


Keywords: corticosterone, environmental stress, genetics, obesity, rat model
Abstract
We previously identified keratinocyte-associated protein 3, Krtcap3, as an obesity-related gene in female rats where a whole body Krtcap3 knockout (KO) led to increased adiposity compared to wild-type (WT) controls when fed a high-fat diet (HFD). We sought to replicate this work to better understand the function of Krtcap3 but were unable to reproduce the adiposity phenotype. In the current work, WT female rats ate more compared to WT in the prior study, with corresponding increases in body weight and fat mass, while there were no changes in these measures in KO females between the studies. The prior study was conducted before the COVID-19 pandemic, while the current study started after initial lockdown orders and was completed during the pandemic in a generally less stressful environment. We hypothesize that the environmental changes impacted stress levels and may explain the failure to replicate our results. Analysis of corticosterone (CORT) at euthanasia showed a significant study-by-genotype interaction where WT had significantly higher CORT relative to KO in study 1, with no differences in study 2. These data suggest that decreasing Krtcap3 expression may alter the environmental stress response to influence adiposity. We also found that KO rats in both studies, but not WT, experienced a dramatic increase in CORT after their cage mate was removed, suggesting a separate connection to social behavioral stress. Future work is necessary to confirm and elucidate the finer mechanisms of these relationships, but these data indicate the possibility of Krtcap3 as a novel stress gene.
NEW & NOTEWORTHY Obesity is linked to both genetics and environmental factors such as stress. Krtcap3 has previously been identified as a gene associated with adiposity, and our work here demonstrates that environmental stress may influence the role of Krtcap3 on both food intake and adiposity. Obesity is strongly influenced by stress in humans, so the identification of novel genes that link stress and obesity will greatly advance our understanding of the disease.
INTRODUCTION
Obesity rates continue to climb in the United States. The 2017–2020 prepandemic National Health and Nutrition Examination Survey found that rates of childhood obesity were just under 20%, and adult obesity was just over 40% (1). However, early studies during the COVID-19 pandemic examining obesity rates in children and adults showed that obesity prevalence increased ∼3% in each group in just 1 year alone (2, 3). The obesity epidemic continues to accelerate, and this highlights the severe need in the medical community to better understand the underlying causes of obesity and develop more effective treatment options for patients.
Obesity is a complex disease, influenced by both genetics and environment. Although environmental components such as physical activity, socioeconomic status, and stress exposure are well recognized (4), the genetic susceptibility to obesity remains poorly understood (5–7). Hundreds of genetic loci for obesity have been identified in human genome-wide association studies (GWAS), but they still only explain a small portion of the heritability (6–8). Furthermore, many of the causal genes within these loci are unknown. Notably, most common obesity variants will impact weight subtly, so as more variants are found researchers should expect diminishing effect sizes (9). While there is much work to be done in understanding the genetics of obesity and no gene alone may be a cure-all for patients, it is important to continue to advance the field to improve treatment options and personalize approaches for patients suffering from obesity and its related complications.
Previously, we identified keratinocyte-associated protein 3 (Krtcap3) as a candidate gene for visceral fat mass in rats (10, 11). At the same time, a different group found that Krtcap3 may be a pleiotropic gene for obesity, type 2 diabetes, and dyslipidemia in humans (12). Our laboratory conducted the first published study to investigate Krtcap3 as an adiposity gene and was able to validate these GWAS findings (13). Specifically, we developed a Krtcap3 knockout (KO) rat model on the inbred Wistar-Kyoto background strain. We showed that KO rats had increased body weight in both sexes relative to wild-type (WT) rats and that female KO rats had increased eating and fat mass relative to WT rats (13). Despite this validation, very little is known about the underlying molecular mechanisms of how Krtcap3 impacts adiposity.
The initial purpose of the current work was to conduct a second study to replicate our previous findings and to probe the functional aspect of Krtcap3 in obesity. We chose to focus on female rats in this study to better understand the role of Krtcap3 and then later apply that knowledge to studies with male rats. Surprisingly, we failed to replicate the adiposity difference we had previously seen in the female rats, as there were no longer any significant differences in body weight, food intake, or fat mass between WT and KO females. Vitally, between the first study (study 1) and this second study (study 2), there was a large environmental change due to the COVID-19 shutdown and the completion of a nearby construction project. Our results support the importance of environmental factors in contributing to gene-by-environment interactions and suggest that Krtcap3 may interact in the stress response pathway, thereby indirectly affecting adiposity in rats.
METHODS
Animals
As previously described, we generated a whole-body in vivo Krtcap3 knockout (KO) on the Wistar-Kyoto (WKY/NCrl; RGD_1358112) inbred rat strain (WKY-Krtcap3em3Mcwi) and established a breeding colony at Wake Forest University School of Medicine (WFSoM) in 2019 (13). Rats were housed in the same conditions previously described (13). Rats were housed in standard caging at 22°C in a 12:12-h light-dark cycle (dark from 18:00 to 6:00) at standard temperature and humidity conditions and given ad libitum access to water. The breeding colony has been maintained at WFSoM since 2019, with WT and KO experimental rats weaned from heterozygous breeders. Experimental rats for study 1 (13) were weaned from July 2019 through February 2020, during which time the building was fully occupied and there was a neighboring construction project. Experimental rats for study 2 (described below) were weaned from May 2020 through November 2020, when the facility operated at minimum capacity and construction had finished. Breeders and experimental rats before the study start were given a standard chow diet (Lab Diet, Prolab RMH 3000, cat. no. 5P00). At the study start, experimental WT (n = 11) and KO (n = 18) rats were placed on an experimental diet as described below.
To conduct analyses of Krtcap3 expression in multiple tissues, we ordered WKY/NCrl rats from Charles River (male n = 10; female n = 3). Rats remained at WFSoM for at least 2 wk in the same housing conditions as described above on a standard chow diet and housed two to three rats per cage. At 12 wk of age, the rats were euthanized, and their tissues were dissected for expression analyses. Tissues were collected from multiple groups of males from 2019 to 2022 while tissues were collected from only one group of females in 2022.
Genotyping
Experimental rats were genotyped by fluorescent-based fragment analysis at the Medical College of Wisconsin using the ABI 3730 capillary sequencer, followed by analysis in Genemapper software.
Study Design
Female WT and KO experimental rats were weaned at 3 wk of age and placed two per cage in same-sex, same-genotype cages. We housed WT rats separately from KO rats unless the same genotype cage-mate was not available. Before starting a high-fat diet (HFD), as described below, experimental rats were maintained on the same diet as breeders (see above). At 6 wk of age, rats were weighed, analyzed by EchoMRI analysis, and began a HFD (60% kcal fat; Research Diet D12492). Rats were allowed access to diet ad libitum with body weight and cage food intake recorded weekly starting at 6 wk of age. Weekly caloric intake and cumulative caloric intake were calculated as previously described (13). Rats were on a diet for 13 wk, with metabolic phenotyping tests beginning after 10 wk on the diet and euthanasia after 13 wk on the diet (Fig. 1). All experiments were performed using a protocol approved by the Institutional Animal Care and Use Committee at WFSoM.
Figure 1.

Study timeline. Timeline outlining study design, with weeks relative to diet start shown. Metabolic phenotyping included EchoMRI analysis (EchoMRI), individual food intake (Food Intake, shaded gray), TSE metabolic phenotyping systems (TSE), an intraperitoneal glucose tolerance test (IPGTT), and euthanasia (Sac). Body weight and cage-wide food intake were recorded weekly.
Metabolic Phenotyping
At 0 and 10 wk on diet, rats went through EchoMRI (EchoMRI LLC, Houston, TX) analysis as previously described (13) to precisely measure the total fat and lean mass of live rats.
Immediately following the second EchoMRI analysis, rats began a 5- to 7-day-long period of individual housing to measure individual food intake and to measure metabolism. To measure individual food intake, rats were housed in the same standard caging, with the addition of a tube for enrichment, and still allowed access to diet ad libitum. Body weight was recorded at the start and the end, and food remaining was recorded every day between 11:00 AM and 1:00 PM.
Rats were then transferred to TSE Phenomaster chambers (TSE Systems, Berlin, Germany) to measure metabolism. TSE Phenomaster chambers use indirect gas calorimetry to measure the volume of oxygen consumed by the rat and the volume of carbon dioxide produced. The system’s climate chamber controls temperature (22°C), humidity, and lighting conditions, which are held constant in the rat’s home environment. Rats were given a 24-h adaptation period, and then data were recorded for at least an additional 24 h. Total energy expenditure (TEE) was calculated by the indirect calorimetry chamber and normalized by individual body weight. The respiratory exchange ratio (RER), a measure of fuel utilization, was calculated by dividing the volume of carbon dioxide produced (V̇co2) by the volume of oxygen used (V̇o2). Food intake was also measured during this time period.
After 12 wk on the diet, rats were fasted for 16 h overnight before being administered an intraperitoneal glucose tolerance test (IPGTT). Rats were transferred to a procedure room away from the housing room 30 min before the test began. We measured blood glucose (Contour Next EZ) and collected serum (Sarstedt Inc., MicrovetteCB 300 LH) for subsequent insulin analysis from a tail nick at fasting and 15, 30, 60, 90, and 120 min after a 1 mg/kg glucose injection. To calculate a rat’s response to the glucose challenge, the glucose area under the curve (AUC), we used the following equation:
where serum glucose (SG) was measured at time t = 0, 15, 30, 60, 90, and 120 min.
Tissue Harvest
After 13 wk on the diet, rats were euthanized via decapitation after either no fast or a 4-h morning fast. Statistical analysis confirmed that the length of fast did not affect adiposity measures. Rats were transferred to the anteroom of the necropsy suite 30 min before the start of the euthanasia protocol (9:00 AM or 11:00 AM, respective to fast length) and were euthanized one at a time. Weight gain was calculated as the difference between the final body weight of the rats following the fast and the body weight at the start of the diet. Trunk blood was collected, and serum was saved and stored at −80°C. Body length from nose to anus and tail length from anus to tail tip were measured with a ruler. The brain, visceral fat pad tissues [retroperitoneal (RetroFat), omental/mesenteric fat (OmenFat), and parametrial fat (ParaFat)], liver, kidneys, and heart were dissected, weighed, and snap-frozen. Sections of the ileum and colon were also dissected and snap-frozen.
WKY/NCrl tissues were collected for quantitative PCR analyses, and rats were euthanized via decapitation after no fast. Rats were transferred to the anteroom of the necropsy suite 30 min before the start of euthanasia (9:00) and were euthanized one at a time. The brain, visceral fat pad tissues, liver, kidneys, and heart were dissected and snap-frozen in both male and female rats. We also collected pituitary, adrenals, sections of the ileum and colon, subcutaneous white and brown adipose tissue, and soleus muscle from male inbred rats, but based on findings in male rats, additional tissues collected in the females included the pituitary, adrenals, ileum, and colon.
Insulin
We used ultrasensitive ELISA kits (Alpco, ref. no. 80-INSTRU-E10) to analyze fasting serum insulin collected from the IPGTT.
Corticosterone
We used a corticosterone (CORT) competitive ELISA kit (ThermoFisher, ref. no. EIACORT) to analyze serum corticosterone from trunk blood collected at euthanasia. Samples from HFD females in study 1 were sent to the Redei laboratory at Northwestern University, while all other samples were run in-house at WFSoM. As guided by kit instructions, samples were diluted at least 1:100 and analyzed at 450 nm against a four-parameter standard curve. The ELISA kit is confirmed to preferentially bind the active corticosterone molecule.
RNA Extraction
For both experimental animals and inbred animals, RNA was extracted from fatty tissues using the RNeasy Lipid Tissue Mini Kit (Qiagen, cat. no. 74804). Nonfatty tissues, such as the liver or intestine, were extracted by TRIzol.
Real-Time Quantitative PCR
We assessed expression of Krtcap3 and adenylate cyclase 3 (Adcy3) (Table 1) between study 1 and study 2 rats to determine if there had been changes in gene expression that could contribute to the change in phenotype we saw. We examined Krtcap3 expression in the liver only in WT animals but examined Adcy3 expression in the RetroFat for both studies and genotypes. We chose to investigate expression of Adcy3 in addition to Krtcap3 because of its well-known influence on adiposity (14, 15) and the close proximity of the genes to each other (10).
Table 1.
Primer sequences
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| Adcy3 | AGAAGACCAAGACCGGAGTG | CATCTAGGTAGTCGCAGCGA |
| β-Actin | TGAGGTAGTCTGTCAGGTCCCG | ACCACTGGCATTGTGATGGACT |
| Gapdh | CATGGAGAAGGCTGGGGCTC | AACGGATACATTGGGGGTAG |
| Hsd11β1 | GCAGACCGATTTGTTGTTGA | GTGGATATCATCGTGGAAGAGAG |
| Hsd11β2 | TCCATCACCGGTTGTGACACT | CACGCAGTTCTAGAGCACCA |
| Krtcap3 | GTTACTGTTGTGTGGCTGCA | AGCACCTCCTGTCCTAAACC |
| Nr3c1 | GAAGGGAACTCCAGTCAGAAC | AATGTCTGGAAGCAGTAGGTAAG |
We examined expression of several stress-related genes in liver and RetroFat between study 1 and study 2 WT and KO to evaluate if genotype or environment affected gene expression. Liver and fat were selected because both tissues had been collected in both studies. We analyzed Nr3c1, Hsd11β1, and Hsd11β2 (Table 1) in both sets of tissues. Nuclear receptor subfamily 3 group C member 1 (NR3C1) is a glucocorticoid (GC) receptor, and the 11β-hydroxysteroid dehydrogenases (11β-HSDs) are responsible for catalyzing corticosterone, where isoform 1 (11β-HSD1) primarily activates the pathway and isoform 2 (11β-HSD2) deactivates the pathway.
To measure gene expression, either Gapdh or β-actin were used as housekeeping genes (Table 1). Fold change of the corresponding gene of interest transcript was calculated by the following equation:
where ΔCt is the difference between the crossing threshold (Ct) of the gene of interest and the housekeeping gene, and ΔΔCt is the difference between each sample ΔCt and the average control ΔCt.
To measure gene expression in the diet study rats, the average control ΔCt was the S1 WT mean ΔCt values, respective to each gene and tissue. To measure Krtcap3 expression in WKY/NCrl rats, expression of Krtcap3 in the WKY male pituitary was used as the average control ΔCt.
RNA Sequencing and Analysis
Sections of the hypothalamus were dissected from the frozen brain of select female rats from study 1 and study 2 (n = 4/study and genotype). Although Krtcap3 has low expression in the hypothalamus, the hypothalamus is well connected to satiety signaling (16) and the hypothalamic-pituitary-adrenal (HPA) axis of stress response (17), and we aimed to identify changes in these pathways. We chose rats that were the first rat of their cage to be euthanized and were representative of phenotype differences between WT and KO rats. Brains had been snap-frozen at euthanasia and stored at −80 C, were dissected at −20 C, and then the tissue was stored again at −80 C until they were shipped to Azenta (Genomics from Azenta Life Sciences, South Plainfield, NJ).
RNA extraction.
Total RNA was extracted from fresh frozen tissue samples using Qiagen RNeasy Plus Universal mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
Library preparation with PolyA selection.
RNA samples were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA), and RNA integrity was checked using Agilent TapeStation 4200 (Agilent Technologies, Palo Alto, CA). For all samples, the RNA concentration was at least 200 ng/µL, and the RIN was at least 7.9.
RNA sequencing libraries were prepared using the NEBNext Ultra II RNA Library Prep for Illumina using the manufacturer’s instructions (NEB, Ipswich, MA). Briefly, mRNAs were initially enriched with Oligod(T) beads. Enriched mRNAs were fragmented for 15 min at 94°C. First-strand and second-strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3′-ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing libraries were validated on the Agilent TapeStation and quantified by using Qubit 2.0 Fluorometer as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA).
Illumina sequencing.
The sequencing libraries were clustered on a lane of a HiSeq flowcell. After clustering, the flowcell was loaded on the Illumina instrument (4,000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2 × 150 bp paired-end configuration. Image analysis and base calling were conducted by the Control software. Raw sequence data (.bcl files) generated by the sequencer were converted into fastq files and demultiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Differentially expressed gene analysis.
After investigating the quality of the raw data, sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Rattus norvegicus reference genome RN6 available on ENSEMBL using the STAR aligner v.2.5.2b. BAM files were generated as a result of this step. Unique gene hit counts were calculated by using feature Counts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted. After extraction of gene hit counts and normalization, the gene hit counts table was used for downstream differential expression analysis. Using DESeq2, comparisons of gene expression between the groups of samples (WT vs. KO and study 1 vs. study 2) were performed (Table 2). The Wald test was used to generate P values and log2 fold changes. In our analysis, genes with P < 0.05 were called as significantly differentially expressed genes for each comparison.
Table 2.
Comparison groups for differentially expressed genes in the RNA-seq analysis
| Group 1 | Group 2 | |
|---|---|---|
| Comparison 1 | Study 1 WT | Study 1 KO |
| Comparison 2 | Study 2 WT | Study 2 KO |
| Comparison 3 | Study 1 WT | Study 2 WT |
| Comparison 4 | Study 1 KO | Study 2 KO |
We analyzed differences between wild-type (WT) and Krtcap3 knockout (KO) for each study and then differences by study for each genotype. RNA-seq, RNA sequencing.
Ingenuity Pathway Analysis.
We used Ingenuity Pathway Analysis (IPA) software (Qiagen Digital Insights) to analyze differentially expressed genes in the context of known biological responses and regulatory networks. Differentially expressed genes in each comparison were uploaded to the IPA software. We identified canonical pathways that differed between each comparison and networks that included both direct and indirect relationships, with significance determined in IPA by Fisher’s exact test and the threshold set at –log(P value) > 1.3. We then mapped upstream regulators that were activated with a P value of overlap < 0.05 and removed indirect relationships as well as orphan nodes.
Statistical Analysis
All data were analyzed in R (1.4.1103), with the exception of RNA sequencing (RNA-seq) data, which was analyzed as described above, and cumulative energy intake and WKY/NCrl tissue expression data, which were analyzed in GraphPad PRISM [9.4.1(681)]. Outliers according to Grubb’s test were removed, distribution was assessed by the Shapiro-Wilks test, and data were transformed to reflect a normal distribution if necessary. The homogeneity of variance was assessed by Levene’s test.
In examining data collected from study 2, we first analyzed the effect of genotype. All single-point adiposity measurements were analyzed by Student’s t test. Growth, food/energy consumption, and energy expenditure curves were assessed with a repeated-measures ANOVA, where the effect of genotype was examined over time.
When analyzing phenotypes between study 1 and study 2, most of the metabolic data were analyzed by a two-way ANOVA, where the factors were genotype and study. Please see Supplemental Table S1 for adiposity measures from study 1 rats. Growth and food consumption curves were analyzed by a two-way repeated measures ANOVA, examining the effects of genotype and study over time. If there was a significant interaction, the analysis was split by genotype, and either a Student’s t test or a repeated measures ANOVA (respective to the phenotype) assessed the effect of the study. Slopes of the cumulative energy intake curves were tested in PRISM to determine if they were significantly different and then split by genotype to examine slopes between studies.
When examining the results from the CORT assays, we noticed the data were spread in an almost bimodal fashion, particularly for KO animals. Upon further investigation, the factor that most likely explained these differences was euthanasia order within the cage: which rat had been euthanized first and which rat had been euthanized second. Due to this, we included euthanasia order as a third factor in the analyses, in addition to genotype and study. The data were analyzed by a three-way ANOVA and split according to interactions that were significant.
In the data comparing male and female Krtcap3 expression in multiple tissues from naïve WKY/NCrl rats, male and female data were compared for each tissue individually using multiple comparisons t tests corrected with the Holm-Šidák test.
RESULTS
No Difference in Adiposity Measures Between WT and KO Rats
In contrast to our previous findings (13), there were no differences in body weight between WT and KO females at wean or 6 wk of age nor were there differences in adiposity (Table 3). Unexpectedly, no differences ever developed in the weight between WT and KO females (Fig. 2A) or in food consumption (Fig. 2B). During the week of individual housing, WT and KO females consumed similar amounts of calories per day (Table 3). We ultimately did not see any differences in weight gain between WT and KO females (Table 3), and at euthanasia, KO females were not heavier than WT females (Table 3).
Table 3.
Adiposity measures between wild-type and Krtcap3 knockout female rats from study 2
| Phenotype | WT | KO | Significance |
|---|---|---|---|
| Wean weight, g | 36.5 ± 1.43 | 35.2 ± 0.86 | NS |
| Diet start weight, g | 106.8 ± 3.73 | 104.5 ± 2.94 | NS |
| Diet start fat mass, g | 5.5 ± 0.38 | 5.6 ± 0.27 | NS |
| Weight gain, g | 149.4 ± 3.82 | 145.1 ± 3.90 | NS |
| Final weight, g | 256.2 ± 2.93 | 249.6 ± 4.01 | NS |
| Daily individual food intake, g/day | 9.5 ± 0.27 | 9.8 ± 0.25 | NS |
| Fasting glucose, mg/dL | 88.8 ± 2.57 | 86.1 ± 1.92 | NS |
| Glucose AUC | 18,788.9 ± 841.59 | 16,725.4 ± 606.35 | P = 0.052 |
| Fasting insulin, ng | 1.4 ± 0.13 | 1.3 ± 0.08 | NS |
| 10 WoD lean mass, g | 194.2 ± 2.61 | 189.4 ± 2.77 | NS |
| 10 WoD fat mass, g | 36.6 ± 2.15 | 34.7 ± 2.29 | NS |
| RetroFat mass, g | 7.8 ± 0.32 | 7.3 ± 0.42 | NS |
| ParaFat mass, g | 7.0 ± 0.31 | 6.5 ± 0.38 | NS |
Values are given as the mean average ± SE. Significance is indicated in the final column. WT, wild type; KO, Krtcap3 knockout; WoD, weeks on diet; AUC, area under the curve; RetroFat, retroperitoneal fat; ParaFat, parametrial fat; NS, not significant.
Figure 2.

Body weight growth and food consumption in wild-type (WT) and Krtcap3 knockout (KO) rats in study 2. Rats were weighed weekly throughout the study, and food consumption per cage was recorded for the first 10 wk on diet. A: there were no significant differences between the growth curve of WT (black circle) and KO (gray square) female rats. B: there were also no differences in the food consumption patterns between WT and KO cages over the course of the study. HFD, high-fat diet.
WT and KO females had comparable fasting glucose levels (Table 3) although unlike the prior study, WT females did have a slightly increased glucose AUC compared to KO females (T27 = 2.03, P = 0.052; Table 3). Surprisingly, there were no differences in fasting insulin between WT and KO females (Table 3).
Over the 24-h measurement period, there were no differences in total energy expenditure between WT and KO rats when normalized to body weight (Supplemental Fig. S1A). There were also no differences in the RER between the genotypes (Supplemental Fig. S1B), with comparable usages of fats and carbohydrates as fuel sources. Finally, while a visual difference develops in food intake between WT and KO rats (Supplemental Fig. S1C), this difference is ultimately not significant, corroborating the other measures of food intake throughout the study.
At EchoMRI analysis after 10 wk on the diet, WT and KO females had similar amounts of total lean mass and fat mass (Table 3). At euthanasia, there were no differences in RetroFat mass or ParaFat mass between WT and KO females (Table 3). Although these data contradict our previous findings (13), the adiposity and metabolic measures do support the lack of body weight differences seen between WT and KO females in this study. There were also no noteworthy differences in body size or non-adipose organ weights between WT and KO rats (Table 4).
Table 4.
Body size and nonadipose organ weights between wild-type and Krtcap3 knockout female rats in study 2
| Phenotype | WT | KO | Significance |
|---|---|---|---|
| Body length, cm | 21.4 ± 0.13 | 21.2 ± 0.12 | NS |
| Tail length, cm | 16.9 ± 0.19 | 16.4 ± 0.6 | P = 0.09 |
| Brain weight, g | 1.9 ± 0.02 | 1.9 ± 0.02 | NS |
| Liver weight, g | 7.6 ± 0.11 | 7.7 ± 0.16 | NS |
| Heart weight, g | 1.1 ± 0.09 | 1.0 ± 0.03 | P = 0.092 |
| Kidney weight, g | 0.8 ± 0.02 | 0.7 ± 0.01 | NS |
Values are given as the mean average ± SE. Body length was measured from nose to anus, and tail length was measured from anus to end of tail. Significance is indicated in the final column. WT, wild type; KO, Krtcap3 knockout; NS, not significant.
WT Rats in Study 2 Are Heavier and Eating More Than in Study 1, With No Change in KO
When examining differences between study 1 and study 2, we found no differences in body weight at study start between study 1 and study 2 females of either genotype (Fig. 3A). However, when we examined body weight over time, we saw a significant interaction between genotype and study (Fig. 3B; F1,44 = 10.3, P = 0.002) where study 2 WT females were heavier over time compared to study 1 WT females (F2.46,44.22 = 8.02, P = 5.05e-4), with no effect in KO females.
Figure 3.
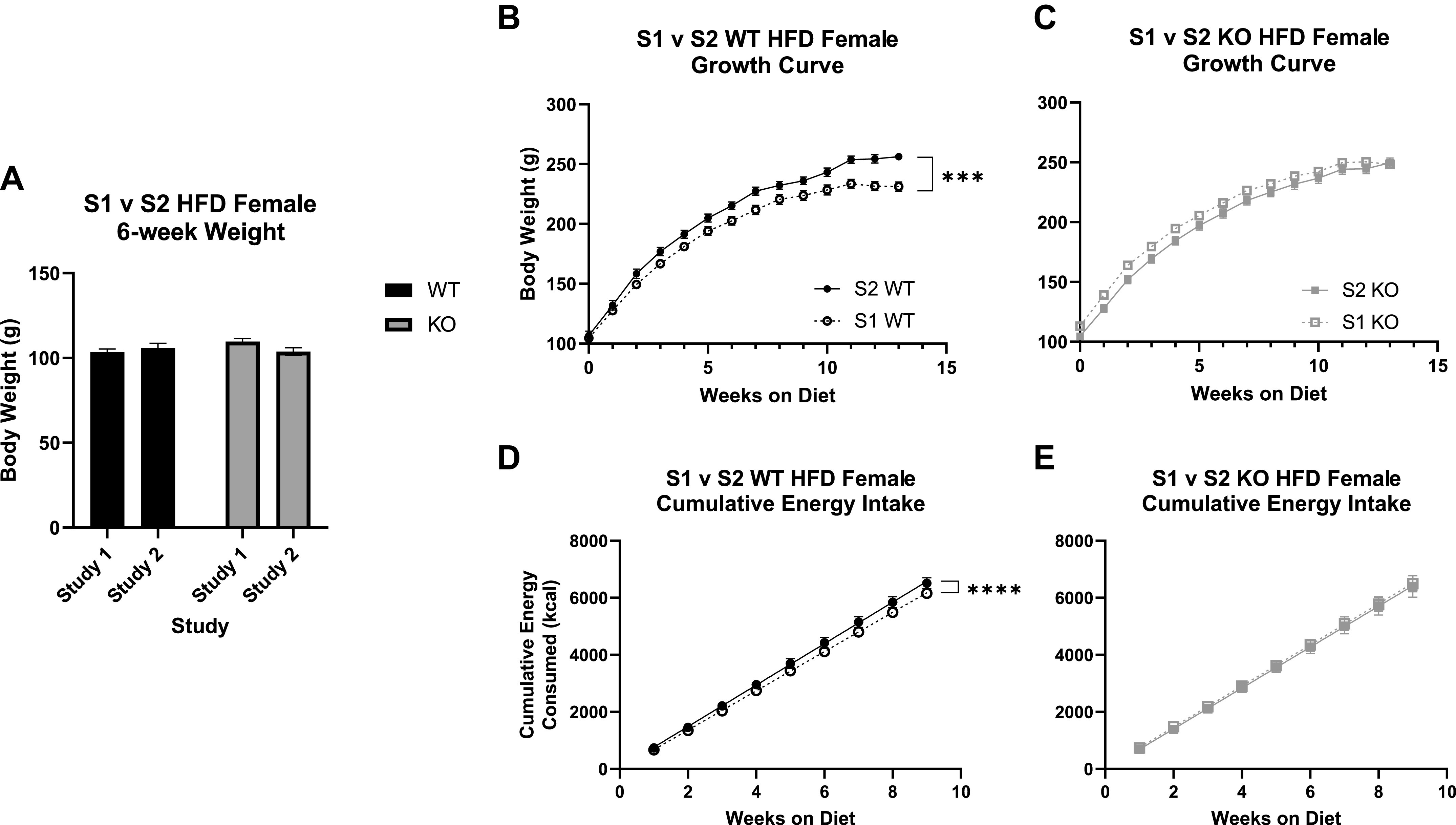
Differences in select adiposity phenotypes between study 1 (S1) and study 2 (S2) in wild-type (WT) and Krtcap3 knockout (KO) rats. A: comparing body weight at diet start, or 6 wk of age, between the 2 studies, there are no differences in the starting weight of the WT rats (black) or the KO rats (gray). B and D: study 2 WT rats (filled circle, solid line) gained significantly more weight over the course of the study compared to study 1 WT rats (open circle, dashed line; B), while there were no comparable differences between the growth curves of study 1 KO rats (open square, dashed line; D) and study 2 KO rats (filled square, solid line). C and E: perhaps explaining these differences in body weight curves, study 2 WT rats had a greater cumulative energy consumption compared to study 1 WT rats (C), while there were no differences between the KO rats (E). HFD, high-fat diet. ***P < 0.001, ****P < 0.0001, effect of study and weeks on diet respective to each genotype.
We then compared food consumption between the two studies. There was a significant interaction between study and genotype (F1,17 = 4.55, P = 0.048). WT rats in study 2 consumed significantly more food than WT rats from study 1 (F1,7 = 6.72, P = 0.036; Supplemental Fig. S2), with no changes between studies in KO rats. When confirming caloric intake over the study, there was a near interaction between genotype and study (F1,19 = 3.24, P = 0.088) and a main effect of week (F9,171 = 3.36, P = 8.14e-4). To better examine the difference in energy intake over time between study and genotype, we calculated cumulative energy intake and compared the slopes of the curves. While study 2 WT females had a greater caloric intake than study 1 WT females (F1, 198 = 6.94e14, P < 0.0001; Fig. 3C), there were no changes between studies in KO caloric intake.
We next compared different adiposity measures between study 1 and study 2 in WT and KO rats, namely the measurements recorded by EchoMRI analysis and those collected at euthanasia. At EchoMRI analysis after 10 wk on the diet, the spread of the data for total fat mass was too large to see statistically significant effects, but there was a visual increase in study 2 WT rats compared to study 1 that is not present in the KO rats (Fig. 4A). There was, however, a significant interaction between genotype and study for total lean mass (F1,45 = 5.96, P = 0.019) where study 2 WT rats had significantly greater lean mass compared to study 1 counterparts (T19 = 2.20, P = 0.041; Fig. 4B), with no difference in KO rats.
Figure 4.
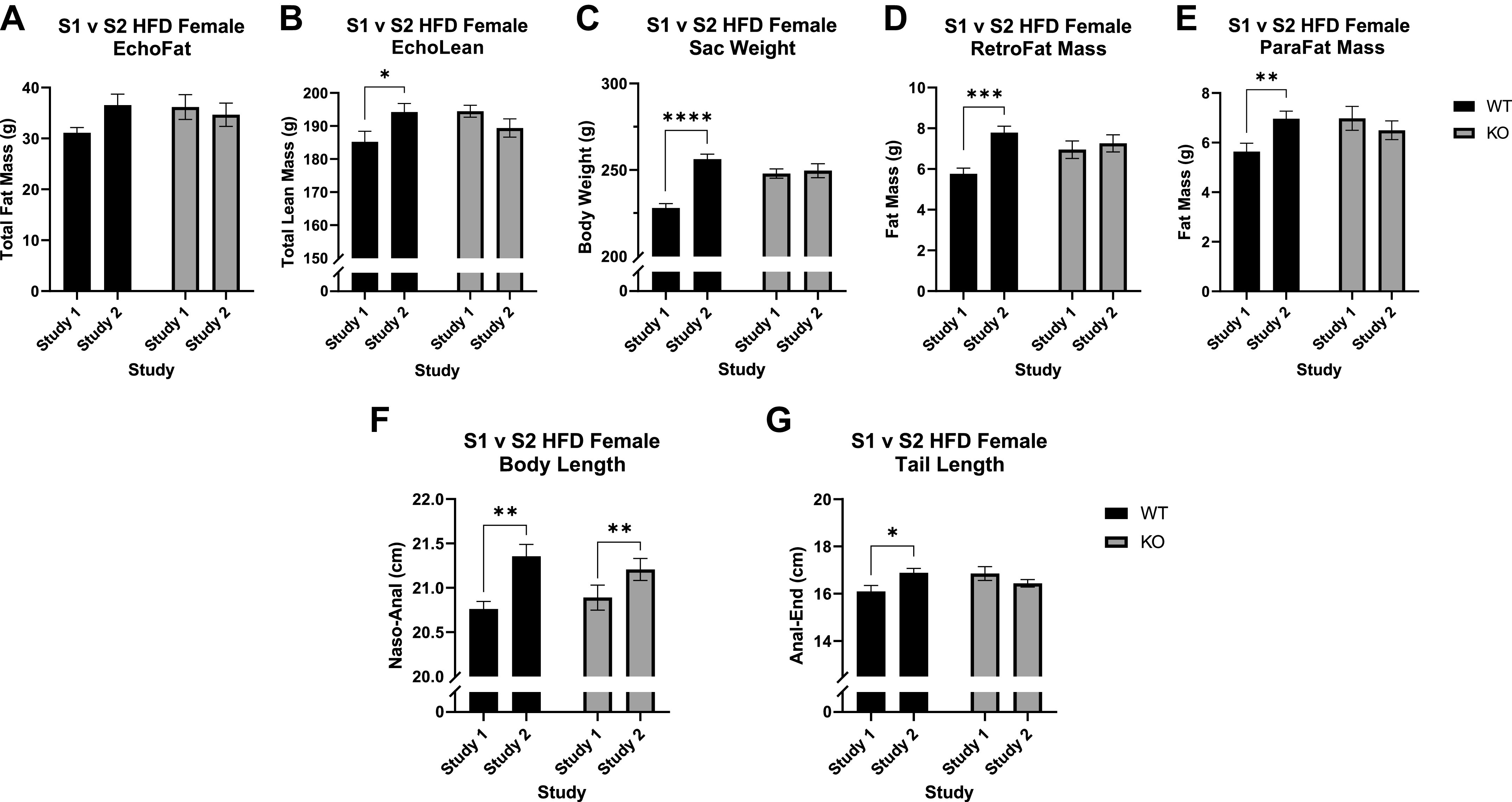
Differences in size between study 1 (S1) and study 2 (S2) wild-type (WT) and Krtcap3 knockout (KO) rats. Total fat mass (A) and total lean mass (B) were recorded after 10 wk on the diet. At euthanasia 3 wk later body weight (C) plus retroperitoneal fat (RetroFat; D) and parametrial fat (ParaFat; E) mass were recorded. Body length measured from nose to anus (F) and tail length measured from anus to end of tail (G) were also recorded at this time. A: there were no differences in total fat mass by study in either WT (black) or KO (gray) rats at EchoMRI analysis, though there was a visual increase in S2 WT rats. B: S2 WT rats did have a greater total lean mass compared to S1 counterparts, with no differences in KO rats. C: in WT rats, study 2 rats had an increased final body weight compared to study 1 rats, with no changes between the final body weights of KO rats. D and E: study 2 WT rats had greater RetroFat mass and ParaFat mass compared to study 1 WT rats, while there were no significant differences in the KO rats. F and G: there was a significant increase in the body length of study 2 WT and KO rats (F) but there was only a significant increase in tail length in study 2 WT animals and no difference in KO rats (G). HFD, high-fat diet. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, effect of study respective to each genotype, with the exception of the body length phenotype, which represents the main effect of study.
The adiposity differences were more prominent 3 wk later at euthanasia, likely because WT rats in study 2 continued to grow in size while WT rats in study 1 appeared to plateau (Fig. 3B). Specifically, there was a significant interaction between study and genotype (F1,44 = 12.5, P = 9.72e-4) where study 2 WT rats were much heavier than study 1 WT rats (T18 = 7.19, P = 1.09e-6; Fig. 4C), with no differences in KO rats. Similar trends were seen in the measures for visceral fat mass: there was an interaction between study and genotype for RetroFat mass (F1,45 = 4.34, P = 0.043) where study 2 WT females had a significant increase in RetroFat mass compared to study 1 WT females (T19 = 4.71, P = 1.5e-4; Fig. 4D) but no differences in KO females. This was also true for ParaFat mass: there was an interaction between study and genotype (F1,45 = 4.92, P = 0.032), but ultimately the only difference was in WT rats (T19 = 2.89, P = 9.3e-3; Fig. 4E). Interestingly, when examining the data from EchoMRI analysis normalized to body weight, there were no differences in total body fat percentage or total body lean percentage (data not shown).
There was a statistically significant increase in the body length of study 2 rats compared to study 1 rats for both genotypes (F1,44 = 11.6, P = 0.001; Fig. 4F). There was also a significant interaction between study and genotype for tail length (F1,45 = 7.4, P = 0.009), where only study 2 WT rats had a longer tail length compared to study 1 (T19 = 2.53, P = 0.021; Fig. 4G). There were no statistical differences in brain weight (Supplemental Fig. S3A) or in heart weight (Supplemental Fig. S3B) between the studies, but study 2 rats had an increased average kidney weight compared to study 1 rats for both genotypes (F1,45 = 8.76, P = 0.005; Supplemental Fig. S3C).
No Changes in Krtcap3 or Adcy3 Expression
We measured liver Krtcap3 between study 1 and study 2 WT rats to confirm expression had not diminished and found no differences (Supplemental Fig. S4A). We did not include KO rats in this analysis as we previously demonstrated KO rats had significantly reduced Krtcap3 expression in the liver (13). We also measured RetroFat Adcy3 expression to verify its expression was not interfering with the phenotype but found no differences by genotype or by study (Supplemental Fig. S4B).
Differences in Basal and Stress CORT Between WT and KO Rats Is Dependent on Study and Cage Order
When we initially assessed CORT collected from trunk blood at euthanasia, there were no significant differences by either genotype or study (Fig. 5A), though there was a slight trend toward an interaction between study and genotype (F1,38 = 2.95, P = 0.094). A closer investigation of the data revealed a bimodal pattern to the data that we determined was related to the order of euthanasia in the cage. When we accounted for order as a factor, there were interactions between study and genotype (F1,34 = 5.49, P = 0.025) and between genotype and order (F1,34 = 12.68, P = 0.001).
Figure 5.
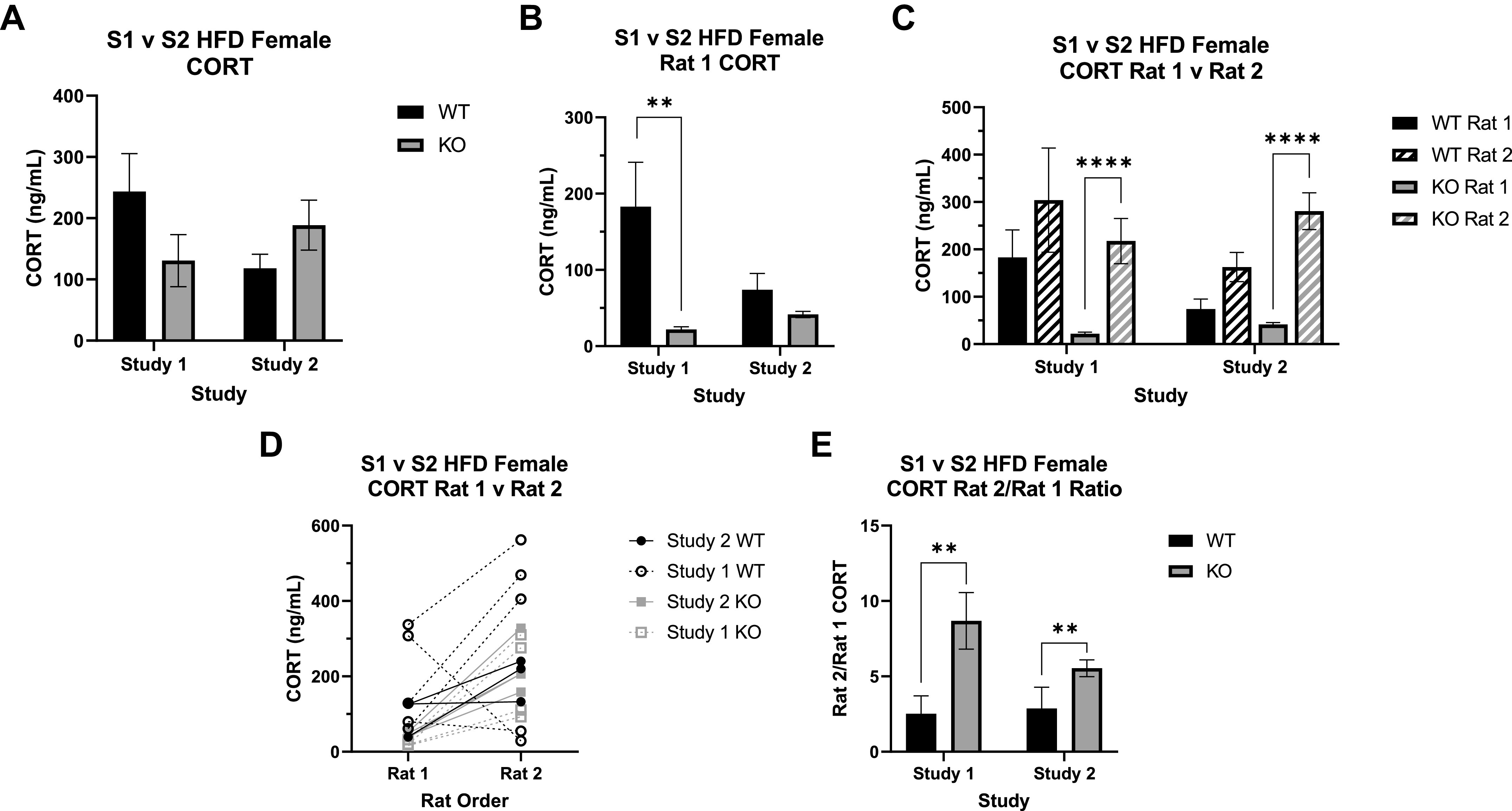
Corticosterone (CORT) measurements between study 1 (S1) and study 2 (S2) wild-type (WT) and Krtcap3 knockout (KO) rats. CORT was measured in the trunk blood collected at euthanasia. A: all rats evaluated together. When grouped only by study and genotype, there are no significant differences between WT (black) and KO (gray) CORT in either study 1 or study 2. Further assessment of the data revealed a third factor to consider, the order within the cage the rats were euthanized, either first (rat 1) or second (rat 2). Reanalysis with this additional variable revealed a significant interaction between study and genotype as well as one between genotype and order. HFD, high-fat diet. B: assessing only rats euthanized first. KO rats had lower CORT than WT rats in both studies, but this was statistically significant only in study 1, not study 2. **P < 0.01, effect of genotype respective to each study. C: comparing rats euthanized first and second. In both studies, KO CORT significantly increases in rat 2 of the cage, an effect that is not seen in the WT rats. ****P < 0.0001, effect of order respective to each genotype. D and E: rat 2/rat 1 CORT ratio (E). There was a significant main effect of genotype, where KO rats of both studies had a greater ratio than WT rats, indicating a greater CORT response. **P < 0.01, main effect of genotype.
We first split the data by euthanasia order: analyzing rats euthanized first (rat 1) separately from those euthanized second (rat 2). For rats euthanized first, what we consider to be a true basal measure, there was a main effect of genotype (F1,15 = 21.2, P = 3.4e-4) where KO rats had lower CORT than WT rats (Fig. 5B). There was also an interaction between study and genotype (F1,15 = 8.69, P = 0.01). Specifically, WT rats from study 1 showed significantly higher basal CORT relative to KO rats (T5.21 = 5.16, P = 3.17e-3; Fig. 5B), with no significant difference by genotype in study 2. Additionally, WT rats in study 2 had lower CORT than those in study 1 (T7.79 = 1.88, P = 0.098); while CORT in KO rats did increase between the two studies (T5.99 = 3.71, P = 0.01), the difference is likely not physiologically meaningful. There were no differences by study or by genotype in the rats euthanized second.
We then included both rat 1 and rat 2 and analyzed the data by genotype to assess the effect of study and order. We found that study (F1,18 = 8.08, P = 0.011) and euthanasia order (F1,18 = 143.15, P = 5.29e-10; Fig. 5C) strongly impacted CORT in the KO rats but had no effect in the WT rats. Specifically, study 2 KO rats had a slightly higher basal CORT compared to study 1 counterparts, while in both studies there was a major CORT increase in the KO rats that were euthanized second.
Finally, to continue evaluating the differences in CORT response between WT and KO rats at euthanasia, we plotted the change in CORT between rat 1 and rat 2 for each cage and then calculated the ratio increase of rat 2 CORT compared to rat 1. There was a main effect of genotype, where KO rats of both studies had a significantly higher CORT ratio compared to WT rats (F1,14 = 12.58, P = 0.003; Fig. 5D). These data demonstrate a stronger acute social stress response in KO relative to WT rats, specifically in the response of rat 2 to the removal of rat 1.
Changes in Expression of Genes Associated With Glucocorticoid Processing Indicate Different Stress Environments Between Study 1 and Study 2
We measured the expression of genes associated with CORT function and metabolism in liver and fat from study 1 and study 2 rats to evaluate if expression was different by genotype or by study.
In liver tissue, there was higher expression of the glucocorticoid receptor Nr3c1 in study 1 compared to study 2 (F1,22 = 65.1, P = 7.18e-8; Fig. 6A) but no difference by genotype in either study. Expression of the hydroxysteroid dehydrogenases followed a similar pattern, with expression elevated in study 1 for both Hsd11β1 (F1,22 = 19.35, P = 2.3e-4; Fig. 6B) and Hsd11β2 (F1,20 = 24.35, P = 97.99e-5; Fig. 6C), although ultimately no genotype differences in either.
Figure 6.
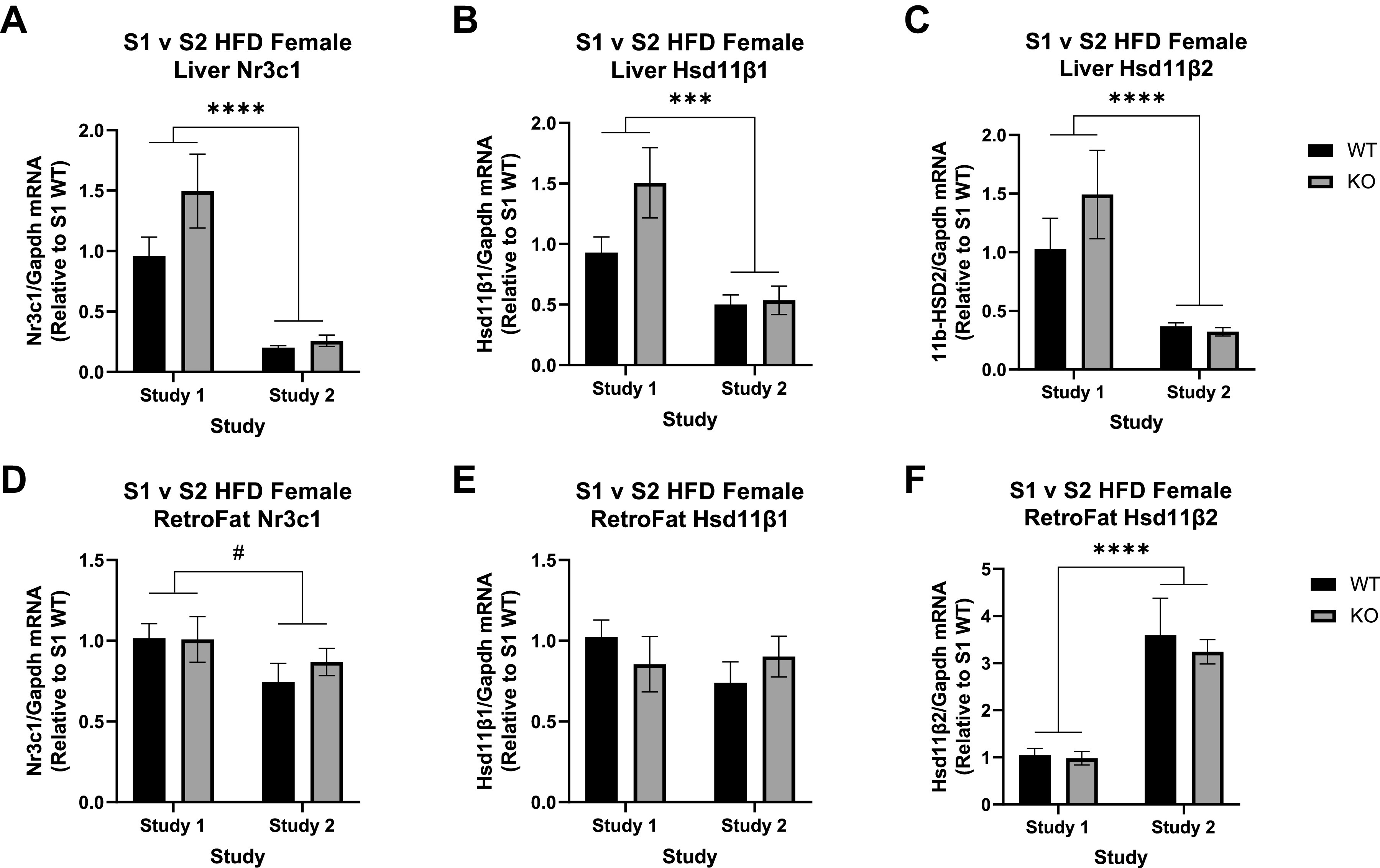
Expression of the glucocorticoid receptor and genes associated with corticosteroid processing between study 1 (S1) and study 2 (S2) in wild-type (WT) and Krtcap3 knockout (KO) rats. We measured expression of nuclear receptor subfamily 3 group C member 1 (Nr3c1), 11β-hydroxysteroid dehydrogenase isoform 1 (Hsd11β1), and 11β-hydroxysteroid dehydrogenase isoform 2 (Hsd11β2) in liver and retroperitoneal fat (RetroFat) tissue of rats from both studies. A: in the liver, study 1 WT (black) and KO (gray) rats had significantly higher expression of NR3C1 compared to study 2 rats, with no differences by genotype. B and C: similarly, study 1 rats of both genotypes had higher expression of Hsd11β1 (B) and Hsd11β2 (C) when compared to study 2 rats. D: in the RetroFat, study 1 WT and KO rats had slightly higher Nr3c1 when compared to study 2 rats. E and F: there were no differences in Hsd11β1 expression between the 2 studies (E), but study 2 rats had significantly higher Hsd11β2 compared to study 1 rats (F). HFD, high-fat diet. #P < 0.1, ***P < 0.001, ****P < 0.0001, main effect of study.
In RetroFat tissue Nr3c1 expression was also slightly higher in study 1 compared to study 2 (F1,18 = 3.54, P = 0.076; Fig. 6D), although still no differences by genotype. The hydroxysteroid dehydrogenases, on the other hand, displayed different patterns in RetroFat tissue compared to liver. While there were no differences in Hsd11β1 expression by study or by genotype (Fig. 6E), Hsd11β2 expression was lower in study 1 compared to study 2 (F1,17 = 47.55, P = 2.59e-6; Fig. 6F).
Krtcap3 Is Highly Expressed in Both Sexes Along the Gastrointestinal Tract and in the Pituitary Gland, With Differences in Expression Between Males and Females in the Gonads and Pituitary
Using naïve WKY/NCrl male rats, Krtcap3 had low expression in all the different fat pads, the adrenals, the heart, the muscle, and different regions of the hypothalamus (Fig. 7A). Expression between the liver and kidney was comparable while expression in the pituitary, testes, and gastrointestinal tract was high.
Figure 7.

Krtcap3 expression in multiple tissues between male and female naïve Wistar-Kyoto (WKY/NCrl) rats ordered from Charles-River and euthanized for tissue collection. Tissues include liver, adrenal, nucleus accumbens (NAcc), retroperitoneal fat (RetroFat), subcutaneous white fat (SubQ Fat), omental/mesenteric fat (OmenFat), arcuate nucleus (ArcN), heart, Epididymal fat (EpiFat), soleus muscle, subcutaneous brown fat (BAT), kidney, pituitary, gonads (testes or ovaries, respectively), whole hypothalamus, ileum, and colon. A: comparing Krtcap3 expression in male WKY rats across multiple tissues, nearly all tissues had lower expression when compared to pituitary Krtcap3 expression, particularly the adrenal gland, brain regions, different fat pads, and liver. Noteworthy is that tissues with higher Krtcap3 expression than pituitary are the male gonad and the intestines. The dotted line across the plot represents a fold change of one or Krtcap3 expression in the pituitary. B: when comparing Krtcap3 expression between male and female WKY rats in select tissues, there were significant sex differences in two tissues: the pituitary where females had higher Krtcap3 expression than males, and the gonads where females had lower Krtcap3 expression than males. For intertissue visual comparison, all tissues were normalized to Krtcap3 expression in the WKY male pituitary. *P < 0.05, **P < 0.01, for sex differences, adjusted for multiple comparisons.
We then assessed Krtcap3 expression in select tissues between naïve male and female WKY rats to establish any differences in expression by sex (Fig. 7B). Female rats had significantly higher Krtcap3 expression in the pituitary (T4 = 7.05, P = 0.015) compared to male rats. Male rats, on the other hand, had much higher Krtcap3 expression in the testes than females did in the ovaries (T4 = 13.41, P = 1.43e-3). There was an additional visual difference between the sexes for Krtcap3 expression in the colon, but it did not meet the significance threshold.
Pathway Analysis of Hypothalamic RNA-Seq Data Indicate Improved Cholesterol Biosynthesis and Myelination in Study 2 Relative to Study 1
We assessed changes in gene expression in the hypothalamus of WT and KO rats of both study 1 and study 2. Although we have shown that Krtcap3 has very low expression in the hypothalamus, we aimed to identify changes in either satiety signaling or stress response signaling pathways that could indicate which pathways Krtcap3 may be involved in. We identified 132 significant differentially expressed genes (DEGs) between study 1 WT and KO female rats (Supplemental Table S2) and 290 significant DEGs between study 2 WT and KO rats (Supplemental Table S3), with only 3 genes in common. ATPase phospholipid transporting 8B3 (Atp8b3) was upregulated in KO rats in both studies, while receptor for activated C kinase 1 (Rack1) and SRY-box transcription factor 14 (Sox14) were downregulated in KO rats in both studies.
There were 301 significant DEGs between WT rats of study 1 and study 2 (Supplemental Table S4), and 230 significant DEGs between KO rats of study 1 and study 2 (Supplemental Table S5), with four genes in common. Interferon induced protein with tetratricopeptide repeats 3 (Ifit3) and arginine and serine rich protein 1 (Rsrp1) were both upregulated in study 2 rats, regardless of genotype; Kelch domain containing 7 A (Klhdc7a) and Tribbles homolog 2 (Trib2) were both downregulated.
As our focus here was on differences between the studies, we focused IPA analyses on differences between rats of the two studies separately for WT and KO rats. Between WT rats of study 1 and study 2, the top difference was an upregulation in study 2 of the cholesterol biosynthesis superpathway (Z = 2.24, P = 2e-5; Supplemental Fig. S5A). Notably, sterol regulatory element binding factors 1 and 2 (Srebf1, Srebf2) were predicted to be activated in study 2 rats (Fig. 8). In addition, glucagon like peptide 2 receptor (Glp2r) was significantly upregulated in study 2 rats (log2 = 1.58, P = 0.039; Supplemental Table S4).
Figure 8.

Canonical pathways and upstream regulators between study 1 and study 2 in wild-type (WT) rats support changes in environmental stress between the 2 studies. The top canonical pathway in WT rats was the superpathway for cholesterol biosynthesis, although some genes had connection to the myelination signaling pathway. Notably, there is predicted activation of sterol regulatory element-binding factors 1 and 2 (SREBF1, SREBF2) in study 2 relative to study 1, which have sterol sensing functions. Red and green indicate up- and downregulated genes in study 2 relative to study 1, respectively. Orange and blue indicate Ingenuity Pathway Analysis predictions of activity, where orange indicates predicted activation while blue indicates predicted inhibition. Correspondingly, orange arrows suggest activation while blue arrows suggest inhibition; yellow arrows indicate that the prediction is inconsistent with the state of the downstream molecule.
Between KO rats of the studies, the top finding was an upregulation of the myelination signaling pathway (Z = 1.81, P = 3.59e-7; Supplemental Fig. S5B). Analysis examining the effect of genotype for each study suggests possible differences in inflammatory response (Supplemental Fig. S6), but the evidence suggests that the hypothalamus is not the correct tissue to understand genotype-driven differences.
DISCUSSION
In the current study, there were no differences in the adiposity measures between WT and KO females, contradicting our previous results (13). Upon closer inspection of the data, we determined that in study 2 WT rats consumed a greater amount of food, which led to an increased body weight and fat mass: WT rats had increased in size without a corresponding change in KO rats. We hypothesize that this difference is due to a significant change in the environment between the two studies, where the environment in study 1 was likely more stressful. Our CORT data support this hypothesis, showing that WT rats had increased basal CORT relative to KO rats in study 1, with no differences in study 2. Changes in the expression of CORT metabolism genes and the hypothalamic RNA-seq data further support the argument for environmental differences between the studies. These findings suggest a role of Krtcap3 in the stress response. Importantly, this work highlights the need to more broadly consider environmental factors in animal research and how those factors may impact replication and translation to human research.
A Change in Environment and Circulating CORT Levels May Have Altered the Adiposity Phenotype
As previously described, Krtcap3 was first identified as a potential adiposity gene through GWAS analyses (10–12, 18) and we confirmed in vivo that decreased expression of Krtcap3 led to increased adiposity (13). We sought here to repeat the in vivo study with additional metabolic phenotyping and tissue collection to better understand the function of Krtcap3 in adiposity but failed to replicate our results. There were significant changes in the adiposity measures of WT rats in the current study relative to the previous study but no changes in Krtcap3 expression or the nearby obesity-related gene Adcy3. Furthermore, the data imply that study 2 WT rats were proportionally larger, instead of merely fatter, suggesting that study 1 WT rats had possibly failed to grow appropriately. This prompted us to wonder what caused such a shift between the two studies. We determined that there were two large environmental changes that we had not considered during study design: the COVID-19 pandemic and associated facility shutdowns plus the completion of a nearby construction project. These changes meant that the environment for the rats in study 2 was much quieter than it had been in study 1. Environmental noise is known to impact phenotype in rodent studies (19–21), leading us to hypothesize that the change in environment may have affected the adiposity phenotype by impacting the stress levels of the rats.
We first examined serum CORT in the rats and identified several interesting findings. First, in both studies, KO rats had lower serum CORT relative to WT rats, but the difference was highly significant only in study 1. This supported our hypothesis that stress levels may be related to the adiposity phenotype: differences in basal CORT between WT and KO correlated with differences in adiposity. It was initially surprising that a decrease in CORT was associated with an increase in body weight, as most research has correlated high CORT with obesity and poor metabolic health (22, 23). However, rodents are known to exhibit stress-induced hypophagia depending on factors such as sex, stress type and intensity, and age at stress exposure (24–33). Furthermore, substrains of the WKY, the background strain of the Krtcap3-KO rats, differ by body weight and plasma CORT levels in an inverse manner. Specifically, Wistar-Kyoto More Immobile (WMI) females have lower body weight and higher basal CORT compared to Wistar-Kyoto Less Immobile females (34, 35). Given this, we suggest that study 1 rats were smaller due to increased stress and hypophagia while KO rats were unaffected, presenting an adiposity phenotype in study 1 that did not manifest when neither genotype was exposed to a stressful environment in study 2. We propose that the genotype-driven difference we previously saw in adiposity was rather a genotype-driven difference in environmental stress response that had an indirect impact on body weight and fat mass.
This difference in stress response is supported by the distinct CORT response at euthanasia between WT and KO rats: when euthanized second, WT rats have a minimal CORT increase, but KO rats experience a spike in their CORT. We initially presumed this was evidence of chronic stress altering CORT secretion in response to acute stress in the study 1 WT rats (28, 32, 33), but replicating this pattern in study 2 rats indicated the effect was not related to environment. We propose that Krtcap3-KO rats exhibit low basal CORT and are relatively unaffected by low-grade chronic environmental stress relative to the stress-responsive WKY rat (36) but exhibit a heightened response to acute social stress relative to WT rats. Studies in rodents have previously shown strain-specific differences in CORT response following restraint stress plus differences in glucocorticoid receptor (GR) expression (37–39), supporting a role of genetics in basal CORT levels and stress response. Few causal genes, however, have been found. The current work suggests that Krtcap3 may be a novel stress gene.
Changes in CORT Processing Further Indicate a Change in Environment Between Study 1 and Study 2
We also considered if there were changes in gene expression that would further support our hypothesis of an environmental change between the studies. We examined expression of genes related to GC function and processing: Nr3c1, Hsd11β1, and Hsd11β2. Nr3c1 encodes the GR while the hydroxysteroid dehydrogenases isoforms respectively catalyze the activation or deactivation of CORT (40). Specifically, HSD11β1 is a bidirectional low-affinity enzyme that predominantly reactivates 11-dehydrocorticosterone to corticosterone (41), while HSD11β2 is a high affinity enzyme that exclusively oxidizes the biologically active corticosterone to inactive cortisone (42). Alterations of GC metabolism via these genes have been linked to obesity (43–47). We found that in liver tissue, the expression of the genes Nr3c1, Hsd11β1, and Hsd11β2 was higher in study 1 than study 2, with no genotype-driven differences. Since increased GCs have been shown to upregulate expression of both Hsd11β1 and Hsd11β2 (48), these findings are in agreement with increased prolonged environmental stress-induced levels of CORT in study 1. In RetroFat, the most striking difference was the higher expression of Hsd11β2 in study 2 relative to study 1, with again no differences by genotype, suggesting a decreased availability of the local bioactive glucocorticoids in study 2. Whether the increased Nr3c1 expression in both liver and fat tissue in study 1 compared to study 2 is in response to the increased stress environment in study 1 cannot be determined from this study. However, the lack of genotype difference in these findings suggests the suspected effects of Krtcap3 on stress regulation are not related to GC processing in the liver or adipose.
Tissue Expression Supports a Possible Role of Krtcap3 in Stress Response
By examining Krtcap3 expression in multiple tissues between male and female WKY rats, we have identified several tissues of interest for future investigations into the stress hypothesis and for evaluation of the sex differences we had previously identified (13). Krtcap3 was highly expressed in the pituitary gland and gastrointestinal tract of both male and female rats but only had high expression in the male gonads. The high expression of Krtcap3 in the pituitary gland supports the possibility of a direct role in the HPA axis and stress response. Krtcap3 is also highly expressed in the gastrointestinal tract of rats, which supports our previously suggested role along the gut-brain axis (13). The gut-brain axis has repeatedly been linked to behavior in preclinical and clinical models, germ-free status can negatively impact basal and stimulated HPA activity (49), and stress can disrupt intestinal barrier integrity (50, 51). These findings offer promising new leads for Krtcap3’s tissue of action, but additional work is still needed.
RNA-Seq Results Further Suggest the Effect of Environmental Stress on WT Rats
Despite low expression of Krtcap3 in the hypothalamus of WKY/NCrl rats, we chose to perform RNA-seq analysis on hypothalamus tissue from rats from study 1 and study 2. There were two reasons for this choice: we had limited tissues that had been collected between both studies and of those tissues the hypothalamus is connected to satiety and obesity (16) and to the stress response (17).
Overall, the results from RNA-seq analysis also support our hypothesis that there was a significant difference in the environments between study 1 and study 2 but do not shed much light on the pathways Krtcap3 acts in. In WT rats, IPA analysis demonstrated upregulation in cholesterol biosynthesis in study 2 WT. Cholesterol is vital for neuronal signaling, as it is involved in pre- and postsynaptic neurotransmitter signaling and serves as insulation for myelin sheaths (52). It is of real interest that the major hubs of the pathway are involved in the positive regulation of cholesterol biosynthesis and storage, specifically as Srebf1 and Srebf2 have sterol sensing functions. An additional interest is that there was a major expression difference in Glp2r between WT rats of the two studies, as Glp2r has been implicated in the regulation of food intake (53).
There was an upregulation of myelination signaling pathways in study 2 compared to study 1 KOs, and reduced myelin thickness has been well-connected to chronic stress (54, 55). Overall, the RNA-seq data demonstrate an increase in neural health in study 2 relative to study 1. The data also suggest that Krtcap3 is likely not acting at the level of the hypothalamus. Differences in basal and stress CORT between WT and KO rats, coupled with the high levels of Krtcap3 in the pituitary gland, suggest Krtcap3 may be acting at the level of the pituitary. Future studies are necessary for confirmation.
Conclusions
While we sought to replicate the previous adiposity phenotype between WT and Krtcap3-KO rats and identify the mechanism by which Krtcap3 is influencing adiposity, this study instead demonstrates a possible connection between Krtcap3 and stress response. In the environment of the second study, WT and KO rats had comparable levels of adiposity due to an increase in eating, body weight, and fat mass in the WT rats relative to the first study, without any corresponding changes in the KO rats. Analyses of CORT indicate that KO rats were able to maintain low CORT despite the potentially stressful environment of study 1, unlike WT rats, and that decreased Krtcap3 expression may impart resistance to low-grade chronic environmental stress. This may also account for the lack of change in eating behavior in the KO rats between studies. Expression of GR and the hydroxysteroid dehydrogenases in the liver and visceral fat also support significant differences in CORT processing between the two studies but do not explain the differences we see between WT and KO rats. The results from RNA-seq analysis also indicate a change in environment between the two studies that primarily had an impact on the neuronal health of WT rats. That Krtcap3 is highly expressed in both the pituitary and the gastrointestinal tract further supports a potential role of Krtcap3 in stress. In total, these data suggest that decreased expression of Krtcap3 may protect against low-grade, chronic environmental stress and prevent alterations to eating behavior. WT and KO rats also respond to the euthanasia protocol differently, indicating that KO rats may have a larger response to acute psychosocial stressors than WT rats.
Despite these intriguing findings, there are limitations to this study. We cannot entirely eliminate the possibility that the differences we see are noise. Ours would not be the first laboratory to see phenotypic differences emerge under different environmental conditions or at a different time (56, 57), and many in vivo studies have been marked by a reproducibility crisis (58–60). However, we argue that seeing a different phenotype under different environmental conditions does not mean either phenotype is false or that the variation is random. Rather, gene-by-environment interactions influencing phenotype should be expected, as organisms without such phenotypic plasticity would not survive. Failing to consider environmental factors that do not fall directly under experimental design (19, 61–63), and rigorously standardizing animal experiments (64, 65) fails to consider this plasticity.
An additional limitation is that we currently have not identified where Krtcap3 is acting in the body to influence CORT nor are we able to draw conclusions on its mechanistic role in the HPA axis. Finally, although previous work demonstrated sex differences in adiposity (13), the current study was conducted only in female rats and we are unable to make any claims on the role of Krtcap3 in the stress response of male rats.
While we have identified a potential connection between Krtcap3 and stress that may play a role in obesity, additional studies are required to confirm this connection. Studies are currently underway to clarify the relationship between Krtcap3 expression and stress response and to confirm if it has downstream consequences for obesity. Future studies will also identify the tissue of action for Krtcap3, elucidate the mechanism of Krtcap3 in the stress response pathway, identify potential protein interaction partners, and investigate the role of Krtcap3 in stress in male rats.
Broader Impacts
These data point to an important role of Krtcap3 in the stress response pathway. Serum CORT measurements indicate that decreased Krtcap3 expression is associated with low basal CORT and resistance to low-grade chronic environmental stress but an enhanced response to acute social behavioral stressors. High Krtcap3 expression in the pituitary and gastrointestinal tract further reflects potential roles in the stress response pathway either by the HPA axis, the gut-brain axis, or both.
The possible role of Krtcap3 in the stress response is an important consideration for the translational aspect of this research, as the environment for human patients is much more complex than the laboratory environment and plays a significant role in obesity development and treatment. Genes for complex diseases such as obesity must be considered within the environment that human patients exist in. Better consideration of the genetics of stress response (66–69) will improve understanding of stress-related diseases, such as obesity (4, 70–73). Although the underlying molecular mechanisms are still unclear, genetic variation in GC sensitivity in patients has been shown to alter obesity progression and sequelae when patients were exposed to stress (74). Obesity results from a complex web of genetic and environmental factors that are difficult to untangle, and it is necessary to understand how adiposity-related genes function in a translationally relevant environment. Realizing that the role of Krtcap3 in growth and adiposity may be dependent on its function in the stress response opens up a new avenue of research for this gene in particular but also broadens our understanding of how to investigate the genetics of obesity moving forward.
In addition to our specific findings for Krtcap3, an important takeaway from the current study is what it reveals about animal research. Many animal studies only describe the factors that investigators are able to control: the type of housing, the day/night cycles, the type of food, etc. Factors that are not commonly described in published research, but have been shown to have an impact, include who was handling the animals (63), the noise levels within and outside of the facility (19, 20, 61, 75, 76), the light intensity (61), etc. Failing to fully consider the environment can lead investigators to inadvertently misinterpret results and hinder scientific progress (77) and contributes to science’s reproducibility crisis. Moving forward, genetic research using animal models ought to address these issues, as it is only when we consider the phenotype within the broader context of gene-by-environment interactions that we can fully understand the role of that gene. For example, without this in vivo study, we would not have made the connection between Krtcap3 and stress. How many other genes might have been ignored because of phenotypic plasticity?
DATA AVAILABILITY
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (https://doi.org/10.1093/nar/30.1.207) and are accessible through GEO Series accession number GSE236873 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE236873).
SUPPLEMENTAL MATERIAL
Supplemental Tables S1–S5: https://doi.org/10.6084/m9.figshare.23751456.v1.
Supplemental Figs. S1–S6: https://doi.org/10.6084/m9.figshare.23751450.v1.
GRANTS
This work was supported by NIH Grants R01DK106386, T32DA041349, and R01DK120667.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M.S. and L.C.S.W. conceived and designed research; A.M.S., G.G., E.G., and O.S. performed experiments; A.M.S. analyzed data; A.M.S., E.E.R., and L.C.S.W. interpreted results of experiments; A.M.S. prepared figures; A.M.S. drafted manuscript; A.M.S., E.E.R., and L.C.S.W. edited and revised manuscript; A.M.S., G.G., E.G., O.S., M.G., J.K., A.M.G., E.E.R., and L.C.S.W. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Aaron Deal for assisting with feed and weigh, Bailey McDonald for assisting with IPGTT experiments, Katherine Przybyl for conducting initial analyses of corticosterone in the rats, Mackenzie Fitzpatrick for helping dissect inbred rats, and Dr. Laura Cox for guidance on RNA-seq analysis. The authors also acknowledge Azenta for running RNA-seq analysis and analyzing differentially expressed genes. As always, the authors thank the Medical College of Wisconsin Genotyping Core for assistance in genotyping the rats. Graphical abstract image created with BioRender.com and published with permission.
Preprint is available at https://doi.org/10.1101/2023.03.15.532439.
REFERENCES
- 1. Stierman BA, JosephCarroll MD, Chen TC, Davy O, Fink S, Fryar CD, Gu Q, Hales CM, Hughes JP, Ostchega Y, Storandt RJ, Akinbami LJ. National Health and Nutrition Examination Survey 2017–March 2020 Prepandemic Data Files Development of Files and Prevalence Estimates for Selected Health Outcomes. Hyattsville, MD: National Center for Health Statistics, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Restrepo BJ. Obesity prevalence among U.S. adults during the COVID-19 pandemic. Am J Prev Med 63: 102–106, 2022. doi: 10.1016/j.amepre.2022.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lange SJ, Kompaniyets L, Freedman DS, Kraus EM, Porter R, Blanck HM, Goodman AB, DNP3. Longitudinal trends in body mass index before and during the COVID-19 pandemic among persons aged 2–19 years–United States, 2018-2020. MMWR Morb Mortal Wkly Rep 70: 1278–1283, 2021. [Erratum in MMWR Morb Mortal Wkly Rep 70: 1355, 2021]. doi: 10.15585/mmwr.mm7037a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kahan LG, Mehrzad R. Environmental factors related to the obesity epidemic. In: Obesity: Global Impact and Epidemiology, edited by Mehrzad R. Amsterdam, The Netherlands: Elsevier, 2020, p. 117–139. [Google Scholar]
- 5. Abadi A, Alyass A, Robiou Du Pont S, Bolker B, Singh P, Mohan V, Diaz R, Engert JC, Yusuf S, Gerstein HC, Anand SS, Meyre D. Penetrance of polygenic obesity susceptibility loci across the body mass index distribution. Am J Hum Genet 101: 925–938, 2017. doi: 10.1016/j.ajhg.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, Giant Consortium. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet 27: 3641–3649, 2018. doi: 10.1093/hmg/ddy271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, Yengo L, Ferreira T, Marouli E, Ji Y, Yang J, Jones S, Beaumont R, Croteau-Chonka DC, Winkler TW, Consortium G, Hattersley AT, Loos RJ, Hirschhorn JN, Visscher PM, Frayling TM, Yaghootkar H, Lindgren CM, GIANT Consortium. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet 28: 166–174, 2019. doi: 10.1093/hmg/ddy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rohde K, Keller M, la Cour Poulsen L, Bluher M, Kovacs P, Bottcher Y. Genetics and epigenetics in obesity. Metabolism 92: 37–50, 2019. doi: 10.1016/j.metabol.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 9. Bouchard C. Genetics of obesity: what we have learned over decades of research. Obesity (Silver Spring) 29: 802–820, 2021. doi: 10.1002/oby.23116. [DOI] [PubMed] [Google Scholar]
- 10. Keele GR, Prokop JW, He H, Holl K, Littrell J, Deal A, Francic S, Cui L, Gatti DM, Broman KW, Tschannen M, Tsaih SW, Zagloul M, Kim Y, Baur B, Fox J, Robinson M, Levy S, Flister MJ, Mott R, Valdar W, Solberg Woods LC. Genetic fine-mapping and identification of candidate genes and variants for adiposity traits in outbred rats. Obesity (Silver Spring) 26: 213–222, 2018. doi: 10.1002/oby.22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chitre AS, Polesskaya O, Holl K, Gao J, Cheng R, Bimschleger H, Garcia Martinez A, George T, Gileta AF, Han W, Horvath A, Hughson A, Ishiwari K, King CP, Lamparelli A, Versaggi CL, Martin C, St Pierre CL, Tripi JA, Wang T, Chen H, Flagel SB, Meyer P, Richards J, Robinson TE, Palmer AA, Solberg Woods LC. Genome-wide association study in 3,173 outbred rats identifies multiple loci for body weight, adiposity, and fasting glucose. Obesity (Silver Spring) 28: 1964–1973, 2020. doi: 10.1002/oby.22927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen YC, Xu C, Zhang JG, Zeng CP, Wang XF, Zhou R, Lin X, Ao ZX, Lu JM, Shen J, Deng HW. Multivariate analysis of genomics data to identify potential pleiotropic genes for type 2 diabetes, obesity and dyslipidemia using Meta-CCA and gene-based approach. PLoS One 13: e0201173, 2018. doi: 10.1371/journal.pone.0201173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szalanczy AM, Goff E, Seshie O, Deal A, Grzybowski M, Klotz J, Chuang Key CC, Geurts AM, Solberg Woods LC. Keratinocyte-associated protein 3 plays a role in body weight and adiposity with differential effects in males and females. Front Genet 13: 942574, 2022. doi: 10.3389/fgene.2022.942574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tian Y, Peng B, Fu X. New ADCY3 variants dance in obesity etiology. Trends Endocrinol Metab 29: 361–363, 2018. doi: 10.1016/j.tem.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 15. Wu L, Shen C, Seed Ahmed M, Ostenson CG, Gu HF. Adenylate cyclase 3: a new target for anti-obesity drug development. Obes Rev 17: 907–914, 2016. doi: 10.1111/obr.12430. [DOI] [PubMed] [Google Scholar]
- 16. Ahima RS, Antwi DA. Brain regulation of appetite and satiety. Endocrinol Metab Clin North Am 37: 811–823, 2008. doi: 10.1016/j.ecl.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci 8: 383–395, 2006. doi: 10.31887/DCNS.2006.8.4/ssmith. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Le TH, Crouse WL, Keele GR, Holl K, Seshie O, Tschannen M, Craddock A, Das SK, Szalanczy AM, McDonald B, Grzybowski M, Klotz J, Sharma NK, Geurts AM, Chuang Key CC, Hawkins G, Valdar W, Mott R, Solberg Woods LC. Genetic mapping of multiple metabolic traits identifies novel genes for adiposity, lipids and insulin secretory capacity in outbred rats. Diabetes 72: 135–148, 2022. doi: 10.2337/db22-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zymantiene J, Zelvyte R, Pampariene I, Aniuliene A, Juodziukyniene N, Kantautaite J, Oberauskas V. Effects of long-term construction noise on health of adult female Wistar rats. Pol J Vet Sci 20: 155–165, 2017. doi: 10.1515/pjvs-2017-0020. [DOI] [PubMed] [Google Scholar]
- 20. Toukh M, Gordon SP, Othman M. Construction noise induces hypercoagulability and elevated plasma corticosteroids in rats. Clin Appl Thromb Hemost 20: 710–715, 2014. doi: 10.1177/1076029613483168. [DOI] [PubMed] [Google Scholar]
- 21. Shepherd EJ, Helliwell PA, Mace OJ, Morgan EL, Patel N, Kellett GL. Stress and glucocorticoid inhibit apical GLUT2-trafficking and intestinal glucose absorption in rat small intestine. J Physiol 560: 281–290, 2004. doi: 10.1113/jphysiol.2004.072447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Campbell JE, Peckett AJ, D'Souza AM, Hawke TJ, Riddell MC. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am J Physiol Cell Physiol 300: C198–C209, 2011. doi: 10.1152/ajpcell.00045.2010. [DOI] [PubMed] [Google Scholar]
- 23. Roberge C, Carpentier AC, Langlois MF, Baillargeon JP, Ardilouze JL, Maheux P, Gallo-Payet N. Adrenocortical dysregulation as a major player in insulin resistance and onset of obesity. Am J Physiol Endocrinol Metab 293: E1465–E1478, 2007. doi: 10.1152/ajpendo.00516.2007. [DOI] [PubMed] [Google Scholar]
- 24. Kinlein SA, Shahanoor Z, Romeo RD, Karatsoreos IN. Chronic corticosterone treatment during adolescence has significant effects on metabolism and skeletal development in male C57BL6/N mice. Endocrinology 158: 2239–2254, 2017. doi: 10.1210/en.2017-00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oliver DK, Intson K, Sargin D, Power SK, McNabb J, Ramsey AJ, Lambe EK. Chronic social isolation exerts opposing sex-specific consequences on serotonin neuronal excitability and behaviour. Neuropharmacology 168: 108015, 2020. doi: 10.1016/j.neuropharm.2020.108015. [DOI] [PubMed] [Google Scholar]
- 26. Yamada C, Iizuka S, Nahata M, Hattori T, Takeda H. Vulnerability to psychological stress-induced anorexia in female mice depends on blockade of ghrelin signal in nucleus tractus solitarius. Br J Pharmacol 177: 4666–4682, 2020. doi: 10.1111/bph.15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Francois M, Canal Delgado I, Shargorodsky N, Leu CS, Zeltser L. Assessing the effects of stress on feeding behaviors in laboratory mice. Elife 11: e70271, 2022. doi: 10.7554/eLife.70271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui DH, Tabira T. Chronic stress differentially regulates glucocorticoid negative feedback response in rats. Psychoneuroendocrinology 26: 443–459, 2001. doi: 10.1016/s0306-4530(01)00004-x. [DOI] [PubMed] [Google Scholar]
- 29. Santha P, Veszelka S, Hoyk Z, Meszaros M, Walter FR, Toth AE, Kiss L, Kincses A, Olah Z, Seprenyi G, Rakhely G, Der A, Pakaski M, Kalman J, Kittel A, Deli MA. Restraint stress-induced morphological changes at the blood-brain barrier in adult rats. Front Mol Neurosci 8: 88, 2015. doi: 10.3389/fnmol.2015.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shoji H, Miyakawa T. Differential effects of stress exposure via two types of restraint apparatuses on behavior and plasma corticosterone level in inbred male BALB/cAJcl mice. Neuropsychopharmacol Rep 40: 73–84, 2020. doi: 10.1002/npr2.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Olave FA, Aguayo FI, Roman-Albasini L, Corrales WA, Silva JP, Gonzalez PI, Lagos S, Garcia MA, Alarcon-Mardones M, Rojas PS, Xu X, Cidlowski JA, Aliaga E, Fiedler J. Chronic restraint stress produces sex-specific behavioral and molecular outcomes in the dorsal and ventral rat hippocampus. Neurobiol Stress 17: 100440, 2022. doi: 10.1016/j.ynstr.2022.100440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gilles EE, Schultz L, Baram TZ. Abnormal corticosterone regulation in an immature rat model of continuous chronic stress. Pediatr Neurol 15: 114–119, 1996. doi: 10.1016/0887-8994(96)00153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wulsin AC, Wick-Carlson D, Packard BA, Morano R, Herman JP. Adolescent chronic stress causes hypothalamo-pituitary-adrenocortical hypo-responsiveness and depression-like behavior in adult female rats. Psychoneuroendocrinology 65: 109–117, 2016. doi: 10.1016/j.psyneuen.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo W, Lim PH, Wert SL, Gacek SA, Chen H, Redei EE. Hypothalamic gene expression and postpartum behavior in a genetic rat model of depression. Front Behav Neurosci 14: 589967, 2020. doi: 10.3389/fnbeh.2020.589967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lim PH, Wert SL, Tunc-Ozcan E, Marr R, Ferreira A, Redei EE. Premature hippocampus-dependent memory decline in middle-aged females of a genetic rat model of depression. Behav Brain Res 353: 242–249, 2018. doi: 10.1016/j.bbr.2018.02.030. [DOI] [PubMed] [Google Scholar]
- 36. Redei EE, Udell ME, Solberg-Woods LC, Chen H. The Wistar Kyoto rat: a model of depression traits. Curr Neuropharmacol 21: 1884–1905, 2023. doi: 10.2174/1570159X21666221129120902. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCutcheon JE, Fisher AS, Guzdar E, Wood SA, Lightman SL, Hunt SP. Genetic background influences the behavioural and molecular consequences of neurokinin-1 receptor knockout. Eur J Neurosci 27: 683–690, 2008. doi: 10.1111/j.1460-9568.2008.06043.x. [DOI] [PubMed] [Google Scholar]
- 38. Terenina EE, Cavigelli S, Mormede P, Zhao W, Parks C, Lu L, Jones BC, Mulligan MK. Genetic factors mediate the impact of chronic stress and subsequent response to novel acute stress. Front Neurosci 13: 438, 2019. doi: 10.3389/fnins.2019.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Solberg LC, Baum AE, Ahmadiyeh N, Shimomura K, Li R, Turek FW, Takahashi JS, Churchill GA, Redei EE. Genetic analysis of the stress-responsive adrenocortical axis. Physiol Genomics 27: 362–369, 2006. doi: 10.1152/physiolgenomics.00052.2006. [DOI] [PubMed] [Google Scholar]
- 40. Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Front Immunol 10: 1545, 2019. doi: 10.3389/fimmu.2019.01545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1–a tissue-specific amplifier of glucocorticoid action. Endocrinology 142: 1371–1376, 2001. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- 42. Draper N, Stewart PM. 11β-Hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol 186: 251–271, 2005. doi: 10.1677/joe.1.06019. [DOI] [PubMed] [Google Scholar]
- 43. Majer-Łobodzińska A, Adamiec-Mroczek J. Glucocorticoid receptor polymorphism in obesity and glucose homeostasis. Adv Clin Exp Med 26: 143–148, 2017. doi: 10.17219/acem/41231. [DOI] [PubMed] [Google Scholar]
- 44. Wamil M, Seckl JR. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discov Today 12: 504–520, 2007. doi: 10.1016/j.drudis.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 45. Pereira CD, Azevedo I, Monteiro R, Martins MJ. 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab 14: 869–881, 2012. doi: 10.1111/j.1463-1326.2012.01582.x. [DOI] [PubMed] [Google Scholar]
- 46. Morton NM, Seckl JR. 11β-Hydroxysteroid dehydrogenase type 1 and obesity. Front Horm Res 36: 146–164, 2008. doi: 10.1159/000115363. [DOI] [PubMed] [Google Scholar]
- 47. Mussig K, Remer T, Haupt A, Gallwitz B, Fritsche A, Haring HU, Maser-Gluth C. 11β-Hydroxysteroid dehydrogenase 2 activity is elevated in severe obesity and negatively associated with insulin sensitivity. Obesity (Silver Spring) 16: 1256–1260, 2008. doi: 10.1038/oby.2008.218. [DOI] [PubMed] [Google Scholar]
- 48. Chapman K, Holmes M, Seckl J. 11β-Hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev 93: 1139–1206, 2013. doi: 10.1152/physrev.00020.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martin CR, Mayer EA. Gut-brain axis and behavior. Nestle Nutr Inst Workshop Ser 88: 45–53, 2017. doi: 10.1159/000461732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shukla PK, Meena AS, Pierre JF, Rao R. Central role of intestinal epithelial glucocorticoid receptor in alcohol- and corticosterone-induced gut permeability and systemic response. FASEB J 36: e22061, 2022. doi: 10.1096/fj.202101424R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bahlouli W, Breton J, Lelouard M, L'Huillier C, Tirelle P, Salameh E, Amamou A, Atmani K, Goichon A, Bole-Feysot C, Ducrotte P, Ribet D, Dechelotte P, Coeffier M. Stress-induced intestinal barrier dysfunction is exacerbated during diet-induced obesity. J Nutr Biochem 81: 108382, 2020. doi: 10.1016/j.jnutbio.2020.108382. [DOI] [PubMed] [Google Scholar]
- 52. Petrov AM, Kasimov MR, Zefirov AL. Brain cholesterol metabolism and its defects: linkage to neurodegenerative diseases and synaptic dysfunction. Acta Naturae 8: 58–73, 2016. doi: 10.32607/20758251-2016-8-1-58-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tang-Christensen M, Larsen PJ, Thulesen J, Romer J, Vrang N. The proglucagon-derived peptide, glucagon-like peptide-2, is a neurotransmitter involved in the regulation of food intake. Nat Med 6: 802–807, 2000. doi: 10.1038/77535. [DOI] [PubMed] [Google Scholar]
- 54. Forbes TA, Gallo V. All wrapped up: environmental effects on myelination. Trends Neurosci 40: 572–587, 2017. doi: 10.1016/j.tins.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Antontseva E, Bondar N, Reshetnikov V, Merkulova T. The effects of chronic stress on brain myelination in humans and in various rodent models. Neuroscience 441: 226–238, 2020. doi: 10.1016/j.neuroscience.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 56. Crabbe JC, Wahlsten D, Dudek BC. Genetics of mouse behavior: interactions with laboratory environment. Science 284: 1670–1672, 1999. doi: 10.1126/science.284.5420.1670. [DOI] [PubMed] [Google Scholar]
- 57. Karp NA, Speak AO, White JK, Adams DJ, Hrabe de Angelis M, Herault Y, Mott RF. Impact of temporal variation on design and analysis of mouse knockout phenotyping studies. PLoS One 9: e111239, 2014. doi: 10.1371/journal.pone.0111239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov 10: 712, 2011. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 59. Hunter P. The reproducibility “crisis”: reaction to replication crisis should not stifle innovation. EMBO Rep 18: 1493–1496, 2017. doi: 10.15252/embr.201744876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Drucker DJ. Never waste a good crisis: confronting reproducibility in translational research. Cell Metab 24: 348–360, 2016. doi: 10.1016/j.cmet.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 61. Castelhano-Carlos MJ, Baumans V. The impact of light, noise, cage cleaning and in-house transport on welfare and stress of laboratory rats. Lab Anim 43: 311–327, 2009. doi: 10.1258/la.2009.0080098. [DOI] [PubMed] [Google Scholar]
- 62. Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 56: 1129–1139, 2013. doi: 10.1007/s00125-013-2846-8. [DOI] [PubMed] [Google Scholar]
- 63. Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, Wieskopf JS, Acland EL, Dokova A, Kadoura B, Leger P, Mapplebeck JC, McPhail M, Delaney A, Wigerblad G, Schumann AP, Quinn T, Frasnelli J, Svensson CI, Sternberg WF, Mogil JS. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods 11: 629–632, 2014. doi: 10.1038/nmeth.2935. [DOI] [PubMed] [Google Scholar]
- 64. Voelkl B, Vogt L, Sena ES, Wurbel H. Reproducibility of preclinical animal research improves with heterogeneity of study samples. PLoS Biol 16: e2003693, 2018. doi: 10.1371/journal.pbio.2003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. von Kortzfleisch VT, Karp NA, Palme R, Kaiser S, Sachser N, Richter SH. Improving reproducibility in animal research by splitting the study population into several ‘mini-experiments’. Sci Rep 10: 16579, 2020. doi: 10.1038/s41598-020-73503-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ising M, Holsboer F. Genetics of stress response and stress-related disorders. Dialogues Clin Neurosci 8: 433–444, 2006. doi: 10.31887/DCNS.2006.8.4/mising. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van den Bos R, Harteveld M, Stoop H. Stress and decision-making in humans: performance is related to cortisol reactivity, albeit differently in men and women. Psychoneuroendocrinology 34: 1449–1458, 2009. doi: 10.1016/j.psyneuen.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 68. Henckens MJ, Klumpers F, Everaerd D, Kooijman SC, van Wingen GA, Fernandez G. Interindividual differences in stress sensitivity: basal and stress-induced cortisol levels differentially predict neural vigilance processing under stress. Soc Cogn Affect Neurosci 11: 663–673, 2016. doi: 10.1093/scan/nsv149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Flati T, Gioiosa S, Chillemi G, Mele A, Oliverio A, Mannironi C, Rinaldi A, Castrignano T. A gene expression atlas for different kinds of stress in the mouse brain. Sci Data 7: 437, 2020. doi: 10.1038/s41597-020-00772-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xiao Y, Liu D, Cline MA, Gilbert ER. Chronic stress, epigenetics, and adipose tissue metabolism in the obese state. Nutr Metab (Lond) 17: 88, 2020. doi: 10.1186/s12986-020-00513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Torres SJ, Nowson CA. Relationship between stress, eating behavior, and obesity. Nutrition 23: 887–894, 2007. doi: 10.1016/j.nut.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 72. Lee A, Cardel M, Donahoo WT. Social and environmental factors influencing Obesity. In: Endotext, edited by Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP.. South Dartmouth, MA: MDText.com, Inc., 2000. [Google Scholar]
- 73. Scott KA, Melhorn SJ, Sakai RR. Effects of chronic social stress on obesity. Curr Obes Rep 1: 16–25, 2012. doi: 10.1007/s13679-011-0006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van der Valk ES, Savas M, van Rossum EF. Stress and obesity: are there more susceptible individuals? Curr Obes Rep 7: 193–203, 2018. doi: 10.1007/s13679-018-0306-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dallman MF, Akana SF, Bell ME, Bhatnagar S, Choi S, Chu A, Gomez F, Laugero K, Soriano L, Viau V. Warning! Nearby construction can profoundly affect your experiments. Endocrine 11: 111–113, 1999. doi: 10.1385/ENDO:11:2:111. [DOI] [PubMed] [Google Scholar]
- 76. Raff H, Bruder ED, Cullinan WE, Ziegler DR, Cohen EP. Effect of animal facility construction on basal hypothalamic-pituitary-adrenal and renin-aldosterone activity in the rat. Endocrinology 152: 1218–1221, 2011. doi: 10.1210/en.2010-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Voelkl B, Wurbel H. Reproducibility crisis: are we ignoring reaction norms? Trends Pharmacol Sci 37: 509–510, 2016. doi: 10.1016/j.tips.2016.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Tables S1–S5: https://doi.org/10.6084/m9.figshare.23751456.v1.
Supplemental Figs. S1–S6: https://doi.org/10.6084/m9.figshare.23751450.v1.
Data Availability Statement
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (https://doi.org/10.1093/nar/30.1.207) and are accessible through GEO Series accession number GSE236873 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE236873).