

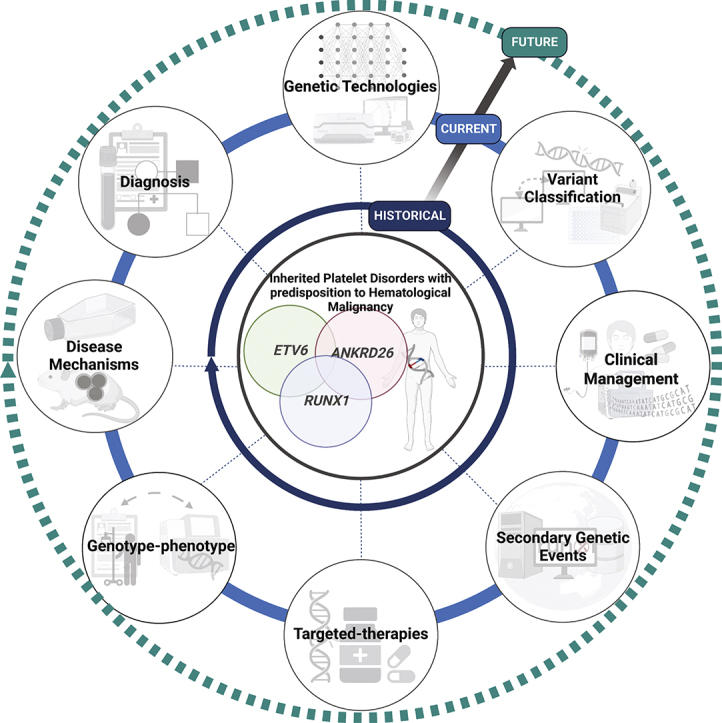
Visual Abstract
Abstract
Hereditary platelet disorders (HPDs) are a group of blood disorders with variable severity and clinical impact. Although phenotypically there is much overlap, known genetic causes are many, prompting the curation of multigene panels for clinical use, which are being deployed in increasingly large-scale populations to uncover missing heritability more efficiently. For some of these disorders, in particular RUNX1, ETV6, and ANKRD26, pathogenic germ line variants in these genes also come with a risk of developing hematological malignancy (HM). Although they may initially present as similarly mild-moderate thrombocytopenia, each of these 3 disorders have distinct penetrance of HM and a different range of somatic alterations associated with malignancy development. As our ability to diagnose HPDs has improved, we are now faced with the challenges of integrating these advances into routine clinical practice for patients and how to optimize management and surveillance of patients and carriers who have not developed malignancy. The volume of genetic information now being generated has created new challenges in how to accurately assess and report identified variants. The answers to all these questions involve international initiatives on rare diseases to better understand the biology of these disorders and design appropriate models and therapies for preclinical testing and clinical trials. Partnered with this are continued technological developments, including the rapid sharing of genetic variant information and automated integration with variant classification relevant data, such as high-throughput functional data. Collective progress in this area will drive timely diagnosis and, in time, leukemia preventive therapeutic interventions.
Routine use of next-generation sequencing of hematologic malignancies has greatly expanded the representation of hereditary predisposition syndromes among patients with leukemia. Introduced by Associate Editor Mario Cazzola, this Review Series highlights 4 such genetic predisposition syndromes and provides strong support for the need to include germ line genetic testing for patients with myelodysplasia and myeloid leukemia.
Introduction
Hereditary platelet disorders (HPDs) encompass a large spectrum of rare disorders that demonstrate variable severity and clinical impact. HPDs affect the number and/or function of platelets, resulting in defects in hemostasis in patients, ranging from mild to severe.1 Although bleeding is a frequent complication, other clinical manifestations, including kidney disease, deafness, immunodeficiencies, and malignancies, can occur. Diagnosis remains a challenge because of the heterogeneity in penetrance, clinical presentation, and many causative genes, with many patients still not receiving a genetic diagnosis. Currently, there are >60 types of HPD caused by 75 known HPD genes,1,2 some of which also predispose to hematological malignancy (HM), most frequently myeloid neoplasms (MNs).3,4
Since 2016, the World Health Organization classification of MNs has included “MNs with germ line predisposition and preexisting platelet disorders.”5 This category recognized germ line RUNX1, ETV6, and ANKRD26 as causing HPDs with a significant risk of developing a MN. Subsequently, several other bodies have also included HPD in updated guidelines, including the European LeukemiaNet and the recent International Consensus Classification (ICC) of MNs and acute leukemias.6,7
Given the large number of HPDs and other recent comprehensive reviews,1,2 this review will focus primarily on 3 HPDs that also predispose to hematological neoplasms, caused by variants in RUNX1, ETV6, and ANKRD26. Here, we will compare clinical phenotypic and genetic differences with historical and future facing perspectives of where we have come from and where the field is heading.
HPDs due to germ line variants in RUNX1, ETV6, and ANKRD26
Historically, the diagnosis of HPDs has proved challenging because of the considerable heterogeneity in clinical presentation and laboratory tests. Typically, the diagnosis is made when patients present with easy bruising, low platelet counts, and a family history. These 3 syndromes largely phenocopy each other in terms of platelet defects and the predisposition to HMs. The 3 syndromes are described below, and key aspects of the disorders are summarized in Table 1 and a timeline of important events in Figure 1.
Table 1.
Characteristics relevant to RUNX1-, ETV6-, and ANKRD26-driven HPDs
| RUNX1 | ETV6 | ANKRD26 | |
|---|---|---|---|
| Full gene name | RUNX family transcription factor 1 | ETS variant transcription factor 6 | Ankyrin-repeat domain 26 |
| No. of exons | 9 | 8 | 34 |
| Transcript (HGVS reference) | NM_001754.4 | NM_001987.5 | NM_014915.3 |
| Protein (HGVS reference) | NP_001745.2 | NP_001978.1 | NP_055730.2 |
| Protein length (HGVS reference), amino acids | 480∗ | 452 | 1710 |
| Gene function | Transcription factor: DNA binding, activator, repressor | Transcription factor: DNA binding, repressor | Ankyrin-repeat protein-protein interactions: MAPK/extracellular signal-regulated kinase signaling |
| Disease association | OMIM:601399; MONDO:0011071 | OMIM 616216; MONDO:0014536 | OMIM:188000, MONDO:0008555 |
| First phenotype description | 1969 | Unknown | 1965 |
| First genetic description | 1999 | 2015 | 2011 |
| Mode of inheritance | AD | AD | AD |
| Germ line variants | Missense, nonsense, frameshift, deletions, duplications, splicing, structural variants | Missense, nonsense, frameshift, deletions, structural variants | 5′ UTR variants |
| Disease mechanism | Haploinsufficiency and dominant-negative | Haploinsufficiency and dominant-negative | Loss of RUNX1-, FLI1-, and ETS-binding sites in 5′ UTR, gene derepression |
| Platelet levels, ×109/L | 70-145 | >75 | <150 |
| Platelet function | Aspirin-like defect on platelet aggregometry and α/δ-granule deficits | Normal platelet size and variable mean platelet volume. Abnormal platelet aggregation, α granules deficits | Platelet size is often unchanged |
| Non-HM hematological phenotypes | Thrombocytopenia | Thrombocytopenia | Thrombocytopenia, some reports of red cell macrocytosis, erythrocytosis, neutropenia |
| HMs | MDS, AML, T-cell ALL, T-NHL, CLL, HCL |
B-ALL, MPAL, MN, MDS, AML, multiple myeloma, CMML | MDS, AML, CML, CLL |
| HM cumulative risk | 43% by 50 y | 30% | 8% lifetime risk |
| HM median age of onset, y | 29 | 11 | 33 |
| HM age range, y | 2-72 | 2-82 | 1-84 |
| Other phenotypes | Eczema/psoriasis | Not reported | Not available |
| Non-HM malignancies | Breast, prostate, bone, gastric, pancreas, and skin cancer | Melanoma, colorectal, breast, kidney, and skin and meningioma cancer | Prostate, breast, and colon cancer |
| Somatic variant pattern | Loss of RUNX1 WT allele in myeloid. Lymphoid driver variants in lymphoid disease | Hyperdiploid. Loss of ETV6 WT allele | Not well described, add more detail |
| Mouse models and phenotypes | LOF and FPD-MM missense variants/homozygous KO and FPD-MM are embryonic lethal/Runx1R188Q/+ mice: reduced platelet function, defect DNA-damage response, myeloid expansion in the BM | LOF allele, Etv6 P214L knockin mice/KO embryonic lethal/Etv6 P214L homozygous mice: decreased platelet counts, impaired megakaryocyte maturation, and defect in platelet activation | KO allele (not appropriate for disease). No hematological abnormalities |
AD, autosomal dominant; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; HCL, hairy cell leukemia; HGVS, Human Genome Variation Society; LOF, loss-of-function; MPAL, mixed phenotype leukemia; T-NHL, T-cell non-Hodgkin lymphoma; WT, wild-type; y, year.
Also known as RUNX1c isoform and is the predominant isoform in hematopoietic cells.
Figure 1.

Milestones and future perspectives in the discovery and treatment of HPDs with predisposition to HM. Although these disorders have been recognized phenotypically for decades, the advent and availability of NGS technologies have improved diagnosis, gene discovery, and understanding of disease progression in the last decade. We predict that with the development and accessibility of genomic technologies, including single-cell sequencing, PacBio optical genome mapping, the integration of artificial intelligence technologies, international standardization, and data sharing initiatives (ie, MatchMaker exchange and Shariant), and consortium-based projects to develop high-throughput functional studies (ie, Impact of Genomic Variation on Function [IGVF] Consortium, multiplexed assays of variant effect), this will rapidly advance the discovery of novel phenotypic-genotypic relationships in these disorders, identify novel pathogenic genes and disease mechanisms, and the much anticipated development of targeted therapies and clinical management guidelines for these patients. Created with BioRender.com. NIH, National Institutes of Health; WHO, World Health Organization.
Initial descriptions
Pathogenic germ line RUNX1 variants are causative of a familial platelet disorder with a predisposition to myeloid malignancy (FPD-MM). The first documented phenotypic description of a family was >50 years ago: 4 generations with autosomal dominant hereditary platelet defects.8 An additional large 7-generation family with multiple generations of HM and platelet disorders revealed the linkage region to 21q22.9,10 Thirty years after the initial description, germ line RUNX1 variants were identified (Figure 1),11 with the original family confirmed to have a germ line heterozygous RUNX1 p.Tyr260∗ (RUNX1b annotation, Tyr287∗ in RUNX1c; ClinGen allele ID CA248619) in 2002.12 With the next-generation sequencing (NGS) era and the increased accessibility and reduced costs of these technologies, >259 FPD-MM families with 164 unique variants in RUNX1 have been identified.13
The first family with ANKRD26-related thrombocytopenia and leukemia predisposition disorder/thrombocytopenia 2 (THC2) was documented as early as 1965, when a multigenerational family was clinically recognized as having mild thrombocytopenia in 11 members across 3 generations.14 It took another 34 years before a second family was documented and the linkage region of THC2 was mapped to locus 10p11.2-12,15 and another 12 years for 5′ untranslated region (UTR) variants in ANKRD26 to be defined as causative (Figure 1).16 More than 70 families have now been reported as having THC2.17
ETV6-related THC5 are caused by germ line pathogenic variants in ETV6.18, 19, 20, 21 In contrast to the long RUNX1 history, the discovery of ETV6 as cause of THC5 can be largely attributed to the NGS era and availability of whole-exome sequencing (WES) technologies. Within the space of a year (2015), 4 publications confirmed ETV6 as the causative gene in 9 pedigrees (Figure 1).18, 19, 20, 21 Since these first reported publications, 100 individuals have been identified in 26 families.22
Germ line variants and biological functions of RUNX1, ANKRD26, and ETV6
RUNX1, ANKRD26, and ETV6 are crucial regulators of different stages of hematopoiesis, with interplay observed at different stages among all 3 proteins. In the following text and in Table 1, we highlight some of the key biological functions, cofactors, and the association with altered platelet biogenesis and HM.
RUNX1 encodes a transcription factor and is considered a master regulator of hematopoiesis required for definitive hematopoiesis, as seen in homozygous knockout (KO) mouse models.23 RUNX1 mediates its transcriptional effects through heterodimerization with the core binding factor subunit β, which interacts with the DNA-binding RUNT homology domain.24 Germ line RUNX1 variants encompass partial and whole gene deletions, frameshift, stopgain, and missense variants.13,25 Frameshift and stopgain variants occur throughout the protein. Missense variants are mostly confined to the RUNT homology domain and frequently affect DNA-binding residues, in some cases displaying loss of DNA-binding activity while retaining core binding factor subunit β binding, suggesting potential for dominant-negative activity.25,26 The location or type of variants have not been demonstrated to associate with different disease phenotypes or malignancy penetrance, and heterogeneity is often observed among family members carrying the same germ line lesion. In some cases, this may be associated with uncaptured additional germ line variation, although this is currently uncertain and requires directed research (Figure 2).25 Frequent germ line deletions associated with a FPD-MM phenotype are predicted to affect only the RUNX1c isoform, suggesting that this is the predominant “oncogenic” isoform.13 Homozygous conditional Runx1-deficient mice over time display age-related stem cell exhaustion and greater susceptibility to leukemia induced by secondary variants, including modeling of variants found recently in collective studies of human germ line RUNX1 tumors.4,25,27, 28, 29
Figure 2.

Additional genetic findings in a family initially diagnosed with RUNX1 FPD-MM. The original diagnosis was obtained using Sanger sequencing of the RUNX1 gene and identified RUNX1 R320∗ (red +). Subsequent NGS revealed an additional pathogenic DDX41 variant K108Sfs∗3 (blue +) and an ANKRD26 M1I VUS (green +) in some family members.
ETV6 is a member of the ETS factor transcription factor family required for bone marrow (BM) hematopoiesis and megakaryopoiesis, as demonstrated in mouse KO models.30,31 ETV6 functions predominantly in a complex with SIN3A, N-COR, and HDAC3 as a transcriptional repressor.32 For ETV6, most of the pathogenic variants reside within the ETS domain, required for DNA binding, which leads to a dominant-negative effect via the loss of transcriptional repression.33,34 Most variants are missense, although nonsense, frameshift, deletion, and structural variants leading to loss of function have also been observed.22,35 Thrombocytopenia is observed in Etv6 megakaryocyte-erythroid progenitor conditional KO mice because of an increase in megakaryocytic colony-forming cells, suggesting a terminal defect in megakaryocyte maturation with the loss of Etv6.36 Interestingly, it has been reported that ETV6 interacts with the FLI1 oncoprotein to inhibit its transcriptional activity.37 FLI1 is also a platelet and megakaryocyte ETS domain transcription factor and is also implicated in hereditary thrombocytopenia.2,38
ANKRD26 encodes an ankyrin-repeat domain protein, which mediates protein-protein interactions. Expression is tightly regulated during hematopoiesis, with high expression in the hematopoietic stem cells and downregulation during megakaryopoiesis, with expression almost undetectable in megakaryocytes and platelets. ANKRD26 is involved in regulating thrombopoietin (TPO)–dependent signaling, with ANKRD26 expression in megakaryocytes leading to activation of TPO-dependent signaling pathways, including the MAPK/extracellular signal-regulated kinase pathway.39 Most ANKRD26 pathogenic variants are located within the 5′ UTR of ANKRD26 in a highly conserved region (c.-116 to c.-134). Both RUNX1 and FLI1 transcription factors have been shown to bind motifs in the 5′ UTR region of ANKRD26, negatively regulating ANKRD26 expression during megakaryopoiesis. Pathogenic ANKRD26 variants result in the loss of this repression and persistent ANKRD26 expression during megakaryopoiesis, leading to increased extracellular signal-regulated kinase phosphorylation and elevated MAPK signaling during early platelet development.39 As inhibition of the MAPK pathway is required for platelet formation, sustained activation of this pathway is the likely cause of thrombocytopenia in these patients.39 In addition, recent studies have also identified a role for ANKRD26 in maintaining centriole copy number, dysregulation of which may affect megakaryocyte polyploidization and subsequent platelet production.40, 41, 42 Variants outside of the 5′ UTR region, including a start-loss p.M1I/p.M1? variant, have been described (Figure 2) but are not currently considered to be pathogenic.43
Germ line variant–associated phenotypes
FPD-MM, THC2, and THC5 often present with mild-to-moderate platelet counts and mild-to-moderate bleeding risk. Specific details are described in Table 1. Briefly, platelet counts are most often mildly affected but can be lower or within the normal range in variant carriers.12,44, 45, 46 Platelet function studies frequently show an aspirin-like defect on platelet aggregometry and α/δ-granule deficits in germ line RUNX1 carriers.12,47,48 Platelets in ETV6 and ANKRD26 carriers are typically normal size.22,49,50
Germ line RUNX1 variants most frequently predispose to myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML) and are present in ∼12% of families with hereditary MDS and AML.51,52 Lymphoid malignancy is seen at lower frequency, most commonly childhood T-cell acute lymphoblastic leukemia (ALL),53 and rarely lymphoma and B-cell ALL (B-ALL).25,54,55 RUNX1 carriers have a median age of HM onset of ∼30 years and a cumulative incidence of 43% by 50 years.4,56, 57, 58 ETV6 most frequently gives rise to ALL, commonly B-ALL, with biphenotypic leukemia, chronic myelomonocytic leukemia, myeloma, and MDS/AML are sometimes observed.59 In childhood B-ALL cohorts, pathogenic germ line ETV6 variants were found in 0.8% of cases, typically in older children with hyperdiploid karyotypes.21 Penetrance is incomplete, with only ∼30% of carriers developing a HM; however, the penetrance of thrombocytopenia is >90%.22 ANKRD26-mutated individuals have an increased risk of developing most commonly MDS and AML; however, lymphoid malignancies have been reported, with chronic lymphocytic leukemia being the most frequent.59 There is an estimated 8% lifetime risk of progression to MDS, AML, or chronic myeloid leukemia.3,16,60,61 In addition, some individuals can develop erythrocytosis and leukocytosis.50,60 In a recent study of a pediatric cohort of individuals with germ line RUNX1, ANKRD26, or ETV6 variants, clinical presentation of mild-to-moderate thrombocytopenia and mild bleeding phenotypes was found, with HM also coexistent with RUNX1 (10/14) and ETV6 (1/2) variants but not ANKRD26 (0/8 patients), consistent with the reduced penetrance of ANKRD26 for HM.62
Molecular harbingers of progression and malignancy
Because of the relative rarity of these germ line predisposition syndromes, it is difficult for individual centers to accumulate sufficient cases to examine patterns in secondary somatic variants that may drive HM from the predisposed state. To address this for RUNX1, over the past several years, we have established an international data sharing consortium and centralized, standardized, and reviewed data.4,25 Data analysis from this consortium shows that somatic mutation of the second RUNX1 allele is frequently associated with malignancy (including duplication of the germ line variant through trisomy 21 or uniparental disomy). Other recurrent somatically mutated genes include PHF6, BCOR, and TET2.4,25,63 In contrast, tumors from germ line RUNX1 carriers with a lymphoid phenotype have different somatic cooperating and phenotype-driving variants; for example, childhood ETP T-cell ALL frequently acquires activating variants in JAK3.53 For ETV6, a childhood ALL cohort with damaging germ line ETV6 variants, frequently identified gross chromosomal gains, 70% having hyperdiploid ALL.33 Hyperdiploid ALL also commonly included RAS pathway variants with second hits in NRAS, KRAS, and PTPN11. Diploid ETV6-driven ALL had a high prevalence of PAX5 (86%) and ETV6 deletions (57%).33 ANKRD26 less frequently transforms to HM, limiting the opportunity to profile somatic variants, although several patients have now been identified with somatic variants in ASXL1, KRAS, and SF3B1.64, 65, 66
Premalignant somatic variants have been observed and occur frequently in germ line RUNX1 carriers, most commonly in BCOR, TET2, DNMT3A, KRAS, and SRSF2, occurring as early-onset clonal hematopoiesis of indeterminate potential (CHIP) in carriers as young as 16 years.4,25,67 It is now clear that this represents a common state, with initial studies showing 67% of carriers (<50 years) harboring a clonal somatic variant profile.67 Our recent studies have confirmed variants occur in clinically relevant genes, indicating CHIP is present in one-third of unselected RUNX1 carriers before any development of malignancy.63 We recently showed that CHIP can also occur in preleukemic ANKRD26 carriers (14%) and has not yet been observed in ETV6 carriers; however, larger patient numbers are required to define the risk for CHIP in these cohorts (Figure 1).68 As discussed in more detail in ‟Clinical managment and treatment,” longitudinal data will be essential to connect these observations directly to increased risk of HM and determine the interplay of acquired variants on phenotypic heterogeneity observed within families.
Approaches and challenges for diagnosis of HPDs
There are many good resources for understanding the clinical risk of a germ line predisposition for patients presenting with either thrombocytopenia or a MN, including a recent companion paper to the new ICC of MNs and acute leukemia.3,6,26,69 Key points include a careful assessment of family history, consideration of the age of malignancy development, and the diagnosis of multiple malignancies in an individual. These parameters may enrich the identification of individuals with HPDs; however, it is becoming increasingly clear that cases are missed because of confounding factors, such as de novo variants and a malignancy diagnosis in the nonpredisposed age range.70 In this section, we summarize different processes that may lead to the positive identification of a variant associated with genetic predisposition to HPD, including predisposition to HM.
Sample screening type
Once suspicion of predisposition triggers a decision for germ line testing, several other technical considerations come into play. These include the use of an appropriate sample type for screening, which minimizes hematopoietic cell contamination. All tissue types have their own limitations, and we and others have also discussed this previously.3,4,69,71 Although cultured skin fibroblasts are a “gold standard” source of germ line material, we find them less frequently used at our center because of the requirement for an extra procedure, laboratory resources, and time taken to get a test-ready sample. Despite the perceived limitations of DNA quantity from hair bulbs and nail clippings, as well as the potential for monocyte contamination in the latter, we find these options more acceptable to the patient, including the option of self-collection, and we routinely obtain sufficient DNA for germ line NGS. Our experience is that saliva and/or buccal swabs have a greater potential for high leukocyte content and should be avoided. Inappropriate source material for germ line screening risks false-positive or false-negative results, either of which will confound a genetic diagnosis. Examples include attribution of germ line status to a somatic variant for a patient with an active malignancy or the lack of identification of a germ line variant due to somatic reversion in blood cells correcting the germ line defect. Somatic reversion is recently described for RUNX1 and is likely to be frequently overlooked (Figure 1).72
Panels, gene lists, and precision medicine
Once an appropriate sample type has been acquired, the next decision pertains to the type of technology to be used for testing. In many cases, there may be limited choices, influenced by regulatory, geographical, financial, and other aspects. A common starting point for many clinicians may be the use of NGS panels. As such, appropriate physical or virtual (eg, on the backbone of a whole-genome sequencing [WGS] platform) gene lists for diagnostic screening are a topic of much discussion. For platelet disorders, the International Society on Thrombosis and Hemostasis has curated a diagnostic gene list that is updated annually.73 Other guidelines, such as World Health Organization, European LeukemiaNet, ICC, and numerous reviews, discuss genes of importance to malignancy predisposition without necessarily specifying a recommended gene list, acknowledging the rapid changes in this area and the speed at which lists may become outdated. Mechanisms for gene list compilation with regular expert review exist, facilitating a broad range of gene lists for hereditary disorders as part of the broader “Gene Curation Coalition.”74,75
The hit rates for NGS panels in populations with relevant phenotypes can vary. Recently, the clinical utility of a thrombogenomics high-throughput sequencing test (Figure 1) was assessed on 2396 patients identified through clinical and laboratory phenotypes and classified into 5 broad disease groups: thrombotic, platelet count, platelet function, coagulation, and unexplained bleeding, based on human phenotype ontology (HPO) terms.76 A variant was detected in ∼50% of patients with a clear clinical phenotype, whereas the diagnostic rate was much lower with ambiguous unexplained bleeding (3.2%). This supports the need for comprehensive, standardized phenotyping to increase the molecular diagnosis rate in selected population screens. As a genetic finding, likely pathogenic or pathogenic variants in ANKRD26 were more frequent than in either RUNX1 or ETV6, suggesting that the prevalence of ANKRD26 is higher than previously thought.76
Precision medicine clinics have been successful in ending the “diagnostic odyssey” for some patients with unexplained cytopenias and bleeding disorders. Algorithm-based decision-making in these clinics use collaborations among clinicians, bioinformaticians, and molecular biologists to identify patients with HPDs.77 Using this approach, 40% of prospectively identified patients obtained a genetic diagnosis, resulting in 91% receiving altered clinical care.66 Patients with a suspected hematological germ line predisposition syndrome, including ∼19% with a history of thrombocytopenia, were identified, including germ line variants in RUNX1 (10%), ANKRD26 (6%), and ETV6 (1%).
Somatic tumor panels
The increased routine use of somatic cancer gene panels for tumor screening in relevant patient populations has paved the way for more frequent detection and clinical recognition of germ line predisposition variants.78,79 This provides both an opportunity and a challenge because matched testing of tumor/germ line samples is not routinely performed in most laboratories. When a variant suspicious of being germ line is found, it triggers additional workloads of germ line confirmation and genetics referral for patient and family. This may be urgent, because cancer panels are often run when patients have an acute clinical presentation, such as overt neoplasia possibly requiring identification of donors for hematopoietic stem cell transplantation. The American College of Medical Genetics has addressed this with general guidelines across all tumor types80, 81, 82 and for MNs, expert opinion has suggested genes that should have germ line scrutiny for variants detected at high variant allele frequency, which include RUNX1 and ETV6.78,79 For RUNX1, the same variants have been detected in both a somatic and germ line context in HM, highlighting the challenge of when to employ germ line confirmation.
Limitations of genetic testing, wrong gene, wrong place, wrong time?
Despite the now impressive pick-up rates that can be achieved in clinics with defined gene panels for both thrombocytopenia and malignancy predisposition, there remain individuals for whom a genetic diagnosis is not obtained with first-pass testing. For this, there can be many reasons, including sample type selection (discussed in “Sample screening type”), gene list selection (the variant is in another gene, maybe one yet to be recognized), or technology selection (the variant is there in the gene of interest, but our technology could not detect it). ETV6 germ line variants are present in the coding region and detectable using simple panels. Germ line copy number variations frequently occur in RUNX1,25,52,83 with ∼30% of cases harboring deletions and may be missed without genome array technology or WGS and specialized bioinformatic pipelines. Good custom panel designs now include the 5′ UTR of ANKRD26 as a known variant hot spot. However, in some cases, even the full gamut of new and emerging technologies may be needed to secure a genetic diagnosis. For example, for a thrombocytopenia family that remained unresolved after conventional WES, WGS, and Sanger sequencing, the use of PacBio long-read genome sequencing and RNA sequencing eventually identified a paired-duplication inversion resulting in a gain-of-function WAC-ANKRD26 fusion transcript.84
The period in which the diagnosis was initially obtained and the technology used also matter. For example, in the Australian Familial Haematological Conditions Study (running for over 2 decades), we recently presented germ line RUNX1 families identified over time and using different technologies, ranging from single-gene Sanger sequencing to WES.25 In 1 family, diagnosed decades earlier with a pathogenic RUNX1 variant, subsequent studies using NGS panels/WES revealed individuals also with a germ line pathogenic DDX41 variant or a start-loss variant of uncertain significance in ANKRD26 (Figure 2). Because both of these genes are also associated with relevant phenotypes seen in the family, this case study highlights the requirement for periodic review of individuals and families to keep pace with new discoveries, technologies, and consideration of potential additional genotype-phenotype correlations.
Assessment of variant pathogenicity: variant classification and resolution of the VUS
As germ line genetic testing has grown, ClinGen has responded both by forming specific rules for the classification of germ line variants85 and disease- and gene-focused committees. Gene-specific variant curation is a specialized and dynamic process, with regular reviews of classification codes needed as more information about disorders becomes available. The ClinGen MM variant curation expert panel, cofounded by the American Society of Hematology, focuses on the predisposition area relevant to this review and, to date, has released 2 versions of germ line classification rules specifically for RUNX1.86,87 Version 1 (V1) modified 19 classification codes specifically for RUNX1, with the most recent V2 of the guidelines further modifying 7 classification codes relative to V1 rules.87 This has assisted in centralizing disease focus and rule modification that promotes accurate classification and strategies for the resolution of variants of uncertain significance (VUS). Novel variants can be difficult to accurately assess to determine variant pathogenicity and often sit as a VUS. These VUSs can be resolved by the identification of additional families/probands or family members with phenotypes and functional analysis of variants. Our research group has multiple initiatives to assist with these processes. These include a centralized location for germ line RUNX1 genomics, clinical, and classification data (RUNX1db) to assist with classification and other research efforts,13 and a variant sharing platform (Shariant), allowing clinical genetics laboratories to contribute variant classifications with automated tracking and classification discordance notifications (Figure 1).88
The American College of Medical Genetics has guidelines for functional assays for use in clinical classification, and recommendations for RUNX1 exist, which include a luciferase-based transactivation assay with a known RUNX1 target gene.86,89 These are relevant to RUNX1-specific modifications to code PS3/BS3,87 and recent studies have focused on validating these assays for clinical use, allowing more accurate application of these codes to resolve VUSs.89 Luciferase assays are also established for the assessment of variants in ETV6 and ANKRD2639 and will be assessed for clinical use for these disorders in the near future. As the speed of variant identification increases, the task of individually testing hundreds of variants of interest, even in a well-validated assay, is imposing. As for other cancer predisposition genes, such as TP53 and BRCA1, high-throughput screening technologies such as multiplexed assays of variant effect90 that generate functional data for all possible missense variants in a protein or all possible nucleotide substitutions in a promoter can provide an invaluable resource.91 If integrated with variant sharing initiatives, this will automate the variant curation workflow.92
Clinical management and treatment
There are no official evidence-based guidelines for the surveillance of unaffected carriers of germ line variants in RUNX1, ETV6, and ANKRD26, with standard of care informed by a consensus of expert opinion.3,79,93 Frequently, this approach involves an initial baseline BM and full blood count examination followed by full blood count examination periodically (3-6 months). Blood counts that deviate from the baseline values will trigger further investigation with a repeat BM biopsy. Progression to malignancy may occur in the absence of cytopenias or other blood count abnormalities, so we and others also recommend the use of somatic NGS panels to detect somatic variants (CHIP) in peripheral blood at regular intervals. Standard HM somatic NGS panels are likely to cover many genes known to be somatically mutated in tumors associated with RUNX1, ETV6, and ANKRD26 predisposition disorders;33,63,66 however, periodic gene list review is recommended. Although somatic variant information in unaffected carriers is not currently validated with longitudinal data for risk ratios and time frame to malignancy, this is being addressed, for RUNX1, through longitudinal studies such as the RUNX1 natural history study at the National Institutes of Health and our ongoing studies through RUNX1db among others (Figure 1). Notwithstanding the time needed to accumulate longitudinal data, current knowledge suggests that progression to malignancy is less likely to occur in the absence of additional somatic variants, and the detection of a somatic variant provides another biomarker that can be monitored for changes.
Therapeutic management of patients with HPD is largely limited to strategies to circumvent low platelet numbers and manage the risk of bleeding in patients during surgeries or childbirth, with antifibrinolytics and platelet transfusion recommended as needed.60 TPO receptor agonists (ie, eltrombopag and romiplostim) have shown clinical utility in some HPDs to increase platelet count before surgery.94,95
There are currently no specific guidelines for the treatment of HM within predisposition disorders, and a lack of information exists on how these populations respond to well-established standard-of-care approaches, with the exception of emerging data for some of the more prevalent disorders, such as those caused by DDX41 variants.96 In general, for germ line predisposition disorders, allogeneic hematopoietic stem cell transplantation (HSCT) may be the only curative option for these patients, which both treats HM and reduces HM recurrence risk. For example, even an initial good response to chemotherapy, such as is seen in germ line CEBPA patients with AML, does not prevent the development of new, independent leukemias that may occur even decades later.97 In addition, for RUNX1 carriers, who likely have defects in homologous recombination DNA repair pathways,4 there are emerging concerns that an initial chemotherapy approach may increase the risk of secondary or therapy-related leukemias. Given the improvements in HSCT success and risk modulation (ie, customized conditioning regimens), it is topical to try and define a risk ratio that allows intervention with HSCT before the development of malignancy. In some cases, such as germ line GATA2-associated immunodeficiency, it is increasingly implemented to rescue morbidity associated with immunodeficiency.98 However, for predisposition disorders without the preleukemic phenotype severity, the risk calculations are complicated.99
New approaches to therapy for germ line carriers would be a welcome addition to the clinics, with several in development, particularly for RUNX1 because of specific research program support. Some seek to correct the underlying molecular defect directly. For example, seeking to increase the amount of RUNX1 activity in the cell by inhibiting the degradation of the remaining wild-type allele.100 Permanent correction through gene therapy or editing is under investigation, and recently a Rhesus macaque CRISPR RUNX1 gene-editing model has been established, providing insights into the growth dynamics of RUNX1-mutant cells, as well as the potential suitability of this therapy approach.101 Targeted therapies aimed at preventing the clonal growth of cells with a detected CHIP variant may also show utility through a mechanism of “resetting” the preleukemic landscape of predisposed individuals. In contrast, once the development of malignancy has occurred, the survival of RUNX1-mutated leukemia cells in both the hereditary and sporadic contexts are dependent on residual RUNX1 activity for survival, which has been exploited in vitro by approaches to remove remaining RUNX1 activity, such as small interfering RNA.102,103
Conclusions and future directions
With the increased cost-effectiveness of high-throughput NGS technologies, the diagnosis of HPD is steadily improving. However, there remain large differences in access to genetic technologies that modulate many of the considerations discussed herein. In centers where access to WGS is becoming commonplace, more time must be spent ascertaining the pathogenicity of VUS, compared with situations where access to NGS is restricted or absent. Soon, machine learning and artificial intelligence algorithms will likely improve clinical diagnosis, treatment, workflows, and outcomes based on clinical and laboratory data in real time. Examples of advances in blood smear and genomic analysis through artificial intelligence showcased at the recent American Society of Hematology annual meeting may also lead to machine-learning algorithms to identify patterns associated with predisposition. Key challenges to this vision for these relatively rare disorders include aggregation of sufficient quality standardized (across laboratories) data for use as training and testing data sets.
Patient support groups and dedicated research initiatives for rare diseases, such as the RUNX1 Research Program, also have a powerful role to play. They can fulfill multiple roles, including directly supporting researchers, acting as a lobby point with government and other organizations to create further research opportunities, as well as advocating for patients and giving them a voice. In all cases, it is clear for the “rare” disease portfolio that international cooperative data sharing is the most efficient way to make rapid progress toward a timely genetic diagnosis and to continue driving and advocating for research-informed care, with the eventual aim of safely reducing, or ideally removing, cancer risk for these individuals and their families.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank Christopher N. Hahn for critical review of the manuscript. The authors also thank the patients and their family members for participating in this research program.
This work is supported by a grant from the RUNX1 Research Program. This project is also proudly supported by funding from the Leukemia Foundation of Australia and grants from the National Health and Medical Research Council of Australia (project grants APP1145278 and APP1164601). This work was produced with the financial and other support of Cancer Council SA’s Beat Cancer Project on behalf of its donors and the State Government of South Australia through the Department of Health, PRF Fellowship (H.S.S.) and project grants (APP1163447 and APP1145385) (A.L.B.).
Authorship
Contribution: All authors performed background research; analyzed and interpreted data; and wrote, critically reviewed, and approved the manuscript.
References
- 1.Palma-Barqueros V, Revilla N, Sánchez A, et al. Inherited platelet disorders: an updated overview. Int J Mol Sci. 2021;22(9):4521–4552. doi: 10.3390/ijms22094521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warren JT, Di Paola J. Genetics of inherited thrombocytopenias. Blood. 2022;139(22):3264–3277. doi: 10.1182/blood.2020009300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tawana K, Brown AL, Churpek JE. Integrating germline variant assessment into routine clinical practice for myelodysplastic syndrome and acute myeloid leukaemia: current strategies and challenges. Br J Haematol. 2022;196(6):1293–1310. doi: 10.1111/bjh.17855. [DOI] [PubMed] [Google Scholar]
- 4.Brown AL, Hahn CN, Scott HS. Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA) Blood. 2020;136(1):24–35. doi: 10.1182/blood.2019000937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 6.Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–1377. doi: 10.1182/blood.2022016867. [DOI] [PubMed] [Google Scholar]
- 7.Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200–1228. doi: 10.1182/blood.2022015850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss HJ, Chervenick PA, Zalusky R, Factor A. A familial defect in platelet function associated with XXXm paired release of adenosine diphosphate. N Engl J Med. 1969;281(23):1264–1270. doi: 10.1056/NEJM196912042812303. [DOI] [PubMed] [Google Scholar]
- 9.Dowton SB, Beardsley D, Jamison D, Blattner S, Li FP. Studies of a familial platelet disorder. Blood. 1985;65(3):557–563. [PubMed] [Google Scholar]
- 10.Ho CY, Otterud B, Legare RD, et al. Linkage of a familial platelet disorder with a propensity to develop myeloid malignancies to human chromosome 21q22.1-22.2. Blood. 1996;87(12):5218–5224. [PubMed] [Google Scholar]
- 11.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 12.Michaud J, Wu F, Osato M, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood. 2002;99(4):1364–1372. doi: 10.1182/blood.v99.4.1364. [DOI] [PubMed] [Google Scholar]
- 13.Homan CC, King-Smith SL, Lawrence DM, et al. The RUNX1 database (RUNX1db): establishment of an expert curated RUNX1 registry and genomics database as a public resource for familial platelet disorder with myeloid malignancy. Haematologica. 2021;106(11):3004–3007. doi: 10.3324/haematol.2021.278762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bithell TC, Didisheim P, Cartwright GE, Wintrobe MM. Thromobocytopenia inherited as an autosomal dominant trait. Blood. 1965;25(2):231–240. [PubMed] [Google Scholar]
- 15.Savoia A, Del Vecchio M, Totaro A, et al. An autosomal dominant thrombocytopenia gene maps to chromosomal region 10p. Am J Hum Genet. 1999;65(5):1401–1405. doi: 10.1086/302637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pippucci T, Savoia A, Perrotta S, et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. 2011;88(1):115–120. doi: 10.1016/j.ajhg.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan MJ, Palmer EL, Botero JP. ANKRD26-related thrombocytopenia and predisposition to myeloid neoplasms. Curr Hematol Malig Rep. 2022;17(5):105–112. doi: 10.1007/s11899-022-00666-4. [DOI] [PubMed] [Google Scholar]
- 18.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. doi: 10.1038/ng.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47(5):535–538. doi: 10.1038/ng.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topka S, Vijai J, Walsh MF, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PloS Genet. 2015;11(6):e1005262. doi: 10.1371/journal.pgen.1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriyama T, Metzger ML, Wu G, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015;16(16):1659–1666. doi: 10.1016/S1470-2045(15)00369-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter CC, Di Paola J, Pencheva B. In: GeneReviews®. Adam MP, Everman DB, Mirzaa GM, et al., editors. University of Washington; 2020. ETV6 thrombocytopenia and predisposition to leukemia. [Google Scholar]
- 23.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 24.Ogawa E, Inuzuka M, Maruyama M, et al. Molecular cloning and characterization of PEBP2 beta, the heterodimeric partner of a novel Drosophila runt-related DNA binding protein PEBP2 alpha. Virology. 1993;194(1):314–331. doi: 10.1006/viro.1993.1262. [DOI] [PubMed] [Google Scholar]
- 25.Brown AL, Arts P, Carmichael CL, et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020;4(6):1131–1144. doi: 10.1182/bloodadvances.2019000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown AL, Churpek JE, Malcovati L, Dohner H, Godley LA. Recognition of familial myeloid neoplasia in adults. Semin Hematol. 2017;54(2):60–68. doi: 10.1053/j.seminhematol.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Growney JD, Shigematsu H, Li Z, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106(2):494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y-J, Chen J-Y, Yan M, et al. RUNX1 deficiency cooperates with SRSF2 mutation to induce multilineage hematopoietic defects characteristic of MDS. Blood Adv. 2022;6(23):6078–6092. doi: 10.1182/bloodadvances.2022007804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmad MH, Hegde M, Wong WJ, et al. Germline Runx1 mutations induce inflammation in hematopoietic stem and progenitor cells and predispose to hematologic malignancies [abstract] Blood. 2021;138(suppl 1) Abstract 2201. [Google Scholar]
- 30.Wang LC, Swat W, Fujiwara Y, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12(15):2392–2402. doi: 10.1101/gad.12.15.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang LC, Kuo F, Fujiwara Y, et al. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J. 1997;16(14):4374–4383. doi: 10.1093/emboj/16.14.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Hiebert SW. TEL contacts multiple co-repressors and specifically associates with histone deacetylase-3. Oncogene. 2001;20(28):3716–3725. doi: 10.1038/sj.onc.1204479. [DOI] [PubMed] [Google Scholar]
- 33.Nishii R, Baskin-Doerfler R, Yang W, et al. Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood. 2021;137(3):364–373. doi: 10.1182/blood.2020006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faleschini M, Ammeti D, Papa N, et al. ETV6-related thrombocytopenia: dominant negative effect of mutations as common pathogenic mechanism. Haematologica. 2022;107(9):2249–2254. doi: 10.3324/haematol.2022.280729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21(2):122–137. doi: 10.1038/s41568-020-00315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hock H, Meade E, Medeiros S, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18(19):2336–2341. doi: 10.1101/gad.1239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwiatkowski BA, Bastian LS, Bauer TR, Jr., et al. The ets family member Tel binds to the Fli-1 oncoprotein and inhibits its transcriptional activity. J Biol Chem. 1998;273(28):17525–17530. doi: 10.1074/jbc.273.28.17525. [DOI] [PubMed] [Google Scholar]
- 38.Stevenson WS, Rabbolini DJ, Beutler L, et al. Paris-Trousseau thrombocytopenia is phenocopied by the autosomal recessive inheritance of a DNA-binding domain mutation in FLI1. Blood. 2015;126(17):2027–2030. doi: 10.1182/blood-2015-06-650887. [DOI] [PubMed] [Google Scholar]
- 39.Bluteau D, Balduini A, Balayn N, et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest. 2014;124(2):580–591. doi: 10.1172/JCI71861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans LT, Anglen T, Scott P, et al. ANKRD26 recruits PIDD1 to centriolar distal appendages to activate the PIDDosome following centrosome amplification. EMBO J. 2021;40(4):e105106. doi: 10.15252/embj.2020105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burigotto M, Mattivi A, Migliorati D, et al. Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. EMBO J. 2021;40(4):e104844. doi: 10.15252/embj.2020104844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowler M, Kong D, Sun S, et al. High-resolution characterization of centriole distal appendage morphology and dynamics by correlative STORM and electron microscopy. Nat Commun. 2019;10(1):993. doi: 10.1038/s41467-018-08216-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marconi C, Canobbio I, Bozzi V, et al. 5’UTR point substitutions and N-terminal truncating mutations of ANKRD26 in acute myeloid leukemia. J Hematol Oncol. 2017;10(1):18–21. doi: 10.1186/s13045-016-0382-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Latger-Cannard V, Philippe C, Bouquet A, et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J Rare Dis. 2016;11:49–64. doi: 10.1186/s13023-016-0432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chisholm KM, Denton C, Keel S, et al. Bone marrow morphology associated with germline RUNX1 mutations in patients with familial platelet disorder with associated myeloid malignancy. Pediatr Dev Pathol. 2019;22(4):315–328. doi: 10.1177/1093526618822108. [DOI] [PubMed] [Google Scholar]
- 46.Deuitch N, Broadbridge E, Cunningham L, Liu P. In: GeneReviews®. Adam MP, Everman DB, Mirzaa GM, et al., editors. University of Washington; 2021. RUNX1 familial platelet disorder with associated myeloid malignancies. [Google Scholar]
- 47.Marneth AE, van Heerde WL, Hebeda KM, et al. Platelet CD34 expression and α/δ-granule abnormalities in GFI1B- and RUNX1-related familial bleeding disorders. Blood. 2017;129(12):1733–1736. doi: 10.1182/blood-2016-11-749366. [DOI] [PubMed] [Google Scholar]
- 48.Glembotsky AC, Bluteau D, Espasandin YR, et al. Mechanisms underlying platelet function defect in a pedigree with familial platelet disorder with a predisposition to acute myelogenous leukemia: potential role for candidate RUNX1 targets. J Thromb Haemost. 2014;12(5):761–772. doi: 10.1111/jth.12550. [DOI] [PubMed] [Google Scholar]
- 49.Feurstein S, Godley LA. Germline ETV6 mutations and predisposition to hematological malignancies. Int J Hematol. 2017;106(2):189–195. doi: 10.1007/s12185-017-2259-4. [DOI] [PubMed] [Google Scholar]
- 50.Perez Botero J, Dugan SN, Anderson MW. In: GeneReviews®. Adam MP, Everman DB, Mirzaa GM, et al., editors. University of Washington; 2018. ANKRD26-related thrombocytopenia. [Google Scholar]
- 51.Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717–732. doi: 10.1182/blood-2017-09-806489. [DOI] [PubMed] [Google Scholar]
- 52.Rio-Machin A, Vulliamy T, Hug N, et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun. 2020;11(1):1044. doi: 10.1038/s41467-020-14829-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Yang W, Devidas M, et al. Germline RUNX1 variation and predisposition to childhood acute lymphoblastic leukemia. J Clin Invest. 2021;131(17):e147898. doi: 10.1172/JCI147898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Six KA, Gerdemann U, Brown AL, et al. B-cell acute lymphoblastic leukemia in patients with germline RUNX1 mutations. Blood Adv. 2021;5(16):3199–3202. doi: 10.1182/bloodadvances.2021004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karki N, Savage N, Kutlar A. Novel germline RUNX1 mutation associated with familial thrombocytopenia as well as B-acute lymphoblastic leukemia: a case report and review of the literature. Case Rep. Oncol. 2021;14(1):439–445. doi: 10.1159/000512016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ganly P, Walker LC, Morris CM. Familial mutations of the transcription factor RUNX1 (AML1, CBFA2) predispose to acute myeloid leukemia. Leuk Lymphoma. 2004;45(1):1–10. doi: 10.1080/1042819031000139611. [DOI] [PubMed] [Google Scholar]
- 57.Liew E, Owen C. Familial myelodysplastic syndromes: a review of the literature. Haematologica. 2011;96(10):1536–1542. doi: 10.3324/haematol.2011.043422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayashi Y, Harada Y, Huang G, Harada H. Myeloid neoplasms with germ line RUNX1 mutation. Int J Hematol. 2017;106(2):183–188. doi: 10.1007/s12185-017-2258-5. [DOI] [PubMed] [Google Scholar]
- 59.Vyas H, Alcheikh A, Lowe G, et al. Prevalence and natural history of variants in the ANKRD26 gene: a short review and update of reported cases. Platelets. 2022;33(8):1107–1112. doi: 10.1080/09537104.2022.2071853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noris P, Perrotta S, Seri M, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117(24):6673–6680. doi: 10.1182/blood-2011-02-336537. [DOI] [PubMed] [Google Scholar]
- 61.Noris P, Favier R, Alessi M-C, et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood. 2013;122(11):1987–1989. doi: 10.1182/blood-2013-04-499319. [DOI] [PubMed] [Google Scholar]
- 62.Ovsyannikova GS, Fedorova DV, Tesakov IP, et al. Platelet functional abnormalities and clinical presentation in pediatric patients with germline RUNX1, ANKRD26, and ETV6 mutations. Haematologica. 2022;107(10):2511–2516. doi: 10.3324/haematol.2022.281340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown AL, Homan CC, Drazer M, et al. Somatic mutational landscape of hereditary hematopoietic malignancies associated with germline variants in RUNX1, GATA2 and DDX41 [abstract] Blood. 2022;140(suppl 1) doi: 10.1182/bloodadvances.2023010045. Abstract 1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perez Botero J, Oliveira JL, Chen D, et al. ASXL1 mutated chronic myelomonocytic leukemia in a patient with familial thrombocytopenia secondary to germline mutation in ANKRD26. Blood Cancer J. 2015;5:e315. doi: 10.1038/bcj.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perez Botero J, Ho TP, Hogan WJ, et al. Clinical spectrum and clonal evolution in germline syndromes with predisposition to myeloid neoplasms. Br J Haematol. 2018;182(1):141–145. doi: 10.1111/bjh.14746. [DOI] [PubMed] [Google Scholar]
- 66.Martin ES, Ferrer A, Mangaonkar AA, et al. Spectrum of hematological malignancies, clonal evolution and outcomes in 144 Mayo Clinic patients with germline predisposition syndromes. Am J Hematol. 2021;96(11):1450–1460. doi: 10.1002/ajh.26321. [DOI] [PubMed] [Google Scholar]
- 67.Churpek JE, Pyrtel K, Kanchi KL, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood. 2015;126(22):2484–2490. doi: 10.1182/blood-2015-04-641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drazer MW, Homan CC, Yu K, et al. Clonal hematopoiesis in patients with ANKRD26 or ETV6 germline mutations. Blood Adv. 2022;6(15):4357–4359. doi: 10.1182/bloodadvances.2022007211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duncavage EJ, Bagg A, Hasserjian RP, et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood. 2022;140(21):2228–2247. doi: 10.1182/blood.2022015853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feurstein S, Trottier AM, Estrada-Merly N, et al. Germ line predisposition variants occur in myelodysplastic syndrome patients of all ages. Blood. 2022;140(24):2533–2548. doi: 10.1182/blood.2022015790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.DeRoin L, Cavalcante de Andrade Silva M, Petras K, et al. Feasibility and limitations of cultured skin fibroblasts for germline genetic testing in hematologic disorders. Hum Mutat. 2022;43(7):950–962. doi: 10.1002/humu.24374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Glembotsky AC, Marin Oyarzún CP, De Luca G, et al. First description of revertant mosaicism in familial platelet disorder with predisposition to acute myelogenous leukemia: correlation with the clinical phenotype. Haematologica. 2020;105(10):e535. doi: 10.3324/haematol.2020.253070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Megy K, Downes K, Simeoni I, et al. Curated disease-causing genes for bleeding, thrombotic, and platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17(8):1253–1260. doi: 10.1111/jth.14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DiStefano MT, Goehringer S, Babb L, et al. The Gene Curation Coalition: a global effort to harmonize gene–disease evidence resources. Genet Med. 2022;24(8):1732–1742. doi: 10.1016/j.gim.2022.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stark Z, Foulger RE, Williams E, et al. Scaling national and international improvement in virtual gene panel curation via a collaborative approach to discordance resolution. Am J Hum Genet. 2021;108(9):1551–1557. doi: 10.1016/j.ajhg.2021.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Downes K, Megy K, Duarte D, et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134(23):2082–2091. doi: 10.1182/blood.2018891192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mangaonkar AA, Ferrer A, Pinto E Vairo F, et al. Clinical applications and utility of a precision medicine approach for patients with unexplained cytopenias. Mayo Clin Proc. 2019;94(9):1753–1768. doi: 10.1016/j.mayocp.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brown AL, Hiwase DK. What’s germane in the germline? Finding clinically relevant germline variants in myeloid neoplasms from tumor only screening. Leuk Res. 2020;96:106431. doi: 10.1016/j.leukres.2020.106431. [DOI] [PubMed] [Google Scholar]
- 79.Roloff GW, Drazer MW, Godley LA. Inherited susceptibility to hematopoietic malignancies in the era of precision oncology. JCO Precis Oncol. 2021;5:107–122. doi: 10.1200/PO.20.00387. [DOI] [PubMed] [Google Scholar]
- 80.Trottier AM, Cavalcante de Andrade Silva M, Li Z, Godley LA. Somatic mutation panels: time to clear their names. Cancer Genet. 2019;235-236:84–92. doi: 10.1016/j.cancergen.2019.04.065. [DOI] [PubMed] [Google Scholar]
- 81.Li MM, Chao E, Esplin ED, et al. Points to consider for reporting of germline variation in patients undergoing tumor testing: a statement of the American College of Medical Genetics and Genomics (ACMG) Genet Med. 2020;22(7):1142–1148. doi: 10.1038/s41436-020-0783-8. [DOI] [PubMed] [Google Scholar]
- 82.Miller DT, Lee K, Abul-Husn NS, et al. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet Med. 2022;24(7):1407–1414. doi: 10.1016/j.gim.2022.04.006. [DOI] [PubMed] [Google Scholar]
- 83.Jongmans MC, Kuiper RP, Carmichael CL, et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: clues for improved identification of the FPD/AML syndrome. Leukemia. 2010;24(1):242–246. doi: 10.1038/leu.2009.210. [DOI] [PubMed] [Google Scholar]
- 84.Wahlster L, Verboon JM, Ludwig LS, et al. Familial thrombocytopenia due to a complex structural variant resulting in a WAC-ANKRD26 fusion transcript. J Exp Med. 2021;218(6):e20210444. doi: 10.1084/jem.20210444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo X, Feurstein S, Mohan S, et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 2019;3(20):2962–2979. doi: 10.1182/bloodadvances.2019000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feurstein S, Luo X, Shah M, et al. Revision of RUNX1 variant curation rules. Blood Adv. 2022;6(16):4726–4730. doi: 10.1182/bloodadvances.2022008017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tudini E, Andrews J, Lawrence DM, et al. Shariant platform: enabling evidence sharing across Australian clinical genetic-testing laboratories to support variant interpretation. Am J Hum Genet. 2022;109(11):1960–1973. doi: 10.1016/j.ajhg.2022.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brnich SE, Abou Tayoun AN, Couch FJ, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3. doi: 10.1186/s13073-019-0690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Esposito D, Weile J, Shendure J, et al. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019;20(1):223. doi: 10.1186/s13059-019-1845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fayer S, Horton C, Dines JN, et al. Closing the gap: systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN. Am J Hum Genet. 2021;108(12):2248–2258. doi: 10.1016/j.ajhg.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.National Human Genome Research Institute Impact of Genomic Variation on Function (IGVF) Consortium. National Institutes of Health. 2021. https://www.genome.gov/Funded-Programs-Projects/Impact-of-Genomic-Variation-on-Function-Consortium
- 93.Baliakas P, Tesi B, Wartiovaara-Kautto U, et al. Nordic guidelines for germline predisposition to myeloid neoplasms in adults: recommendations for genetic diagnosis, clinical management and follow-up. Hemasphere. 2019;3(6):e321. doi: 10.1097/HS9.0000000000000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zaninetti C, Gresele P, Bertomoro A, et al. Eltrombopag for the treatment of inherited thrombocytopenias: a phase II clinical trial. Haematologica. 2020;105(3):820–828. doi: 10.3324/haematol.2019.223966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bastida JM, Gonzalez-Porras JR, Rivera J, Lozano ML. Role of thrombopoietin receptor agonists in inherited thrombocytopenia. Int J Mol Sci. 2021;22(9):4330. doi: 10.3390/ijms22094330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saygin C, Roloff G, Hahn CN, et al. Allogeneic hematopoietic stem cell transplant outcomes in adults with inherited myeloid malignancies. Blood Adv. 2023;7(4):549–554. doi: 10.1182/bloodadvances.2022008172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tawana K, Rio-Machin A, Preudhomme C, Fitzgibbon J. Familial CEBPA-mutated acute myeloid leukemia. Semin Hematol. 2017;54(2):87–93. doi: 10.1053/j.seminhematol.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 98.Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant. 2014;20(12):1940–1948. doi: 10.1016/j.bbmt.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hamilton KV, Maese L, Marron JM, et al. Stopping leukemia in its tracks: should preemptive hematopoietic stem-cell transplantation be offered to patients at increased genetic risk for acute myeloid leukemia? J Clin Oncol. 2019;37(24):2098–2104. doi: 10.1200/JCO.19.00181. [DOI] [PubMed] [Google Scholar]
- 100.Krutein MC, Hart MR, Anderson DJ, et al. Restoring RUNX1 deficiency in RUNX1 familial platelet disorder by inhibiting its degradation. Blood Adv. 2021;5(3):687–699. doi: 10.1182/bloodadvances.2020002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee B-C, Zhou Y, Bresciani E, et al. A RUNX1-FPDMM rhesus macaque model reproduces the human phenotype and predicts challenges to curative gene therapies. Blood. 2023;141(3):231–237. doi: 10.1182/blood.2022018193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mill CP, Fiskus W, DiNardo CD, et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood. 2019;134(1):59–73. doi: 10.1182/blood.2018893982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iida K, Tsuchiya A, Tamura M, et al. RUNX1 inhibition using lipid nanoparticle-mediated silencing RNA delivery as an effective treatment for acute leukemias. Exp Hematol. 2022;112:1–8. doi: 10.1016/j.exphem.2022.05.001. [DOI] [PubMed] [Google Scholar]