140-6736(19)32977-0. [DOI] [PubMed] [Google Scholar]
- 2. Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol 16: 269–288, 2020. doi: 10.1038/s41581-019-0248-y. [DOI] [PubMed] [Google Scholar]
- 3. Dendooven A, Ishola DA Jr, Nguyen TQ, Van der Giezen DM, Kok RJ, Goldschmeding R, Joles JA. Oxidative stress in obstructive nephropathy. Int J Exp Pathol 92: 202–210, 2011. doi: 10.1111/j.1365-2613.2010.00730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am J Physiol Renal Physiol 283: F861–F875, 2002. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- 5. Hinman F. Experimental hydronephrosis—repair following uretero cystoneostomy in white rats with complete ureteral obstruction. J Urol 3: 147–174, 1919. doi: 10.1016/S0022-5347(17)74173-0. [DOI] [Google Scholar]
- 6. Klahr S. Obstructive nephropathy. Intern Med 39: 355–361, 2000. doi: 10.2169/internalmedicine.39.355. [DOI] [PubMed] [Google Scholar]
- 7. Singh P, Harris PC, Sas DJ, Lieske JC. The genetics of kidney stone disease and nephrocalcinosis. Nat Rev Nephrol 18: 224–240, 2022. doi: 10.1038/s41581-021-00513-4. [DOI] [PubMed] [Google Scholar]
- 8. Gaujoux S, Couchard AC, Al Youssef J, Paquet JC. Ureteral obstruction and Crohn’s disease. Gastroenterol Clin Biol 31: 611–613, 2007. doi: 10.1016/s0399-8320(07)89439-5. [DOI] [PubMed] [Google Scholar]
- 9. Michelassi F, Sultan S. Surgical treatment of complex small bowel Crohn disease. Ann Surg 260: 230–235, 2014. doi: 10.1097/SLA.0000000000000697. [DOI] [PubMed] [Google Scholar]
- 10. Vaglio A, Salvarani C, Buzio C. Retroperitoneal fibrosis. Lancet 367: 241–251, 2006. doi: 10.1016/S0140-6736(06)68035-5. [DOI] [PubMed] [Google Scholar]
- 11. Fiadjoe P, Kannan K, Rane A. Maternal urological problems in pregnancy. Eur J Obstet Gynecol Reprod Biol 152: 13–17, 2010. doi: 10.1016/j.ejogrb.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 12. Bell ET. Obstruction of the urinary tract—hydronephrosis. In: Renal Diseases. Philadelphia, PA: Lea & Febiger, 1947, p. 113–139. [Google Scholar]
- 13. Low ZY, Allen SE, Arumuham V, Davis LM, Allen C, Bomanji J, Smith RD. Does relative renal function improve after intervention for chronic ureteric obstruction? Cent European J Urol 74: 64–70, 2021. doi: 10.5173/ceju.2021.0274.R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manucha W, Oliveros L, Carrizo L, Seltzer A, Vallés P. Losartan modulation on NOS isoforms and COX-2 expression in early renal fibrogenesis in unilateral obstruction. Kidney Int 65: 2091–2107, 2004. doi: 10.1111/j.1523-1755.2004.00643.x. [DOI] [PubMed] [Google Scholar]
- 15. de Jong MA, Mirkovic K, Mencke R, Hoenderop JG, Bindels RJ, Vervloet MG, Hillebrands JL, van den Born J, Navis G, de Borst MH; NIGRAM consortium. Fibroblast growth factor 23 modifies the pharmacological effects of angiotensin receptor blockade in experimental renal fibrosis. Nephrol Dial Transplant 32: 73–80, 2017. doi: 10.1093/ndt/gfw105. [DOI] [PubMed] [Google Scholar]
- 16. Song J, Xia Y, Yan X, Luo J, Jiang C, Zhang M, Shi GP, Zhu W. Losartan accelerates the repair process of renal fibrosis in UUO mouse after the surgical recanalization by upregulating the expression of Tregs. Int Urol Nephrol 51: 2073–2081, 2019. doi: 10.1007/s11255-019-02253-8. [DOI] [PubMed] [Google Scholar]
- 17. Elkappany S, Hashem A, Elkarta A, Sheashaa H, Osman Y, Shokeir AA. Effect of losartan on the recoverability of renal function in anuric and oliguric patients with a solitary obstructed kidney: a double-blind randomized placebo-controlled trial. BJU Int 126: 715–721, 2020. doi: 10.1111/bju.15168. [DOI] [PubMed] [Google Scholar]
- 18. Ren H, Zuo S, Hou Y, Shang W, Liu N, Yin Z. Inhibition of α1-adrenoceptor reduces TGF-β1-induced epithelial-to-mesenchymal transition and attenuates UUO-induced renal fibrosis in mice. FASEB J 34: 14892–14904, 2020. doi: 10.1096/fj.202000737RRR. [DOI] [PubMed] [Google Scholar]
- 19. Pathan SA, Mitra B, Cameron PA. A systematic review and meta-analysis comparing the efficacy of nonsteroidal anti-inflammatory drugs, opioids, and paracetamol in the treatment of acute renal colic. Eur Urol 73: 583–595, 2018. doi: 10.1016/j.eururo.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 20. Baker M, Perazella MA. NSAIDs in CKD: are they safe? Am J Kidney Dis 76: 546–557, 2020. doi: 10.1053/j.ajkd.2020.03.023. [DOI] [PubMed] [Google Scholar]
- 21. Dixit M, Doan T, Kirschner R, Dixit N. Significant acute kidney injury due to non-steroidal anti-inflammatory drugs: inpatient setting. Pharmaceuticals (Basel) 3: 1279–1285, 2010. doi: 10.3390/ph3041279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. LaForge JM, Urso K, Day JM, Bourgeois CW, Ross MM, Ahmadzadeh S, Shekoohi S, Cornett EM, Kaye AM, Kaye AD. Non-steroidal anti-inflammatory drugs: clinical implications, renal impairment risks, and AKI. Adv Ther 40: 2082–2096, 2023. doi: 10.1007/s12325-023-02481-6. [DOI] [PubMed] [Google Scholar]
- 23. Frøkiaer J, Marples D, Knepper MA, Nielsen S. Bilateral ureteral obstruction downregulates expression of vasopressin-sensitive AQP-2 water channel in rat kidney. Am J Physiol Renal Physiol 270: F657–F668, 1996. doi: 10.1152/ajprenal.1996.270.4.F657. [DOI] [PubMed] [Google Scholar]
- 24. Li C, Wang W, Kwon TH, Isikay L, Wen JG, Marples D, Djurhuus JC, Stockwell A, Knepper MA, Nielsen S, Frøkiaer J. Downregulation of AQP1, -2, and -3 after ureteral obstruction is associated with a long-term urine-concentrating defect. Am J Physiol Renal Physiol 281: F163–F171, 2001. doi: 10.1152/ajprenal.2001.281.1.F163. [DOI] [PubMed] [Google Scholar]
- 25. Li C, Wang W, Kwon TH, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of major renal Na transporters in rats with bilateral ureteral obstruction and release of obstruction. Am J Physiol Renal Physiol 285: F889–F901, 2003. doi: 10.1152/ajprenal.00170.2003. [DOI] [PubMed] [Google Scholar]
- 26. Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int 75: 1145–1152, 2009. doi: 10.1038/ki.2009.86. [DOI] [PubMed] [Google Scholar]
- 27. Thornhill BA, Burt LE, Chen C, Forbes MS, Chevalier RL. Variable chronic partial ureteral obstruction in the neonatal rat: a new model of ureteropelvic junction obstruction. Kidney Int 67: 42–52, 2005. doi: 10.1111/j.1523-1755.2005.00052.x. [DOI] [PubMed] [Google Scholar]
- 28. Thornhill BA, Chevalier RL. Variable partial unilateral ureteral obstruction and its release in the neonatal and adult mouse. Methods Mol Biol 886: 381–392, 2012. doi: 10.1007/978-1-61779-851-1_33. [DOI] [PubMed] [Google Scholar]
- 29. Chen CO, Park MH, Forbes MS, Thornhill BA, Kiley SC, Yoo KH, Chevalier RL. Angiotensin-converting enzyme inhibition aggravates renal interstitial injury resulting from partial unilateral ureteral obstruction in the neonatal rat. Am J Physiol Renal Physiol 292: F946–F955, 2007. doi: 10.1152/ajprenal.00287.2006. [DOI] [PubMed] [Google Scholar]
- 30. Ucero AC, Benito-Martin A, Izquierdo MC, Sanchez-Niño MD, Sanz AB, Ramos AM, Berzal S, Ruiz-Ortega M, Egido J, Ortiz A. Unilateral ureteral obstruction: beyond obstruction. Int Urol Nephrol 46: 765–776, 2014. doi: 10.1007/s11255-013-0520-1. [DOI] [PubMed] [Google Scholar]
- 31. Higgins CE, Tang J, Higgins SP, Gifford CC, Mian BM, Jones DM, Zhang W, Costello A, Conti DJ, Samarakoon R, Higgins PJ. The genomic response to TGF-β1 dictates failed repair and progression of fibrotic disease in the obstructed kidney. Front Cell Dev Biol 9: 678524, 2021. doi: 10.3389/fcell.2021.678524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martínez-Klimova E, Aparicio-Trejo OE, Tapia E, Pedraza-Chaverri J. Unilateral ureteral obstruction as a model to investigate fibrosis-attenuating treatments. Biomolecules 9: 141, 2019. doi: 10.3390/biom9040141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grande MT, López-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol 5: 319–328, 2009. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- 34. Eddy AA, López-Guisa JM, Okamura DM, Yamaguchi I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr Nephrol 27: 1233–1247, 2012. doi: 10.1007/s00467-011-1938-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Z, Li Y, Yuan Y, Lai K, Ye K, Lin Y, Lan R, Chen H, Xu Y. Single-cell sequencing reveals homogeneity and heterogeneity of the cytopathological mechanisms in different etiology-induced AKI. Cell Death Dis 14: 318, 2023. doi: 10.1038/s41419-023-05830-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Becker GJ, Hewitson TD. Animal models of chronic kidney disease: useful but not perfect. Nephrol Dial Transplant 100: 1004–1014, 2016. doi: 10.1093/ndt/gft071. [DOI] [PubMed] [Google Scholar]
- 37. Nogueira A, Pires MJ, Oliveira PA. Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo 31: 1–22, 2017. doi: 10.21873/invivo.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abbas NA, El Salem A, Awad MM. Empagliflozin, SGLT2 inhibitor, attenuates renal fibrosis in rats exposed to unilateral ureteric obstruction: potential role of klotho expression. Naunyn Schmiedebergs Arch Pharmacol 391: 1347–1360, 2018. doi: 10.1007/s00210-018-1544-y. [DOI] [PubMed] [Google Scholar]
- 39. Xuan MY, Piao SG, Ding J, Nan QY, Piao MH, Jiang YJ, Zheng HL, Jin JZ, Li C. Dapagliflozin alleviates renal fibrosis by inhibiting RIP1-RIP3-MLKL-mediated necroinflammation in unilateral ureteral obstruction. Front Pharmacol 12: 798381, 2021. doi: 10.3389/fphar.2021.798381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu F, Wang L, Qi H, Wang J, Wang Y, Jiang W, Xu L, Liu N, Zhuang S. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin Sci (Lond) 131: 2125–2143, 2017. doi: 10.1042/CS20170134. [DOI] [PubMed] [Google Scholar]
- 41. Shimizu T, Kuroda T, Hata S, Fukagawa M, Margolin SB, Kurokawa K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int 54: 99–109, 1998. doi: 10.1046/j.1523-1755.1998.00962.x. [DOI] [PubMed] [Google Scholar]
- 42. Cochrane AL, Kett MM, Samuel CS, Campanale NV, Anderson WP, Hume DA, Little MH, Bertram JF, Ricardo SD. Renal structural and functional repair in a mouse model of reversal of ureteral obstruction. J Am Soc Nephrol 16: 3623–3630, 2005. doi: 10.1681/ASN.2004090771. [DOI] [PubMed] [Google Scholar]
- 43. Conway BR, O’Sullivan ED, Cairns C, O'Sullivan J, Simpson DJ, Salzano A, Connor K, Ding P, Humphries D, Stewart K, Teenan O, Pius R, Henderson NC, Bénézech C, Ramachandran P, Ferenbach D, Hughes J, Chandra T, Denby L. Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. J Am Soc Nephrol 31: 2833–2854, 2020. doi: 10.1681/ASN.2020060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hesketh EE, Vernon MA, Ding P, Clay S, Borthwick G, Conway B, Hughes J. A murine model of irreversible and reversible unilateral ureteric obstruction. J Vis Exp 94: 52559, 2014. doi: 10.3791/52559-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Narváez Barros A, Guiteras R, Sola A, Manonelles A, Morote J, Cruzado JM. Reversal unilateral ureteral obstruction: a mice experimental model. Nephron 142: 125–134, 2019. doi: 10.1159/000497119. [DOI] [PubMed] [Google Scholar]
- 46. Ito K, Chen J, El Chaar M, Stern JM, Seshan SV, Khodadadian JJ, Richardson I, Hyman MJ, Vaughan ED Jr, Poppas DP, Felsen D. Renal damage progresses despite improvement of renal function after relief of unilateral ureteral obstruction in adult rats. Am J Physiol Renal Physiol 287: F1283–F1293, 2004. doi: 10.1152/ajprenal.00441.2003. [DOI] [PubMed] [Google Scholar]
- 47. Manson SR, Niederhoff RA, Hruska KA, Austin PF. Endogenous BMP-7 is a critical molecular determinant of the reversibility of obstruction-induced renal injuries. Am J Physiol Renal Physiol 301: F1293–F1302, 2011. doi: 10.1152/ajprenal.00071.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Puri TS, Shakaib MI, Chang A, Mathew L, Olayinka O, Minto AW, Sarav M, Hack BK, Quigg RJ. Chronic kidney disease induced in mice by reversible unilateral ureteral obstruction is dependent on genetic background. Am J Physiol Renal Physiol 298: F1024–F1032, 2010. doi: 10.1152/ajprenal.00384.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tapmeier TT, Brown KL, Tang Z, Sacks SH, Sheerin NS, Wong W. Reimplantation of the ureter after unilateral ureteral obstruction provides a model that allows functional evaluation. Kidney Int 73: 885–889, 2008. doi: 10.1038/sj.ki.5002797. [DOI] [PubMed] [Google Scholar]
- 50. Kaeidi A, Maleki M, Shamsizadeh A, Fatemi I, Hakimizadeh E, Hassanshahi J. The therapeutic approaches of renal recovery after relief of the unilateral ureteral obstruction: a comprehensive review. Iran J Basic Med Sci 23: 1367–1373, 2020. doi: 10.22038/ijbms.2020.41984.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heikkilä J, Holmberg C, Kyllönen L, Rintala R, Taskinen S. Long-term risk of end stage renal disease in patients with posterior urethral valves. J Urol 186: 2392–2396, 2011. doi: 10.1016/j.juro.2011.07.109. [DOI] [PubMed] [Google Scholar]
- 52. Ravanan R, Tomson CR. Natural history of postobstructive nephropathy: a single-center retrospective study. Nephron Clin Pract 105: c165–c170, 2007. doi: 10.1159/000099007. [DOI] [PubMed] [Google Scholar]
- 53. Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311: F145–F161, 2016. doi: 10.1152/ajprenal.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Klahr S, Morrison A, Buerkert J. Effects of urinary tract obstruction on renal function. Contrib Nephrol 23: 34–46, 1980. doi: 10.1159/000389997. [DOI] [PubMed] [Google Scholar]
- 55. Klahr S, Harris K, Purkerson ML. Effects of obstruction on renal functions. Pediatr Nephrol 2: 34–42, 1988. doi: 10.1007/BF00870378. [DOI] [PubMed] [Google Scholar]
- 56. Moody TE, Vaughn ED Jr, Gillenwater JY. Relationship between renal blood flow and ureteral pressure during 18 hours of total unilateral uretheral occlusion. Implications for changing sites of increased renal resistance. Invest Urol 13: 246–251, 1975. [PubMed] [Google Scholar]
- 57. Vaughan ED Jr, Shenasky JH Jr, Gillenwater JY. Mechanism of acute hemodynamic response to ureteral occlusion. Invest Urol 9: 109–118, 1971. [PubMed] [Google Scholar]
- 58. Suki WN, Guthrie AG, Martinez-Maldonado M, Eknoyan G. Effects of ureteral pressure elevation on renal hemodynamics and urine concentration. Am J Physiol 220: 38–43, 1971. doi: 10.1152/ajplegacy.1971.220.1.38. [DOI] [PubMed] [Google Scholar]
- 59. Oliw E. Acute unilateral ureteral occlusion increases plasma renin activity and contralateral urinary prostaglandin excretion in rabbits. Eur J Pharmacol 53: 95–102, 1978. doi: 10.1016/0014-2999(78)90271-6. [DOI] [PubMed] [Google Scholar]
- 60. Dal Canton A, Stanziale R, Corradi A, Andreucci VE, Migone L. Effects of acute ureteral obstruction on glomerular hemodynamics in rat kidney. Kidney Int 12: 403–411, 1977. doi: 10.1038/ki.1977.131. [DOI] [PubMed] [Google Scholar]
- 61. Sjödin JG, Häggmark S, Reiz S. Effects of indomethacin on central, renal and coronary hemodynamics. An experimental study in swine with unilateral ureteral obstruction. Scand J Urol Nephrol 17: 73–79, 1983. doi: 10.3109/00365598309179786. [DOI] [PubMed] [Google Scholar]
- 62. Bugge JF, Stokke ES, Vikse A, Kiil F. Stimulation of renin release by PGE2 and PGI2 infusion in the dog: enhancing effect of ureteral occlusion or administration of ethacrynic acid. Acta Physiol Scand 138: 193–201, 1990. doi: 10.1111/j.1748-1716.1990.tb08833.x. [DOI] [PubMed] [Google Scholar]
- 63. Chevalier RL, Thornhill BA. Ureteral obstruction in the neonatal rat: renal nerves modulate hemodynamic effects. Pediatr Nephrol 9: 447–450, 1995. doi: 10.1007/BF00866725. [DOI] [PubMed] [Google Scholar]
- 64. Chevalier RL, Thornhill BA. Ureteral obstruction in the neonatal guinea pig: interaction of sympathetic nerves and angiotensin. Pediatr Nephrol 9: 441–446, 1995. doi: 10.1007/BF00866723. [DOI] [PubMed] [Google Scholar]
- 65. Persson AE, Wahlberg J, Safirstein R, Wright FS. The effect of 2 hours of complete unilateral ureteral obstruction on tubuloglomerular feedback control. Acta Physiol Scand 122: 35–43, 1984. doi: 10.1111/j.1748-1716.1984.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 66. Wahlberg J, Stenberg A, Wilson DR, Persson AE. Tubuloglomerular feedback and interstitial pressure in obstructive nephropathy. Kidney Int 26: 294–301, 1984. doi: 10.1038/ki.1984.172. [DOI] [PubMed] [Google Scholar]
- 67. Yarger WE, Griffith LD. Intrarenal hemodynamics following chronic unilateral ureteral obstruction in the dog. Am J Physiol 227: 816–826, 1974. doi: 10.1152/ajplegacy.1974.227.4.816. [DOI] [PubMed] [Google Scholar]
- 68. Blantz RC, Konnen KS, Tucker BJ. Glomerular filtration response to elevated ureteral pressure in both the hydropenic and the plasma-expanded rat. Circ Res 37: 819–829, 1975. doi: 10.1161/01.res.37.6.819. [DOI] [PubMed] [Google Scholar]
- 69. Kim KM, Bogaert GA, Nguyen HT, Borirakchanyavat S, Kogan BA. Hemodynamic changes after complete unilateral ureteral obstruction in the young lamb. J Urol 158: 1090–1093, 1997. doi: 10.1016/S0022-5347(01)64395-7. [DOI] [PubMed] [Google Scholar]
- 70. Frøkiaer J, Knudsen L, Nielsen AS, Pedersen EB, Djurhuus JC. Enhanced intrarenal angiotensin II generation in response to obstruction of the pig ureter. Am J Physiol Renal Physiol 263: F527–F533, 1992. doi: 10.1152/ajprenal.1992.263.3.F527. [DOI] [PubMed] [Google Scholar]
- 71. Hvistendahl JJ, Pedersen TS, Jørgensen HH, Rehling M, Frøkiaer J. Renal hemodynamic response to gradated ureter obstruction in the pig. Nephron 74: 168–174, 1996. doi: 10.1159/000189297. [DOI] [PubMed] [Google Scholar]
- 72. Eide I, Loyning E, Langård O, Kiil F. Mechanism of renin release during acute ureteral constriction in dogs. Circ Res 40: 293–299, 1977. doi: 10.1161/01.res.40.3.293. [DOI] [PubMed] [Google Scholar]
- 73. Wahlberg J. The renal response to ureteral obstruction. Scand J Urol Nephrol Suppl 73: 1–30, 1983. [PubMed] [Google Scholar]
- 74. Frøkiaer J, Nielsen AS, Knudsen L, Djurhuus JC, Pedersen EB. The effect of indomethacin infusion on renal hemodynamics and on the renin-angiotensin system during unilateral ureteral obstruction of the pig. J Urol 150: 1557–1563, 1993. doi: 10.1016/s0022-5347(17)35841-x. [DOI] [PubMed] [Google Scholar]
- 75. Harris RH, Yarger WE. Renal function after release of unilateral ureteral obstruction in rats. Am J Physiol 227: 806–815, 1974. doi: 10.1152/ajplegacy.1974.227.4.806. [DOI] [PubMed] [Google Scholar]
- 76. Arendshorst WJ, Finn WF, Gottschalk CW. Nephron stop-flow pressure response to obstruction for 24 hours in the rat kidney. J Clin Invest 53: 1497–1500, 1974. doi: 10.1172/JCI107699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dal Canton A, Corradi A, Stanziale R, Maruccio G, Migone L. Effects of 24-hour unilateral ureteral obstruction on glomerular hemodynamics in rat kidney. Kidney Int 15: 457–462, 1979. doi: 10.1038/ki.1979.61. [DOI] [PubMed] [Google Scholar]
- 78. Yarger WE, Schocken DD, Harris RH. Obstructive nephropathy in the rat: possible roles for the renin-angiotensin system, prostaglandins, and thromboxanes in postobstructive renal function. J Clin Invest 65: 400–412, 1980. doi: 10.1172/JCI109683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McDougal WS. Pharmacologic preservation of renal mass and function in obstructive uropathy. J Urol 128: 418–421, 1982. doi: 10.1016/s0022-5347(17)52950-x. [DOI] [PubMed] [Google Scholar]
- 80. Chevalier RL, Jones CE. Contribution of endogenous vasoactive compounds to renal vascular resistance in neonatal chronic partial ureteral obstruction. J Urol 136: 532–535, 1986. doi: 10.1016/s0022-5347(17)44938-x. [DOI] [PubMed] [Google Scholar]
- 81. Katzberg RW, DiMarco PL, Morris TW, Harnish PP, Graves RF, Merguerian PA, Ventura JA, Proskin HM. Role of angiotensin II in renal hemodynamic functions during the initial stages of acute ureteral obstruction. Life Sci 42: 1963–1971, 1988. doi: 10.1016/0024-3205(88)90495-x. [DOI] [PubMed] [Google Scholar]
- 82. Loo MH, Egan D, Vaughan ED Jr, Marion D, Felsen D, Weisman S. The effect of the thromboxane A2 synthesis inhibitor OKY-046 on renal function in rabbits following release of unilateral ureteral obstruction. J Urol 137: 571–576, 1987. doi: 10.1016/s0022-5347(17)44108-5. [DOI] [PubMed] [Google Scholar]
- 83. Yarger WE, Newman WJ, Klotman PE. Renal effects of aprotinin after 24 hours of unilateral ureteral obstruction. Am J Physiol Renal Physiol 253: F1006–F1014, 1987. doi: 10.1152/ajprenal.1987.253.5.F1006. [DOI] [PubMed] [Google Scholar]
- 84. Bhangdia DK, Gulmi FA, Chou SY, Mooppan UM, Kim H. Alterations of renal hemodynamics in unilateral ureteral obstruction mediated by activation of endothelin receptor subtypes. J Urol 170: 2057–2062, 2003. doi: 10.1097/01.ju.0000081956.16457.67. [DOI] [PubMed] [Google Scholar]
- 85. Shenasky JH Jr, Gillenwater JY, Graham SD Jr, Wooster LD. Effects of vasoactive drugs on renal vascular resistance in obstructive disease. J Urol 106: 355–359, 1971. doi: 10.1016/s0022-5347(17)61287-4. [DOI] [PubMed] [Google Scholar]
- 86. Hammad FT, Wheatley AM, Davis G. Long-term renal effects of unilateral ureteral obstruction and the role of endothelin. Kidney Int 58: 242–250, 2000. doi: 10.1046/j.1523-1755.2000.00159.x. [DOI] [PubMed] [Google Scholar]
- 87. Vander AJ, Miller R. Control of renin secretion in the anesthetized dog. Am J Physiol 207: 537–546, 1964. doi: 10.1152/ajplegacy.1964.207.3.537. [DOI] [PubMed] [Google Scholar]
- 88. Chevalier RL, Peach MJ. Hemodynamic effects of enalapril on neonatal chronic partial ureteral obstruction. Kidney Int 28: 891–898, 1985. doi: 10.1038/ki.1985.215. [DOI] [PubMed] [Google Scholar]
- 89. Frøkiaer J, Djurhuus JC, Nielsen M, Pedersen EB. Renal hemodynamic response to ureteral obstruction during converting enzyme inhibition. Urol Res 24: 217–227, 1996. doi: 10.1007/BF00295895. [DOI] [PubMed] [Google Scholar]
- 90. Frøkiaer J, Sørensen SS. Eicosanoid excretion from the contralateral kidney in pigs with complete unilateral ureteral obstruction. J Urol 154: 1205–1209, 1995. doi: 10.1016/S0022-5347(01)67032-0. [DOI] [PubMed] [Google Scholar]
- 91. Allen JT, Vaughan ED Jr, Gillenwater JY. The effect of indomethacin on renal blood flow and uretral pressure in unilateral ureteral obstruction in awake dogs. Invest Urol 15: 324–327, 1978. [PubMed] [Google Scholar]
- 92. Ichikawa I, Brenner BM. Local intrarenal vasoconstrictor-vasodilator interactions in mild partial ureteral obstruction. Am J Physiol Renal Physiol 236: F131–F140, 1979. doi: 10.1152/ajprenal.1979.236.2.F131. [DOI] [PubMed] [Google Scholar]
- 93. Perlmutter A, Miller L, Trimble LA, Marion DN, Vaughan ED Jr, Felsen D. Toradol, an NSAID used for renal colic, decreases renal perfusion and ureteral pressure in a canine model of unilateral ureteral obstruction. J Urol 149: 926–930, 1993. doi: 10.1016/s0022-5347(17)36261-4. [DOI] [PubMed] [Google Scholar]
- 94. Buerkert J, Martin D, Head M, Prasad J, Klahr S. Deep nephron function after release of acute unilateral ureteral obstruction in the young rat. J Clin Invest 62: 1228–1239, 1978. doi: 10.1172/JCI109243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hanley MJ, Davidson K. Isolated nephron segments from rabbit models of obstructive nephropathy. J Clin Invest 69: 165–174, 1982. doi: 10.1172/jci110427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Li C, Wang W, Norregaard R, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of epithelial sodium channel in rats with bilateral or unilateral ureteral obstruction. Am J Physiol Renal Physiol 293: F333–F341, 2007. doi: 10.1152/ajprenal.00372.2006. [DOI] [PubMed] [Google Scholar]
- 97. Li C, Klein JD, Wang W, Knepper MA, Nielsen S, Sands JM, Frøkiaer J. Altered expression of urea transporters in response to ureteral obstruction. Am J Physiol Renal Physiol 286: F1154–F1162, 2004. doi: 10.1152/ajprenal.00453.2003. [DOI] [PubMed] [Google Scholar]
- 98. Li C, Wang W, Knepper MA, Nielsen S, Frøkiaer J. Downregulation of renal aquaporins in response to unilateral ureteral obstruction. Am J Physiol Renal Physiol 284: F1066–F1079, 2003. doi: 10.1152/ajprenal.00090.2002. [DOI] [PubMed] [Google Scholar]
- 99. Li C, Wang W, Kwon TH, Knepper MA, Nielsen S, Frøkiaer J. Altered expression of major renal Na transporters in rats with unilateral ureteral obstruction. Am J Physiol Renal Physiol 284: F155–F166, 2003. doi: 10.1152/ajprenal.00272.2002. [DOI] [PubMed] [Google Scholar]
- 100. Frøkiaer J, Christensen BM, Marples D, Djurhuus JC, Jensen UB, Knepper MA, Nielsen S. Downregulation of aquaporin-2 parallels changes in renal water excretion in unilateral ureteral obstruction. Am J Physiol Renal Physiol 273: F213–F223, 1997. doi: 10.1152/ajprenal.1997.273.2.F213. [DOI] [PubMed] [Google Scholar]
- 101. Bedford JJ, Leader JP, Walker RJ. Aquaporin expression in normal human kidney and in renal disease. J Am Soc Nephrol 14: 2581–2587, 2003. doi: 10.1097/01.asn.0000089566.28106.f6. [DOI] [PubMed] [Google Scholar]
- 102. Cheng X, Zhang H, Lee HL, Park JM. Cyclooxygenase-2 inhibitor preserves medullary aquaporin-2 expression and prevents polyuria after ureteral obstruction. J Urol 172: 2387–2390, 2004. doi: 10.1097/01.ju.0000143882.52960.ee. [DOI] [PubMed] [Google Scholar]
- 103. Nørregaard R, Jensen BL, Li C, Wang W, Knepper MA, Nielsen S, Frøkiaer J. COX-2 inhibition prevents downregulation of key renal water and sodium transport proteins in response to bilateral ureteral obstruction. Am J Physiol Renal Physiol 289: F322–F333, 2005. doi: 10.1152/ajprenal.00061.2005. [DOI] [PubMed] [Google Scholar]
- 104. Nørregaard R, Jensen BL, Topcu SO, Wang G, Schweer H, Nielsen S, Frøkiaer J. Urinary tract obstruction induces transient accumulation of COX-2-derived prostanoids in kidney tissue. Am J Physiol Regul Integr Comp Physiol 298: R1017–R1025, 2010. doi: 10.1152/ajpregu.00336.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Nørregaard R, Jensen BL, Topcu SO, Diget M, Schweer H, Knepper MA, Nielsen S, Frøkiaer J. COX-2 activity transiently contributes to increased water and NaCl excretion in the polyuric phase after release of ureteral obstruction. Am J Physiol Renal Physiol 292: F1322–F1333, 2007. doi: 10.1152/ajprenal.00394.2006. [DOI] [PubMed] [Google Scholar]
- 106. Liu M, Sun Y, Xu M, Yu X, Zhang Y, Huang S, Ding G, Zhang A, Jia Z. Role of mitochondrial oxidative stress in modulating the expressions of aquaporins in obstructive kidney disease. Am J Physiol Renal Physiol 314: F658–F666, 2018. doi: 10.1152/ajprenal.00234.2017. [DOI] [PubMed] [Google Scholar]
- 107. Schreiner GF, Harris KP, Purkerson ML, Klahr S. Immunological aspects of acute ureteral obstruction: immune cell infiltrate in the kidney. Kidney Int 34: 487–493, 1988. doi: 10.1038/ki.1988.207. [DOI] [PubMed] [Google Scholar]
- 108. Nørregaard R, Kwon TH, Frøkiær J. Physiology and pathophysiology of cyclooxygenase-2 and prostaglandin E2 in the kidney. Kidney Res Clin Pract 34: 194–200, 2015. doi: 10.1016/j.krcp.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nilsson L, Madsen K, Topcu SO, Jensen BL, Frøkiær J, Nørregaard R. Disruption of cyclooxygenase-2 prevents downregulation of cortical AQP2 and AQP3 in response to bilateral ureteral obstruction in the mouse. Am J Physiol Renal Physiol 302: F1430–F1439, 2012. doi: 10.1152/ajprenal.00682.2011. [DOI] [PubMed] [Google Scholar]
- 110. el-Dahr SS, Gee J, Dipp S, Hanss BG, Vari RC, Chao J. Upregulation of renin-angiotensin system and downregulation of kallikrein in obstructive nephropathy. Am J Physiol Renal Physiol 264: F874–F881, 1993. doi: 10.1152/ajprenal.1993.264.5.F874. [DOI] [PubMed] [Google Scholar]
- 111. Jensen AM, Li C, Praetorius HA, Nørregaard R, Frische S, Knepper MA, Nielsen S, Frøkiaer J. Angiotensin II mediates downregulation of aquaporin water channels and key renal sodium transporters in response to urinary tract obstruction. Am J Physiol Renal Physiol 291: F1021–F1032, 2006. doi: 10.1152/ajprenal.00387.2005. [DOI] [PubMed] [Google Scholar]
- 112. Lee YJ, Song IK, Jang KJ, Nielsen J, Frøkiaer J, Nielsen S, Kwon TH. Increased AQP2 targeting in primary cultured IMCD cells in response to angiotensin II through AT1 receptor. Am J Physiol Renal Physiol 292: F340–F350, 2007. doi: 10.1152/ajprenal.00090.2006. [DOI] [PubMed] [Google Scholar]
- 113. Kwon TH, Nielsen J, Knepper MA, Frøkiaer J, Nielsen S. Angiotensin II AT1 receptor blockade decreases vasopressin-induced water reabsorption and AQP2 levels in NaCl-restricted rats. Am J Physiol Renal Physiol 288: F673–F684, 2005. doi: 10.1152/ajprenal.00304.2004. [DOI] [PubMed] [Google Scholar]
- 114. Li C, Wang W, Rivard CJ, Lanaspa MA, Summer S, Schrier RW. Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am J Physiol Renal Physiol 300: F1255–F1261, 2011. doi: 10.1152/ajprenal.00469.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wang W, Li C, Summer S, Falk S, Schrier RW. Interaction between vasopressin and angiotensin II in vivo and in vitro: effect on aquaporins and urine concentration. Am J Physiol Renal Physiol 299: F577–F584, 2010. doi: 10.1152/ajprenal.00168.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Chou SY, Cai H, Pai D, Mansour M, Huynh P. Regional expression of cyclooxygenase isoforms in the rat kidney in complete unilateral ureteral obstruction. J Urol 170: 1403–1408, 2003. doi: 10.1097/01.ju.0000082964.24635.15. [DOI] [PubMed] [Google Scholar]
- 117. Carlsen I, Donohue KE, Jensen AM, Selzer AL, Chen J, Poppas DP, Felsen D, Frøkiær J, Nørregaard R. Increased cyclooxygenase-2 expression and prostaglandin E2 production in pressurized renal medullary interstitial cells. Am J Physiol Regul Integr Comp Physiol 299: R823–R831, 2010. doi: 10.1152/ajpregu.00544.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ostergaard M, Christensen M, Nilsson L, Carlsen I, Frøkiær J, Nørregaard R. ROS dependence of cyclooxygenase-2 induction in rats subjected to unilateral ureteral obstruction. Am J Physiol Renal Physiol 306: F259–F270, 2014. doi: 10.1152/ajprenal.00352.2013. [DOI] [PubMed] [Google Scholar]
- 119. Choi HJ, Jung HJ, Kwon TH. Extracellular pH affects phosphorylation and intracellular trafficking of AQP2 in inner medullary collecting duct cells. Am J Physiol Renal Physiol 308: F737–F748, 2015. doi: 10.1152/ajprenal.00376.2014. [DOI] [PubMed] [Google Scholar]
- 120. Choi HJ, Yoon YJ, Kwon YK, Lee YJ, Chae S, Hwang D, Hwang GS, Kwon TH. Patterns of gene and metabolite define the effects of extracellular osmolality on kidney collecting duct. J Proteome Res 11: 3816–3828, 2012. doi: 10.1021/pr300309d. [DOI] [PubMed] [Google Scholar]
- 121. Jang KJ, Cho HS, Kang DH, Bae WG, Kwon TH, Suh KY. Fluid-shear-stress-induced translocation of aquaporin-2 and reorganization of actin cytoskeleton in renal tubular epithelial cells. Integr Biol (Camb) 3: 134–141, 2011. doi: 10.1039/c0ib00018c. [DOI] [PubMed] [Google Scholar]
- 122. Hwang GS, Yang JY, Ryu DH, Kwon TH. Metabolic profiling of kidney and urine in rats with lithium-induced nephrogenic diabetes insipidus by 1H-NMR-based metabonomics. Am J Physiol Renal Physiol 298: F461–F470, 2010. doi: 10.1152/ajprenal.00389.2009. [DOI] [PubMed] [Google Scholar]
- 123. Kim JA, Choi HJ, Kwon YK, Ryu DH, Kwon TH, Hwang GS. 1H NMR-based metabolite profiling of plasma in a rat model of chronic kidney disease. PLoS One 9: e85445, 2014. doi: 10.1371/journal.pone.0085445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Weiss RH, Kim K. Metabolomics in the study of kidney diseases. Nat Rev Nephrol 8: 22–33, 2011. doi: 10.1038/nrneph.2011.152. [DOI] [PubMed] [Google Scholar]
- 125. Wikoff WR, Nagle MA, Kouznetsova VL, Tsigelny IF, Nigam SK. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J Proteome Res 10: 2842–2851, 2011. doi: 10.1021/pr200093w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chen DQ, Cao G, Chen H, Argyopoulos CP, Yu H, Su W, Chen L, Samuels DC, Zhuang S, Bayliss GP, Zhao S, Yu XY, Vaziri ND, Wang M, Liu D, Mao JR, Ma SX, Zhao J, Zhang Y, Shang YQ, Kang H, Ye F, Cheng XH, Li XR, Zhang L, Meng MX, Guo Y, Zhao YY. Identification of serum metabolites associating with chronic kidney disease progression and anti-fibrotic effect of 5-methoxytryptophan. Nat Commun 10: 1476, 2019. doi: 10.1038/s41467-019-09329-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jung JW, Lee MS, Choi HJ, Jung S, Lee YJ, Hwang GS, Kwon TH. Mass spectrometric imaging of metabolites in kidney tissues from rats treated with furosemide. Am J Physiol Renal Physiol 310: F1317–F1327, 2016. doi: 10.1152/ajprenal.00524.2015. [DOI] [PubMed] [Google Scholar]
- 128. Liu H, Li W, He Q, Xue J, Wang J, Xiong C, Pu X, Nie Z. Mass Spectrometry imaging of kidney tissue sections of rat subjected to unilateral ureteral obstruction. Sci Rep 7: 41954, 2017. doi: 10.1038/srep41954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tannenbaum J, Purkerson ML, Klahr S. Effect of unilateral ureteral obstruction on metabolism of renal lipids in the rat. Am J Physiol Renal Physiol 245: F254–F262, 1983. doi: 10.1152/ajprenal.1983.245.2.F254. [DOI] [PubMed] [Google Scholar]
- 130. Niles DJ, Gordon JW, Huang G, Reese S, Adamson EB, Djamali A, Fain SB. Evaluation of renal metabolic response to partial ureteral obstruction with hyperpolarized 13C MRI. NMR Biomed 31: 10.1002/nbm.3846, 2018. doi: 10.1002/nbm.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, Venkatachalam MA. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27: 3356–3367, 2016. doi: 10.1681/ASN.2015020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lee M, Harley G, Katerelos M, Gleich K, Sullivan MA, Laskowski A, Coughlan M, Fraser SA, Mount PF, Power DA. Mutation of regulatory phosphorylation sites in PFKFB2 worsens renal fibrosis. Sci Rep 10: 14531, 2020. doi: 10.1038/s41598-020-71475-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Yin XN, Wang J, Cui LF, Fan WX. Enhanced glycolysis in the process of renal fibrosis aggravated the development of chronic kidney disease. Eur Rev Med Pharmacol Sci 22: 4243–4251, 2018. doi: 10.26355/eurrev_201807_15419. [DOI] [PubMed] [Google Scholar]
- 134. Yu H, Zhu J, Chang L, Liang C, Li X, Wang W. 3-Bromopyruvate decreased kidney fibrosis and fibroblast activation by suppressing aerobic glycolysis in unilateral ureteral obstruction mice model. Life Sci 272: 119206, 2021. doi: 10.1016/j.lfs.2021.119206. [DOI] [PubMed] [Google Scholar]
- 135. Wei Q, Su J, Dong G, Zhang M, Huo Y, Dong Z. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am J Physiol Renal Physiol 316: F1162–F1172, 2019. doi: 10.1152/ajprenal.00422.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, Luo J, Zen K, Yang J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol 313: F561–F575, 2017. doi: 10.1152/ajprenal.00036.2017. [DOI] [PubMed] [Google Scholar]
- 137. Miguel V, Tituaña J, Herrero JI, Herrero L, Serra D, Cuevas P, Barbas C, Puyol DR, Márquez-Expósito L, Ruiz-Ortega M, Castillo C, Sheng X, Susztak K, Ruiz-Canela M, Salas-Salvadó J, González MA, Ortega S, Ramos R, Lamas S. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest 131: e140695, 2021. doi: 10.1172/JCI140695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jogl G, Hsiao YS, Tong L. Structure and function of carnitine acyltransferases. Ann NY Acad Sci 1033: 17–29, 2004. doi: 10.1196/annals.1320.002. [DOI] [PubMed] [Google Scholar]
- 139. Houten SM, Violante S, Ventura FV, Wanders RJ. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu Rev Physiol 78: 23–44, 2016. doi: 10.1146/annurev-physiol-021115-105045. [DOI] [PubMed] [Google Scholar]
- 140. Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med (Lausanne) 2: 52, 2015. doi: 10.3389/fmed.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chung KW, Lee EK, Lee MK, Oh GT, Yu BP, Chung HY. Impairment of PPARα and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol 29: 1223–1237, 2018. doi: 10.1681/ASN.2017070802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhu X, Jiang L, Long M, Wei X, Hou Y, Du Y. Metabolic reprogramming and renal fibrosis. Front Med (Lausanne) 8: 746920, 2021. doi: 10.3389/fmed.2021.746920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Peng H, Wang Q, Lou T, Qin J, Jung S, Shetty V, Li F, Wang Y, Feng XH, Mitch WE, Graham BH, Hu Z. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat Commun 8: 1493, 2017. doi: 10.1038/s41467-017-01646-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhao X, Kwan JY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov 19: 57–75, 2020. doi: 10.1038/s41573-019-0040-5. [DOI] [PubMed] [Google Scholar]
- 145. Console L, Scalise M, Giangregorio N, Tonazzi A, Barile M, Indiveri C. The link between the mitochondrial fatty acid oxidation derangement and kidney injury. Front Physiol 11: 794, 2020. doi: 10.3389/fphys.2020.00794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chevalier RL. Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat Clin Pract Nephrol 2: 157–168, 2006. doi: 10.1038/ncpneph0098. [DOI] [PubMed] [Google Scholar]
- 147. Wu B, Brooks JD. Gene expression changes induced by unilateral ureteral obstruction in mice. J Urol 188: 1033–1041, 2012. doi: 10.1016/j.juro.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 148. Arvaniti E, Moulos P, Vakrakou A, Chatziantoniou C, Chadjichristos C, Kavvadas P, Charonis A, Politis PK. Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci Rep 6: 26235, 2016. doi: 10.1038/srep26235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Silverstein DM, Travis BR, Thornhill BA, Schurr JS, Kolls JK, Leung JC, Chevalier RL. Altered expression of immune modulator and structural genes in neonatal unilateral ureteral obstruction. Kidney Int 64: 25–35, 2003. doi: 10.1046/j.1523-1755.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 150. Higgins DF, Lappin DW, Kieran NE, Anders HJ, Watson RW, Strutz F, Schlondorff D, Haase VH, Fitzpatrick JM, Godson C, Brady HR. DNA oligonucleotide microarray technology identifies fisp-12 among other potential fibrogenic genes following murine unilateral ureteral obstruction (UUO): modulation during epithelial-mesenchymal transition. Kidney Int 64: 2079–2091, 2003. doi: 10.1046/j.1523-1755.2003.00306.x. [DOI] [PubMed] [Google Scholar]
- 151. Gerarduzzi C, Kumar RK, Trivedi P, Ajay AK, Iyer A, Boswell S, Hutchinson JN, Waikar SS, Vaidya VS. Silencing SMOC2 ameliorates kidney fibrosis by inhibiting fibroblast to myofibroblast transformation. JCI Insight 2: e90299, 2017. doi: 10.1172/jci.insight.90299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Sun J, Zhang S, Shi B, Zheng D, Shi J. Transcriptome identified lncRNAs associated with renal fibrosis in UUO rat model. Front Physiol 8: 658, 2017. doi: 10.3389/fphys.2017.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Pavkovic M, Pantano L, Gerlach CV, Brutus S, Boswell SA, Everley RA, Shah JV, Sui SH, Vaidya VS. Multi omics analysis of fibrotic kidneys in two mouse models. Sci Data 6: 92, 2019. doi: 10.1038/s41597-019-0095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Forbes MS, Thornhill BA, Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol 301: F110–F117, 2011. doi: 10.1152/ajprenal.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Chevalier RL, Forbes MS. Generation and evolution of atubular glomeruli in the progression of renal disorders. J Am Soc Nephrol 19: 197–206, 2008. doi: 10.1681/ASN.2007080862. [DOI] [PubMed] [Google Scholar]
- 156. Oliver J. Architecture of the Kidney in Chronic Bright’s Disease. New York: P.B. Hoeber, 1939. [Google Scholar]
- 157. Marcussen N. Atubular glomeruli in chronic renal disease. Curr Top Pathol 88: 145–174, 1995. doi: 10.1007/978-3-642-79517-6_6. [DOI] [PubMed] [Google Scholar]
- 158. Marcussen N. Atubular glomeruli and the structural basis for chronic renal failure. Lab Invest 66: 265–284, 1992. [PubMed] [Google Scholar]
- 159. Marcussen N. Tubulointerstitial damage leads to atubular glomeruli: significance and possible role in progression. Nephrol Dial Transplant 15, Suppl 6: 74–75, 2000. doi: 10.1093/ndt/15.suppl_6.74. [DOI] [PubMed] [Google Scholar]
- 160. Damadian RV, Shwayri E, Bricker NS. On the existence of non-urine forming nephrons in the diseased kidney of the dog. J Lab Clin Med 65: 26–39, 1965. [PubMed] [Google Scholar]
- 161. Konda R, Orikasa S, Sakai K, Ota S, Kimura N. The distribution of renin containing cells in scarred kidneys. J Urol 156: 1450–1454, 1996. doi: 10.1016/S0022-5347(01)65627-1. [DOI] [PubMed] [Google Scholar]
- 162. Wang K, Kestenbaum B. Proximal tubular secretory clearance: a neglected partner of kidney function. Clin J Am Soc Nephrol 13: 1291–1296, 2018. doi: 10.2215/CJN.12001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R, Argiles A; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol 23: 1258–1270, 2012. doi: 10.1681/ASN.2011121175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Masereeuw R, Mutsaers HA, Toyohara T, Abe T, Jhawar S, Sweet DH, Lowenstein J. The kidney and uremic toxin removal: glomerulus or tubule? Semin Nephrol 34: 191–208, 2014. doi: 10.1016/j.semnephrol.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 165. Cha RH, Kang SW, Park CW, Cha DR, Na KY, Kim SG, Yoon SA, Kim S, Han SY, Park JH, Chang JH, Lim CS, Kim YS. Sustained uremic toxin control improves renal and cardiovascular outcomes in patients with advanced renal dysfunction: post-hoc analysis of the Kremezin Study against renal disease progression in Korea. Kidney Res Clin Pract 36: 68–78, 2017. doi: 10.23876/j.krcp.2017.36.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Liabeuf S, Cheddani L, Massy ZA. Uremic Toxins and Clinical Outcomes: The Impact of Kidney Transplantation. Toxins (Basel) 10: 229, 2018. doi: 10.3390/toxins10060229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Lim YJ, Sidor NA, Tonial NC, Che A, Urquhart BL. Uremic toxins in the progression of chronic kidney disease and cardiovascular disease: mechanisms and therapeutic targets. Toxins (Basel) 13: 142, 2021. doi: 10.3390/toxins13020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Veldeman L, Vanmassenhove J, Van Biesen W, Massy ZA, Liabeuf S, Glorieux G, Vanholder R. Evolution of protein-bound uremic toxins indoxyl sulphate and p-cresyl sulphate in acute kidney injury. Int Urol Nephrol 51: 293–302, 2019. doi: 10.1007/s11255-018-2056-x. [DOI] [PubMed] [Google Scholar]
- 169. Wang W, Hao G, Pan Y, Ma S, Yang T, Shi P, Zhu Q, Xie Y, Ma S, Zhang Q, Ruan H, Ding F. Serum indoxyl sulfate is associated with mortality in hospital-acquired acute kidney injury: a prospective cohort study. BMC Nephrol 20: 57, 2019. doi: 10.1186/s12882-019-1238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Chen L, Chen DQ, Liu JR, Zhang J, Vaziri ND, Zhuang S, Chen H, Feng YL, Guo Y, Zhao YY. Unilateral ureteral obstruction causes gut microbial dysbiosis and metabolome disorders contributing to tubulointerstitial fibrosis. Exp Mol Med 51: 1–18, 2019. doi: 10.1038/s12276-019-0234-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Mihajlovic M, Krebber MM, Yang Y, Ahmed S, Lozovanu V, Andreeva D, Verhaar MC, Masereeuw R. Protein-bound uremic toxins induce reactive oxygen species-dependent and inflammasome-mediated IL-1β production in kidney proximal tubule cells. Biomedicines 9: 1326, 2021. doi: 10.3390/biomedicines9101326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Mutsaers HA, Wilmer MJ, Reijnders D, Jansen J, van den Broek PH, Forkink M, Schepers E, Glorieux G, Vanholder R, van den Heuvel LP, Hoenderop JG, Masereeuw R. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim Biophys Acta 1832: 142–150, 2013. doi: 10.1016/j.bbadis.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 173. Diamond JR, van Goor H, Ding G, Engelmyer E. Myofibroblasts in experimental hydronephrosis. Am J Pathol 146: 121–129, 1995. [PMC free article] [PubMed] [Google Scholar]
- 174. Maxwell PH, Ferguson DJ, Nicholls LG, Johnson MH, Ratcliffe PJ. The interstitial response to renal injury: fibroblast-like cells show phenotypic changes and have reduced potential for erythropoietin gene expression. Kidney Int 52: 715–724, 1997. doi: 10.1038/ki.1997.387. [DOI] [PubMed] [Google Scholar]
- 175. Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol 130: 247–262, 2008. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Kaissling B, Le Hir M. Characterization and distribution of interstitial cell types in the renal cortex of rats. Kidney Int 45: 709–720, 1994. doi: 10.1038/ki.1994.95. [DOI] [PubMed] [Google Scholar]
- 177. Kaissling B, Hegyi I, Loffing J, Le Hir M. Morphology of interstitial cells in the healthy kidney. Anat Embryol (Berl) 193: 303–318, 1996. doi: 10.1007/BF00186688. [DOI] [PubMed] [Google Scholar]
- 178. Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol 130: 141–155, 2008. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Sato Y, Yanagita M. Renal anemia: from incurable to curable. Am J Physiol Renal Physiol 305: F1239–F1248, 2013. doi: 10.1152/ajprenal.00233.2013. [DOI] [PubMed] [Google Scholar]
- 180. Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, Yamamura KI, Nagoshi N, Shibata S, Rao TN, Fehling HJ, Fukatsu A, Minegishi N, Kita T, Kimura T, Okano H, Yamamoto M, Yanagita M. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 121: 3981–3990, 2011. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Souma T, Yamazaki S, Moriguchi T, Suzuki N, Hirano I, Pan X, Minegishi N, Abe M, Kiyomoto H, Ito S, Yamamoto M. Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol 24: 1599–1616, 2013. doi: 10.1681/ASN.2013010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kratofil RM, Kubes P, Deniset JF. Monocyte conversion during inflammation and injury. Arterioscler Thromb Vasc Biol 37: 35–42, 2017. doi: 10.1161/ATVBAHA.116.308198. [DOI] [PubMed] [Google Scholar]
- 184. Djudjaj S, Chatziantoniou C, Raffetseder U, Guerrot D, Dussaule JC, Boor P, Kerroch M, Hanssen L, Brandt S, Dittrich A, Ostendorf T, Floege J, Zhu C, Lindenmeyer M, Cohen CD, Mertens PR. Notch-3 receptor activation drives inflammation and fibrosis following tubulointerstitial kidney injury. J Pathol 228: 286–299, 2012. doi: 10.1002/path.4076. [DOI] [PubMed] [Google Scholar]
- 185. Ma FY, Blease K, Nikolic-Paterson DJ. A role for spleen tyrosine kinase in renal fibrosis in the mouse obstructed kidney. Life Sci 146: 192–200, 2016. doi: 10.1016/j.lfs.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 186. Tan TK, Zheng G, Hsu TT, Lee SR, Zhang J, Zhao Y, Tian X, Wang Y, Wang YM, Cao Q, Wang Y, Lee VW, Wang C, Zheng D, Alexander SI, Thompson E, Harris DC. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab Invest 93: 434–449, 2013. doi: 10.1038/labinvest.2013.3. [DOI] [PubMed] [Google Scholar]
- 187. Rovin BH, Harris KP, Morrison A, Klahr S, Schreiner GF. Renal cortical release of a specific macrophage chemoattractant in response to ureteral obstruction. Lab Invest 63: 213–220, 1990. [PubMed] [Google Scholar]
- 188. Pan B, Liu G, Jiang Z, Zheng D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem 35: 1062–1069, 2015. doi: 10.1159/000373932. [DOI] [PubMed] [Google Scholar]
- 189. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol 15: 144–158, 2019. doi: 10.1038/s41581-019-0110-2. [DOI] [PubMed] [Google Scholar]
- 191. Lin L, Hu K. Tissue-type plasminogen activator modulates macrophage M2 to M1 phenotypic change through annexin A2-mediated NF-κB pathway. Oncotarget 8: 88094–88103, 2017. doi: 10.18632/oncotarget.21510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Shen B, Liu X, Fan Y, Qiu J. Macrophages regulate renal fibrosis through modulating TGFβ superfamily signaling. Inflammation 37: 2076–2084, 2014. doi: 10.1007/s10753-014-9941-y. [DOI] [PubMed] [Google Scholar]
- 193. Eitner F, Bücher E, van Roeyen C, Kunter U, Rong S, Seikrit C, Villa L, Boor P, Fredriksson L, Bäckström G, Eriksson U, Ostman A, Floege J, Ostendorf T. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol 19: 281–289, 2008. doi: 10.1681/ASN.2007030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Henderson NC, Mackinnon AC, Farnworth SL, Kipari T, Haslett C, Iredale JP, Liu FT, Hughes J, Sethi T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 172: 288–298, 2008. doi: 10.2353/ajpath.2008.070726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Roufosse C, Bou-Gharios G, Prodromidi E, Alexakis C, Jeffery R, Khan S, Otto WR, Alter J, Poulsom R, Cook HT. Bone marrow-derived cells do not contribute significantly to collagen I synthesis in a murine model of renal fibrosis. J Am Soc Nephrol 17: 775–782, 2006. doi: 10.1681/ASN.2005080795. [DOI] [PubMed] [Google Scholar]
- 196. Sakai N, Wada T, Yokoyama H, Lipp M, Ueha S, Matsushima K, Kaneko S. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci USA 103: 14098–14103, 2006. doi: 10.1073/pnas.0511200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Göbel N, Talke Y, Schweda F, Mack M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci USA 106: 17892–17897, 2009. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Reich B, Schmidbauer K, Rodriguez Gomez M, Johannes Hermann F, Göbel N, Brühl H, Ketelsen I, Talke Y, Mack M. Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int 84: 78–89, 2013. doi: 10.1038/ki.2013.84. [DOI] [PubMed] [Google Scholar]
- 199. Buchtler S, Grill A, Hofmarksrichter S, Stöckert P, Schiechl-Brachner G, Rodriguez Gomez M, Neumayer S, Schmidbauer K, Talke Y, Klinkhammer BM, Boor P, Medvinsky A, Renner K, Castrop H, Mack M. Cellular origin and functional relevance of collagen I production in the kidney. J Am Soc Nephrol 29: 1859–1873, 2018. doi: 10.1681/ASN.2018020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Kramann R, Machado F, Wu H, Kusaba T, Hoeft K, Schneider RK, Humphreys BD. Parabiosis and single-cell RNA sequencing reveal a limited contribution of monocytes to myofibroblasts in kidney fibrosis. JCI Insight 3: e99561, 2018. doi: 10.1172/jci.insight.99561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y, To KF, Nikolic-Paterson DJ, Lan HY. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis 7: e2495, 2016. doi: 10.1038/cddis.2016.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Tang PM, Zhang YY, Xiao J, Tang PC, Chung JY, Li J, Xue VW, Huang XR, Chong CC, Ng CF, Lee TL, To KF, Nikolic-Paterson DJ, Lan HY. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc Natl Acad Sci USA 117: 20741–20752, 2020. doi: 10.1073/pnas.1917663117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Yang Y, Feng X, Liu X, Wang Y, Hu M, Cao Q, Zhang Z, Zhao L, Zhang J, Guo R, Wang H, Qiao X, Wang L, Zheng G. Fate alteration of bone marrow-derived macrophages ameliorates kidney fibrosis in murine model of unilateral ureteral obstruction. Nephrol Dial Transplant 34: 1657–1668, 2019. doi: 10.1093/ndt/gfy381. [DOI] [PubMed] [Google Scholar]
- 204. Tang PM, Zhou S, Li CJ, Liao J, Xiao J, Wang QM, Lian GY, Li J, Huang XR, To KF, Ng CF, Chong CC, Ma RC, Lee TL, Lan HY. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int 93: 173–187, 2018. doi: 10.1016/j.kint.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 205. Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Patón J, Jansen J, Reimer KC, Smith JR, Dobie R, Wilson-Kanamori JR, Halder M, Xu Y, Kabgani N, Kaesler N, Klaus M, Gernhold L, Puelles VG, Huber TB, Boor P, Menzel S, Hoogenboezem RM, Bindels EM, Steffens J, Floege J, Schneider RK, Saez-Rodriguez J, Henderson NC, Kramann R. Decoding myofibroblast origins in human kidney fibrosis. Nature 589: 281–286, 2021. doi: 10.1038/s41586-020-2941-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420–1428, 2009. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Sheng L, Zhuang S. New insights into the role and mechanism of partial epithelial-mesenchymal transition in kidney fibrosis. Front Physiol 11: 569322, 2020. doi: 10.3389/fphys.2020.569322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 130: 393–405, 1995. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest 121: 468–474, 2011. doi: 10.1172/jci44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97, 2010. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Li L, Zepeda-Orozco D, Black R, Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 176: 1767–1778, 2010. doi: 10.2353/ajpath.2010.090345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Wu H, Kirita Y, Donnelly EL, Humphreys BD. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30: 23–32, 2019. doi: 10.1681/ASN.2018090912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21: 998–1009, 2015. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Li J, Bertram JF. Review: Endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrology (Carlton) 15: 507–512, 2010. doi: 10.1111/j.1440-1797.2010.01319.x. [DOI] [PubMed] [Google Scholar]
- 216. Strutz F, Zeisberg M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J Am Soc Nephrol 17: 2992–2998, 2006. doi: 10.1681/ASN.2006050420. [DOI] [PubMed] [Google Scholar]
- 217. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 19: 2282–2287, 2008. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Hung TW, Chu CY, Yu CL, Lee CC, Hsu LS, Chen YS, Hsieh YH, Tsai JP. Endothelial cell-specific molecule 1 promotes endothelial to mesenchymal transition in renal fibrosis. Toxins (Basel) 12: 506, 2020. doi: 10.3390/toxins12080506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Zhao Y, Qiao X, Tan TK, Zhao H, Zhang Y, Liu L, Zhang J, Wang L, Cao Q, Wang Y, Wang Y, Wang YM, Lee VW, Alexander SI, Harris DC, Zheng G. Matrix metalloproteinase 9-dependent Notch signaling contributes to kidney fibrosis through peritubular endothelial-mesenchymal transition. Nephrol Dial Transplant 32: 781–791, 2017. doi: 10.1093/ndt/gfw308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16: 51–66, 2015. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Hiatt MJ, Ivanova L, Trnka P, Solomon M, Matsell DG. Urinary tract obstruction in the mouse: the kinetics of distal nephron injury. Lab Invest 93: 1012–1023, 2013. doi: 10.1038/labinvest.2013.90. [DOI] [PubMed] [Google Scholar]
- 223. Butt MJ, Tarantal AF, Jimenez DF, Matsell DG. Collecting duct epithelial-mesenchymal transition in fetal urinary tract obstruction. Kidney Int 72: 936–944, 2007. doi: 10.1038/sj.ki.5002457. [DOI] [PubMed] [Google Scholar]
- 224. Fujiu K, Manabe I, Nagai R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J Clin Invest 121: 3425–3441, 2011. doi: 10.1172/JCI57582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Ivanova L, Butt MJ, Matsell DG. Mesenchymal transition in kidney collecting duct epithelial cells. Am J Physiol Renal Physiol 294: F1238–F1248, 2008. doi: 10.1152/ajprenal.00326.2007. [DOI] [PubMed] [Google Scholar]
- 226. Mamuya FA, Xie D, Lei L, Huang M, Tsuji K, Capen DE, Yang B, Weissleder R, Păunescu TG, Lu HA. Deletion of β1-integrin in collecting duct principal cells leads to tubular injury and renal medullary fibrosis. Am J Physiol Renal Physiol 313: F1026–F1037, 2017. doi: 10.1152/ajprenal.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Choi A, Nam SA, Kim WY, Park SH, Kim H, Yang CW, Kim J, Kim YK. Notch signaling in the collecting duct regulates renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction in mice. Korean J Intern Med 33: 774–782, 2018. doi: 10.3904/kjim.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Zhang L, Chen L, Gao C, Chen E, Lightle AR, Foulke L, Zhao B, Higgins PJ, Zhang W. Loss of histone H3 K79 methyltransferase Dot1l facilitates kidney fibrosis by upregulating endothelin 1 through histone deacetylase 2. J Am Soc Nephrol 31: 337–349, 2020. doi: 10.1681/ASN.2019070739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Voelker J, Berg PH, Sheetz M, Duffin K, Shen T, Moser B, Greene T, Blumenthal SS, Rychlik I, Yagil Y, Zaoui P, Lewis JB. Anti-TGF-β1 antibody therapy in patients with diabetic nephropathy. J Am Soc Nephrol 28: 953–962, 2017. doi: 10.1681/ASN.2015111230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P, Praga M, Radhakrishnan J, Sellin L, Singh A, Thornley-Brown D, Veronese FV, Accomando B, Engstrand S, Ledbetter S, Lin J, Neylan J, Tumlin J; Focal Segmental Glomerulosclerosis Study Group. A phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int Rep 2: 800–810, 2017. doi: 10.1016/j.ekir.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Balasubramaniam G, Parker T, Turner D, Parker M, Scales J, Harnett P, Harrison M, Ahmed K, Bhagat S, Marianayagam T, Pitzalis C, Mallen C, Roddy E, Almond M, Dasgupta B. Feasibility randomised multicentre, double-blind, double-dummy controlled trial of anakinra, an interleukin-1 receptor antagonist versus intramuscular methylprednisolone for acute gout attacks in patients with chronic kidney disease (ASGARD): protocol study. BMJ Open 7: e017121, 2017. doi: 10.1136/bmjopen-2017-017121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Nowak KL, Chonchol M, Ikizler TA, Farmer-Bailey H, Salas N, Chaudhry R, Wang W, Smits G, Tengesdal I, Dinarello CA, Hung AM. IL-1 inhibition and vascular function in CKD. J Am Soc Nephrol 28: 971–980, 2017. doi: 10.1681/ASN.2016040453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Trachtman H, Vento S, Herreshoff E, Radeva M, Gassman J, Stein DT, Savin VJ, Sharma M, Reiser J, Wei C, Somers M, Srivastava T, Gipson DS. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol 16: 111, 2015. doi: 10.1186/s12882-015-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Don BR, Kim K, Li J, Dwyer T, Alexander F, Kaysen GA. The effect of etanercept on suppression of the systemic inflammatory response in chronic hemodialysis patients. Clin Nephrol 73: 431–438, 2010. doi: 10.5414/cnp73431. [DOI] [PubMed] [Google Scholar]
- 235. Nakamoto H, Yu XQ, Kim S, Origasa H, Zheng H, Chen J, Joo KW, Sritippayawan S, Chen Q, Chen HC, Tsubakihara Y, Tamai H, Song SH, Vaithilingam I, Lee KW, Shu KH, Hok-King Lo S, Isono M, Kurumatani H, Okada K, Kanoh H, Kiriyama T, Yamada S, Fujita T. Effects of sustained-release beraprost in patients with primary glomerular disease or nephrosclerosis: CASSIOPEIR Study results. Ther Apher Dial 24: 42–55, 2020. doi: 10.1111/1744-9987.12840. [DOI] [PubMed] [Google Scholar]
- 236. Heerspink HJ, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou FF, Kitzman DW, Kohan D, Makino H, McMurray JJ, Melnick JZ, Miller MG, Pergola PE, Perkovic V, Tobe S, Yi T, Wigderson M, de Zeeuw D; SONAR Committees and Investigators. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet 393: 1937–1947, 2019. doi: 10.1016/S0140-6736(19)30772-X. [DOI] [PubMed] [Google Scholar]
- 237. Waijer SW, Gansevoort RT, Bakris GL, Correa-Rotter R, Hou FF, Kohan DE, Kitzman DW, Makino H, McMurray JJ, Perkovic V, Tobe S, Parving HH, de Zeeuw D, Heerspink HJ. The effect of atrasentan on kidney and heart failure outcomes by baseline albuminuria and kidney function: a post hoc analysis of the SONAR randomized trial. Clin J Am Soc Nephrol 16: 1824–1832, 2021. doi: 10.2215/CJN.07340521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Mann JF, Green D, Jamerson K, Ruilope LM, Kuranoff SJ, Littke T, Viberti G; ASCEND Study Group. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol 21: 527–535, 2010. doi: 10.1681/ASN.2009060593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Wang X, Zhang H, Zhang Q, Guan M, Sheng S, Mo W, Zou M, Li J, Bi J, Tang X, Zeng H, He J, Xu G, Li P, Xue Y. Exenatide and renal outcomes in patients with type 2 diabetes and diabetic kidney disease. Am J Nephrol 51: 806–814, 2020. doi: 10.1159/000510255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Heerspink HJ, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, Mann JF, McMurray JJ, Lindberg M, Rossing P, Sjöström CD, Toto RD, Langkilde AM, Wheeler DC; DAPA-CKD Trial Committees and Investigators. Dapagliflozin in patients with chronic kidney disease. N Engl J Med 383: 1436–1446, 2020. doi: 10.1056/NEJMoa2024816. [DOI] [PubMed] [Google Scholar]
- 241. Herrington WG, Staplin N, Wanner C, Green JB, Hauske SJ, Emberson JR, , et al. Empagliflozin in patients with chronic kidney disease. N Engl J Med 388: 117–127, 2023. doi: 10.1056/NEJMoa2204233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Samarakoon R, Overstreet JM, Higgins SP, Higgins PJ. TGF-β1 → SMAD/p53/USF2 → PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res 347: 117–128, 2012. doi: 10.1007/s00441-011-1181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Diamond JR, Kees-Folts D, Ding G, Frye JE, Restrepo NC. Macrophages, monocyte chemoattractant peptide-1, and TGF-beta 1 in experimental hydronephrosis. Am J Physiol Renal Physiol 266: F926–F933, 1994. doi: 10.1152/ajprenal.1994.266.6.F926. [DOI] [PubMed] [Google Scholar]
- 244. Miyajima A, Chen J, Lawrence C, Ledbetter S, Soslow RA, Stern J, Jha S, Pigato J, Lemer ML, Poppas DP, Vaughan ED, Felsen D. Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int 58: 2301–2313, 2000. doi: 10.1046/j.1523-1755.2000.00414.x. [DOI] [PubMed] [Google Scholar]
- 245. Kaneto H, Morrissey J, Klahr S. Increased expression of TGF-beta 1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int 44: 313–321, 1993. doi: 10.1038/ki.1993.246. [DOI] [PubMed] [Google Scholar]
- 246. Isaka Y, Tsujie M, Ando Y, Nakamura H, Kaneda Y, Imai E, Hori M. Transforming growth factor-beta 1 antisense oligodeoxynucleotides block interstitial fibrosis in unilateral ureteral obstruction. Kidney Int 58: 1885–1892, 2000. doi: 10.1111/j.1523-1755.2000.00360.x. [DOI] [PubMed] [Google Scholar]
- 247. Prieto M, Rodríguez-Peña AB, Düwel A, Rivas JV, Docherty N, Pérez-Barriocanal F, Arévalo M, Vary CP, Bernabeu C, López-Novoa JM, Eleno N. Temporal changes in renal endoglin and TGF-beta1 expression following ureteral obstruction in rats. J Physiol Biochem 61: 457–467, 2005. doi: 10.1007/BF03168452. [DOI] [PubMed] [Google Scholar]
- 248. Fukuda K, Yoshitomi K, Yanagida T, Tokumoto M, Hirakata H. Quantification of TGF-beta1 mRNA along rat nephron in obstructive nephropathy. Am J Physiol Renal Physiol 281: F513–F521, 2001. doi: 10.1152/ajprenal.2001.281.3.F513. [DOI] [PubMed] [Google Scholar]
- 249. Sager C, Lopez JC, Duran V, Burek C, Perazzo E. Transforming growth factor-beta1 in congenital ureteropelvic junction obstruction: diagnosis and follow-up. Int Braz J Urol 35: 315–323, 2009. doi: 10.1590/s1677-55382009000300008. [DOI] [PubMed] [Google Scholar]
- 250. El-Sherbiny MT, Mousa OM, Shokeir AA, Ghoneim MA. Role of urinary transforming growth factor-beta1 concentration in the diagnosis of upper urinary tract obstruction in children. J Urol 168: 1798–1800, 2002. doi: 10.1097/01.ju.0000027231.84450.8f. [DOI] [PubMed] [Google Scholar]
- 251. Furness PD 3rd, Maizels M, Han SW, Cohn RA, Cheng EY. Elevated bladder urine concentration of transforming growth factor-beta1 correlates with upper urinary tract obstruction in children. J Urol 162: 1033–1036, 1999. doi: 10.1016/S0022-5347(01)68056-X. [DOI] [PubMed] [Google Scholar]
- 252. Almodhen F, Loutochin O, Capolicchio JP, Jednak R, El-Sherbiny M. The role of bladder urine transforming growth factor-beta1 concentrations in diagnosis and management of unilateral prenatal hydronephrosis. J Urol 182: 292–298, 2009. doi: 10.1016/j.juro.2009.02.132. [DOI] [PubMed] [Google Scholar]
- 253. Merrikhi A, Bahraminia E. Association of urinary transforming growth factor-β1 with the ureteropelvic junction obstruction. Adv Biomed Res 3: 123–123, 2014. doi: 10.4103/2277-9175.133196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 92: 102–107, 2016. doi: 10.1016/j.diff.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 255. Stahl PJ, Felsen D. Transforming growth factor-beta, basement membrane, and epithelial-mesenchymal transdifferentiation: implications for fibrosis in kidney disease. Am J Pathol 159: 1187–1192, 2001. doi: 10.1016/s0002-9440(10)62503-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Meng X-M, Tang PM-K, Li J, Lan HY. TGF-β/Smad signaling in renal fibrosis. Front Physiol 6: 82, 2015. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Moon JA, Kim HT, Cho IS, Sheen YY, Kim DK. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int 70: 1234–1243, 2006. doi: 10.1038/sj.ki.5001775. [DOI] [PubMed] [Google Scholar]
- 258. Shindo T, Doi S, Nakashima A, Sasaki K, Arihiro K, Masaki T. TGF-β1 promotes expression of fibrosis-related genes through the induction of histone variant H3.3 and histone chaperone HIRA. Sci Rep 8: 14060, 2018. doi: 10.1038/s41598-018-32518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Mizuno S, Matsumoto K, Nakamura T. Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int 59: 1304–1314, 2001. doi: 10.1046/j.1523-1755.2001.0590041304.x. [DOI] [PubMed] [Google Scholar]
- 260. Yokoi H, Mukoyama M, Sugawara A, Mori K, Nagae T, Makino H, Suganami T, Yahata K, Fujinaga Y, Tanaka I, Nakao K. Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. Am J Physiol Renal Physiol 282: F933–F942, 2002. doi: 10.1152/ajprenal.00122.2001. [DOI] [PubMed] [Google Scholar]
- 261. Gwon MG, An HJ, Kim JY, Kim WH, Gu H, Kim HJ, Leem J, Jung HJ, Park KK. Anti-fibrotic effects of synthetic TGF-β1 and Smad oligodeoxynucleotide on kidney fibrosis in vivo and in vitro through inhibition of both epithelial dedifferentiation and endothelial-mesenchymal transitions. FASEB J 34: 333–349, 2020. doi: 10.1096/fj.201901307RR. [DOI] [PubMed] [Google Scholar]
- 262. Meng XM, Chung AC, Lan HY. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond) 124: 243–254, 2013. doi: 10.1042/CS20120252. [DOI] [PubMed] [Google Scholar]
- 263. Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, Ju W, Bottinger EP, Lan HY. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol 21: 1477–1487, 2010. doi: 10.1681/ASN.2009121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112: 1486–1494, 2003. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Resveratrol inhibits renal fibrosis in the obstructed kidney: potential role in deacetylation of Smad3. Am J Pathol 177: 1065–1071, 2010. doi: 10.2353/ajpath.2010.090923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Ji X, Wang H, Wu Z, Zhong X, Zhu M, Zhang Y, Tan R, Liu Y, Li J, Wang L. Specific Inhibitor of Smad3 (SIS3) attenuates fibrosis, apoptosis, and inflammation in unilateral ureteral obstruction kidneys by inhibition of transforming growth factor β (TGF-β)/Smad3 signaling. Med Sci Monit 24: 1633–1641, 2018. doi: 10.12659/msm.909236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Li RX, Yiu WH, Tang SC. Role of bone morphogenetic protein-7 in renal fibrosis. Front Physiol 6: 114–114, 2015. doi: 10.3389/fphys.2015.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Hruska KA, Guo G, Wozniak M, Martin D, Miller S, Liapis H, Loveday K, Klahr S, Sampath TK, Morrissey J. Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am J Physiol Renal Physiol 279: F130–F143, 2000. doi: 10.1152/ajprenal.2000.279.1.F130. [DOI] [PubMed] [Google Scholar]
- 269. Manson SR, Song JB, Guo Q, Liapis H, Austin PF. Cell type specific changes in BMP-7 expression contribute to the progression of kidney disease in patients with obstructive uropathy. J Urol 193: 1860–1869, 2015. doi: 10.1016/j.juro.2014.10.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Ji X, Cao J, Zhang L, Zhang Z, Shuai W, Yin W. Kaempferol protects renal fibrosis through activating the BMP-7-Smad1/5 signaling pathway. Biol Pharm Bull 43: 533–539, 2020. doi: 10.1248/bpb.b19-01010. [DOI] [PubMed] [Google Scholar]
- 271. Morrissey J, Hruska K, Guo G, Wang S, Chen Q, Klahr S. Bone morphogenetic protein-7 improves renal fibrosis and accelerates the return of renal function. J Am Soc Nephrol 13: S14–S21, 2002. doi: 10.1681/ASN.V13suppl_1s14. [DOI] [PubMed] [Google Scholar]
- 272. Manson SR, Niederhoff RA, Hruska KA, Austin PF. The BMP-7-Smad1/5/8 pathway promotes kidney repair after obstruction induced renal injury. J Urol 185: 2523–2530, 2011. doi: 10.1016/j.juro.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Higgins DF, Ewart LM, Masterson E, Tennant S, Grebnev G, Prunotto M, Pomposiello S, Conde-Knape K, Martin FM, Godson C. BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim Biophys Acta Mol Basis Dis 1863: 3095–3104, 2017. doi: 10.1016/j.bbadis.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 274. Sureshbabu A, Muhsin SA, Choi ME. TGF-β signaling in the kidney: profibrotic and protective effects. Am J Physiol Renal Physiol 310: F596–F606, 2016. doi: 10.1152/ajprenal.00365.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Cho ME, Smith DC, Branton MH, Penzak SR, Kopp JB. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2: 906–913, 2007. doi: 10.2215/CJN.01050207. [DOI] [PubMed] [Google Scholar]
- 276. Sharma K, Ix JH, Mathew AV, Cho M, Pflueger A, Dunn SR, Francos B, Sharma S, Falkner B, McGowan TA, Donohue M, Ramachandrarao S, Xu R, Fervenza FC, Kopp JB. Pirfenidone for diabetic nephropathy. J Am Soc Nephrol 22: 1144–1151, 2011. doi: 10.1681/ASN.2010101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687, 2002. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 278. Nishimura SL. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol 175: 1362–1370, 2009. doi: 10.2353/ajpath.2009.090393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Worthington JJ, Klementowicz JE, Travis MA. TGFβ: a sleeping giant awoken by integrins. Trends Biochem Sci 36: 47–54, 2011. doi: 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 280. Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM, Sheppard D, Fogo AB. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6-/- mice. Am J Pathol 163: 1261–1273, 2003. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Chang Y, Lau WL, Jo H, Tsujino K, Gewin L, Reed NI, Atakilit A, Nunes AC, DeGrado WF, Sheppard D. Pharmacologic blockade of αvβ1 integrin ameliorates renal failure and fibrosis in vivo. J Am Soc Nephrol 28: 1998–2005, 2017. doi: 10.1681/ASN.2015050585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Li S, Jiang S, Zhang Q, Jin B, Lv D, Li W, Zhao M, Jiang C, Dai C, Liu Z. Integrin β3 induction promotes tubular cell senescence and kidney fibrosis. Front Cell Dev Biol 9: 733831, 2021. doi: 10.3389/fcell.2021.733831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Zhou J, Li R, Zhang J, Liu Q, Wu T, Tang Q, Huang C, Zhang Z, Huang Y, Huang H, Zhang G, Zhao Y, Zhang T, Mo L, Li Y, He J. Targeting interstitial myofibroblast-expressed integrin αvβ3 alleviates renal fibrosis. Mol Pharm 18: 1373–1385, 2021. doi: 10.1021/acs.molpharmaceut.0c01182. [DOI] [PubMed] [Google Scholar]
- 284. Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature 379: 91–96, 1996. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- 285. Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest 112: 503–516, 2003. doi: 10.1172/JCI200317913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Li M, Zhou H, Di J, Yang M, Jia F. ILK participates in renal interstitial fibrosis by altering the phenotype of renal tubular epithelial cells via TGF-β1/smad pathway. Eur Rev Med Pharmacol Sci 23: 289–296, 2019. doi: 10.26355/eurrev_201901_16775. [DOI] [PubMed] [Google Scholar]
- 287. Li Y, Tan X, Dai C, Stolz DB, Wang D, Liu Y. Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol 20: 1907–1918, 2009. doi: 10.1681/ASN.2008090930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Zhou X, Zhang J, Haimbach R, Zhu W, Mayer-Ezell R, Garcia-Calvo M, Smith E, Price O, Kan Y, Zycband E, Zhu Y, Hoek M, Cox JM, Ma L, Kelley DE, Pinto S. An integrin antagonist (MK-0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmacol Res Perspect 5: e00354, 2017. doi: 10.1002/prp2.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Cox JM, Ma L, Zhou X, Haimbach RE, Coleman PJ, Zhou H, Kelley DE, Stoch SA, Duong LT, Hoek M. Composition and Methods for Treating Chronic Kidney Disease. US Patent US20180110762. March 24, 2018.
- 290. Distler JH, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol 15: 705–730, 2019. doi: 10.1038/s41584-019-0322-7. [DOI] [PubMed] [Google Scholar]
- 291. Lv W, Booz GW, Wang Y, Fan F, Roman RJ. Inflammation and renal fibrosis: recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharmacol 820: 65–76, 2018. doi: 10.1016/j.ejphar.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Klinkhammer BM, Floege J, Boor P. PDGF in organ fibrosis. Mol Aspects Med 62: 44–62, 2018. doi: 10.1016/j.mam.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 293. Sommer M, Eismann U, Deuther-Conrad W, Wendt T, Mohorn T, Fünfstück R, Stein G. Time course of cytokine mRNA expression in kidneys of rats with unilateral ureteral obstruction. Nephron 84: 49–57, 2000. doi: 10.1159/000045538. [DOI] [PubMed] [Google Scholar]
- 294. Taneda S, Hudkins KL, Topouzis S, Gilbertson DG, Ophascharoensuk V, Truong L, Johnson RJ, Alpers CE. Obstructive uropathy in mice and humans: potential role for PDGF-D in the progression of tubulointerstitial injury. J Am Soc Nephrol 14: 2544–2555, 2003. doi: 10.1097/01.asn.0000089828.73014.c8. [DOI] [PubMed] [Google Scholar]
- 295. Buhl EM, Djudjaj S, Babickova J, Klinkhammer BM, Folestad E, Borkham-Kamphorst E, Weiskirchen R, Hudkins K, Alpers CE, Eriksson U, Floege J, Boor P. The role of PDGF-D in healthy and fibrotic kidneys. Kidney Int 89: 848–861, 2016. doi: 10.1016/j.kint.2015.12.037. [DOI] [PubMed] [Google Scholar]
- 296. Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ, Duffield JS, Lin SL. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 80: 1170–1181, 2011. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 297. Poosti F, Bansal R, Yazdani S, Prakash J, Post E, Klok P, van den Born J, de Borst MH, van Goor H, Poelstra K, Hillebrands JL. Selective delivery of IFN-γ to renal interstitial myofibroblasts: a novel strategy for the treatment of renal fibrosis. FASEB J 29: 1029–1042, 2015. doi: 10.1096/fj.14-258459. [DOI] [PubMed] [Google Scholar]
- 298. Perbal B, Tweedie S, Bruford E. The official unified nomenclature adopted by the HGNC calls for the use of the acronyms, CCN1-6, and discontinuation in the use of CYR61, CTGF, NOV and WISP 1-3 respectively. J Cell Commun Signal 12: 625–629, 2018. doi: 10.1007/s12079-018-0491-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 130: 2779–2791, 2003. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Chen Z, Zhang N, Chu HY, Yu Y, Zhang ZK, Zhang G, Zhang BT. Connective tissue growth factor: from molecular understandings to drug discovery. Front Cell Dev Biol 8: 593269, 2020. doi: 10.3389/fcell.2020.593269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Rayego-Mateos S, Campillo S, Rodrigues-Diez RR, Tejera-Muñoz A, Marquez-Exposito L, Goldschmeding R, Rodríguez-Puyol D, Calleros L, Ruiz-Ortega M. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin Sci (Lond) 135: 1999–2029, 2021. doi: 10.1042/CS20201016. [DOI] [PubMed] [Google Scholar]
- 302. Kaasbøll OJ, Gadicherla AK, Wang JH, Monsen VT, Hagelin EM, Dong MQ, Attramadal H. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J Biol Chem 293: 17953–17970, 2018. doi: 10.1074/jbc.RA118.004559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Mason RM. Fell-Muir lecture: Connective tissue growth factor (CCN2)—a pernicious and pleiotropic player in the development of kidney fibrosis. Int J Exp Pathol 94: 1–16, 2013. doi: 10.1111/j.1365-2613.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Ramazani Y, Knops N, Elmonem MA, Nguyen TQ, Arcolino FO, van den Heuvel L, Levtchenko E, Kuypers D, Goldschmeding R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol 68-69: 44–66, 2018. doi: 10.1016/j.matbio.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 305. Yin Q, Liu H. Connective tissue growth factor and renal fibrosis. Adv Exp Med Biol 1165: 365–380, 2019. doi: 10.1007/978-981-13-8871-2_17. [DOI] [PubMed] [Google Scholar]
- 306. Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 53: 853–861, 1998. doi: 10.1111/j.1523-1755.1998.00820.x. [DOI] [PubMed] [Google Scholar]
- 307. Yokoi H, Mukoyama M, Nagae T, Mori K, Suganami T, Sawai K, Yoshioka T, Koshikawa M, Nishida T, Takigawa M, Sugawara A, Nakao K. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol 15: 1430–1440, 2004. doi: 10.1097/01.asn.0000130565.69170.85. [DOI] [PubMed] [Google Scholar]
- 308. Adler SG, Schwartz S, Williams ME, Arauz-Pacheco C, Bolton WK, Lee T, Li D, Neff TB, Urquilla PR, Sewell KL. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin J Am Soc Nephrol 5: 1420–1428, 2010. doi: 10.2215/CJN.09321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Rubartelli A, Lotze MT, Latz E, Manfredi A. Mechanisms of sterile inflammation. Front Immunol 4: 398, 2013. doi: 10.3389/fimmu.2013.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Chen WY, Chang YJ, Su CH, Tsai TH, Chen SD, Hsing CH, Yang JL. Upregulation of Interleukin-33 in obstructive renal injury. Biochem Biophys Res Commun 473: 1026–1032, 2016. doi: 10.1016/j.bbrc.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 311. Braga TT, Forni MF, Correa-Costa M, Ramos RN, Barbuto JA, Branco P, Castoldi A, Hiyane MI, Davanso MR, Latz E, Franklin BS, Kowaltowski AJ, Camara NO. Soluble uric acid activates the NLRP3 inflammasome. Sci Rep 7: 39884, 2017. doi: 10.1038/srep39884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Tammaro A, Florquin S, Brok M, Claessen N, Butter LM, Teske GJ, de Boer OJ, Vogl T, Leemans JC, Dessing MC. S100A8/A9 promotes parenchymal damage and renal fibrosis in obstructive nephropathy. Clin Exp Immunol 193: 361–375, 2018. doi: 10.1111/cei.13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Tian S, Zhang L, Tang J, Guo X, Dong K, Chen SY. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol 308: F69–F75, 2015. doi: 10.1152/ajprenal.00484.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Wyczanska M, Lange-Sperandio B. DAMPs in unilateral ureteral obstruction. Front Immunol 11: 581300, 2020. doi: 10.3389/fimmu.2020.581300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol 25: 1387–1400, 2014. doi: 10.1681/ASN.2014010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Solini A, Usuelli V, Fiorina P. The dark side of extracellular ATP in kidney diseases. J Am Soc Nephrol 26: 1007–1016, 2015. doi: 10.1681/ASN.2014070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Bignold R, Johnson JR. Effects of cytokine signaling inhibition on inflammation-driven tissue remodeling. Curr Res Pharmacol Drug Discov 2: 100023, 2021. doi: 10.1016/j.crphar.2021.100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Bani-Hani AH, Leslie JA, Asanuma H, Dinarello CA, Campbell MT, Meldrum DR, Zhang H, Hile K, Meldrum KK. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int 76: 500–511, 2009. doi: 10.1038/ki.2009.216. [DOI] [PubMed] [Google Scholar]
- 319. Hung AM, Ellis CD, Shintani A, Booker C, Ikizler TA. IL-1β receptor antagonist reduces inflammation in hemodialysis patients. J Am Soc Nephrol 22: 437–442, 2011. doi: 10.1681/ASN.2010070760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Masola V, Carraro A, Granata S, Signorini L, Bellin G, Violi P, Lupo A, Tedeschi U, Onisto M, Gambaro G, Zaza G. In vitro effects of interleukin (IL)-1 beta inhibition on the epithelial-to-mesenchymal transition (EMT) of renal tubular and hepatic stellate cells. J Transl Med 17: 12, 2019. doi: 10.1186/s12967-019-1770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Corden B, Adami E, Sweeney M, Schafer S, Cook SA. IL-11 in cardiac and renal fibrosis: Late to the party but a central player. Br J Pharmacol 177: 1695–1708, 2020. doi: 10.1111/bph.15013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Gonçalves RG, Gabrich L, Rosário A Jr, Takiya CM, Ferreira ML, Chiarini LB, Persechini PM, Coutinho-Silva R, Leite M Jr.. The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Kidney Int 70: 1599–1606, 2006. doi: 10.1038/sj.ki.5001804. [DOI] [PubMed] [Google Scholar]
- 323. Pereira JMS, Barreira AL, Gomes CR, Ornellas FM, Ornellas DS, Miranda LC, Cardoso LR, Coutinho-Silva R, Schanaider A, Morales MM, Leite M Jr, Takiya CM. Brilliant blue G, a P2X7 receptor antagonist, attenuates early phase of renal inflammation, interstitial fibrosis and is associated with renal cell proliferation in ureteral obstruction in rats. BMC Nephrol 21: 206, 2020. doi: 10.1186/s12882-020-01861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Vergani A, Tezza S, Fotino C, Visner G, Pileggi A, Chandraker A, Fiorina P. The purinergic system in allotransplantation. Am J Transplant 14: 507–514, 2014. doi: 10.1111/ajt.12567. [DOI] [PubMed] [Google Scholar]
- 325. Cronstein BN. Adenosine receptors and fibrosis: a translational review. F1000 Biol Rep 3: 21, 2011. doi: 10.3410/B3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Dwyer KM, Kishore BK, Robson SC. Conversion of extracellular ATP into adenosine: a master switch in renal health and disease. Nat Rev Nephrol 16: 509–524, 2020. doi: 10.1038/s41581-020-0304-7. [DOI] [PubMed] [Google Scholar]
- 327. Roberts VS, Cowan PJ, Alexander SI, Robson SC, Dwyer KM. The role of adenosine receptors A2A and A2B signaling in renal fibrosis. Kidney Int 86: 685–692, 2014. doi: 10.1038/ki.2014.244. [DOI] [PubMed] [Google Scholar]
- 328. Vallon V, Mühlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev 86: 901–940, 2006. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 329. Antonioli L, Fornai M, Blandizzi C, Pacher P, Haskó G. Adenosine signaling and the immune system: when a lot could be too much. Immunol Lett 205: 9–15, 2019. doi: 10.1016/j.imlet.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 330. Ohta A, Sitkovsky M. The adenosinergic immunomodulatory drugs. Curr Opin Pharmacol 9: 501–506, 2009. doi: 10.1016/j.coph.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Xiao H, Shen HY, Liu W, Xiong RP, Li P, Meng G, Yang N, Chen X, Si LY, Zhou YG. Adenosine A2A receptor: a target for regulating renal interstitial fibrosis in obstructive nephropathy. PLoS One 8: e60173, 2013. doi: 10.1371/journal.pone.0060173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Xiao H, Si LY, Liu W, Li N, Meng G, Yang N, Chen X, Zhou YG, Shen HY. The effects of adenosine A2A receptor knockout on renal interstitial fibrosis in a mouse model of unilateral ureteral obstruction. Acta Histochem 115: 315–319, 2013. doi: 10.1016/j.acthis.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 333. Lange-Sperandio B, Forbes MS, Thornhill B, Okusa MD, Linden J, Chevalier RL. A2A adenosine receptor agonist and PDE4 inhibition delays inflammation but fails to reduce injury in experimental obstructive nephropathy. Nephron Exp Nephrol 100: e113–e123, 2005. doi: 10.1159/000085057. [DOI] [PubMed] [Google Scholar]
- 334. Dai Y, Zhang W, Wen J, Zhang Y, Kellems RE, Xia Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD. J Am Soc Nephrol 22: 890–901, 2011. doi: 10.1681/ASN.2010080890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Pak ES, Jeong LS, Hou X, Tripathi SK, Lee J, Ha H. Dual actions of A2A and A3 adenosine receptor ligand prevents obstruction-induced kidney fibrosis in mice. Int J Mol Sci 22: 5667, 2021. doi: 10.3390/ijms22115667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10: 417–426, 2002. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 337. Zhang WJ, Chen SJ, Zhou SC, Wu SZ, Wang H. Inflammasomes and fibrosis. Front Immunol 12: 643149, 2021. doi: 10.3389/fimmu.2021.643149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Huang G, Zhang Y, Zhang Y, Ma Y. Chronic kidney disease and NLRP3 inflammasome: Pathogenesis, development and targeted therapeutic strategies. Biochem Biophys Rep 33: 101417, 2023. doi: 10.1016/j.bbrep.2022.101417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Mulay SR. Multifactorial functions of the inflammasome component NLRP3 in pathogenesis of chronic kidney diseases. Kidney Int 96: 58–66, 2019. doi: 10.1016/j.kint.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 340. Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, Hemmelgarn BR, Beck PL, Muruve DA. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 21: 1732–1744, 2010. doi: 10.1681/ASN.2010020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Guo H, Bi X, Zhou P, Zhu S, Ding W. NLRP3 deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators Inflamm 2017: 8316560, 2017. doi: 10.1155/2017/8316560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Pulskens WP, Butter LM, Teske GJ, Claessen N, Dessing MC, Flavell RA, Sutterwala FS, Florquin S, Leemans JC. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One 9: e85775, 2014. doi: 10.1371/journal.pone.0085775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Seo JB, Choi YK, Woo HI, Jung YA, Lee S, Lee S, Park M, Lee IK, Jung GS, Park KG. Gemigliptin attenuates renal fibrosis through down-regulation of the NLRP3 inflammasome. Diabetes Metab J 43: 830–839, 2019. doi: 10.4093/dmj.2018.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, Wang A, Tao J, Wang C, Liu Q, Jin T, Jiang W, Deng X, Zhou R. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214: 3219–3238, 2017. doi: 10.1084/jem.20171419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, Carta S, Tengesdal I, Nemkov T, D’Alessandro A, Henry C, Jones GS, Goodrich SA, St Laurent JP, Jones TM, Scribner CL, Barrow RB, Altman RD, Skouras DB, Gattorno M, Grau V, Janciauskiene S, Rubartelli A, Joosten LAB, Dinarello CA. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA 115: E1530–E1539, 2018. doi: 10.1073/pnas.1716095115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Elsayed MS, Abu-Elsaad NM, Nader MA. The NLRP3 inhibitor dapansutrile attenuates folic acid induced nephrotoxicity via inhibiting inflammasome/caspase-1/IL axis and regulating autophagy/proliferation. Life Sci 285: 119974, 2021. doi: 10.1016/j.lfs.2021.119974. [DOI] [PubMed] [Google Scholar]
- 347. Yang M, Zhao L. The selective NLRP3-inflammasome inhibitor CY-09 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3-inflammasome activation. Curr Med Chem 30: 3261–3270, 2023. doi: 10.2174/0929867329666220922104654. [DOI] [PubMed] [Google Scholar]
- 348. Klahr S, Morrissey JJ. The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int Suppl 75: S7–S14, 2000. [PubMed] [Google Scholar]
- 349. Kaneto H, Morrissey JJ, McCracken R, Ishidoya S, Reyes AA, Klahr S. The expression of mRNA for tumour necrosis factor-α increases in the obstructed kidney of rats soon after unilateral ureteral ligation. Nephrology 2: 161–166, 1996. doi: 10.1111/j.1440-1797.1996.tb00082.x. [DOI] [Google Scholar]
- 350. Guo G, Morrissey J, McCracken R, Tolley T, Klahr S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am J Physiol Renal Physiol 277: F766–F772, 1999. doi: 10.1152/ajprenal.1999.277.5.F766. [DOI] [PubMed] [Google Scholar]
- 351. Guo G, Morrissey J, McCracken R, Tolley T, Liapis H, Klahr S. Contributions of angiotensin II and tumor necrosis factor-alpha to the development of renal fibrosis. Am J Physiol Renal Physiol 280: F777–F785, 2001. doi: 10.1152/ajprenal.2001.280.5.F777. [DOI] [PubMed] [Google Scholar]
- 352. Moriwaki Y, Inokuchi T, Yamamoto A, Ka T, Tsutsumi Z, Takahashi S, Yamamoto T. Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic rats. Acta Diabetol 44: 215–218, 2007. doi: 10.1007/s00592-007-0007-6. [DOI] [PubMed] [Google Scholar]
- 353. Omote K, Gohda T, Murakoshi M, Sasaki Y, Kazuno S, Fujimura T, Ishizaka M, Sonoda Y, Tomino Y. Role of the TNF pathway in the progression of diabetic nephropathy in KK-Ay mice. Am J Physiol Renal Physiol 306: F1335–F1347, 2014. doi: 10.1152/ajprenal.00509.2013. [DOI] [PubMed] [Google Scholar]
- 354. Therrien FJ, Agharazii M, Lebel M, Larivière R. Neutralization of tumor necrosis factor-alpha reduces renal fibrosis and hypertension in rats with renal failure. Am J Nephrol 36: 151–161, 2012. doi: 10.1159/000340033. [DOI] [PubMed] [Google Scholar]
- 355. Aranda-Rivera AK, Cruz-Gregorio A, Aparicio-Trejo OE, Ortega-Lozano AJ, Pedraza-Chaverri J. Redox signaling pathways in unilateral ureteral obstruction (UUO)-induced renal fibrosis. Free Radic Biol Med 172: 65–81, 2021. doi: 10.1016/j.freeradbiomed.2021.05.034. [DOI] [PubMed] [Google Scholar]
- 356. Manucha W. Biochemical-molecular markers in unilateral ureteral obstruction. Biocell 31: 1–12, 2007. [PubMed] [Google Scholar]
- 357. Nakatani T, Tamada S, Asai T, Iwai Y, Kim T, Tsujino T, Kumata N, Uchida J, Tashiro K, Kuwabara N, Komiya T, Sumi T, Okamura M, Miura K. Role of renin-angiotensin system and nuclear factor-kappaB in the obstructed kidney of rats with unilateral ureteral obstruction. Jpn J Pharmacol 90: 361–364, 2002. doi: 10.1254/jjp.90.361. [DOI] [PubMed] [Google Scholar]
- 358. Hosseinian S, Rad AK, Bideskan AE, Soukhtanloo M, Sadeghnia H, Shafei MN, Motejadded F, Mohebbati R, Shahraki S, Beheshti F. Thymoquinone ameliorates renal damage in unilateral ureteral obstruction in rats. Pharmacol Rep 69: 648–657, 2017. doi: 10.1016/j.pharep.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 359. Esteban V, Lorenzo O, Rupérez M, Suzuki Y, Mezzano S, Blanco J, Kretzler M, Sugaya T, Egido J, Ruiz-Ortega M. Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol 15: 1514–1529, 2004. doi: 10.1097/01.asn.0000130564.75008.f5. [DOI] [PubMed] [Google Scholar]
- 360. Morrissey JJ, Klahr S. Enalapril decreases nuclear factor kappa B activation in the kidney with ureteral obstruction. Kidney Int 52: 926–933, 1997. doi: 10.1038/ki.1997.414. [DOI] [PubMed] [Google Scholar]
- 361. Ishidoya S, Morrissey J, McCracken R, Reyes A, Klahr S. Angiotensin II receptor antagonist ameliorates renal tubulointerstitial fibrosis caused by unilateral ureteral obstruction. Kidney Int 47: 1285–1294, 1995. doi: 10.1038/ki.1995.183. [DOI] [PubMed] [Google Scholar]
- 362. Klahr S, Ishidoya S, Morrissey J. Role of angiotensin II in the tubulointerstitial fibrosis of obstructive nephropathy. Am J Kidney Dis 26: 141–146, 1995. doi: 10.1016/0272-6386(95)90167-1. [DOI] [PubMed] [Google Scholar]
- 363. Kaneto H, Morrissey J, McCracken R, Reyes A, Klahr S. Enalapril reduces collagen type IV synthesis and expansion of the interstitium in the obstructed rat kidney. Kidney Int 45: 1637–1647, 1994. doi: 10.1038/ki.1994.215. [DOI] [PubMed] [Google Scholar]
- 364. Satoh M, Kashihara N, Yamasaki Y, Maruyama K, Okamoto K, Maeshima Y, Sugiyama H, Sugaya T, Murakami K, Makino H. Renal interstitial fibrosis is reduced in angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol 12: 317–325, 2001. doi: 10.1681/ASN.V122317. [DOI] [PubMed] [Google Scholar]
- 365. Fern RJ, Yesko CM, Thornhill BA, Kim HS, Smithies O, Chevalier RL. Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest 103: 39–46, 1999. doi: 10.1172/JCI4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Nishida M, Fujinaka H, Matsusaka T, Price J, Kon V, Fogo AB, Davidson JM, Linton MF, Fazio S, Homma T, Yoshida H, Ichikawa I. Absence of angiotensin II type 1 receptor in bone marrow-derived cells is detrimental in the evolution of renal fibrosis. J Clin Invest 110: 1859–1868, 2002. doi: 10.1172/JCI15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Martínez-Klimova E, Aparicio-Trejo OE, Gómez-Sierra T, Jiménez-Uribe AP, Bellido B, Pedraza-Chaverri J. Mitochondrial dysfunction and endoplasmic reticulum stress in the promotion of fibrosis in obstructive nephropathy induced by unilateral ureteral obstruction. BioFactors 46: 716–733, 2020. doi: 10.1002/biof.1673. [DOI] [PubMed] [Google Scholar]
- 368. Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol 302: 148–158, 2009. doi: 10.1016/j.mce.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Kim S, Kim SJ, Yoon HE, Chung S, Choi BS, Park CW, Shin SJ. Fimasartan, a novel angiotensin-receptor blocker, protects against renal inflammation and fibrosis in mice with unilateral ureteral obstruction: the possible role of Nrf2. Int J Med Sci 12: 891–904, 2015. doi: 10.7150/ijms.13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Nlandu Khodo S, Dizin E, Sossauer G, Szanto I, Martin PY, Feraille E, Krause KH, de Seigneux S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol 23: 1967–1976, 2012. doi: 10.1681/ASN.2012040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Wang H, Qian J, Zhao X, Xing C, Sun B. β-Aminoisobutyric acid ameliorates the renal fibrosis in mouse obstructed kidneys via inhibition of renal fibroblast activation and fibrosis. J Pharmacol Sci 133: 203–213, 2017. doi: 10.1016/j.jphs.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 372. Cheng X, Zheng X, Song Y, Qu L, Tang J, Meng L, Wang Y. Apocynin attenuates renal fibrosis via inhibition of NOXs-ROS-ERK-myofibroblast accumulation in UUO rats. Free Radic Res 50: 840–852, 2016. doi: 10.1080/10715762.2016.1181757. [DOI] [PubMed] [Google Scholar]
- 373. Sugiyama H, Kobayashi M, Wang DH, Sunami R, Maeshima Y, Yamasaki Y, Masuoka N, Kira S, Makino H. Telmisartan inhibits both oxidative stress and renal fibrosis after unilateral ureteral obstruction in acatalasemic mice. Nephrol Dial Transplant 20: 2670–2680, 2005. doi: 10.1093/ndt/gfi045. [DOI] [PubMed] [Google Scholar]
- 374. Figueroa SM, Lozano M, Lobos C, Hennrikus MT, Gonzalez AA, Amador CA. Upregulation of cortical renin and downregulation of medullary (pro)renin receptor in unilateral ureteral obstruction. Front Pharmacol 10: 1314, 2019. doi: 10.3389/fphar.2019.01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 376. Bian X, Bai Y, Su X, Zhao G, Sun G, Li D. Knockdown of periostin attenuates 5/6 nephrectomy-induced intrarenal renin-angiotensin system activation, fibrosis, and inflammation in rats. J Cell Physiol 234: 22857–22873, 2019. doi: 10.1002/jcp.28849. [DOI] [PubMed] [Google Scholar]
- 377. Shin GT, Kim WH, Yim H, Kim MS, Kim H. Effects of suppressing intrarenal angiotensinogen on renal transforming growth factor-beta1 expression in acute ureteral obstruction. Kidney Int 67: 897–908, 2005. doi: 10.1111/j.1523-1755.2005.00154.x. [DOI] [PubMed] [Google Scholar]
- 378. Kalaitzidis RG, Elisaf MS. Treatment of hypertension in chronic kidney disease. Curr Hypertens Rep 20: 64, 2018. doi: 10.1007/s11906-018-0864-0. [DOI] [PubMed] [Google Scholar]
- 379. Shabaka A, Cases-Corona C, Fernandez-Juarez G. Therapeutic insights in chronic kidney disease progression. Front Med (Lausanne) 8: 645187, 2021. doi: 10.3389/fmed.2021.645187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Nørregaard R, Jensen BL, Topcu SO, Nielsen SS, Walter S, Djurhuus JC, Frøkiaer J. Cyclooxygenase type 2 is increased in obstructed rat and human ureter and contributes to pelvic pressure increase after obstruction. Kidney Int 70: 872–881, 2006. doi: 10.1038/sj.ki.5001616. [DOI] [PubMed] [Google Scholar]
- 381. Whinnery MA, Shaw JO, Beck N. Thromboxane B2 and prostaglandin E2 in the rat kidney with unilateral ureteral obstruction. Am J Physiol Renal Physiol 242: F220–F225, 1982. doi: 10.1152/ajprenal.1982.242.3.F220. [DOI] [PubMed] [Google Scholar]
- 382. Honma S, Shinohara M, Takahashi N, Nakamura K, Hamano S, Mitazaki S, Abe S, Yoshida M. Effect of cyclooxygenase (COX)-2 inhibition on mouse renal interstitial fibrosis. Eur J Pharmacol 740: 578–583, 2014. doi: 10.1016/j.ejphar.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 383. Miyajima A, Ito K, Asano T, Seta K, Ueda A, Hayakawa M. Does cyclooxygenase-2 inhibitor prevent renal tissue damage in unilateral ureteral obstruction? J Urol 166: 1124–1129, 2001. doi: 10.1016/S0022-5347(05)65933-2. [DOI] [PubMed] [Google Scholar]
- 384. Nawroth I, Alsner J, Deleuran BW, Dagnaes-Hansen F, Yang C, Horsman MR, Overgaard J, Howard KA, Kjems J, Gao S. Peritoneal macrophages mediated delivery of chitosan/siRNA nanoparticle to the lesion site in a murine radiation-induced fibrosis model. Acta Oncol 52: 1730–1738, 2013. doi: 10.3109/0284186X.2012.726373. [DOI] [PubMed] [Google Scholar]
- 385. Yang C, Nilsson L, Cheema MU, Wang Y, Frøkiær J, Gao S, Kjems J, Nørregaard R. Chitosan/siRNA nanoparticles targeting cyclooxygenase type 2 attenuate unilateral ureteral obstruction-induced kidney injury in mice. Theranostics 5: 110–123, 2015. doi: 10.7150/thno.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Yang C, Vu-Quang H, Husum DM, Tingskov SJ, Vinding MS, Nielsen T, Song P, Nielsen NC, Nørregaard R, Kjems J. Theranostic poly(lactic-co-glycolic acid) nanoparticle for magnetic resonance/infrared fluorescence bimodal imaging and efficient siRNA delivery to macrophages and its evaluation in a kidney injury model. Nanomedicine 13: 2451–2462, 2017. doi: 10.1016/j.nano.2017.08.007. [DOI] [PubMed] [Google Scholar]
- 387. Kamata M, Hosono K, Fujita T, Kamata K, Majima M. Role of cyclooxygenase-2 in the development of interstitial fibrosis in kidneys following unilateral ureteral obstruction in mice. Biomed Pharmacother 70: 174–180, 2015. doi: 10.1016/j.biopha.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 388. Nilsson L, Madsen K, Krag S, Frøkiær J, Jensen BL, Nørregaard R. Disruption of cyclooxygenase type 2 exacerbates apoptosis and renal damage during obstructive nephropathy. Am J Physiol Renal Physiol 309: F1035–F048, 2015. doi: 10.1152/ajprenal.00253.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Luo R, Kakizoe Y, Wang F, Fan X, Hu S, Yang T, Wang W, Li C. Deficiency of mPGES-1 exacerbates renal fibrosis and inflammation in mice with unilateral ureteral obstruction. Am J Physiol Renal Physiol 312: F121–F133, 2017. doi: 10.1152/ajprenal.00231.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Mutsaers HA, Nørregaard R. Prostaglandin E2 receptors as therapeutic targets in renal fibrosis. Kidney Res Clin Pract 41: 4–13, 2022. doi: 10.23876/j.krcp.21.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Sun Y, Zhang Y, Zhu Y, Zhang A, Huang S, Yin X, Ding G, Liu M, Jia Z. Inhibition of mitochondrial complex-1 restores the downregulation of aquaporins in obstructive nephropathy. Am J Physiol Renal Physiol 311: F777–F786, 2016. doi: 10.1152/ajprenal.00215.2015. [DOI] [PubMed] [Google Scholar]
- 392. Jensen MS, Mutsaers HA, Tingskov SJ, Christensen M, Madsen MG, Olinga P, Kwon TH, Nørregaard R. Activation of the prostaglandin E2 EP2 receptor attenuates renal fibrosis in unilateral ureteral obstructed mice and human kidney slices. Acta Physiol (Oxf) 227: e13291, 2019. doi: 10.1111/apha.13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Stribos EG, Luangmonkong T, Leliveld AM, de Jong IJ, van Son WJ, Hillebrands JL, Seelen MA, van Goor H, Olinga P, Mutsaers HA. Precision-cut human kidney slices as a model to elucidate the process of renal fibrosis. Transl Res 170: 8–16.e11, 2016. doi: 10.1016/j.trsl.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 394. Nakagawa N, Yuhki K, Kawabe J, Fujino T, Takahata O, Kabara M, Abe K, Kojima F, Kashiwagi H, Hasebe N, Kikuchi K, Sugimoto Y, Narumiya S, Ushikubi F. The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int 82: 158–171, 2012. doi: 10.1038/ki.2012.115. [DOI] [PubMed] [Google Scholar]
- 395. Kresse JC, Mutsaers HA, Jensen MS, Tingskov SJ, Madsen MG, Nejsum LN, Praetorius H, Nørregaard R. EP1 receptor antagonism mitigates early and late stage renal fibrosis. Acta Physiol (Oxf) 234: e13780, 2022. doi: 10.1111/apha.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Li S, Wang Y, Chen L, Wang Z, Liu G, Zuo B, Liu C, Sun D. Beraprost sodium mitigates renal interstitial fibrosis through repairing renal microvessels. J Mol Med (Berl) 97: 777–791, 2019. doi: 10.1007/s00109-019-01769-x. [DOI] [PubMed] [Google Scholar]
- 397. Nasu T, Kinomura M, Tanabe K, Yamasaki H, Htay SL, Saito D, Hinamoto N, Watatani H, Ujike H, Suzuki Y, Sugaya T, Sugiyama H, Sakai Y, Matsumoto K, Maeshima Y, Makino H. Sustained-release prostacyclin analog ONO-1301 ameliorates tubulointerstitial alterations in a mouse obstructive nephropathy model. Am J Physiol Renal Physiol 302: F1616–F1629, 2012. doi: 10.1152/ajprenal.00538.2011. [DOI] [PubMed] [Google Scholar]
- 398. Li Y, Xia W, Zhao F, Wen Z, Zhang A, Huang S, Jia Z, Zhang Y. Prostaglandins in the pathogenesis of kidney diseases. Oncotarget 9: 26586–26602, 2018. doi: 10.18632/oncotarget.25005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Ito H, Yan X, Nagata N, Aritake K, Katsumata Y, Matsuhashi T, Nakamura M, Hirai H, Urade Y, Asano K, Kubo M, Utsunomiya Y, Hosoya T, Fukuda K, Sano M. PGD2-CRTH2 pathway promotes tubulointerstitial fibrosis. J Am Soc Nephrol 23: 1797–1809, 2012. doi: 10.1681/ASN.2012020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Harirforoosh S, Asghar W, Jamali F. Adverse effects of nonsteroidal antiinflammatory drugs: an update of gastrointestinal, cardiovascular and renal complications. J Pharm Pharm Sci 16: 821–847, 2013. doi: 10.18433/j3vw2f. [DOI] [PubMed] [Google Scholar]
- 401. Harris RC, Breyer MD. Update on cyclooxygenase-2 inhibitors. Clin J Am Soc Nephrol 1: 236–245, 2006. doi: 10.2215/CJN.00890805. [DOI] [PubMed] [Google Scholar]
- 402. Gooch K, Culleton BF, Manns BJ, Zhang J, Alfonso H, Tonelli M, Frank C, Klarenbach S, Hemmelgarn BR. NSAID use and progression of chronic kidney disease. Am J Med 120: 280.e1–280.e7, 2007. doi: 10.1016/j.amjmed.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 403. Whelton A, Maurath CJ, Verburg KM, Geis GS. Renal safety and tolerability of celecoxib, a novel cyclooxygenase-2 inhibitor. Am J Ther 7: 159–175, 2000. doi: 10.1097/00045391-200007030-00004. [DOI] [PubMed] [Google Scholar]
- 404. Kohan DE, Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 86: 896–904, 2014. doi: 10.1038/ki.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Wendel M, Knels L, Kummer W, Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J Histochem Cytochem 54: 1193–1203, 2006. doi: 10.1369/jhc.5A6888.2006. [DOI] [PubMed] [Google Scholar]
- 406. Hegarty NJ, Young LS, O’Neill AJ, Watson RW, Fitzpatrick JM. Endothelin in unilateral ureteral obstruction: vascular and cellular effects. J Urol 169: 740–744, 2003. doi: 10.1097/01.ju.0000036813.52746.89. [DOI] [PubMed] [Google Scholar]
- 407. Chang YK, Choi H, Jeong JY, Na KR, Lee KW, Choi DE. Co-inhibition of angiotensin II Receptor and endothelin-1 attenuates renal injury in unilateral ureteral obstructed mice. Kidney Blood Press Res 41: 450–459, 2016. doi: 10.1159/000443446. [DOI] [PubMed] [Google Scholar]
- 408. Moridaira K, Morrissey J, Fitzgerald M, Guo G, McCracken R, Tolley T, Klahr S. ACE inhibition increases expression of the ETB receptor in kidneys of mice with unilateral obstruction. Am J Physiol Renal Physiol 284: F209–F217, 2003. doi: 10.1152/ajprenal.00352.2001. [DOI] [PubMed] [Google Scholar]
- 409. Neder TH, Schrankl J, Fuchs MA, Broeker KA, Wagner C. Endothelin receptors in renal interstitial cells do not contribute to the development of fibrosis during experimental kidney disease. Pflugers Arch 473: 1667–1683, 2021. doi: 10.1007/s00424-021-02604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Feldman DL, Mogelesky TC, Chou M, Jeng AY. Enhanced expression of renal endothelin-converting enzyme-1 and endothelin-A-receptor mRNA in rats with interstitial fibrosis following ureter ligation. J Cardiovasc Pharmacol 36: S255–S259, 2000. doi: 10.1097/00005344-200036051-00075. [DOI] [PubMed] [Google Scholar]
- 411. Goddard J, Johnston NR, Hand MF, Cumming AD, Rabelink TJ, Rankin AJ, Webb DJ. Endothelin-A receptor antagonism reduces blood pressure and increases renal blood flow in hypertensive patients with chronic renal failure: a comparison of selective and combined endothelin receptor blockade. Circulation 109: 1186–1193, 2004. doi: 10.1161/01.CIR.0000118499.69469.51. [DOI] [PubMed] [Google Scholar]
- 412. Dhaun N, Macintyre IM, Melville V, Lilitkarntakul P, Johnston NR, Goddard J, Webb DJ. Blood pressure-independent reduction in proteinuria and arterial stiffness after acute endothelin-a receptor antagonism in chronic kidney disease. Hypertension 54: 113–119, 2009. doi: 10.1161/HYPERTENSIONAHA.109.132670. [DOI] [PubMed] [Google Scholar]
- 413. Dhaun N, MacIntyre IM, Kerr D, Melville V, Johnston NR, Haughie S, Goddard J, Webb DJ. Selective endothelin-A receptor antagonism reduces proteinuria, blood pressure, and arterial stiffness in chronic proteinuric kidney disease. Hypertension 57: 772–779, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167486. [DOI] [PubMed] [Google Scholar]
- 414. Kohan DE, Pritchett Y, Molitch M, Wen S, Garimella T, Audhya P, Andress DL. Addition of atrasentan to renin-angiotensin system blockade reduces albuminuria in diabetic nephropathy. J Am Soc Nephrol 22: 763–772, 2011. doi: 10.1681/ASN.2010080869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Leipziger J, Praetorius H. Renal autocrine and paracrine signaling: a story of self-protection. Physiol Rev 100: 1229–1289, 2020. doi: 10.1152/physrev.00014.2019. [DOI] [PubMed] [Google Scholar]
- 417. Hewitson TD, Holt SG, Smith ER. Progression of tubulointerstitial fibrosis and the chronic kidney disease phenotype—role of risk factors and epigenetics. Front Pharmacol 8: 520, 2017. doi: 10.3389/fphar.2017.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Jung HJ, Park EJ, Choi HJ, Kwon TH. Regulation of aquaporin-2 by RNA interference. Vitam Horm 112: 119–145, 2020. doi: 10.1016/bs.vh.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 419. Park EJ, Jung HJ, Choi HJ, Cho JI, Park HJ, Kwon TH. miR-34c-5p and CaMKII are involved in aldosterone-induced fibrosis in kidney collecting duct cells. Am J Physiol Renal Physiol 314: F329–F342, 2018. doi: 10.1152/ajprenal.00358.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Kim JE, Jung HJ, Lee YJ, Kwon TH. Vasopressin-regulated miRNAs and AQP2-targeting miRNAs in kidney collecting duct cells. Am J Physiol Renal Physiol 308: F749–F764, 2015. doi: 10.1152/ajprenal.00334.2014. [DOI] [PubMed] [Google Scholar]
- 421. Ranieri M, Zahedi K, Tamma G, Centrone M, Di Mise A, Soleimani M, Valenti G. CaSR signaling down-regulates AQP2 expression via a novel microRNA pathway in pendrin and NaCl cotransporter knockout mice. FASEB J 32: 2148–2159, 2018. doi: 10.1096/fj.201700412RR. [DOI] [PubMed] [Google Scholar]
- 422. Fu H, Gu YH, Yang YN, Liao S, Wang GH. MiR-200b/c family inhibits renal fibrosis through modulating epithelial-to-mesenchymal transition via targeting fascin-1/CD44 axis. Life Sci 252: 117589, 2020. doi: 10.1016/j.lfs.2020.117589. [DOI] [PubMed] [Google Scholar]
- 423. Yanai K, Kaneko S, Ishii H, Aomatsu A, Ito K, Hirai K, Ookawara S, Ishibashi K, Morishita Y. Quantitative real-time PCR evaluation of microRNA expressions in mouse kidney with unilateral ureteral obstruction. J Vis Exp 162: 61383, 2020. doi: 10.3791/61383. [DOI] [PubMed] [Google Scholar]
- 424. Zhao S, Li W, Yu W, Rao T, Li H, Ruan Y, Yuan R, Li C, Ning J, Li S, Chen W, Cheng F, Zhou X. Exosomal miR-21 from tubular cells contributes to renal fibrosis by activating fibroblasts via targeting PTEN in obstructed kidneys. Theranostics 11: 8660–8673, 2021. doi: 10.7150/thno.62820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Ståhl AL, Johansson K, Mossberg M, Kahn R, Karpman D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr Nephrol 34: 11–30, 2019. doi: 10.1007/s00467-017-3816-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Liu Y, Bi X, Xiong J, Han W, Xiao T, Xu X, Yang K, Liu C, Jiang W, He T, Yu Y, Li Y, Zhang J, Zhang B, Zhao J. MicroRNA-34a promotes renal fibrosis by downregulation of klotho in tubular epithelial cells. Mol Ther 27: 1051–1065, 2019. doi: 10.1016/j.ymthe.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Zhou X, Zhao S, Li W, Ruan Y, Yuan R, Ning J, Jiang K, Xie J, Yao X, Li H, Li C, Rao T, Yu W, Cheng F. Tubular cell-derived exosomal miR-150-5p contributes to renal fibrosis following unilateral ischemia-reperfusion injury by activating fibroblast in vitro and in vivo. Int J Biol Sci 17: 4021–4033, 2021. doi: 10.7150/ijbs.62478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Qu NY, Zhang ZH, Zhang XX, Xie WW, Niu XQ. Microvesicles containing microRNA-216a secreted by tubular epithelial cells participate in renal interstitial fibrosis through activating PTEN/AKT pathway. Eur Rev Med Pharmacol Sci 23: 6629–6636, 2019. doi: 10.26355/eurrev_201908_18552. [DOI] [PubMed] [Google Scholar]
- 429. Zhang XF, Yang Y, Zhang J, Cao W. Microvesicle-containing miRNA-153-3p induces the apoptosis of proximal tubular epithelial cells and participates in renal interstitial fibrosis. Eur Rev Med Pharmacol Sci 23: 10065–10071, 2019. doi: 10.26355/eurrev_201911_19574. [DOI] [PubMed] [Google Scholar]
- 430. Yang R, Xu X, Li H, Chen J, Xiang X, Dong Z, Zhang D. p53 induces miR199a-3p to suppress SOCS7 for STAT3 activation and renal fibrosis in UUO. Sci Rep 7: 43409, 2017. doi: 10.1038/srep43409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Wang H, Wang B, Zhang A, Hassounah F, Seow Y, Wood M, Ma F, Klein JD, Price SR, Wang XH. Exosome-mediated miR-29 transfer reduces muscle atrophy and kidney fibrosis in mice. Mol Ther 27: 571–583, 2019. doi: 10.1016/j.ymthe.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Wang B, Wang J, He W, Zhao Y, Zhang A, Liu Y, Hassounah F, Ma F, Klein JD, Wang XH, Wang H. Exogenous miR-29a attenuates muscle atrophy and kidney fibrosis in unilateral ureteral obstruction mice. Hum Gene Ther 31: 367–375, 2020. doi: 10.1089/hum.2019.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Fierro-Fernández M, Miguel V, Márquez-Expósito L, Nuevo-Tapioles C, Herrero JI, Blanco-Ruiz E, Tituaña J, Castillo C, Cannata P, Monsalve M, Ruiz-Ortega M, Ramos R, Lamas S. MiR-9-5p protects from kidney fibrosis by metabolic reprogramming. FASEB J 34: 410–431, 2020. doi: 10.1096/fj.201901599RR. [DOI] [PubMed] [Google Scholar]
- 434. Bai L, Lin Y, Xie J, Zhang Y, Wang H, Zheng D. MiR-27b-3p inhibits the progression of renal fibrosis via suppressing STAT1. Hum Cell 34: 383–393, 2021. doi: 10.1007/s13577-020-00474-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Aghajani Nargesi A, Lerman LO, Eirin A. Mesenchymal stem cell-derived extracellular vesicles for kidney repair: current status and looming challenges. Stem Cell Res Ther 8: 273, 2017. doi: 10.1186/s13287-017-0727-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436. He J, Wang Y, Lu X, Zhu B, Pei X, Wu J, Zhao W. Micro-vesicles derived from bone marrow stem cells protect the kidney both in vivo and in vitro by microRNA-dependent repairing. Nephrology (Carlton) 20: 591–600, 2015. doi: 10.1111/nep.12490. [DOI] [PubMed] [Google Scholar]
- 437. Choi HY, Lee HG, Kim BS, Ahn SH, Jung A, Lee M, Lee JE, Kim HJ, Ha SK, Park HC. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res Ther 6: 18, 2015. doi: 10.1186/s13287-015-0012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438. Park HJ, Kong MJ, Jang HJ, Cho JI, Park EJ, Lee IK, Frøkiær J, Norregaard R, Park KM, Kwon TH. A nonbiodegradable scaffold-free cell sheet of genome-engineered mesenchymal stem cells inhibits development of acute kidney injury. Kidney Int 99: 117–133, 2021. doi: 10.1016/j.kint.2020.07.043. [DOI] [PubMed] [Google Scholar]
- 439. Nowak N, Yamanouchi M, Satake E. The nephroprotective properties of extracellular vesicles in experimental models of chronic kidney disease: a systematic review. Stem Cell Rev Rep 18: 902–932, 2022. doi: 10.1007/s12015-021-10189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Wagner MC, Sandoval RM, Campos-Bilderback SB, Molitoris BA. Using 2-photon microscopy to quantify the effects of chronic unilateral ureteral obstruction on glomerular processes. J Vis Exp 181: 63329, 2022. doi: 10.3791/63329-v. [DOI] [PubMed] [Google Scholar]
- 441. Berchtold L, Friedli I, Vallée JP, Moll S, Martin PY, de Seigneux S. Diagnosis and assessment of renal fibrosis: the state of the art. Swiss Med Wkly 147: w14442, 2017. doi: 10.4414/smw.2017.14442. [DOI] [PubMed] [Google Scholar]
- 442. Bülow RD, Boor P. Extracellular matrix in kidney fibrosis: more than just a scaffold. J Histochem Cytochem 67: 643–661, 2019. doi: 10.1369/0022155419849388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Genovese F, Kàrpàti ZS, Nielsen SH, Karsdal MA. Precision-cut kidney slices as a tool to understand the dynamics of extracellular matrix remodeling in renal fibrosis. Biomark Insights 11: 77–84, 2016. doi: 10.4137/BMI.S38439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Papasotiriou M, Genovese F, Klinkhammer BM, Kunter U, Nielsen SH, Karsdal MA, Floege J, Boor P. Serum and urine markers of collagen degradation reflect renal fibrosis in experimental kidney diseases. Nephrol Dial Transplant 30: 1112–1121, 2015. doi: 10.1093/ndt/gfv063. [DOI] [PubMed] [Google Scholar]
- 445. Sun Q, Baues M, Klinkhammer BM, Ehling J, Djudjaj S, Drude NI, Daniel C, Amann K, Kramann R, Kim H, Saez-Rodriguez J, Weiskirchen R, Onthank DC, Botnar RM, Kiessling F, Floege J, Lammers T, Boor P. Elastin imaging enables noninvasive staging and treatment monitoring of kidney fibrosis. Sci Transl Med 11: eaat4865, 2019. doi: 10.1126/scitranslmed.aat4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Jafar TH. FDA approval of dapagliflozin for chronic kidney disease: a remarkable achievement? Lancet 398: 283–284, 2021. doi: 10.1016/S0140-6736(21)01242-3. [DOI] [PubMed] [Google Scholar]
- 447. Nastase MV, Zeng-Brouwers J, Wygrecka M, Schaefer L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv Drug Deliv Rev 129: 295–307, 2018. doi: 10.1016/j.addr.2017.12.019. [DOI] [PubMed] [Google Scholar]