

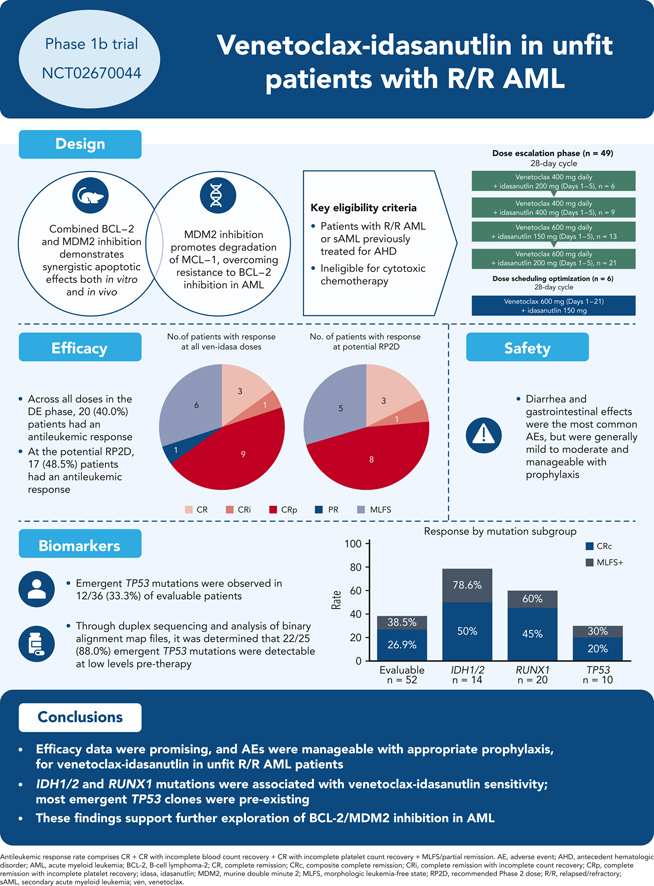
Key Points
-
•
Manageable safety and encouraging preliminary efficacy support extra evaluation of BCL-2/MDM2 inhibition in AML.
-
•
IDH1/2 and RUNX1 mutations were associated with ven-idasa sensitivity; TP53 mutations were unfavorable.
Visual Abstract
Abstract
This phase 1b trial (NCT02670044) evaluated venetoclax-idasanutlin in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML) ineligible for cytotoxic chemotherapy. Two-dimensional dose escalation (DE, n = 50) was performed for venetoclax daily with idasanutlin on days 1 to 5 in 28-day cycles, followed by dosing schedule optimization (n = 6) to evaluate reduced venetoclax schedules (21-/14-day dosing). Common adverse events (occurring in ≥40% of patients) included diarrhea (87.3% of patients), nausea (74.5%), vomiting (52.7%), hypokalemia (50.9%), and febrile neutropenia (45.5%). During DE, across all doses, composite complete remission (CRc; CR + CR with incomplete blood count recovery + CR with incomplete platelet count recovery) rate was 26.0% and morphologic leukemia-free state (MLFS) rate was 12%. For anticipated recommended phase 2 doses (venetoclax 600 mg + idasanutlin 150 mg; venetoclax 600 mg + idasanutlin 200 mg), the combined CRc rate was 34.3% and the MLFS rate was 14.3%. Pretreatment IDH1/2 and RUNX1 mutations were associated with higher CRc rates (50.0% and 45.0%, respectively). CRc rate in patients with TP53 mutations was 20.0%, with responses noted among those with co-occurring IDH and RUNX1 mutations. In 12 out of 36 evaluable patients, 25 emergent TP53 mutations were observed; 22 were present at baseline with low TP53 variant allele frequency (median 0.0095% [range, 0.0006-0.4]). Venetoclax-idasanutlin showed manageable safety and encouraging efficacy in unfit patients with R/R AML. IDH1/2 and RUNX1 mutations were associated with venetoclax-idasanutlin sensitivity, even in some patients with co-occurring TP53 mutations; most emergent TP53 clones were preexisting. Our findings will aid ongoing/future trials of BCL-2/MDM2 inhibitor combinations. This trial was registered at www.clinicaltrials.gov as #NCT02670044.
Daver and colleagues report on a phase 1b trial combining venetoclax and the MDM inhibitor idasanutlin in patients with relapsed/refractory acute myeloid leukemia (AML) who are ineligible for cytotoxic chemotherapy. Composite complete remissions are seen in 25-40% of patients; however, response duration is short. Patients with AML bearing IDH1/2 and RUNX1 mutations respond better, and those with TP53 mutations do less well. Of note, 12 of 36 evaluable patients developed emergent TP53 mutation-bearing clones, almost all of which had been present at baseline but at very low variant allele frequency, suggesting a possible need to screen and select patients best suited for this combination.
Introduction
Therapies for relapsed or refractory (R/R) acute myeloid leukemia (AML) remain limited, with poor outcomes, especially for patients ineligible for cytotoxic chemotherapy or targeted therapy.1, 2, 3, 4, 5 Treatments targeting survival pathways that are dysregulated or aberrant in AML may improve outcomes, particularly if less toxic than current regimens.
Venetoclax (ven), an oral B-cell lymphoma-2 (BCL-2) inhibitor,6 and idasanutlin (idasa), a second generation p53-murine double minute 2 (MDM2) inhibitor,7 have shown limited single-agent activity in R/R AML. Ven monotherapy showed a complete remission (CR)/CR with incomplete blood count recovery (CRi) rate of 19% in both R/R AML and newly diagnosed patients with AML unfit for intensive chemotherapy.8 Idasa monotherapy demonstrated a CR/CRi/morphologic leukemia-free state (MLFS) rate of 21%.9
Combined BCL-2 and MDM2 inhibition, however, has demonstrated synergistic apoptotic effects in vitro and in vivo.10, 11, 12, 13 In mouse models of resistant AML, concomitant p53 activation and BCL-2 inhibition overcame apoptosis resistance.10 Further, ven-idasa demonstrated synergistic antitumor activity in p53 wild-type AML cell lines and led to superior efficacy and survival relative to either single agent in AML models.10,14 The synergistic mechanism of ven-idasa may involve upregulation of proapoptotic proteins (BAX, BIM, and PUMA) by p53 activation, which inactivates myeloid cell leukemia 1 (MCL-1) and BCL-extra large (xL), and concomitant degradation of MCL-1 via dual MCL-1 phosphorylation. MCL-1 is upregulated in AML, particularly during relapse, and is a major contributor to AML progression.15,16 Thus, indirect targeting of MCL-1 may synergize with ven.
Given the promising preclinical synergy of ven + MDM2 inhibition in the chemoresistant setting, we initiated this study to assess the safety, tolerability, and preliminary efficacy of the oral, novel-novel combination, ven-idasa, in older patients with R/R AML, ineligible for cytotoxic chemotherapy.
Methods
Study design
This open-label, multicenter phase 1b trial evaluated ven-idasa and ven-cobimetinib (ClinicalTrials.gov identifier: NCT02670044). We present results for the ven-idasa arm. The primary objectives were to evaluate the safety profile and determine the maximum tolerated dose (MTD) and recommended phase 2 dose (RP2D). Secondary objectives were preliminary efficacy and pharmacokinetics evaluation. Exploratory objectives included assessing biomarkers related to drug targets and disease biology.
Ven was administered daily on days 1 to 28 with idasa daily or twice daily on days 1 to 5 of each 28-day cycle (C). Enrollment was nonrandomized and based on slot availability and sponsor/investigator choice, with multiple cohorts open simultaneously. Patients were treated on only 1 combination arm. Dose escalation (DE) followed a 2-dimensional, 3 + 3 + 3 design (supplemental Figure 1, available on the Blood website). Ven initiation included a 3 to 5-day ramp-up to 400, 600, or 800 mg daily with idasa (150, 200, 400 daily or 300 mg twice daily) on days 1 to 5. Patients were hospitalized during ramp-up in C1 and received tumor lysis syndrome (TLS) prophylaxis.
A dosing schedule optimization (DSO) phase evaluated whether a reduced ven schedule could mitigate myelosuppression and increase CR/CRi rates. Two cohorts evaluating a ven schedule of 21 days, and if indicated, 14 days, at the anticipated RP2D of ven 600 mg + idasa 150 mg were added.
Safety monitoring criteria and stopping rules for toxicity were protocol-defined (details in supplemental Table 1).
The study protocol was approved by the institutional review board or ethics committees at participating institutions in accordance with the International Conference on Harmonization guidelines, including Good Clinical Practice and the ethical principles originating from the Declaration of Helsinki. Informed consent was obtained from all patients. The authors had access to and reviewed the clinical trial results.
Patient population
Eligible patients had R/R AML by World Health Organization criteria17 or newly diagnosed secondary AML (sAML) after prior treatment for an antecedent hematological disorder. In the initial protocol versions, during which the DE stages were enrolled, patients were deemed ineligible for cytotoxic chemotherapy based on age (≥60 years) and investigator opinion. Following a protocol amendment for the DSO stage, patients were ineligible if they were ≥18 years old with documented comorbidities by modified Ferrara criteria.18 Key exclusion criteria included the use of strong or moderate cytochrome P450-3A inducers or inhibitors ≤7 days before study drug administration, and prior BCL-2 inhibitor or MDM2 antagonist.
Assessments
Adverse events (AEs) were reported by the treating physician throughout the study and for ≥30 days after the last dose or initiation of another anticancer therapy. MTD was the highest dose at which less than one-third of ≥6 patients experienced a dose-limiting toxicity.
Response was assessed by routine laboratory tests and bone marrow (BM) examinations and evaluated per the International Working Group 2003 AML response criteria.19 Composite CR (CRc) was defined as CR + CRi + CR with incomplete platelet count recovery (CRp) and antileukemic response as CRc + MLFS/partial response. CRp and CRi were considered mutually exclusive.
Pharmacokinetic assessments
Plasma concentration-vs-time data for ven were analyzed using noncompartmental methods. Summary statistics of pharmacokinetic parameters, such as maximum plasma concentration at steady state (Cmax) and trough/minimum plasma concentration at steady state, were computed.
Biomarker assessments
Baseline and end-of-study mutational analyses were performed on BM-derived mononuclear cells using the FoundationOne Heme Panel (465 gene mutation panel, Roche Foundation Medicine, Grenzach-Wyhlen, Germany) as previously published.20 TP53 mutation (TP53-mut) sensitivity was at 1% variant allele frequency (VAF). Duplex sequencing of TP53 was performed on select baseline samples by TwinStrand Biosciences (Seattle, WA) as previously published.21 TP53 sensitivity was dependent on sequencing depth and varied by sample (mean 0.0212%, range, 0.0111-0.04). Single-cell DNA sequencing (scDNA-seq) used the Tapestri platform (Mission Bio, South San Francisco, CA), per the manufacturer’s instructions, on BM aspirate samples. TP53-muts were evaluated for functional impact at https://tp53.isb-cgc.org.
Minimal residual disease (MRD) and expression of MDM2 and BCL-2 family proteins in peripheral blood were assessed by multiparameter flow cytometry centrally by LabCorp Central Laboratory (Burlington, NC). MRD was used to evaluate response depth in BM aspirates of patients with CR/CRi/CRp using markers per the European LeukemiaNet consensus for flow cytometry–based MRD assessments in AML.22 Integrated leukemia–associated immunophenotypes and different-than-normal procedures were used.22,23 Assay validation established the MRD panel analytical sensitivity as 0.0027% to 0.0037%. Intracellular MDM2, BCL-2, MCL-1, and BCL-xL expression was evaluated on blast cells, myeloid/monocytes, and lymphocytes using surface backbone lineage markers to identify cell populations (CD45, CD34, CD117, and HLA-DR).
Macrophage inhibitory cytokine-1 (MIC-1) concentration in human serum was determined by enzyme-linked immunosorbent assay (ELISA) using the Quantikine ELISA Human GDF-15 ELISA Immunoassay from R&D Systems (distributed by Bio-Techne, Wiesbaden, Germany; catalog number DGD150).
Additional details are in the supplemental Methods.
Statistical methods
Safety and efficacy were summarized by descriptive statistics. The efficacy population was the intent-to-treat population; the safety-evaluable population included all patients who received ≥1 dose of study drug. Time-to-event analyses were conducted using the Kaplan-Meier method. Fisher exact test compared response rates between treatment groups. TP53-muts were compared between treatment groups using Pearson chi-squared test with Yates’ continuity correction.
Results
A total of 56 patients were enrolled for ven-idasa (DE, n = 50; DSO, n = 6) across 17 centers (United States, Italy, Canada, and France) from March 2016 to July 2020 (supplemental Figure 2). Data cutoff date was 10 December 2020. Overall, 55 patients received ≥1 dose of study drug (DE, n = 49; DSO, n = 6; supplemental Figure 1).
Patient characteristics
In the DE stage, the median age was 72 years (range, 62-93) (Table 1). Overall, 55% of patients had refractory disease, 35% had relapsed disease, 10% had newly diagnosed AML transformed from previously treated antecedent hematological disorder, and 49% had sAML. Patients had a median of 1 (range, 1-4) prior line of therapy (details are in supplemental Table 2). In the DSO stage, the median age was 69 years (range, 41-77); 67% of patients had refractory disease, 33% had relapsed AML, and 17% had sAML. Patients had a median of 3 (range, 1-4) prior lines of therapy. Baseline cytogenetics included 11 (20%) patients with complex karyotype. Overall, 10 (18%) patients had TP53-mut (all loss of function; median number of mutations, 1; median VAF, 14.50 [range, 1.24-91.95]; supplemental Table 3). Three patients with TP53-mut had complex karyotype and 1 had therapy-related AML. Complex karyotype was considered separate from TP53-mut, given the availability of next-generation sequencing.
Table 1.
Patient demographics and baseline characteristics of the safety population
| ven-idasa, n = 55 |
|||
|---|---|---|---|
| DE, n = 49 | DSO, n = 6 | All patients, n = 55 | |
| Median age, y (range) | 72.0 (62-93) | 69.0 (41-77) | 72.0 (41-93) |
| Male sex, n (%) | 32 (65) | 2 (33) | 34 (62) |
| ECOG PS, n (%) | |||
| 0 | 15 (31) | 2 (33) | 17 (31) |
| 1 | 26 (53) | 1 (17) | 27 (49) |
| 2 | 8 (16) | 3 (50) | 11 (20) |
| Disease status, n (%) | |||
| Refractory | 27 (55) | 4 (67) | 31 (56) |
| Relapse | 17 (35) | 2 (33) | 19 (35) |
| Newly diagnosed, transformed from AHD (previously treated) | 5 (10) | 0 (0) | 5 (9) |
| AML type, n (%) | |||
| De novo | 25 (51) | 5 (83) | 30 (55) |
| Secondary | 24 (49) | 1 (17) | 25 (45) |
| Median prior therapies, n (range) | 1 (1-4) | 3 (1-4) | 1 (1-4) |
| Previous HSCT, n (%) | 1 (2) | 1 (17) | 2 (4) |
| WBC at baseline, 109/L, median (range) | 2.2 (0.5-27.1) | 3.4 (1.4-8.4) | 2.4 (0.5-27.1) |
| Aspirate BM blast %, median (range) | 34.0 (7.0-100.0) | 88.0 (22.0-97.0) | 34.5 (7.0-100.0) |
| Cytogenetics, n (%) | |||
| TP53 mutation status | |||
| Mutation detected∗ | 9 (18) | 1 (17) | 10 (18) |
| Mutation undetected | 37 (76) | 5 (83) | 42 (76) |
| Not evaluable | 3 (6) | 0 (0) | 3 (5) |
| t(9;11)(p22;q23);MLLT3-MLL | 1 (2) | 0 (0) | 1 (2) |
| inv(3)(q21q26.2) or t(3;3)(q21;q26.2);RPN1-EVI1 | 1 (2) | 0 (0) | 1 (2) |
| −5 or del(5q) | 3 (6) | 0 (0) | 3 (5) |
| −7 | 4 (8) | 0 (0) | 4 (7) |
| Cytogenetic abnormalities not classified as favorable or adverse | 15 (31) | 1 (17) | 16 (29) |
| Complex karyotype | 11 (22) | 0 (0) | 11 (20) |
AHD, antecedent hematological disorder; ECOG PS, Eastern Cooperative Oncology Group performance status; HSCT, hematopoietic stem cell transplantation; WBC, white blood cells.
Using FM Heme cutoff of 1%.
The median number of cycles received was 3.7 (range, 1-15) for DE and 2.8 (range, 1-5) for DSO. Reasons for treatment discontinuation are in supplemental Table 1. The study was terminated early in July 2020 while the DSO stage was enrolling, based on the sponsor’s decision to discontinue idasa development for adults after the MIRROS trial (NCT02545283) failed to meet its primary end point of survival.
Safety
The most common (≥40.0%) all-grade treatment-emergent AEs were diarrhea (87.3%), nausea (74.5%), vomiting (52.7%), hypokalemia (50.9%), and febrile neutropenia (45.5%) (Table 2). Mandatory antidiarrheal prophylaxis was implemented for the ven-idasa arm as a protocol amendment during DE due to the frequent occurrence of diarrhea with ven-cobimetinib (data not shown); following this, 1 patient out of 31 who received mandatory prophylaxis had grade 3 to 4 diarrhea. Serious AEs (SAEs) were reported in 81.8% of patients. The most commonly reported (≥10.0%) SAEs were febrile neutropenia (36.4%), sepsis (16.4%), and pneumonia (14.5%) (supplemental Table 4). Overall, 21.8% (12/55) and 16.4% (9/55) of patients had an AE or SAE, respectively, that resulted in treatment withdrawal. No AEs resulted in treatment discontinuation in more than 1 patient (supplemental Table 1).
Table 2.
Most common treatment-emergent all-grade and grade 3 to 4 AEs
| AE (MedDRA preferred term), n (%) | ven-idasa, n = 55 |
|
|---|---|---|
| All-grade AE∗ | Grade 3 to 4 AE† | |
| Diarrhea | 48 (87.3) | 3 (5.5) |
| Nausea | 41 (74.5) | 1 (1.8) |
| Vomiting | 29 (52.7) | 0 (0.0) |
| Hypokalemia | 28 (50.9) | 10 (18.2) |
| Febrile neutropenia | 25 (45.5) | 25 (45.5) |
| Decreased appetite | 18 (32.7) | 5 (9.1) |
| Hypomagnesemia | 17 (30.9) | 1 (1.8) |
| Neutropenia | 17 (30.9) | 17 (30.9) |
| Thrombocytopenia | 17 (30.9) | 16 (29.1) |
| Anemia | 16 (29.1) | 12 (21.8) |
| Asthenia | 14 (25.5) | 4 (7.3) |
| Fatigue | 14 (25.5) | 5 (9.1) |
| Constipation | 13 (23.6) | 0 (0.0) |
| Edema, peripheral | 12 (21.8) | 0 (0.0) |
| Pneumonia | 12 (21.8) | 10 (18.2) |
AEs were reported by the treating physician, categorized according to the Medical Dictionary for Regulatory Activities version 23.1, and graded per the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
MedDRA, Medical Dictionary for Regulatory Activities.
Any grade AEs occurring in ≥25% of patients.
Grade 3 to 4 AEs occurring in ≥15% of patients.
Laboratory TLS occurred in 3 patients. Clinical TLS (grade 3; increased creatinine) was noted in 1 patient who had baseline renal insufficiency and IDH1 and RUNX1 mutations on day 2 after ven 100 mg; this was reversible with supportive measures. Thirty- and 60-day mortality rates (Kaplan-Meier estimates) were 5.6% (3 patient deaths) and 16.9% (9 patient deaths), respectively, causes of which are in supplemental Table 5. Dose-limiting toxicity included neutropenia (2 patients [3.6%]), and muscular weakness, diarrhea, blood bilirubin increase, and acute coronary syndrome (1 patient [1.8%] each). MTD was ven 600 mg + idasa 200 mg. The DSO stage was not completed at study termination, and the RP2D was not determined.
Efficacy
In the DE stage, across all dose levels, CRc and antileukemic response rates were 26% (13/50) and 40% (20/50), respectively (Table 3). The median time to best CRc response was 1.4 months (range, 0.8-4.3) and the median duration of response (DoR) was 3.9 months (range, 0.7-12.5). Encouraging blast count reduction was seen across doses (Figure 1). For doses under consideration for the RP2D (ven 600 mg + idasa 150 mg and ven 600 mg + idasa 200 mg), the combined rates for antileukemic response and CRc were 48.5% (17/35) and 34.3% (12/35), respectively. The median (range) time to best CRc response and the duration of CRc response were 1.5 (0.8-4.3) and 5.5 (2.3-8.8) months, respectively (responses reported by dose are in supplemental Table 6). Thirteen patients with MLFS in the DE stage ultimately achieved CR/CRi/CRp after ven dose interruption or reduction, suggesting a reduced ven schedule when in remission may facilitate count recovery (supplemental Figure 3). In the DSO stage, 6 patients were treated with a 21-day schedule of ven 600 mg + idasa 150 mg before study termination. CRc and antileukemic response rates were both 33.3% (2/6). Responses were consistent across doses in patients aged ≥75 years, patients with sAML, and patients with prior hypomethylating agent (HMA) treatment for AML (supplemental Figure 4).
Table 3.
Response outcomes for ven-idasa
| n (%) | DE |
DSO |
Total, n = 56 | |||
|---|---|---|---|---|---|---|
| ven 400 mg + idasa 200 mg, n = 6 |
ven 600 mg + idasa 150 mg and ven 600 mg + idasa 200 mg, n = 35 |
ven 400 mg + idasa 400 mg, n = 9 |
Total, n = 50 | ven 600 mg (D1–21) + idasa 150 mg, n = 6 | ||
| Antileukemic responders (CRc/PR/MLFS) | 1 (16.7) | 17 (48.5) | 2 (22.2) | 20 (40.0) | 2 (33.3) | 22 (39.3) |
| CRc (CR/CRi/CRp) | 1 (16.7) | 12 (34.3) | 0 (0.0) | 13 (26.0) | 2 (33.3) | 15 (26.8) |
| CR | 0 (0.0) | 3 (8.6) | 0 (0.0) | 3 (6.0) | 1 (16.7) | 4 (7.1) |
| CRi | 0 (0.0) | 1 (2.9) | 0 (0.0) | 1 (2.0) | 0 (0.0) | 1 (1.8) |
| CRp | 1 (16.7) | 8 (22.9) | 0 (0.0) | 9 (18.0) | 1 (16.7) | 10 (17.9) |
| PR | 0 (0.0) | 0 (0.0) | 1 (11.1) | 1 (2.0) | 0 (0.0) | 1 (1.8) |
| MLFS | 0 (0.0) | 5 (14.3) | 1 (11.1) | 6 (12.0) | 0 (0.0) | 6 (10.7) |
| Time to best CRc response, median (range) | 1.3 (1.3-1.3) | 1.5 (0.8-4.3) | NE | 1.4 (0.8-4.3) | 3.2 (1.6-4.8) | 1.6 (0.8-4.8) |
| Median DoR (CRc), median (range) | NE (0.7∗–0.7∗) | 5.5 (2.3, 8.8) | NE | 3.9 (0.7∗–12.5) | NE (1.0-1.2∗) | 3.0 (0.7∗–12.5) |
| Median follow-up, median (range) | 2.3 (1.6-21.9) | 5.3 (0.0-23.3) | 2.8 (0.4-14.4) | 3.9 (0.0-23.3) | 4.4 (3.0-5.8) | 4.0 (0.0-23.3) |
D, day; NE, not evaluable; PR, partial response.
Censored, response is ongoing before patient dropout. Efficacy data are presented for the intent-to-treat population.
Figure 1.

Best percent BM blast reduction and individual patient best responses. ∗Best percent change from baseline in BM blasts is >100. PR, partial response; RD, refractory disease; SD, stable disease.
Median duration of follow-up was 4.0 months (range, 0.0-23.3), the median overall survival (OS) was 5.1 months (95% confidence interval, 3.4-7.3) (Figure 2). No patients had a subsequent hematopoietic stem cell transplantation. Patients who achieved CRc had an improved median OS over nonresponders (13.8 vs 3.0 months, respectively; P < .0001). Achievement of partial response/MLFS showed a trend toward improved median OS vs nonresponders (5.7 vs 3.0 months, respectively, P = .09). MRD negativity (<0.1%23) was achieved in 42.9% (6/14) of evaluable patients who achieved CRc. Association between MRD negativity and OS was not observed (supplemental Figure 5).
Figure 2.

Kaplan-Meier curves for OS in the ven-idasa arm. Median OS was 5.1 months among all patients, 13.8 months patients with a CRc response, and 5.7 months in patients with a PR/MLFS response. Data for patients without an event were censored at the date of the last study visit or the last known date to be alive, whichever was later. ∗Other = stable disease, refractory disease, relapse, missing or not done. CI, confidence interval; PR, partial response.
Pharmacokinetics
Pharmacokinetic parameters were available for 49 patients across the 5 cohorts (supplemental Table 7). Mean Cmax for ven was 1.11, 1.04, 1.45, 1.47, and 1.15 μg/mL, and mean Cmax for idasa was 3.77, 7.11, 3.22, 3.76, and 4.13 μg/mL in cohorts B1 (n = 6), B4 (n = 8), B1.2.1 (n = 5), B1.2 (n = 11), and B2 (n = 19), respectively. Pharmacokinetics of ven in this combination were comparable to ven monotherapy (supplemental Figure 6).
Pharmacodynamic biomarker analysis
Ven-idasa treatment induced serum MIC-1 levels that correlated with idasa dose and plasma idasa concentration (area under the plasma concentration-vs-time curve within a 24-hour dosing interval; supplemental Figure 7). At C1 day 5, following idasa 150 or 200 mg, the median of the highest fold increase was 10.19; following idasa 400 mg, the median of the highest fold increase was 21.98. MIC-1 induction was not associated with response. Ven-idasa reduced MCL-1 and BCL-xL expression in myeloid blast cells within 15 days of treatment in 64.5% (median, 0.85-fold relative to baseline; range, 0.30-2.55; n = 31) and 48.5% (median, 0.95-fold relative to baseline; range, 0.01-52.9; n = 33) of patients, respectively. These reductions trended with both idasa dose and response (Figure 3).
Figure 3.

MCL-1 and BCL-xL PD data. MCL-1 and BCL-xL were assessed by flow cytometry in blood at C1D1, C1D5 pretreatment, and C1D15 pretreatment. Ven-idasa treatment resulted in modest decreases in MCL-1, and to a lesser degree BCL-xL, that trend with dose and response. Groups by treatment include patients who received indicated idasa dose, regardless of ven dose received. D, day; MFI, mean fluorescence intensity; ND, no data; PD, progressive disease; PR, partial response; RD, refractory disease; SD, stable disease.
Correlative biomarker analysis
Antileukemic responses were achieved in 52.6% (10/19) of patients with high (>1.5) baseline BCL-2:BCL-xL or BCL-2:MCL-1 ratio vs 23.5% (4/17) with low (≤1.5) baseline BCL-2:BCL-xL and BCL-2:MCL-1 ratio (supplemental Figure 8, P = .097). No specific association with or impact on predictability of the BCL-2 family ratios was observed in TP53-muts vs TP53-wild-type. Individual analysis of BCL-2 family proteins revealed a trend for high BCL-xL, but not MCL-1, in nonresponders (supplemental Figure 9). MDM2 expression was not associated with response (data not shown).
Baseline mutation profiling was available for 52 of 55 patients (Figure 4A; supplemental Table 8; supplemental Figure 10). Patients with IDH1/2, RUNX1, or IDH1/2 and/or RUNX1 mutations had antileukemic response rates of 78.6% (11/14), 60.0% (12/20), and 64.3% (18/28), respectively; median DoR was 4.86, 5.30, and 5.17 months, respectively, for these patients. Median OS was highest in patients with IDH1/2 mutation, compared with the evaluable study population, (7.64 vs 3.95 months, respectively). In patients with any RAS signaling pathway genes (FLT3, RAS, NF1, CBL, and PTPN11) and ≥2 RAS signaling mutations, antileukemic response rates were 26.7% (8/30) and 20.0% (2/10), respectively. Hierarchy of responses in the IDH1/2 and RUNX1 subgroups and how co-occurring mutations affected response and survival are presented in supplemental Figure 11.
Figure 4.

Mutational analysis describing (A) heat map of mutations and (B) outgrowth of TP53 mutations on study. In the heat map, detected mutations are indicated by shaded boxes. ∗Mutations that emerged on treatment and were detected at last BM aspirate sampling before therapy discontinuation. Green, favorable mutations; Red, unfavorable mutations (dark red, TP53; light red, RAS signaling mutations). The outgrowth graph indicates the VAF of TP53 mutations that emerged on therapy plotted over time. A total of 25 emergent TP53 mutations in 12 patients were noted on study. Most (22 of 25) emergent TP53 mutations were detectable at low levels in baseline samples either using duplex sequencing or by evaluating binary alignment map files provided by Foundation Medicine. Three were not detected at baseline (red). PR, partial response; ND, no data; RD, refractory disease; SD, stable disease.
In patients with TP53-mut, antileukemic response rate was 30.0% (3/10: 2 CRp, 1 MLFS), median DoR was 2.3 months, and median OS was 3.67 months. Analysis of these 3 responders revealed co-occurring mutations in IDH1 in 1 patient and RUNX1 in 1 patient (supplemental Table 8), with similar or higher VAFs for these mutations (35.6 baseline TP53 VAF co-occurred with 52.83 IDH VAF; 13.09 baseline TP53 VAF co-occurred with 10.0 RUNX1 VAF; the third patient had a baseline TP53 VAF of 15.9 with complex karyotype, del7, and no known ven-sensitizing mutations, but achieved a short-lived [1.18 months] best response of MLFS). The CRc rate was 20.0% (2/10) in patients with TP53 mutations and 28.6% (12/42) in patients with TP53 wild-type. In nonresponders, TP53-muts were mutually exclusive with IDH1/2 and RUNX1 mutations. Twelve baseline TP53-muts (>1% VAF) were detected in 10 patients (2 patients had 2 mutations), all of which were reported to be loss of function.
Analysis of mutations at treatment discontinuation revealed the emergence of TP53 and RAS signaling mutations in 33.3% (12/36) and 27.8% (10/36) of evaluable patients, respectively. All emergent TP53-muts were loss-of-function except 1, which was partially functional and co-occurred with 3 other loss-of-function TP53-muts (4 emergent mutations total). Emergent TP53-muts were more common in responders (40%, 8/20) vs nonresponders (12.5%, 4/32) presumably owing to the longer time on treatment allowing outgrowth. In IDH1/2-, RUNX1- and IDH1/2- and/or RUNX1-mutated subsets, the frequencies of emergent TP53 clones were 36.4%, 33.3%, and 30.4%, respectively, similar to the overall population (33.3%). Emergent TP53-muts had variable kinetics of outgrowth (Figure 4B; supplemental Table 9).
Twenty-five emergent mutations in 12 patients were detected using the FoundationOne Heme mutation panel. Through duplex sequencing and analysis of binary alignment map files, it was determined that 22 of 25 (88.0%) emergent TP53-muts were present at low levels pretherapy (median 0.0095% [range, 0.0006-0.4]). Of the low level (<1% VAF) mutations at baseline (median, 33 [range, 12-93]), most did not expand to detectable levels. All patients with >1% VAF at baseline had expansion of these VAFs on study.
scDNA-seq was performed on longitudinal samples on 4 representative patients to explore on-treatment clonal dynamics. Emergent clones were consistent with bulk sequencing, with demonstrable outgrowth of clones harboring mutations in TP53 and signaling genes (RAS, NF1, and EPHA3; supplemental Figure 12).
Discussion
In this phase 1b trial, supported by preclinical data, ven-idasa had a manageable safety profile at dose levels evaluated for the RP2D, in patients with R/R AML, ineligible for cytotoxic chemotherapy. No new safety signals were identified for either of the drugs or the combination.
Diarrhea and gastrointestinal effects were the most common AEs but were generally mild to moderate and manageable with implementation of prophylaxis. The observed gastrointestinal toxicity rates with ven-idasa were similar to those reported with idasa monotherapy and increased compared with ven monotherapy. All-grade gastrointestinal toxicity rates for ven-idasa compared with idasa or ven monotherapy, respectively, were 87.3% vs 91.3% or 56% for diarrhea; 74.0% vs 73.9% or 59% for nausea; and 52.7% vs 39.1% or 41% for vomiting.24 Rates of grade ≥3 diarrhea and nausea for ven-idasa were decreased vs idasa or ven monotherapy (respectively, 5.5% vs 23.9% or 6% for diarrhea and 1.8% vs 0 or 6% for nausea). Caveats for crosstrial comparisons include differences in the idasa dosing and formulation across studies (idasa 150-200 mg daily spray-dried powder formulation in this study vs 400-800 mg twice daily microprecipitated bulk powder formulation in the idasa monotherapy study). Furthermore, antidiarrheal prophylaxis was not mandatory in both monotherapy studies. The frequency of hematological toxicities and infections was consistent with the known myelosuppressive effects of the therapies and within the previously reported ranges for a similar R/R population.1,3, 4, 5,8,25,26 Mandatory gastrointestinal prophylaxis and DSO are encouraged in future studies to render the combination more tolerable. As reported in other ven AML trials, TLS was not frequent with appropriate ramp-up and TLS prophylaxis. Clinical TLS was noted in 1 patient per arm, suggesting TLS prophylaxis and monitoring should continue when using ven-based combinations in AML.
Clinical activity with ven-idasa was encouraging, particularly at the 2 doses under consideration for the RP2D (n = 35; combined rates: CRc, 34.3%; antileukemic response, 48.5%). Response rates were better than those seen with either agent as monotherapy (ven: CRc, 19%; idasa: antileukemic response, 21%),8,9 although crosstrial comparisons are limited by differences in the patient populations and response rate definitions. Myelosuppression responded to dosing interruptions, thus future evaluation of reduced ven schedules may optimize combined ven and MDM2 inhibition. Although durability remained short, CRc and OS rates were comparable with those with noncytotoxic treatment options (eg, low-intensity therapy, such as HMA alone, ven with HMA, or low-dose cytarabine).5,8,25,27, 28, 29, 30, 31 Furthermore, the 42.9% MRD negativity rate among CRc responders suggests deep responses could be achieved in some patients. Although CRc was associated with OS, an association between MRD negativity and OS was not observed in the small number of patients in our study. Our findings support CRc as a more established end point in R/R AML until more data regarding the type, timing, and prognostic relevance of MRD in R/R AML can be generated.31,32
There was no significant impact of the coadministration of idasa on ven pharmacokinetics or vice versa. Overall, the pharmacokinetic drug-drug interactions between ven and idasa were minimal. Pharmacodynamic biomarker analyses demonstrated that idasa dosing was sufficient to robustly induce MIC-1, an MDM2 inhibition biomarker, confirming on-target activity. In contrast to the 10- to 20-fold increases on day 5 seen in this study, in the idasa polycythemia vera (PV) study,33 plasma MIC-1 levels were increased by 4.8-fold on day 5 of treatment. The differences in MIC-1 induction may be due to the doses used, 100 mg vs 150 to 400 mg daily for 5 days in the PV vs AML study, respectively. As MIC-1 is not tumor-specific, differences in the cellular composition and activation of TP53 between PV and AML may have also contributed. MCL-1/BCL-xL was downregulated in some, but not all patients, suggesting clinical synergy is variable. Longitudinal flow assessments were limited in this study; similar biomarker studies in future MDM2 inhibitor trials may help further characterize and inform the variable downregulation observed.
Patients with low (≤1.5) BCL-2:BCL-xL and BCL-2:MCL-1 ratios had a lower ven-idasa response rate. Individually, BCL-xL was most aligned with nonresponse to ven-idasa, indicating that BCL-xL may be a resistance factor. The ability of idasa to inactivate and degrade MCL-1 was observed in some patients and may explain why MCL-1 was not predictive of response to this combination. TP53-mut status did not appear to affect the predictive value of BCL-2 family ratios, nor did TP53-mut appear to be linked to lower or higher ratios.
Patients with IDH mutations were more responsive to ven-idasa than those without, consistent with previous reports that IDH mutations sensitize AML cells to ven treatment.8,27,34 Response rates were higher with ven-idasa both in IDH1/2-mutated and wild-type subsets (CRc 50% [7/14] and 18% [7/38], respectively) compared with those reported for ven monotherapy (CRc 33% [4/12] and 10% [2/20], respectively). With the caveat of small numbers and crosstrial comparison, these results suggest that idasa contributes to the combination’s activity. RUNX1 mutations were also associated with an improved response rate, consistent with prior observations that RUNX1 mutations are linked to improved outcomes with ven-based regimens. RUNX1 mutations may sensitize AML cells by reducing the apoptotic threshold via dysregulation of BCL-2 expression.28,35, 36, 37 Mutations in the RAS signaling pathway (N/KRAS, FLT3, CBL, NF1, PTPN11) were associated with poor responsiveness to ven-idasa, consistent with prior studies demonstrating lower response rates and/or short DoR to ven-based therapies.25,28,38,39 Although interpretation is limited owing to small numbers, a proposed hierarchy of responses in the setting of comutations demonstrates that IDH1/2 and RUNX1 were favorable prognostic mutations unless they co-occurred with RAS signaling mutations. In the absence of IDH1/2 or RUNX1 mutations, TP53 was unfavorable. In the absence of IDH1/2, RUNX1, or TP53-muts, any RAS signaling mutations indicated poor prognosis.
Antileukemic response rate was 30% in patients with TP53-mut. Although an association between VAF and response was not observed, co-occurring mutations in IDH1 and RUNX1 in 2 of the 3 responders were noted, with similar or higher VAFs for these sensitizing mutations. Though the numbers are small, this suggests that some patients with TP53-mut could still benefit from ven + MDM2 inhibitor therapy and that the presence of co-occurring mutations is a potentially important selection factor. Larger data sets are needed to confirm these observations and to delineate the ideal patient population for these regimens.
An important question relating to the use of MDM2 antagonists is whether they select for outgrowth of TP53-mut clones or even induce de novo TP53-muts. Through deep sequencing analysis, we determined that most (88% [22/25]) newly detected TP53-muts were pre-existing before therapy, indicating ven-idasa likely applies selective pressure for pre-existing TP53 clones in most cases, rather than inducing de novo TP53-muts. TP53-muts have been shown to emerge over the course of nontargeted therapies in AML40 and are associated with ven resistance,41 thus they may not necessarily be MDM2 inhibitor-specific. Furthermore, the frequency of TP53-mut expansion is difficult to compare across crosstrial comparisons using different sequencing panels. Most low-level TP53-muts detected at baseline did not expand to standard detectable levels (>1% VAF), whereas expansion was noted in patients with baseline VAF >1%. It is possible that the low-level TP53-muts are CHiP clones or transient in nature, as reported in idasa-treated PV.42 Further evaluation is needed to inform the role of ultradeep sequencing for TP53-muts in directing therapy with MDM2 inhibitors. TP53-muts should be longitudinally tracked with high-sensitivity monitoring techniques in MDM2 inhibitor trials in AML to understand the biology of low-level mutations and the clinical significance of TP53-mut expansion/emergence.
As this study was designed and initiated before ven approval for first-line treatment of unfit patients with AML, a limitation of the study was the evaluation of ven retreatment. Recent data of ven in combination with gilteritinib in R/R FLT3-mutated AML, demonstrated durable responses and survival benefit in patients with prior ven treatment,43 suggesting that potential unique synergies may exist with different ven combinations rendering retreatment effective. Future studies should be directed toward better understanding the outcomes of ven with MDM2 inhibitors in both prior ven-exposed and ven-naïve patients.
Although a number of MDM2 inhibitors have been under investigation, the largest trial to date has been of idasa in combination with cytarabine in R/R AML in the phase 3 MIRROS trial.44 MIRROS failed to demonstrate improved OS compared with cytarabine, but the ven-idasa combination in this study was based on a different rationale, with synergies suggested by preclinical work. The clinical activity observed with ven-idasa, although moderate, was encouraging, particularly considering the unfit R/R population, most of whom had failed other low-intensity treatments and had limited alternative treatment options.
In summary, ven-idasa demonstrated manageable safety and encouraging preliminary efficacy in a difficult-to-treat unfit R/R AML population, supporting further evaluation of combined BCL-2 and MDM2 inhibition in AML. Correlatives demonstrated that IDH and RUNX1 mutations were associated with sensitivity to ven-idasa, including in patients with co-occurring TP53-muts, and that MDM2 inhibition was associated with the emergence of pre-existing TP53 clones. Other MDM2 inhibitors (eg, siremadlin, APG-115) are still in development. Trials of ven with other MDM2 inhibitors as doublets (siremadlin-ven; NCT03940352)45 or triplets (siremadlin-ven-azacitidine; NCT05155709) are ongoing and are anticipated to further expand on these findings. The dosing optimization/myelosuppression approaches, response, molecular, and pharmacodynamic learnings from this study may guide optimization of future trial design and patient selection for BCL-2 and MDM2 inhibitor combinations.
Conflict-of-interest disclosure: N.G.D. has received research funding from AbbVie and Genentech. M.D., M.O. J.W., and W.H. are employees of Genentech and may hold Roche stock or stock options. J.S.G. has received research funding (for trials) from AbbVie, Genentech, Pfizer, Prelude, and AstraZeneca; and has served on advisory board for AbbVie, Astellas, and Takeda. B.A.J. is a consultant/advisor for AbbVie, BMS, Celgene, Genentech, Gilead, GlycoMimetics, Jazz, Pfizer, Servier, Takeda, Tolero, and Treadwell; has received travel reimbursement from AbbVie; and research funding to his institution from 47, AbbVie, Accelerated Medical Diagnostics, Amgen, AROG, BMS, Celgene, Daiichi Sankyo, F. Hoffmann-La Roche, Forma Therapeutics, Genentech/Roche, Gilead, GlycoMimetics, Hanmi, Immune-Onc, Incyte, Jazz, Loxo, LP Therapeutics, Pfizer, Pharmacyclics, Sigma-Tau and Treadwell. K.W.L.Y. is a consultant for Astex, BMS/Celgene, F. Hoffmann-La Roche, Novartis, Otsuka, Paladin, Pfizer, Shattuck Labs, Taiho, and Takeda; has received honorarium from AbbVie and Novartis; and has received research funding from Astex, Forma Therapeutics, F. Hoffmann-La Roche, Genentech, Geron, Janssen, Jazz, MedImmune, Novartis, Onconova, and Tolero. K.R.K. received honoraria from Janssen, Novartis, Celgene, Epizyme, Pharmacyclics, Karyopharm, GSK, Bristol-Myers Squibb, Seattle Genetics, and Gilead, consulting fees from Sanofi, AstraZeneca, Sanofi-Aventis, Denovo Biopharma and Amgen and received research funding from Takeda and Oncolytics Biotech Inc. S.A. reports nonfinancial support from Roche/Genentech during the conduct of the study; and has received personal fees from Roche Canada, Pfizer, BMS, Paladin, and Lundbeck outside the submitted work. G.J.R. consultancy AbbVie, Agios, Amgen, Amphivena, Astex, AstraZeneca, Celator, Celgene, Clovis Oncology, CTI BioPharma, Genoptix, Immune Pharmaceuticals, Janssen Pharmaceuticals, Juno, MedImmune, MEI Pharma, Novartis, Onconova, Pfizer, Roche, Sunesis, and Jazz; and has received research funding from Cellectis. D.A.P. serves on a data safety monitoring board for GlycoMimetics and Aptevo; has received research funding from AbbVie, BMS, Teva and Karyopharm; and is an advisory board member or consultant for Novartis, AbbVie, BeiGene, BerGenBio, Arcellx, Jazz, Syros, BMS, Genentech, ImmunoGen, AstraZeneca, Kura, Ryvu, Magenta, and Qihan Zentalis. J.M.B. consultancy AbbVie, BMS, Astex, Pfizer, Astellas, Taiho, and Jazz. R.L.O. has received research support for trials from Astellas, Pfizer, Genentech, Daiichi Sankyo, and Cellectis; and consulting from AbbVie, Astellas, and Actinium. G.M. is a consultant for AbbVie, Celgene, Roche, Janssen, Astellas, Pfizer, and Incyte. B.L.P. has received research funding from Ambit Biosciences, Genentech, F. Hoffmann-La Roche, Jazz, Novartis, Pfizer, and Rafael Pharmaceuticals; and is a consultant for Rafael Pharmaceuticals. W-J.H. is a former employee of Genentech and may hold Roche stock; is a former employee of Imago BioSciences and may hold stock; and is a current employee of Prelude Therapeutics. M.Y.K. is a consultant for AbbVie, Genentech, and F. Hoffmann-La Roche; is an advisory board member for F. Hoffmann-La Roche; holds shares from Reata; has received honoraria from Amgen, AbbVie, and Genentech; and has received research funding from AbbVie, Genentech, Eli Lilly, Cellectis, Calithera, Stemline, Threshold, Flexus Biosciences, Novartis, Ablynx, and Agios. M.A. consulted for Amgen, AstraZeneca, Daiichi Sankyo, Syndax, GlycoMimetics, Oncoceutics, and Aptose; has received research funding from F. Hoffmann-La Roche, AstraZeneca, Amgen, Daiichi Sankyo, Jazz, GlycoMimetics; and holds stock from Reata, Oncoceutics/Chimerix and Aptose. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank the participating patients and their families, investigators, and trial staff. Support in analysis of the data was provided by Denise Cheung, Diana Dunshee, and Connie Ma of Genentech, Inc, Fang Yin Lo of TwinStrand Biosciences, Inc, and Dexter Jin of Foundation Medicine. Third-party medical writing assistance, under the direction of Maika Onishi, was provided by Roisin Weaver, and Sinéad Holland of Ashfield MedComms, an Inizio company, and was funded by Genentech Inc.
Venetoclax is being developed in a collaboration between Genentech and AbbVie. Genentech provided financial support for the study and participated in the design, study conduct, analysis, and interpretation of data, as well as the writing, review, and approval of the manuscript.
Authorship
Contribution: N.G.D., M.D., M.O., J.W., M.G.O., W.-J.H., M.Y.K., and M.A. designed the study; N.G.D., J.S.G., B.A.J., K.W.L.Y., K.R.K., N.V., S.A., G.J.R., S.P., D.A.P., A.T., J.M.B., A.P., B.L.P., P.F., R.L.O., G.V., G.M., M.O., W.-J.H., M.Y.K., and M.A. conducted the study; N.G.D., J.S.G., B.A.J., K.W.L.Y., K.R.K., N.V., S.A., G.J.R., S.P., D.A.P., A.T., J.M.B., A.P., B.L.P., P.F., R.L.O., G.V., G.M., M.O., M.Y.K., and M.A. carried out recruitment and follow-up of patients; N.G.D., J.S.G., B.A.J., K.W.L.Y., K.R.K., N.V., S.A., G.J.R., S.P., D.A.P., A.T., J.M.B., A.P., B.L.P., P.F., R.L.O., G.V., G.M., M.O., M.Y.K., and M.A. collected data; N.G.D., M.D., J.S.G., B.A.J., K.W.L.Y., K.R.K., N.V., S.A., G.J.R., S.P., D.A.P., A.T., J.M.B., A.P., B.L.P., P.F., R.L.O., G.V., G.M., M.O., J.W., W.H., C.G., M.G.O., W.-J.H., M.Y.K., and M.A. carried out data analysis; and all authors assisted in the development of the manuscript and read and approved the submitted version of the manuscript.
Footnotes
As a phase 1 trial, this study is not in scope per the Roche global policy on data sharing. The decision to share patient-level data needs to be handled on a case-by-case basis to determine if the data can be adequately anonymized to give an acceptably low risk of patient reidentification. Owing to technical limitations, scDNA-seq and flow cytometry data cannot be shared. Qualified researchers may submit an enquiry through the data request platform, Vivli, https://vivli.org/ourmember/roche/, but this does not guarantee that the data can be shared. For up-to-date details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing. Anonymized records for individual patients across more than 1 data source external to Roche cannot, and should not, be linked owing to a potential increase in risk of patient reidentification.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Naval G. Daver, Email: ndaver@mdanderson.org.
Marina Y. Konopleva, Email: mkonople@mdanderson.org.
Michael Andreeff, Email: mandreef@mdanderson.org.
Supplementary Material
References
- 1.Appelbaum FR, Gundacker H, Head DR, et al. Age and acute myeloid leukemia. Blood. 2006;107(9):3481–3485. doi: 10.1182/blood-2005-09-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ganzel C, Sun Z, Cripe LD, et al. Very poor long-term survival in past and more recent studies for relapsed AML patients: the ECOG-ACRIN experience. Am J Hematol. 2018;93(8):1074–1081. doi: 10.1002/ajh.25162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravandi F, Ritchie EK, Sayar H, et al. Vosaroxin plus cytarabine versus placebo plus cytarabine in patients with first relapsed or refractory acute myeloid leukaemia (VALOR): a randomised, controlled, double-blind, multinational, phase 3 study. Lancet Oncol. 2015;16(9):1025–1036. doi: 10.1016/S1470-2045(15)00201-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roboz GJ, Rosenblat T, Arellano M, et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol. 2014;32(18):1919–1926. doi: 10.1200/JCO.2013.52.8562. [DOI] [PubMed] [Google Scholar]
- 5.Stahl M, DeVeaux M, Montesinos P, et al. Hypomethylating agents in relapsed and refractory AML: outcomes and their predictors in a large international patient cohort. Blood Adv. 2018;2(8):923–932. doi: 10.1182/bloodadvances.2018016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 7.Higgins B, Glenn K, Walz A, et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin Cancer Res. 2014;20(14):3742–3752. doi: 10.1158/1078-0432.CCR-14-0460. [DOI] [PubMed] [Google Scholar]
- 8.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase ii study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yee K, Martinelli G, Vey N, et al. Phase 1/1b study of RG7388, a potent MDM2 antagonist, in acute myelogenous leukemia (AML) patients (Pts) Blood. 2014;124(21):116. [Google Scholar]
- 10.Pan R, Ruvolo V, Mu H, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell. 2017;32(6):748–760.e746. doi: 10.1016/j.ccell.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kojima K, Konopleva M, Samudio IJ, Schober WD, Bornmann WG, Andreeff M. Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle. 2006;5(23):2778–2786. doi: 10.4161/cc.5.23.3520. [DOI] [PubMed] [Google Scholar]
- 12.Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106(9):3150–3159. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan R, Kojima K, Zheng Z, et al. Activation of p53 by novel MDM2 antagonist RG7388 overcomes AML inherent and acquired resistance to Bcl-2 inhibitor ABT-199 (GDC-0199) Blood. 2014;124(21):2162. [Google Scholar]
- 14.Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol. 2016;9(1):50. doi: 10.1186/s13045-016-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufmann SH, Karp JE, Svingen PA, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91(3):991–1000. [PubMed] [Google Scholar]
- 16.Xiang Z, Luo H, Payton JE, et al. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest. 2010;120(6):2109–2118. doi: 10.1172/JCI39964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 18.Ferrara F, Barosi G, Venditti A, et al. Consensus-based definition of unfitness to intensive and non-intensive chemotherapy in acute myeloid leukemia: a project of SIE, SIES and GITMO group on a new tool for therapy decision making. Leukemia. 2013;27(5):997–999. doi: 10.1038/leu.2012.303. [DOI] [PubMed] [Google Scholar]
- 19.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in acute myeloid leukemia. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2003;21(24):4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 20.He J, Abdel-Wahab O, Nahas MK, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127(24):3004–3014. doi: 10.1182/blood-2015-08-664649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salk JJ, Loubet-Senear K, Maritschnegg E, et al. Ultra-sensitive TP53 sequencing for cancer detection reveals progressive clonal selection in normal tissue over a century of human lifespan. Cell Rep. 2019;28(1):132–144.e133. doi: 10.1016/j.celrep.2019.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131(12):1275–1291. doi: 10.1182/blood-2017-09-801498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yee K, Papayannidis C, Vey N, et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study⋆. Leuk Res. 2021;100 doi: 10.1016/j.leukres.2020.106489. [DOI] [PubMed] [Google Scholar]
- 25.Aldoss I, Yang D, Aribi A, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica. 2018;103(9):e404–e407. doi: 10.3324/haematol.2018.188094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tenold ME, Moskoff BN, Benjamin DJ, et al. Outcomes of adults with relapsed/refractory acute myeloid leukemia treated with venetoclax plus hypomethylating agents at a comprehensive cancer center. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.649209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiNardo CD, Maiti A, Rausch CR, et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: a single-centre, phase 2 trial. Lancet Haematol. 2020;7(10):e724–e736. doi: 10.1016/S2352-3026(20)30210-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiNardo CD, Rausch CR, Benton C, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93(3):401–407. doi: 10.1002/ajh.25000. [DOI] [PubMed] [Google Scholar]
- 29.Itzykson R, Thépot S, Berthon C, et al. Azacitidine for the treatment of relapsed and refractory AML in older patients. Leuk Res. 2015;39(2):124–130. doi: 10.1016/j.leukres.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Sarkozy C, Gardin C, Gachard N, et al. Outcome of older patients with acute myeloid leukemia in first relapse. Am J Hematol. 2013;88(9):758–764. doi: 10.1002/ajh.23498. [DOI] [PubMed] [Google Scholar]
- 31.Short NJ, Rafei H, Daver N, et al. Prognostic impact of complete remission with MRD negativity in patients with relapsed or refractory AML. Blood Adv. 2020;4(24):6117–6126. doi: 10.1182/bloodadvances.2020002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aitken M.J.L., Ravandi F, Patel KP, Short NJ. Prognostic and therapeutic implications of measurable residual disease in acute myeloid leukemia. J Hematol Oncol. 2021;14(1):137. doi: 10.1186/s13045-021-01148-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mascarenhas J, Lu M, Kosiorek H, et al. Oral idasanutlin in patients with polycythemia vera. Blood. 2019;134(6):525–533. doi: 10.1182/blood.2018893545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chow S, Tang K, Al-Abri M, et al. RUNX1 mutations correlate with response to venetoclax combination therapies in relapsed/refractory acute myeloid leukemia. Leuk Res. 2021;111 doi: 10.1016/j.leukres.2021.106735. [DOI] [PubMed] [Google Scholar]
- 36.Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123(9):3876–3888. doi: 10.1172/JCI68557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherry EM, Abbott D, Amaya M, et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021;5(24):5565–5573. doi: 10.1182/bloodadvances.2021005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chyla B, Daver N, Doyle K, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am J Hematol. 2018;93(8):E202–E205. doi: 10.1002/ajh.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alwash Y, Khoury JD, Tashakori M, et al. Development of TP53 mutations over the course of therapy for acute myeloid leukemia. Am J Hematol. 2021;96(11):1420–1428. doi: 10.1002/ajh.26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollyea DA, Pratz KW, Wei AH, et al. Outcomes in patients with poor-risk cytogenetics with or without TP53 mutations treated with venetoclax combined with hypomethylating agents. Blood. 2021;138(Suppl. 1):224. [Google Scholar]
- 42.Marcellino BK, Farnoud N, Cassinat B, et al. Transient expansion of TP53 mutated clones in polycythemia vera patients treated with idasanutlin. Blood Adv. 2020;4(22):5735–5744. doi: 10.1182/bloodadvances.2020002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daver N, Perl AE, Maly J, et al. Venetoclax plus gilteritinib for FLT3-mutated relapsed/refractory acute myeloid leukemia. J Clin Oncol. 2022;40(35):4048–4059. doi: 10.1200/JCO.22.00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konopleva MY, Röllig C, Cavenagh J, et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 2022;6(14):4147–4156. doi: 10.1182/bloodadvances.2021006303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stein EM, DeAngelo DJ, Chromik J, et al. Results from a first-in-human phase i study of siremadlin (HDM201) in patients with advanced wild-type TP53 solid tumors and acute leukemia. Clin Cancer Res. 2022;28(5):870–881. doi: 10.1158/1078-0432.CCR-21-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.