

Abstract
The antigens that trigger the pathogenic immune response in rheumatoid arthritis (RA) remain unknown. Until recently it was assumed that either viral or microbial antigens, or joint-specific antigens were the target of arthritogenic T and B lymphocytes in RA. Consequently, murine models of arthritis are induced by immunization with either joint-specific antigens such as type II collagen or microbial products such as streptococcal cell wall. In the K/B×N T-cell receptor transgenic mouse model arthritis is caused by a systemic autoimmune response to the ubiquitously expressed glycolytic enzyme glucose-6-phosphate isomerase (G6PI). The autoreactive transgenic T cells recognize G6PI and provide help for the production of arthritogenic IgG antibodies against G6PI. More recently it was shown that G6PI immunization induces severe symmetrical peripheral polyarthritis in genetically unaltered DBA/I mice. In that model CD4+ T cells are necessary not only for the induction but also for the effector phase of arthritis. Here we review the pathomechanisms that lead from systemic autoreactivity to arthritis in these models, consider the relevance of anti-G6PI immune reactivity for RA, and discuss the insights into the pathogenesis of RA and possibly other autoimmune conditions that can be gained from these models.
Keywords: arthritis, CD4+ T lymphocytes, DBA/I mice, FCγ receptors, glucose-6-phosphate-isomerase
Introduction
The aetiology of rheumatoid arthritis (RA), which affects approximately 1% of the population, remains obscure. There is considerable evidence suggesting that RA is an autoimmune disease in which autoreactive lymphocytes trigger macrophages, synoviocytes and other effector cells that mediate synovitis and the destruction of cartilage and bone [1-7].
B and T lymphocytes in rheumatoid arthritis and experimental models
Approximately two-thirds of RA patients produce rheumatoid factors – autoantibodies that are directed against IgG [8]. Because of this strong and diagnostically relevant association, B lymphocytes were long suspected to be the main culprits in RA pathogenesis [1,8]. RA susceptibility and severity are strongly associated with certain HLA-DR haplotypes in Caucasians [9]. The discovery of this linkage led to a more T-cell centred view [3,9-13] because antigen presentation to T lymphocytes is the only known immunological function of MHC class II molecules such as HLA-DR. The difficulty in detecting cellular immune responses against autoantigens in RA patients [14-16], together with the failure of some T-cell directed immunomodulatory treatment strategies [17-22] and impressive successes of therapeutic tumour necrosis factor (TNF)-α blockade in RA, appeared to implicate macrophages as the major effector cells in the clinically overt stages of RA [7,23].
Most recently, however, two different lines of evidence reassert the importance of T cells. First, a large clinical trial [24] showed clear clinical benefits from treating active RA by blocking T-cell costimulation and activation. Second, a spontaneous point mutation in the gene encoding an Src homology 2 (SH2) domain of ZAP-70, a key signal transduction molecule in T cells, causes chronic autoimmune arthritis in mice that resembles human RA in many respects [25]. Moreover, the pathogenic importance of B lymphocytes is again becoming appreciated [26,27], partly because depletion of these cells has been shown to be a successful treatment for RA patients [28]. Taken together, a consensus is beginning to emerge that many different cell types, both from the innate and the adaptive immune systems, are crucial to the pathogenesis of RA [4].
Arthritogenic cartilage antigens?
Although some autoantibodies, such as rheumatoid factors that recognize IgG and antibodies against citrullinated antigens, have diagnostic significance [8,29,30], the autoantigen(s) that are recognized in chronic inflammatory arthritides such as RA are unknown [5,16,22,31,32]. Collagen type II (CII) is the major protein in articular cartilage. It is a candidate autoantigen for RA because antibodies and perhaps T cells against CII occur in patients with RA [5,33-35] and because it is arthritogenic in animals [36]. Collagen-induced arthritis (CIA) has thus become the most intensively studied murine model for human inflammatory arthritides [37].
Autoantibodies are important players in CIA. Adoptive transfer of either polyclonal IgG antibodies purified from the sera of arthritic mice [38-40] or combinations of monoclonal antibodies against CII [41] can induce arthritis even in mouse strains that are not susceptible to actively induced CIA [38]. This form of adoptively transferred arthritis has been called CII antibody-induced arthritis [42]. Antibodies against CII are also found in the blood and joints of some RA patients [33,34,43,44]. In contrast, the role of T lymphocytes in the pathogenesis of CIA is less clear. Collagen-specific proinflammatory T cells can be demonstrated in the blood and synovial fluid of mice with CIA [45]. However, most attempts to induce CIA in mice by T-cell transfer have been unsuccessful [46] and CD4-deficient mice develop CIA with unaltered incidence and severity [47]. Mice lacking α/β T cells are resistant to CIA, whereas γ/δ T cells are neither necessary nor protective. A single report on CIA, albeit at reduced severity as compared with wild-type littermates, in rag-deficient DBA/1 mice [48] has not been corroborated by others to date. Taken together, the question regarding how T cells operate in the pathogenesis of CIA has not yet been answered definitively.
CII-specific T cells have also been difficult to demonstrate in the blood or synovial fluid of RA patients [15,49-51]. Moreover, attempts to treat RA by inducing T-cell tolerance to CII have yielded disappointing results [18,19,22,52]. Taken together, there is little solid evidence that CII or any other single joint-specific antigen such as collagen type XI [53], gp39 [54], cartilage oligomeric matrix protein [55], or cartilage proteoglycan (aggrecan) [56] is a diagnostically or pathogenetically significant autoantigen in all RA patients. Given the complexity and clinical and pathological diversity of RA, it seems more likely that different autoantigens are important in different subsets of RA patients.
Arthritogenic noncartilagenous antigens?
Some noncartilagenous antigens have been used to induce and study arthritis in mice and rats [37]. These are either various microbial compounds with adjuvant effects and/or antigenic properties as in adjuvant arthritis, CpG induced arthritis, or streptococcal cell wall induced arthritis [57-61]; or antigens directly injected into the joints of experimental animals following systemic immunization (antigen-induced arthritis) [62,63]. These arthritides are not the subject of this review because the inciting noncartilagenous antigens are non-self antigens. The importance of noncartilagenous self-antigens to the pathophysiology of arthritis had not been considered until recently.
Autoreactivity against a systemically expressed antigen causes symmetrical peripheral polyarthritis in TCR transgenic K/B×N mice
A T-cell receptor (TCR)-transgenic mouse model of arthritis has challenged the concept that arthritis necessarily results from an autoimmune attack against joint-specific antigens. When C57BL/6 mice expressing a transgene-encoded TCR recognizing amino acids 41–61 of bovine ribonuclease bound to the MHC molecule I-Ak (the 'KRN' receptor) were inadvertently crossed with diabetes-susceptible NOD mice, all of the F1 offspring (the K/B×N mice) spontaneously developed peripheral symmetrical polyarthritis [64]. Arthritis in the K/B×N mice resembles RA in that it symmetrically affects the small peripheral joints. In contrast to RA, the distal interphalangeal joints are regularly affected in K/B×N mice, there are no systemic manifestations, the mice do not produce rheumatoid factors, and the arthritis does not remit [64]. Thus, K/B×N mice spontaneously develop peripheral polyarthritis that resembles human RA in many clinical and pathological respects. This surprising finding induced intense research into the pathophysiology of arthritis in the K/B×N mice.
KRN T cells and I-Aβg7 molecules are necessary for the induction but not the effector phase of arthritis in K/B×N mice
Both the KRN TCR and one copy of the NOD I-Aβg7 MHC molecule are necessary for development of arthritis in K/B×N mice. Neither KRN TCR transgenic C57BL/6 mice nor the F1 from crosses of the transgene-expressing C57BL/6 mice with strains other than the I-Aβg7-bearing NOD mice develop arthritis. KRN T cells proliferate in response to I-Aβg7APC in the absence of experimentally added antigens [65]. Importantly, Th cells are only necessary in the induction phase of arthritis. Once the pathogenesis has passed a certain point, Th cells are dispensable. Treatment with anti-CD4 antibodies is ineffective if it is started less than 5 days before the onset of arthritis. What, then, are the effector mechanisms that induce arthritis in K/B×N mice? Using adoptive transfer experiments and a variety of knockout mice, Mathis and colleagues [65] demonstrated that immunoglobulin is responsible for arthritis induction in K/B×N mice. Transfer of serum (as little as 100 μl) or IgG antibodies from arthritic K/B×N mice induced arthritis in recipient mice of different strains, even in rag-2-/- mice that lack T and B lymphocytes [65]. What do these autoantibodies recognize?
The pathogenic autoantibodies recognize a ubiquitously expressed glycolytic enzyme
Quite surprisingly, the target antigen recognized by both the transgenic T cells and the pathogenic autoantibodies was not joint-specific but the ubiquitously expressed glycolytic enzyme glucose-6-phosphate isomerase (G6PI, or GPI) [66]. G6PI, also known as phosphohexose isomerase, catalyzes the interconversion of fructose-6-phosphate and glucose-6-phosphate [67]. It is an essential glycolytic enzyme, expressed by all cells, and G6PI deficiency is lethal at the two-cell stage [68].
Arthritis can be induced in recipient mice by transfer of polyclonal IgG1 or combinations of at least two different monoclonal IgG1 antibodies against G6PI [69]. Mice that lack the activating FcγRIII are less susceptible to arthritis induced by transfer of K/B×N serum than are normal mice, pointing to FcγRIII+ effector cells in arthritis pathogenesis [70,71]. K/B×N serum transfer arthritis in mice that lack the inhibitory FcγRII has been reported to be similar [70,71] or more severe than in wild-type littermates [72].
Complement, neutrophils and mast cells are all indispensable for arthritis development
In the K/B×N transfer arthritis IgG1 antibodies against G6PI induce several different effector functions of the innate immune system; the alternative pathway of the complement cascade is triggered, resulting in chemotactic activity (but not the membrane attack complex) [70,71]. Neutrophils [73] and mast cells [74] are both required as effector cells to mediate joint destruction.
IL-1 is important but neither TNF-α or IL-6 is needed for arthritis development in K/B×N mice
In contrast to RA in humans and most other murine models of arthritis, neither TNF-α or IL-6 is needed for arthritis development in K/B×N mice [75,76]. TNF-α is an important mediator of joint destruction in RA and several murine models of it [7]. Therefore, the K/B×N model yielded another surprising finding when it turned out that TNF-α blockade had no effect on the development and progression of arthritis [75]. Moreover, K/B×N serum induced arthritis in mice that were deficient for both TNF receptor-1 and TNF receptor-2, or lymphotoxin-α with the same incidence and severity as in normal littermates [76]. In that same study a somewhat reduced incidence of arthritis was noted upon serum transfer in TNF-α-deficient mice obtained from one particular colony as compared with wild-type controls. However, that difference was not found with TNF-α-deficient mice obtained from a different colony [76]. Similarly, and again in contrast to previous findings in other murine models of arthritis [77], IL-6 deficiency had no influence on the development of K/B×N serum transfer arthritis [76].
Taken together, the above findings indicate that recognition of a ubiquitously expressed self-antigen by T cells that bear a transgenically encoded receptor and escape negative selection in the thymus [78] induces an arthritogenic autoantibody response, which then triggers innate immune effector mechanisms to induce arthritis.
Autoreactivity against G6PI in the pathogenesis of RA?
This perplexing and informative model raises the question of whether autoreactivity against G6PI is relevant to the pathogenesis of human RA or other chronic inflammatory arthritides. One initial report [79] indicated that IgG antibodies against G6PI were detectable at low dilution (1:50) in the serum of 64% of RA patients but not in control individuals.
However, a number of other investigators did not find increased levels of α-G6PI antibodies in the serum of patients with RA [80-83], collagen tissue disease [81-83], or other chronic arthritides [81,83,84]. Moreover, the commercial G6PI preparation used in the initial study was found to be contaminated with other proteins, and RA sera contained antibodies against some of these other proteins [81]. Taken together, these findings indicate that antibodies against G6PI are not diagnostic markers for RA. An interesting further twist in the story was added by a recent report. van Gaalen and coworkers [85] found that whereas only one of 55 RA patients who did not have systemic manifestations of the disease produced antibodies against G6PI, seven of 22 patients with systemic manifestations (nodules or vasculitis) and 12 of 13 patients with Felty's syndrome had detectable α-G6PI antibodies in their sera [85]. Thus, the possibility remains that antibodies against G6PI occur frequently in Felty's syndrome. However, it is currently not yet clear whether the increased seropositivity is specific for antibodies against G6PI or a sign of generally dysregulated autoantibody production in patients with Felty's syndrome, and the data must be independently confirmed. Another group [86] compared α-G6PI antibody titres in serum and synovial fluid and found increased concentrations of α-G6PI antibodies in the synovial fluid of RA patients. Taken together, the currently available data argue against a pathogenic role for anti-G6PI immune responses in RA.
Are the immunological events that induce arthritis in the K/B×N model relevant to RA?
Even if G6PI is not a relevant autoantigen to RA pathogenesis, the question remains of whether the immunological events that lead to the development of arthritis in the K/B×N model are involved in human RA. Both experimental and clinical data strongly support the possibility that autoreactivity against systemically expressed autoantigens may result in organ-specific autoimmune disease.
Systemic autoreactivity causes peripheral neuritis in TCR transgenic mice
The K/B×N model is not the only transgenic model in which organ-specific autoimmunity develops as a consequence of systemic self-reactivity of T cells. Oono and coworkers [87] produced transgenic mice that express Eα52–68 covalently bound to the I-Ab molecule as their only MHC peptide complex. These mice spontaneously develop a CD4+ Th cell dependent peripheral nervous system-specific autoimmune disease. Neuritis in these TCR transgenic mice shares many of the histopathological features found in experimental autoimmune neuritis, including demyelination and axon degeneration [87]. Serum from these transgenic mice did not stain peripheral nerves and could not transfer the disease to other animals [87].
Autoantibodies against systemically expressed autoantigens are diagnostically important in organ-specific autoimmune diseases
There are several clinical examples of autoantibodies against systemically expressed autoantigens that are highly sensitive and specific diagnostic markers for certain organ-specific autoimmune diseases. These include the anti-Jo-1 autoantibodies that bind to and inhibit activity of histidyl-tRNA synthetase, and the autoantibodies that recognize proteasomes that are found in different but overlapping subsets of myositis patients [88-90]; the antimitochondrial antibodies (AMA-2) that recognize the E2 subunit of mitochondrial pyruvate dehydrogenise [91], which are found in patients with primary biliary cirrhosis; the autoantibodies against proteinase 3 (c-ANCAs) in Wegener's granulomatosis [92]; and, of course, the rheumatoid factors that recognize IgG antibodies [8]. All of these autoantibodies are directed against systemically expressed autoantigens, yet they are highly specific and sensitive markers for the respective diseases. However, their pathogenic significance remains unknown, partly because of the lack of suitable animal models.
Systemic autoreactivity causes severe peripheral symmetrical polyarthritis in genetically unaltered mice
To bridge the gap between the potentially very informative yet rather artificial transgenic mouse models and the situation in patients, we considered whether a systemic immune response against G6PI could induce joint-specific pathology in genetically unaltered mice. Of several inbred strains tested, DBA/1 mice, the same inbred strain of mice that is also susceptible to CIA, develop severe symmetrical peripheral polyarthritis arthritis following one single immunization with recombinant human or murine G6PI in adjuvant [93] (Fig. 1). The incidence of arthritis upon immunization is in excess of 90%, and the time course is uniform and highly predictable. Clinical signs of arthritis are first visible 9 days after immunization; the arthritis then rapidly progresses, reaches its maximum at about day 14 after immunization, and then slowly resolves. The pattern of joints affected is similar to but not identical to that observed in RA; wrists, metacarpal joints, proximal and distal interphalangeal joints are affected at the front limbs, and the tarsal, ankle and knee joints at the rear limbs. Neither the spine, nor the hip, elbow, or shoulder were affected in any of the animals analyzed [93]. Histologically, there are no further signs of inflammation past day 21. Instead, reorganization and fibrosis become visible. Importantly, there were no pathological findings in any other organs apart from the joints. The sudden onset, and high incidence and severity of arthritis distinguishes this model from CIA. The spontaneous remission of G6PI-induced arthritis in genetically unaltered DBA/1 mice is one important clinical difference from the transgenic K/B×N model. This allows for the study of the immunological mechanisms that modulate the autoimmune response and induce remission of the disease.
Figure 1.
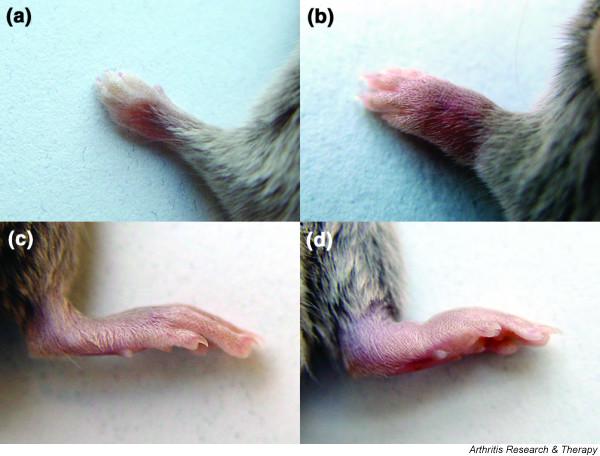
(a, b) Front and (c, d) hind limbs from DBA/1 mice that had been immunized with glucose-6-phosphate isomerase (G6PI) subcutaneously (panels b and d) or administered phosphate-buffered saline subcutaneously (panels a and c) 14 days earlier. From [93], © 2004. The American Association of Immunologists, Inc. Reprinted with permission.
Continuous requirement for Th cells in the pathogenesis of G6PI-induced arthritis
Th cells are needed throughout the effector cells in G6PI-induced arthritis in normal mice. Depletion of CD4+ cells immediately after immunization prevents arthritis. More importantly, depletion of CD4+ cell on days 11 and 14 (i.e. when clinical symptoms are at their maximum) induces rapid remission of arthritis both clinically and histologically [93]. Thus, in G6PI-induced arthritis in normal mice, CD4+ cells are not only important in the induction but also throughout the effector phase of the disease. This is in contrast to the K/B×N model, in which Th cells are only necessary to provide help to B cells that produce the arthritogenic antibodies against G6PI. Once these antibodies are produced, T cells are no longer necessary for arthritis development in the K/B×N model, and transfer of serum or antibodies from arthritic K/B×N mice [66,69] can transfer arthritis to naïve recipients of almost any mouse strain. Similarly, CIA can be transferred by antibodies or serum [38-41] and it can be induced even in the absence of CD4+ cells [47,48].
IgG antibodies are necessary but not sufficient for G6PI-induced arthritis in normal mice
DBA/1 mice that lack the FcγR common γ chain and thus cannot signal through the activating FcγRI and FcγRIII are protected from G6PI-induced arthritis. In addition, mice that lack the inhibitory FcγRIIB develop severe and prolonged G6PI-induced arthritis (Fig. 2) [93]. Therefore, IgG antibodies and FcγR+effector cells are necessary for the development of G6PI-induced arthritis. Nevertheless, arthritis cannot be induced in naïve recipients by transfer of serum or antibodies from arthritic DBA/1 mice [93]. It is currently unclear why G6PI-induced arthritis cannot be transferred with serum from arthritic animals; it is possible that the antibodies present in the serum some 14 days after immunization lack the necessary affinity for G6PI. Thus, unlike the CIA and K/B×N models, both CD4+ T cells and antibodies are necessary for the development of G6PI-induced arthritis, and neither transfer of T cells nor transfer of antibodies alone can induce arthritis in recipient mice. TNF-α is indispensable for the development of G6PI-induced arthritis in normal mice; treatment of mice with the soluble p75 TNF receptor (etanercept) completely prevents the development of arthritis [93].
Figure 2.
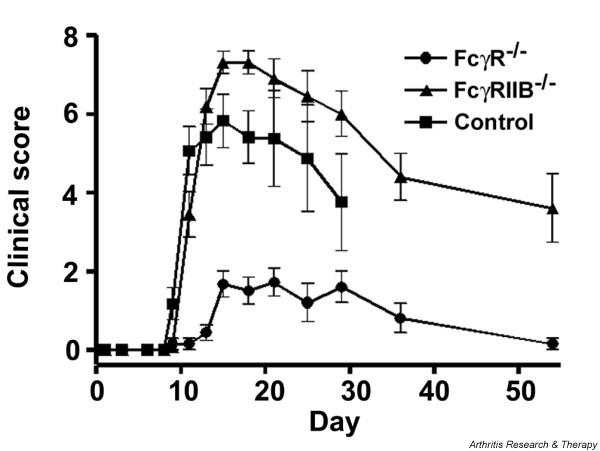
Arthritis scores of (■) DBA/1 wild-type, (●) DBA/1 FcγR common γ-chain deficient, and (▲) DBA/1 FcγRIIB deficient mice. Data are presented as mean clinical scores ± standard error of the mean only for those mice that developed arthritis. Arthritis incidence was 10/11 in DBA/1 wild-type, 8/24 in DBA/1 FcγR common γ-chain deficient, and 16/16 in DBA/1 FcγRIIB deficient mice. From [93], © 2004. The American Association of Immunologists, Inc. Reprinted with permission.
Taken together, the above findings indicate that G6PI-induced arthritis in genetically unaltered mice provides a reliable and robust model in which the induction, effector phase and modulation of organ-specific disease induced by systemic autoimmunity can be dissected and therapeutically targeted. Thus, G6PI-induced arthritis narrows the gap between the TCR transgenic K/B×N model and the situation in patients. Some of the major clinical and immunological similarities and differences between CIA, the K/B×N model and G6PI-induced arthritis are summarized in Table 1. Several important questions remain unanswered.
Table 1.
Clinical and pathological characteristics of different arthritis models in mice
| Arthritis model | |||
| Characteristics | Collagen-induced arthritis | K/B×N arthritis | G6PI-induced arthritis |
| Arthritogenic antigen | Cartilage specific (CII) | Systemic (G6PI) | Systemic (G6PI) |
| Susceptible strain | DBA/1 | KRN TCR transgenic × NOD F1 mice | DBA/1 |
| Arthritis induction | CII immunization and boost | Spontaneous | G6PI immunization |
| Pathogenic cells and effector mechanisms | |||
| T cells | ? | Induction phase | Induction and effector phases |
| Antibodies | Necessary and sufficient | Necessary and sufficient | Necessary |
| FcγRI or FcγRIII necessary | + | + | + |
| FcγRII | + | + or - in different studies | + |
| C5 | + | + | + |
| Mast cells | Uncertain | + | Unknown |
| Neutrophils | + | + | Unknown |
| Macrophages | Uncertain | Unknown | Unknown |
| TNF-α | + | (±) | + |
| IL-1 | + | + | Unknown |
| IL-6 | + | + | Unknown |
| Spontaneous remission | + | - | + |
CII, type II collagen; G6PI, glucose-6-phosphate isomerase; TNF, tumor necrosis factor.
Why the joint?
It is currently unclear why systemic autoreactivity against the ubiquitously expressed glycolytic enzyme G6PI specifically induces arthritis, with no other symptoms of organ-specific or systemic autoimmune disease. Interestingly, although GPI is ubiquitously expressed in the body, the immune response against GPI appears to initiate in the draining lymph nodes of peripheral joints in K/B×N mice [94]. The reasons for this early localized immune response are currently unclear but these findings suggest that something unique to the joints initiates a local immune response to a systemic autoantigen. This contention is further supported by positron emission tomography studies [95,96] that demonstrate rapid localization of adoptively transferred antibodies against G6PI to the peripheral joints of the recipient mice.
It is known that cationic antigens such as G6PI bind well to cartilage [62,97]. However, systemically expressed antigens such as G6PI are not only presented in the joint, and not every cationic antigen induces arthritis upon immunization. Immune complexes are a critical component in the pathogenesis of K/B×N serum transfer arthritis [96,98] and for G6PI-induced arthritis in normal mice (unpublished observations). G6PI deposits, together with IgG and C3, are detectable in the joints of arthritic mice in the K/B×N serum transfer model [98]. G6PI–IgG immune complexes are also visible in the glomeruli of arthritic K/B×N mice. In contrast to the joints, however, the immune complexes in the kidney were not colocalized with C3 [98]. It is has therefore been suggested that G6PI–immunoglobulin immune complex trigger the complement cascade exclusively in the joints. This would be explained by the absence of membrane-bound C3 inhibitors from chondrocytes [98]. The question remains as to why only a particular pattern of joints is arthritic in the K/B×N model and in G6PI-induced arthritis. Furthermore, immune complex diseases do not necessarily induce erosive arthritis. Systemic lupus erythematosus provides a classical example; there, the immune complex induces glomerulonephritis but not erosive arthritis.
Why G6PI?
Currently, there is no mechanistic explanation for the association between autoantibodies against certain systemically expressed antigens and particular autoimmune diseases [8,88-92]. Similarly, it is not clear why G6PI becomes the target of an arthritogenic autoimmune response in K/B×N and DBA/1 mice. In addition to its function as a glycolytic enzyme, G6PI can be secreted and serves a variety of other physiological functions [99]. G6PI is identical to neuroleukin, a neurotrophic factor for spinal and sensory neurones [100,101], which is associated with terminal sprouting; autocrine motility factor [102], which stimulates motility via a receptor-mediated pathway [103]; and maturation factor, which mediates the differentiation of human myeloid leukemic HL-60 calls to terminal monocytic cells [104].
A receptor for G6PI, namely gp78, has been identified. Gp78 is a transmembrane protein, a RING finger-dependent ubiquitin protein ligase (E3) of the endoplasmatic reticulum [105,106]. It remains to be investigated whether one or more of these physiological functions of G6PI contribute to its immunogenicity and arthritogenicity.
Conclusion
The search for arthritogenic autoantigens has long focused on joint-specific antigens. The TCR transgenic K/B×N model of arthritis and more recently G6PI-induced arthritis in genetically unaltered mice have demonstrated that a noncartilagenous systemically expressed self-antigen can be the target of an arthritogenic immune response. Although the two models exhibit clinical and pathophysiological differences, a complex interplay between cells and effector mechanisms of both the adaptive and innate immune system is necessary in each model. Whereas autoreactivity against G6PI does not seem to play a role in RA, there are well known clinical examples of autoantibodies that are directed against autoantigens that are systemically expressed but pathognomonic for organ-specific diseases, and the possibility that arthritis may be induced by systemic autoreactivity remains interesting and plausible. G6PI-induced arthritis provides a model in which both the induction and modulation of arthritis induced by autoreactivity against noncartilagenous antigens can be studied.
Competing interests
The author(s) declare that they have no competing interests.
Abbreviations
CIA = collagen-induced arthritis; CII = collagen type II; G6PI = glucose-6-phosphate isomerase; IL = interleukin; MHC = major histocompatibility complex; RA = rheumatoid arthritis; TCR = T-cell receptor; Th = T helper; TNF = tumour necrosis factor.
Acknowledgments
Acknowledgements
The authors' work on G6PI-induced arthritis has been supported by the DFG (SFB 421 TPC2), the BMBF (Kompetenznetz Rheuma) and the IZKF Jena.
References
- Zvaifler NJ. The immunopathology of joint inflammation in rheumatoid arthritis. Adv Immunol. 1973;16:265–336. doi: 10.1016/s0065-2776(08)60299-0. [DOI] [PubMed] [Google Scholar]
- Van Boxel JA, Paget SA. Predominantly T cell infiltrate in rheumatoid synovial membranes. N Engl J Med. 1975;293:517–520. doi: 10.1056/NEJM197509112931101. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis. Arthritis Rheum. 1990;33:768–773. doi: 10.1002/art.1780330602. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Steiner G. Rheumatoid arthritis is more than cytokines: autoimmunity and rheumatoid arthritis. Arthritis Rheum. 2001;44:2218–2220. doi: 10.1002/1529-0131(200110)44:10<2218::AID-ART382>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Steiner G, Smolen J. Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res. 2002;(Suppl 2):S1–S5. doi: 10.1186/ar551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis?: II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 2002;46:298–308. doi: 10.1002/art.502. [DOI] [PubMed] [Google Scholar]
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- Dorner T, Egerer K, Feist E, Burmester GR. Rheumatoid factor revisited. Curr Opin Rheumatol. 2004;16:246–253. doi: 10.1097/00002281-200405000-00013. [DOI] [PubMed] [Google Scholar]
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- Winchester R. The molecular basis of susceptibility to rheumatoid arthritis. Adv Immunol. 1994;56:389–466. doi: 10.1016/s0065-2776(08)60456-3. [DOI] [PubMed] [Google Scholar]
- Hammer J, Gallazi F, Bono E, Karr RW, Guenot J, Valsasnini P, Nagy ZA, Sinigaglia F. Peptide binding specificity of HLA-DR4 molecules: correlation with rheumatoid arthritis association. J Exp Med. 1995;181:1847–1855. doi: 10.1084/jem.181.5.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner JH, Nepom GT. Genetics of rheumatoid arthritis: is there a scientific explanation for the human leukocyte antigen association? Curr Opin Rheumatol. 2002;14:254–259. doi: 10.1097/00002281-200205000-00011. [DOI] [PubMed] [Google Scholar]
- Nepom GT. Major histocompatibility complex-directed susceptibility to rheumatoid arthritis. Adv Immunol. 1998;68:315–332. doi: 10.1016/s0065-2776(08)60563-5. [DOI] [PubMed] [Google Scholar]
- Berg L, Ronnelid J, Sanjeevi CB, Lampa J, Klareskog L. Interferon-gamma production in response to in vitro stimulation with collagen type II in rheumatoid arthritis is associated with HLA-DRB1(*)0401 and HLA-DQ8. Arthritis Res. 2000;2:75–84. doi: 10.1186/ar71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzin BL, Falta MT, Crawford F, Rosloniec EF, Bill J, Marrack P, Kappler J. Use of soluble peptide-DR4 tetramers to detect synovial T cells specific for cartilage antigens in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:291–296. doi: 10.1073/pnas.97.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SR, Falta MT, Bill J, Kotzin BL. Antigen-specific T cells in rheumatoid arthritis. Curr Rheumatol Rep. 2003;5:255–263. doi: 10.1007/s11926-003-0003-y. [DOI] [PubMed] [Google Scholar]
- Horneff G, Burmester GR, Emmrich F, Kalden JR. Treatment of rheumatoid arthritis with an anti-CD4 monoclonal antibody. Arthritis Rheum. 1991;34:129–140. doi: 10.1002/art.1780340202. [DOI] [PubMed] [Google Scholar]
- Trentham DE, Dynesius-Trentham RA, Orav EJ, Combitchi D, Lorenzo C, Sewell KL, Hafler DA, Weiner HL. Effects of oral administration of type II collagen on rheumatoid arthritis. Science. 1993;261:1727–1730. doi: 10.1126/science.8378772. [DOI] [PubMed] [Google Scholar]
- Sieper J, Kary S, Sorensen H, Alten R, Eggens U, Huge W, Hiepe F, Kuhne A, Listing J, Ulbrich N, et al. Oral type II collagen treatment in early rheumatoid arthritis. A double-blind, placebo-controlled, randomized trial. Arthritis Rheum. 1996;39:41–51. doi: 10.1002/art.1780390106. [DOI] [PubMed] [Google Scholar]
- Kalden JR, Sieper J. Oral collagen in the treatment of rheumatoid arthritis. Arthritis Rheum. 1998;41:191–194. doi: 10.1002/1529-0131(199802)41:2<191::AID-ART2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Kalden JR, Breedveld FC, Burkhardt H, Burmester GR. Immunological treatment of autoimmune diseases. Adv Immunol. 1998;68:333–361. doi: 10.1016/s0065-2776(08)60564-7. [DOI] [PubMed] [Google Scholar]
- Kamradt T, Mitchison NA. Tolerance and autoimmunity. N Engl J Med. 2001;344:655–664. doi: 10.1056/NEJM200103013440907. [DOI] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/S0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349:1907–1915. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]
- Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- Arce S, Cassese G, Hauser A, Dorner T, Odendahl M, Manz R, Radbruch A, Hiepe F. The role of long-lived plasma cells in autoimmunity. Immunobiology. 2002;206:558–562. doi: 10.1078/0171-2985-00204. [DOI] [PubMed] [Google Scholar]
- Dorner T, Burmester GR. The role of B cells in rheumatoid arthritis: mechanisms and therapeutic targets. Curr Opin Rheumatol. 2003;15:246–252. doi: 10.1097/00002281-200305000-00011. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- van Boekel MA, Vossenaar ER, van den Hoogen FH, van Venrooij WJ. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 2002;4:87–93. doi: 10.1186/ar395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, Toes RE, Huizinga TW. Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum. 2004;50:709–715. doi: 10.1002/art.20044. [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R, Lee J, McCoy A, Hoxworth J, Yarboro C, Smolen JS, Steiner G, Rosen A, Zhang C, Menard HA, et al. Rheumatoid arthritis associated autoantibodies in patients with synovitis of recent onset. Arthritis Res. 2000;2:236–243. doi: 10.1186/ar93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigall VM, Panayi GS. Autoantigens and immune pathways in rheumatoid arthritis. Crit Rev Immunol. 2002;22:281–293. [PubMed] [Google Scholar]
- Wooley PH, Luthra HS, O'Duffy JD, Bunch TW, Moore SB, Stuart JM. Anti-type II collagen antibodies in rheumatoid arthritis. The influence of HLA phenotype. Tissue Antigens. 1984;23:263–269. doi: 10.1111/j.1399-0039.1984.tb00043.x. [DOI] [PubMed] [Google Scholar]
- Ronnelid J, Lysholm J, Engstrom-Laurent A, Klareskog L, Heyman B. Local anti-type II collagen antibody production in rheumatoid arthritis synovial fluid. Evidence for an HLA-DR4-restricted IgG response. Arthritis Rheum. 1994;37:1023–1029. doi: 10.1002/art.1780370707. [DOI] [PubMed] [Google Scholar]
- Backlund J, Carlsen S, Hoger T, Holm B, Fugger L, Kihlberg J, Burkhardt H, Holmdahl R. Predominant selection of T cells specific for the glycosylated collagen type II epitope (263–270) in humanized transgenic mice and in rheumatoid arthritis. Proc Natl Acad Sci USA. 2002;99:9960–9965. doi: 10.1073/pnas.132254199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Andersson M, Goldschmidt TJ, Gustafsson K, Jansson L, Mo JA. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- Jirholt J, Lindqvist AB, Holmdahl R. The genetics of rheumatoid arthritis and the need for animal models to find and understand the underlying genes. Arthritis Res. 2001;3:87–97. doi: 10.1186/ar145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J Exp Med. 1983;158:378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson WC, Brown PS, Pitcock JA, Townes AS. Passive transfer studies with type II collagen antibody in B10.D2/old and new line and C57Bl/6 normal and beige (Chediak–Higashi) strains: evidence of important roles for C5 and multiple inflammatory cell types in the development of erosive arthritis. Arthritis Rheum. 1987;30:460–465. doi: 10.1002/art.1780300418. [DOI] [PubMed] [Google Scholar]
- Holmdahl R, Jansson L, Larsson A, Jonsson R. Arthritis in DBA/1 mice induced with passively transferred collagen type II immune serum. Immunohistopathology and serum levels of anti-type II collagen autoantibodies. Scand J Immunol. 1990;31:147–157. doi: 10.1111/j.1365-3083.1990.tb02754.x. [DOI] [PubMed] [Google Scholar]
- Terato K, Hasty KA, Reife RA, Cremer MA, Kang AH, Stuart JM. Induction of arthritis with monoclonal antibodies to collagen. J Immunol. 1992;148:2103–2108. [PubMed] [Google Scholar]
- Nandakumar KS, Svensson L, Holmdahl R. Collagen type II-specific monoclonal antibody-induced arthritis in mice: description of the disease and the influence of age, sex, and genes. Am J Pathol. 2003;163:1827–1837. doi: 10.1016/S0002-9440(10)63542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski A, Klareskog L, Carlsten H, Herberts P, Koopman WJ. Secretion of antibodies to types I and II collagen by synovial tissue cells in patients with rheumatoid arthritis. Arthritis Rheum. 1989;32:1087–1092. doi: 10.1002/anr.1780320906. [DOI] [PubMed] [Google Scholar]
- Cook AD, Rowley MJ, Mackay IR, Gough A, Emery P. Antibodies to type II collagen in early rheumatoid arthritis. Correlation with disease progression. Arthritis Rheum. 1996;39:1720–1727. doi: 10.1002/art.1780391015. [DOI] [PubMed] [Google Scholar]
- Svendsen P, Andersen CB, Willcox N, Coyle AJ, Holmdahl R, Kamradt T, Fugger L. Tracking of proinflammatory collagen-specific T cells in early and late collagen-induced arthritis in humanized mice. J Immunol. 2004;173:7037–7045. doi: 10.4049/jimmunol.173.11.7037. [DOI] [PubMed] [Google Scholar]
- Taurog JD, Kerwar SS, McReynolds RA, Sandberg GP, Leary SL, Mahowald ML. Synergy between adjuvant arthritis and collagen-induced arthritis in rats. J Exp Med. 1985;162:962–978. doi: 10.1084/jem.162.3.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada Y, Ho A, Koh DR, Mak TW. Collagen-induced arthritis in CD4- or CD8-deficient mice: CD8+ T cells play a role in initiation and regulate recovery phase of collagen-induced arthritis. J Immunol. 1996;156:4520–4526. [PubMed] [Google Scholar]
- Plows D, Kontogeorgos G, Kollias G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J Immunol. 1999;162:1018–1023. [PubMed] [Google Scholar]
- Londei M, Savill CM, Verhoef A, Brennan F, Leech ZA, Duance V, Maini RN, Feldmann M. Persistence of collagen type II-specific T-cell clones in the synovial membrane of a patient with rheumatoid arthritis. Proc Natl Acad Sci USA. 1989;86:636–640. doi: 10.1073/pnas.86.2.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden N, Reynolds I, Morgan K, Holt L. T cell responses to human type II collagen in patients with rheumatoid arthritis and healthy controls. Arthritis Rheum. 1997;40:1210–1218. doi: 10.1002/1529-0131(199707)40:7<1210::AID-ART4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Kim HY, Kim WU, Cho ML, Lee SK, Youn J, Kim SI, Yoo WH, Park JH, Min JK, Lee SH, et al. Enhanced T cell proliferative response to type II collagen and synthetic peptide CII (255–274) in patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:2085–2093. doi: 10.1002/1529-0131(199910)42:10<2085::AID-ANR8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- McKown KM, Carbone LD, Kaplan SB, Aelion JA, Lohr KM, Cremer MA, Bustillo J, Gonzalez M, Kaeley G, Steere EL, et al. Lack of efficacy of oral bovine type II collagen added to existing therapy in rheumatoid arthritis. Arthritis Rheum. 1999;42:1204–1208. doi: 10.1002/1529-0131(199906)42:6<1204::AID-ANR17>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Cremer MA, Ye XJ, Terato K, Owens SW, Seyer JM, Kang AH. Type XI collagen-induced arthritis in the Lewis rat. Characterization of cellular and humoral immune responses to native types XI, V, and II collagen and constituent alpha-chains. J Immunol. 1994;153:824–832. [PubMed] [Google Scholar]
- Cope AP, Patel SD, Hall F, Congia M, Hubers HA, Verheijden GF, Boots AM, Menon R, Trucco M, Rijnders AW, et al. T cell responses to a human cartilage autoantigen in the context of rheumatoid arthritis-associated and nonassociated HLA-DR4 alleles. Arthritis Rheum. 1999;42:1497–1507. doi: 10.1002/1529-0131(199907)42:7<1497::AID-ANR25>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Carlsen S, Hansson AS, Olsson H, Heinegard D, Holmdahl R. Cartilage oligomeric matrix protein (COMP)-induced arthritis in rats. Clin Exp Immunol. 1998;114:477–484. doi: 10.1046/j.1365-2249.1998.00739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glant TT, Finnegan A, Mikecz K. Proteoglycan-induced arthritis: immune regulation, cellular mechanisms, and genetics. Crit Rev Immunol. 2003;23:199–250. doi: 10.1615/CritRevImmunol.v23.i3.20. [DOI] [PubMed] [Google Scholar]
- Pearson CM. Development of arthritis, periarthritis, and periostitis in rats given adjuvants. Proc Soc Exp Biol Med. 1956;91:95–101. doi: 10.3181/00379727-91-22179. [DOI] [PubMed] [Google Scholar]
- van Eden W, Thole JE, van der Zee R, Noordzij A, van Embden JD, Hensen EJ, Cohen IR. Cloning of the mycobacterial epitope recognized by T lymphocytes in adjuvant arthritis. Nature. 1988;331:171–173. doi: 10.1038/331171a0. [DOI] [PubMed] [Google Scholar]
- Holmdahl R, Lorentzen JC, Lu S, Olofsson P, Wester L, Holmberg J, Pettersson U. Arthritis induced in rats with nonimmunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev. 2001;184:184–202. doi: 10.1034/j.1600-065x.2001.1840117.x. [DOI] [PubMed] [Google Scholar]
- Deng GM, Nilsson IM, Verdrengh M, Collins LV, Tarkowski A. Intra-articularly localized bacterial DNA containing CpG motifs induces arthritis. Nat Med. 1999;5:702–705. doi: 10.1038/9554. [DOI] [PubMed] [Google Scholar]
- Kimpel D, Dayton T, Kannan K, Wolf RE. Streptococcal cell wall arthritis: kinetics of immune cell activation in inflammatory arthritis. Clin Immunol. 2002;105:351–362. doi: 10.1006/clim.2002.5305. [DOI] [PubMed] [Google Scholar]
- van den Berg WB, van de Putte LB, Zwarts WA, Joosten LA. Electrical charge of the antigen determines intraarticular antigen handling and chronicity of arthritis in mice. J Clin Invest. 1984;74:1850–1859. doi: 10.1172/JCI111604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Banchet GS, Petrow PK, Brauer R, Schaible HG. Monoarticular antigen-induced arthritis leads to pronounced bilateral upregulation of the expression of neurokinin 1 and bradykinin 2 receptors in dorsal root ganglion neurons of rats. Arthritis Res. 2000;2:424–427. doi: 10.1186/ar121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/S0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- Gracy RW, Tilley BE. Phosphoglucose isomerase of human erythrocytes and cardiac tissue. Methods Enzymol. 1975;41:392–400. doi: 10.1016/s0076-6879(75)41086-2. [DOI] [PubMed] [Google Scholar]
- West JD, Flockhart JH, Peters J, Ball ST. Death of mouse embryos that lack a functional gene for glucose phosphate isomerase. Genet Res. 1990;56:223–236. doi: 10.1017/s0016672300035321. [DOI] [PubMed] [Google Scholar]
- Maccioni M, Zeder-Lutz G, Huang H, Ebel C, Gerber P, Hergueux J, Marchal P, Duchatelle V, Degott C, van Regenmortel M, et al. Arthritogenic monoclonal antibodies from K/B×N mice. J Exp Med. 2002;195:1071–1077. doi: 10.1084/jem.20011941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/S1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- Ji H, Gauguier D, Ohmura K, Gonzalez A, Duchatelle V, Danoy P, Garchon HJ, Degott C, Lathrop M, Benoist C, et al. Genetic influences on the end-stage effector phase of arthritis. J Exp Med. 2001;194:321–330. doi: 10.1084/jem.194.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corr M, Crain B. The role of FcgammaR signaling in the K/B × N serum transfer model of arthritis. J Immunol. 2002;169:6604–6609. doi: 10.4049/jimmunol.169.11.6604. [DOI] [PubMed] [Google Scholar]
- Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- Kyburz D, Carson D, Corr M. The role of CD40 ligand and tumor necrosis factor alpha signaling in the transgenic K/B×N mouse model of rheumatoid arthritis. Arthritis Rheum. 2000;43:2571–2577. doi: 10.1002/1529-0131(200011)43:11<2571::AID-ANR26>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih FF, Mandik-Nayak L, Wipke BT, Allen PM. Massive thymic deletion results in systemic autoimmunity through elimination of CD4+CD25+ T regulatory cells. J Exp Med. 2004;199:323–335. doi: 10.1084/jem.20031137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M, Burton DR, Ditzel HJ. Autoantibodies to GPI in rheumatoid arthritis: linkage between an animal model and human disease. Nat Immunol. 2001;2:746–753. doi: 10.1038/90696. [DOI] [PubMed] [Google Scholar]
- Kassahn D, Kolb C, Solomon S, Bochtler P, Illges H. Few human autoimmune sera detect GPI [letter] Nat Immunol. 2002;3:411–412. doi: 10.1038/ni0502-411b. [DOI] [PubMed] [Google Scholar]
- Schubert D, Schmidt M, Zaiss D, Jungblut PR, Kamradt T. Autoantibodies against GPI and creatine kinase in rheumatoid arthritis [letter] Nat Immunol. 2002;3:411. doi: 10.1038/ni0502-411a. [DOI] [PubMed] [Google Scholar]
- Herve CA, Wait R, Venables PJ. Glucose-6-phosphate isomerase is not a specific autoantigen in rheumatoid arthritis. Rheumatology (Oxford) 2003;8:986–988. doi: 10.1093/rheumatology/keg271. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Lee DM, Goldbach-Mansky R, Sumida T, Hitchon CA, Schur PH, Anderson RJ, Coblyn JS, Weinblatt ME, Brenner M, et al. Low prevalence of antibodies to glucose-6-phosphate isomerase in patients with rheumatoid arthritis and a spectrum of other chronic autoimmune disorders. Arthritis Rheum. 2003;48:944–954. doi: 10.1002/art.10898. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Schubert D, Sengler C, Kamradt T. Autoantibodies against glucose-6-phosphate-isomerase are not a diagnostic marker for juvenile idiopathic arthritis [letter] Ann Rheum Dis. 2004;63:463. doi: 10.1136/ard.2003.014225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gaalen FA, Toes REM, Ditzel HJ, Schaller M, Breedveld FC, Verweij CL, Huizinga TWJ. Association of autoantibodies to glucose-6-phosphate isomerase with extraarticular complications in rheumatoid arthritis. Arthritis Rheum. 2004;50:395–399. doi: 10.1002/art.20028. [DOI] [PubMed] [Google Scholar]
- Cha H-S, Kim TJ, Kim J-Y, Lee M-H, Jeon CH, Kim J, Bae E-K, Ahn K-S, Koh E-M. Autoantibodies to glucose-6-phosphate isomerase are elevated in the synovial fluid of rheumatoid arthritis patients. Scand J Rheumatol. 2004;33:179–184. doi: 10.1080/03009740310004757. [DOI] [PubMed] [Google Scholar]
- Oono T, Fukui Y, Masuko S, Hashimoto O, Ueno T, Sanui T, Inayoshi A, Noda M, Sata M, Sasazuki T. Organ-specific autoimmunity in mice whose T cell repertoire is shaped by a single antigenic peptide. J Clin Invest. 2001;108:1589–1596. doi: 10.1172/JCI200113256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MB, Bernstein RM. Myositis autoantibody inhibits histidyl-tRNA synthetase: a model for autoimmunity. Nature. 1983;304:177–179. doi: 10.1038/304177a0. [DOI] [PubMed] [Google Scholar]
- Feist E, Dorner T, Kuckelkorn U, Schmidtke G, Micheel B, Hiepe F, Burmester GR, Kloetzel PM. Proteasome alpha-type subunit C9 is a primary target of autoantibodies in sera of patients with myositis and systemic lupus erythematosus. J Exp Med. 1996;184:1313–1318. doi: 10.1084/jem.184.4.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotz PH. The autoantibody repertoire: searching for order. Nat Rev Immunol. 2003;3:73–78. doi: 10.1038/nri976. [DOI] [PubMed] [Google Scholar]
- Kaplan MM. Primary biliary cirrhosis. N Engl J Med. 1996;335:1570–1580. doi: 10.1056/NEJM199611213352107. [DOI] [PubMed] [Google Scholar]
- van der Geld YM, Limburg PC, Kallenberg CG. Proteinase 3, Wegener's autoantigen: from gene to antigen. J Leukoc Biol. 2001;69:177–190. [PubMed] [Google Scholar]
- Schubert D, Maier B, Morawietz L, Krenn V, Kamradt T. Immunization with glucose-6-phosphate isomerase induces T-cell dependent peripheral polyarthritis in genetically unaltered mice. J Immunol. 2004;172:4503–4509. doi: 10.4049/jimmunol.172.7.4503. [DOI] [PubMed] [Google Scholar]
- Mandik-Nayak L, Wipke BT, Shih FF, Unanue ER, Allen PM. Despite ubiquitous autoantigen expression, arthritogenic autoantibody response initiates in the local lymph node. Proc Natl Acad Sci USA. 2002;99:14368–14373. doi: 10.1073/pnas.182549099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipke BT, Wang Z, Kim J, McCarthy TJ, Allen PM. Dynamic visualization of a joint-specific autoimmune response through positron emission tomography. Nat Immunol. 2002;3:366–372. doi: 10.1038/ni775. [DOI] [PubMed] [Google Scholar]
- Wipke BT, Wang Z, Nagengast W, Reichert DE, Allen PM. Staging the initiation of autoantibody-induced arthritis: a critical role for immune complexes. J Immunol. 2004;172:7694–7702. doi: 10.4049/jimmunol.172.12.7694. [DOI] [PubMed] [Google Scholar]
- Gondolf KB, Mihatsch M, Curschellas M, Dunn JJ, Batsford SR. Induction of experimental allergic arthritis with outer surface proteins of Borrelia burgdorferi. Arthritis Rheum. 1994;37:1070–1077. doi: 10.1002/art.1780370713. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3:360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- Jeffery CJ. Moonlighting proteins. Trends Biochem Sci. 1999;24:8–11. doi: 10.1016/S0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Heinrich SP, Lee MR, Yin HS. Molecular cloning and expression of neuroleukin, a neurotrophic factor for spinal and sensory neurons. Science. 1986;234:566–574. doi: 10.1126/science.3764429. [DOI] [PubMed] [Google Scholar]
- Chaput M, Claes V, Portetelle D, Cludts I, Cravador A, Burny A, Gras H, Tartar A. The neurotrophic factor neuroleukin is 90% homologous with phosphohexose isomerase. Nature. 1988;332:454–455. doi: 10.1038/332454a0. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Mandler R, Murano G, Katz DA, Gordon RK, Chiang PK, Schiffmann E. Tumor cell autocrine motility factor. Proc Natl Acad Sci USA. 1986;83:3302–3306. doi: 10.1073/pnas.83.10.3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silletti S, Raz A. Regulation of autocrine motility factor receptor expression in tumor cell locomotion and metastasis. Curr Top Microbiol Immunol. 1996;213:137–169. doi: 10.1007/978-3-642-61109-4_7. [DOI] [PubMed] [Google Scholar]
- Xu W, Seiter K, Feldman E, Ahmed T, Chiao JW. The differentiation and maturation mediator for human myeloid leukemia cells shares homology with neuroleukin or phosphoglucose isomerase. Blood. 1996;87:4502–4506. [PubMed] [Google Scholar]
- Nabi IR, Raz A. Cell shape modulation alters glycosylation of a metastatic melanoma cell-surface antigen. Int J Cancer. 1987;40:396–402. doi: 10.1002/ijc.2910400319. [DOI] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmatic reticulum. Proc Natl Acad Sci USA. 2001;98:14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]