

Key Points
-
•
Heptad TF occupancy is highly dynamic across HSPC subsets and associated with cell-type–specific gene expression.
-
•
Enhancers with cell-type–specific heptad occupancy share a common grammar with respect to TF binding motifs.
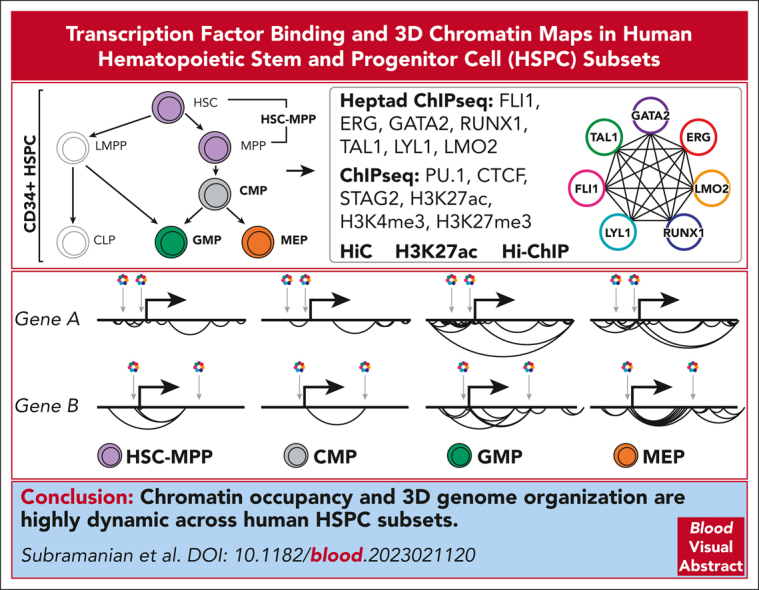
Visual Abstract
Abstract
Hematopoietic stem and progenitor cells (HSPCs) rely on a complex interplay among transcription factors (TFs) to regulate differentiation into mature blood cells. A heptad of TFs (FLI1, ERG, GATA2, RUNX1, TAL1, LYL1, LMO2) bind regulatory elements in bulk CD34+ HSPCs. However, whether specific heptad-TF combinations have distinct roles in regulating hematopoietic differentiation remains unknown. We mapped genome-wide chromatin contacts (HiC, H3K27ac, HiChIP), chromatin modifications (H3K4me3, H3K27ac, H3K27me3) and 10 TF binding profiles (heptad, PU.1, CTCF, STAG2) in HSPC subsets (stem/multipotent progenitors plus common myeloid, granulocyte macrophage, and megakaryocyte erythrocyte progenitors) and found TF occupancy and enhancer-promoter interactions varied significantly across cell types and were associated with cell-type–specific gene expression. Distinct regulatory elements were enriched with specific heptad-TF combinations, including stem-cell–specific elements with ERG, and myeloid- and erythroid-specific elements with combinations of FLI1, RUNX1, GATA2, TAL1, LYL1, and LMO2. Furthermore, heptad-occupied regions in HSPCs were subsequently bound by lineage-defining TFs, including PU.1 and GATA1, suggesting that heptad factors may prime regulatory elements for use in mature cell types. We also found that enhancers with cell-type–specific heptad occupancy shared a common grammar with respect to TF binding motifs, suggesting that combinatorial binding of TF complexes was at least partially regulated by features encoded in DNA sequence motifs. Taken together, this study comprehensively characterizes the gene regulatory landscape in rare subpopulations of human HSPCs. The accompanying data sets should serve as a valuable resource for understanding adult hematopoiesis and a framework for analyzing aberrant regulatory networks in leukemic cells.
Subramanian and colleagues report on a map of genome-wide chromatin contacts of canonical transcription factor complexes that elucidates the determinants of cell-type–specific gene expression and maturation. This represents a valuable resource database for analysis of the regulatory networks of normal hematopoietic stem cell maturation and their disruption in leukemic cells.
Introduction
Hematopoietic stem cells (HSCs) maintain production of circulating blood cells via their capacity to either self-renew or differentiate into mature cell types.1 The most primitive HSCs have multilineage potential but give rise to progenitor cells with increasing lineage restriction. Although single-cell analyses have suggested that differentiation occurs over a continuum rather than in discrete leaps,2, 3, 4 relatively pure populations which correspond to intermediate progenitor stages can be prospectively isolated based on cell-surface markers.4
Changes in cell identity across differentiation trajectories are directly related to altered transcriptional programs5 which are controlled by lineage-specific gene regulatory networks (GRNs).6 At the simplest level, GRNs comprise genes, their associated regulatory elements (promoters and cis-regulatory elements [CREs], such as enhancers), and transcriptional regulators, including transcription factors (TF), which bind these elements.7,8 Accessibility of regulatory elements is controlled by various chromatin modifications, and the DNA sequence of such elements at least partially determines which TFs can bind.9,10 Further control is imposed by chromatin organization into topologically associated domains.11 Interactions between promoters and their CREs, mediated by chromatin loops and complexes of transcriptional regulators, modulate GRNs and therefore cell identity.12
We previously showed that 7 TFs (heptad: FLI1, ERG, GATA2, RUNX1, TAL1, LYL1, and LMO2), all known regulators of healthy and leukemic haematopoiesis,13, 14, 15, 16, 17, 18, 19, 20 bind combinatorically in bulk CD34+ hematopoietic stem and progenitor cells (HSPCs)21 and leukemias.22, 23, 24 In healthy HSPCs, heptad combinatorial binding occurs at regulatory regions associated with genes involved in stem-cell maintenance and function and also at heptad CREs such that heptad genes form a highly interconnected regulatory circuit.21,25 However, study of GRNs in HSPCs is hindered by heterogeneity within the CD34+ population and lack of experimental evidence linking promoters to distal regulatory elements.
To address these issues and further our understanding of heptad centered GRNs in blood development, we sorted CD34+ HSPCs into HSC/multipotent progenitor (MPP, collectively HSC-MPP), common myeloid progenitor (CMP), granulocyte macrophage progenitor (GMP), and megakaryocyte erythrocyte progenitor (MEP) cell types. We then used chromatin immunoprecipitation followed by sequencing (ChIP-seq) targeting 10 TFs (heptad, PU.1, CTCF, and STAG2) and 3 histone modifications (H3K27ac, H3K4me3, and H3K27me3), Hi-C, and H3K27ac-HiChIP in each of these cell types to chart the regulatory landscape of human HSPC differentiation. Combinatorial binding of heptad TFs was observed in all sorted populations, although specific patterns of chromatin occupancy differed among cell types. Heptad promoter looping to putative enhancers was variable across cell types, and in many cases combinatorial binding was observed at CREs in immature cells before formation of loops in more mature progenitors. Genome-wide occupancy of heptad TFs was also variable across cell types, with distinct sets of CREs enriched for heptad binding in MEP compared with that in GMP. This variation was at least partially due to sequence motifs in the CREs, with motif composition sufficient to predict cell type with high sensitivity and specificity.
Methods
Mobilized peripheral blood was collected with patient consent in accordance with the Declaration of Helsinki and used with institutional ethics approval ref:08/190 from the South-Eastern Sydney Local Health District.
Cell sorting, preparation of nuclei, and ChIP were performed essentially as described4,22,26 (detailed in supplemental Methods, available on the Blood website). HiC and HiChIP libraries were generated using the Arima Genomics HiC+ kit (Arima catalog no. A101020).
Bioinformatic analysis used standard pipelines for ChIP21 and HiC/HiChIP data.27 Machine learning models were trained using XGBoost.28 A UCSC browser session is provided at http://genome.ucsc.edu/s/PimandaLab/Heptad_Regulome. We also provide a tool for data exploration (http://unsw-data-analytics.shinyapps.io/CD34_Heptad_Regulome).
Details of standard experimental and analysis techniques are provided in the supplemental Methods.
Results
Genome-wide heptad factor binding in HSPCs
Primary mobilized human CD34+ HSPCs were sorted (Figure 1A; supplemental Figure 1A), purity checked with colony assays (supplemental Figure 1B), and fixed for downstream assays (Figure 1B; supplemental Table 1). Cell populations were chosen to span the trajectory from early multipotent stem cells (HSC-MPP) to progenitors committed to the myeloid (GMP) or erythroid (MEP) lineages.
Figure 1.

Genome-wide patterns of heptad TF binding in fractionated primary human HSPCs. (A) Human MNCs were isolated from granulocyte-colony stimulating factor–stimulated donors or patients with a nonhematologic malignancy before being enriched for CD34 expression using magnetic-activated cell sorting (MACS) and further subfractionated into individual stem and progenitor cells based on surface marker expression using fluorescence-activated cell sorting (FACS) (colored cells are those studied in this manuscript). (B) The workflow and analysis pipeline followed for ChIPmentation and ChIP-seq experiments. (C) UCSC browser track at the RUNX1 locus (GRCh38 chr21:34,627,969-35,209,177) showing the reads per kilobase of transcript, per million mapped reads (RPKM)-normalized signal from FLI1, ERG, GATA2, RUNX1, TAL1, LYL1, and LMO2, along with H3K4me3, H3K27ac, H3K27me3, immunoglobulin G (IgG) (control), and publicly available RNA-seq tracks (GSE75384) for the 4 cell types. Full UCSC browser tracks are available http://genome.ucsc.edu/s/PimandaLab/Heptad_Regulome. (D) Characterization of identified peaks. Number of TF peaks were identified by macs2 (P value ≤ 1e−5) and their overall distribution along the genome (as percentages of total peaks identified) is shown. Each peak was assigned as either promoter-like (proximal [orange] or adjacent [blue], based on its distance from the TSS), intragenic [green], or intergenic [red], and enrichment (fraction of peaks containing that motif) calculated for the known ETS, GATA, RUNX, and E-Box motifs.
High-quality ChIP data were obtained for all heptad factors (Figure 1C-D; supplemental Table 2; http://genome.ucsc.edu/s/PimandaLab/Heptad_Regulome). Total peak numbers were highly variable between heptad factors and across cell types (Figure 1D). We observed cell-type–specific trends consistent with known expression patterns and biology. For example, ERG peaks were most abundant in HSC-MPPs, consistent with its role in maintaining the stem cell state,29,30 whereas TAL1 peaks were most abundant in MEP, consistent with its role in erythroid development (Figure 1D). Distributions of TFs across genome features were generally conserved across cell types but differed among heptad factors. For example, FLI1, ERG, and RUNX1 peaks were located at both promoter and nonpromoter regions (Figure 1D), whereas GATA2, TAL1, LYL1, and LMO2 peaks were predominantly located at nonpromoter regions. TAL1 peak distribution in MEP was unique, with many peaks found at intergenic regions. Motif enrichment analysis showed ETS (GGAA) and E-Box (CANNTG) motifs in TF-occupied regions from all factors. FLI1, ERG, and RUNX1 peaks were highly enriched for the ETS motif, whereas GATA2, TAL1, LYL1, and LMO2 peaks showed additional enrichment for the GATA motif (GATA), particularly for GATA2 and TAL1 in MEP. Consistent with observations in bulks HSPCs21 enrichment of RUNX1 motifs was minimal. Overall, we observed not only conserved patterns of heptad binding, but also distinct differences between factors and across cell types, consistent with dynamic remodeling of the heptad network across the HSPC differentiation trajectory.
Combinatorial binding of heptad TFs is cell-type–specific
We previously described combinatorial binding of heptad factors in bulk human HSPCs21 and now extend these observations to specific cell populations. We quantified ChIP peaks containing all possible combinations of ≥2 heptad factors (Figure 2Ai-ii), and evaluated the probability of these occurring by random chance in each cell type (Figure 2Aiii). As in bulk HSPCs,21 combinatorial binding of all 7 factors was the most significant event in any of the 4 cell types (z scores for all 7 factors; HSC-MPP: 8199.94, CMP:6314.92, GMP:3543.93, and MEP:1877.48). Pairwise combinations had low significance scores, with exceptions such as ERG/RUNX in HSC-MPPs. Higher order complexes generally had higher significance scores, with 5 of 7 possible 6-factor complexes showing highly significant binding in at least 1 cell type. Specific combinations broadly matching known TF function were also observed. For example, 5- or 6-factor combinations lacking GATA2, TAL1, or both, had low significance scores in MEPs (Figure 2A, stars). We also asked whether PU.1 showed combinatorial binding with heptad factors. As previously observed,25 co-binding of single heptad factors with PU.1 was minimal (supplemental Figure 2A), whereas addition of PU.1 to 6 of 7 TF combinations modestly accentuated existing patterns (supplemental Figure 2B). Overall, we find that combinatorial binding of heptad TFs is a general feature of stem and progenitor cells, with some combinations restricted to specific cell types.
Figure 2.

Combinatorial binding of heptad TFs is cell-type specific. (A) A composite graph with 3 components: (i) number of combinatorial binding peaks identified in the 4 cell types, for (ii) combinations of 2, 5, 6, and 7 heptad factors and (iii) heatmap showing z scores for the combinations presented in panel Aii. Star indicates combinations lacking GATA2 and/or TAL1. (B-C) UCSC browser tracks showing RPKM-normalized signal tracks of the heptad factors, H3K4me3, H3K27ac, H3K27me3, RNA-seq (public data: GSE75384), and IgG (control) in HSC-MPP, CMP, GMP, and MEP (left to right), at (B) the GATA1 locus (GRCh38 chrX:48,724,037-48,839,866), a gene vital for erythroid lineage specification, and at (C) the MPO locus (GRCh38 chr17:58,238,087-58,348,896), a gene specific to the monocytic lineage.
Given the cell-type specificity of some TF combinations we predicted that dynamic formation of TF complexes might play a role in priming CREs for subsequent activation. We explored heptad factor binding at promoter regions of 2 lineage-specific genes: the erythroid regulator GATA1 and the monocyte gene MPO, neither of which showed heptad binding in HSC-MPPs. At the GATA1 promoter (Figure 2B; yellow region), GATA2, RUNX1, TAL1, LYL1, and LMO2 binding was observed in CMP. As cells transitioned to MEP, binding peaks became more prominent and now included FLI1 and ERG. However, there was essentially no heptad binding at the GATA1 promoter in GMP. At the MPO promoter (Figure 2C; yellow region), FLI1, ERG, TAL1, LYL1, and LMO2 binding was observed in CMP. Binding peaks became more prominent as cells transitioned to GMP, with RUNX1 also bound, but no TF binding was observed at the MPO promoter in MEP. Taken together our data suggest that distinct patterns of heptad TF binding may prime the genomic landscape of human blood stem cells toward either an erythroid or myeloid fate.
Heptad regulatory circuits are remodeled during myeloid progenitor development
Genes encoding heptad TFs form a densely interconnected regulatory circuit in bulk HSPCs,21 and chromatin accessibility at heptad gene CREs is sufficient to predict blood-cell identity.23 To better understand genome organization and connectivity at heptad loci during HSPC development we performed HiC and H3K27ac HiChIP experiments on HSC-MPP, CMP, GMP, and MEP. Although the majority of genome compartments remained stable across all cell types, some compartments underwent B to A or A to B switching upon transition from CMP to lineage-committed progenitor (supplemental Figure 3Ai). Notably, some regions underwent compartment switching only in GMP, with corresponding changes in H3K27ac signal (supplemental Figure 3Aii-iii). Topologically associated domain boundaries were highly conserved between cell types (supplemental Figure 3B), and HiC contact matrixes around heptad gene loci were also highly similar (supplemental Figure 4). Global CTCF and STAG2 binding was also conserved across cell types (supplemental Figure 5). Overall, consistent with previous reports,31 we observed minimal variation in high-level genome organization among HSPC subpopulations.
H3K27ac HiChIP experiments generated thousands of significant interactions with false discovery rate ≤0.01 (HSC-MPP: 26210, CMP: 8170, GMP: 43448, and MEP: 32773; supplemental Table 3). We focused on loops in which at least 1 interacting region was annotated as a promoter (P). Because this experiment enriched fragments with the H3K27ac active enhancer mark, we consider looped CREs as putative enhancers (E). We filtered promoter-enhancer (P-E) loops based on presence of a transposase-accessible chromatin with sequencing (ATACseq) peak at the distal enhancer and integrated these with ChIP-seq data to create regulatory network maps at each heptad gene locus for each cell type.
Promoter-enhancer loops corresponding to the heptad spanned wide genomic regions; ∼500 kb for FLI1, GATA2, and TAL1 and from 1 to 2 Mb for ERG, RUNX1, LYL1, and LMO2 (Figure 3Ai-ii; supplemental Figure 6). Overall, we detected multiple putative enhancers for the heptad genes ERG (−610/−410/−230/+85/+88/+191/+1200), FLI1 (+27/+32/+64), GATA2 (−123/−92/+4), RUNX1 (−880/+22/+100/+110/+141/+161), TAL1 (−101/−82/−25/+0.5/+14/+45), LYL1 (−744/−50/+165/+310), and LMO2 (−570/−100/−67/−61/−51/−23/−22/−15/−12) (Figure 3Ai-ii; supplemental Figure 6; supplemental Table 4). Some regions have been described in humans and/or in mice 17,21,32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 42, 43, 44, 45 whereas others were novel (supplemental Table 4). Two known heptad enhancers were not directly looped to their corresponding promoter (FLI1-15, GATA2-117; supplemental Figure 6Ai,Bi),21,44 although we did observe looping between GATA2-117 and other putative enhancers in MEPs (supplemental Figure 6Bi). Furthermore, looping at heptad genes generally increased in complexity in GMP and MEP compared to HSC-MPP, with the sparsest looping observed in CMP (Figure 3Ai; supplemental Figure 6). This additional complexity often included extensive looping to CREs not directly connected to promoters, suggesting that heptad gene expression is fine-tuned by highly interconnected cell-specific enhancer communities (eg, TAL1 locus in MEP [supplemental Figure 6Di], LYL1 locus in GMP and MEP [supplemental Figure 6Ei]).
Figure 3.

Heptad regulatory circuits are remodeled during myeloid progenitor development. (A) Stepwise identification of potential regulatory regions interacting with the ERG promoter. (i) Raw HiChIP contact matrix, CTCF, H3K4me3, H3K27ac, IgG, RNA-seq, and significant H3K27ac HiChIP interactions (false discovery rate [FDR] ≤ 0.01) at the ERG locus (GRCh38 chr21:37370238-39198738). The ERG promoter is indicated by the green arrow (only those HiChIP interactions where both interacting ends were found at the given locus are shown). (ii) Magnified view of the ERG locus, with regulators identified to loop to the ERG proximal promoter shown as red triangles. (iii) FLI1, ERG, GATA2, RUNX1, TAL1, LYL1, and LMO2 peaks at the defined regulators in each individual cell type. The peaks shown are RPKM-normalized and white boxes indicate presence of a computationally called ChIP-seq peak at the specific region. (B) Summary plot of gene regulatory interactions across the heptad genes. (i) Individual heptad gene loci with identified regulators indicated by red markers. (ii) Dot plots showing regulatory regions as rows and the 4 cell types as columns, with size of the dot indicating number of heptad factors bound and black color indicating the presence of an active regulatory link to the promoter (using H3K27ac HiChIP). Promoters are underlined. (iii) Bar plots with individual replicates showing average log2 counts of relevant heptad gene expression in the 4 cell types (GSE75384).
Most directly looped elements showed heptad factor binding in at least 1 cell type (Figure 3Aiii; supplemental Figure 6). Some core CREs showed extensive heptad binding in all cell types, including known and functionally validated enhancers, such as ERG+85, GATA2+4, RUNX1+22, and TAL+45 (Figure 3Aiii; supplemental Figures 6Biii,Ciii,Diii; supplemental Table 4), plus novel CREs, such as ERG-410 and LMO2-570 that can now be linked to heptad genes (Figure 3Aiii; supplemental Figure 6Fiii). Integration of HiChIP and ChIP data at heptad gene loci showed diverse patterns of looping and TF binding across the 4 cell types (Figure 3Bi-ii). At the ERG locus, the ERG+85 enhancer was linked to the proximal promoter in HSC-MPP, GMP, and MEP, whereas other ERG elements showed promoter looping in GMP and/or MEP. (Figure 3Bii). Furthermore, heptad TF binding often occurred in HSC-MPPs with subsequent promoter looping of that element in more differentiated cell types (eg, GATA2−123, TAL1+45, and LMO2−570; Figure 3Bii). However, these epigenetic changes did not directly correspond to the steady state transcriptional output of heptad genes, which was relatively stable across the 4 cell types (Figure 3Biii).
Analysis of bulk HSPCs found that heptad genes regulate themselves and each other via a densely interconnected autoregulatory circuit.21 We used our expanded set of heptad CREs to construct network connectivity maps for each cell type (supplemental Figure 7). These maps show that although the transcriptional output of the heptad genes was relatively stable across HSC-MPP, CMP, GMP, and MEP, there were cell-type–specific differences in cis- and trans-regulatory mechanisms by which stable expression was maintained. Overall, our data set allowed us to observe extensive remodeling of the regulatory connections within and among individual heptad genes during hematopoiesis and increases our understanding of the complex network regulating heptad genes during hematopoiesis.
Heptad TFs and regulation of lineage-specific gene expression
We next asked whether heptad factor chromatin occupancy was associated with cell-type–specific transcriptional output. We identified genomic regions with cell-type–enriched binding of at least 2 heptad TFs (differentially enriched for heptad [DEH]) and linked these to genes using HiChIP data (genes associated with DEH [DEHG]) (Figure 4A; supplemental Table 5A). Approximately half of the regions were promoter-like (up to 10 kb upstream of a transcription start site [TSS]). To characterize candidate regulatory elements and their associated genes, we conducted gene set enrichment analysis, ingenuity pathway analysis, and single-cell analysis. DEHG in HSC-MPP/GMP/MEP had greater expression in their respective cell types compared to other cell types (Figure 4B) including previously reported lineage-specific genes such as SLAMF1 and MPO in GMPs46,47 and GATA1 and KLF1 in MEPs.48, 49, 50 Furthermore, genes differentially bound in GMP were enriched for pathways linked to myelopoiesis and granulopoiesis (supplemental Figure 8A) whereas genes linked to differentially bound regions in MEP were enriched for pathways linked to erythropoiesis (supplemental Figure 8B). Only 16 HSC-MPP–specific genes were identified, precluding pathway analysis. However, these genes included stem-cell regulators,51 such as HOXB1, HOXB2, and HOXB4 (supplemental Table 5A).
Figure 4.

The role of heptad TFs in regulating lineage-specific gene expression. (A) Schematic of the bioinformatic strategy used to derive regions showing differential heptad factor binding: (i) Candidate regulatory elements (REs) with binding of at least 2 heptad factors were chosen in the 4 cell types; and (ii) DiffBind was used to filter for regions showing differential enrichment for heptad factors with an FDR <0.05. To perform DiffBind analysis only HSC-MPP (HSC), GMP, and MEP populations were chosen. (iii) These DEH regions were linked to genes either directly (present across a 10 kb promoter region) or indirectly (distal links using significant [FDR <0.01] H3K27ac- HiChIP interactions), and (iv) used as input for multiple characterization assays. (B) Gene set enrichment analysis (GSEA) plots showing enrichment of derived gene sets in pairwise gene expression comparisons: (i) DEHGHSC (genes linked to DEH regions in HSC-MPP) enriched in HSC-MPP with respect to GMP, (ii) DEHGGMP enriched in GMP with respect to HSC-MPP, (iii) DEHGHSC enriched in HSC-MPP with respect to MEP, and (iv) DEHGMEP enriched in MEP with respect to HSC-MPP. (C) Scoring cell-specific DEHGs along a (i) single-cell expression map reveals localized enrichment of expression: (ii) DEHGHSC, (iii) DEHGGMP, and (iv) DEHGMEP. ES, enrichment score; NES, normalized enrichment score; q, FDR q value from GSEA.
We also explored expression of DEHG in single cells across hematopoietic differentiation (Figure 4Ci).3 HSC-MPP–associated DEHG (DEHGHSC) were enriched in cells annotated as HSC clusters (Figure 4Cii), whereas DEHGGMP were enriched in monocyte clusters (Figure 4Ciii) and DEHGMEP in erythroid lineage cells (Figure 4Civ). Together these data support our hypothesis that heptad occupancy at regulatory elements can be linked to cell-specific transcriptional output.
Heptad TF binding at regulators of genes crucial for myeloid and erythroid cell development
We next asked whether we could detect specific patterns of heptad occupancy at known lineage-specific genes with roles in mature monocyte and granulocyte maintenance (myeloid cell development), at genes linked to erythroid cell development and heme metabolism (erythroid cell development), or at genes linked to stem cell function (supplemental Table 5B). From these lists we focused on genes whose promoters were looped to a putative enhancer in any of our HiChIP data sets. We identified 40 P-E pairs from the myeloid genes, 91 from the erythroid genes, and 81 from the stem cell genes (supplemental Table 5B) and used k-means clustering of TF binding signals to compare heptad occupancy patterns at each P-E pair in each cell type. We observed variable TF binding across associated promoter and enhancer regions. Genes in cluster 1 (C1) showed TF enrichment at promoters, genes in cluster 2 (C2) showed TF enrichment at enhancers, and genes in clusters 3 and 4 (C3 and C4) had TF enrichment at both promoters and enhancers (Figure 5A-B; supplemental Figure 9A). Furthermore, several TF-specific observations were evident. First, ERG occupancy at stem cell, myeloid, and erythroid genes was generally highest in HSC-MPPs and to a lesser extent GMPs, consistent with its role in maintaining stem cells (supplemental Figure 9B; supplemental Table 6).23,29,30 Second, FLI1 and RUNX1 were bound across both myeloid and erythroid gene–associated regions across all differentiation stages (supplemental Figure 9C). Third, GATA2, LYL1, and LMO2 have increased occupancy at myeloid- and erythroid-specific regulatory regions during lineage commitment (supplemental Figure 9D; supplemental Table 6). Fourth, high TAL1 occupancy was observed in MEPs and their precursor CMPs, and this occurred at regions linked to erythroid genes and regions linked to myeloid genes (supplemental Figure 9E; supplemental Table 6). Binding in CMPs may indicate a role for TAL1 in priming regulatory regions and recruiting activators or repressors in downstream cell types. Finally, there was a general pattern of heptad TF occupancy at promoters and enhancers of lineage-specific genes even in the earliest stem cells, suggesting that heptad factors bind lineage-specific regulatory regions before lineage commitment and subsequent differentiation.
Figure 5.

Heptad TFs at promoters and distal regulators of genes crucial for myeloid and erythroid cell development. (A) Genes associated with myeloid development. Left: k-means clustered heatmaps of TF binding intensity at promoters and distal regulatory regions. Profile plots show normalized signal for each TF in each cell type at the regions depicted in the heatmap. Right: z score normalized heatmaps of RNA-seq counts (GSE75384) for the corresponding gene in each cell type. White rows are genes with no expression values in the data set. (B) Genes associated with erythroid development. Left: k-means clustered heatmaps of TF binding intensity at promoters and distal regulatory regions. Profile plots show normalized signal for each TF in each cell type at the regions depicted in the heatmap. Right: z score normalized heatmaps of RNA-seq counts (GSE75384) for the corresponding gene in each cell type.
To further explore whether heptad binding in differentiated cell types is initiated in early stem cells, we identified ChIP-seq peaks bound by a single heptad factor in HSC-MPPs and compared TF binding density at these regions across cell types. Regions with peaks in HSC-MPPs had additional heptad factors bound in committed progenitors. For example, peaks bound by ERG in HSC-MPPs (supplemental Figure 10A) showed prominent binding of FLI1 and RUNX1 in GMPs, and binding of all heptad factors, except for ERG, in MEPs (supplemental Figure 10B-C). Furthermore, regions bound by single heptad TFs in HSC-MPPs were subsequently bound by PU.1 in dendritic cells or GATA1 in proerythroblasts (supplemental Figure 10D). Overall, these data support dynamic assembly of heptad factor complexes at sites subsequently occupied by lineage-determining TFs. Further experimentation will resolve whether heptad factors actively prime CREs for use in more mature cell types.
Regulatory regions with cell-type–specific heptad occupancy have distinct epigenetic features
To better understand the underlying mechanisms regulating dynamic heptad occupancy during blood formation, we used our ChIP data sets to annotate and cluster ∼85 000 regions previously shown to be accessible in any of our 4 cell types.4 The resulting Uniform Manifold Approximation and Projection (UMAP) of the accessibility landscape could be segmented into 13 clusters (Figure 6A). Clusters 1 to 3 had characteristics of promoters, including high H3K4me3 signal (Figure 6B). We could further classify cluster 1 as active promoters (H3K4me3 and H3K27ac), cluster 2 as bivalent promoters (H3K4me3 and H3K27me3 enriched), and cluster 3 as active promoters which were also bound by CTCF. These classifications aligned with ChromHMM annotation of the same regions (Figure 6C).52 Cluster 4 was enriched for CTCF alone (Figure 6B) and may contain regions involved in 3-dimensional genome organization. Clusters 5 to 11 could be broadly characterized as nonpromoter regulatory regions which again aligned with their ChromHMM annotations (Figure 6C). Most of the variations among these regions mapped to variable TF occupancy in specific cell types (supplemental Figure 11A), which was particularly pronounced in clusters 8 to 11 which also showed cell-type–specific changes in chromatin accessibility (Figure 6B). For example, cluster 9 and 10 showed high accessibility in MEP compared to GMP, accompanied by high TAL1 and LYL1 occupancy (supplemental Figure 11A), suggesting that regions in these clusters may function in the erythroid lineage.
Figure 6.

Regulatory regions with cell-type-specific heptad occupancy have distinct epigenetic features. (A) A Uniform Manifold Approximation and Projection (UMAP) depicting the result of clustering 85 100 accessible regions in HSPCs annotated with ChIPmentation/ChIP-seq signal strengths using the Louvain algorithm. (B) Individual violin plots of log normalized signal derived from ATAC, 3 histone marks (H3K27ac, H3K4me3, and H3K27me3), and CTCF, accompanied by a bar plot showing the number of regions in each cluster. Intercluster signal variability allows annotation of individual clusters based on their regulatory potential. (C) UMAPs overlaid with ChromHMM annotation of 85 100 individual regions show striking similarity to annotations shown in Figure 6B. (D) UMAPs colored based on log2 fold change of binding of the heptad TFs in pairwise comparisons between GMP and MEP. MEP- and GMP-specific enrichment of TF binding is identified, and borders demarcated by dashed lines: black (enriched in MEP) or gray (enriched in GMP). (E) Signal of PU.1 in dendritic cells (DC) (GSE58864) across the clustered regions. PU.1 signal enrichment in dendritic cells mirrors heptad factor enrichment patterns in GMP. (F) Signal of GATA1 in proerythroblasts (ProE) (GSE36985) across the clustered regions. GATA1 signal enrichment in proerythroblasts mirrors heptad factor enrichment patterns at these regions in MEP.
Visualizing TF signal across the accessibility landscape revealed variable occupancy patterns across cell types (supplemental Figure 11B). FLI1, RUNX1, and ERG had similar distributions across all cell types, with highest FLI1 and RUNX1 occupancy in CMP and GMP, and ERG occupancy reduced in MEP. LYL1 and LMO2 had similar distributions with enrichment in clusters 5, 6, or 7 in GMP and clusters 9 or 10 in MEP (supplemental Figure 11B). TAL1 was also enriched in clusters 9 or 10 in MEP with lower binding in other cell types, whereas PU.1 was enriched in cluster 5, 6, or 7 in CMP and GMP (supplemental Figure 11B). GATA2 had a unique occupancy pattern with enrichment in cluster 5 or 6 in HSC-MPP and GMP and additional occupancy in cluster 9 in CMP and MEP (supplemental Figure 11B) which may reflect distinct roles of GATA2 in early hematopoiesis and subsequent erythroid specification.53
We then made pairwise comparisons of TF binding signals in GMP vs MEP, HSC-MPP vs GMP, and HSC-MPP vs MEP (Figure 6D; supplemental Figure 11C). Comparing GMP (Figure 6D, green) to MEP (Figure 6D, orange), there were distinct zones of TF enrichment in each cell type (Figure 6D, gray dotted line [GMP], black dotted line [MEP]) except for TAL1 which showed no region of enrichment in GMP. To confirm that regions enriched for heptad TF binding in GMP and MEP represent lineage-specific regulators, we mapped ChIP-seq signal from lineage-defining TFs in 2 mature cell types (Figure 6E and F). Regions with high heptad occupancy in GMP showed similar occupancy of PU.1 in dendritic cells (Figure 6E),54 whereas regions with high heptad occupancy in MEP showed similar occupancy of GATA1 in proerythroblasts (Figure 6F).6 Taken together these data show that regulatory regions with heptad occupancy in progenitor populations are regions occupied by lineage-specific TFs in more mature cells.
Cell-type specificity of regulatory elements is encoded in the underlying motif composition
TFs bind DNA through consensus binding motifs whose sequence and relative locations in each regulator determine which TF complexes can potentially bind. To better understand enhancer features underpinning lineage-specific TF occupancy, we selected regions with differential accessibility and TF occupancy in HSC-MPP (Figure 7Ai, 3992 regions), GMP (Figure 7Bi, 4395 regions), and MEP (Figure 7Ci, 3469 regions) and developed machine learning models to predict cell-type associations based on DNA sequence motifs. All models showed high sensitivity and specificity to associate regions with cell types (Figure 7Aii,Bii,Cii; supplemental Table 7). To understand how specific motifs contribute to assigning regulatory elements to cell types, we calculated SHapley Additive exPlanations (SHAP) values for each cell type (Figure 7Aiii,Biii,Ciii; supplemental Table 8). Distinct combinations of motifs were found to contribute to each model, many of which fit the expected profile for cell-specific regulatory regions. For example, ETS motifs had positive SHAP values in the GMP model but negative values in the MEP model, consistent with known roles for ETS factors, such as PU.1, in driving myeloid differentiation, whereas GATA motifs had positive SHAP values in HSC-MPP and MEP models but negative values in the GMP model, consistent with GATA factors, such as GATA1, driving the erythroid lineage.55,56 Motifs not corresponding to heptad factors had high SHAP scores in all models, consistent with heptad factors binding at lineage-specific enhancers in the context of larger regulatory protein complexes. We explored expression of nonheptad TFs corresponding to motifs predicted to positively affect SHAP scores for each model (supplemental Table 8). GATA3 (HSC-MPP model), PU.1, and SPI-B (GMP model), and GATA1 (MEP model) had lineage-specific expression patterns consistent with known expression and activity of these factors49,53,56,57,58,59,60 (supplemental Figure 12A-C). All other motif-associated TFs had minimal expression variation across cell types.
Figure 7.

Cell-type specificity of regulatory elements is encoded in the underlying motif composition. (A) (i) UMAP representation of ATAC-seq regions in CD34+ cells (gray) with heptad TF bound HSC-MPP specific regions colored in purple. (ii) An XGBoost machine learning model was trained and tested with motif counts from a mixture of regions specified in panel Ai and background regions, to predict cell type with high accuracy. The receiver operating characteristic (ROC) curve shows the predictive performance of the constructed model to predict HSC-MPP specific regions. (iii) A beeswarm plot depicting the top 12 representative motifs in HSC-MPP specific regions, ranked based on their absolute importance in contributing to the predictive model. Each row shows the motif (and canonical TF family if known), and the corresponding SHAP values for the cell type in question (right) and the others (left). The feature count indicates the normalized motif counts with a range of 0 to 1. (B) (i) UMAP representation of ATAC-seq regions in CD34+ cells (gray) with heptad TF bound GMP specific regions colored in green. (ii) ROC curve showing the performance of the model to predict GMP specific regions. (iii) A beeswarm plot depicting the top 12 representative motifs in GMP specific regions, ranked based on their absolute importance in contributing to the predictive model. (C) (i) UMAP representation of ATAC-seq regions in CD34+ cells (gray) with heptad TF bound MEP specific regions colored in orange. (ii) ROC curve showing the performance of the model to predict MEP specific regions. (iii) A beeswarm plot depicting the top 12 representative motifs in MEP specific regions, ranked based on their absolute importance in contributing to the predictive model.
Finally, we used our models to classify functional HSC regulatory elements identified through analysis of γ-retroviral integration sites (γRV-IS) in patients undergoing gene therapy.61 γRV-IS had similar features to HSC-MPP–specific regions (supplemental Figures 7A and 12D). Importantly, our models scored γRV-IS as most likely to be HSC-MPP–specific, confirming the validity of our analyses (supplemental Figure 12E).
Discussion
In this study we explored genome-wide dynamics of chromatin occupancy and structure in 4 cell types along the HSC to myeloid/erythroid differentiation axis. Analysis of putative CREs with direct looping to heptad promoter regions revealed greater regulatory complexity than previously appreciated. Previous work identified a set of 9 regions with combinatorial heptad binding in human HSPCs whose relative accessibility is sufficient to predict cell identity.23 This study identified >30 putative CREs directly looped to heptad promoters in at least 1 cell type (Figure 3B). Although most previously described heptad regulatory elements have been tested in functional assays (supplemental Table 4), further validation is required to precisely understand individual and cooperative roles of specific CREs for each heptad gene.62,63 Surprisingly, some previously identified enhancers were not directly looped to heptad promoters in this analysis. For example GATA2−117, an enhancer dysregulated in inv(3) acute myeloid leukemia,64,65 was only indirectly looped to the GATA2 promoter, and therefore excluded from our network model (supplemental Figure 6B). However, lack of direct looping does not preclude a role for this enhancer in regulating GATA2. Indeed, previous studies have found that enhancer-enhancer interactions can stabilize and amplify TF binding, and the resulting enhancer communities can drive and coordinate cell-type–specific gene expression.66,67 Consistent with this model, we observed a trend of increasingly complex chromatin looping interactions involving heptad promoters and putative enhancers as cells committed to either erythroid or myeloid lineages (Figure 3B). Increased looping may restrict committed progenitors from accessing alternate fates or participate in shutting off stem cell programs. Intriguingly, the stem cell gene ERG23,29,30 acquired loops to a +1200 kb region in MEPs (Figure 3Ai), whereas increased long range looping and heptad binding occurred at the +45 enhancer of the erythroid gene TAL114,68 (supplemental Figure 6Di). Thus, increased looping can be associated with genes subsequently down or upregulated. Further experimentation is needed to fully understand rapidly increased chromatin looping in committed progenitors.
Motif-based analysis showed that heptad-occupied CREs contain information encoding cell-type specificity. Among motifs that contributed to cell-type specificity, we found motifs for additional factors including STAT, interferon-regulatory factor, and homeodomain TFs (Figure 7; supplemental Table 8). Although our focus was on heptad factors, it is clear that additional regulators co-occupy CREs and contribute to enhancer specificity.69 To date these studies have focused on transcriptional cofactors with broad function, such as P53 and MED14,70 and further work is required to understand how heptad factors, general cofactors, and intrinsic features of enhancers and promoters cooperate to regulate transcriptional programs throughout hematopoiesis.
Numerous bone-marrow failure syndromes have an underlying germ line genetic cause and may be amenable to gene therapy approaches targeting primitive HSCs to enable long-term benefits. However, ectopic gene expression may lead to lineage skewing and other unwanted outcomes. Cell-type–specific enhancers are conserved across evolution and can potentially be used to restrict transgene expression.6,71,72 Indeed, erythroid-specific GATA1 enhancers can drive restricted expression of GATA1 to rescue erythroid differentiation in Diamond-Blackfan anemia.73 However, identifying enhancers with appropriate spatiotemporal activity is not trivial. Although experimental techniques for massively parallel interrogation of enhancer activity and specificity have been developed,63,74 our data provide a catalog of putative enhancers that could fast track functional studies.
GRNs are significantly perturbed in multiple acute myeloid leukemia subtypes,8,75 often as a result of specific translocation events, including RUNX1-RUNX1T176,77 and inv(3).64,65 Recent sequencing studies found that small structural variants (SVs) can mediate enhancer hijacking and subsequent oncogenic transformation.63,78 However, understanding the effects of SVs requires significant knowledge of cell-specific enhancers; our data set provides extensive annotation of regulatory regions that could be used to guide functional experiments exploring the effects of specific SVs.
Although extensive, our data set is limited to myeloid cell types. Sorted HSPCs, particularly CMPs, are also inherently heterogenous79, 80, 81 which may limit our ability to resolve differences among adjacent cell types. Furthermore, although capable of reconstituting patients who underwent transplantation, mobilized cells may have subtle epigenomic differences to their unstimulated counterparts. Finally, even using low cell input techniques, our data sets represent averaged binding and looping probabilities captured across a shifting epigenetic landscape, rather than truly capturing GRNs, which operate at the level of single cells.7 Nonetheless, our data set represents the most extensive epigenetic characterization of primary human blood progenitors to date and provides important insights for future studies.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors acknowledge members of the Pimanda group for assistance in thawing apheresis bags. The authors thank the South Australian Cancer Research Biobank and all patients who donated samples.
J.A.I.T. was supported by the Anthony Rothe Memorial Trust. B.G. acknowledges funding from Wellcome (206328/Z/17/Z). J.W.H.W was supported by the Research Grants Council, HK (17100920). J.E.P. was supported by grants from the National Health and Medical Research Council of Australia (GNT1139787, GNT2011627, MRF1200271), a translational program grant from the Leukemia and Lymphoma Society-Snowdome Foundation-Leukaemia Foundation (6620-21), and the Anthony Rothe Memorial Trust.
Authorship
Contribution: S. Subramanian, J.A.I.T., J.W.H.W., and J.E.P. conceptualized the study; S. Subramanian, J.A.I.T., Y.H., S. Jacquelin, S. Shen, E.S., C.B., P.S.W., D.C-F., D.B., A.T., and S.W.L. developed the methodology; S. Subramanian, J.A.I.T., Y.H., S. Shen, E.S., S. Joshi, D.J.C., K.Y., V.A., T.O., J.A.P., I.D.L., S.M.P., and M.K.G. carried out investigation; S. Subramanian, J.A.I.T., P.C-P., F.C.K., F.V., E.S.W., B.G., H.A.R., J.W.H.W., and J.E.P. did formal analysis; J.A.I.T., J.W.H.W., and J.E.P. supervised the study; S. Subramanian, J.A.I.T., and J.E.P. contributed to writing original draft; J.A.I.T. and J.E.P. reviewed and edited the manuscript; and J.E.P. contributed in funding acquisition.
Footnotes
∗S. Subramanian and J.A.I.T. contributed equally to this study.
Sequencing data have been uploaded to Gene Expression Omnibus with accession number GSE231486.
Any original data not included herewith will be shared upon reasonable request from the corresponding author, John E. Pimanda (jpimanda@unsw.edu.au).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Julie A. I. Thoms, Email: j.thoms@unsw.edu.au.
Jason W. H. Wong, Email: jwhwong@hku.hk.
John E. Pimanda, Email: jpimanda@unsw.edu.au.
Supplementary Material
References
- 1.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Laurenti E, Gottgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553(7689):418–426. doi: 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Setty M, Kiseliovas V, Levine J, Gayoso A, Mazutis L, Pe'er D. Characterization of cell fate probabilities in single-cell data with Palantir. Nat Biotechnol. 2019;37(4):451–460. doi: 10.1038/s41587-019-0068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corces MR, Buenrostro JD, Wu B, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novershtern N, Subramanian A, Lawton LN, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Shao Z, Glass K, et al. Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev Cell. 2012;23(4):796–811. doi: 10.1016/j.devcel.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson EH. Emerging properties of animal gene regulatory networks. Nature. 2010;468(7326):911–920. doi: 10.1038/nature09645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thoms JAI, Beck D, Pimanda JE. Transcriptional networks in acute myeloid leukemia. Genes Chromosomes Cancer. 2019;58(12):859–874. doi: 10.1002/gcc.22794. [DOI] [PubMed] [Google Scholar]
- 9.Nasrallah R, Fast EM, Solaimani P, et al. Identification of novel regulators of developmental hematopoiesis using Endoglin regulatory elements as molecular probes. Blood. 2016;128(15):1928–1939. doi: 10.1182/blood-2016-02-697870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345(6199):943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon JR, Jung I, Selvaraj S, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oram SH, Thoms JA, Pridans C, et al. A previously unrecognized promoter of LMO2 forms part of a transcriptional regulatory circuit mediating LMO2 expression in a subset of T-acute lymphoblastic leukaemia patients. Oncogene. 2010;29(43):5796–5808. doi: 10.1038/onc.2010.320. [DOI] [PubMed] [Google Scholar]
- 14.Curtis DJ, Salmon JM, Pimanda JE. Concise review: blood relatives: formation and regulation of hematopoietic stem cells by the basic helix-loop-helix transcription factors stem cell leukemia and lymphoblastic leukemia-derived sequence 1. Stem Cells. 2012;30(6):1053–1058. doi: 10.1002/stem.1093. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Luo H, Liu T, Zacksenhaus E, Ben-David Y. The ets transcription factor Fli-1 in development, cancer and disease. Oncogene. 2015;34(16):2022–2031. doi: 10.1038/onc.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pimanda JE, Ottersbach K, Knezevic K, et al. Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc Natl Acad Sci U S A. 2007;104(45):17692–17697. doi: 10.1073/pnas.0707045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai Z, de Bruijn M, Ma X, et al. Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity. 2000;13(4):423–431. doi: 10.1016/s1074-7613(00)00042-x. [DOI] [PubMed] [Google Scholar]
- 19.Mangan JK, Speck NA. RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog. 2011;16(1-2):77–91. doi: 10.1615/critrevoncog.v16.i1-2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thoms JA, Birger Y, Foster S, et al. ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood. 2011;117(26):7079–7089. doi: 10.1182/blood-2010-12-317990. [DOI] [PubMed] [Google Scholar]
- 21.Beck D, Thoms JA, Perera D, et al. Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood. 2013;122(14):e12–22. doi: 10.1182/blood-2013-03-490425. [DOI] [PubMed] [Google Scholar]
- 22.Diffner E, Beck D, Gudgin E, et al. Activity of a heptad of transcription factors is associated with stem cell programs and clinical outcome in acute myeloid leukemia. Blood. 2013;121(12):2289–2300. doi: 10.1182/blood-2012-07-446120. [DOI] [PubMed] [Google Scholar]
- 23.Thoms JAI, Truong P, Subramanian S, et al. Disruption of a GATA2-TAL1-ERG regulatory circuit promotes erythroid transition in healthy and leukemic stem cells. Blood. 2021;138(16):1441–1455. doi: 10.1182/blood.2020009707. [DOI] [PubMed] [Google Scholar]
- 24.Mandoli A, Singh AA, Jansen PW, et al. CBFB-MYH11/RUNX1 together with a compendium of hematopoietic regulators, chromatin modifiers and basal transcription factors occupies self-renewal genes in inv(16) acute myeloid leukemia. Leukemia. 2014;28(4):770–778. doi: 10.1038/leu.2013.257. [DOI] [PubMed] [Google Scholar]
- 25.Wilson NK, Foster SD, Wang X, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7(4):532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 26.Schmidl C, Rendeiro AF, Sheffield NC, Bock C. ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods. 2015;12(10):963–965. doi: 10.1038/nmeth.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alinejad-Rokny H, Ghavami Modegh R, Rabiee HR, et al. MaxHiC: a robust background correction model to identify biologically relevant chromatin interactions in Hi-C and capture Hi-C experiments. PLoS Comput Biol. 2022;18(6) doi: 10.1371/journal.pcbi.1010241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen T, Guestrin C. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Association for Computing Machinery. 2016. XGBoost: a scalable tree boosting system; pp. 785–794. [Google Scholar]
- 29.Tursky ML, Beck D, Thoms JA, et al. Overexpression of ERG in cord blood progenitors promotes expansion and recapitulates molecular signatures of high ERG leukemias. Leukemia. 2015;29(4):819–827. doi: 10.1038/leu.2014.299. [DOI] [PubMed] [Google Scholar]
- 30.Knudsen KJ, Rehn M, Hasemann MS, et al. ERG promotes the maintenance of hematopoietic stem cells by restricting their differentiation. Genes Dev. 2015;29(18):1915–1929. doi: 10.1101/gad.268409.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takayama N, Murison A, Takayanagi SI, et al. The transition from quiescent to activated states in human hematopoietic stem cells is governed by dynamic 3D genome reorganization. Cell Stem Cell. 2021;28(3):488–501.e10. doi: 10.1016/j.stem.2020.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Schutte J, Wang H, Antoniou S, et al. An experimentally validated network of nine haematopoietic transcription factors reveals mechanisms of cell state stability. Elife. 2016;5 doi: 10.7554/eLife.11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson NK, Miranda-Saavedra D, Kinston S, et al. The transcriptional program controlled by the stem cell leukemia gene Scl/Tal1 during early embryonic hematopoietic development. Blood. 2009;113(22):5456–5465. doi: 10.1182/blood-2009-01-200048. [DOI] [PubMed] [Google Scholar]
- 34.Sinclair AM, Gottgens B, Barton LM, et al. Distinct 5' SCL enhancers direct transcription to developing brain, spinal cord, and endothelium: neural expression is mediated by GATA factor binding sites. Dev Biol. 1999;209(1):128–142. doi: 10.1006/dbio.1999.9236. [DOI] [PubMed] [Google Scholar]
- 35.Moignard V, Macaulay IC, Swiers G, et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol. 2013;15(4):363–372. doi: 10.1038/ncb2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landry JR, Bonadies N, Kinston S, et al. Expression of the leukemia oncogene Lmo2 is controlled by an array of tissue-specific elements dispersed over 100 kb and bound by Tal1/Lmo2, Ets, and Gata factors. Blood. 2009;113(23):5783–5792. doi: 10.1182/blood-2008-11-187757. [DOI] [PubMed] [Google Scholar]
- 37.Gottgens B, Broccardo C, Sanchez MJ, et al. The scl +18/19 stem cell enhancer is not required for hematopoiesis: identification of a 5' bifunctional hematopoietic-endothelial enhancer bound by Fli-1 and Elf-1. Mol Cell Biol. 2004;24(5):1870–1883. doi: 10.1128/MCB.24.5.1870-1883.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gottgens B, Ferreira R, Sanchez MJ, et al. cis-regulatory remodeling of the SCL locus during vertebrate evolution. Mol Cell Biol. 2010;30(24):5741–5751. doi: 10.1128/MCB.00870-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottgens B, Nastos A, Kinston S, et al. Establishing the transcriptional programme for blood: the SCL stem cell enhancer is regulated by a multiprotein complex containing Ets and GATA factors. EMBO J. 2002;21(12):3039–3050. doi: 10.1093/emboj/cdf286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanchez M, Gottgens B, Sinclair AM, et al. An SCL 3' enhancer targets developing endothelium together with embryonic and adult haematopoietic progenitors. Development. 1999;126(17):3891–3904. doi: 10.1242/dev.126.17.3891. [DOI] [PubMed] [Google Scholar]
- 41.Bee T, Ashley EL, Bickley SR, et al. The mouse Runx1 +23 hematopoietic stem cell enhancer confers hematopoietic specificity to both Runx1 promoters. Blood. 2009;113(21):5121–5124. doi: 10.1182/blood-2008-12-193003. [DOI] [PubMed] [Google Scholar]
- 42.Nottingham WT, Jarratt A, Burgess M, et al. Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood. 2007;110(13):4188–4197. doi: 10.1182/blood-2007-07-100883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan WY, Follows GA, Lacaud G, et al. The paralogous hematopoietic regulators Lyl1 and Scl are coregulated by Ets and GATA factors, but Lyl1 cannot rescue the early Scl-/- phenotype. Blood. 2007;109(5):1908–1916. doi: 10.1182/blood-2006-05-023226. [DOI] [PubMed] [Google Scholar]
- 44.Johnson KD, Kong G, Gao X, et al. Cis-regulatory mechanisms governing stem and progenitor cell transitions. Sci Adv. 2015;1(8) doi: 10.1126/sciadv.1500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wozniak RJ, Boyer ME, Grass JA, Lee Y, Bresnick EH. Context-dependent GATA factor function: combinatorial requirements for transcriptional control in hematopoietic and endothelial cells. J Biol Chem. 2007;282(19):14665–14674. doi: 10.1074/jbc.M700792200. [DOI] [PubMed] [Google Scholar]
- 46.Beerman I, Bhattacharya D, Zandi S, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107(12):5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strobl H, Takimoto M, Majdic O, et al. Myeloperoxidase expression in CD34+ normal human hematopoietic cells. Blood. 1993;82(7):2069–2078. [PubMed] [Google Scholar]
- 48.Pevny L, Simon MC, Robertson E, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature. 1991;349(6306):257–260. doi: 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki M, Kobayashi-Osaki M, Tsutsumi S, et al. GATA factor switching from GATA2 to GATA1 contributes to erythroid differentiation. Genes Cells. 2013;18(11):921–933. doi: 10.1111/gtc.12086. [DOI] [PubMed] [Google Scholar]
- 50.Rossmann MP, Zon LI. 'Enhancing' red cell fate through epigenetic mechanisms. Curr Opin Hematol. 2021;28(3):129–137. doi: 10.1097/MOH.0000000000000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cabezas-Wallscheid N, Klimmeck D, Hansson J, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15(4):507–522. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 52.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9(3):215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J Biol Chem. 2010;285(41):31087–31093. doi: 10.1074/jbc.R110.159079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seguin-Estevez Q, Dunand-Sauthier I, Lemeille S, et al. Extensive remodeling of DC function by rapid maturation-induced transcriptional silencing. Nucleic Acids Res. 2014;42(15):9641–9655. doi: 10.1093/nar/gku674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36(6):624–630. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 56.Zhang P, Zhang X, Iwama A, et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood. 2000;96(8):2641–2648. [PubMed] [Google Scholar]
- 57.Dahl R, Ramirez-Bergeron DL, Rao S, Simon MC. Spi-B can functionally replace PU.1 in myeloid but not lymphoid development. EMBO J. 2002;21(9):2220–2230. doi: 10.1093/emboj/21.9.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ku CJ, Hosoya T, Maillard I, Engel JD. GATA-3 regulates hematopoietic stem cell maintenance and cell-cycle entry. Blood. 2012;119(10):2242–2251. doi: 10.1182/blood-2011-07-366070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frelin C, Herrington R, Janmohamed S, et al. GATA-3 regulates the self-renewal of long-term hematopoietic stem cells. Nat Immunol. 2013;14(10):1037–1044. doi: 10.1038/ni.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nerlov C, Graf T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998;12(15):2403–2412. doi: 10.1101/gad.12.15.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wunsche P, Eckert ESP, Holland-Letz T, et al. Mapping active gene-regulatory regions in human repopulating long-term HSCs. Cell Stem Cell. 2018;23(1):132–146.e9. doi: 10.1016/j.stem.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Canver MC, Smith EC, Sher F, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527(7577):192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu J, Song F, Lyu H, et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature. 2022;611(7935):387–398. doi: 10.1038/s41586-022-05365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamazaki H, Suzuki M, Otsuki A, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25(4):415–427. doi: 10.1016/j.ccr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Groschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157(2):369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- 66.Madsen JGS, Madsen MS, Rauch A, et al. Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells. Nat Genet. 2020;52(11):1227–1238. doi: 10.1038/s41588-020-0709-z. [DOI] [PubMed] [Google Scholar]
- 67.Zuin J, Roth G, Zhan Y, et al. Nonlinear control of transcription through enhancer-promoter interactions. Nature. 2022;604(7906):571–577. doi: 10.1038/s41586-022-04570-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiu SK, Orive SL, Moon MJ, et al. Shared roles for Scl and Lyl1 in murine platelet production and function. Blood. 2019;134(10):826–835. doi: 10.1182/blood.2019896175. [DOI] [PubMed] [Google Scholar]
- 69.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13(9):613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 70.Neumayr C, Haberle V, Serebreni L, et al. Differential cofactor dependencies define distinct types of human enhancers. Nature. 2022;606(7913):406–413. doi: 10.1038/s41586-022-04779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cornejo-Paramo P, Roper K, Degnan SM, Degnan BM, Wong ES. Distal regulation, silencers, and a shared combinatorial syntax are hallmarks of animal embryogenesis. Genome Res. 2022;32(3):474–487. doi: 10.1101/gr.275864.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goode DK, Obier N, Vijayabaskar MS, et al. Dynamic gene regulatory networks drive hematopoietic specification and differentiation. Dev Cell. 2016;36(5):572–587. doi: 10.1016/j.devcel.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voit RA, Liao X, Cohen B, et al. Regulated expression of GATA1 as a gene therapy cure for Diamond-Blackfan Anemia. Blood. 2022;140(suppl 1):986–987. [Google Scholar]
- 74.Edginton-White B, Maytum A, Kellaway SG, et al. A genome-wide relay of signalling-responsive enhancers drives hematopoietic specification. Nat Commun. 2023;14(1):267. doi: 10.1038/s41467-023-35910-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Assi SA, Imperato MR, Coleman DJL, et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet. 2019;51(1):151–162. doi: 10.1038/s41588-018-0270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loke J, Assi SA, Imperato MR, et al. RUNX1-ETO and RUNX1-EVI1 differentially reprogram the chromatin landscape in t(8;21) and t(3;21) AML. Cell Rep. 2017;19(8):1654–1668. doi: 10.1016/j.celrep.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ptasinska A, Assi SA, Mannari D, et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia. 2012;26(8):1829–1841. doi: 10.1038/leu.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Botten GA, Zhang Y, Dudnyk K, et al. Structural variation cooperates with permissive chromatin to control enhancer hijacking-mediated oncogenic transcription. Blood. 2022;140(suppl 1):1007–1008. doi: 10.1182/blood.2022017555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buenrostro JD, Corces MR, Lareau CA, et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. 2018;173(6):1535–1548.e16. doi: 10.1016/j.cell.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karamitros D, Stoilova B, Aboukhalil Z, et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat Immunol. 2018;19(1):85–97. doi: 10.1038/s41590-017-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Drissen R, Thongjuea S, Theilgaard-Monch K, Nerlov C. Identification of two distinct pathways of human myelopoiesis. Sci Immunol. 2019;4(35) doi: 10.1126/sciimmunol.aau7148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.