

Abstract
Activation and transdifferentiation of hepatic stellate cell (HSC) into migratory myofibroblasts is a key process in liver fibrogenesis. Cell migration requires an active remodeling of the cytoskeleton, which is a tightly regulated process coordinated by Rho-specific guanine nucleotide exchange factors (GEFs) and Rho family of small GTPases. Rho-associated kinase (ROCK) promotes assembly of focal adhesions and actin stress fibers by regulating cytoskeleton organization. GEF exchange protein directly activated by cAMP 1 (EPAC1), has been implicated in modulating TGFβ1 and Rho signaling; however, its role in HSC migration has never been examined. The aim of this study was to evaluate the role of cAMP-degrading phosphodiesterase 4 (PDE4) enzymes in regulating EPAC1 signaling, HSC migration and fibrogenesis. We show that PDE4 protein expression is increased in activated HSCs expressing alpha smooth muscle actin and active myosin light chain (MLC) in fibrotic tissues of human NASH cirrhosis livers and mouse livers exposed to carbon tetrachloride (CCl4). In human livers, TGFβ1 levels were highly correlated with PDE4 expression. TGFβ1 treatment of LX2 HSCs decreased levels of cAMP and EPAC1 and increased PDE4D expression. PDE4 specific inhibitor, rolipram, and an EPAC specific agonist decreased TGFβ1 mediated cell migration in vitro. In vivo, targeted delivery of rolipram to the liver prevented fibrogenesis, collagen deposition, decreased expression of several fibrosis related genes and HSC activation. Proteomic analysis of mouse liver tissues identified regulation of actin cytoskeleton by the kinase effectors of Rho GTPases as a major pathway impacted by rolipram. Western blot analyses confirmed that PDE4 inhibition decreased active MLC and endothelin 1 levels, key proteins involved in cytoskeleton remodeling and contractility. The current study, for the first time, demonstrates that PDE4 enzymes are expressed in hepatic myofibroblasts and promote cytoskeleton remodeling and HSC migration.
Keywords: Liver fibrosis, hepatic stellate cells, cyclic AMP signaling, proteomics, cytoskeleton remodeling, cell migration, phosphodiesterase 4
Introduction
Liver fibrosis occurs in multiple types of chronic liver injury, and unfortunately, there is no FDA-approved therapy for fibrosis [1]. Hepatic stellate cells are major contributors to liver fibrosis, as they are predominant cell type in the liver to produce extracellular matrix (ECM) proteins, including collagens [2, 3]. Upon activation, HSCs undergo transdifferentiation into proliferative, contractile, and migratory myofibroblasts which is a key process in liver fibrogenesis. Cell migration requires active cytoskeleton remodeling, which is tightly regulated process coordinated by Rho-specific guanine nucleotide exchange factors (GEFs) and Rho family of small GTPases [4–7]. Rho-associated kinase (ROCK) promotes assembly of focal adhesions and actin stress fibers by regulating cytoskeleton organization. Importantly, cytoskeleton remodeling plays a critical role in activation and motility of HSCs. Transforming growth factor 1 beta (TGFβ1) is a significant profibrotic mediator and HSC activator which triggers canonical (SMAD) and noncanonical pathways (Rho GTPase) in HSCs. Rho kinase 1 (ROCK1) is a mediator of RhoA, a small GTPase of Rho family of GTPases. Activation of ROCK1 has been shown to play a role in HSC activation and fibrosis [8, 9] and cell contraction/migration by regulating actin cytoskeleton reorganization [5, 10].
The phosphodiesterase 4 (PDE4) sub-family is the largest among 11 PDEs and is responsible for maintaining cAMP homeostasis. PDE4 is a complex family with 4 genes (PDE4A/B/C/D) encoding over 20 distinct PDE4 isoforms. Distinct tissue and subcellular expression of PDE4 isoforms ensures the specificity of cAMP signaling to various stimuli. cAMP regulates the activity of several effectors, including protein kinase A (PKA) and exchange protein activated by cAMP (EPAC) [11, 12]. Increases in both cAMP and EPAC signaling have been shown to have anti-inflammatory and anti-fibrotic effects (reviewed in [13, 14]). Indeed, it has been suggested that EPAC expression and activation is critical for the regulation of fibroblast function during fibrogenesis in various tissues [15, 16]. One study reported decreased EPAC expression in human and mouse fibrotic livers which was associated with increased activation of MLC, a key regulator of cytoskeleton remodeling[16]. However, the mechanisms of diminished levels of EPAC1 during fibrogenesis have never been examined. Our previous work demonstrated that quiescent HSCs do not express high levels of PDE4 enzymes; however, upon HSC activation, PDE4 enzyme expression increases and contributes to spontaneous activation of HSCs into alpha smooth muscle actin (αSMA) expressing HSCs[17]. These observations indicated that PDE4 enzymes might play a role in HSC transdifferentiating into myofibroblasts. In this study we aimed to further understand the role of PDE4 enzymes in regulating signaling pathways contributing to fibrogenesis in vivo and in vitro. We show for the first time that PDE4 enzymes are expressed in hepatic myofibroblasts in human and mouse fibrosis livers. We also show that TGFβ1 increases PDE4 expression and decreases cAMP and EPAC1 levels in HSCs. PDE4 inhibition decreased fibrogenesis largely by affecting cytoskeleton remodeling and HSC migration in vitro and in vivo.
Materials and methods
Mouse model of carbon tetrachloride (CCl4)-induced liver fibrosis:
Male C57BL/6J mice (25–30 g body weight) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). CCl4 dissolved in corn oil was administered intraperitoneally (i.p.) at a dose of 1 mg/kg body weight twice a week for 4 weeks. The same volume of corn oil was administered to control mice (vehicle control). Another group of mice was treated with the PDE4 specific inhibitor, rolipram (Biomol, Enzo Life Sciences, Farmingdale, NY, USA) encapsulated into fusogenic lipid vesicles for targeted delivery to the liver. Preparation of FLVs-Rol has been previously described [18, 19]. Rolipram was administered 24 h after CCl4 at 3.3 mg/kg body weight to avoid any interference with CCl4 metabolism. Mice were killed 48 h after CCl4 administration.
Liver tissue proteomic and bioinformatics analyses.
Proteomic experiments were conducted as described previously [20]. Hepatic proteins that had significant abundance were imported into Ingenuity Pathways Analysis (www.ingenuity.com version 65367011) for canonical pathways analysis (CPA) and upstream activator analysis (UAA). Clarivate analytics software MetaCore was also used for specific pathway and network analysis.
In vitro studies.
LX2 human hepatic stellate cell line (HSC) was obtained from Sigma-Aldrich (Burlington, MA, USA). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (ATCC, Manassas, VA, USA) with 2% fetal bovine serum (FBS) and 1% penicillin/streptomycin (10000 U/ml stock solution, Gibco, Billings, MT, USA) at 37 °C in 5% CO2. Cells were cultured in serum-free DMEM overnight and then treated with 2.5 ng/ml human recombinant TGFβ1 (R&D Systems, a Bio-techne brand, Minneapolis, MN USA) with and without rolipram (10 μM) and an EPAC specific agonist, 8-pCPT-2’-O-Me-cAMP (50 μM) (BIOLOG Life Science Institute, Bremen, Germany). Cells were collected at various times after TGFβ1 treatment.
cAMP measurement:
cAMP levels were measured using a cAMP complete ELISA kit from ENZO (Enzo Life Sciences, Farmingdale, NY, USA, cat# ADI-900–163). Levels were normalized by protein content as described [18].
Scratch (wound healing) assay:
LX2 cells were plated at 0.2 million cells/well in 6-well plates in DMEM with 2% FBS and 1% penicillin/streptomycin (10000 U/ml stock solution, Gibco) at 37 °C in 5% CO2 for 24 h. The next day, the cell-coated surface was scraped with a 1 ml pipette tip in a single stripe and the medium changed to serum free DMEM. After treatment, cells were allowed to heal at 37 °C in 5% CO2 for 24 h. Migration of cells was observed using an inverted microscope and photographed at 0 and 24 h. The average extent of wound closure was evaluated by multiple measurements of the width of the wound space.
Cell proliferation assay:
LX2 cells were plated in 96-well plates at 5,000 cells/well. Cells were treated with rolipram (10 μM) and EPAC agonist (50 μM) followed by TGFβ1 stimulation for 24 h as described above. Then, 10 μl of WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) dye (Sigma-Aldrich) was added to 100 μl cell medium for 4 h and Absorbance was measured at 450 nm. Absorbance values for TGFβ1-treated cell are set at 100% (absorbance OD/average absorbance ODx100%). Absorbance values for rolipram and EPAC agonist treated cells are compared to those of TGFβ1-treated cells and data are presented as %TGFβ1.
Hematoxylin and Eosin (H&E), Sirius red and immunostaining, RNA isolation, and RT-qPCR analysis:
These were performed according to standard procedures [17, 18, 21]. Images were acquired and analyzed using BZ-X810 All-in-One Fluorescence Microscope (Keyence, Itasca, IL, U.S.A.). Immunofluorescence (IF) staining was performed on formalin-fixed paraffin embedded tissues. Tissue de-paraffinization/rehydration and antigen retrieval procedures were the same as described in our earlier publications [17, 18]. Mouse liver tissues were blocked with M.O.M. blocking reagent for 30 min at room temperature (RT) before they were further blocked using 10% normal donkey serum for 1 h at RT. Tissues were incubated with primary antibody for overnight at 4 °C followed by a secondary antibody for 1 h at RT. DAPI was used for 5 min before slides were mounted. Antibodies were diluted in 1% BSA in TRIS-buffered saline containing 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA ). Isotype rabbit and mouse IgG were used as negative controls for immunostaining. Antibody and reagent information is provided in supplementary material, Table S1.
Western blotting.
Lysates were prepared by using RIPA buffer [18, 22] and 25–30 μg total protein was used per lane. GAPDH and α-tubulin were used as loading controls. Densitometric analyses and ratios of target proteins to loading controls were performed as previously described [18, 23]. Densitometric ratios and P values are indicated on western blot images. Antibody information is provided in supplementary material, Table S1.
Human liver tissue.
The University of Louisville Hospital provided explant liver tissues from non-alcoholic steatohepatitis (NASH) cirrhosis patients undergoing liver transplant (n=5). Samples from five donor livers were acquired from the Resource Center at John Hopkins University. All studies were approved by the respective institutional review boards and written consent was obtained from all participants.
Statistical analysis
Pearson’s Chi-squared and Student’s t-test or ANOVA was used [24, 25]. The correlation heatmap was generated by SAS statistical software (SAS Institute Inc., Cary, NC, USA). The proteomic data were analyzed using SATP (Statistical Analysis Tool for Proteomics) [26, 27], after making the logarithm transformation and using the KNN imputation method. The LIMMA/Moderated t-test was selected to perform differential expression analysis of proteomics for each protein. The p-values were adjusted for multiple comparisons using False Discovery Rate (FDR, or Benjamini–Hochberg, BH) method and the volcano plots were generated from SATP[24]. The Venn diagrams were generated using statistical software R (R Core Team, 2020, R: A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org).
For animal experiments statistical analyses were performed using GraphPad Prism, version 9.1.2. for Windows (GraphPad Software Inc., La Jolla, CA, USA). Differences between two groups were analyzed using the unpaired t-test. Multiple group analyses were performed using one-way analysis of variance followed by Tukey’s post hoc multiple comparison test. Results were expressed as mean ± SD. P < 0.05 was considered statistically significant.
Results
Activated hepatic stellate cells express PDE4A and PDE4D enzymes in carbon tetrachloride treated mouse livers.
Repeated administration of CCl4 led to the development of fibrosis in mice as demonstrated by collagen deposition by Sirius Red staining and increased hydroxyproline levels (Figure 1A, B). Correspondingly, increased mRNA levels of Acta2, encoding the hepatic stellate activation marker αSMA were observed as shown by RT-qPCR (Figure 1C). Importantly, significant number of αSMA positive HSCs were co-stained with phosphorylated myosin light chain (pMLC) (Figure 1D), a marker of HSC contractility. To examine whether activated HSCs express PDE4 enzymes, we performed double immunofluorescence staining with αSMA and Pde4a, b and d antibodies. We did not observe any co-staining with Pde4b (data not shown); however, we saw that significant number of αSMA-positive cells co-stained with Pde4d (Figure 1G). We also observed increased Pde4d protein expression in CCl4 treated mouse livers (Figure 1H). Interestingly, although we did not see a significant increase in Pde4a protein expression in CCl4 treated mouse livers (Figure 1F), we did observe accumulation of Pde4a expressing cells in fibrotic areas which co-stained with αSMA (Figure 1E). However, these cells were markedly less in numbers in comparison to Pde4d-aSMA double-positive cells. These results demonstrate that activated HSCs in fibrotic areas express Pde4a and d enzymes.
Figure 1. Increased Pde4 expression in αSMA-positive activated hepatic stellate cells in carbon tetrachloride (CCl4) treated mouse livers.
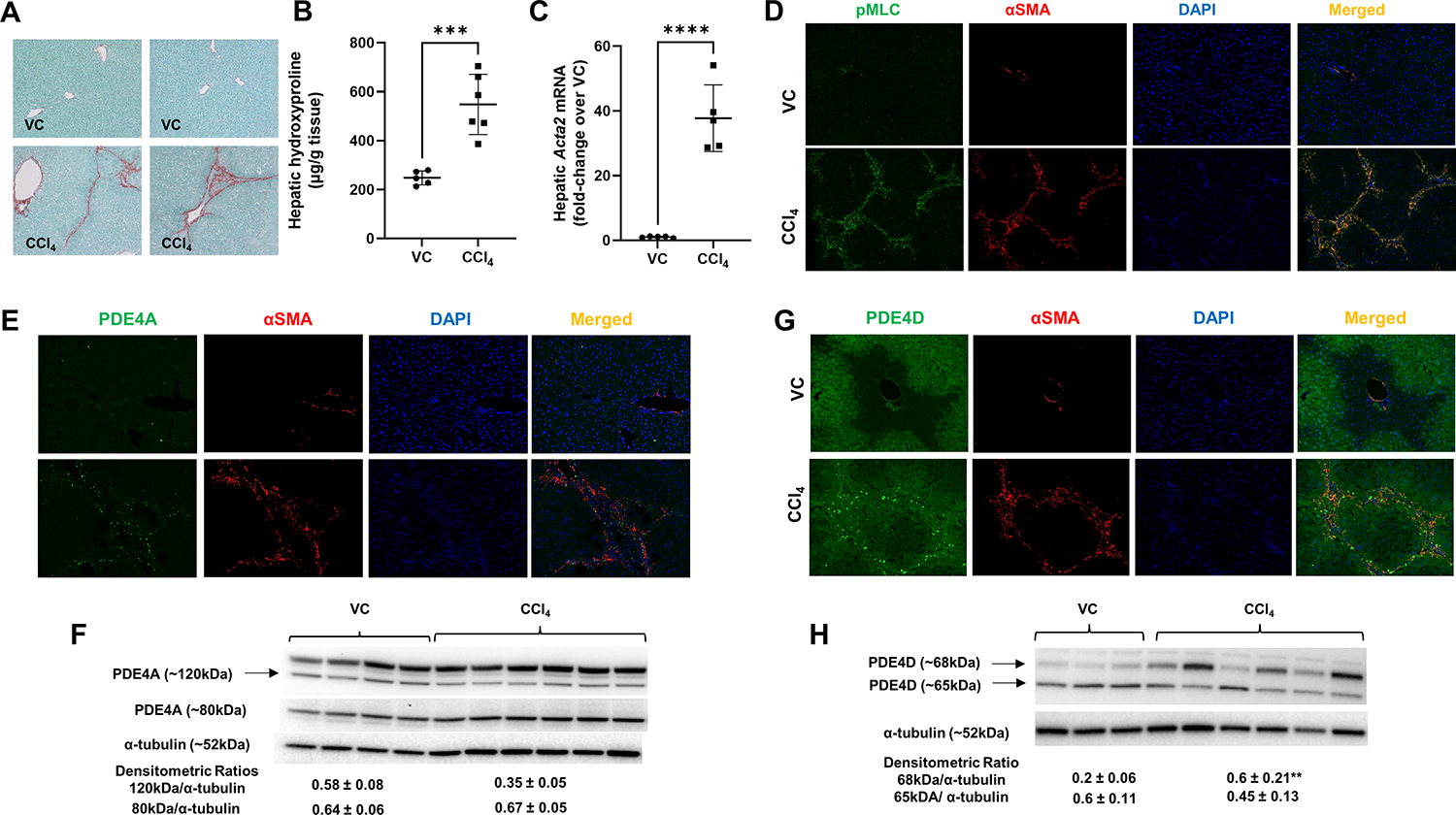
(A) Representative images of Sirius Red staining of control (vehicle control, VC) and CCl4 treated mouse livers, (B) Increased hepatic hydroxyproline levels in CCl4 treated mice, (C) Increased αSMA (Acta2) mRNA expression in CCl4 treated mouse livers, (D) No significant expression of pMLC and αSMA in VC mice, while αSMA positive HSCs express activated MLC (pMLC) in CCl4 treated mice, representative images, DAPI- nuclear staining, original magnification 20x. (E) Accumulation of Pde4a- αSMA-positive HSCs in fibrotic areas of CCl4 treated fibrotic livers. (F) Western blot analysis of whole liver Pde4a protein expression. (G) Increased accumulation of Pde4d-aSMA double positive cells in fibrotic area of CCl4 treated mice. (H) Western blot analysis of whole liver Pde4d protein expression. Densitometric ratios to α-tubulin (loading control) are shown. Unpaired t-test **p<0.01, ***p<0.001, ****p<0.0001.
PDE4 inhibition attenuates fibrogenic process and collagen deposition in a chronic CCl4 induced fibrosis mouse model.
We next evaluated the effect of PDE4 inhibition on HSC activation and fibrogenesis. In one group of mice, every dose of CCl4 was followed by administration of targeted hepatic delivery of rolipram to inhibit PDE4 activity [18, 19]. As previous studies have showed that cAMP signaling affects activity of CYP2E1, an enzyme which metabolizes CCl4 [28–33], we administered rolipram after 24 h of CCl4 to avoid any interference with CCl4 metabolism. Rolipram attenuated SMAD3 signaling and stellate cell activation marker αSMA (Figure 2A,B), mRNA levels for matrix metalloproteinase-2 (Mmp2) and tissue inhibitor of metalloproteinase 2 (Timp2) (Figure 2C, D). Rolipram also decreased mRNA levels for chaperone protein for collagen, Hsp47 (Serpinh1) (Figure 2E) and lysyl oxidases Lox and Loxl1 (Figure 2F). As a result, we observed a significantly lower deposition of collagen as shown by Sirius red staining and hydroxyproline levels (Figure 2G,H).
Figure 2. Decreased fibrogenesis and collagen deposition in mice treated with a PDE4 inhibitor, rolipram.
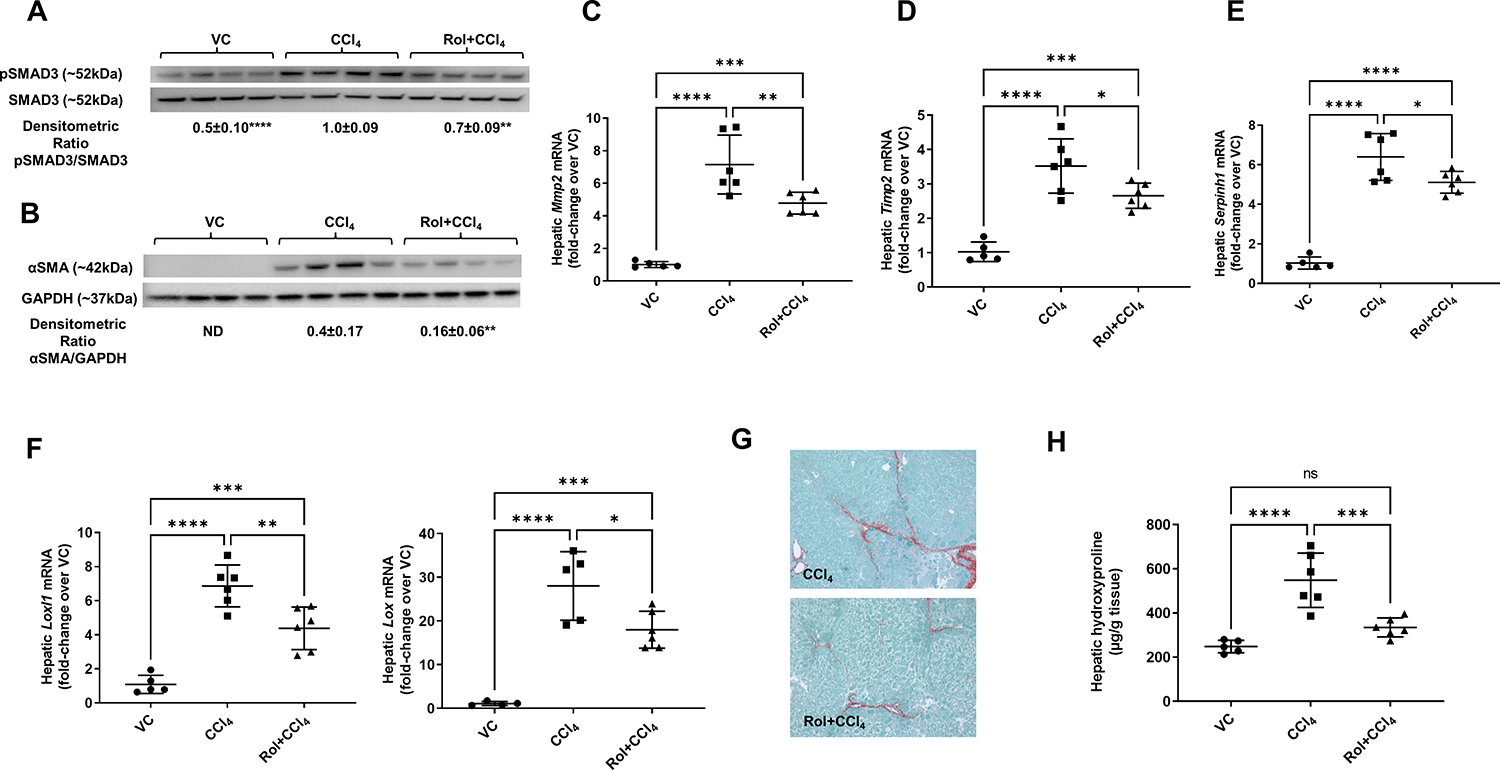
(A) pSMAD3 and (B) αSMA protein levels. Western blot, SMAD3 and GAPDH are loading controls, densitometric ratios are shown. (C) Hepatic mRNA levels of Mmp2, (D) Timp2, (E) Serpinh1 (Hsp47), (F) Loxl1 and Lox. (G) Collagen deposition, Sirius red staining and (H) Hydroxyproline levels. One-way ANOVA with Tukey’s post hoc test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Liver proteomic analysis.
To better understand the effect of PDE4 inhibition on fibrogenic processes in this model, we performed liver proteome analysis. A total of 3,884 enriched proteins at P<0.05 were identified in all three treatment groups (CCl4 in comparison to vehicle control (VC), rolipram+CCl4 in comparison to VC, and rolipram+CCl4 in comparison to CCl4) (Volcano plots, Figure 3); 2,437 proteins were significantly changed by CCl4 when compared to VC group (P<0.05), and only 2,251 proteins were significantly changed in the CCl4 group treated with rolipram (Venn diagrams, Figure 3B). Ingenuity pathway analysis (IPA) identified 10 top downregulated pathways in the rolipram treated group when compared to VC. Among these pathways, we found inflammatory pathways (fMLP and IL-8 signaling, Figure 3C) and pathways related to the development of fibrosis: Ephrin receptor signaling, Signaling by Rho family GTPases, Integrin, and Actin cytoskeleton signaling (Figure 3C). Among fibrosis related pathways, Cytoskeleton remodeling, regulation of actin cytoskeleton by the kinase effectors of Rho GTPases had the highest number of significantly decreased proteins in rolipram treated CCl4 group when compared to CCl4 group alone (Table 1). Notably, expression of Rho GTPases, RhoA and Cdc42, which regulate focal adhesions and cytoskeleton remodeling, was among the attenuated proteins. Rho GDP-dissociation inhibitor 1 (Arhgdia), which interacts with RhoA and CDC42 and inhibits their activation [34, 35], was also downregulated (Table 1). Cdc42bpb, Pkn1 and Mylk kinases playing a role in the regulation of cytoskeleton reorganization and cell motility/migration were also downregulated by rolipram treatment. Other downregulated proteins included Myosin 6 and 10, Moesin, Add1, Actn4, Cfl1, Dstn and Vcl all of which are involved in the regulation of migration and adhesion. IPA upstream regulator analysis showed that among upstream regulators which were downregulated by rolipram were 11 cytokines, three G protein coupled receptors and seven growth factors (Figure 3D). Among these regulators were TGFβ, TNF, oncostatin M (OSM), endothelin 1, and adenosine a2 receptor (ADORA2A) which have been shown to promote fibrogenesis[36–41]. Western blot analyses confirmed that indeed rolipram co-treatment attenuated pMLC and endothelin 1 protein levels induced by CCl4 treatment (Figure 3E,F).
Figure 3. Proteomic analysis of differentially expressed proteins between experimental groups.
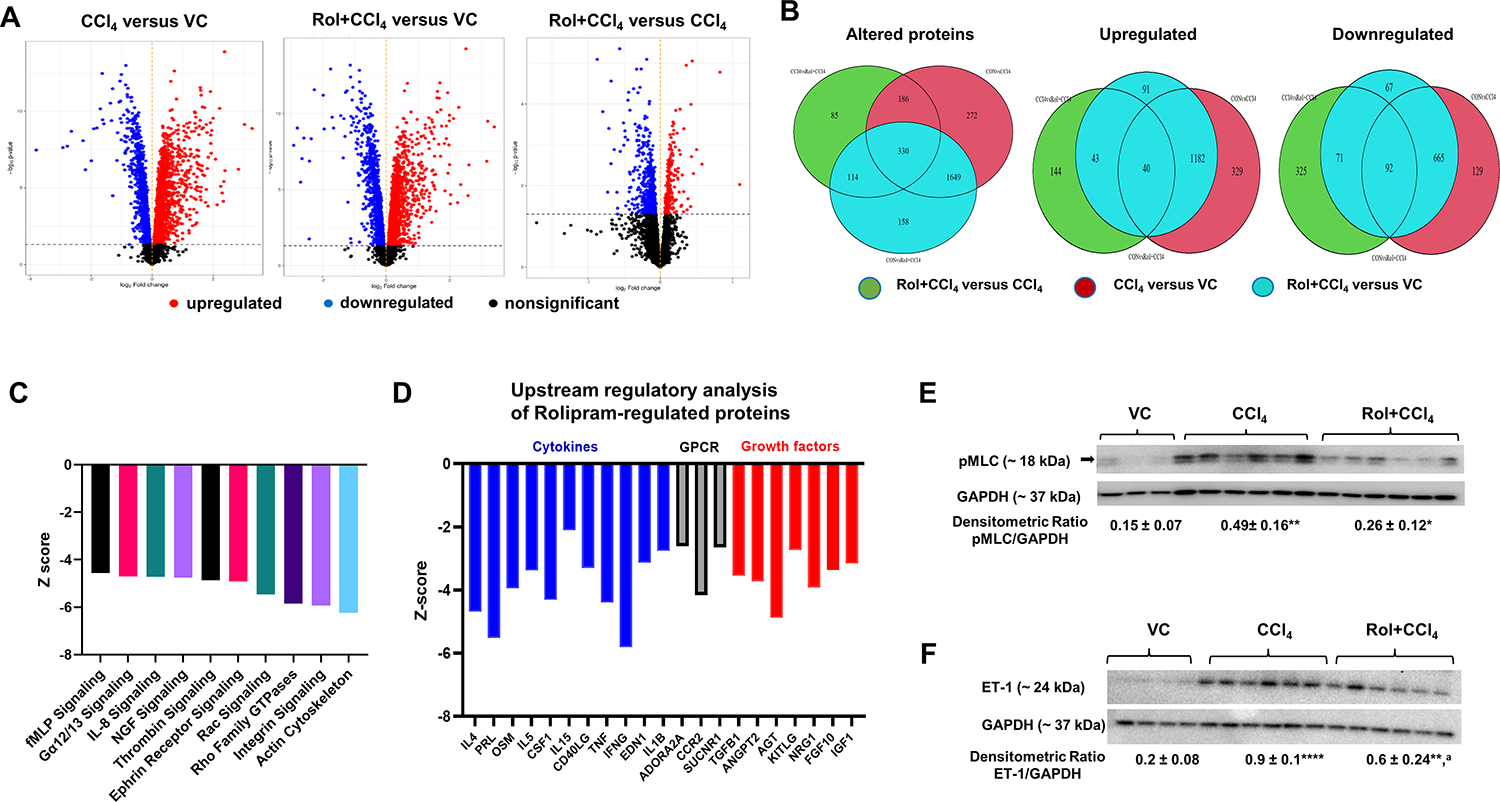
(A) Volcano Plot. Proteins are plotted as -log10(P-value) versus log2 Fold-Change. (B) Venn diagrams showing common and unique altered proteins between experimental groups. (C) Top 10 pathways downregulated by rolipram, (D) upstream regulatory analysis of rolipram-regulated proteins, (E) Western blot for liver pMLC and (F) Endothelin 1 (ET-1). VC – vehicle control, CCl4 – carbon tetrachloride, Rol+CCl4 – Rolipram-cotreated CCl4 mice. GAPDH – loading control. One-way ANOVA with Tukey’s post hoc test, *p<0.05, **p<0.01, ****p<0.0001 compared to vehicle control group (VC), ap<0.05 compared to CCl4.
Table 1.
Cytoskeleton remodeling, regulation of actin cytoskeleton by the kinase effectors of Rho GTPases
| Gene symbol | Object type | Description | p-value |
|---|---|---|---|
| Arhgdia | Regulators (GDI, GAP, GEF) | Rho GDP-dissociation inhibitor 1 | 0.004 |
| Cdc42bpb | Protein kinase | Serine/threonine-protein kinase MRCK beta | 0.006 |
| Arpc1b | Generic binding protein | Actin-related protein 2/3 complex subunit 1B | 0.008 |
| Pkn1 | Protein kinase | Serine/threonine-protein kinase N1 | 0.009 |
| Dstn | Generic binding protein | Destrin, actin depolymerizing protein | 0.014 |
| Cfl1 | Generic binding protein | Cofilin-1 | 0.014 |
| Add1 | Generic binding protein | Alpha-adducin | 0.035 |
| Myl6 | Generic binding protein | Myosin, light polypeptide 6 | 0.036 |
| Msn | Generic binding protein | Moesin | 0.039 |
| Myh10 | Generic binding protein | Myosin, heavy polypeptide 10 | 0.041 |
| Rhoa | RAS - superfamily | Transforming protein RhoA | 0.042 |
| Mylk | Protein kinase | Myosin, light polypeptide kinase | 0.045 |
| Myl12b | Generic binding protein | Myosin regulatory light chain 12b | 0.047 |
| Actn4 | Generic binding protein | Alpha-actinin 4, F-actin cross-linking protein | 0.053 |
| Cdc42 | RAS - superfamily | Cell division control protein 42 homolog | 0.053 |
| Vcl | Generic binding protein | Vinculin, F-actin binding protein | 0.055 |
Increased PDE4 expression in activated HSCs in human NASH cirrhosis liver tissues.
To evaluate the clinical relevance of our findings, we obtained liver tissues from healthy donors and patients with NASH cirrhosis who underwent liver transplantation at University of Louisville Hospital. As expected, NASH cirrhosis livers showed extensive collagen deposition (Figure 4A). mRNA, immunostaining, and western blot analyses of PDE4 expression confirmed that NASH cirrhosis livers had much higher expression of all three PDE4 enzymes (Figure 4B–D). Notably, correlation analysis showed a significant strong correlation of all PDE4 mRNAs with TGFB1, which was higher in NASH cirrhosis livers (Figure 4E,F).
Figure 4. Increased PDE4 expression in NASH patient livers strongly correlates with TGFβ1.
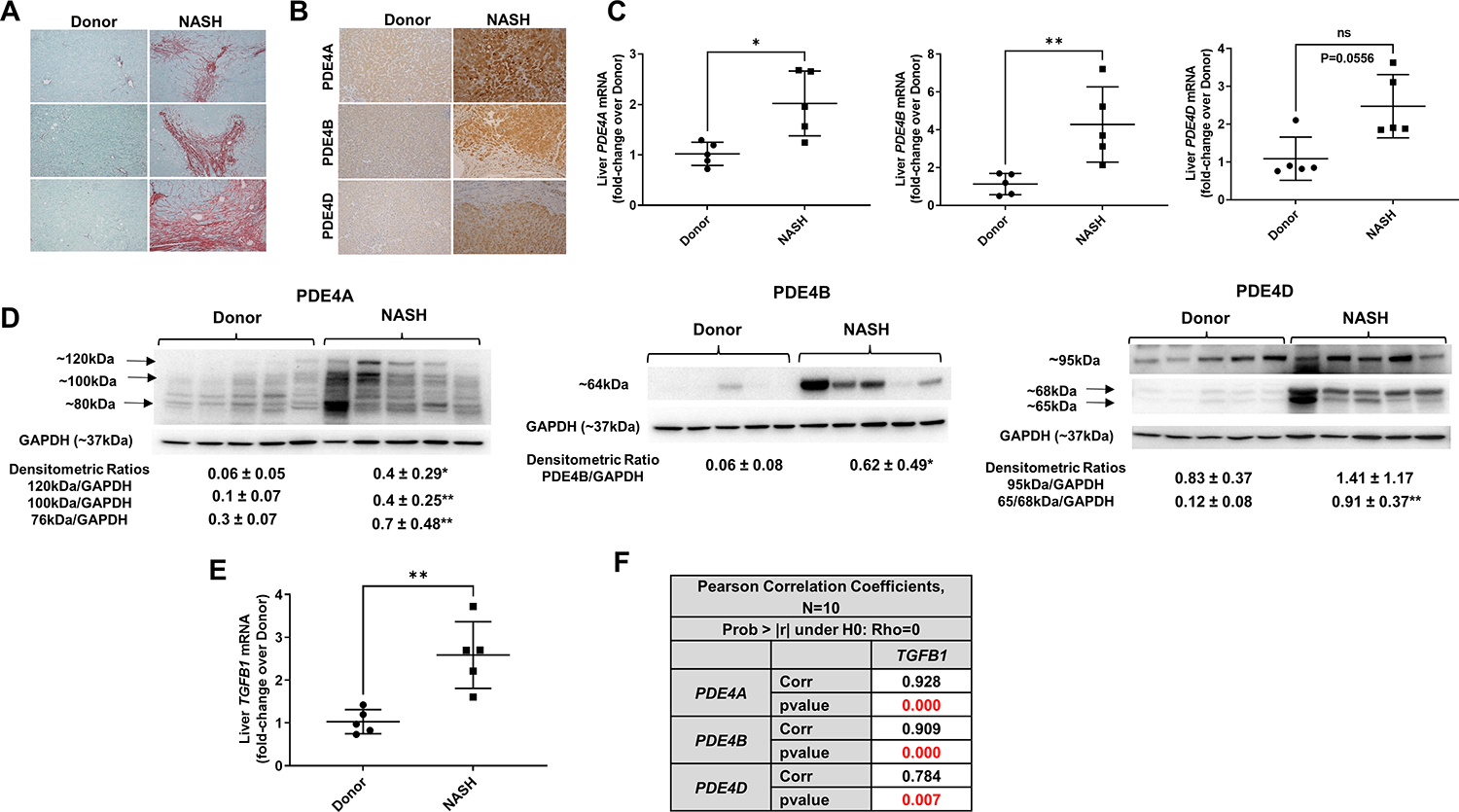
(A) representative images of Sirius red staining documenting a significant collagen deposition in livers of NASH patients. Original magnification 10x. (B) Increased PDE4A, B and D protein immunostaining in NASH livers, magnification 10x. (C) Liver PDE4A, B and D mRNA. (D) Liver PDE4A, B and D protein levels by Western blot, densitometric ratios to GAPDH- loading control are shown (from top to bottom bands). E. Liver TGFB1 mRNA, (F) Pearson correlation analysis shows a positive strong correlation of TGFβ1 with PDE4. The P values were computed using t-statistic under , where is the correlation coefficient. *p<0.05, **p<0.01.
We further performed double-immunofluorescence staining using antibodies against αSMA and the PDE4 subfamily (PDE4A, B and D). We found that all three PDE4 subfamily of enzymes were co-stained with αSMA-positive cells in in the nodule (Figure 5A–C). We also found that αSMA-positive HSCs were co-stained with pMLC in NASH cirrhosis livers (Figure 5D).
Figure 5. PDE4 enzymes are expressed in activated hepatic stellate cells in livers from patients with NASH cirrhosis.
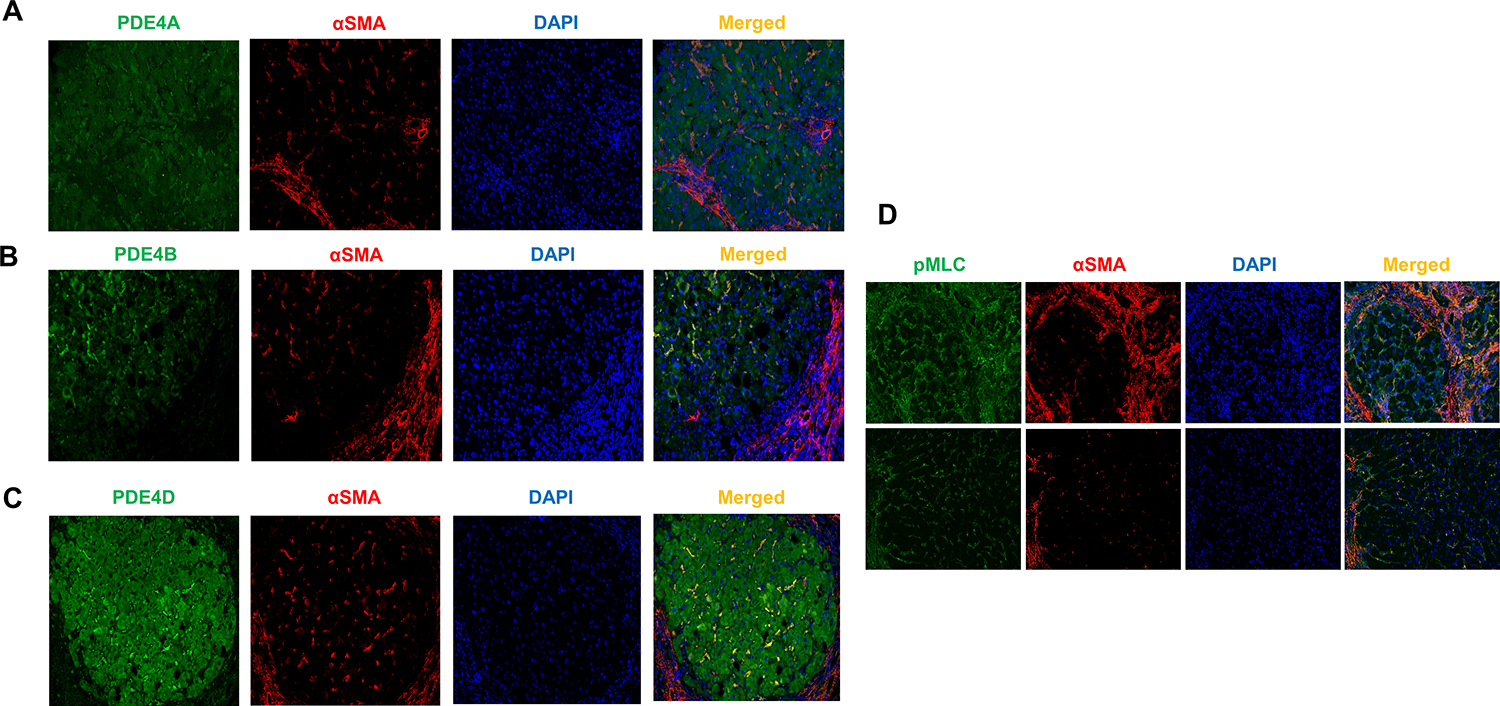
(A–C) representative images of αSMA and PDE4A, B and D double immunofluorescence staining of NASH cirrhosis livers. DAPI- nuclear staining. (D) αSMA positive HSCs express activated MLC. Original magnification 20x.
PDE4 inhibition decreases TGFβ1 mediated HSC migration.
We performed in vitro studies using the human HSC cell line LX2 stimulated with TGFβ1. As expected TGFβ1 induced activation of LX2 cells as shown by increased levels ACTA2 (Figure 6A). Notably, TGFβ1 treated cells showed a time dependent decrease in cAMP levels (Fig. 6B) which was accompanied by decreased EPAC1 (RAPGEF3) (Fig. 6C) and increased PDE4D mRNA and PDE4D protein expression (Fig. 6D, E). Since EPAC1 (guanine-nucleotide-exchange factor (GEF)) has been linked to RhoA kinase signaling and MLC activation, we examined whether PDE4 inhibition influenced LX2 cell motility using a scratch assay. LX2 cells treated with TGFβ1 were able to close 80% of the wound width in 24 h. Rolipram and EPAC specific agonist significantly attenuated TGFβ1-induced cell migration/wound closure in LX2 cells by half in 24 h (Figure 6F,G). Importantly, this effect was not mediated by their effect on cell proliferation as shown by WST-8 cell proliferation assay (Figure 6H).
Figure 6. EPAC activation and PDE4 inhibition decrease HSC migration in vitro.
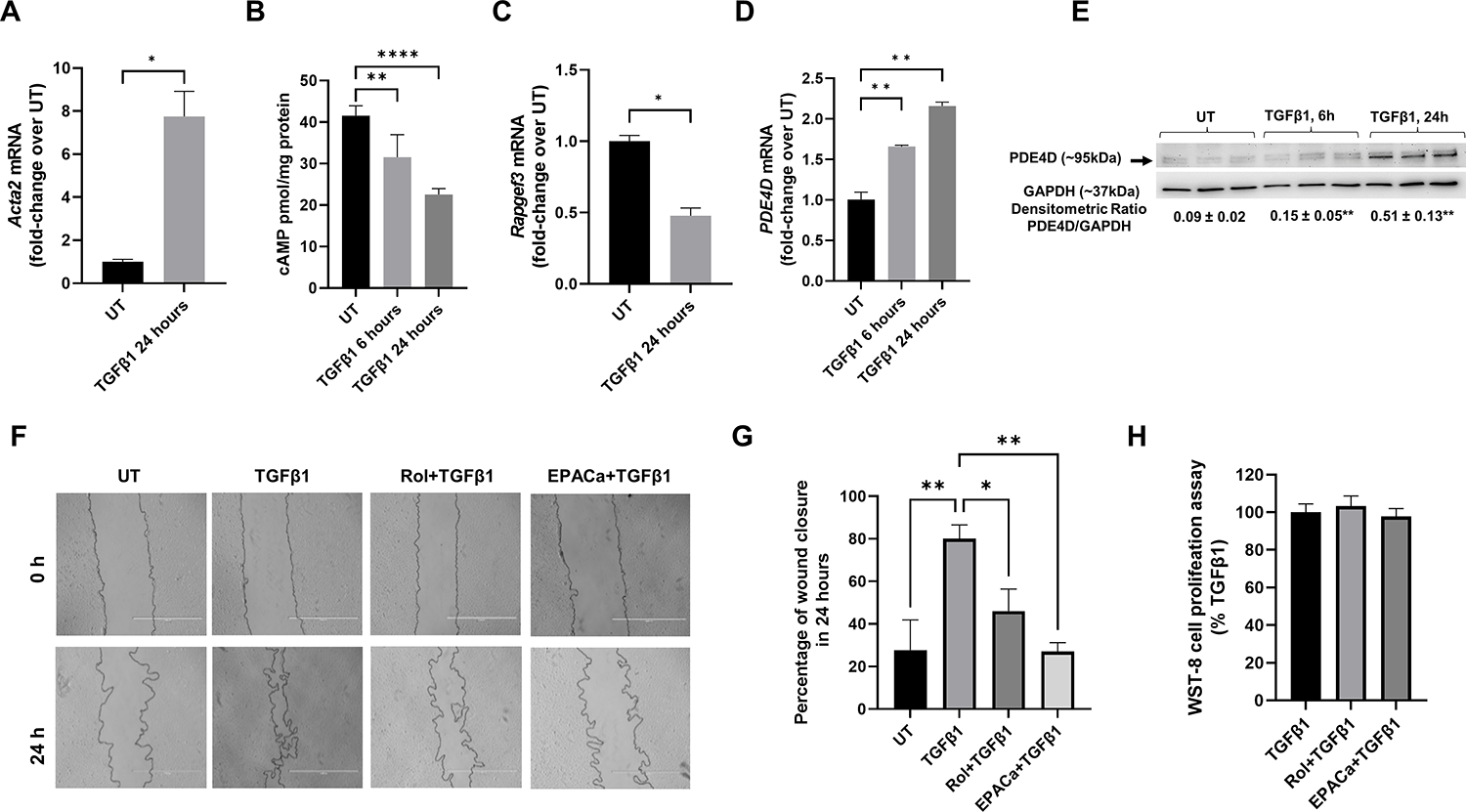
(A) TGFβ1 induces LX2 cell activation as shown by increased αSMA (ACTA2) mRNA expression, (B) TGFβ1 decreases cAMP levels in LX2 cells, (C) TGFβ1 increased PDE4D expression, (D) TGFβ1 decreases EPAC1 (RAPGEF3) mRNA expression, (E) Representative images of scratch assay of LX2 cells before and after 24-h treatment with TGFβ1 with and without PDE4 specific inhibitor rolipram and EPAC specific agonist, (F) Quantification of average wound closure percentage measured at 0 and 24 h, (G) WST-8 cell proliferation assay of LX2 cells treated with TGFβ1 with and without rolipram and EPAC specific agonist for 24 h. One-way ANOVA with Tukey’s post hoc test, *p<0.05, **p<0.01.
Discussion
Activation of quiescent hepatic stellate cells into proliferative, contractile, and chemotactic myofibroblasts is a key process in fibrogenesis [42–45]. During hepatic fibrogenesis, myofibroblasts migrate and accumulate at the site of injury and produce increasing amounts of extracellular matrix (ECM) components such as collagens and fibronectin. Excessive scar deposition results in a significant deterioration of liver function, altered blood flow and eventually liver failure [46]. Because transdifferentiation of HSCs plays a key role in the development of liver fibrosis, targeting HSC activation has become a focal point in treating liver fibrosis [47]. Relevant to this study, it has been shown that activation of cAMP/PKA pathway inhibits HSC proliferation [48] and cAMP/PKA/pCREB signaling is essential to maintain quiescence in HSCs [48]. In agreement with these studies, we later showed that upon spontaneous activation, primary HSCs start expressing cAMP degrading PDE4 enzymes and PDE4 inhibition prevented their phenotypic change into αSMA and Col1a1expressing myofibroblasts [17]. Several other studies also demonstrated antifibrotic properties of PDE inhibition[13, 14, 49], however the role of PDE4 and PDE4 regulated cAMP signaling in TGFβ1 mediated profibrogenic processes in HSCs is still not clear.
Cytoskeleton remodeling is an active, well controlled process which is required for cell migration [4]. Small G-protein RhoA and its mediator Rho kinase (ROCK) promote assembly of focal adhesions and actin stress fibers by regulating cytoskeleton organization in hepatic stellate cells (HSCs). Rho-kinase signaling has been identified as a regulator of cytoskeleton reorganization [5, 10, 50]. Inhibition of Rho kinase has been shown to attenuate HSC transdifferentiation into myofibroblasts and fibrosis [8, 9, 51–55]. A key molecule downstream of ROCK signaling is myosin light chain (MLC), which mediates stress fiber formation and contractility [16, 56]. Importantly, cAMP signaling has been shown to regulate ROCK/MLC signaling in various cells. Specifically, the critical role of cAMP effector EPAC1, a guanine-nucleotide-exchange factor, in HSC function and fibrosis has been demonstrated [16, 57]. It has been also proposed that decreased expression of EPAC1 serves as a profibrogenic signal in fibroblasts [15]. Our results showed that TGFβ1, a potent fibrogenic cytokine decreased cellular levels of cAMP and EPAC1 expression and increased PDE4D in HSCs. These decreases were associated with increased expression of HSC activation marker αSMA and migration. We also found that increased expression of TGFβ1 in NASH fibrosis livers was positively correlated with PDE4 expression. These results strongly suggest that EPAC/PDE4 is involved in TGFβ1 mediated phenotypic changes of HSCs. This notion was further supported by our observation that PDE4 inhibitor and EPAC specific agonist decreased TGFβ1 mediated migration of HSCs in vitro. These results agree with our previous work that quiescent HSCs express very low levels of cAMP degrading phosphodiesterase enzymes PDE4A, B and D [17]. However, upon spontaneous activation, HSCs rapidly expressed PDE4 proteins even before expressing αSMA (HSC activation marker), and inhibition of PDE4 attenuated spontaneous activation of HSCs and expression levels of αSMA (protein and mRNA) and Col1a1 mRNA [17]. Our current results uncovered a novel role of PDE4 enzymes in TGFβ1-induced HSC activation and migration, which has not been shown before.
The role of PDE4 enzymes in cytoskeleton remodeling was further demonstrated by our proteomic studies. Our results showed that modulation of PDE4 activity led to decreased expression of Rho GTPases, RhoA and cell division cycle 42 (Cdc42), which regulate focal adhesions, cytoskeleton remodeling and cell movement [58]. Notably, expression of Rho GDP dissociation inhibitor alpha (Arhgdia), which regulates their activation by regulating GDP/GTP exchange on these Rho proteins, was also downregulated. These results suggest that rolipram treatment also decreased activation of these Rho GTPases, which was confirmed by western blot analyses revealing lower phosphorylation levels of their mediator, MLC. Additionally, we observed decreased expression of myosin kinase Mylk, which phosphorylates MLC and is implicated in smooth muscle contraction [59]. Serine/threonine protein kinases Cdc42bpb (also known as MRCK beta) and protein kinase N1 (Pkn1) were on the list of proteins downregulated by rolipram. These kinases are involved in the regulation of cytoskeleton reorganization and cell migration [60–63]. Other downregulated proteins involved in regulation of migration and adhesion included Myosin 6 and 10, Moesin, adducin 1 (Add1), α-actinin 4 (Actn4), cofilin 1 (Cfl1), Destrin (Dstn) and vinculin (Vcl). Importantly, ECM binding to integrin receptors results in the formation of focal adhesions, which involves recruitment of focal adhesion kinase (FAK), talin, paxillin, actinin and vinculin, and serves a critical site of signal transduction [50, 64]. These focal adhesion proteins and focal adhesion assembly have been shown to play significant role in HSC activation and fibrosis [65, 66]. Signaling via focal adhesions also induces cytoskeleton reorganization, which is critical for cell motility and contraction [10, 50]. Notably, it was reported previously that increased pMLC levels in fibrotic livers from humans and rodents were negatively correlated with levels of EPAC1 (cAMP effector) [16]. Our results demonstrate that PDE4 inhibitor reduced RhoA-MLC signaling, and decreased levels of proteins involved in cytoskeleton remodeling (e.g., beta actin, cofilin 1, paxillin and myosin). These finding suggest that increased PDE4 activity downregulates cAMP/EPAC1 signaling during HSC activation and fibrogenesis. However, the exact mechanisms of this regulation, as well as the transcriptional drivers of PDE4 expression are still not clear and need to be investigated.
Our current study also demonstrates that modulation of PDE4 activity reduces fibrogenic signaling as demonstrated by decreased markers of HSC activation, ECM processing and deposition. Notably, we observed decreases in mRNA levels of lysyl oxidases (Lox and Loxl1) and Serpinh1 by rolipram treatment in mice, which are important enzymes in collagen crosslinking and collagen synthesis, respectively. Importantly, lysyl oxidases and HSP47 have been the targets in clinical trials for liver fibrosis (reviewed [67, 68]).
It is noteworthy that PDE4 inhibitor has been evaluated in a proof-of-concept phase 2 clinical trial in biopsy-confirmed NASH fibrosis patients. This 12-week trial did not show any effect of PDE4 inhibitor on biochemical end points[69]. The clinical utility of PDE4 inhibition as a therapy for NASH was questioned after this trial because it failed to decrease inflammation and liver injury markers. Authors suggested that the role and expression patterns of PDE4 family in the pathogenesis of NASH should be established. However, their focus was more on steatohepatitis and inflammatory cells. Results of our study show for the first time that PDE4 enzymes are expressed in activated HSCs/myofibroblasts in fibrotic livers from humans and mice. We also show that TGFβ1, a potent fibrogenic cytokine, correlates with increased PDE4 expression in livers from patients with NASH cirrhosis, and TGFβ1 induces PDE4D expression in human HSC cells. This increase results in downregulation of cAMP and EPAC1 expression, which contributes to cytoskeleton remodeling and increased migration of HSCs. PDE4 inhibition and EPAC activation decrease Rho signaling leading to impaired cytoskeleton remodeling and HSC contractility and migration. Further studies are needed to determine specific roles of PDE4A, B and D in hepatic stellate cell function which could lead to the development of subtype specific PDE4 inhibitors for the treatment of hepatic fibrosis, including NASH.
Supplementary Material
Table S1. Antibody and reagent information
Acknowledgments
We thank the Resource Center at John Hopkins University for providing human liver tissues for our studies (R24AA025017). We also thank Marion McClain for editing the manuscript. This study was supported by grants from the National Institutes of Health: R01AA029798 (LG), P20GM113226 (CJM), P50AA024337 (CJM), R44AA021331 (LG, CM), and Jewish Heritage Fund for Excellence (LG).
Footnotes
Conflict of interests: CM and PB declare a conflict of interest in employment and having equity in EndoProtech, Inc. No other conflicts of interest were decalred
Data availability statement
Proteomic files have been deposited in MassIVE (http://massive.ucsd.edu/) for this study. Data files for the following have been included in the MassIVE submission: (A) the primary data files (.RAW), (B) sample key, (C) the sequence databases (Mouse reviewed FASTA dated Feb 28, 2020), (D) TMT isotopic purity correction factors and (E) PeaksX results as excel files. The shared data will be released from private embargo for public access upon final acceptance for publication.
References
- 1.Allen AM, Lazarus JV, and Younossi ZM, Healthcare and socioeconomic costs of NAFLD: a global framework to navigate the uncertainties. J Hepatol, 2023. Volume 79, Issue 1, Pages 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parola M and Pinzani M, Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med, 2019. 65: p. 37–55. [DOI] [PubMed] [Google Scholar]
- 3.Kisseleva T and Brenner D, Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol, 2021. 18(3): p. 151–166. [DOI] [PubMed] [Google Scholar]
- 4.Lawson CD and Ridley AJ, Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol, 2018. 217(2): p. 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amano M, Nakayama M, and Kaibuchi K, Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken), 2010. 67(9): p. 545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciszewski WM, et al. , Cytoskeleton Reorganization in EndMT-The Role in Cancer and Fibrotic Diseases. Int J Mol Sci, 2021. 22(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Devreotes P and Horwitz AR, Signaling networks that regulate cell migration. Cold Spring Harb Perspect Biol, 2015. 7(8): p. a005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwamoto H, et al. , A p160ROCK-specific inhibitor, Y-27632, attenuates rat hepatic stellate cell growth. J Hepatol, 2000. 32(5): p. 762–70. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, et al. , Hypoxia-inducible factor 1α and ROCK1 regulate proliferation and collagen synthesis in hepatic stellate cells under hypoxia. Mol Med Rep, 2018. 18(4): p. 3997–4003. [DOI] [PubMed] [Google Scholar]
- 10.Fukata Y, Amano M, and Kaibuchi K, Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci, 2001. 22(1): p. 32–9. [DOI] [PubMed] [Google Scholar]
- 11.Zaccolo M, Zerio A, and Lobo MJ, Subcellular Organization of the cAMP Signaling Pathway. Pharmacol Rev, 2021. 73(1): p. 278–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng X, et al. , Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai), 2008. 40(7): p. 651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wahlang B, et al. , Role of cAMP and phosphodiesterase signaling in liver health and disease. Cell Signal, 2018. 49: p. 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elnagdy M, et al. , cAMP Signaling in Pathobiology of Alcohol Associated Liver Disease. Biomolecules, 2020. 10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoyama U, et al. , The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc Natl Acad Sci U S A, 2008. 105(17): p. 6386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schippers M, et al. , Upregulation of Epac-1 in Hepatic Stellate Cells by Prostaglandin E(2) in Liver Fibrosis Is Associated with Reduced Fibrogenesis. J Pharmacol Exp Ther, 2017. 363(2): p. 126–135. [DOI] [PubMed] [Google Scholar]
- 17.Gobejishvili L, et al. , Rolipram attenuates bile duct ligation-induced liver injury in rats: a potential pathogenic role of PDE4. J Pharmacol Exp Ther, 2013. 347(1): p. 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez WE, et al. , Phosphodiesterase 4 Inhibition as a Therapeutic Target for Alcoholic Liver Disease: From Bedside to Bench. Hepatology, 2019. 70(6): p. 1958–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gobejishvili L, et al. , Novel Liposomal Rolipram Formulation for Clinical Application to Reduce Emesis. Drug Des Devel Ther, 2022. 16: p. 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y, et al. , Redox States of Protein Cysteines in Pathways of Protein Turnover and Cytoskeleton Dynamics Are Changed with Aging and Reversed by Slc7a11 Restoration in Mouse Lung Fibroblasts. Oxid Med Cell Longev, 2020. 2020: p. 2468986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gobejishvili L, et al. , S-adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via cAMP/protein kinase A pathway. J Pharmacol Exp Ther, 2011. 337(2): p. 433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avila DV, et al. , Dysregulation of hepatic cAMP levels via altered Pde4b expression plays a critical role in alcohol-induced steatosis. J Pathol, 2016. 240(1): p. 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avila DV, et al. , Phosphodiesterase 4b expression plays a major role in alcohol-induced neuro-inflammation. Neuropharmacology, 2017. 125: p. 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y and Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological), 1995. 57(1): p. 289–300. [Google Scholar]
- 25.Agresti A, Categorical data analysis. 2 ed, ed. Agresti A. 2002, New York: Wiley. [Google Scholar]
- 26.Srivastava S, et al. , Standardizing Proteomics Workflow for Liquid Chromatography-Mass Spectrometry: Technical and Statistical Considerations. J Proteomics Bioinform, 2019. 12(3): p. 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srivastava S, et al. , Interactive Web Tool for Standardizing Proteomics Workflow for Liquid Chromatography-Mass Spectrometry Data. J Proteomics Bioinform, 2019. 12(4): p. 85–88. [PMC free article] [PubMed] [Google Scholar]
- 28.Gouillon ZQ, et al. , Role of CYP2E1 in the pathogenesis of alcoholic liver disease: modifications by cAMP and ubiquitin-proteasome pathway. Front Biosci, 1999. 4: p. A16–25. [DOI] [PubMed] [Google Scholar]
- 29.Menez JF, et al. , Phosphorylation of cytochrome P4502E1 (CYP2E1) by calmodulin dependent protein kinase, protein kinase C and cAMP dependent protein kinase. Alcohol Alcohol, 1993. 28(4): p. 445–51. [PubMed] [Google Scholar]
- 30.Oesch-Bartlomowicz B, et al. , Differential modulation of CYP2E1 activity by cAMP-dependent protein kinase upon Ser129 replacement. Exp Cell Res, 1998. 242(1): p. 294–302. [DOI] [PubMed] [Google Scholar]
- 31.Wong FW, Chan WY, and Lee SS, Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol, 1998. 153(1): p. 109–18. [DOI] [PubMed] [Google Scholar]
- 32.Scholten D, et al. , The carbon tetrachloride model in mice. Lab Anim, 2015. 49(1 Suppl): p. 4–11. [DOI] [PubMed] [Google Scholar]
- 33.Constandinou C, Henderson N, and Iredale JP, Modeling liver fibrosis in rodents. Methods Mol Med, 2005. 117: p. 237–50. [DOI] [PubMed] [Google Scholar]
- 34.Takai Y, Sasaki T, and Matozaki T, Small GTP-binding proteins. Physiol Rev, 2001. 81(1): p. 153–208. [DOI] [PubMed] [Google Scholar]
- 35.Liang L, et al. , Loss of ARHGDIA expression is associated with poor prognosis in HCC and promotes invasion and metastasis of HCC cells. Int J Oncol, 2014. 45(2): p. 659–66. [DOI] [PubMed] [Google Scholar]
- 36.Chiang DJ, et al. , Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One, 2013. 8(7): p. e69114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan ES, et al. , Adenosine A(2A) receptors play a role in the pathogenesis of hepatic cirrhosis. Br J Pharmacol, 2006. 148(8): p. 1144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stawski L and Trojanowska M, Oncostatin M and its role in fibrosis. Connect Tissue Res, 2019. 60(1): p. 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dichtel LE, Cordoba-Chacon J, and Kineman RD, Growth Hormone and Insulin-Like Growth Factor 1 Regulation of Nonalcoholic Fatty Liver Disease. J Clin Endocrinol Metab, 2022. 107(7): p. 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinzani M, et al. , Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology, 1996. 110(2): p. 534–48. [DOI] [PubMed] [Google Scholar]
- 41.Daher Z, Noel J, and Claing A, Endothelin-1 promotes migration of endothelial cells through the activation of ARF6 and the regulation of FAK activity. Cell Signal, 2008. 20(12): p. 2256–65. [DOI] [PubMed] [Google Scholar]
- 42.Friedman SL, Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem, 2000. 275(4): p. 2247–50. [DOI] [PubMed] [Google Scholar]
- 43.Kisseleva T and Brenner DA, Mechanisms of fibrogenesis. Exp Biol Med (Maywood), 2008. 233(2): p. 109–22. [DOI] [PubMed] [Google Scholar]
- 44.Aoyama T, Paik YH, and Seki E, Toll-like receptor signaling and liver fibrosis. Gastroenterol Res Pract, 2010. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kisseleva T and Brenner DA, Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol, 2011. 25(2): p. 305–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mallat A and Lotersztajn S, Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol, 2013. 305(8): p. C789–99. [DOI] [PubMed] [Google Scholar]
- 47.Li JT, et al. , Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol, 2008. 43(6): p. 419–28. [DOI] [PubMed] [Google Scholar]
- 48.Houglum K, Lee KS, and Chojkier M, Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. J Clin Invest, 1997. 99(6): p. 1322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El Awdan SA, et al. , Regression of fibrosis by cilostazol in a rat model of thioacetamide-induced liver fibrosis: Up regulation of hepatic cAMP, and modulation of inflammatory, oxidative stress and apoptotic biomarkers. PLoS One, 2019. 14(5): p. e0216301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burridge K, Focal adhesions: a personal perspective on a half century of progress. FEBS J, 2017. 284(20): p. 3355–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Görtzen J, et al. , Interplay of Matrix Stiffness and c-SRC in Hepatic Fibrosis. Front Physiol, 2015. 6: p. 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okimoto S, et al. , Vitamin A-coupled liposomal Rho-kinase inhibitor ameliorates liver fibrosis without systemic adverse effects. Hepatol Res, 2019. 49(6): p. 663–675. [DOI] [PubMed] [Google Scholar]
- 53.van Beuge MM, et al. , Reduction of fibrogenesis by selective delivery of a Rho kinase inhibitor to hepatic stellate cells in mice. J Pharmacol Exp Ther, 2011. 337(3): p. 628–35. [DOI] [PubMed] [Google Scholar]
- 54.van Dijk F, et al. , The antifibrotic potential of a sustained release formulation of a PDGFβ-receptor targeted rho kinase inhibitor. J Control Release, 2019. 296: p. 250–257. [DOI] [PubMed] [Google Scholar]
- 55.Zhou W, et al. , Inhibition of Rho-Kinase Downregulates Th17 Cells and Ameliorates Hepatic Fibrosis by Schistosoma japonicum Infection. Cells, 2019. 8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kureishi Y, et al. , Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem, 1997. 272(19): p. 12257–60. [DOI] [PubMed] [Google Scholar]
- 57.Yang Y, et al. , EPAC activation inhibits acetaldehyde-induced activation and proliferation of hepatic stellate cell via Rap1. Can J Physiol Pharmacol, 2016. 94(5): p. 498–507. [DOI] [PubMed] [Google Scholar]
- 58.Fu J, et al. , The role of cell division control protein 42 in tumor and non-tumor diseases: A systematic review. J Cancer, 2022. 13(3): p. 800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin J, et al. , MYLK promotes hepatocellular carcinoma progression through regulating cytoskeleton to enhance epithelial-mesenchymal transition. Clin Exp Med, 2018. 18(4): p. 523–533. [DOI] [PubMed] [Google Scholar]
- 60.Dong LQ, et al. , Phosphorylation of protein kinase N by phosphoinositide-dependent protein kinase-1 mediates insulin signals to the actin cytoskeleton. Proc Natl Acad Sci U S A, 2000. 97(10): p. 5089–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lachmann S, et al. , Regulatory domain selectivity in the cell-type specific PKN-dependence of cell migration. PLoS One, 2011. 6(7): p. e21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kale VP, et al. , A novel selective multikinase inhibitor of ROCK and MRCK effectively blocks cancer cell migration and invasion. Cancer Lett, 2014. 354(2): p. 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huo L, et al. , Cdc42-dependent formation of the ZO-1/MRCKbeta complex at the leading edge controls cell migration. EMBO J, 2011. 30(4): p. 665–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanchanawong P, et al. , Nanoscale architecture of integrin-based cell adhesions. Nature, 2010. 468(7323): p. 580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao XK, et al. , Focal Adhesion Kinase Regulates Hepatic Stellate Cell Activation and Liver Fibrosis. Sci Rep, 2017. 7(1): p. 4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar P, et al. , Adiponectin modulates focal adhesion disassembly in activated hepatic stellate cells: implication for reversing hepatic fibrosis. FASEB J, 2014. 28(12): p. 5172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee YA and Friedman SL, Inflammatory and fibrotic mechanisms in NAFLD-Implications for new treatment strategies. J Intern Med, 2021. 291:11–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen W, et al. , Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology, 2020. 72(2): p. 729–741. [DOI] [PubMed] [Google Scholar]
- 69.Ratziu V, et al. , Lack of efficacy of an inhibitor of PDE4 in phase 1 and 2 trials of patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol, 2014. 12(10): p. 1724–30 e5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibody and reagent information
Data Availability Statement
Proteomic files have been deposited in MassIVE (http://massive.ucsd.edu/) for this study. Data files for the following have been included in the MassIVE submission: (A) the primary data files (.RAW), (B) sample key, (C) the sequence databases (Mouse reviewed FASTA dated Feb 28, 2020), (D) TMT isotopic purity correction factors and (E) PeaksX results as excel files. The shared data will be released from private embargo for public access upon final acceptance for publication.