

Abstract
Transition state analogue inhibitor design (TSID) and fragment-based drug design (FBDD) are drug design approaches typically used independently. Methylthio-DADMe-Immucillin-A (MTDIA) is a tight-binding transition state analogue of bacterial 5′-methylthioadenosine nucleosidases (MTANs). Previously, Salmonella enterica MTAN structures were found to bind MTDIA and ethylene glycol fragments, but MTDIA modified to contain similar fragments did not enhance affinity. Seventy-five published MTAN structures were analyzed, and co-crystallization fragments were found that might enhance the binding of MTDIA to other bacterial MTANs through contacts external to MTDIA binding. The fragment-modified MTDIAs were tested with Helicobacter pylori MTAN and Staphylococcus aureus MTANs (HpMTAN and SaMTAN) as test cases to explore inhibitor optimization by potential contacts beyond the transition state contacts. Replacement of a methyl group with a 2′-ethoxyethanol group in MTDIA improved the dissociation constant 14-fold (0.09 nM vs 1.25 nM) for HpMTAN and 81-fold for SaMTAN (0.096 nM vs 7.8 nM). TSID combined with FBDD can be useful in enhancing already powerful inhibitors.
Stable analogues resembling the transition state are often tight-binding inhibitors and therefore are attractive as drug prospects. Design of these analogues requires the elucidation of transition state information, often using kinetic isotope effects and computational chemistry. (1–5) Inhibitor design based on a transition state structure is called transition state analogue inhibitor design (TSID) (Figure 1A). (5–8) Transition state analogues are designed by replicating the geometry and electrostatic potential of the reactants at the transition state as mimics of the normal substrate or substrates of the physiological reaction. We considered the use of structure-based enhancement for complexes of the target enzyme and the initial transition state analogue family. Using this approach, it has been possible to identify structurally favorable sites to enhance binding of the original analogues.
Figure 1.

Schematic view of TSID and FBDD in enzyme inhibitor design. (A) TSID designs inhibitors after elucidation of the transition state structure. (B) FBDD identifies and tailors the binding fragments to generate inhibitors.
The concept of fragment-based drug discovery (FBDD) was introduced decades ago and is now well developed in the discovery of lead compounds. (9–12) FBDD screens provide structure–activity relationship (SAR) information for small fragments, usually having low binding affinities. Potent inhibitors are designed by integrating multiple active fragments, producing enhanced affinity (Figure 1B). (11–13) Often, several iterative cycles of optimization are required to achieve tighter binding.
Bacterial 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidases (MTANs) hydrolyze 5′-methylthioadenosine (MTA), S-adenosylhomocysteine (SAH), and 5′-deoxyadenosine to produce adenine and the respective ribose derivatives. MTAN plays important roles in bacterial quorum sensing, adenine recycling, polyamine synthesis, and the methionine cycle. (14–16) Recently, this enzyme was also discovered to be essential for menaquinone synthesis in Helicobacter pylori, where aminofutalosine is hydrolyzed to produce dehypoxanthinyl-futalosine and adenine. (17) Consequently, inhibiting H. pylori MTAN (HpMTAN) prevents bacterial growth in vitro. (17) The transition state of HpMTAN has been reported to be a neutral adenine leaving group and a ribose cation, with the N-glycosidic bond extended to nearly 3 Å. (18,19) An original transition state analogue of HpMTAN was methylthio-DADMe-Immucillin-A (MTDIA) (Figure S1). This compound mimics the MTA substrate at the transition state and has a nanomolar to picomolar affinity for MTANs isolated from various bacterial species. (6,19) A chemical library based on MTDIA has been synthesized. (6) When assayed against HpMTAN, some compounds from this library demonstrated affinities equivalent to or better than that of MTDIA. (20,21) MTAN is not an essential enzyme for Staphylococcus aureus, but it is involved in the pathway of quorum sensing, linked to biofilm production, a major virulence factor of S. aureus. The deletion of MTAN led to a decreased level of biofilm formation of S. aureus in vitro and reduced pathogenicity of S. aureus in mouse and zebrafish models, (22) suggesting SaMTAN is a potential target for S. aureus infection. SaMTAN has been suggested to have a transition state structure similar to that of HpMTAN. (19) Consistently, MTDIA also binds to SaMTAN with a picomolar affinity. (19)
Here we use X-ray crystallography to explore the interactions of low-molecular weight compounds that might improve the potency of MTDIA in MTANs other than that from Salmonella enterica. (23) We reviewed 75 crystal structures of MTANs deposited in the Protein Data Bank (Table S1). Like S. enterica, crystal structures of several bacterial MTANs were found to contain small chemical fragments originating from the crystallization medium. These include acetate, glycine, ethanediol, isopropanol, glycerol, triethylene glycol (PEG3, C6H14O4), tetraethylene glycol (PEG4, C8H18O5), and di(hydroxyethyl)ether (PEG2.5, C5H12O3) (Table S1). Fragment-modified MTDIAs do not improve the binding of transition state analogues to the S. enterica MTAN; (23) thus, we wanted to determine if the fragment-modified MTDIAs improved the binding of analogues to other MTANs, using HpMTAN and SaMTAN to exemplify the approach.
The amino acid sequence of HpMTAN is >70% similar to those of both Escherichia coli and S. enterica MTANs (EcMTAN and SeMTAN, respectively). In structural alignment, MTDIA-bound HpMTAN has a root-mean-square deviation (RMSD) of <1 Å from EcMTAN and SeMTAN. (21,23,24) Seven of the available structures are MTAN–MTDIA complexes, and the MTDIA moieties overlap closely with an RMSD of <0.1 Å. Testing of small related fragments, incorporated into MTDIA, provided ligands to be tested in fragment-based drug design (FBDD) against additional MTANs.
The conformation of bacterial MTANs is well conserved across all available crystal structures, especially with respect to the active sites. (23–26) The active site of MTAN includes an adenine binding site, a ribose binding site, and the 5′-methylthio binding site in a hydrophobic channel oriented toward the solvent. The transition state analogue MTDIA bound to HpMTAN occupies all of these sites (Figure 2A). (21) While the adenine binding site and ribose binding site are fully occupied, there is an unoccupied space adjacent to the 5′-methylthio binding site (Figure 2A). Thus, MTDIA could be tailored with fragments that bind to the open space of the 5′-binding site in attempts to improve affinity.
Figure 2.

(A) Binding of MTDIA, (B) PEG3, and (C) compound 2 in the active site of MTANs. In panel A, there is an extra space next to the 5′-region of MTDIA for HpMTAN [Protein Data Bank (PDB) entry 4WKN]. (21) In panel B, PEG3 binds to the extra space next to the 5′-binding site (PDB entry 4F1W). (23) In panel C, two conformations of compound 2 both bind at MTDIA and PEG3 sites (PDB entry 4F2P). (23) MTAN is shown in a van der Waals surface view (gray), and MTDIA, PEG3, and compound 2 are shown as colored sticks.
Crystal structures of MTDIA bound to MTANs confirm that an open space to accept an added fragment is at the active site adjacent to the 5′-methylthio group of MTDIA (Figure 2A). Among the 75 crystal structures of MTANs (Table S1), the EcMTAN and SeMTAN structures include a polyethylene glycol fragment occupying the space adjacent to the 5′-binding site (Figure S2). Notably, this space is occupied by two glycol repeating units from PEG2.5 in EcMTAN and PEG3 in SeMTAN, crystallized in the absence of MTDIA (Figure S2). The presence of PEG2.5 and PEG3 fragments indicates favorable binding energetics in the crystal and suggests the addition of ethylene glycol units to MTDIA (Figure S2). Specifically, two ethylene glycol units interact with MTANs using both hydrophobic and hydrophilic interactions (Figure 2B).
On the basis of the diglycol interactions, compounds 2–6 were developed and originally tested for transition state analogue enhancement for SeMTAN. (23) These 5′-substituents made an only 3-fold difference in the affinity for SeMTAN (comparing MTDIA with 2–6). (23) In contrast, the compounds gave ≤14-fold increased affinity relative to that of MTDIA for HpMTAN (Figure 3). The metabolic role of HpMTAN differs from that of the MTANs of most enteric bacteria in its essential contribution to the synthesis of menaquinone via the futalosine pathway. (27) Compound 7 was designed by the addition of a 5′-aminofultalosine substituent onto the scaffold of MTDIA and gave a dissociation constant of 1.08 nM. The six 5′-substituted compounds had dissociation constants from 0.09 to 1.08 nM at 37 °C, demonstrating excellent mimicry of the HpMTAN transition state structure. Slow-onset inhibition was not apparent for any transition state analogue in this study at 37 °C, and the Ki value, a dissociation constant for competitive inhibitors, is reported here (Figure 3). Four compounds (3–6) had Ki values up to 4-fold lower than that of MTDIA; one (7, with a bulky aromatic group) had a comparable Ki value, and one (2) had a Ki 14-fold lower than that of MTDIA (Figure 3).
Figure 3.

Km and Ki values for transition state analogues of HpMTAN (dark gray) and SaMTAN (light gray). (20,21) The top scale indicates values (nanomolar) for the Ki and Km values at 37 °C. Note that the values for MTA are Km values.
The affinity of PEG small fragments alone was tested in direct inhibiton experiments as indicated in the Supporting Information. Compared to the nanomolar inhibition of MTDIA, the fragments bound at least 6 orders of magnitude weaker, in the millimolar range (Table 1 and Tables S2 and S3).
Table 1.
Inhibition of MTANs by PEG Fragments
| Ki for HpMTAN (mM) | |||
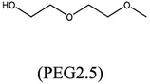
|
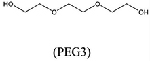
|
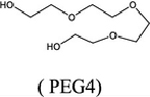
|
|
| 25 °C | 16 ± 3 | 2.3 ± 0.2 | 7.1 ± 3.5 |
| 37 °C | > 100 | 13 ± 1 | 54 ± 19 |
| Ki for SaMTAN (mM) | |||
| 25 °C | 6.2 ± 0.8 | 2.6 ± 0.5 | 2.8 ± 0.3 |
| 37 °C | > 100 | 27 ± 8 | 41 ± 9 |
The PEG2.5-bound and PEG3-bound structures suggested binding of two ethylene glycol units in the 5′-region of the active site; thus, inhibitors containing one ethylene glycol unit (compounds 3–6) are suboptimal on the basis of the fragment interactions and show less improved Ki values. Compound 7 was designed to reflect the specific catalytic function of HpMTAN with its action on aminofutalosine. Compound 7 had a Ki value comparable to that of MTDIA. Compound 2 is the most successful design, using a combination of TSID and FBDD; the MTDIA moiety mimics the transition state in TSID, while a substituent similar in size to two ethylene glycol units as an additional 5′-substituent correspond directly to enzyme-bound fragments identified in crystal structures. Compound 2 has an affinity 14-fold better than that of MTDIA for HpMTAN. Considering that MTDIA is a 1 nM inhibitor, the improvement brings the inhibitor into the picomolar range for its dissociation constant. In the co-crystal structures (Figure 2C), one confirmation of the ethylene glycol mimic of compound 2 is similar to that of ethylene glycol units of PEG3 while the other conformation partially resembles the binding of PEG3, supporting the ability of covalent ethylene glycol mimics to assist in inhibitor binding. Combined with the high solubility for compounds 3–6, this structure-based FBDD has produced inhibitors with improved affinities and drug properties.
Using MTDIA for comparison, the improvements in Ki for compounds 2–5 are >10-fold for binding to SaMTAN (Figure 3). Compound 2 was the most successful design for HpMTAN and also gave the most significant improvement for SaMTAN, a factor of 81-fold. Interestingly, compounds 6 and 7 bind more weakly than MTDIA to SaMTAN, while they bind with an affinity similar to that of MTDIA for HpMTAN. Both compounds 6 and 7 bind to HpMTAN 20-fold tighter than to SaMTAN. Compounds 6 and 7 have an ethylene group connected to C4′ of the ribose, while all other compounds, including substrate MTA, have a methylene group at the same position.
HpMTAN, but not SaMTAN, plays a physiological role in hydrolysis of aminofutalosine, which is similar to that of compound 7 with an ethylene group connected to C4′ of the ribose. As such, the differences in the Ki values of compounds 6 and 7 highlight the distinct physiological roles of MTAN in H. pylori and S. aureus. Collectively, the results indicate that the affinity of compound 2 increases by combining TSID and FBDD and also suggest that this approach may be useful for other enzymes (Figure 4).
Figure 4.

Proposed procedures of drug discovery using TSID and FBDD. The last step (gray) may or may not be needed depending on the enzymes studied.
Supplementary Material
Acknowledgements
The authors thank Mr. Lakshitha Prabath Balapiti Modarage of the University of Arkansas for Medical Science, Mr. Mu Feng of Albert Einstein College of Medicine, and Dr. Zeid Nima and Dr. Bao Vang-Dings of the University of Arkansas at Little Rock for technical support.
Funding
This work was supported in part by a CALS Summer Research fellowship and a 2018 Seed Grant (S.W.), Grant 5R01GM041916 (V.L.S.), and Grant 5R01CA197178 (H.L.).
Abbreviations
- MTDIA,
5′-methylthio-DADMe-Immucillin-A
- MTAN,
methylthioadenosine nucleosidase
- MTA,
5′-methylthioadenosine
- SAH,
S-adenosylhomocysteine
- TSID,
transition state inhibitor design
- FBDD,
fragment-based drug design
- SAR,
structure–activity relationship
- HTS,
high-throughput screening
Footnotes
Notes
The authors declare no competing financial interest.
Contributor Information
Di Zhang, College of Animal Science and Technology, Jilin Agricultural University, Changchun 130118, China; Department of Chemistry, University of Arkansas at Little Rock, Little Rock, Arkansas 72204, United States.
Brandon E. Burdette, Department of Chemistry, University of Arkansas at Little Rock, Little Rock, Arkansas 72204, United States.
Zhengyu Wang, Department of Pharmaceutical Sciences, University of Arkansas for Medical Science, Little Rock, Arkansas 72205, United States.
Kumari Karn, Department of Chemistry, University of Arkansas at Little Rock, Little Rock, Arkansas 72204, United States.
Hong-yu Li, Department of Pharmaceutical Sciences, University of Arkansas for Medical Science, Little Rock, Arkansas 72205, United States.
Vern L. Schramm, Department of Biochemistry, Albert Einstein College of Medicine, New York, New York 10461, United States.
Peter C. Tyler, Ferrier Research Institute, Victoria University of Wellington, Wellington 5040, New Zealand
Gary B. Evans, Ferrier Research Institute, Victoria University of Wellington, Wellington 5040, New Zealand
Shanzhi Wang, Department of Chemistry, University of Arkansas at Little Rock, Little Rock, Arkansas 72204, United States.
References
- 1.Wolfenden R (1969) Transition state analogues for enzyme catalysis. Nature 223, 704–705, DOI: 10.1038/223704a0 [DOI] [PubMed] [Google Scholar]
- 2.Wolfenden R (1976) Transition state analog inhibitors and enzyme catalysis. Annu. Rev. Biophys. Bioeng 5, 271–306, DOI: 10.1146/annurev.bb.05.060176.001415 [DOI] [PubMed] [Google Scholar]
- 3.Schramm VL (2005) Enzymatic transition states: thermodynamics, dynamics and analogue design. Arch. Biochem. Biophys. 433, 13–26, DOI: 10.1016/j.abb.2004.08.035 [DOI] [PubMed] [Google Scholar]
- 4.Schramm VL (2005) Enzymatic transition states and transition state analogues. Curr. Opin. Struct. Biol. 15, 604–613, DOI: 10.1016/j.sbi.2005.10.017 [DOI] [PubMed] [Google Scholar]
- 5.Schramm VL (2011) Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu. Rev. Biochem. 80, 703–732, DOI: 10.1146/annurev-biochem-061809-100742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh V, Evans GB., Lenz DH., Mason JM., Clinch K., Mee S., Painter GF., Tyler PC., Furneaux RH., Lee JE., Howell PL., and Schramm VL. (2005) Femtomolar transition state analogue inhibitors of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli. J. Biol. Chem. 280, 18265–18273, DOI: 10.1074/jbc.M414472200 [DOI] [PubMed] [Google Scholar]
- 7.Schramm VL (2009) Transition States. J. Biol. Chem. 284, 32201–32208, DOI: 10.1074/jbc.X109.060111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho MC, Shi W., Rinaldo-Matthis A., Tyler PC., Evans GB., Clinch K., Almo SC., and Schramm VL. (2010) Four generations of transition-state analogues for human purine nucleoside phosphorylase. Proc. Natl. Acad. Sci. U. S. A. 107, 4805–4812, DOI: 10.1073/pnas.0913439107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke MD, Berger EM., and Schreiber SL. (2003) Generating diverse skeletons of small molecules combinatorially. Science 302, 613–618, DOI: 10.1126/science.1089946 [DOI] [PubMed] [Google Scholar]
- 10.Babaoglu K and Shoichet BK. (2006) Deconstructing fragment-based inhibitor discovery. Nat. Chem. Biol. 2, 720–723, DOI: 10.1038/nchembio831 [DOI] [PubMed] [Google Scholar]
- 11.Murray CW and Rees DC. (2009) The rise of fragment-based drug discovery. Nat. Chem. 1, 187–192, DOI: 10.1038/nchem.217 [DOI] [PubMed] [Google Scholar]
- 12.Erlanson DA, Wells JA., and Braisted AC. (2004) Tethering: fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct. 33, 199–223, DOI: 10.1146/annurev.biophys.33.110502.140409 [DOI] [PubMed] [Google Scholar]
- 13.Erlanson DA (2011) Introduction to fragment-based drug discovery. Top. Curr. Chem. 317, 1–32, DOI: 10.1007/128_2011_180 [DOI] [PubMed] [Google Scholar]
- 14.Miller MB and Bassler BL. (2001) Quorum sensing in bacteria. Annu. Rev. Microbiol. 55, 165–199, DOI: 10.1146/annurev.micro.55.1.165 [DOI] [PubMed] [Google Scholar]
- 15.Parveen N and Cornell, K. A. (2011) Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 79, 7–20, DOI: 10.1111/j.1365-2958.2010.07455.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutierrez JA, Crowder T., Rinaldo-Matthis A., Ho MC., Almo SC., and Schramm VL. (2009) Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 5, 251–257, DOI: 10.1038/nchembio.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang S, Haapalainen AM., Yan F., Du Q., Tyler PC., Evans GB., Rinaldo-Matthis A., Brown RL., Norris GE., Almo SC., and Schramm VL. (2012) A picomolar transition state analogue inhibitor of MTAN as a specific antibiotic for Helicobacter pylori. Biochemistry 51, 6892–6894, DOI: 10.1021/bi3009664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh V, Lee JE., Nunez S., Howell PL., and Schramm VL. (2005) Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry 44, 11647–11659, DOI: 10.1021/bi050863a [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez JA, Luo M., Singh V., Li L., Brown RL., Norris GE., Evans GB., Furneaux RH., Tyler PC., Painter GF., Lenz DH., and Schramm VL. (2007) Picomolar inhibitors as transition-state probes of 5′-methylthioadenosine nucleosidases. ACS Chem. Biol. 2, 725–734, DOI: 10.1021/cb700166z [DOI] [PubMed] [Google Scholar]
- 20.Schramm VL, Wang S., Haapalainen AM., Evans GB., Furneaux RH., Clinch K., Tyler PC., and Gulab SA. (2015) Treatment of Helicobacter pylori infections. US Patent Appl. US20150210701A1. [Google Scholar]
- 21.Wang S, Cameron SA., Clinch K., Evans GB., Wu Z., Schramm VL., and Tyler PC. (2015) New Antibiotic Candidates against Helicobacter pylori. J. Am. Chem. Soc. 137, 14275–14280, DOI: 10.1021/jacs.5b06110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao Y, Li Y., Jiang Q., Zhao L., Xue T., Hu B., and Sun B. (2013) Methylthioadenosine/S-adenosylhomocysteine nucleosidase (Pfs) of Staphylococcus aureus is essential for the virulence independent of LuxS/AI-2 system. Int. J. Med. Microbiol. 303, 190–200, DOI: 10.1016/j.ijmm.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 23.Haapalainen AM, Thomas K., Tyler PC., Evans GB., Almo SC., and Schramm VL. (2013) Salmonella enterica MTAN at 1.36 A resolution: a structure-based design of tailored transition state analogs. Structure 21, 963–974, DOI: 10.1016/j.str.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JE, Singh V., Evans GB., Tyler PC., Furneaux RH., Cornell KA., Riscoe MK., Schramm VL., and Howell PL. (2005) Structural rationale for the affinity of pico- and femtomolar transition state analogues of Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. J. Biol. Chem. 280, 18274–18282, DOI: 10.1074/jbc.M414471200 [DOI] [PubMed] [Google Scholar]
- 25.Ronning DR, Iacopelli NM., and Mishra V. (2010) Enzyme-ligand interactions that drive active site rearrangements in the Helicobacter pylori 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Protein Sci. 19, 2498–2510, DOI: 10.1002/pro.524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra V and Ronning DR. (2012) Crystal structures of the Helicobacter pylori MTAN enzyme reveal specific interactions between S-adenosylhomocysteine and the 5′-alkylthio binding subsite. Biochemistry 51, 9763–9772, DOI: 10.1021/bi301221k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogasawara Y and Dairi, T. (2019) Searching for potent and specific antibiotics against pathogenic Helicobacter and Campylobacter strains. J. Ind. Microbiol. Biotechnol. 46, 409–414, DOI: 10.1007/s10295-018-2108-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.