

Abstract

Antibody–drug conjugates (ADCs) are targeted immunoconjugate constructs that integrate the potency of cytotoxic drugs with the selectivity of monoclonal antibodies, minimizing damage to healthy cells and reducing systemic toxicity. Their design allows for higher doses of the cytotoxic drug to be administered, potentially increasing efficacy. They are currently among the most promising drug classes in oncology, with efforts to expand their application for nononcological indications and in combination therapies. Here we provide a detailed overview of the recent advances in ADC research and consider future directions and challenges in promoting this promising platform to widespread therapeutic use. We examine data from the CAS Content Collection, the largest human-curated collection of published scientific information, and analyze the publication landscape of recent research to reveal the exploration trends in published documents and to provide insights into the scientific advances in the area. We also discuss the evolution of the key concepts in the field, the major technologies, and their development pipelines with company research focuses, disease targets, development stages, and publication and investment trends. A comprehensive concept map has been created based on the documents in the CAS Content Collection. We hope that this report can serve as a useful resource for understanding the current state of knowledge in the field of ADCs and the remaining challenges to fulfill their potential.
1. Introduction
Antibody–drug conjugates (ADCs) are currently among the most promising drug classes in oncology because of their ability to deliver cytotoxic agents to specific tumor sites, combining the selectivity of monoclonal antibodies (mAbs) and the efficacy of chemotherapeutic drugs.1−4 Cancer is a major global health threat, causing millions of fatalities yearly.5,6 Chemotherapies based on cytotoxic drugs have been the main strategy for treating of a wide assortment of cancers for decades.7,8 Cytotoxic drugs include a large variety of compounds such as alkylating agents, antimetabolites, antitumor antibiotics, topoisomerase and mitotic inhibitors, and corticosteroids among many others.9−14 Most of these antitumor drugs, however, exhibit low therapeutic index and cause severe side effects due to nonspecific drug exposure to off-target tissues.15 To address these challenges, intense research has been carried out, aimed at developing novel cancer therapeutics with better targeting ability and less harmful side effects.
ADCs are targeted immunoconjugate constructs that integrate the potency of cytotoxic drugs with the selectivity of monoclonal antibodies (Figure 1), minimizing damage to healthy cells and reducing systemic toxicity. The structure of an ADC consists of three main components: a monoclonal antibody, a cytotoxic drug payload, and a linker molecule.16−19 The monoclonal antibody is engineered to bind specifically to a target antigen that is overexpressed on cancer cells. This allows the ADC to selectively target cancer cells while sparing normal cells. The cytotoxic drug payload is a potent chemotherapeutic agent that is highly effective in killing cancer cells. The linker molecule attaches the cytotoxic drug to the antibody, and its stability is crucial in controlling the release of the drug within the target cell.16,20−22 Once the ADC binds to the cancer cell surface, it is internalized through receptor-mediated endocytosis. Within the cancer cell, the linker molecule is designed to release the cytotoxic drug payload either by enzymatic cleavage or through chemical degradation. Once released, the cytotoxic drug exerts its therapeutic effect by disrupting key cellular processes and inducing cancer cell death.
Figure 1.

Structure and mechanism of action of ADCs. (A) Scheme of antibody structure including heavy chains, light chains, constant region, variable region, and antigen binding site. (B) Antitumor ADCs combine three key elements: a monoclonal antibody moiety that binds to an antigen preferentially expressed on the tumor cell surface, thereby ensuring specific binding to tumor cells; a covalent linker that warrants that the payload drug is not released in blood ahead of time, but is released within the tumor cell; and a cytotoxic payload that causes tumor cell apoptosis through targeting of key components such as DNA, microtubules. (C) ADC mechanism of action includes key sequential steps: binding to cell surface antigen; internalization of the ADC–antigen complex through endocytosis; lysosomal degradation; release of the cytotoxic payload within the cytoplasm; and interaction with target cell components. A fraction of the payload may be released in the extracellular environment and taken up by neighboring cells, known as the bystander effect.
ADCs offer several advantages over conventional chemotherapy. By selective targeting of cancer cells, ADCs reduce damage to healthy tissues and minimize side effects. This allows for higher doses of the cytotoxic drug to be delivered to the tumor, potentially increasing efficacy. Additionally, the antibody component of ADCs can elicit an immune response against the cancer cells, further enhancing their antitumor activity. Therefore, ADCs have become a major approach in the research and development of antitumor drugs.16,21,22
The German scientist Paul Ehrlich is credited with coining the term chemotherapy to indicate the use of chemical compounds to treat disease.23 Following his experience with antibodies, Ehrlich also proposed the concept of a “magic bullet” (Figure 2) that would bring about a selective targeting of pathogens without injuring the rest of the organism, one of the most notable notions of modern medicine.24 Ehrlich’s idea of targeted therapy was first materialized almost 50 years later, with methotrexate linked to an antibody targeting leukemia cells.25 The innovative development of hybridoma technology to produce mouse mAbs greatly advanced the field.26 It took nearly eight decades until Ehrlich’s visionary concept was advanced to achieve the first human trial of ADC therapy using an anticarcinoembryonic antigen antibody-vindesine conjugate.27 Subsequent advances in technologies for the ADC constituent building blocks–the antibody, the linker, and the payload–have resulted in the development of greater number of ADCs with enhanced potency, improved pharmacokinetics, reduced immunogenicity and overall toxicity, and upgraded specificity for cancer cells.28 Early ADC prototypes were created but they suffered from issues such as limited stability and inadequate target specificity. In the 1990s preclinical studies demonstrated the potential of ADCs in improving the therapeutic index of cytotoxic drugs by targeting specific antigens expressed on cancer cells.29−31 However, early clinical trials encountered challenges including toxicity and insufficient efficacy. In the late 1990s to early 2000s advances in antibody engineering and linker technologies contribute to the development of more stable and effective ADCs.32−35 Several ADC candidates have entered clinical trials, showing promising results in terms of efficacy and safety.
Figure 2.

Timeline of key events and discoveries in the antibody–drug conjugate research and development.21,24−27,29−34,36−80
The early, first-generation, ADCs used clinically approved drugs with well-documented mechanisms of action, including the antimetabolites methotrexate and 5-fluorouracil, the DNA cross-linker mitomycin and the antimicrotubule agent vinblastine.30 First-generation ADC, however, showed insufficient drug efficacy, linker instability, targeted antigens expressed in both normal and cancerous cells, and triggered undesired immune responses.29 Further developments, including higher drug efficacy and thoroughly selected targets, eventually led to the first ADC, Mylotarg (gemtuzumab ozogamicin), to get approval from the US Food and Drug Administration (FDA) in 2000 for the treatment of CD33-positive acute myelogenous leukemia.33,34 Regardless of the hopeful clinical results, Mylotarg was withdrawn from the market because it did not exhibit progress in overall survival. The second ADC to be developed, Adcetris (brentuximab vedotin), received approval from the US FDA in 2011 for the treatment of Hodgkin’s lymphoma and anaplastic large-cell lymphoma.43,44 The next ADC, Kadcyla (trastuzumab emtansine), used a construct combining the humanized antibody trastuzumab with a powerful microtubule inhibitor cytotoxic drug via a highly stable linker. It was approved for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer in 2013.45,46
Recent advancements have resulted in a new generation of ADCs with better chemistry, manufacturing, and control (CMC) properties, including linkers with optimized stability and powerful cytotoxic agents. Currently, there are 15 approved ADCs, which have received market endorsement worldwide, with over 150 candidates being investigated in various stages of clinical trials at present. Out of these, ∼12% are in the late-phase (Phase III/IV) owing to rapid advancements in technology required for generating ADCs. Thus, ADCs as an anticancer treatment strategy is leading a new era of targeted cancer defeat. Additionally, ADCs are being increasingly applied in combination with other agents. The growing diversification of target antigens as well as bioactive cytotoxic payloads is rapidly extending the range and scope of tumors targeted by ADCs. However, toxicity continues to remain a key issue in the development of these agents, and a better understanding and management of ADC-related toxicities will be essential for further optimization. Although numerous challenges remain, recent clinical accomplishments have produced intense interest in this therapeutic class reflected in a rapidly growing number of publications (Figure 3). The development of ADCs continues to be a particularly active area of research and development, with ongoing efforts to optimize the design of antibodies, linkers, and cytotoxic drug payloads. The goal is to improve the efficacy and safety profile of ADCs and apply them to a wider range of cancers and to other diseases.
Figure 3.

Yearly growth of the number of documents (journal articles and patents) in the CAS Content Collection related to antibody–drug conjugate research and development.
This report provides a detailed overview of the recent advances in antibody–drug conjugate research and considers future directions and challenges in advancing this promising platform to widespread therapeutic use. We examine data from the CAS Content Collection,81 the largest human-curated collection of published scientific information, and analyzed the ADC publication landscape of recent research (2000 onward) to uncover the trends in published documents (both journals as well as patents) and to provide insights into scientific advances in the area. We also discuss the evolution of key concepts in the field as well as the major technologies and the development pipelines of ADCs with a particular focus/emphasis on company research, disease targets, and development stages. We hope this report can serve as a useful resource for understanding the current state of the field of ADCs and the remaining challenges to fulfill and achieve their full potential.
2. ADC Basics
2.1. Selection/Optimization of Antibodies
Antibodies are an essential building block of ADCs (Figure 1) that possess characteristic requirements. It should have a high affinity and avidity for the target antigen but no or insignificant binding to off-target sites and it is important to have selective binding to the target antigen leading to the accumulation and retention of ADCs at the target site.82−84 In addition, the antibody should have low immunogenicity, low cross-reactivity, optimum-linker binding, and a long half-life.85,86 It is interesting to note that the antibody component of many ADCs retains its activity profile and can therefore interfere with target cell function, alter downstream signaling, and/or cause immune effector cells to elicit payload-independent antitumor immunity, thereby acting beyond mere payload carrier. For example, the Fab region of the antibody in ADCs can disrupt target function by blocking ligand binding, interfering with dimerization and/or endocytosis, and target protein degradation.84,87 Likewise, the Fc region of the antibody can induce antibody-dependent cell cytotoxicity (ADCC). Most ADCs such as Kadcyla (T-DM1), Enhertu (T-DXd), Polivy, Padcev, Trodelvy, just to mention a few, rely upon the ADCC-competent IgG1 backbone.59,61,65,85,88 The Fc region is also responsible for complement-dependent cytotoxicity and antibody-dependent cellular phagocytosis.
A significant and long-recognized challenge in the field is the heterogeneous distribution of the antibodies when administered systemically.89 Antibody internalization and clearance obstruct uptake in solid tumors, limited by tumor vascular permeability and diffusion.90 Mathematical analysis of antibody distribution through tumors applying simple scaling relationships can help understanding the tumor physiology and antibody tumor penetration.90,91 Fundamental understanding of the mechanisms and time scales of antibody transport and clearance are essential for the prediction of the distance an antibody may permeate through tumor tissue, with the balance between these processes controlling the antibody penetration into a tumor and therefore its optimal efficacy.91 Thus, for solid tumors, optimal binding affinity has been suggested to depend on the level of target expression.91
Initially, first-generation ADCs used murine antibodies, but they elicited a robust immune response with some patients producing antihuman antibodies, leading to the generation of second-generation chimeric antibodies. These mouse/human chimeric antibodies contain the variable regions of mouse light- and heavy-chains linked to human constant regions. Recent developments in the field have moved toward fully humanized mAbs which do not produce immunogenic responses.88,92 Many innovative strategies are being used to maximize the efficacy of ADCs including the use of biparatopic antibodies. These antibodies target two different epitopes of the same target antigen and can help cluster the antigenic receptors, leading to the rapid internalization of ADCs.
For designing present-day ADCs, immunoglobulin G (IgG) is the most widely used antibody isotype. ADCs must have similar pharmacokinetic properties to those of normal human IgGs. Human IgG comprises four subclasses: IgG1, IgG2, IgG3, and IgG4, which differ in their constant and hinge regions. Despite being the most immunogenic, use of IgG3 is avoided in ADC design due to its short serum circulating half-life (∼7 days) when compared to other subclasses (∼21 days).88,93 Though IgG1, IgG2, and IgG4 have suitable serum half-lives, IgG2 is rarely used owing to its tendency to dimerize and aggregate in vivo.94 Most ADCs are developed on IgG1 platforms because of improved solubility, greater complement-fixation, low nonspecific immunity, and better immune effector cell receptor (FcγR)-binding efficiencies of IgG1, which can play a crucial role in anticancer activity of ADCs.3,84 Though IgG4 can also induce antibody-dependent phagocytosis (ADCP), its dynamic nature due to Fab-arm exchange contributes to reduced efficacy. Despite this, a few ADCs, such as Gemtuzumab ozogamicin and Inotuzumab ozogamicin, use IgG4 as the platform to target CD33 and CD22, respectively.95−97 Gemtuzumab ozogamicin contains an IgG4 core-hinge mutation that blocks Fab-arm exchange.98
″Biparatopic” or “bispecific antibodies” are antibodies designed to simultaneously bind to two different epitopes (binding sites) on the same target antigen or two different antigens.99,100 This dual binding capacity can offer several advantages in terms of therapeutic applications. Thus, having two antigen-binding sites allows them to target either two distinct epitopes on the same antigen or two different antigens altogether. Their advantages include (i) enhanced targeting–bispecific antibodies can engage two binding sites on the same antigen, potentially increasing binding affinity and specificity; (ii) versatility–they can target multiple antigens simultaneously, which is particularly valuable in cancer therapy or immunological diseases; (iii) cross-linking–in the context of cancer therapy, bispecific antibodies can cross-link cancer cells and immune cells, such as T cells, leading to targeted cell killing.100
Antibodies with masked binding domains have been engineered to have one or more of their antigen-binding sites temporarily inactivated or “masked”.101,102 These antibodies can be designed to change their binding properties under specific conditions. Their applications include conditional activation, for instance, an antibody may only become active when exposed to specific environmental factors or cellular cues; reduced off-target effects—in some cases, masking can be used to prevent antibody binding to nontarget tissues or cells until it reaches the desired site. CytomX’s Probody technology is one example where the binding domains of antibodies are masked by a peptide, which is selectively cleaved by proteases present at the tumor site.103 This allows for the activation of the antibody’s binding and therapeutic functions specifically within the tumor microenvironment.
Both biparatopic/bispecific antibodies and antibodies with masked binding domains are innovative strategies in antibody engineering, offering greater control and versatility in targeting diseases while minimizing off-target effects. These approaches continue to advance in the field of immunotherapy and targeted therapies with potential applications in cancer, autoimmune diseases, and other medical conditions.
The antibodies used in ADC design are large compared to the actual cytotoxic payload, which implies that much of the actual formulation is being utilized to deliver the antigen to the target rather than to execute its pharmacological activity. The large size of the antibody (∼150 kDa) can also cause issues with target penetration in solid tumors. The targeting capacity of antibodies is achieved through its small variable loop structures (VH) present at the terminal portion of Fab fragments; therefore, fragments of native antibodies can be used to develop smaller binding motifs such as F(ab)2, Fab′, Fab, and Fv fragments. In addition, engineered scaffolds, such as single-chain variable fragments (scFv-Fc), single domain antibodies (sdAbs), and diabodies, are being explored. Furthermore, humanized fragments of unusual IgGs, such as heavy chain variable domain(VHH) and heavy chain variable domain-based antibody (VNAR) fragments from camelids and shark antibodies, respectively known as nanobodies, are being studied.104 Owing to their small size, ease of production, manipulation, conjugation, high solubility, stability, and delayed serum clearance, these antibody fragment conjugates or FDCs have various advantages compared to their antibody precursors.105
For example, Fan et al. developed an epidermal growth factor (EGFR)-targeting nanobody-drug conjugate that displays potent anticancer activity in solid tumor models.104 In recent years, ASN004, an scFv-Fc ADC, has been developed that targets 5T4 oncofetal antigens expressed on a wide range of malignant tumors. It is designed by conjugating a novel scFv-Fc antibody with an Auristatin F hydroxypropylamide (AF-HPA) payload using Dohtlexin drug-linker technology. The developed ADC has a high drug-antigen ratio (DAR) of approximately 10–15 and has shown tumor repression in preclinical models.106 ANT-045 and ANT-043 are other scFv-Fc conjugated ADCs being developed by Antikor Biopharma that have shown successful results against solid tumors. While ANT-043 has tumor ablation effects in gastric, breast, and cancer xenograft models, ANT-045 has high stability, excellent in vitro cell-kill potency, and is successful in in vivo tumor cures.107
With the advancement in molecular biology techniques, site-specific antibody conjugation is being introduced into ADC development to increase the therapeutic efficacy. Antibodies can be engineered using genetic engineering, chemoenzymatic modifications, or metabolic labeling through their Fab or Fc region to introduce specific reaction sites for ease of conjugation. A few modifications include introducing natural amino acids such as cysteine (Cys) or glutamine (Gln).108−110 Apart from natural amino acids, unnatural amino acids (UAA) (also referred to as noncanonical amino acids) containing orthogonal side-chain functional groups are also introduced at different positions in the antibody for generating sites for stable conjugation. For example, tubulin inhibitor payload AS269 is conjugated to a HER2- targeted mAb incorporated with a UAA, pAF. The resulting anti-HER2 ADC (ARX788) has a DAR of 1.9 and exhibits a high serum stability and half-life. ARX788 showed strong antitumor activity in mice and is currently undergoing Phase III clinical trials.111,112 Recently, other ligands such as short peptide tags, modified glycans, and small molecule-based affinity ligands are also used to generate conjugation sites for antibody payloads.109,113,114 Some ADCs in preclinical/clinical development have attenuated effector functional activity, with the intention for reducing off-target off-tumor toxicity.115−117
Similar to ADCs, peptide-drug conjugates (PDCs) consist of a peptide two-five amino acids in length bound to the cytotoxic payload via a linker. A key difference between ADCs and PDCs is the substantially smaller size of the peptide component (2–20 kDa) of PDCs than the antibody component (∼150 kDa) of ADCs (5–25 amino acids vs ∼1000 amino acids in peptides and antibodies, respectively)118,119 This allows for better absorption and uptake than conventional ADCs, which is especially important for drugs that need to cross the BBB.118,119 ADCs and PDCs show a great degree of overlap in terms of linker chemistry and types of cytotoxic payloads. A variety of peptides have been utilized in PDCs, including bicyclic toxin peptides and dendritic peptides.119 Bicyclic peptides are small, constrained peptides, many of which have high affinity for target antigens. An example of a PDC is BT8009, which consists of a bicyclic peptide linked to MMAE. BT8009 binds cell adhesion molecule, nectin-4 with high affinity (∼3 nM) and specificity and has shown promise in NSCLC and pancreatic ductal xenograft models.120 Currently, several PDCs are in clinical trials or being actively explored.119
2.2. Linkers for Use in Antibody–Drug Conjugates
Linkers connect the active molecule in an ADC to the antibody that determines the target for the drug. To be effective, a linker should be stable in plasma.121 It should not alter the behavior of either the drug or the antibody to which it is attached. The linker should be sufficiently hydrophilic to moderate or mitigate the solubility effects of warheads, which, in most cases, are lipophilic. The linkers should not aggregate; since aggregation is likely to impair the activity of the antibody and may reduce the stability of the ADC. Finally, the linker should release the drug completely and selectively under appropriate conditions. The choice of linker will be determined by the functionality present on the drug and the antibody.
2.2.1. Linker Types
Linkers can be divided into two major classes: cleavable and noncleavable. Cleavable linkers allow the drug to be separated from the antibody without proteolytic cleavage of the antibody, while noncleavable linkers require proteolysis of the antibody for the drug to diffuse to its site of action.
2.2.1.1. Cleavable Linkers
Cleavable linkers allow the drug attached to an antibody to be freed from the ADC without destroying the antibody.17 Most commonly, cleavable linkers can be severed by acid, reducing agents, or enzymes.
Acid-sensitive linkers are chosen because the microenvironment of tumors is often acidic and hypoxic;122 with parenteral administration ensuring that the drug is released selectively only in the tumor. Acid-cleavable linkers are most commonly hydrazones; for example, a hydrazone is used in the linker for the first USFDA-approved ADC Mylotarg.123 One disadvantage of acid-sensitive linkers is premature or nonselective release; Mylotarg was withdrawn in part because of nonselective toxicity (which was mitigated by changes in ADC dosing).124
Reductive linkers allow the release of the drug under reducing conditions; the hypoxic environment of tumors makes release of the drug at the tumor site more likely than elsewhere. Reductive linkers have most commonly been disulfides; disulfides can be cleaved either by reduction of the sulfur–sulfur bond or by exchange of thiols (such as glutathione, present in high concentrations in cancer cells) with disulfides to release a thiol (either the antibody or the drug).121 Cysteines or other thiols comprise the linker, but when the disulfide is attached to unhindered carbon atoms, nonselective cleavage of the disulfide is more likely. This can be mitigated significantly by two α-methyl groups on one of the carbons at the disulfide.125,126
Finally, as the name suggests, enzyme-cleavable linkers incorporate linkages that are selectively cleaved by enzymes. Enzyme-cleavable linkers most commonly use amides as the linking moieties because of the prevalence of enzymes in organisms that process and cleave amide bonds in proteins. Amides are thermally and hydrolytically stable, reducing the likelihood of premature or nonselective drug cleavage. The availability of a wide variety of enzymes and enzyme targets allows linkers to be tuned to avoid hydrolysis by enzymes in normal cells and to facilitate hydrolysis in tumor cells. Initially, di- and tripeptide linkers were used; one of the most common linkers contain the valine-citrulline (Val-Cit) linkage, a target for cathepsin B which is overexpressed in some tumor cells.127 Phenylalaninylvaline, valinylalanine, and tetrapeptide linkers have been deployed as well.128 The valine-citrulline linker is used in the approved ADC Adcetris, while the terminated clinical candidate T-Rova uses a valine-alanine linker.129 While amides are the most common in enzyme-cleavable linkers, other motifs have also been used. For example, substituted ortho-hydroxybenzyl β-glucuronides17 and β-galactosides121 are susceptible to hydrolysis by β-glucuronidase and β-galactosidase, respectively. Alternatively, substitution of the hydroxybenzyl phosphate-containing linkers yields linkers susceptible to hydrolysis by phosphatases. Cleavage of the acetal or phosphate linkage generates an ortho-hydroxybenzyl group, which subsequently undergoes dealkylative cleavage via an ortho-quinone methide to split the linker. The protected ortho-quinone methide linker is termed a self-immolative linker;130 while it is stable under physiological conditions, rapid cleavage occurs when the hydroxy group is unveiled. The self-immolative linker can be altered to incorporate a variety of groups sensitive to various enzymes, allowing linker cleavage to be tailored to the necessary selectivity.
2.2.1.2. Noncleavable Linkers
Noncleavable linkers are designed to remain intact until the antibody is proteolyzed in lysosomes after internalization. One example of a noncleavable linker is the maleimido thioether linker in Kadcyla;88,131 while retro-Michael reaction of the β-thioether amide is possible, the modes of cleavage common under physiological conditions are limited. Since conditional cleavage of ADC containing noncleavable linkers is rare to nonexistent, premature release of the drug moiety should be limited to cleavage of the antibody itself, an unlikely event. The off-target toxicity of ADC using noncleavable linkers should thus be minimal. In addition, proteolytic cleavage of the ADC yields a drug moiety substituted with a remaining fragment of the antibody; the peptide fragment is likely polar and charged, preventing the escape of the drug from the cell. The peptide-substituted drug is thus retained within the cell, limiting its ability to kill neighboring cells or to enter circulation and kill nontarget cells; the antibody fragment may also limit transporter-mediated resistance to the drug (though it is unlikely to reduce the effects of other resistance pathways).
Exemplary cleavable and noncleavable linkers used in ADCs are shown in Figure 4.
Figure 4.

Exemplary ADC linkers: (A) acid labile hydrazone linker; (B) enzyme cleavable Val-Cit linker; (C) glutathione-sensitive disulfide linker; (D) noncleavable maleimidomethyl cyclohexane-1-carboxylate (MCC) linker.
2.2.1.3. Branched Linkers
Branched linkers have been devised for ADC to access ADC with higher DAR.132 The closest technology to clinical use is that developed by Mersana Therapeutics in which a glycerol-glycolaldehyde condensation polymer (Fleximer) with pendant esters of mercaptocarboxylic acids and drug moieties is prepared and attached to an arylmaleimide-substituted antibody.133,134 The method can produce ADC with a DAR of 10–15. A vincristine-functionalized trastuzumab using the technology had antitumor activity similar to and lower toxicity than the corresponding conventionally generated ADC. Two ADC using this technology have entered clinical trials. Upifitomab rilsodotum135,136 uses MMAF as the warhead; in a Phase 1/2 trial, it did not show improvement over control against platinum-resistant ovarian cancer. A second ADC, XMT 2056, uses the same platform but uses an STING agonist as the warhead. It is in Phase 1 clinical trials for treating metastatic HER2-positive tumors; unfortunately, its trials are in clinical hold due to a severe adverse event.137 Other branched linker methodologies use transglutaminase-mediated coupling of trisubstituted piperidine-containing amines with sequence-modified antibodies,134,138 the preparation and use of carbamoylethyl- and carbamoylethoxy arylmaleimides by Firefly Bio as branched linkers for ADC,139 the use of pentaerythritol-derived linkers containing amine or oxime moieties for antibody conjugation, fluorescent linkers, and three azide moieties for attachment pf payloads by Sapozhnikova et al.,140 and the preparation and use of disubstituted tetrahydropyrazinoindoles as antibody linkers via condensation of aldehyde-containing antibodies with trisubstituted indolemethanehydrazines by R.P. Scherer Technologies.141 Linkers that carry two drug units per linker have been also reported for typical hinge cysteine-based conjugation.142,143
2.2.2. What Types of Linkers Are Used in ADCs? Why?
Of the currently approved ADCs, cleavable linkers (particularly enzyme-cleavable linkers) predominate, with 11 out of 13 USFDA-approved ADCs having cleavable linkers19 (and eight of the ADCs having enzyme-cleavable linkers). Thus, while premature release of the payload (as seen in Mylotarg) in ADC possessing cleavable linkers can lead to unacceptable off-target toxicity and reduced exposure that potentially affects efficacy, the benefits of selective linker cleavage appear to outweigh the liabilities. One way in which this may occur is by the “bystander effect”.127 If the payload is released either near a target cell or is transported from (or diffuses from if the drug lacks charged groups such as carboxylates), then the drug can be taken up by neighboring cells, resulting in their death. In most cases, cells in the vicinity of a tumor cell are likely to be tumor cells; thus, the bystander effect allows the ADC to affect cells in the vicinity of an antigen-presenting cell. Since one of the many ways that cancer cells evade treatment is to downregulate the production and expression of surface antigens, ADCs using cleavable linkers can take advantage of the bystander effect and circumvent some of the resistance modes of tumor cells.
2.3. Drugs Incorporated into ADCs
2.3.1. What Are the Requirements for Drugs To Be Incorporated as Payloads into ADCs?
First, antibodies such as IgG (approximately 150 kDa) are large; attachment of many drug molecules to an antibody often makes it more lipophilic, causing aggregation of the ADC and subsequent degradation and inactivation. The number of drug molecules linked to an antibody is small (in most cases, four or fewer), making the effective dose of the drug in an ADC small. In addition, the low numbers of antigens per cell and the imperfect delivery of ADC to cells further reduces the likely dose of drug from ADC.131 Thus, because only a small dose of drug is possible, the drug chosen for use in an ADC needs to be highly potent (exhibiting its effects at nM to pM concentrations) to boost efficacy. However, the effective dosage of drug to cells from ADCs has shown a plateau in mice, indicating that a sufficient ADC can be delivered to a tumor to oversaturate it with the drug (with the caveat that the drugs delivered in this case were highly potent and adapted for the ADC).144 Second, drugs in ADCs are attached to antibodies and are not free to diffuse into cells. If the linker between a drug and an antibody remains intact until delivery, the antibody controls where the drug is released, allowing less selective drugs to be used in ADCs than those given systemically. Finally, the drug needs to be stable both to storage and to administration; since ADCs are administered parentally, the drug in an ADC needs to be stable in blood and plasma (though attachment to an antibody may provide steric shielding for a drug and thus may reduce its reactivity and increase its stability). A secondary consideration is if the ADC is being used in combination with other drugs, the ADC warhead should act on a different target, a different biological pathway, or at a different phase of the cell cycle than the other drugs being used.3
2.3.2. What Types of Drugs Are Used as Payloads for ADCs?
Auristatins are tubulin polymerization inhibitors derived from the marine natural product dolastatin 10, with potencies of 50–100 pM.88 Monomethyl auristatins F and E have been used as warheads for ADCs such as Adcetris, Polivy, and Blenrep.145
Maytansinoids are ansa-macrolide natural products isolated from the shrub Maytenus serrata.146 They bind tubulin and prevent its assembly into microtubules, thus inhibiting mitosis and cell replication.147 Maytansinoids were tried as antitumor agents but were not effective at tolerable concentrations. The maytansine DM1 is the warhead of the ADC Kadcyla.88
Camptothecin and its analogs such as irinotecan, topotecan, govitecan, SN-38, exatecan, and deruxtecan inhibit topoisomerase 1, an enzyme that unwinds and cuts a single strand of supercoiled DNA, allowing it to be repaired and replicated;148 its inhibition leads to DNA cleavage and cell death. A variety of camptothecin analogs have been tried as antitumor agents, but their aqueous solubilities and side effects have hindered their use as antitumor agents.149 Some of the camptothecins have also been susceptible to export by ABC transporters; annulation of an additional ring as in exatecan prevented transport but caused myelotoxicity, which was reduced further by addition of a maleimide-terminated peptide to form deruxtecan. Camptothecin analogs are warheads in the USFDA-approved ADC Enhertu and Trodelvy.88
Pyrrolobenzodiazepines such as tesirine and talirine were derived from the natural product anthramycin.150 They alkylate DNA with extremely high potencies (as low as 100 fM).88 One pyrrolobenzodiazepine (SJG-136) was tried as an antitumor agent but progressed only to Phase I trials due to significant toxicity with no antitumor response.151 Their high potency makes them attractive warheads for ADCs.152 A variety of ADCs with pyrrolobenzodiazepine warheads have been tried, with one (Zylonta) having been approved as of December 2021 for the treatment of B-cell lymphomas. Their dimeric nature and the presence of two alkylating moieties allows them to cross-link DNA, which creates DNA damage that is difficult to repair. However, these same features are also likely responsible for undesired off-target toxicity. Structurally related indolinobenzodiazepine dimers have also been studied as warheads for ADC;153 incorporation of monoamine indolinobenzodiazepines has been used to generate antitumor ADC with reduced off-target toxicities.131,154
Calicheamicin γ1 and related natural products such as dynemicin contain strained enediyne moieties. Under reductive conditions, DNA-bound calicheamicin undergoes Bergmann cyclization to generate diradicals which cleave both strands of DNA, leading to damage which is difficult or impossible for cells to repair. It inhibits DNA replication at pg/mL concentrations155 but is also toxic as a result. The combination of potency and toxicity suggested the potential use of calicheamicin γ1 in ADCs. The first approved ADC, Mylotarg, incorporated calicheamicin as a warhead; it was approved in 2001 but withdrawn in 2010 because of its toxicity. Development of modified dosing allowed Mylotarg to be reapproved in 2017 with an expanded patient population.124 The ADC Besponsa also uses a calicheamicin derivative as a warhead.
Many other compounds have been tried as warheads for ADC. Duocarmycins such as CC-1065 and seco-DUBA are DNA-alkylating agents effective at nanomolar to picomolar concentrations.156 Tubulysins are noncanonical peptides containing thiazole moieties that act as microtubule polymerization inhibitors and are active against cancer cells at nanomolar to picomolar concentrations,157 making them attractive candidates for ADCs.158 However, some of the tubulysins are unstable in aqueous environment and can show unselective toxicity in cancer cells. Cryptophycins are macrolides which inhibit tubulin polymerization at picomolar concentrations; however, trials against cancer showed toxicity but not efficacy.159,160 Despite this, their potency has made them attractive payloads for ADCs.161 The spliceostatins and thailanostatins are natural products that inhibit the spliceosome, modifying mRNA sequences and thus influencing protein expression. They have shown inhibition of cancer cells at nanomolar concentrations162 and hence are potential ADC payloads.163 Doxorubicin is an intercalating agent for modification of DNA which was used as one of the first payloads for ADCs but was not effective because of its low potency. However, if the DAR of doxorubicin conjugates can be increased by newer conjugation methods, it may prove to be an effective warhead for ADCs. Alternatively, more potent anthracycline drugs have been developed. For example, PNU-159682 is an anthracycline acting by a similar mechanism to doxorubicin which was found to be nearly 1000 times more potent than doxorubicin.164,165 This enhanced potency enables ADCs incorporating it to be highly effective.166 α-Amanitin, a fungal toxin which inhibits RNA polymerase II, is a significant cause of liver failure and death from toxic mushroom ingestion.167 Its toxicity, robustness, and moderate size make it a reasonable choice as a warhead for ADC.168 Protein toxins have been used as warheads for ADCs; for example, Pseudomonas exotoxin A169 is the warhead for the USFDA-approved ADC Lumoxiti.170 Diphtheria exotoxin171 has also been used as a payload for ADC.172
Finally, immunomodulating agents have been tested as antibody payloads, either to suppress immune responses for anti-inflammatory or immunosuppressant activities or to enhance the immune response to cancer.173−177 In addition, an ADC with antibiotic warheads have been designed for use as an antibacterial agent.178
2.4. Conjugation Methods for Antibody–Drug Conjugates
2.4.1. What Are the Important Features of a Conjugation Method?
As with the linking moiety, it is important that the method of attaching a drug to an antibody through a linker does not alter the activity or stability of the drug or the antibody. In addition, conjugation should be efficient, proceed in high yield (so that as little as possible of the reagents are used per unit of ADC) and proceed as rapidly as possible. It should also be selective and predictable so that the locations of attachment of a drug to the antibody are controllable, known, and consistent.179 While it would be optimal to have a single species generated by a method, it is not necessary as long as the ADC is composed of a consistent mixture of species with consistent stability, biological and physical properties, and biological activity.
2.4.2. What Is the Structure of an Antibody? Where Can It Be Functionalized?
Most antibodies used for ADC are immunoglobulins, of which the most commonly used is immunoglobulin G (IgG) (Figure 1).127 IgG contains two heavy chains and two light chains. The heavy chains are attached to each other and to the light chains through four (interchain) disulfide S–S bonds that hold the antibody together. Twelve other intrachain disulfide bonds control the tertiary structures of the light and heavy chains. There are also roughly 80 lysine residues in a typical antibody, of which 40 may be functionalized. In addition, one of the heavy chain glutamine residues is substituted with a branched-chain heptasaccharide which improves the ability of the antibody to trigger an immune response.180
2.4.3. Conjugation Methods to Native Antibodies
2.4.3.1. Lysine
Under most circumstances, however, the reactivity of specific residues is rarely controlled, unless the basicity of a residue is significantly altered by its position in the protein sequence or by the side chains of nearby residues. In most cases, the average number of residues functionalized can be controlled by stoichiometry but not the position of functionalization. The lysine ε-amine forms stable amides with acylating agents such as N-hydroxysuccinimidyl esters (particularly sulfonated N-hydroxysuccinimidyl esters,181 which have better aqueous solubilities), benzoyl fluorides,182 and acid anhydrides.183 Acylating agents can form esters with tyrosine, threonine, or serine residues, but the esters are not stable; thus, if not prevented, some of the acylating agents will be consumed, decreasing the amount of drug attached to the antibody. Affinity peptides have been used to control the location of conjugation with antibodies;184 for example, binding of a peptide-containing acylating agent to the Fc domain of an antibody directs the acylation reaction to nearby residues185 and the conjugation method has been performed on gram scale using a peptide-substituted thioester.186 Cleavage of the peptide yielded a thiol available for further reaction.
2.4.3.2. Cysteine
Cysteine is the most common residue functionalized in antibodies because the sulfur atom of its side chain is highly nucleophilic. Cysteine’s thiol moiety is acidic enough for its anion to be accessible under physiological conditions; the resultant anion is more reactive toward electrophiles than other residues but is not basic enough to cause side reactions. IgG does not natively contain free cysteine residues, requiring some of the disulfide bonds of the antibody to be cleaved in order to allow functionalization. The four interchain disulfide bonds are most often used;187 while this preserves the structure and function of the individual antibody fragments, the stability of the antibody may be compromised. Selectivity among the cysteine residues is difficult; while a single species can be formed if all eight cysteines are functionalized (as for the approved ADC Troldelvy and Enhertu), most methods yield mixtures of products.
The most common monofunctionalization reagent used is the maleimide. Maleimides undergo Michael additions of thiols readily under ambient conditions to yield alkylthiosuccinimides.188 The thiol-succinimide adducts, however, can undergo retro-Michael addition, which could lead to either incomplete functionalization of the antibody or premature release of the drug from the antibody and undesired toxicity. Ring opening of the imide to form a carboxylate-containing amide reduces retro-Michael reactions substantially.189 Exposure of the imide product to mildly basic conditions can be used to form the carboxylate;190 the presence of nearby positively charged residues facilitates imide ring opening and stabilizes the maleimide adducts. Intramolecular reactions can also be used to stabilize the cysteine adducts.191 Other monofunctionalization reagents for cysteine include palladium aryl complexes to yield aryl thioethers,192 disulfides derived from exogenous thiols (particularly acidic thiols),193 and alkenyl and alkynyl phosphorus194,195 and iodine reagents.196 The known reactions of haloacetamides with cysteine residues can also be used;96 while stable thioether linkages are formed, their reactivity may lead to difficulty in controlling the number of appended drug moieties or their residue selectivities.
Disulfide rebridging can be used to reduce the potential instability caused by complete functionalization of the cysteines generated by reduction of the four interchain disulfide bonds.179 Reaction of the intermediate octathiols with biselectrophiles yields bisthioethers in which two of the cysteines are connected by alkyl or aryl groups; the thioether linkages are robust and can stabilize the dimeric antibody structure. The number of bridges formed (and thus the number of drugs incorporated) is easily controlled; the structure of the alkylating agent and the number of available sites for drug conjugation on it determine the carrying capacity of the ADC. One disadvantage of the method is that the biselectrophiles are likely to not be commercially available and thus require synthesis. A variety of reagents has been developed for disulfide rebridging. Abzena developed bis(sulfonylmethyl)methyl ketones (ThioBridge) which undergo sequential elimination and Michael reactions to yield thioether-substituted ketones.197 The payload can then be attached with alkoxyamines or by reductive amination. A similar method for rebridging has been used by Novartis with dichloroacetone as the biselectrophile.198 Maleimides with bromo-, iodo-, or arylthiol-substituents undergo Michael/retro-Michael reactions to yield substituted maleimides;199−201 incubation at pH 8.4 yields maleimidic acids which are resistant to Michael and retro-Michael reactions at the linker. Dihalo- or bis(phenylthio)pyridazines undergo analogous reactions to dihalo- or bis(arylthiol)maleimides but are resistant to linker cleavage by retro-Michael reactions.202 The pyridazines can incorporate multiple drug moieties; in addition, their reactivity can be controlled by temperature. Other biselectrophiles used for disulfide rebridging are aryl di(bromomethyl)quinoxalines (C-Lock, Concortis Therapeutics203), cis-platinum diamine complexes (Invictus Oncology),204 and divinylpyrimidines.205
2.4.3.3. Other Residues
The N-terminal amino group of peptides is less basic than the ε-amino groups of lysine residues, making selective reactions at the N-terminus of the antibody heavy chains possible. The proximity of the N-termini to the receptor binding site of the antibody may complicate functionalization. While reaction at the C-terminus would be preferable (because the C-terminus of the heavy chains is distant from the antigen- or receptor-binding sites of the antibody), chemical methods for doing so are uncommon. Transamination at an N-terminal glutamine residue with a formylpyridinium salt followed by reaction with an alkoxyamine yielded a stable N-terminal oxime ether.206 Oxidation of an N-terminal serine moiety yields a formylglycine residue, which can react with an alkoxyamine to form an oxime ether or can form nitrogen heterocycles by condensation with carbonyl compounds.
Arginine residues are unreactive under physiological conditions because the guanidine conjugate base of the native guanidinium ion is highly basic, inhibiting reaction. However, dicarbonyl compounds can react with amidines or guanidines to yield stable imidazoles. An azidophenylglyoxal was designed for reaction with arginine residues to form azidophenylaminoimidazole moieties; azide–alkyne cycloaddition with a terminal alkyne-substituted warhead yields the functionalized antibody.207 Finally, Ugi reactions of a lysine residue, a nearby aspartate or glutamate residue, an azidoaldehyde, and an isonitrile formed azide-functionalized macrocycles amenable to drug attachment via azide–alkyne cycloaddition.208
2.4.4. Conjugation Methods for Non-Native Antibodies
If an ADC with a precisely known structure is desired, native antibodies may not allow sufficient selectivity. Incorporating non-native functional groups by modification of the heavy chain sequence makes selective antibody conjugation with existing chemistry possible. Antibodies can be produced either by inoculation of mice with an antigen, by phage display, or by biopanning,209 separation of the cells producing the antibody and hybridization with cancer cells, cloning of the antibody sequence, and insertion into CHO cells.210 Since the antibody DNA and protein sequences are known, they can be modified to obtain antibodies with non-native sequences.210 Multiple methods of sequence modification may be useful.
2.4.4.1. Cysteine Incorporation
IgG normally does not possess free cysteine residues; the cysteines are connected by oxidation to intra- and interchain disulfides. Because the reactivity of cysteine is distinct from other residues, a non-native cysteine residue should be readily functionalizable, and has been used in technologies such as THIOMAB.211 Incorporation of an N-terminal cysteine allows reaction with aldehydes to form thiazolidines212 which slowly releases the aldehyde payload and enables the adducts to be effective as ADC. The position of insertion of the cysteine into the antibody sequence, however, needs to be chosen to minimize perturbation of antibody structure; in addition, as noted earlier, the presence of positively charged residues nearby facilitates ring opening of maleimide conjugates and improves their stabilities to deconjugation. Another caveat of cysteine incorporation is that cysteines form disulfides with glutathione during antibody production which must be reductively cleaved, but methods have been developed to address this issue.213 Recent studies have shown that antibody engineering methods are used to add two or three unpaired Cys residues, boosting the DAR of ADCs to >2 per antibody.109
2.4.4.2. Noncanonical Amino Acids
The use of amino acids containing functionality not normally present in peptides or antibodies allows facile, biocompatible, and selective reactions such as azide–alkyne cycloaddition and oxime ether formation to be used for linking to drugs. However, the biosynthesis of antibodies containing noncanonical amino acids (NCAA) is difficult. NCAA incorporation normally requires one of the stop codons in translation (most commonly, the amber codon UAG) to be suppressed and instead be used to encode the desired amino acid. Amber codon suppression, however, is believed to harm the cells in which it is deployed,214 requiring alterations to the expression methods. Cell-free systems can be used to generate NCAA-containing antibodies, but they lack the ability to glycosylate the finished antibody,215 which reduces its ability to summon an immune response. In addition, the site of incorporation requires optimization to avoid altering the stability or function of the antibody. Antigen- and receptor-binding sites and the hinge regions must be avoided, while solvent-exposed sites are preferred to increase the rate of drug attachment.
p-Acetylphenylalanine,216,217 p-azidophenylalanine,218,219 p-azidomethylphenylalanine,220,221 and azidolysine222−224 are stable in cells and do not alter protein structure significantly. p-Acetylphenylalanine reacts readily with alkoxyamines, while p-azidophenylalanine, p-azidomethylphenylalanine, and azidolysine undergo either copper-catalyzed or strain-promoted azide–alkyne cycloadditions with terminal alkynes or cyclic alkynes, respectively. All can be incorporated into peptides in reasonable yields. N-Propargyllysine is stable and amenable to copper-catalyzed azide–alkyne cycloaddition225 but differs enough in structure from lysine to inhibit its incorporation into peptides.226 Cyclopropene- and cyclopentadiene-substituted amino acids undergo cycloaddition reactions with tetrazines or maleimides to form stable pyrazine or bicycloheptene adducts.227,228 Selenocysteine is a rare but natural amino acid; the selenol group is more acidic and yields an even better nucleophile than a cysteine residue. Incorporation of selenocysteine into an antibody was achieved,229 despite low yields because of undesired termination at the erstwhile stop codon. Finally, suppression of two of the three stop codons allows peptides to be translated with two NCAA, which allows two different warheads to be attached to a single ADC.230 The difficulty of incorporating NCAA into an antibody likely means that the use of NCAA-based conjugation reduces the possible drug capacity of the ADC.
2.4.4.3. Incorporation of Additional Amino Acids
Addition of amino acid sequences to the C-terminus of the heavy chain of an antibody can be used to selectively conjugate drugs. (While the addition of amino acids increases the size of the ADC, large addends are required to perturb the antibody’s movement because it is already large, 150 kDa in the case of IgG). The cysteine in a π-clamp sequence (FCPF) selectively reacts with perfluorinated benzenes to yield substituted fluoroaryl thioethers.231 The cysteine in a different cysteine-containing sequence (LCYPWVY) undergoes reaction with dibenzocyclooctynes to yield stable dibenzocyclooctenyl thioethers.232 Both reactions can thus be used to attach drugs to an antibody. Appending a receptor to the C-terminus of an antibody can be used to attach drugs to the antibody if a covalent or irreversible inhibitor of the receptor exists; this method was used with CD38.233 Finally, a catalytic antibody sequence (38C2) was incorporated into an IgG1 antibody (without significant alteration of its properties); the antibody alters the basicity of nearby lysine and arginine residues, making their selective functionalization possible.207 The size and position of added sequences are likely to limit the drug loading of an antibody.
2.4.5. Enzymatic Methods for Conjugation
Enzymatic methods can circumvent some of the limitations that exist in chemical methods to conjugate drugs to antibodies. The evolutionary constraints for enzyme function are well-suited for chemoselective attachment of substrates to antibodies, and the development of bioorthogonal chemistries to interrogate protein function has further enhanced their capabilities.
Some enzymic modifications allow moieties to be directly attached to an antibody; while they require specific chemical matter to be present or may require mutant or additional sequences to be added to an antibody, the methods need only one step to functionalize antibodies. In most cases, the need for a single residue or functional handle for specificity limits the number of warheads that can be easily attached to an antibody. Transglutaminases exchange the amino group of a glutamine’s amide moiety for another amine, allowing for facile attachment of amino-containing payloads or linkers. A glutamine residue (Q295) in the heavy chain possesses a carbohydrate moiety that helps to determine the physical properties and immune responses of the antibody; removal of the carbohydrate can have negative consequences for ADC performance234 but provides a reliable attachment point for payloads.235 Alternatively, inclusion of glutamine residues (either by extension or by mutation of the antibody sequence) allows transglutaminase-mediated coupling reactions to attach payloads without perturbing the immune effects of the antibody.236 A large variety of amine-containing linkers can be used; with mutant transglutaminases, hydrazones (acyl hydrazines) can also be exchanged with glutamine residues.237 Prenyltransferases attach farnesyl or geranylgeranyl (15- or twenty-carbon) pyrophosphates to cysteine residues followed by two aliphatic amino acid residues.238 One of the prenyl methyl groups can be substituted; when a ketone or azide moiety is included, bioorthogonal coupling methods are applicable to attachment of linkers or payloads.239 Sortases attach N-terminal substituents to peptides or antibodies with the C-terminal sequence LPXTG–OH (effectively coupling an amine to the C-terminal glycine), allowing attachment of N-terminal substituents to an antibody where they are less likely to interfere with other functions.240 Butelase 1 appends dipeptides to C-terminal asparaginylhistidylvaline moieties to form dipeptidyl asparagine amides; the enzyme has been used with sortase A and modification of the antibody light chains to provide doubly modified antibodies.241 SpyLigase attaches a peptide containing an N-terminal tag to a peptide with a corresponding C-terminal tag, eliding the intervening peptide and forming a new substituted antibody.242 Phosphopanteinyltransferases acylate serine residues with CoA thioesters;243 while the functionality that can be incorporated is broader (requiring only a CoA thioester, the ester linkage formed may not be sufficiently stable or persistent.
Other enzymic methods convert native peptides or amino acid residues to reactive moieties that are amenable to conjugation with a variety of linkers or warheads but do not directly attach substituents to antibodies. Formylglycine-generating enzyme (FGE) reacts with peptides with the N-terminal sequence H-CXPXR (X = any amino acid), again requiring mutation of the antibody sequence. FGE generates formylglycine residues from the N-terminal cysteine;244 the aldehyde moiety is amenable to condensation with oxime ethers or with electron-rich arylmethylhydrazines in iso-Pictet-Spengler reactions to form aryl-fused tetrahydropyridazines.245 FGE-mediated conjugation may be useful when conjugation with cysteines is not compatible with the linker chemistry or when different requirements for linker stability are necessary. Tyrosinases and horseradish peroxidases oxidize tyrosine residues to form ortho-quinones which can undergo either Michael addition of amines to the quinones and rearomatization to yield stable 3-aminotyrosine residues246 or Diels–Alder reactions.247 While aminotyrosine residues are potentially oxidizable, the carbon–nitrogen bond formed is robust.
Finally, the sugar moieties present in the antibodies can be remodeled to yield attachment points for conjugation. Fucose, sialic acid, and galactose moieties contain vicinal cis-hydroxyl groups which can be oxidatively cleaved by sodium periodate to yield dialdehydes; condensation with oxime ethers or hydrazines yields oxime ethers and hydrazones.248 While the oxidation provides multiple attachment points for payloads, it also can oxidize methionine residues of the antibody, which increases clearance and decreases efficacy.249 An alternative method is to alter the sugar moieties by incorporating sugars with non-native functionalities into the pendant sugar moieties. For example, endoglycosidases catalyze the exchange of the terminal aminosugar moieties of saccharides, incorporating 2-N-acetylglycosamines into saccharides via oxazoline intermediates.250 Glucosyl-, galactosyl-, and thiofucosylamines with azide or ketone substituents (or, with thiofucosylamine, a thiol group) can thus be swapped into the sugar moieties of antibodies;251 reaction of azides with alkynes (using copper catalysis or strained alkynes), of ketones with alkoxyamines, or of thiofucose moieties with maleimides immobilizes payloads onto antibodies. Both methods may alter the immune functions of the antibody–drug conjugate by sugar modification and thus the activity of the conjugate.
2.4.6. Drug–Antibody Ratio
Drug–antibody ratio (DAR) characterizes how many drug molecules an ADC can carry; theoretically, it should characterize the ability of an ADC to deliver drug to a tumor and thus positively correlate to effectiveness.19 However, the presence of large numbers of drugs (and thus large numbers of linkers) on an ADC perturbs the properties of the antibody. Lipophilic drugs and linkers increase the aggregation of ADC, preventing them from reaching their site of action; they may also hinder access to binding sites necessary for antigen recognition or binding, reducing the activity of the ADC. Reducing the lipophilicity of linker moieties has been used to reduce the negative consequences of antibody functionalization,121 but not the alteration in ADC properties. In addition, significant toxicity has been noted for ADC with high DAR (DAR ≥ 8) and high DAR may increase the clearance of ADC (reducing their residence time and effectiveness).252 DAR is an important analytical property of ADC; the ability to produce ADC with consistent DAR is likely to lead to ADC with consistent properties and biological activity and thus is likely a critical attribute for ADC synthesis and production.
2.4.7. What Types of Drugs, Linkers, and Conjugation Methods Have Been Used for Approved ADCs?
A variety of types of drugs are used in clinically approved ADCs and in ADCs in clinical trials (discussed further in the text). Calicheamicin, maytansinoids, auristatins, duocarmycins, and pyrrolobenzodiazepines have been used, while tubulysin-, eribulin-, and amberstatin-containing ADCs are being researched in clinical trials.19 The drugs in ADCs are highly potent (effective at nM to pM concentrations); the difficulty in delivering significant amounts of drug to tumors with an ADC (as noted earlier) may explain the prevalence of potent drugs in ADC.
Most of the currently approved drugs and nearly all ADCs in clinical trials use cleavable linkers, and in most cases, enzyme-cleavable linkers.19 The preference for cleavable linkers indicates (as noted) that the contribution of bystander effects to the ADC clinical effectiveness is critical. In addition, the continued development of enzyme-cleavable linkers is likely important. While the toxicity of Mylotarg and its subsequent withdrawal and reapproval provided concern for chemically reactive linkers, the incorporation of chemically stable linkers allows the best of both worlds. The controlled drug release provides safety assurance as well as increased antitumor response via the bystander effect. The use of cleavable linkers also may prevent or reduce the resistance of tumors to ADCs by decreasing the effect of antigen loss on toxicity and releasing ADCs from the requirement for lysosomal cleavage.
As of mid-2023, all of the approved ADCs used conjugation to lysine or cysteine residues; none of the currently approved ADC used site-selective functionalization methods,19 while few of the ADCs use site-selective conjugation methods. It is unclear why site-selective methods have not been more effective; knowledge of antibody structure and function should be sufficient to avoid antibody inactivation or a loss of function. Methods to site-specifically conjugate drugs to antibodies were less advanced and may have been insufficient for incorporation into a drug (or may have had insufficient data and thus too much risk to incorporate into a clinical candidate). The reasons for the failures in the current trials, however, appear unclear.
ADCs currently approved and in clinical trials vary significantly in their DAR as well; DAR between 1.8 and 8 have been tried (see Section 7.3 below enlisting approved ADCs), with most possessing DAR around 4. The DAR may be an artifact of the conjugation method; immobilization using the interchain bridging disulfides should yield ADC with a DAR of roughly 4. The presence of larger linker and drug moieties may also require a lower DAR to avoid aggregation or inactivation of the ADC. ADCs with higher DARs and less-potent drugs have not yet reached clinical trials; it is not clear whether this is due to limitations on DAR, ineffectiveness in previously tried high-DAR ADCs with less potent payloads, or some other reason.
2.5. Selection/Optimization of Target Antigen Moiety
The efficacy of an ADC depends on the expression levels of target antigens. ADCs are designed to release the payload upon interaction with cognate antigens.88,253 As the field continues to grow, over 50 antigens have been identified as successful ADC targets under various clinical evaluation stages.253,254 To achieve optimal therapeutic efficiency, antibody–antigen binding affinity can be optimized on a case-by-case basis depending on the tumor size, target antigen concentration, and receptor-mediated internalization kinetics.
The most used antigenic targets are CD19, erb-b2 receptor tyrosine kinase 2 (ERBB2), HER2, CD22, CD30, CD33, CD79b, and Mesothelin (MSLN).46 These antigenic markers vary depending on the tumor type. Antigens such as HER2, EGFR, 5T4, trophoblast cell-surface antigen 2 (TROP2), and nectin 4 are commonly used ADC targets in solid tumors due to their higher level of expression in malignant cells when compared to the nonmalignant ones.46,255,256 For hematological cancers, markers such as CD30, CD22, CD79b, CD19, CD138, and B-cell maturation antigen (BCMA), which are distinct from solid tumor markers, are commonly used.88 For example, CD30, the target of brentuximab vedotin (Adcetris by Seattle Genetics), is mainly expressed by the malignant lymphoid cells of Hodgkin lymphoma and anaplastic large cell lymphoma (ALCL). Likewise, for specifically targeting B cell lineages, markers such as CD22, CD79b, and BCMA are used. These markers have successfully been used as the targets of inotuzumab ozogamicin (for the treatment of relapsed or refractory (R/R) B cell acute lymphoblastic leukemia), polatuzumab vedotin (for R/R diffuse large B cell lymphoma), and belantamab mafodotin (for R/R multiple myeloma), respectively.257,258 Apart from these well-known targets, work to identify suitable antigenic targets in the tumor microenvironment, such as in the stroma and vasculature, is ongoing.
A successful antigenic target of an ADC should be uniformly and heterogeneously expressed on the surface of target cells or other components of the tumor microenvironment and have minimal to no expression in off-target sites.259,260 Other essential factors include the internalization and processing of ADCs that help in their cellular uptake and increase the efficiency of the cytotoxic drug. In addition, it is advantageous that ADCs are designed against functional/oncogenic targets as they can have higher antitumor activity; for example, data from preclinical studies suggest that HER2-targeted ADCs T-DM1 and T-DXd having anti-HER2 mAb trastuzumab’s Fab region prevents ligand-independent HER2 dimerization, inhibiting HER2 downstream signaling.
Once the antibody in an ADC binds to the target antigen, it is internalized via the early endosome, and the internalization rate and efficiency depend on the target antigen and the payload. Affinity correlates well with internalization with higher affinity often resulting in rapid internalization up to a ceiling limit.88 However, a very strong binding affinity between the antibody and target antigen can lead to uneven distribution of ADCs in solid tumors due to the presence “binding site barrier”. This leads to stronger binding of antibodies with the antigens presents on cells near blood vessels and less penetration away from the tumors.261,262
Once ADCs are inside the cell, the endosomes containing them mature into late endosomes and finally fuse with lysosomes. This is followed by the release of cytotoxic payloads in the lysosome upon linker cleavage or antibody degradation. The payload eventually escapes into the cytosol to exert its effects. ADCs with cleavable linkers can release drug to neighboring cells both with and without ADC internalization.263,264 ADC with noncleavable linkers require proteolysis of the ADC for drug release. Proteolysis yields drugs with an attached amino acid residue (lysine or cysteine) which is charged at cellular pH.265 The ability of drugs to diffuse out of the cell depends on their lipophilicity; charge impairs their ability to diffuse across the membrane and thus leave the cell through passive transport. ABC transporters prefer neutral and hydrophobic compounds and export neutral hydrophobic drugs out of the cell, so drugs with amino acid residues derived from ADC catabolism are poor substrates for ABC266 and require help to leave the lysosome267 and the cell. The susceptibility of payloads to active transport decreases the effectiveness of ADC but also makes cells that do not take up the antibody subject to its effects. In particular, the bystander effect depends on how much drug can escape a cell and then accumulate in neighboring cells and if it is sufficient to kill them.265
ADCs are used to treat solid tumors, but a heterogeneous expression of antigenic targets in these tumors may be overcome by the ’bystander-killing effect’,84,268,269 where the payload is transferred from the antigen-positive cells to the antigen-negative cells in the tumor microenvironment. The lipophilic payload for internalized ADCs diffuses across cell membranes and significantly contributes to ADC activity against tumors. For some ADCs, the payload release might happen extracellularly, killing antigen-negative cells located in proximity.259
Table 1 summarizes antigens used as targets of ADCs in development and in the clinic.4,254,270,271
Table 1. Target Antigens for ADCs in Development and in the Clinic.
| Disease | Target antigens |
|---|---|
| Breast cancer | CD25, CD174, CD197, CD205, CD228, c-MET, CRIPTO, HER2, HER3, FLOR1, Globo H, GPNMB, IGF-1R, integrin β-6, PTK7, nectin-4, ROR2, SLC39A6 |
| Ovarian cancer | CA125, CD142, CD205, FLOR1, Globo H, mesothelin, PTK7, TIM-1 |
| Prostate cancer | CD46, PSMA, STEAP-1, SLC44A4, TENB2 |
| Lung cancer | CD25, CD56, CD71, CD228, CD326, CRIPTO, EGFR, HER3, FAP, Globo H, GD2, IGF-1R, integrin β-6, mesothelin, PTK7, ROR2, SLC34A2, SLC39A6, Axl, αv β6 |
| Pancreatic cancer | CD25, CD71, CD74, CD227, CD228, GRP20, GCC, IGF-1R, integrin β-6, nectin-4, SLC34A2, SLC44A4, αv β6, mesothelin |
| Melanoma | CD276, GD2, GPNMB, ED-B, PMEL 17, endothelin B receptor |
| Gastric cancer | CD25, CD197 (CCR7), CD228 (P79, SEMF), FLOR1(FRα), Globo H, GRP20, GCC, SLC39A6 (LIV1A ZIP6) |
| Colorectal cancer | CD74, CD174, CD166, CD227, CD326, CEACAM5, CRIPTO, FAP, ED-B, HER3 |
| Bladder cancer | CD25, CD205(Ly75) |
| Liver cancer | CD276 (B7–H3), c-MET |
| Renal cancer | AGS-16, EGFR, c-MET, CAIX, CD70, FLOR1, TIM-1 |
| Multiple Myeloma | CD38, CD46, CD56, CD74, CD138, CD269, endothelin B receptor |
| Head and neck cancer | CD71 (transferrin R), CD197 (CCR7), EGFR, SLC39A6 (LIV1A ZIP6) |
| Non-Hodgkin lymphoma | CD19, CD20, CD21, CD22, CD25, CD30, CD37, CD70, CD71, CD72, CD79a/b, CD180, CD205, ROR1 |
| Hodgkin’s lymphoma | CD25, CD30, CD197 |
| Acute myeloid leukemia | CD25, CD33, CD123, FLT3 |
| Gliomas | CD25, EGFR |
| Mesothelioma | mesothelin, CD228 |
3. Landscape View of Antibody–Drug Conjugate Research–insights from the CAS Content Collection
The CAS Content Collection81 is the largest human-compiled collection of published scientific information, which represents a valuable resource to access and keep up to date on the scientific literature all over the world across disciplines including chemistry, biomedical sciences, engineering, materials science, agricultural science, and many more, thus allowing quantitative analysis of global research publications across various parameters including time, geography, scientific area, medical application, disease, and chemical composition. Currently, there are over 25,000 scientific publications (mainly journal articles and patents) in the CAS Content Collection related to ADC research and development. There has been a steady growth of these documents over the last three decades, with an >30% increase in the last three years (Figure 3). Noteworthy, while in the earlier years scientific journal publications dominated, after around the year 2000 the number of patents clearly outnumber them, correlating well with the initial accumulation of scientific knowledge and its subsequent transfer into patentable applications.
United States, China, Japan, United Kingdom, Germany, and South Korea are the leaders with respect to the number of published journal articles and patents related to ADC research with the United States having ∼3- and ∼2.7-fold greater number of journal and patent publications, respectively, as compared to China (Figure 5). Genentech, the Scripps Research Institute, University of California, and the Chinese Academy of Sciences have the largest number of published articles in scientific journals (Figure 6A). The journal Bioconjugate Chemistry publishes the most articles related to ADC research (Figure 7A) and is the most-cited journal for ADC research (Figure 7B). Unsurprisingly, patenting activity is dominated by corporate players as compared to academics (Figure 6B,C). Genentech, Immunomedics, Regeneron Pharmaceuticals, and Seattle Genetics have the highest number of patents among the companies (Figure 6C), while University of California leads among the universities, having nearly double the number of patents as University of Texas, ranked second (Figure 6B).
Figure 5.

Top countries with respect to the number of ADC-related journal articles (blue) and patents (red).
Figure 6.
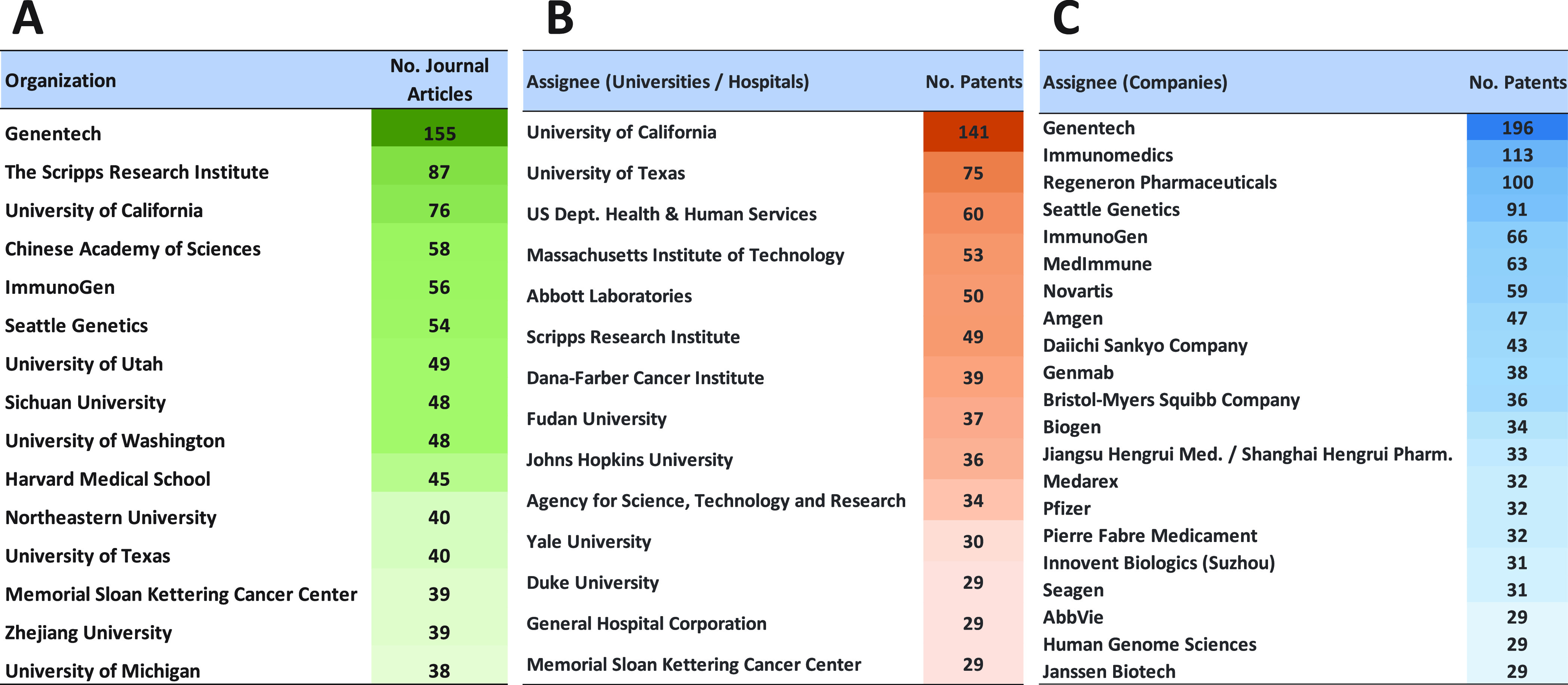
(A) Top organizations publishing ADC-related journal articles. Top patent assignees of ADC-related patents from universities and (B) hospitals and (C) companies.
Figure 7.
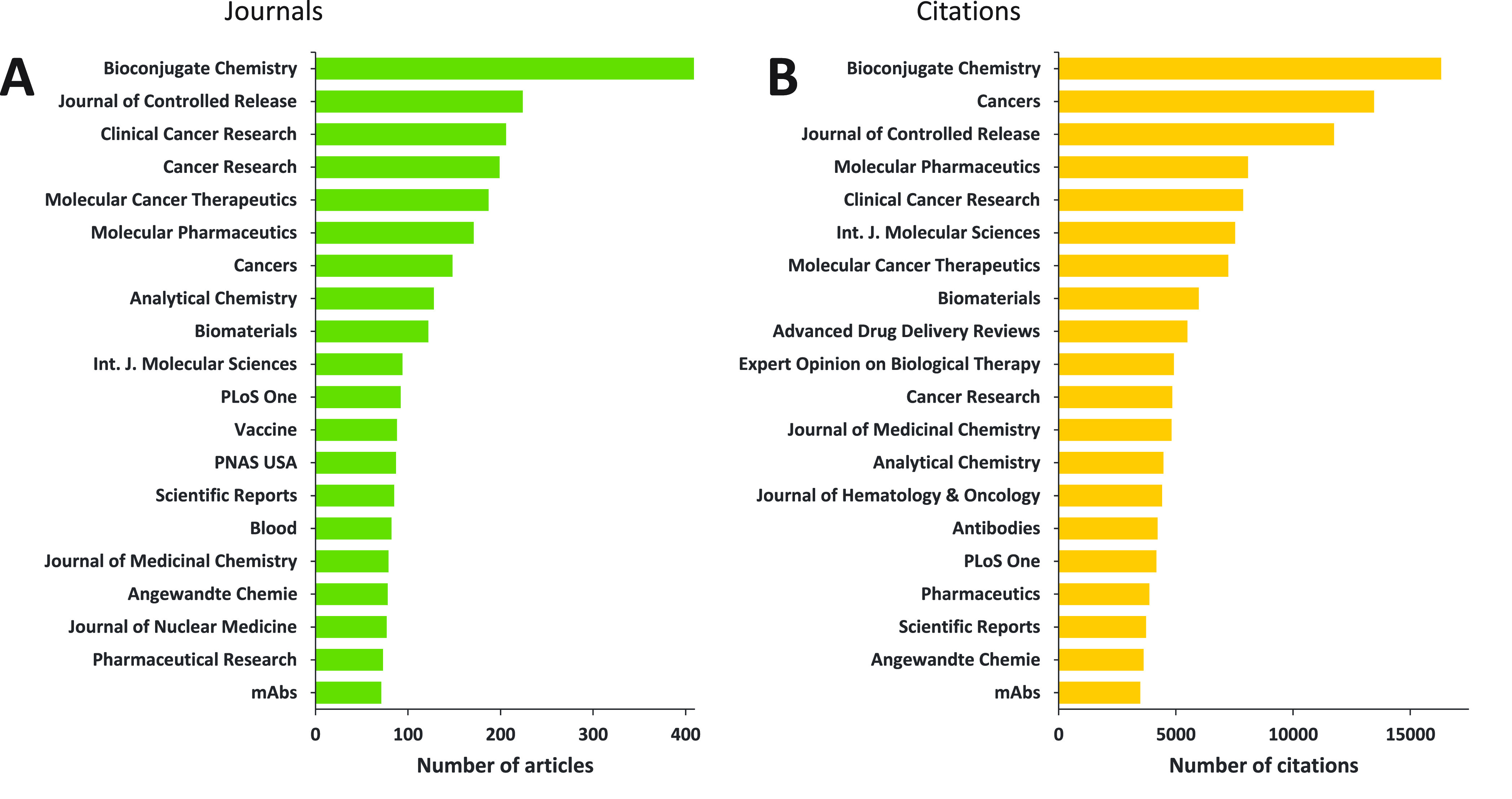
Top scientific journals with respect to the number of ADC-related (A) articles published and the (B) citations they received.
Figure 8A presents the distribution of patents among the top patent offices receiving ADC-related patent applications. The World Intellectual Property Organization (WIPO) patent office clearly dominates accounting for about 2/3 of patents filed, followed by the patent offices of the United States (US), China (CN), Japan (JP), and S. Korea (KR), and the European patent office (EP).
Figure 8.

(A) Top patent offices receiving ADC-related patent applications. (B) Flow of ADC-related patent filings from different patent assignee locations (left) to various patent offices of filing (right). The abbreviations on the right indicate the patent offices of Hong Kong (HK), Australia (AU), Austria (AT), Argentina (AR), Taiwan (TW), Russian Federation (RU), Norway (NO), Colombia (CO), Portugal (PT), Costa Rica (CR), Cyprus (CY), Eurasian Patent Organization (EA), Philippines (PH), United States (US), World Intellectual Property Organization (WO), Israel (IL), Mexico (MX), Spain (ES), Canada (CA), European Patent Office (EP), Japan (JP), Brazil (BR), India (IN), South Korea (KR), Great Britain (GB), China (CN), Germany (DE), and France (FR).
Patent protection is territorial, and therefore the same invention can be filed for patent protection in several jurisdictions. We thus searched for all related files pertaining to ADCs. Certain patent families might be counted multiple times when they have been filed in multiple patent offices. Figure 8B presents the flow of patent filings from various applicant locations to a variety of patent offices of filing. Most of the applicants tend to have a comparable number of filings in their home country and at the WO, while also having a sizable number of filings at other patent offices such as the US, European Patent Office (EP), and others.
We further explored distribution and trends in the published documents (journals and patents) dealing with various ADC-related concepts. Figure 9 presents a number of the ADC-related documents in the CAS Content Collection concerning neoplastic (A) and other diseases (B). The highest number of documents pertain to breast cancer (mammary gland neoplasm) and lymphoma for solid tumors and hematological malignancies, respectively. This data correlates well with approved ADCs used in the treatment of cancers that are currently on the market.53,55 Breast cancer and myeloma exhibit the highest growth rate in the last five years with respect to the number of documents related to them (Figure 9A, inset). Most ADCs developed thus far, aimed at treating various types of cancer (solid and hematological), have nonetheless been restricted to treating cancer. Challenges in designing ADCs for noncancerous diseases include identifying targeting cell types, a specific surface marker expressed on the targeting cells, and an effective payload drug. So far, not many ADCs have been designed for noncancerous indications, with none yet having successfully progressed through clinical trials to market. With the advancement of ADC platforms and technology, more ADCs for nononcology indications are being developed.272 A search in the CAS Content Collection showed that among the noncancerous diseases, autoimmune diseases, inflammations, and infections are the top pathologies with respect to the number of documents related to ADCs (Figure 9B). Despite obvious complications such as difficulty in penetration through the blood-brain barrier (BBB), our data indicate a growing interest in development of ADCs targeting the brain. The number of documents pertaining to ADCs in the context of neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s disease (Figure 9B) are on the rise. Recent approvals of antibody treatments for Alzheimer’s disease by the US FDA273,274 also stimulate ADC development for neurological diseases.
Figure 9.
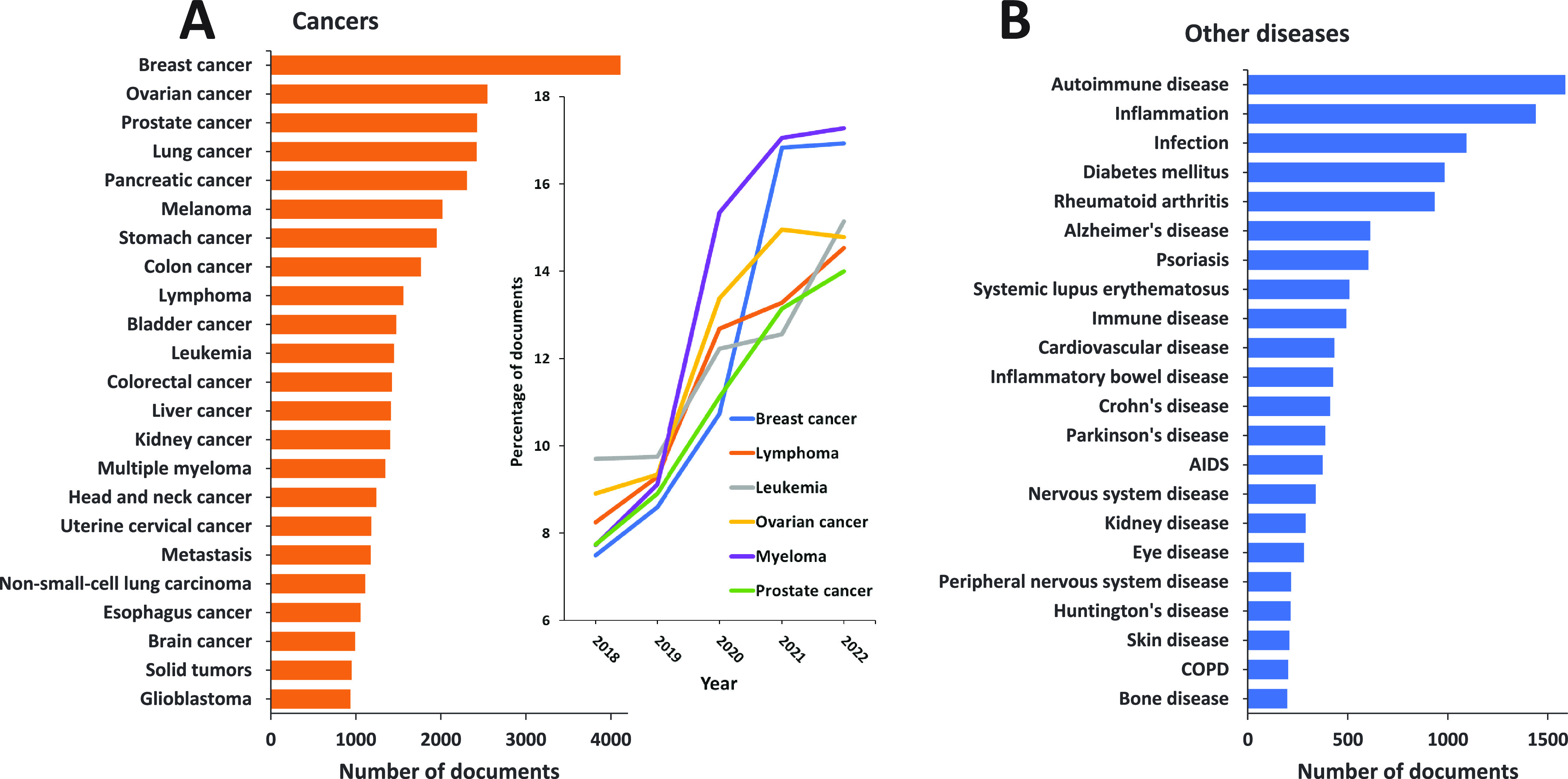
Diseases explored in ADC-related publications: (A) cancers (Inset: Annual growth of the number of documents for the fastest growing solid and hematological cancers for the years 2018–2022); (B) other diseases.
Figure 10 presents the number of ADC-related documents in the CAS Collection concerning various types of therapies. Immunotherapy understandably accounts for the highest number of documents. Indeed, a sound biological rationale supports the research into combining ADCs with immunotherapy to overcome the incidence of resistance and improve patient outcomes.275 Most immunotherapy-related papers involve passive immunotherapy (Figure 10, inset pie chart). Passive immunotherapy agents produce rapid antitumor responses by direct administration of immune-cell factors, such as cytokines or antibodies. With passive immunotherapy, continued dosing may be required for a prolonged response since immune system memory is not engaged.276,277
Figure 10.
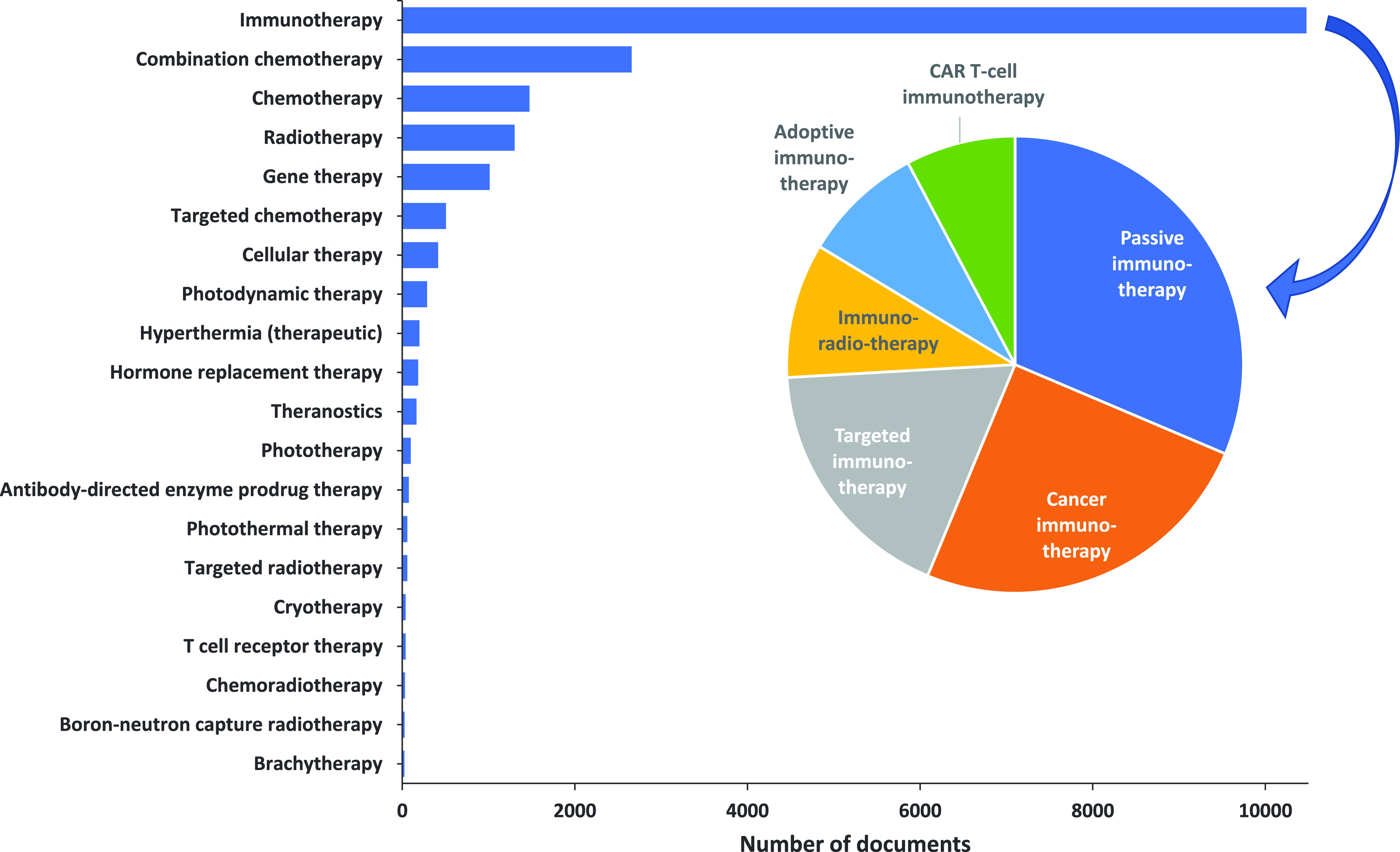
Therapies explored in the ADC-related publications.
Targeted intravenous drug delivery systems for nanoparticles are the most common drug delivery systems explored for ADCs (Figure 11). Indeed, intravenous administration into the bloodstream is the preferred route for ADCs in order to avoid digestion of antibodies by gastric acid and proteolytic enzymes with oral administration.278 At present, all approved ADCs are administered via the intravenous route, and the therapeutic capacity of other routes of ADC administration is rarely explored. Subcutaneous, intramuscular, intravitreal, inhalable, intra-articular, and intratumoral drug delivery methods have all been used to improve the therapeutic indexes of antibodies,279 with subcutaneous and intratumoral routes considered especially promising.280,281 However, with respect to ADCs, there is still a need to explore and evaluate the therapeutic potential of alternative routes of administration.
Figure 11.

Drug delivery systems explored in the ADC-related publications.
Cytotoxic payloads are major components of ADCs, with the tubulin inhibitors and DNA damaging agents being the most widely explored, and until recently the major classes of compounds used in ADCs.17,19,22,88,127 With the approval of trastuzumab deruxtecan (Enhertu) in 2019, the diversification of ADC payloads has been appreciated as a key approach in the ADC development, and a number of new classes of compounds have been examined as potential payloads.282 In the CAS Content Collection, in terms of the number of published documents, auristatins and calicheamicins are the major representatives of tubulin inhibitors and DNA damaging agents, respectively (Figure 12A). As a sign of the payload diversification efforts, STING agonists, glucocorticoid receptor modulators, and topoisomerase I inhibitors exhibit the fastest consistent yearly growth in the number of documents (Figure 12B). The success of immune checkpoint inhibitors targeting the adaptive immune system has greatly stimulated interest in exploring immune-stimulating ADC payloads such as STING agonists and TLR agonists.282 Glucocorticoid receptor modulators are the primary treatment for various immune diseases, and targeted delivery via an ADC may provide significant efficacy at doses that do not lead to unwanted side effects.283 Topoisomerase I inhibitors, particularly camptothecin analogs, constitute a successful ADC payload family, and are a part of two recently FDA approved drugs, trastuzumab deruxtecan, and sacituzumab govitecan.284,285
Figure 12.
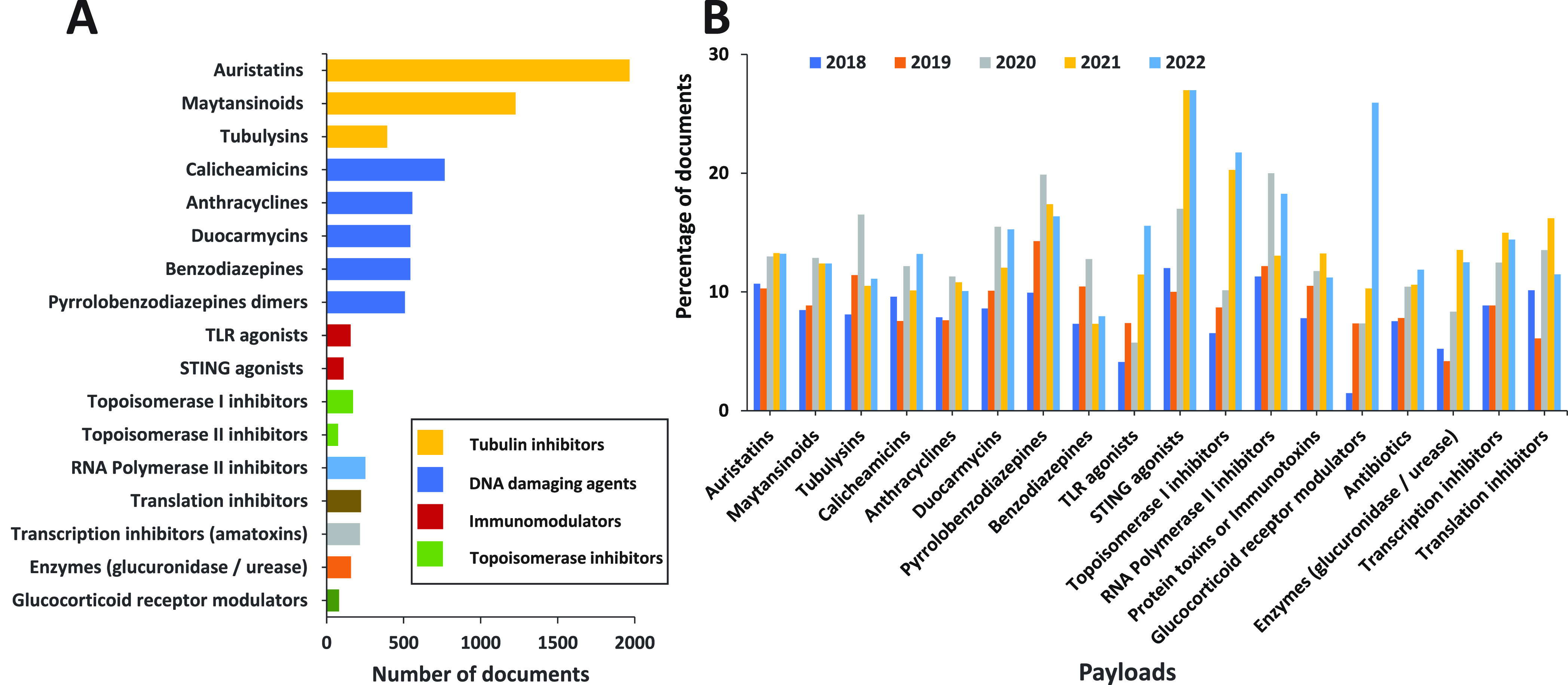
ADC payloads explored in the scientific publications: (A) Number of publications exploring ADC payloads. (B) Trends in number of publications exploring ADC payloads during the years 2018–2022.
The number of documents and growth trends related to target antigens of ADCs according to the CAS Content Collection are listed in Figure 13. While HER2 and EGFR remain the most widely explored target antigens for solid tumors (Figure 13A), Trop-2 and Nectin-4 antigens have exhibited consistent and steady growth in the last five years (Figure 13B).286,287 For hematological malignancies, CD30, CD19, CD22, and CD33 are the most extensively examined with almost triple the number of patents as compared to journal articles (Figure 13A), while CD79B shows the fastest growth in number of documents over the last five years (Figure 13B).
Figure 13.
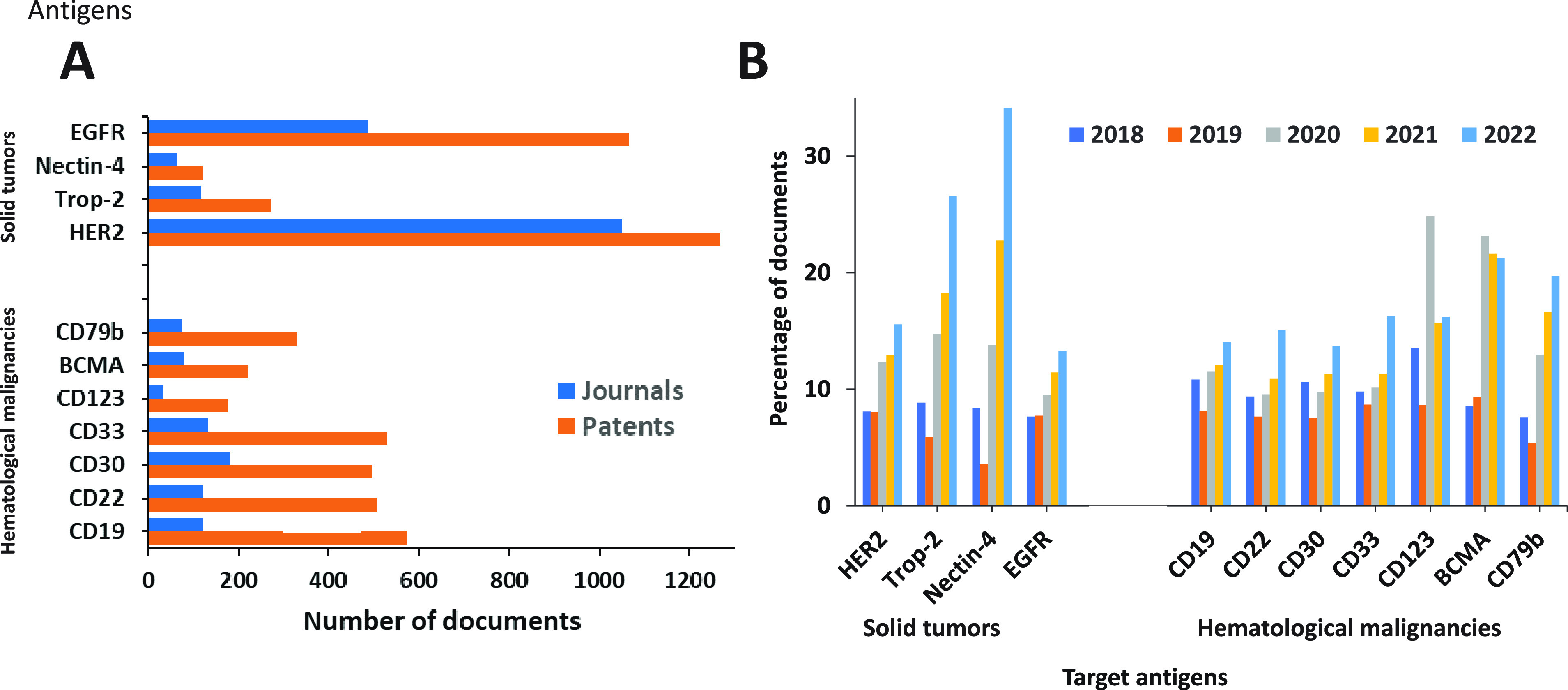
ADC target antigens explored in the scientific publications for solid tumors and hematological malignancies: (A) Number of publications exploring target antigens. (B) Trends in growth of publications exploring target antigens during the years 2018–2022.
We also examined the distribution and trends in published documents pertaining to antibodies used in ADCs, especially the immunoglobulin G (IgG) isotype, which is the most frequently used isotype for cancer immunotherapy (Figure 14). IgG1 and IgG4 are the IgG subtypes that are used in most formulations (and the only ones used in approved ADCs) in line with established knowledge, with the number of patents clearly dominating journal articles by ∼4- and 25-fold, respectively (Figure 14A). Although the four subclasses of IgG have >90% homology, they exhibit distinctive profiles with respect to the hinge region length, the number of interchain disulfide bonds, and Fc-effector functions.288 IgG3 displays the highest affinity binding to most Fc-γ receptors, but is susceptible to proteolysis and aggregation and is avoided in ADC design due to its short circulating half-life.289 Of the remaining subclasses, IgG1 demonstrates the highest affinity for all Fc-γ receptors. Publications discussing the use of IgG2 and IgG3 in ADCs have increased more rapidly in recent years than those discussing the use of IgG1 (Figure 14B).
Figure 14.
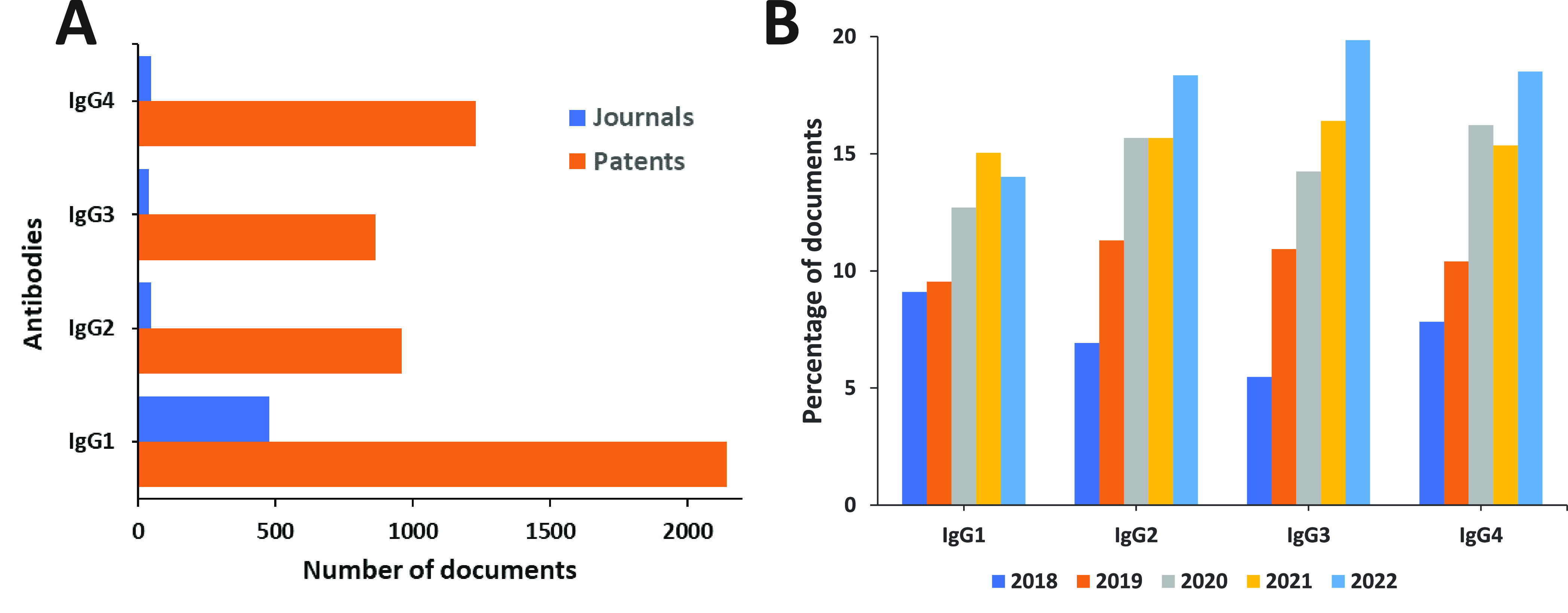
ADC antibodies explored in the scientific publications: (A) Number of publications exploring ADC antibodies. (B) Trends in number of publications exploring ADC antibodies during the years 2018–2022.
Figure 15 shows the antibody-payload linker types as represented in the CAS Content Collection. The cleavable linkers markedly dominate; they are the preferred linker type in the therapeutic ADCs because of their versatility, providing possibility for a controlled payload release.
Figure 15.
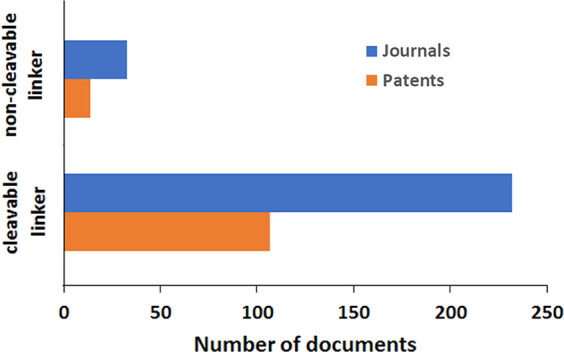
Antibody-payload linker types explored in scientific publications.
Figure 16 presents correlations in the examined documents between various cancers and ADC target antigens (Figure 16A and B), ADC antibodies (Figure 16C), and ADC payloads (Figure 16D), calculated as the percentage of documents related to the given disease. With solid tumors (Figure 16A), the strongest correlation is between breast cancer and HER2 as target antigen, while among hematological malignancies (Figure 16B) lymphoma correlates strongly and comparably with CD19, CD22, and CD30, leukemia–with CD33 and CD19, and myeloma, with BCMA. With respect to ADC payloads (Figure 16D), the strongest correlation is between breast cancer and the class of maytansinoids, followed by auristatins with similar trends observed for lymphoma.
Figure 16.
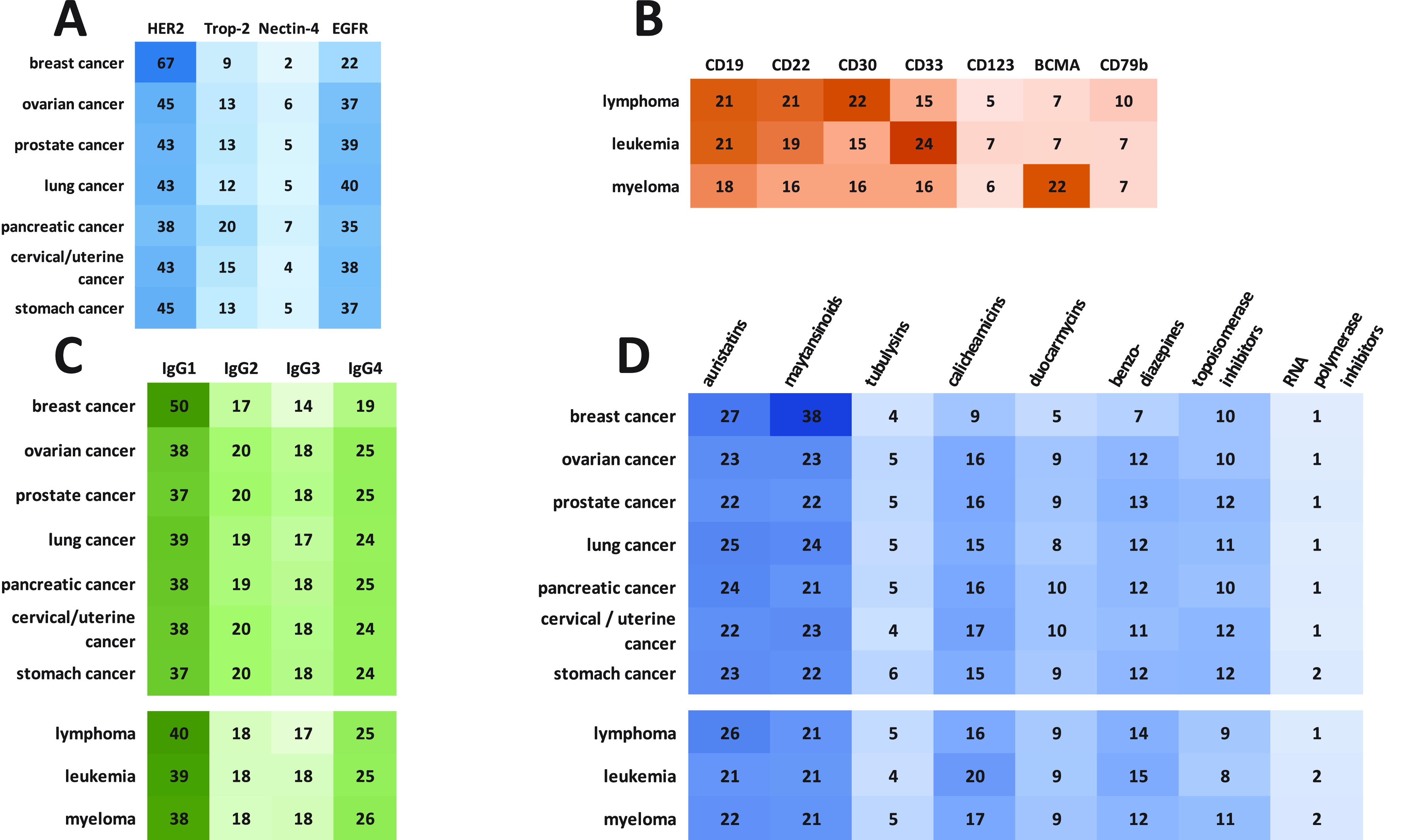
Correlations between different concept pairings are shown as heat maps. ADC target antigens and (A) solid tumors and (B) hematological malignancies. (C) ADC antibodies and types of cancers and (D) ADC payloads and types of cancers (numbers represent percentage of documents related to the given disease). Darker shades correspond to a higher number.
Among the ADC-related concepts in the CAS Content Collection, monoclonal antibodies is clearly the major one, along with antibody–drug conjugates, and antigens (Figure 17A). Bispecific antibodies and nanobodies, along with the antibody–drug conjugates, exhibit the fastest growth rate with respect to the number of published documents (Figure 17B).
Figure 17.

(A) A word cloud of the most widely used ADC-related concepts in the CAS Content Collection. (B) Yearly growth in the number of documents (percentage) over the 2010–2022 period for ADC-related concepts.
Figure 18 represents a map of the ADC-related concepts with an indication of the number of documents in the CAS Content Collection related to the given concept/topic.
Figure 18.

ADC Concept Map. Size of the dot at each concept/topic corresponds to the number of documents (journals and patents) in the CAS Content Collection related to the given concept/topic.
4. ADC-Based Combination Therapies
Development of ADC resistance is a major drawback to their therapeutic potential. Many mechanisms of resistance exist290 including:
Downregulation of/change in antigen expression291,292 – decreases the binding of an ADC to the target antigen and might even increase toxicity due to prolonged presence of ADC in the bloodstream.
Decreased internalization of the ADC-bound antigen291,292 – could result from increased recycling of the target antigen preventing release of cytotoxic load in desired locations.
Inefficient binding of the ADC to its target antigen291,292 – resulting from changes in the target (such as truncation) or increased interactions with other binding partners reducing affinity of ADC for target antigen.
Inefficient/incomplete/improper degradation of ADCs inside lysosomes leading to poor release profiles of cytotoxic payloads and reducing therapeutic effectiveness.293
Poor release from lysosomes292 – many cytotoxic drugs tend to be charged molecules that require active transport from the lysosome into the cytoplasm and change in expression of lysosomal transporters which can affect concentration of payloads achieved in the cytoplasm.267
Overexpression of efflux pumps such as multidrug resistant transporter 1 (MDR1) that actively transport the released cytotoxic payload from the cytoplasm to outside of the tumor cell reducing therapeutic effectiveness of ADC.292
These resistance mechanisms reduce the effectiveness of ADCs in cancer therapy. The simplistic but vital reasoning behind use of combination therapy is to circumvent drawbacks (such as resistance mechanisms) of both types of therapies (including reducing toxicities associated with them) to boost the overall therapeutic efficacy and is especially true for an aggressive illness such as cancer with a high mortality rate.294
4.1. ADC and Conventional Chemotherapy
Conventional cancer therapies tend to include a combination of chemotherapeutic drugs, radiation therapy and surgery.295 Classical chemotherapeutic drugs such as altretamine296 and gemcitabine297 exert their antitumor effects by alkylating or cross-linking DNA.298,299 or by shutting down DNA synthesis by competing with endogenous nucleotides.300 While effective, conventional chemotherapeutic drugs are toxic, show significant side effects because of insufficient selectivity301 and are subject to a variety of resistance mechanisms.302 All of these factors greatly reduce their effectiveness. Combining a targeted approach such as ADCs with standard or conventional untargeted chemotherapeutic approaches has been shown to overcome the drawbacks and improve survival rates.303 Perhaps the most well-known chemotherapeutic-ADC combinations involve gemcitabine, a nucleoside analog that exerts antitumor activity by interfering with DNA synthesis.300 An early study of an ADC-chemotherapeutic drug combination involved gemcitabine and brentuximab vedotin (SGN-35), a CD30 targeted ADC304 and this combination is still being tested for the treatment of various types of cancers.305−307 While no rationale has been given for the effectiveness of the gemcitabine/brentuximab vedotin combination, one hypothesis is that brentuximab vedotin and gemcitabine target neoplastic Reed Sternberg cells and stromal cells, respectively and exert synergistic effects.304 Expression levels of surface antigens are crucial for the success of ADC therapy, and changes in their levels are associated with decreased therapeutic efficacy. Gemcitabine administration has been linked to increased expression of HER2 by as much as 2.5-fold.308 Co-administration of gemcitabine with trastuzumab emtansine, a HER2-specific ADC, boosted the antitumor effect achieved by the ADC in pancreatic ductal adenocarcinoma.308 Other instances of synergistic effects have been observed when gemcitabine was coadministered with camidanlumab tesirinean (ADCT-301),309 an ADC specific for CD25 (interleukin-2 receptor alpha chain)310 or with Oba01 ADC, an ADC targeting death receptor 5 (DR5).311 Other chemotherapeutic drugs that are being explored include alkylating agents cisplatin,312−314 carboplatin315−317 and cyclophosphamide (ClinicalTrials.gov ID: NCT01042379)318 and topoisomerase II inhibitor doxorubicin317 in combination with ADCs whose targets are cell surface glycoproteins such as LYPD3,312 CD205313 and LRG1314 among others.
ADC combinations with not only chemotherapeutic agents but also antibody-based therapy have been also explored. Thus, antiangiogenic agents can stimulate ADC penetration and tumor cell exposure. Combination of anetumab ravtansine or mirvetuximab soravansine with bevacizumab has enhanced efficacy in preclinical models of ovarian cancer. A recent study combined mirvetuximab soravtansine and bevacizumab in ovarian cancer patients with encouraging results in the pivotal AURELIA trial.319 Anetumab ravtansine in combination with bevacizumab has been tested for treatment of ovarian cancer.319 Clinical trials such as KAITLIN, KRISTINE, and MARIANNE were designed on the basis of synergistic antitumor activity with trastuzumab emtansine in combination with pertuzumab.319
4.2. ADC and Immune Checkpoint Inhibitors
Immune checkpoint molecules are proteins that regulate the magnitude, type and duration of immune response320,321 they are classified as either inhibitory or stimulatory depending upon the pathways they influence.322 Meant to act as guards against excessive immune response,323 they can also act as avenues for tumor cells to escape immunosurveillance.320,324 A few well-known examples of inhibitory immune checkpoint molecules include programmed cell death protein-1 (PD-1),325 cytotoxic T lymphocyte-associated antigen-4 (CTLA-4; also known as B7, CD152),326 B and T lymphocyte attenuator (BTLA; also known as CD272),327 and lymphocyte-activation gene 3 (LAG-3; also known as CD223)328,329 among others. For the stimulatory pathways, glycoproteins such as OX40 receptors (also known as CD134 and TNFRSF4)330,331 and 4–1BB (also known as CD137)332 are few well-known examples of immune checkpoint molecules. Expressed on cell surface, immune checkpoint molecules modulate the activity of T cells320 and it is this feature that can be exploited to our advantage. Inhibition of immune checkpoint inhibitors reduces the suppression of immune response to cancers, activating T cells to kill cancer cells.333,334 Alternatively, activating stimulatory immune checkpoint molecules either by preventing interactions with their endogenous ligands or by directly activating them achieves the same effect, i.e., T cell activation and proliferation resulting in increased antitumor effects.335−338 Immune checkpoint molecules are successful targets for FDA approved immunotherapeutic drugs such as the PD-1 inhibitors pembrolizumab (Keytruda)339 and cemiplimab (Libtayo),340 and the CTLA-4 inhibitors ipilimumab (Yervoy)341 and recently approved tremelimumab (Imjudo).342 In addition, CA-170 (a small molecule targeting PD-L1, PD-L2 and VISTA),343 ieramilimab/LAG252 (targeting LAG-3)344 (ClinicalTrials.gov ID: NCT02460224345), and sabatolimab/MBG453 (targeting TIM-3)346,347 (ClinicalTrials.gov ID: NCT04266301348) are examples of immunotherapeutic agents currently in clinical trials.
The development of immune checkpoint modulators has been immensely useful especially in the treatment of solid cancers such as nonsmall cell lung carcinoma (NSCLC)349 and melanoma350,351 among others. Long-term remission, a feat that was considered very hard if not impossible to achieve, has been made possible by immune checkpoint inhibitor (ICI) therapy.352 Unfortunately, the fraction of patients that actually show such long-term remission remains small.353 A recent study354 suggests that immune checkpoint inhibitor therapies become ineffective against chemotherapy-treated triple-negative breast cancer because TP53 mutations alter the expression levels of immune checkpoint molecules, allowing tumor cells to undergo “immune exclusion”. As with other treatment modalities, resistance reduces the effectiveness and utility of immune checkpoint inhibitors.355 As such, they are often used in combination356,357 with either other immune checkpoint modulators358 or conventional chemotherapeutic agents to boost their therapeutic efficacy.359 More recently, there has been an increasing use of ADCs in combination with immunotherapeutic agents.360,361 The ability of ADC to trigger immune responses to cancer cells suggests that they could exhibit synergism with immune checkpoint modulators.360
The most often used immunotherapeutic agents in combination with ADCs appears to be confined to PD-1 inhibitors with pembrolizumab316,362−364 and nivolumab365−370 dominating and followed by atezolizumab,371,372 durvalumab373−375 and toripalimab376−380 and are currently in various stages of clinical trials. A few representative examples of patents for ADC immunotherapy combination therapy are WO2018160538,381 US20220133902,382 WO2022242692,383 WO2018110515.384 Most ADCs used in these combination therapies appear to be directed toward HER2371,380,385−388 with a smaller number directed toward targets such as LIV-1,389 nectin-4,390 TROP2391,392 and B7–H3 (also known as CD276).393 Recently, ADCs have been developed to target immune checkpoint molecules such as PD-L1394 and B7–H3395 by acting both to prevent immune suppression by binding directly to the immune checkpoint molecules and to carry cytotoxic payloads. A recent example of such a bifunctional ADC was a small molecule immunomodulator attached to an anti-PD-L1 antibody via a cleavable disulfide linker, dubbed an immune modulating ADC (IM-ADC).396 The released payload was found to exert its antitumor effect by inducing CD8+ and CD4+ T cytotoxic lymphocyte infiltration.396 In addition, the payload increased PD-L1 expression, especially in tumor cells, thereby increasing the effectiveness of subsequent rounds of treatment with the same ADC.
Tumor associated macrophages (TAMs) are an important part of the tumor microenvironment (TME) and have been linked to protumor effects including initiation397−399 and progression400 among others. The presence of TAMs has been linked to the antitumor efficacy of the anti-CD-30 antibodies, SGN-30401 and SGN-40.402 TAMs have been shown to enhance the uptake of a nontargeted ADC and the release of its payload, leading to an enhanced bystander effect403 which enhances its toxicity to tumor cells with low or variable antigen expression. A major difference between the two macrophage subtypes – M1 and – M2 lies in their functional activities. M1 and M2 macrophages are associated with antitumor (cell death) and pro-tumor (cell proliferation) effects, respectively.404 Efforts have been made to reprogram M2 TAMs to act similar to antitumor M1 TAMs by utilizing a peptide.405
4.3. Sequential/Staggered Therapy
While combination therapies use multiple drugs or therapies simultaneously, sequential or staggered therapies use the sequential administration of multiple anticancer therapies in a specific order interspersed with pauses to maximize their antitumor effects while minimizing their toxicities.406 Growing literature evidence suggests that in some instances sequential therapy may be more effective than combination therapy, especially immunotherapies,407 and that the order of administration plays a crucial role in its success.408 Sequential therapy have shown promise in breast cancers,409 renal cell carcinomas,410 and lung cancers.411 The cBR96-doxorubicin ADC was shown to be more effective when administered before the tubulin inhibitor paclitaxel when compared to coadministration.412 The ADC consisting of a doxorubicin payload attached to the BR96 antibody via a cleavable hydrazone linker, increased the sensitivity of tumor cells to paclitaxel.412 The increased effectiveness of sequential therapy over combination therapy might be attributed to reduced internalization of the ADC by paclitaxel.412
5. ADCs beyond Oncology
Until recently, ADCs in preclinical and clinical development have been applied exclusively for oncology indications, with cytotoxic warheads targeted to antigen-expressing cancer cells. However, the concept of site-specific release of pharmacologically active small molecules for alleviating pathogenic cellular activities with minimal off-target effects by using an ADC-mediated delivery platform might also be effectively applicable for any disease area. Thus, over the recent years, researchers have examined opportunities to develop ADCs beyond cancer, for other disease indications including autoimmune disease, inflammatory and immune disorders, difficult-to-treat bacterial infections, and atherosclerosis, with payloads varying from glucocorticoid receptor modulators and kinase inhibitors to antibiotics and siRNA.413−417 Multiple factors may affect the effective development of an ADC including the choice of target, payload efficiency and mode-of-action, linker design, and conjugation method. With the growing knowledge of the mechanism of action of ADCs, it is highly anticipated that the development of such compounds in therapeutic areas outside of oncology and hematology will rapidly intensify.
5.1. ADCs against Infectious Diseases: Antibody–Antibiotic Conjugates
The effectiveness of antibiotic treatment for bacterial infections has been compromised by the development of widespread drug resistance. In response, antibody–antibiotic conjugates (AAC) were developed as a countermeasure.418 Analogously to ADCs where the antibodies are used to deliver cytotoxic drugs to the antigen-expressing cells, AACs use antibodies to deliver antibiotics to the target bacteria. These AACs combine the specificity of a bacterial antigen-specific monoclonal antibody with the bactericidal capacity of a potent antibiotic419−421 via a linker which ensures an efficient release of antibiotics at the target site. This design takes advantage of the improved absorption, distribution, metabolism, and elimination (ADME) properties of the antibodies.178 Antibiotics used for developing AACs must be highly potent, nonimmunogenic, soluble, and stable under physiological conditions.421
For example, Genentech has developed an AAC to combat hard-to-treat, invasive Staphylococcus aureus infections named DSTA4637S – an IV-administered THIOMAB-type AAC.178 It is composed of an IgG-type b-GlcNAc-WTA monoclonal antibody. The light chain of this antibody is connected to a rifamycin class antibiotic (dmDNA31) via a protease cleavable MC-ValCit-PABQ linker.422,423 This linker has a lysosomal protease-cleavable site, which helps in releasing the antibiotic payload inside the bacteria. Upon administration, the AAC prodrug enters the circulation; subsequently, the antibody portion of the AAC binds to bGlcNAc modification of teichoic acid (a polyanionic glycopolymer present in the peptidoglycan layer of the bacterial cell wall). This leads to the formation of a phagolysosome; the AAC is then opsonized into the intracellular environment. Once inside, host proteases cleave the linker releasing the dmDNA31 antibiotic exhibiting a strong bactericidal action against intracellular S. aureus bacteria (Clinical trial ID#: NCT03162250).422 Unfortunately, this AAC has been discontinued in clinical trials for undisclosed reasons, but provides inspiration for further antibacterial development.
More recently, AACs are being developed against bacterial biofilms,424 the hypothesis being that an antibody can be used to anchor the antibiotic to the surface of the bacteria within the biofilm. Using a trigger, such as an external small molecule, releases the antibiotic at the bacterial cell surface. Studies were done using mitomycin C, an antibiotic with a well-known antitumor effect425,426 which was found effective in controlling S. aureus biofilms in vitro and in vivo. Reduction of biofilms is important in reducing the population of persistent (difficult to kill) bacterial cells (rendering infections more treatable) and in preventing nosocomial infections from medical implants such as i.v. tubing, catheters, and other medical equipment.427 While research in antibody–antibiotic conjugates is limited, they appear to have significant promise as antibacterial agents.
5.2. ADCs as Immunomodulatory Agents
Glucocorticoids are efficient anti-inflammatory drugs, yet their use is dose-limited by systemic toxicity, causing severe side effects such as immunosuppression and metabolic disorders. It has been suggested that applying the ADC strategy developed for oncological malignancies may provide a solution for avoiding glucocorticoid toxicity.
Glucocorticoids exert their anti-inflammatory effects by suppressing the release of tumor-necrosis factor-α (TNF-α) and other cytokines by macrophages.22 Thus, an anti-CD163 dexamethasone conjugate that selectively delivers the glucocorticoid to macrophages has been designed and tested. It elicited reduced TNF-α secretion in vitro and was 50-fold more active in vivo than the nonconjugated dexamethasone in animal models.428−431 Another glucocorticoid, a fluticasone propionate analog, was conjugated onto an anti-CD74 mAb targeting B-cells and was reported to exhibit immuno-suppressive activity in human B cells.173 A glucocorticoid-based ADC, ABBV-3373, has been developed by conjugation of a proprietary dexamethasone derivative on the anti-TNF-α adalimumab, against autoimmune disease432,433 and specifically rheumatoid arthritis.434 The glucocorticoid released after cell internalization and lysosomal escape activates the glucocorticoid receptor pathway and provokes an anti-inflammatory cascade in nucleus.434 The data indicate that anti-TNF ADC delivering a glucocorticoid receptor modulator (GRM) payload into activated immune cells may provide enhanced efficacy against immune mediated diseases while minimizing systemic adverse effects associated with standard glucocorticoid treatment.
Table 2 provides some examples of ADCs tested for nononcological indications, with their targets and payloads.22,131,413−417
Table 2. Examples of ADC Strategy Tested to Modulate Pathogenic Cellular Activity in Non-Oncology Indicationsa.
| Target | Payload | Payload class | Disease | Ref |
|---|---|---|---|---|
| E-selectin (CD62E) | dexamethasone | GRM | inflammatory disorders | (435) |
| CD163 | dexamethasone | GRM | inflammatory disorders | (428) |
| CD70 | budesonide | GRM | immune diseases | (436) |
| CD74 | fluticasone propionate | GRM | autoimmune diseases, systemic lupus erythematosus | (173) |
| Prolactin Receptor (PRLR) | glucocorticoid | GRM | undisclosed | (437) |
| TNFα | proprietary dexamethasone derivative | GRM | autoimmune disease, rheumatoid arthritis | (432−434) |
| CD71 | siRNA | siRNA | muscular diseases | (438) |
| CD19, B220 | siRNA | siRNA | myasthenia gravis | (439) |
| S. aureus | rifampicin analog | antibiotic | infectious disease | (440, 441) |
| β-GlcNAc WTA | ||||
| Chemokine receptor CXCR4 | dasatinib | kinase Inhibitor | hematological disorders | (174) |
| Leukocyte integrin CD11a | LXR agonist | LXR agonist | atherosclerosis, inflammation | (442) |
| Leukocyte integrin CD11a | GSK256066 | PDE4 Inhibitor | chronic inflammation | (175) |
| IL-6 receptor | alendronate | bisphosphonate | rheumatoid arthritis | (443) |
| CD30 | monomethyl auristatin E (MMAE) | microtubule Inhibitor | steroid-refractory acute graft-vs-host disease; severe active diffuse cutaneous systemic sclerosis | (444, 445) |
Abbreviations: GRM, Glucocorticoid Receptor Modulator; LXR, Liver X Receptor; PDE4, Phosphodiesterase 4; TNFα, Tumor Necrosis Factor α.
5.3. ADCs across Different Indications
Aiming at neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases is complicated by the existence of the blood–brain barrier (BBB). To augment brain delivery of antibody therapeutics, endogenous macromolecule transportation pathways such as receptor-mediated transcytosis and carrier-mediated transport have been explored recently.446 Invasive strategies, such as ultrasound, microbubbles, and direct injection into the brain (e.g., intracerebroventricular delivery), have also been used to deliver ADC across the blood-brain barrier.447,448
6. Private Investment
Analyzing the collective international private investments in the ADC sector offers a valuable understanding of the business fascination with the commercial potential of this domain. Conducting a search for ADCs on PitchBook,449 an online platform for investment data, reveals comprehensive capital undertakings in this field. The search revealed that capital investments showed a dramatic and substantial increase after 2017 in this field (Figure 19A). Subsequent years show more or less sustained interest in ADCs with the amount of capital invested being > $600 M (Figure 19A). A breakdown across global regions reveals that the lion’s share of investments are from Asia, followed by United States with Europe and Canada making up the rest (Figure 19B). The venture capital investment data in this area clearly shows significant recent interest in ADCs, endorsing their therapeutic and commercial potential.
Figure 19.

Capital invested by global region for the period 2012–2022 in the antibody–drug conjugate field: (A) Venture capital investment. (B) Total capital investments.
7. ADC Clinical Development
7.1. Preclinical Development
Examination of preclinical ADC development reveals that over 50 worldwide organizations, mainly from the United States, Europe, and Asia, are currently conducting research on over 160 ADC preclinical candidates (Supporting Information, Table S1). Table S1 reveals organizations conducting preclinical ADC research, their ADC drug candidates, and target antigens, along with target indications. Figure 20 is a visual representation of these organizations and their number of ADC candidate drugs, target antigens, and disease indications. A few of these companies have disclosed the target antigens for their ADC. Antigens HER3, Nectin-4, Folate receptor α, B7H3, CD99, and IGF1R are leading the way with the most ADC drug candidates utilizings these targets for disease treatment. Of disclosed preclinical indications ∼50% are categorized into the broader solid tumors and hematological malignancy indication with the other 50% attributed to more specific indications. Solid tumor research is heavily favored currently for preclinical development with the largest number of ADC drug candidates focused on this indication. Disclosed solid tumors with the largest research focus include, lung, breast, gynecologic, brain, pancreatic, and gastric cancers. In general, hematological malignancies appear to have lesser research efforts being directed toward them, with acute myeloid leukemia and lymphoma being the exceptions. Outside of oncology, Duality Biologics is performing preclinical research for the use of ADCs in the treatment of autoimmune disease.
Figure 20.

Organizations conducting preclinical ADC research with the number of ADC candidates in their pipeline (left), target antigen (middle), and disease indication (right).
7.2. Clinical Development
A representative selection of ADC clinical trials has been examined within this section to gain an overall view of the past, present, and future states of clinical development. A selection of approximately 1,500 ADC clinical trials from https://clinicaltrials.gov are examined against time, clinical trial phase, status, and disease indications. These trials reveal that ADCs are slowly growing in clinical development, with Figure 21 showing an oscillating curve starting in 1997 with more consistent and steady growth starting around 2012 and continuing beyond.
Figure 21.

Number of ADC clinical trials by year.
Examination of all ADC clinical trials against their indications further revealed that nearly all clinical trials target the treatment of both solid tumors and hematological malignancies. ADC clinical trials targeting solid tumors make up 55% of all ADC clinical trials, with hematological malignancies making up 44%. Only 1% of all ADC clinical trials target a disease beyond oncology (Figure 22A). With ADCs historically targeting cancers, much room is available for expansion into other disease treatment indications, such as autoimmune diseases, rheumatoid arthritis and diffuse cutaneous systemic sclerosis, which are in current active clinical trials.450,451
Figure 22.

(A) ADC clinical trial indications; (B) Percentage of ADC clinical trials in various phases for the treatment of solid tumors and hematological malignancies; (C) Percentage of ADC clinical trials in various statuses for the treatment of solid tumors and hematological malignancies.
Analysis of ADC clinical trial phases reveals that most oncology clinical trials are in early phase development. Solid tumors have 88% of their trials in early stage clinical trials from early Phase I trials through Phase II trials with hematological malignancies encompassing 86% (Figure 22B). Examining these clinical trials, a step further, by status, shows ADC clinical trials treating solid tumors have a higher percentage of trials in the not yet recruiting, recruiting, and active statuses than trials treating hematological malignancies. 75% of current clinical trials for solid tumor indications are currently active or getting ready to be active in the clinical trial pipeline (Figure 22C) versus 51% for hematological malignancy indications. While ADC clinical trials focused on oncology are largely equal when it comes to phases of study, currently solid tumor indications have a more active presence in the clinical trial pipeline over hematological malignancy indications.
Clinical trials that disclose a specific tumor indication are characterized by the trial phase and stage in Figures 23 and 24. Prostate, melanoma, brain, bone, and pancreatic cancer are the solid tumor indications with the largest percentage of trials in early stage clinical development from early Phase I trials through Phase II trials. On the other hand, breast, gastric, bladder, and lung cancer are more established in the clinical pipeline with the highest percentage of late-stage trials. In respect to hematological malignancies, myeloma has the largest percentage of clinical trials in early phase development with leukemia and lymphoma having the largest percentage of late-stage trials.
Figure 23.

Percentage of ADC clinical trials in various phases for the treatment of specific solid tumors and hematological malignancies.
Figure 24.

Percentage of ADC clinical trials in various statuses for the treatment of specific solid tumors and hematological malignancies.
Examining the above clinical trials, by status, shows ADC clinical trials treating bladder, esophageal, head and neck, melanoma, and gynecologic cancer have the highest percentage of trials in the not yet recruiting, recruiting, and active statuses for solid tumor indications. 79–84% of current clinical trials for solid tumor indications are currently active or getting ready to be active in the clinical trial pipeline (Figure 23). Pancreatic, lung, gastric, bone, and colorectal cancer have the greatest percentage of completed trials ranging from 35 to 65%, respectively. The hematological malignancy indication myeloma has the highest percentage of trials (81%) in the not yet recruiting, recruiting, and active statuses contrasting with myelodysplastic syndrome, which has 44% of its trials in the completed status.
A selection of ADC clinical trials focusing on the treatment of solid tumors is highlighted in Table 3 to display the variety of ADC candidates in clinical development along with their sponsors, targeted solid tumor indications, target antigen, and phases. Only clinical trials currently in recruiting or active status are showcased to reveal the most current and promising ADC candidates. Over 135 ADC candidates are currently in clinical development for solid tumor indications (Supporting Information, Table S2). ADC candidates focusing on the treatment of lung, gastric, pancreatic, colorectal, breast, gynecologic, prostate, and bladder cancers, along with head and neck cancers, are all currently highly represented in the clinical pipeline (Table 3). Bispecific ADC candidates are also included as they have entered Phase I and II clinical trials for the treatment of various solid tumors including breast, lung, and esophageal cancer.
Table 3. Highlighted ADC Clinical Trials with Solid Tumor Indications in the Development Pipeline.
| ADC intervention | Sponsor | Solid tumor indications | Target | NCT number | Clinical trial phase |
|---|---|---|---|---|---|
| ABBV-400 | AbbVie | Non-Small Cell Lung Cancer|Gastresophageal Cancer|Colorectal Cancer | cMET | NCT05029882 | Phase I |
| ADCT-901 | ADC Therapeutics | Advanced Solid Tumors | KAAG1 | NCT04972981 | Phase I |
| ARX517 | Ambrx | Advanced Solid Tumors | PSMA | NCT04662580 | Phase I |
| ASN004 | Kirilys Therapeutics | Solid Tumors | 5T4 | NCT04410224 | Phase I |
| AZD9592 | AstraZeneca | Non-Small Cell Lung Cancer|Head and Neck Cancer | cMET l EGFR(bispecific) | NCT05647122 | Phase I |
| BAY-2315497 | Bayer | Prostate Cancer | PSMA | NCT03724747 | Phase I |
| BYON3521 | Byondis | Solid Tumors | cMET | NCT05323045 | Phase I |
| CMG901 | Keymed Biosciences Co. | Gastric Cancer l Pancreatic Cancer | Claudin 18.2 | NCT04805307 | Phase I |
| DS-6000a | Daiichi Sankyo | Renal Cell Carcinoma| Gynecologic Cancer | CDH6 | NCT04707248 | Phase I |
| HS-20093 | Shanghai Hansoh Biomedical Co. | Advanced Solid Tumors | B7H3 | NCT05276609 | Phase I |
| IBI-343 | Innovent Biologics | Advanced Solid Tumors | Claudin 18.2 | NCT05458219 | Phase I |
| IMGN151 | ImmunoGen | Gynecologic Cancer | FRα | NCT05527184 | Phase I |
| M1231 | EMD Serono Research and Development Institute | Esophageal Cancer| Non-Small Cell Lung Cancer | MUC1-EGFR (bispecific) | NCT04695847 | Phase I |
| MYTX-011 | Mythic Therapeutics | Lung Cancer | cMET | NCT05652868 | Phase I |
| ORM-5029 | Orum Therapeutics | HER2-positive Breast Cancer | HER2 | NCT05511844 | Phase I |
| PYX-201 | Pyxis Oncology | Advanced Solid Tumors | Extradomain B fibronectin | NCT05720117 | Phase I |
| STRO-002 | Sutro Biopharma | Gynecologic Cancer | CD74 | NCT03748186 | Phase I |
| TORL-1–23 | TORL Biotherapeutics | Advanced Solid Tumor| Gynecologic Cancer| Lung Cancer | Claudin 18.2 | NCT05103683 | Phase I |
| XMT-1660 | Mersana Therapeutics | Breast Cancer| Gynecologic Cancer | B7H4 | NCT05377996 | Phase I |
| YL202 | MediLink Therapeutics | Non-Small Cell Lung Cancer| Breast Cancer | HER3 | NCT05653752 | Phase I |
| Zanidatamab Zovodotin | Zymeworks | HER2-expressing Cancers | HER2 domain II l HER2 domain IV (Bispecific) | NCT03821233 | Phase I |
| 9MW2821 | Mabwell | Solid Tumors | Nectin-4 | NCT05216965 | Phase I |Phase II |
| AZD8205 | AstraZeneca | Breast Cancer|Biliary Tract Carcinoma| Gynecologic Cancer | B7H4 | NCT05123482 | Phase I |Phase II |
| BB-1705 | Bliss Biopharmaceutical (Hangzhou) Co. | Solid Tumor | EGFR | NCT05217693 | Phase I |Phase II |
| BDC-1001 | Bolt Biotherapeutics | HER2 Positive Solid Tumors | HER2 | NCT04278144 | Phase I |Phase II |
| BIO-106 | BiOneCure Therapeutics | Advanced Solid Tumor | TROP2 | NCT05320588 | Phase I |Phase II |
| DB-1303 | DualityBio | HER2 Positive Advanced Solid Tumor | HER2 | NCT05150691 | Phase I |Phase II |
| FOR46 | Fortis Therapeutics | Prostate Cancer | CD46 | NCT05011188 | Phase I |Phase II |
| LM-302 | Turning Point Therapeutics | Advanced Solid Tumor | Claudin 18.2 | NCT05001516 | Phase I |Phase II |
| MRG004A | Shanghai Miracogen | Advanced or Metastatic Solid Tumors | Tissue Factor | NCT04843709 | Phase I |Phase II |
| NBE-002 | NBE-Therapeutics | Breast Cancer | ROR1 | NCT04441099 | Phase I |Phase II |
| OBI-999 | OBI Pharma | Advanced Solid Tumor | Globo H | NCT04084366 | Phase I |Phase II |
| Ozuriftamab Vedotin | BioAtla | Non-Small Cell Lung Cancer| Triple Negative Breast Cancer| Melanoma| Head and Neck Cancer | ROR2 | NCT03504488 | Phase I |Phase II |
| PRO1184 | ProfoundBio | Gynecologic Cancer| Lung Cancer | Breast Cancer | FRα | NCT05579366 | Phase I |Phase II |
| REGN5093 | Regeneron Pharmaceuticals | NSCLC | cMET l CMET (Bispecific) | NCT04077099 | Phase I |Phase II |
| SKB264 | Klus Pharma | Gynecologic Cancer| Gastric Cancer| Bladder Cancer| Lung Cancer| Head and Neck Cancer| Breast Cancer | TROP2 | NCT04152499 | Phase I |Phase II |
| SOT102 | SOTIO Biotech | Gastric Cancer| Pancreatic Cancer | Claudin 18.2 | NCT05525286 | Phase I |Phase II |
| W0101 | Pierre Fabre | Advanced or Metastatic Solid Tumors | IGF-1R | NCT03316638 | Phase I |Phase II |
| Zilovertamab vedotin | Merck | Bladder Carcinoma | ROR1 | NCT05562830 | Phase I |Phase II |
| Mecbotamab Vedotin | BioAtla | Non-Small Cell Lung Cancer | AXL | NCT04681131 | Phase II |
| CX-2009 | CytomX Therapeutics | Breast Cancer | CD166 | NCT04596150 | Phase II |
| MORAb-202 | Bristol-Myers Squibb | Non-Small Cell Lung Cancer | Fos-related antigen | NCT05577715 | Phase II |
| RC108 | RemeGen Co. | Gastric Cancer | cMET | NCT05628857 | Phase II |
| Vobramitamab Duocarmazine | MacroGenics | Prostate Cancer | B7H3 | NCT05551117 | Phase II |
| ARX78 | Jiangsu HengRui Medicine Co. | HER2-positive Breast Cancer | HER2 | NCT05426486 | Phase II |Phase III |
| Enfortumab Vedotin | Astellas Pharma | Bladder Cancer | Nectin-4 | NCT03474107 | Phase III |
| Mirvetuximab Soravtansine | ImmunoGen | Gynecologic Cancer | FRα | NCT04209855 | Phase III |
| MRG002 | Shanghai Miracogen | Advanced or Metastatic Urothelium Cancer | HER2 | NCT05754853 | Phase III |
| Patritumab Deruxtecan | Daiichi Sankyo | Nonsmall Cell Lung Cancer | HER3 | NCT05338970 | Phase III |
| RC48 | RemeGen Co. | Bladder Cancer | HER | NCT05302284 | Phase III |
| SAR-408701 | Sanofi | Non-Small Cell Lung Cancer Metastatic | CEACAM5 | NCT04154956 | Phase III |
| Telisotuzumab Vedotin | AbbVie | Non-Small Cell Lung Cancer | cMET | NCT04928846 | Phase III |
| Tisotumab Vedotin | Seagen | Gynecologic Cancer | Tissue Factor | NCT04697628 | Phase III |
| Trastuzumab Emtansine | Hoffmann-La Roche | Breast Cancer | HER2 | NCT01772472 | Phase III |
| Trastuzumab Duocarmazine | Byondis | Metastatic Breast Cancer | HER2 | NCT03262935 | Phase III |
| Upifitimab Rilsodotin | Mersana Therapeutics | Gynecologic Cancer | NaPi2b | NCT05329545 | Phase III |
| Sacituzumab Govitecan | Gilead Sciences | Solid Tumors | TROP2 | NCT04319198 | Phase IV |
A more exhaustive list of ADC candidates in clinical development for all indications, along with their companies and target antigens, can be located in Table S2. The most utilized target antigen for ADCs currently in clinical trials is HER2, followed by TROP2, Claudin 18.2, cMET, and B7H3, respectively. Also, around 40 ADC candidates currently in clinical trials are topoisomerase I inhibitors which impact DNA replication in cancer cells, resulting in cancer cell death.
ADC clinical trials focusing on the treatment of hematological malignancies are highlighted in Table 4, similar to solid tumors. This table showcases the variety of ADC candidates currently being researched for the treatment of hematological malignancies along with their sponsor, hematological malignancy indication, target antigen, and phase. Clinical trials in recruiting or active status are highlighted. The total of ADC candidates currently in clinical development for hematological malignancies is less than solid tumors at around 30 ADC drug candidates compared to greater than 135 for solid tumors (Supporting Information, Table S2). ADC candidates focusing on the treatment of multiple myeloma and various types of leukemia and lymphoma are all currently being researched in the clinical development pipeline (Table 4).
Table 4. Highlighted ADC Clinical Trials with Hematological Malignancies Indications in the Development Pipeline.
| ADC intervention | Sponsor | Hematological malignancy indications | Target | Clinical trial phase | NCT number |
|---|---|---|---|---|---|
| INA03 | INATHERYS | Acute Lymphoblastic Leukemia |Acute Myeloid Leukemia | CD71 | Early Phase I | NCT03957915 |
| ABBV-319 | AbbVie | Diffuse Large B-Cell Lymphoma|Lymphocytic Leukemia|Follicular Lymphoma | CD19 | Phase I | NCT05512390 |
| CC-99712 | Celgene | Multiple Myeloma | BCMA | Phase I | NCT04036461 |
| CS5001 | CStone Pharmaceuticals | Advanced Lymphoma | PTK7 | Phase I | NCT05279300 |
| F0002 | Shanghai Fudan-Zhangjiang Bio-Pharmaceutical Co. | CD30+ hematological malignancies | CD30 | Phase I | NCT03894150 |
| JBH492 | Novartis | Non-Hodgkin’s Lymphoma|Lymphocytic Leukemia | CCR7 | Phase I | NCT04240704 |
| Moxetumomab Pasudotox | National Cancer Institute | Hairy Cell Leukemia | CD22 | Phase I | NCT03805932 |
| MRG001 | Shanghai Miracogen | B-cell Non-Hodgkin Lymphoma | CD20 | Phase I | NCT05155839 |
| TRS005 | Zhejiang Teruisi Pharmaceutical | B-cell Non-Hodgkin’s Lymphoma | CD20 | Phase I | NCT05395533 |
| ADCT-602 | ADC Therapeutics | Acute Lymphoblastic Leukemia | CD22 | Phase I |Phase II | NCT03698552 |
| BN301 | BioNova Pharmaceuticals (Shanghai) | Non Hodgkin’s Lymphoma|Diffuse Large B Cell Lymphoma|Follicular Lymphoma | CD74 | Phase I |Phase II | NCT05611853 |
| CX-2029 | CytomX Therapeutics | Diffuse Large B Cell Lymphoma | CD71 | Phase I |Phase II | NCT03543813 |
| HDP-101 | Heidelberg Pharma | Multiple Myeloma|Plasma Cell Disorder | BCMA | Phase I |Phase II | NCT04879043 |
| PRO1160 | ProfoundBio | Non Hodgkin’s Lymphoma | CD70 | Phase I |Phase II | NCT05721222 |
| STI-6129 | Sorrento Therapeutics | Multiple Myeloma | CD38 | Phase I |Phase II | NCT05308225 |
| Trastuzumab Emtansine | National Cancer Institute | Advanced Lymphoma|Refractory Plasma Cell Myeloma | HER2 | Phase II | NCT04439110 |
| Zilovertamab vedotin | Merck | Diffuse Large B-Cell Lymphoma | ROR1 | Phase II |Phase III | NCT05139017 |
| Belantamab mafodotin | GlaxoSmithKline | Multiple Myeloma | BCMA | Phase III | NCT04162210 |
| Gemtuzumab Ozogamicin | Gruppo Italiano Malattie EMatologiche dell’Adulto | Acute Myeloid Leukemia | CD33 | Phase III | NCT04168502 |
| Loncastuximab Tesirine | ADC Therapeutics | Diffuse Large B-Cell Lymphoma | CD19 | Phase III | NCT04384484 |
| Polatuzumab Vedotin | Hoffmann-La Roche | Diffuse Large B-Cell Lymphoma | CD79 | Phase III | NCT03274492 |
| Brentuximab vedotin | Takeda | Anaplastic Large-cell Lymphoma | CD30 | Phase IV | NCT01909934 |
| Inotuzumab ozogamicin | Pfizer | Leukemia| Precursor B-Cell Lymphoblastic Leukemia-Lymphoma | CD22 | Phase IV | NCT03677596 |
While most ADC clinical trials focus on oncology, there are a few studies exploring research in emerging areas of interest. These include trials looking at the treatment of autoimmune disorders diffuse cutaneous systemic sclerosis and rheumatoid arthritis, along with amyloidosis (Table 5).
Table 5. Highlighted ADC Clinical Trials with Non-Oncology Indications in the Development Pipeline.
| ADC intervention | Sponsor | Indications | Target | Clinical trial phase | NCT number |
|---|---|---|---|---|---|
| Brentuximab vedotin | Seagen | Diffuse Cutaneous Systemic Sclerosis | CD30 | Phase I |Phase II | NCT03222492 |
| STI-6129 | Sorrento Therapeutics | Light Chain Amyloidosis | CD38 | Phase I |Phase II | NCT04316442 |
| ABBV-154 | AbbVie | Polymyalgia Rheumatica | TNF | Phase II | NCT04972968 |
| ABBV-154 | AbbVie | Rheumatoid Arthritis | TNF | Phase II | NCT04888585 |
| Belantamab mafodotin | GlaxoSmithKline | Amyloidosis | BCMA | Phase II | NCT04617925 |
7.3. Approved ADCs
Currently, there are 15 approved ADCs that have received regulatory approval anywhere in the world (Table 6).
Table 6. List of Approved ADCs,21,115,452−456 with the Number of Related Documents in the CAS Content Collectionb.
| Drug | Trade name | CAS REG # | Maker | Condition | Target | Approval year | mAb | Linker | Payload | Payload action | DAR | Conjugation | No. documents |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin51,52 | Mylotarg | 220578–59–6 | Pfizer/Wyeth | relapsed acute myelogenous leukemia | CD33 | 2000; 2017 | humanized IgG4k | hydrazone, acid cleavable | N-acetyl-γ-calicheamicin (ozogamicin) | DNA cleavage | 2–3 | Lys | 2066 |
| Brentuximab vedotin53,54 | Adcetris | 914088–09–8 | Seagen Genetics, Millennium/Takeda | relapsed Hodgkin lymphoma, relapsed anaplastic large cell lymphoma | CD30 | 2011 | chimeric IgG1 | Val-Cit, enzyme cleavable | MMAE/Auristatin | microtubule Inhibitor | 4 | Cys | 1738 |
| Trastuzumab emtansine50,55 | Kadcyla | 1018448–65–1 | Genentech, Roche | metastatic HER2-positive breast cancer | HER2 | 2013 | humanized IgG1 | MCC, noncleavable | DM1/Maytansinoid | microtubule Inhibitor | 3.5 | Lys | 1507 |
| Inotuzumab ozogamicin48,56 | Besponsa | 635715–01–4 | Pfizer/Wyeth | CD22-positive acute lympho-blastic leukemia | CD22 | 2017 | humanized IgG4 | hydrazone, acid cleavable | N-acetyl-γ-calicheamicin (ozogamicin) | DNA cleavage | 6 | Lys | 518 |
| Moxetumomab pasudotox57,58 | Lumoxiti | 1020748–57–5 | AstraZeneca | relapsed or refractory hairy cell leukemia | CD22 | 2018 | - | mc-VC-PABC enzyme cleavable | Pseudomonas Exotoxin A (PE38) | peptide toxin class | - | Cys | 172 |
| Polatuzumab vedotin-piiq59,60 | Polivy | 1313206–42–6 | Genentech, Roche | diffuse large B-cell lymphoma | CD79 | 2019 | humanized IgG1 | Val–Cit, enzyme cleavable | MMAE/Auristatin | microtubule Inhibitor | 3.5 | Cys | 200 |
| Enfortumab vedotin61,62 | Padcev | 1346452–25–2 | Astellas/Seagen Genetics | locally advanced or metastatic urothelial cancer | Nectin-4 | 2019 | humanized IgG1 | Val-Cit, enzyme cleavable | MMAE/Auristatin | microtubule Inhibitor | 3.8 | Cys | 179 |
| Trastuzumab deruxtecan63,64 | Enhertu | 1826843–81–5 | AstraZeneca/Daiichi Sankyo | unresectable or metastatic HER2-positive breast cancer | HER2 | 2019 | humanized IgG1 | maleimide—GGFG enzyme cleavable | DXd/Camptothecin | TOPO1 Inhibitor | 8 | Cys | 291 |
| Sacituzumab govitecan65,66 | Trodelvy | 1491917–83–9 | Immuno-medics | metastatic triple-negative breast cancer | Trop-2 | 2020 | humanized IgG1 | CL2A acid cleavable | SN-38/Camptothecin | TOPO1 Inhibitor | 7.6 | Cys | 237 |
| Belantamab mafodotin-blmf67,68 | Blenrep | 2050232–20–5 | GlaxoSmithKline (GSK) | relapsed or refractory multiple myeloma | BCMA | 2020a | humanized IgG1 | MC noncleavable | MMAF/Auristatin | microtubule Inhibitor | 4 | Cys | 132 |
| Loncastuximab tesirine-lpyl69,70 | Zynlonta | 1879918–31–6 | ADC Therapeutics | large B-cell lymphoma | CD19 | 2021 | IgG1 | enzyme cleavable | SG3199/PBD dimer | DNA cleavage | 2.3 | Cys | 66 |
| Tisotumab vedotin-tftv71,72 | Tivdak | 1418731–10–8 | Seagen Inc. | recurrent or metastatic cervical cancer | Tissue factor | 2021 | IgG1 | enzyme cleavable | MMAE/Auristatin | microtubule Inhibitor | 4 | Cys | 55 |
| Cetuximab Sarotalocan21,73,74 | Akalux | 2166339–33–7 | Rakuten Medical | unresectable locally advanced, recurrent head and neck cancer | EGFR | 2021 | IgG1 | N/A | IRDye700DX | photosensitizer | 1.3–3.8 | Lys | 2 |
| Disitamab Vedotin75,76 | Aidixi | 2136633–23–1 | RemeGen | HER2-overexpressing gastric cancer | HER2 | 2021 | IgG1 | enzyme cleavable | MMAE | microtubule Inhibitor | 4 | Cys | 15 |
| Mirvetuximab soravtansine77,78 | Elahere | 1453084–37–1 | ImmunoGen | platinum-resistant ovarian cancer | FRα | 2022 | IgG1 | enzyme cleavable | DM4 | microtubule inhibitor | 3.4 | Cys | 87 |
Withdrawn on Nov 22, 2022.79
Abbreviations: B cell maturation antigen (BCMA); cluster of differentiation (CD); cleavable PEG8- and triazole-containing PABC–peptide– MC linker (CL2A); derivative of maytansine (DM1); exatecan derivative (DXd); glycine–glycine–phenylalanine–glycine tetrapeptide linker (GGFG); human epidermal growth factor receptor 2 (HER2); maleimidocaproyl (MC); 4-maleimidomethyl cyclohexane-1-carboxylate (MCC); monomethyl auristatin E (MMAE); monomethyl auristatin F (MMAF); active metabolite of the topoisomerase I inhibitor irinotecan (SN-38); tumor-associated calcium signal transducer 2 (TROP-2).
7.3.1. Hematological Malignancies
Gemtuzumab ozogamicin (Mylotarg) contains an anti-CD33 humanized IgG4κ monoclonal antibody connected to ozogamicin, a calicheamicin derivative payload, via a cleavable hydrazone linker.457 It binds preferentially to cells expressing the CD33 surface antigen, leading to the internalization of the gemtuzumab ozogamicin ADC and cleavage of the linker within the lysosomes via acid hydrolysis, with the subsequent calicheamicin reduction by glutathione inducing double-strand DNA breaks, resulting in cell death.458,459 Gemtuzumab ozogamicin was the first ADC to reach the clinic, approved by the FDA in 2000 for the treatment of relapsed or refractory CD33-positive acute myeloid leukemia (AML).460 The accelerated approval required postmarketing trials to be conducted to confirm treatment efficacy. The negative results from those studies (NCT00085709; ISRCTN17161961) prompted Pfizer to withdraw gemtuzumab ozogamicin from the market in 2010.461−463 Later on, based on the positive results of subsequent trials (NCT00927498; NCT00091234), using reduced dosing strategies, gemtuzumab ozogamicin was reapproved by the USFDA in 2017 for treatment of newly diagnosed CD33-positive AML as well as for relapsed or refractory CD33-positive AML.127,463,464
Brentuximab vedotin (Adcetris) was the second ADC to receive accelerated US FDA approval in 2011 based on Phase II trials in patients with relapsed Hodgkin’s lymphoma or systemic anaplastic large cell lymphoma.43,465,466 It comprises MMAE conjugated to an anti-CD30 antibody via an enzyme cleavable Val-Cit linker.44,467 Hodgkin’s lymphoma cells, as well as malignant cells of anaplastic large cell lymphoma express high levels of CD30.44 In 2015, brentuximab vedotin received full approval from the US FDA based on the results of the Phase III trial.468 A recently reported randomized Phase III trial provides compelling evidence in favor of brentuximab vedotin for treating cutaneous T cell lymphoma.469 The benefit of brentuximab vedotin has been examined in randomized studies in combination with approved chemotherapeutic agents (NCT01712490; NCT01777152), as well as in combination with immune checkpoint inhibitors (NCT02684292; NCT03138499).44,88
Inotuzumab ozogamicin (Besponsa) is the second ADC using the calicheamicin ozogamicin payload, linked to a humanized mAb targeting the B cell antigen CD22, with an average DAR of 5–7. CD22 is a cell surface antigen in the majority of B-cell acute lymphoblastic leukemia.470,471 The safety and efficacy of inotuzumab ozogamicin was evaluated in an open-label, randomized, international, multicenter Phase III study (INO-VATE 1022).472 It was approved by the US FDA in 2017 against relapsed or refractory acute lymphoblastic leukemia.127,473,474
Moxetumomab pasudotox (Lumoxiti) consists of moxetumomab targeting CD22 conjugated to a 38kD fragment of Pseudomonas exotoxin A (PE38).58 CD22 is expressed on mature B cells and to a larger extent on 100% of hairy cells, which provides an ideal therapeutic target for the treatment of hairy cell leukemia, a rare hematological malignancy, characterized by splenomegaly, hemorrhage, and an accumulation of abnormal B lymphocytes.475−477 Upon binding to CD22, moxetumomab pasudotox is internalized, its mc-VC-PABC linker cleaved by proteases and the catalytic domain of the exotoxin is released inside cancer cells leading to inhibition of translation of proteins and apoptosis. US FDA approved Lumoxiti of AstraZeneca in 2018 for the treatment of patients with hairy cell leukemia who have previously failed to receive at least two systemic therapies (including purine nucleoside analogs).478 Moxetumomab pasudotox was thus the first new drug approved for the treatment of hairy cell leukemia in the past 20 years.88
Polatuzumab vedotin (Polivy) contains a humanized antibody targeting CD79b antigen, linked to microtubule-disrupting MMAE payload via a protease-cleavable dipeptide linker (mc-VC-PABC) with an average DAR of 3.5.479 CD79b is expressed on >90% of B-cell non-Hodgkin lymphomas and has proven to be a promising antibody target.480,481 Polatuzumab vedotin selectively binds to CD79b upon administration, followed by endocytosis and proteolytic cleavage to release MMAE inducing cell cycle arrest and subsequent cell death. Polivy was approved by the US FDA in 2019, for use in treatment of diffuse large B-cell lymphoma, the most common type of non-Hodgkin lymphomas, in patients who have received at least two prior therapies.482
Belantamab mafodotin (Blenrep) is composed of a humanized anti-B cell maturation antigen (BCMA) mAb coupled with cytotoxic agent MMAF, a mitotic inhibitor, through a noncleavable maleimidocaproyl (MC) linker. Belantamab mafodotin has an average DAR of 4. BCMA is a transmembrane glycoprotein explicitly overexpressed on the surface of multiple myeloma cells.483 Upon internalization and degradation in lysosomes belantamab mafodotin releases MMAF inside multiple myeloma cells, which inhibits cell division by blocking microtubule polymerization, resulting in cell cycle arrest and inducing caspase-3-dependent apoptosis. As a result, belantamab mafodotin is effective at killing cancer cells overexpressing BCMA. The US FDA approved Blenrep in 2020 for the treatment of multiple myeloma, based on the results of the DREAMM-2 clinical trial.484
Loncastuximab tesirine (Zynlonta) comprises a humanized mAb targeting CD19 conjugated to pyrrolobenzodiazepine (PBD) dimer via a cleavable (valine-alanine dipeptide) maleimide type linker, with an average DAR of ∼2.3.485,486 The PBD dimer is a novel generation of cytotoxic payload for ADC development.152 It binds to DNA and causes strong cross-linking that prevents DNA strand separation, thus preventing DNA transcription and replication and killing the cell.487 Zynlonta received accelerated approval by the US FDA in 2021, for the treatment of patients with large B-cell lymphoma after two or more lines of systemic therapy. The approval of Zynlonta was based on data from the LOTIS-2 trial.488
7.3.2. Solid Tumors
Trastuzumab emtansine (Kadcyla) includes an anti-HER2 humanized IgG1 monoclonal antibody connected to a DM1 payload via a noncleavable MCC linker. Because of the noncleavable linker present in trastuzumab emtansine, after entry into the HER2-positive cancer cell, mAb proteolysis inside lysosomes is necessary to release the free DM1 payload.266,489 Upon its release from the lysosome, DM1 binds to tubulin at the vinca-binding site and inhibits tubulin polymerization, inducing mitotic arrest and cell death.490 Trastuzumab emtansine was approved by the US FDA in 2013 as a single-agent treatment for HER2-positive metastatic breast cancer in patients previously administered trastuzumab and a taxane, either separately or in combination.491 In 2019, this was extended to include HER2- positive early breast cancer in patients with residual invasive disease after neoadjuvant taxane-based chemotherapy and trastuzumab-based treatment.127,492
Enfortumab vedotin (Padcev) is approved by the US FDA for the treatment of patients with locally advanced or metastatic urothelial cancer.493 It comprises a fully human antinectin-4 IgG1κ monoclonal antibody (AGS-22C3), linked to MMAE via a protease-cleavable linker (MC-VC-PABC), with an average DAR of ∼3.8.494 Nectin-4 is a transmembrane protein, which is abundantly expressed in several malignancies, especially in urothelial carcinoma, thus being a compelling target for ADC molecular design. An accelerated approval was granted by the FDA in 2019, and a regular approval was further granted in 2021 based on results from an open-label, randomized, multicenter Phase III study (EV-301).495−497
Enfortumab vedotin-ejfv (Padcev, Astellas Pharma) is a member of the first approved ADC-based combination therapy formulation: on April 3, 2023, the US FDA granted accelerated approval to enfortumab vedotin-ejfv with pembrolizumab (Keytruda, Merck) for treatment of locally advanced or metastatic urothelial carcinoma in patients ineligible for cisplatin-containing chemotherapy.80
Trastuzumab deruxtecan (Enhertu) is a HER2-targeted ADC for the treatment of patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2 based regimens in the metastatic setting.498 It includes a humanized HER2 antibody (trastuzumab) conjugated to a topoisomerase I inhibitor (DXd) as a payload through an enzymatically cleavable tetrapeptide-based linker with an average DAR of 7–8. DXd is a very powerful payload,499 and the tetrapeptide-based linker technology stabilizes the ADC in plasma to reduce the risk of systemic toxicity.500 Enhertu was approved by the US FDA in 2019 based on positive results from a single-arm, multicenter, Phase II DESTINY-Breast01 study.501 Subsequently, Enhertu has also been approved for gastric cancer.502
Sacituzumab govitecan (Trodelvy) comprises a humanized monoclonal antibody targeting Trop-2 conjugated to a topoisomerase I inhibitor SN-38 by means of a hydrolyzable linker (CL2A) with an average DAR of ∼7.6. Overexpression of Trop-2, a 40-kDa glycoprotein that plays a role as transducer of intracellular calcium signaling,503,504 was observed in the majority of solid tumors, including triple-negative breast cancer.505 The payload SN-38 inhibits DNA topoisomerase I thus causing DNA single strand breaks and eventually leads to cell death.506 The CL2A linker improves the binding ratio of Trop-2 antibody to SN-38, with higher toxic concentration in tumor but lower concentration in nontarget tissues.507 Linker optimization allows both controlled release of the drug and diffusion through the cell membrane, enabling the drug to kill neighboring tumor cells (the bystander effect).508 Sacituzumab govitecan received accelerated approval by the US FDA in 2020, for the treatment of patients with unresectable locally advanced or metastatic triple-negative breast cancer who have received two or more prior systemic therapies, at least one of them for metastatic disease. The clinical efficacy of sacituzumab govitecan was further confirmed in a multicenter, open-label, randomized trial (ASCENT),509 which promoted the US FDA to grant a regular approval.510,511
Cetuximab sarotalocan (Akalux) includes an anti-EGFR chimeric monoclonal antibody, cetuximab, conjugated with IRDye700DX, a near-infrared photosensitizing dye.512 The average DAR of cetuximab sarotalocan was in the 1.3–3.8 range. EGFR is amply expressed on the surface of multiple kinds of solid tumors, including head and neck squamous cell carcinomas, esophageal cancer, lung cancer, colon cancer, pancreatic cancer and other solid tumors.513 Cetuximab sarotalocan targets EGFR and is locally activated using a laser to accurately induce the rapid death of cancer cells without damaging surrounding normal tissues.514 Cetuximab sarotalocan was approved by the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan in 2019 as a treatment product of near-infrared photoimmunotherapy for unresectable locally advanced or recurrent head and neck squamous cell carcinoma.73 The approval was supported by the positive data from a multicenter, open-label Phase IIa trial.515
Disitamab vedotin (Aidixi) consists of a humanized HER2 antibody, a cathepsin cleavable linker (mc-VC-PABC), and a cytotoxic agent, MMAE, with an average DAR ∼ 4.516 In June 2021, disitamab vedotin was conditionally approved by the National Medical Products Administration (NMPA) of China for the treatment of patients with locally advanced or metastatic gastric cancer, including gastroesophageal junction adenocarcinoma, who have received at least 2 types of systemic chemotherapy. The approval was supported by the results of the RC48-C008 study, which demonstrated a clinically significant response and survival benefit for patients receiving the drug.517 Disitamab vedotin has also conditionally approval by NMPA in December 2021 for treatment of patients with HER2 positive locally advanced or metastatic urothelial cancer who have also previously received platinum-containing chemotherapy treatment. It was also supported by the results of the RC48-C005 study, an open-label, multicenter, single-arm, nonrandomized Phase II study.518
Tisotumab vedotin (Tivdak) contains a fully humanized mAb binding to tissue factor, a cleavable mc-VC-PABC linker, and an antimitotic agent, MMAE, with an average DAR of 4.519 Tissue factor is overexpressed on several solid tumors.520 Bystander effect, and antibody-dependent cellular cytotoxicity and phagocytosis have been also reported to be involved in the mechanism of action of tisotumab vedotin.520 Tivdak was approved by the US FDA in September 2021, for patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy. The approval was supported by findings from the innovaTV 204 study, a multicenter, open-label, single-arm, Phase II trial.521
Mirvetuximab soravtansine (Elahere) comprises a humanized mAb targeting folate receptor alpha (FRα) conjugated to a potent cytotoxic DM4 by a cleavable linker (sulfo-SPDB), for the treatment of ovarian cancer as orphan drug designation.522 FRα is expressed at high levels in most cases of epithelial ovarian cancer as well as in endometrial cancer and lung adenocarcinoma.522 Most normal tissues do not express FRα, making it a promising target for ADC.522 The hydrophilicity of the linker is increased by the introduction of a sulfonate group, while the addition of methyl groups α to the disulfide moiety reduces premature release of the drug in circulation. Mirvetuximab soravtansine was approved by the US FDA in November 2022 for the treatment of patients with FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received one to three prior systemic treatment regimens.78,523
7.3.3. Combination Therapy
In addition to the above approved ADCs, on April 3, 2023, the FDA granted accelerated approval to enfortumab vedotin-ejfv (Padcev, Astellas Pharma) with pembrolizumab (Keytruda, Merck) for treatment of locally advanced or metastatic urothelial carcinoma in patients ineligible for cisplatin-containing chemotherapy.80,524 Another two combination treatments including pembrolizumab (Keytruda, Merck) are in a late stage of clinical trials: datopotamab deruxtecan (Dato-DXd) with pembrolizumab (Keytruda) plus platinum-based chemotherapy,525,526 and sacituzumab govitecan-hziy (Trodelvy, Gilead) and pembrolizumab (Keytruda),527,528 both for treatment of non-small cell lung cancer (NSCLC).
8. Noteworthy Patents
ADC patents within the CAS Content Collection continue to grow not only in numbers but also in ADC technological diversity. The diversity of ADC compounds, linker technology, bioconjugation techniques, target antigen moieties, and diseases treated are highlighted with a selection of notable patents presented in Table 7.
Table 7. Notable ADC Patents.
| Patent number | Year | Assignee, location | Description |
|---|---|---|---|
| WO2009140242 | 2009 | Genentech, USA | Analysis of ADCs by bead-based affinity capture and mass spectrometry |
| US9364554B2 | 2013 | Centrose, USA | Extracellular-targeted drug conjugates (not internalized) in which an antibody or other targeting agent is linked to a drug through a linker |
| WO2017009258 | 2017 | Genmab, Denmark | Axl-specific antibody–drug conjugates for cancer treatment |
| US10772965 | 2018 | RC Biotechnologies, USA | Covalent linkers in ADCs and methods of making and using the same. Provides novel and advantageous compositions having a linker capable of covalently coupling one or more free thiols of an antibody which can be used in ADCs. |
| WO2019219891 | 2019 | Daiichi Sankyo, Japan | Anti-MUC1 ADCs for treating cancer, infection, autoimmune disease, or immunodeficiency. The conjugates consist of exatecan derivatives coupled to anti MUC1 antibodies. |
| US20190099499 | 2019 | Pfizer, USA | Cysteine-engineered ADCs for site-specific conjugation. |
| CN109106951 | 2019 | Sichuan Baili Pharmaceutical, China | Camptothecin-antibody conjugate and application in treating tumors, autoimmune, or infectious diseases |
| WO2019126691 | 2019 | Mersana Therapeutics, USA | ADCs comprising PBD drug moieties and methods of using these conjugates as therapeutics and/or diagnostics. |
| WO2020112588 | 2020 | Bristol-Myers Squibb, USA | Engineering ADCs with glutamine-containing extensions on the C-terminus of the light chain using transglutaminase for improved stability and pharmacokinetics |
| US20200114018 | 2020 | Genentech, USA | Methods of treating residual breast cancer with the ADC trastuzumab emtansine |
| WO2020180121 | 2020 | LegoChem Biosciences, South Korea| Y-Biologics, South Korea | ADC comprising an antibody binding to DLK1 or an antigen-binding fragment, and a pharmaceutical agent |
| WO2020006449 | 2020 | GO Therapeutics, USA | Antiglyco-MUC1 antibodies and antigen-binding fragments that specifically bind to a cancer-specific glycosylation variant of MUC1 treat cancer |
| WO2020075817 | 2020 | Takeda, Japan | Method for manufacturing an ADC by using a microreactor. The method includes mixing, using a microreactor, a solution containing triscarboxyethyl phosphine, and IgG antibody in a reduction reaction initiated by TCEP, and a solution containing a TCEP inhibitor |
| WO2021076196 | 2021 | Genentech, USA| Hoffmann-La Roche, Switzerland | Anti-cd79b immunoconjugates to treat diffuse large b-cell lymphoma comprising anti-CD79b antibodies in combination with an anti-CD20 antibody and one or more chemotherapeutic agents (such as gemcitabine and oxaliplatin) |
| WO2021202984 | 2021 | Mersana Therapeutics, USA | ADCs comprising STING agonists and their use for the treatment of cancer |
| WO2021259928 | 2021 | Sapreme Technologies, Netherlands| Charite - Universitaetsmedizin Berlin, Germany | ADC or an antibody-oligonucleotide conjugate comprising a VHH and a saponin or a ligand-saponin conjugate |
| WO2021222783 | 2021 | Angiex, USA | ADC comprising an anti-TM4SFl antibody or an antigen-binding fragment and a proteasome inhibitor with an anti-TM4SFl antibody or antigen-binding fragment with a modified IgG Fc region with one or more substitutions |
| WO2022199429 | 2022 | Chengdu Scimount Pharmatech, China | Preparation method for dual-drug-linker of ADC and use in cancer treatment |
| US20220226494 | 2022 | AbbVie, USA | Anti-EGFR ADCs which inhibit Bcl-xL |
| WO2022262772 | 2022 | Beijing Sinotau Bio-Pharmaceuticals Technology, China | Engineering of HER3 antibody and use in antibody–drug conjugates for cancer immunotherapy |
| WO2022136642 | 2022 | Sotio Biotech, Czech Republic | Tumor-specific claudin 18.2 ADCs where the antibody or fragment exhibits increased binding to tumor tissue expressing CLDN18.2 over healthy tissue expressing CLDN18.2 |
| WO2022153195 | 2022 | Memorial Sloan Kettering Cancer Center, USA|Tri-Institutional Therapeutics Discovery Institute, USA | Anti-DLL3 antibody–drug conjugate |
| WO2022184082 | 2022 | Sorrento Therapeutics, USA|Levena (Suzhou) Biopharma, China | Antibody–drug conjugates comprising an anti-B-cell maturation antigen antibody |
| US20220162308 | 2022 | Novartis, Switzerland | Engineering anti-CD48 antibody–drug conjugates for use in cancer immunotherapy |
| WO2023274974 | 2023 | ADC Therapeutics, Switzerland| MedImmune, USA | Combination therapy using anti-CD19 ADCs and anti-CD79b conjugates |
| WO2023070125 | 2023 | Academia Sinica, Taiwan| Liu, Fu-Tong, Taiwan | Antibody–drug conjugate for reducing glycosylation of membrane glycoprotein comprising an antibody or antigen-binding fragment, an oligosaccharyltransferase inhibitor, and a linker |
| WO2023275112 | 2023 | Rigshospitalet, Denmark| University of Copenhagen, Denmark | ADCs comprising humanized antibodies targeting uPARAP |
| WO2023281445 | 2023 | TechnoPhage, Portugal|Faculty of Veterinary Medicine of the University of Lisbon, Portugal | Highly specific rabbit anti-cNHL single domain antibodies conjugated with antitumor payload for delivery through the blood brain barrier to the CNS for cancer immunotherapy. |
9. Outlook and Perspectives
Over the past decade, ADCs have made tremendous progress as a result of optimization of the choice of cytotoxic agents, conjugation strategies, better selection of targeted antigens, and improved antibody engineering. However, despite their sophisticated design, ADCs are still associated with certain limitations and the emergence of resistance mechanisms. To overcome these limitations, new antibody formats, new delivery systems, antigenic targets, cytotoxic payloads, and site-specific conjugation methods have continued to be developed and advanced.
9.1. Major Challenges and Perspectives for Antibody–Drug Conjugate Development
The advance of ADCs involves certain challenges that researchers and developers need to overcome in their efforts to design successful ADCs:
Complex design and manufacturing. ADCs are complex molecules that require precise conjugation of the antibody, linker, and cytotoxic drug payload. The manufacturing process can be challenging and involves multiple steps, increasing the complexity and cost of production.
Target selection: Identifying appropriate target antigens that are selectively expressed on cancer cells is a critical challenge. The target antigen should be highly specific to cancer cells to minimize off-target effects and maximize therapeutic efficacy. However, not all cancers have well-defined target antigens, and heterogeneity of antigen expression within tumors can further complicate target selection.
Heterogeneity of target expression. Even when a target antigen is identified, its expression can vary within and between tumors. Heterogeneous antigen expression may result in incomplete target binding and reduce the effectiveness of ADC therapy. Moreover, antigen loss or downregulation can occur during treatment, leading to acquired resistance.
Payload selection and optimization. Selecting an appropriate cytotoxic drug payload is crucial for the potency and effectiveness of ADCs. The cytotoxic drug should have high lethality against cancer cells while maintaining stability during conjugation and circulation. Optimizing the delicate balance between drug potency and linker stability is a complex task that requires careful consideration of the desired mechanism of action and the specific characteristics of the target cancer type.
Linker design and stability. Designing an optimal linker that balances stability, selective cleavage, and efficient drug release is a significant challenge. The linker must be stable during circulation to minimize premature drug release, but it should also be able to efficiently release the cytotoxic drug inside the target cell. Achieving the right balance between linker stability and cleavability is crucial for maximizing therapeutic efficacy.
Pharmacokinetics and biodistribution. Optimizing the pharmacokinetic properties of ADCs is essential for effective drug delivery to tumor sites. ADCs need to have appropriate systemic circulation, tumor penetration, and retention within the tumor microenvironment. Achieving optimal pharmacokinetics and biodistribution can be challenging due to factors such as rapid clearance, limited tumor penetration, and inadequate tumor-specific accumulation.
Maximum tolerated dose (MTD). According to a recent report, the MTDs of ADCs and the related payload small molecule drugs in humans are nearly the same after normalization for cytotoxic agent content,529,530 i.e., current ADCs do not substantially increase the MTDs of their conjugated drugs regardless of their broad diversity. Thus, mechanisms that may provide further improvements in efficacy and tolerability need to be further explored, with in-depth analysis of the pharmacokinetic/pharmacodynamic formulation profiles for optimizing ADC clinical dosing strategies.
Manufacturing complexity and scale-up. ADCs are complex molecules that require precise conjugation of the antibody, linker, and payload. The manufacturing process needs to be scalable, reproducible, and cost-effective. Ensuring consistent quality control throughout the manufacturing process is a significant challenge, especially when dealing with multiple components and their interactions.
DAR heterogeneity. Achieving a consistent and predictable DAR can be difficult for multiple reasons, including manufacturing challenges such as conjugation chemistry and purification; different conjugation techniques leading to higher or lower drug loading on the antibodies; heterogeneity in antibody population, etc.
Immunogenicity and safety. ADCs can induce immune responses, leading to reduced efficacy and potential safety concerns. Minimizing immunogenicity is a challenge that requires careful antibody engineering and testing. Ensuring the safety profile of ADCs, including minimizing off-target effects and potential toxicities, is also a critical consideration.
Off-target effects. While ADCs are designed to target cancer cells selectively, there is a possibility of off-target effects, where the ADC binds to noncancerous cells expressing low levels of the target antigen. This can lead to undesired toxicity in healthy tissues and potential side effects.
Resistance mechanisms. Cancer cells can develop resistance to ADC therapy through various mechanisms, such as antigen loss, altered drug uptake, or drug efflux pumps. These resistance mechanisms can limit the effectiveness of ADCs and reduce treatment efficacy over time.
Regulatory approval. ADCs involve the combination of different components, including an antibody, cytotoxic drug, and linker. Each component may require separate regulatory approval processes, which can be time-consuming and expensive. Meeting regulatory requirements for safety and efficacy is a significant challenge in ADC development.
Cost. ADC therapy can be costly due to the complexity of manufacturing, including the need for highly specialized equipment and processes. The high cost can limit accessibility and affordability for patients.
Addressing these challenges requires continuous advancements in antibody engineering, linker technology, payload design, and the understanding of tumor biology. Collaboration among researchers, pharmaceutical companies, and regulatory agencies is essential to overcome these challenges and bring effective ADC therapies to patients.
An important advance in the development and use of ADC is in combination therapies.3,275,319,531,532 Combining ADCs with other treatment modalities, such as chemotherapy, radiation therapy, or immunotherapies, is an area of active research. Researchers investigate synergistic effects and explore combination strategies to enhance therapeutic outcomes and overcome resistance mechanisms. Noteworthy, on April 3, 2023, the FDA granted accelerated approval to the combination of enfortumab vedotin-ejfv (Padcev, Astellas Pharma) with pembrolizumab (Keytruda, Merck) for treatment of locally advanced or metastatic urothelial carcinoma in patients ineligible for cisplatin-containing chemotherapy.80
Another significant advance in ADC development is companion diagnostics.3,533−536 Biomarker identification and patient stratification based on target expression levels or other predictive factors can help select the most appropriate patients for treatment and improve the clinical outcome of the ADC treatment.
Nanobody-Enhanced ADCs offer another possibility to enhance therapeutic effects. Nanobodies are small, stable, and highly specific, making them suitable for targeting tumor-specific antigens.537,538 When used as the targeting ligands in ADCs, nanobodies offer several advantages: (i) Reduced immunogenicity: nanobodies are derived from camelids or engineered from human antibodies, which can lead to reduced immunogenicity compared to larger antibodies; (ii) Enhanced tissue penetration: their smaller size can improve tissue penetration within solid tumors; (iii) Rapid clearance from circulation: this can reduce off-target effects and systemic toxicity.
Bispecific antibodies are engineered to simultaneously bind to two different antigens or epitopes.539 This enables them to target two distinct molecular targets, often with therapeutic advantages. Bispecific antibodies in ADCs can simultaneously target tumor-specific antigens and immune cells, facilitating immune-mediated killing of cancer cells.540,541 This can enhance therapeutic effects and potentially reduce resistance. For example, a bispecific ADC can bind to a tumor antigen on one arm and CD3 on the other, engaging T cells for tumor cell destruction.
Trispecific antibodies are designed to bind to three different targets or antigens. They offer even greater targeting specificity, potentially allowing for more precise delivery of cytotoxic payloads to tumor cells while sparing healthy tissues.542
By integration of these advanced antibody formats into ADCs, it becomes possible to achieve several goals simultaneously: enhanced target specificity, improved tissue penetration, engagement of the immune system, and reduction in systemic toxicity. These advanced ADC strategies hold great promise for improving the efficacy and safety of cancer therapies and other targeted treatments.
ADC design and use are constantly evolving, driven by advancements in antibody engineering, linker technology, payload development and diversification, and improvements in our understanding of the biology of cancer and other diseases. Enhancements in the specificity, potency, and safety of ADC are necessary to improve their therapeutic indexes and clinical effectiveness.18
In conclusion, ADCs are a promising therapeutic modality, particularly in combination with chemotherapies, immune checkpoint inhibitors, or other therapies. In the past decade, the development of novel targets, linkers, and payloads have fueled advances in ADC, including applications beyond oncology. ADCs provide the possibility of endowing drugs from the past with improved pharmacokinetics and reduced toxicity, making them useful once more.
Acknowledgments
The authors sincerely appreciate the CAS Data, Analytics & Insights team for their assistance in data extraction. The authors are grateful to Manuel Guzman, Gilles Georges, Michael Dennis, Dawn Riedel, Dawn George, and Hong Xie for executive sponsorship. The authors also appreciate Dharmini Patal the rest of the Science Connect team at CAS for the support on project coordination and insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.3c00374.
ADC preclinical trials; ADC clinical trials (PDF)
Author Contributions
§ Authors J.M.S. and R.T. contributed equally to this paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Marei H. E.; Cenciarelli C.; Hasan A. Potential of antibody-drug conjugates (ADCs) for cancer therapy. Cancer Cell Int. 2022, 22, 255. 10.1186/s12935-022-02679-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholap A. D.; Gupta J. S.; Kamandar P. A.; Banchhod G. V.; Hatvate N. T.. Antibody-drug conjugates for cancer therapy: An up-to-date review on the chemistry and pharmacology. In Comprehensive Analytical Chemistry; Elsevier, 2023. [Google Scholar]
- Fuentes-Antrás J.; Genta S.; Vijenthira A.; Siu L. L. Antibody-drug conjugates: in search of partners of choice. Trends in Cancer 2023, 9, 339–354. 10.1016/j.trecan.2023.01.003. [DOI] [PubMed] [Google Scholar]
- Hafeez U.; Parakh S.; Gan H. K.; Scott A. M. Antibody–Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. 10.3390/molecules25204764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Fuchs H. E.; Jemal A. Cancer statistics. 2022. CA: A Cancer Journal for Clinicians 2022, 72, 7–33. 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians 2021, 71, 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Wang H. G. Anti-cancer drug discovery and development: Bcl-2 family small molecule inhibitors. Commun. Integr Biol. 2012, 5, 557–565. 10.4161/cib.21554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loadman P. Anticancer Drug Development. Br. J. Cancer 2002, 86, 1665–1666. 10.1038/sj.bjc.6600309. [DOI] [Google Scholar]
- Gilman A.; Philips F. S. The Biological Actions and Therapeutic Applications of the B-Chloroethyl Amines and Sulfides. Science (New York, N.Y.) 1946, 103, 409–436. 10.1126/science.103.2675.409. [DOI] [PubMed] [Google Scholar]
- Heidelberger C.; Chaudhuri N. K.; Danneberg P.; Mooren D.; Griesbach L.; Duschinsky R.; Schnitzer R. J.; Pleven E.; Scheiner J. Fluorinated Pyrimidines, A New Class of Tumour-Inhibitory Compounds. Nature 1957, 179, 663–666. 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- Norris R. E.; Adamson P. C. Clinical potency of methotrexate, aminopterin, talotrexin and pemetrexed in childhood leukemias. Cancer Chemother Pharmacol 2010, 65, 1125–1130. 10.1007/s00280-009-1120-8. [DOI] [PubMed] [Google Scholar]
- Rosenberg B.; Van Camp L.; Krigas T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- Rowinsky E. K.; Donehower R. C. Paclitaxel (taxol). New England journal of medicine 1995, 332, 1004–1014. 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- Olivier T.; Haslam A.; Prasad V. Anticancer Drugs Approved by the US Food and Drug Administration From 2009 to 2020 According to Their Mechanism of Action. JAMA Network Open 2021, 4, e2138793 10.1001/jamanetworkopen.2021.38793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindley C.; McCune J. S.; Thomason T. E.; Lauder D.; Sauls A.; Adkins S.; Sawyer W. T. Perception of chemotherapy side effects cancer versus noncancer patients. Cancer Pract 1999, 7, 59–65. 10.1046/j.1523-5394.1999.07205.x. [DOI] [PubMed] [Google Scholar]
- Baah S.; Laws M.; Rahman K. M. Antibody-Drug Conjugates-A Tutorial Review. Molecules 2021, 26 (10), 2943. 10.3390/molecules26102943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysz I.; Jackson P. J. M.; Thurston D. E.. Introduction to Antibody–Drug Conjugates (ADCs). In Cytotoxic Payloads for Antibody – Drug Conjugates; Thurston D. E., Jackson P. J. M., Eds.; The Royal Society of Chemistry, 2019. [Google Scholar]
- Gébleux R.; Casi G. Antibody-drug conjugates: Current status and future perspectives. Pharmacology & Therapeutics 2016, 167, 48–59. 10.1016/j.pharmthera.2016.07.012. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Reichert J. M.; Senter P. D.; Lambert J. M.; Beck A. Antibody–drug conjugates come of age in oncology. Nat. Rev. Drug Discovery 2023, 22, 641–661. 10.1038/s41573-023-00709-2. [DOI] [PubMed] [Google Scholar]
- Bruins W. S. C.; Zweegman S.; Mutis T.; van de Donk N. W. C. J. Targeted Therapy With Immunoconjugates for Multiple Myeloma. Frontiers in Immunology 2020, 11, 1155. 10.3389/fimmu.2020.01155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z.; Li S.; Han S.; Shi C.; Zhang Y. Antibody drug conjugate: the ″biological missile″ for targeted cancer therapy. Signal Transduct Target Ther 2022, 7, 93. 10.1038/s41392-022-00947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert N.; Beck A.; Dumontet C.; Denevault-Sabourin C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals (Basel) 2020, 13, 245. 10.3390/ph13090245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita V. T. Jr; Chu E. A history of cancer chemotherapy. Cancer research 2008, 68, 8643–8653. 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- Schwartz R. S. Paul Ehrlich’s magic bullets. New England journal of medicine 2004, 350, 1079–1080. 10.1056/NEJMp048021. [DOI] [PubMed] [Google Scholar]
- Mathe G.; Tran Ba L. O.; Bernard J. [Effect on mouse leukemia 1210 of a combination by diazo-reaction of amethopterin and gamma-globulins from hamsters inoculated with such leukemia by heterografts]. C R Hebd Seances Acad. Sci. 1958, 246, 1626–1628. [PubMed] [Google Scholar]
- Kohler G.; Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Ford C. H.; Newman C. E.; Johnson J. R.; Woodhouse C. S.; Reeder T. A.; Rowland G. F.; Simmonds R. G. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42. 10.1038/bjc.1983.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchikama K.; An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. 10.1007/s13238-016-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B. H.; DeHerdt S. V.; Schneck D. W.; Bumol T. F. The human immune response to KS1/4-desacetylvinblastine (LY256787) and KS1/4-desacetylvinblastine hydrazide (LY203728) in single and multiple dose clinical studies. Cancer research 1991, 51, 2286–2290. [PubMed] [Google Scholar]
- Pietersz G. A.; Krauer K. Antibody-targeted drugs for the therapy of cancer. J. Drug Target 1994, 2, 183–215. 10.3109/10611869408996804. [DOI] [PubMed] [Google Scholar]
- Trail P. A.; Willner D.; Lasch S. J.; Henderson A. J.; Hofstead S.; Casazza A. M.; Firestone R. A.; Hellström I.; Hellström K. E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science (New York, N.Y.) 1993, 261, 212–215. 10.1126/science.8327892. [DOI] [PubMed] [Google Scholar]
- Maloney D. G.; Grillo-López A. J.; Bodkin D. J.; White C. A.; Liles T. M.; Royston I.; Varns C.; Rosenberg J.; Levy R. IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J. Clin Oncol 1997, 15, 3266–3274. 10.1200/JCO.1997.15.10.3266. [DOI] [PubMed] [Google Scholar]
- Linenberger M. L.; Hong T.; Flowers D.; Sievers E. L.; Gooley T. A.; Bennett J. M.; Berger M. S.; Leopold L. H.; Appelbaum F. R.; Bernstein I. D. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994. 10.1182/blood.V98.4.988. [DOI] [PubMed] [Google Scholar]
- Sievers E. L.; Larson R. A.; Stadtmauer E. A.; Estey E.; Löwenberg B.; Dombret H.; Karanes C.; Theobald M.; Bennett J. M.; Sherman M. L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin Oncol 2001, 19, 3244–3254. 10.1200/JCO.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- Nawrat A.Timeline: charting the choppy history of ‘magic bullet’ antibody-drug conjugates. Pharmaceutical Technology; Verdict Media Limited, 2021. https://www.pharmaceutical-technology.com/features/antibody-drug-conjugates-timeline/ (accessed 06-05-2023).
- Ehrlich P.Collected studies on immunity; J. Wiley & Sons: New York, 1906. [Google Scholar]
- Ghose T.; Cerini M.; Carter M.; Nairn R. C. Immunoradioactive agent against cancer. Br Med. J. 1967, 1, 90–93. 10.1136/bmj.1.5532.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose T.; Norvell S. T.; Guclu A.; Cameron D.; Bodurtha A.; MacDonald A. S. Immunochemotherapy of Cancer with Chlorambucil-carrying Antibody. British Medical Journal 1972, 3, 495–499. 10.1136/bmj.3.5825.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland G. F.; O’Neill G. J.; Davies D. A. Suppression of tumour growth in mice by a drug-antibody conjugate using a novel approach to linkage. Nature 1975, 255, 487–488. 10.1038/255487a0. [DOI] [PubMed] [Google Scholar]
- Riechmann L.; Clark M.; Waldmann H.; Winter G. Reshaping human antibodies for therapy. Nature 1988, 332, 323–327. 10.1038/332323a0. [DOI] [PubMed] [Google Scholar]
- Foss F. M. DAB(389)IL-2 (ONTAK): a novel fusion toxin therapy for lymphoma. Clin Lymphoma 2000, 1, 110–116. 10.3816/CLM.2000.n.009. [DOI] [PubMed] [Google Scholar]
- FDA: Pfizer Voluntarily Withdraws Cancer Treatment Mylotarg from U.S. Market. Fierce Pharma; Questex LLC, 2010. https://www.fiercepharma.com/pharma/fda-pfizer-voluntarily-withdraws-cancer-treatment-mylotarg-from-u-s-market (accessed 05-03-2023).
- Younes A.; Bartlett N. L.; Leonard J. P.; Kennedy D. A.; Lynch C. M.; Sievers E. L.; Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. New England journal of medicine 2010, 363, 1812–1821. 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- Senter P. D.; Sievers E. L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- LoRusso P. M.; Weiss D.; Guardino E.; Girish S.; Sliwkowski M. X. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 2011, 17, 6437–6447. 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- Verma S.; Miles D.; Gianni L.; Krop I. E.; Welslau M.; Baselga J.; Pegram M.; Oh D.-Y.; Diéras V.; Guardino E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. New England Journal of Medicine 2012, 367, 1783–1791. 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proposed mechanism of action (MOA) for KADCYLA; Genentech USA, Inc., 2023. https://www.kadcyla-hcp.com/early-breast-cancer/about/how-kadcyla-works.html (accessed 05-03-2023).
- BESPONSA- inotuzumab ozogamicin injection, powder, lyophilized, for solution. DailyMed; NIH, 2020. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=cc7014b1-c775-411d-b374-8113248b4077 (accessed 05-01-2023).
- Mechanism of Action. BESPONSA® (inotuzumab ozogamicin) is the first FDA-approved CD22-directed antibody-drug conjugate (ADC); Pfizer, 2023. https://besponsa.pfizerpro.com/mechanism-of-action (accessed 05-03-2023).
- KADCYLA- ado-trastuzumab emtansine injection, powder, lyophilized, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=23f3c1f4-0fc8-4804-a9e3-04cf25dd302e (accessed 05-01-2023).
- FDA approves Mylotarg for treatment of acute myeloid leukemia; U.S. FDA, 2017. https://www.fda.gov/news-events/press-announcements/fda-approves-mylotarg-treatment-acute-myeloid-leukemia (accessed 05-01-2023).
- MYLOTARG- gemtuzumab ozogamicin injection, powder, lyophilized, for solution. DailyMed; NIH, 2021. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=32fd2bb2-1cfa-4250-feb8-d7956c794e05 (accessed 05-01-2023).
- Brentuximab Vedotin (Adcetris®). ADCReview; Sunvalley Communication, LLC, 2020. https://www.adcreview.com/brentuximab-vedotin-sgn35/ (accessed 05-01-2023).
- ADCETRIS- brentuximab vedotin injection, powder, lyophilized, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=3904f8dd-1aef-3490-e48f-bd55f32ed67f (accessed 05-01-2023).
- Niculescu-Duvaz I. Trastuzumab emtansine, an antibody-drug conjugate for the treatment of HER2+ metastatic breast cancer. Current opinion in molecular therapeutics 2010, 12, 350–360. [PubMed] [Google Scholar]
- Ricart A. D. Antibody-Drug Conjugates of Calicheamicin Derivative: Gemtuzumab Ozogamicin and Inotuzumab Ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. 10.1158/1078-0432.CCR-11-0486. [DOI] [PubMed] [Google Scholar]
- LUMOXITI- moxetumomab pasudotox injection, powder, lyophilized, for solution. DailyMed; NIH, 2022. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=d6510282-1a57-4614-9859-299a227a089c (accessed 05-01-2023).
- Kreitman R. J.; Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clinical cancer research: an official journal of the American Association for Cancer Research 2011, 17, 6398–6405. 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLIVY- polatuzumab vedotin injection, powder, lyophilized, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=20a16ab2-f338-4abb-9dcd-254bd949a2bc (accessed 05-01-2023).
- FDA approves first chemoimmunotherapy regimen for patients with relapsed or refractory diffuse large B-cell lymphoma; U.S. FDA, 2019. fda.gov/news-events/press-announcements/fda-approves-first-chemoimmunotherapy-regimen-patients-relapsed-or-refractory-diffuse-large-b-cell (accessed 05-01-2023).
- PADCEV EJFV- enfortumab vedotin injection, powder, lyophilized, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=b5631d3e-4604-4363-8f20-11dfc5a4a8ed (accessed 05-01-2023).
- FDA approves new type of therapy to treat advanced urothelial cancer; U.S. FDA, 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-new-type-therapy-treat-advanced-urothelial-cancer (accessed 05-01-2023).
- ENHERTU- fam-trastuzumab deruxtecan-nxki injection, powder, lyophilized, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=7e67e73e-ddf4-4e4d-8b50-09d7514910b6 (accessed 05-01-2023).
- FDA approves new treatment option for patients with HER2-positive breast cancer who have progressed on available therapies; U.S. FDA, 2019. https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-option-patients-her2-positive-breast-cancer-who-have-progressed-available (accessed 05-01-2023).
- TRODELVY- sacituzumab govitecan powder, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=57a597d2-03f0-472e-b148-016d7169169d (accessed 05-01-2023).
- FDA grants accelerated approval to sacituzumab govitecan-hziy for metastatic triple negative breast cancer; U.S. FDA, 2020. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sacituzumab-govitecan-hziy-metastatic-triple-negative-breast-cancer (accessed 05-01-2023).
- BLENREP- belantamab injection, powder, lyophilized, for solution. DailyMed; NIH, 2021. 2. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=16a160a4-3ec0-4ddf-99ce-05912dd3382d (accessed 05-01-2023).
- FDA Approves GSK’s BLENREP (belantamab mafodotin-blmf) for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma. BusinessWire; Berkshire Hathaway, 2020. https://www.businesswire.com/news/home/20200805006105/en/ (accessed 05-01-2023).
- ZYNLONTA- loncastuximab tesirine injection, powder, lyophilized, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=af54af12-3edf-4301-8bc5-0446bc813c1d (accessed 05-01-2023).
- Drug Approval Package: ZYNLONTA; U.S. FDA, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761196Orig1s000TOC.cfm (accessed 05-01-2023).
- Seagen and Genmab Announce FDA Accelerated Approval for TIVDAK (tisotumab vedotin-tftv) in Previously Treated Recurrent or Metastatic Cervical Cancer. BusinessWire; Berkshire Hathaway, 2021. https://www.businesswire.com/news/home/20210920005921/en/Seagen-and-Genmab-Announce-FDA-Accelerated-Approval-for-TIVDAK%E2%84%A2-tisotumab-vedotin-tftv-in-Previously-Treated-Recurrent-or-Metastatic-Cervical-Cancer (accessed 05-01-2023).
- TIVDAK- tisotumab vedotin injection, powder, for solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=c9fe3f32-4219-466e-acb9-3f609b4f4df1 (accessed 05-01-2023).
- Gomes-da-Silva L. C.; Kepp O.; Kroemer G. Regulatory approval of photoimmunotherapy: photodynamic therapy that induces immunogenic cell death. Oncoimmunology 2020, 9, 1841393. 10.1080/2162402X.2020.1841393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakuten Medical Japan Announces Marketing Approval of Akalux® IV Infusion 250mg and BioBlade® Laser System from the Ministry of Health, Labour and Welfare in Japan for Locoregional Cancer Treatment. Cision PR Newswire; Cision US Inc., 2020. https://www.prnewswire.com/news-releases/rakuten-medical-japan-announces-marketing-approval-of-akalux-iv-infusion-250mg-and-bioblade-laser-system-from-the-ministry-of-labour-health-and-welfare-in-japan-for-locoregional-cancer-treatment-301138121.html (accessed 05-01-2023).
- Deeks E. D. Disitamab Vedotin: First Approval. Drugs 2021, 81, 1929–1935. 10.1007/s40265-021-01614-x. [DOI] [PubMed] [Google Scholar]
- Disitamab Vedotin Conditionally Approved by Chinese Regulator for treatment of Gastric Cancer. Onco’Zine; Sunvalley Communication, LLC, 2021. https://www.oncozine.com/disitamab-vedotin-conditionally-approved-by-chinese-regulator-for-treatment-of-gastric-cancer/ (accessed 05-01-2023).
- ImmunoGen Announces FDA Accelerated Approval of ELAHERE (mirvetuximab soravtansine-gynx) for the Treatment of Platinum-Resistant Ovarian Cancer. ImmunoGen Inc., 2022. https://investor.immunogen.com/news-releases/news-release-details/immunogen-announces-fda-accelerated-approval-elaheretm (accessed 05-01-2023).
- ELAHERE- mirvetuximab soravtansine injection, solution. DailyMed; NIH, 2023. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=00c424b5-6ccd-48ab-9e88-1986451120e2 (accessed 05-01-2023).
- GSK provides an update on Blenrep (belantamab mafodotin-blmf) US marketing authorization; GSK, 2022. https://us.gsk.com/en-us/media/press-releases/gsk-provides-an-update-on-blenrep-belantamab-mafodotin-blmf-us-marketing-authorization/ (accessed 05-01-2023).
- FDA grants accelerated approval to enfortumab vedotin-ejfv with pembrolizumab for locally advanced or metastatic urothelial carcinoma; U.S. FDA, 2023. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-enfortumab-vedotin-ejfv-pembrolizumab-locally-advanced-or-metastatic (accessed Jun 23, 2023).
- CAS Content Collection; American Chemical Society, 2023. https://www.cas.org/about/cas-content (accessed 01-18-2023).
- Qian L.; Lin X.; Gao X.; Khan R. U.; Liao J.-Y.; Du S.; Ge J.; Zeng S.; Yao S. Q. The Dawn of a New Era: Targeting the “Undruggables” with Antibody-Based Therapeutics. Chem. Rev. 2023, 123, 7782–7853. 10.1021/acs.chemrev.2c00915. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Kyazike J.; Boudanova E.; Drzyzga M.; Honey D.; Cost R.; Hou L.; Duffieux F.; Brun M.-P.; Park A.; et al. Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues. Pharmaceuticals 2021, 14, 672. 10.3390/ph14070672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drago J. Z.; Modi S.; Chandarlapaty S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nature Reviews Clinical Oncology 2021, 18, 327–344. 10.1038/s41571-021-00470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters C.; Brown S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep 2015, 35 (4), e00225. 10.1042/BSR20150089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes B. Antibody-drug conjugates for cancer: poised to deliver? Nature reviews. Drug discovery 2010, 9, 665–667. 10.1038/nrd3270. [DOI] [PubMed] [Google Scholar]
- Redman J. M.; Hill E. M.; AlDeghaither D.; Weiner L. M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol 2015, 67, 28–45. 10.1016/j.molimm.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z.; Li S.; Han S.; Shi C.; Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduction and Targeted Therapy 2022, 7, 93. 10.1038/s41392-022-00947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEINSTEIN J. N.; EGER R. R.; COVELL D. G.; BLACK C. D. V.; MULSHINE J.; CARRASQUILLO J. A.; LARSON S. M.; KEENAN A. M. The Pharmacology of Monoclonal Antibodiesa. Ann. N.Y. Acad. Sci. 1987, 507, 199–210. 10.1111/j.1749-6632.1987.tb45802.x. [DOI] [PubMed] [Google Scholar]
- Thurber G. M.; Zajic S. C.; Wittrup K. D. Theoretic Criteria for Antibody Penetration into Solid Tumors and Micrometastases. J. Nucl. Med. 2007, 48, 995–999. 10.2967/jnumed.106.037069. [DOI] [PubMed] [Google Scholar]
- Thurber G. M.; Schmidt M. M.; Wittrup K. D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Advanced drug delivery reviews 2008, 60, 1421–1434. 10.1016/j.addr.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdollahpour-Alitappeh M.; Lotfinia M.; Gharibi T.; Mardaneh J.; Farhadihosseinabadi B.; Larki P.; Faghfourian B.; Sepehr K. S.; Abbaszadeh-Goudarzi K.; Abbaszadeh-Goudarzi G.; et al. Antibody–drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. Journal of Cellular Physiology 2019, 234, 5628–5642. 10.1002/jcp.27419. [DOI] [PubMed] [Google Scholar]
- Stapleton N. M.; Andersen J. T.; Stemerding A. M.; Bjarnarson S. P.; Verheul R. C.; Gerritsen J.; Zhao Y.; Kleijer M.; Sandlie I.; de Haas M.; et al. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat. Commun. 2011, 2, 599. 10.1038/ncomms1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Woods C.; He F.; Han M.; Treuheit M. J.; Volkin D. B. Structural Changes and Aggregation Mechanisms of Two Different Dimers of an IgG2Monoclonal Antibody. Biochemistry 2018, 57, 5466–5479. 10.1021/acs.biochem.8b00575. [DOI] [PubMed] [Google Scholar]
- Yu J.; Song Y.; Tian W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol Oncol 2020, 13, 45. 10.1186/s13045-020-00876-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; Bertozzi C. R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176–192. 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C.; Bevers J. 3rd; Jackman J.; Chiang N.; Nakamura G.; Dillon M.; Liu H.; Molina P.; Elliott J. M.; Shatz W.; et al. Development of a human IgG4 bispecific antibody for dual targeting of interleukin-4 (IL-4) and interleukin-13 (IL-13) cytokines. J. Biol. Chem. 2013, 288, 26583–26593. 10.1074/jbc.M113.480483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrijn A. F.; Buijsse A. O.; van den Bremer E. T. J.; Verwilligen A. Y. W.; Bleeker W. K.; Thorpe S. J.; Killestein J.; Polman C. H.; Aalberse R. C.; Schuurman J.; et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat. Biotechnol. 2009, 27, 767–771. 10.1038/nbt.1553. [DOI] [PubMed] [Google Scholar]
- Bogen J. P.; Carrara S. C.; Fiebig D.; Grzeschik J.; Hock B.; Kolmar H. Expeditious Generation of Biparatopic Common Light Chain Antibodies via Chicken Immunization and Yeast Display Screening. Frontiers in Immunology 2020, 11, 606878. 10.3389/fimmu.2020.606878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer F.; Gao C.; Coats S.. Bispecific and Biparatopic Antibody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin M., Ed.; Springer International Publishing: Cham, 2018; pp 267–280. [Google Scholar]
- Orozco C. T.; Bersellini M.; Irving L. M.; Howard W. W.; Hargreaves D.; Devine P. W. A.; Siouve E.; Browne G. J.; Bond N. J.; Phillips J. J.; et al. Mechanistic insights into the rational design of masked antibodies. MAbs 2022, 14, 2095701. 10.1080/19420862.2022.2095701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchi R.; Bentanachs J.; Oller-Salvia B. The Masking Game: Design of Activatable Antibodies and Mimetics for Selective Therapeutics and Cell Control. ACS Cent Sci. 2021, 7, 724–738. 10.1021/acscentsci.0c01448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CytomX Therapeutics: Destroying Cancer, Differently; CytomX Therapeutics, 2023. https://cytomx.com/probody-therapeutics/ (accessed 09-26-2023).
- Fan J.; Zhuang X.; Yang X.; Xu Y.; Zhou Z.; Pan L.; Chen S. A multivalent biparatopic EGFR-targeting nanobody drug conjugate displays potent anticancer activity in solid tumor models. Signal Transduction and Targeted Therapy 2021, 6, 320. 10.1038/s41392-021-00666-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikar S. S.; Banu N.; Haramati J.; del Toro-Arreola S.; Riera Leal A.; Salas P. Nanobodies as efficient drug-carriers: Progress and trends in chemotherapy. J. Controlled Release 2021, 334, 389–412. 10.1016/j.jconrel.2021.05.004. [DOI] [PubMed] [Google Scholar]
- Smith R. A.; Zammit D. J.; Damle N. K.; Usansky H.; Reddy S. P.; Lin J. H.; Mistry M.; Rao N. S.; Denis L. J.; Gupta S. ASN004, A 5T4-targeting scFv-Fc Antibody-Drug Conjugate with High Drug-to-Antibody Ratio, Induces Complete and Durable Tumor Regressions in Preclinical Models. Mol. Cancer Ther 2021, 20, 1327–1337. 10.1158/1535-7163.MCT-20-0565. [DOI] [PubMed] [Google Scholar]
- Deonarain M. P.; Yahioglu G.; Stamati I.; Edwards B.; Diez-Posada S.; Pomowski A.; Stewart A.; Perez-Castro I.; Bouche L.; Jurgenson T.; et al. Abstract 909: Antibody fragment drug-conjugates (FDCs)-application of ANT-043 and ANT-045 in solid tumors. Cancer research 2021, 81, 909–909. 10.1158/1538-7445.AM2021-909. [DOI] [Google Scholar]
- Jeffrey S. C.; Burke P. J.; Lyon R. P.; Meyer D. W.; Sussman D.; Anderson M.; Hunter J. H.; Leiske C. I.; Miyamoto J. B.; Nicholas N. D.; et al. A Potent Anti-CD70 Antibody–Drug Conjugate Combining a Dimeric Pyrrolobenzodiazepine Drug with Site-Specific Conjugation Technology. Bioconjugate Chem. 2013, 24, 1256–1263. 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- Zhou Q. Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins. Molecules 2023, 28, 917. 10.3390/molecules28030917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant P.; Pabst M.; Badescu G.; Bird M.; McDowell W.; Jamieson E.; Swierkosz J.; Jurlewicz K.; Tommasi R.; Henseleit K.; et al. In Vitro and In Vivo Evaluation of Cysteine Rebridged Trastuzumab–MMAE Antibody Drug Conjugates with Defined Drug-to-Antibody Ratios. Mol. Pharmaceutics 2015, 12, 1872–1879. 10.1021/acs.molpharmaceut.5b00116. [DOI] [PubMed] [Google Scholar]
- Skidmore L.; Sakamuri S.; Knudsen N. A.; Hewet A. G.; Milutinovic S.; Barkho W.; Biroc S. L.; Kirtley J.; Marsden R.; Storey K.; et al. ARX788, a Site-specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-low and T-DM1-resistant Breast and Gastric Cancers. Mol. Cancer Ther 2020, 19, 1833–1843. 10.1158/1535-7163.MCT-19-1004. [DOI] [PubMed] [Google Scholar]
- ACE-Breast-02 Pivotal Phase 3 Study of Ambrx’s ARX788 for the Treatment of HER2 Positive Metastatic Breast Cancer Achieves Positive Results. BusinessWire; Berkshire Hathaway, 2023. https://www.businesswire.com/news/home/20230301005529/en/ACE-Breast-02-Pivotal-Phase-3-Study-of-Ambrx%E2%80%99s-ARX788-for-the-Treatment-of-HER2-Positive-Metastatic-Breast-Cancer-Achieves-Positive-Results (accessed 07-12-2023).
- Zhang X.; Ou C.; Liu H.; Prabhu S. K.; Li C.; Yang Q.; Wang L.-X. General and Robust Chemoenzymatic Method for Glycan-Mediated Site-Specific Labeling and Conjugation of Antibodies: Facile Synthesis of Homogeneous Antibody–Drug Conjugates. ACS Chem. Biol. 2021, 16, 2502–2514. 10.1021/acschembio.1c00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N.; Ansari A.; Dhoke G. V.; Chilamari M.; Sivaccumar J.; Kumari S.; Chatterjee S.; Goyal R.; Dutta P. K.; Samarla M.; et al. Computationally designed antibody-drug conjugates self-assembled via affinity ligands. Nat. Biomed Eng. 2019, 3, 917–929. 10.1038/s41551-019-0470-8. [DOI] [PubMed] [Google Scholar]
- Tong J. T. W.; Harris P. W. R.; Brimble M. A.; Kavianinia I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26 (19), 5847. 10.3390/molecules26195847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.; Wurst J. M.; Liu T.; Martinez R. M.; Datta-Mannan A.; Feng Y. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies 2020, 9, 2. 10.3390/antib9010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinovsky D. V.; Kibardin A. V.; Kholodenko I. V.; Svirshchevskaya E. V.; Doronin I. I.; Konovalova M. V.; Grechikhina M. V.; Rozov F. N.; Larin S. S.; Deyev S. M.; et al. Therapeutic efficacy of antibody-drug conjugates targeting GD2-positive tumors. Journal for ImmunoTherapy of Cancer 2022, 10, e004646 10.1136/jitc-2022-004646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alas M.; Saghaeidehkordi A.; Kaur K. Peptide–Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. 10.1021/acs.jmedchem.0c01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C.; Yu L.; Miao Y.; Liu X.; Yu Z.; Wei M. Peptide–drug conjugates (PDCs): a novel trend of research and development on targeted therapy, hype or hope?. Acta Pharmaceutica Sinica B 2023, 13, 498–516. 10.1016/j.apsb.2022.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby M.; Bennett G.; Chen L.; Mudd G.; Beswick P.; Harrison H.; Watcham S.; Allen H.; Brown A.; Rietschoten K. V.; et al. Abstract C061: BT8009, a Bicycle Toxin Conjugate targeting Nectin-4, shows target selectivity, and efficacy in preclinical large and small tumor models. Molecular Cancer Therapeutics 2019, 18, C061–C061. 10.1158/1535-7163.TARG-19-C061. [DOI] [Google Scholar]
- Sheyi R.; de la Torre B. G.; Albericio F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14, 396. 10.3390/pharmaceutics14020396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. M.; Simon M. C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. 10.1016/j.cub.2020.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MYLOTARGTM (gemtuzumab ozogamicin) for injection, for intravenous use; U.S. FDA, 2020.https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761060s004lbl.pdf (accessed 07-26-2023).
- Norsworthy K. J.; Ko C. W.; Lee J. E.; Liu J.; John C. S.; Przepiorka D.; Farrell A. T.; Pazdur R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. 10.1634/theoncologist.2017-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostova V.; Désos P.; Starck J.-B.; Kotschy A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 442. 10.3390/ph14050442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg B. A.; Garrett L.; Kovtun Y.; Lai K. C.; Leece B.; Miller M.; Payne G.; Steeves R.; Whiteman K. R.; Widdison W.; et al. Disulfide-Linked Antibody–Maytansinoid Conjugates: Optimization of In Vivo Activity by Varying the Steric Hindrance at Carbon Atoms Adjacent to the Disulfide Linkage. Bioconjugate Chem. 2011, 22, 717–727. 10.1021/bc100480a. [DOI] [PubMed] [Google Scholar]
- Baah S.; Laws M.; Rahman K. M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. 10.3390/molecules26102943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducry L.; Stump B. Antibody–Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjugate Chem. 2010, 21, 5–13. 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- Uprety D.; Remon J.; Adjei A. A. All That Glitters Is Not Gold: The Story of Rovalpituzumab Tesirine in SCLC. Journal of Thoracic Oncology 2021, 16, 1429–1433. 10.1016/j.jtho.2021.07.012. [DOI] [PubMed] [Google Scholar]
- Gavriel A. G.; Sambrook M. R.; Russell A. T.; Hayes W. Recent advances in self-immolative linkers and their applications in polymeric reporting systems. Polym. Chem. 2022, 13, 3188–3269. 10.1039/D2PY00414C. [DOI] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaïa N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Mao S.; Dimasi N.; Gao C. Design and Validation of Linkers for Site-Specific Preparation of Antibody-Drug Conjugates Carrying Multiple Drug Copies Per Cysteine Conjugation Site. International journal of molecular sciences 2020, 21 (18), 6882. 10.3390/ijms21186882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaramella G.; Bouchon A.; Huang E. Y.-C.. NUCLEIC ACID VACCINES. Patent WO2015164674, 2015.
- Strop P.; Delaria K.; Foletti D.; Witt J. M.; Hasa-Moreno A.; Poulsen K.; Casas M. G.; Dorywalska M.; Farias S.; Pios A.; et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol. 2015, 33, 694–696. 10.1038/nbt.3274. [DOI] [PubMed] [Google Scholar]
- Martín-Sabroso C.; Lozza I.; Torres-Suárez A. I.; Fraguas-Sánchez A. I. Antibody-Antineoplastic Conjugates in Gynecological Malignancies: Current Status and Future Perspectives. Pharmaceutics 2021, 13, 1705. 10.3390/pharmaceutics13101705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron J.Mersana halves workforce as shares plummet 75% in wake of lead cancer drug’s failure. Fierce Biotech; Questex LLC, 2023. https://www.fiercebiotech.com/biotech/mersana-halves-workforce-shares-plummet-75-wake-lead-cancer-drugs-fail (accessed 09-27-2023).
- Sava J.FDA Halts Phase 1 Trial of XMT-2056 for HER2+ Recurrent/Metastatic Solid Tumors. Targeted Oncology; MJH Life Sciences, 2023. https://www.targetedonc.com/view/fda-halts-phase-1-trial-of-xmt-2056-for-her2-recurrent-metastatic-solid-tumors (accessed 09-27-2023).
- Strop P.; Delaria K. A.; Dorywalska M.; Foletti D. L.; Dushin R. G.; Shelton D. L.; Rajpal A.. ANTIBODY-DRUG CONJUGATES WITH HIGH DRUG LOADING. Patent WO2015162563, 2015.
- Wesley S. S.; Mitchell S. A.; Hung L. J. C.. PHENYL MALEIMIDE LINKER AGENTS. Patent Application WO/2023/173121, 2023.
- Sapozhnikova K. A.; Gulyak E. L.; Misyurin V. A.; Simonova M. A.; Ryabukhina E. V.; Alexeeva A. V.; Tikhonova N. A.; Lyzhko N. A.; Popova G. P.; Misyurin A. V.; et al. Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies. Molecules 2023, 28, 425. 10.3390/molecules28010425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprakov S.; Ogunkoya A. O.; Drake P. M.. Branched linkers for antibody-drug conjugates and methods of use thereof. Patent WO2022187370, 2022.
- Levengood M. R.; Zhang X.; Hunter J. H.; Emmerton K. K.; Miyamoto J. B.; Lewis T. S.; Senter P. D. Orthogonal Cysteine Protection Enables Homogeneous Multi-Drug Antibody-Drug Conjugates. Angew. Chem., Int. Ed. Engl. 2017, 56, 733–737. 10.1002/anie.201608292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Gianolio D. A.; Stefano J. E.; Manning C. M.; Gregory R. C.; Busch M. M.; Brondyk W. H.; Miller R. J.; Dhal P. K. Design, Synthesis, and in vitro Evaluation of Multivalent Drug Linkers for High-Drug-Load Antibody-Drug Conjugates. ChemMedChem. 2018, 13, 790–794. 10.1002/cmdc.201700722. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Dragovich P. S.; Yu S.-F.; Ma Y.; Pillow T. H.; Sadowsky J. D.; Su D.; Wang W.; Polson A.; Khojasteh S. C.; et al. Exposure-Efficacy Analysis of Antibody-Drug Conjugates Delivering an Excessive Level of Payload to Tissues. Drug Metab. Dispos. 2019, 47, 1146–1155. 10.1124/dmd.119.087023. [DOI] [PubMed] [Google Scholar]
- Akaiwa M.; Dugal-Tessier J.; Mendelsohn B. A. Antibody–Drug Conjugate Payloads; Study of Auristatin Derivatives. Chem. Pharm. Bull. 2020, 68, 201–211. 10.1248/cpb.c19-00853. [DOI] [PubMed] [Google Scholar]
- Kupchan S. M.; Komoda Y.; Branfman A. R.; Sneden A. T.; Court W. A.; Thomas G. J.; Hintz H. P. J.; Smith R. M.; Karim A. Tumor inhibitors. 122. The maytansinoids. Isolation, structural elucidation, and chemical interrelation of novel ansa macrolides. Journal of Organic Chemistry 1977, 42, 2349–2357. 10.1021/jo00434a001. [DOI] [PubMed] [Google Scholar]
- Lopus M.; Oroudjev E.; Wilson L.; Wilhelm S.; Widdison W.; Chari R.; Jordan M. A. Maytansine and Cellular Metabolites of Antibody-Maytansinoid Conjugates Strongly Suppress Microtubule Dynamics by Binding to Microtubules. Molecular Cancer Therapeutics 2010, 9, 2689–2699. 10.1158/1535-7163.MCT-10-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Lim K. S.; Blackburn B. J.; Yun J.; Putnam C. W.; Bull D. A.; Won Y.-W. The Potential of Topoisomerase Inhibitor-Based Antibody—Drug Conjugates. Pharmaceutics 2022, 14, 1707. 10.3390/pharmaceutics14081707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditto V. J.; Simanek E. E. Cancer Therapies Utilizing the Camptothecins: A Review of the in Vivo Literature. Mol. Pharmaceutics 2010, 7, 307–349. 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregson S. J.; Masterson L. A.; Wei B.; Pillow T. H.; Spencer S. D.; Kang G.-D.; Yu S.-F.; Raab H.; Lau J.; Li G.; et al. Pyrrolobenzodiazepine Dimer Antibody–Drug Conjugates: Synthesis and Evaluation of Noncleavable Drug-Linkers. J. Med. Chem. 2017, 60, 9490–9507. 10.1021/acs.jmedchem.7b00736. [DOI] [PubMed] [Google Scholar]
- Janjigian Y. Y.; Lee W.; Kris M. G.; Miller V. A.; Krug L. M.; Azzoli C. G.; Senturk E.; Wade Calcutt M.; Rizvi N. A. A phase I trial of SJG-136 (NSC#694501) in advanced solid tumors. Cancer Chemotherapy and Pharmacology 2010, 65, 833–838. 10.1007/s00280-009-1088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley J. A. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy. Expert Opinion on Biological Therapy 2021, 21, 931–943. 10.1080/14712598.2020.1776255. [DOI] [PubMed] [Google Scholar]
- Singh R.; Reid E. E.; Harris L.; Salomon P. L.; Miller M. L.; Chari R. V. J.; Keating T. A. Antibody–Drug Conjugates with Indolinobenzodiazepine Dimer Payloads: DNA-Binding Mechanism of Indolinobenzodiazepine Dimer Catabolites in Target Cancer Cells. Mol. Pharmaceutics 2020, 17, 50–58. 10.1021/acs.molpharmaceut.9b00675. [DOI] [PubMed] [Google Scholar]
- Miller M. L.; Fishkin N. E.; Li W.; Whiteman K. R.; Kovtun Y.; Reid E. E.; Archer K. E.; Maloney E. K.; Audette C. A.; Mayo M. F.; et al. A New Class of Antibody–Drug Conjugates with Potent DNA Alkylating Activity. Molecular Cancer Therapeutics 2016, 15, 1870–1878. 10.1158/1535-7163.MCT-16-0184. [DOI] [PubMed] [Google Scholar]
- Lee M. D.; Ellestad G. A.; Borders D. B. Calicheamicins: discovery, structure, chemistry, and interaction with DNA. Acc. Chem. Res. 1991, 24, 235–243. 10.1021/ar00008a003. [DOI] [Google Scholar]
- MacMillan K. S.; Boger D. L. Fundamental Relationships between Structure, Reactivity, and Biological Activity for the Duocarmycins and CC-1065. J. Med. Chem. 2009, 52, 5771–5780. 10.1021/jm9006214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courter J. R.; Hamilton J. Z.; Hendrick N. R.; Zaval M.; Waight A. B.; Lyon R. P.; Senter P. D.; Jeffrey S. C.; Burke P. J. Structure-activity relationships of tubulysin analogues. Bioorg. Med. Chem. Lett. 2020, 30, 127241. 10.1016/j.bmcl.2020.127241. [DOI] [PubMed] [Google Scholar]
- Tumey L. N.; Leverett C. A.; Vetelino B.; Li F.; Rago B.; Han X.; Loganzo F.; Musto S.; Bai G.; Sukuru S. C. K.; et al. Optimization of Tubulysin Antibody–Drug Conjugates: A Case Study in Addressing ADC Metabolism. ACS Med. Chem. Lett. 2016, 7, 977–982. 10.1021/acsmedchemlett.6b00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman M. J.; Gandara D. R.; Hausner P.; Israel V.; Thornton D.; DeSanto J.; Doyle L. A. Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer 2003, 39, 197–199. 10.1016/S0169-5002(02)00511-1. [DOI] [PubMed] [Google Scholar]
- Rohr J. Cryptophycin Anticancer Drugs Revisited. ACS Chem. Biol. 2006, 1, 747–750. 10.1021/cb6004678. [DOI] [PubMed] [Google Scholar]
- Verma V. A.; Pillow T. H.; DePalatis L.; Li G.; Phillips G. L.; Polson A. G.; Raab H. E.; Spencer S.; Zheng B. The cryptophycins as potent payloads for antibody drug conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 864–868. 10.1016/j.bmcl.2014.12.070. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Mishevich J. L.; Jurica M. S. Spliceostatins and Derivatives: Chemical Syntheses and Biological Properties of Potent Splicing Inhibitors. J. Nat. Prod. 2021, 84, 1681–1706. 10.1021/acs.jnatprod.1c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveetil S.; He H.; Loganzo F.; Musto S.; Teske J.; Green M.; Tan X.; Hosselet C.; Lucas J.; Tumey L. N.; et al. Multivalent peptidic linker enables identification of preferred sites of conjugation for a potent thialanstatin antibody drug conjugate. PloS one 2017, 12, e0178452 10.1371/journal.pone.0178452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintieri L.; Geroni C.; Fantin M.; Battaglia R.; Rosato A.; Speed W.; Zanovello P.; Floreani M. Formation and Antitumor Activity of PNU-159682, A Major Metabolite of Nemorubicin in Human Liver Microsomes. Clin. Cancer Res. 2005, 11, 1608–1617. 10.1158/1078-0432.CCR-04-1845. [DOI] [PubMed] [Google Scholar]
- Scalabrin M.; Quintieri L.; Palumbo M.; Riccardi Sirtori F.; Gatto B. Virtual Cross-Linking of the Active Nemorubicin Metabolite PNU-159682 to Double-Stranded DNA. Chem. Res. Toxicol. 2017, 30, 614–624. 10.1021/acs.chemrestox.6b00362. [DOI] [PubMed] [Google Scholar]
- Yu S.-F.; Zheng B.; Go M.; Lau J.; Spencer S.; Raab H.; Soriano R.; Jhunjhunwala S.; Cohen R.; Caruso M.; et al. A Novel Anti-CD22 Anthracycline-Based Antibody–Drug Conjugate (ADC) That Overcomes Resistance to Auristatin-Based ADCs. Clin. Cancer Res. 2015, 21, 3298–3306. 10.1158/1078-0432.CCR-14-2035. [DOI] [PubMed] [Google Scholar]
- Le Daré B.; Ferron P.-J.; Gicquel T. Toxic Effects of Amanitins: Repurposing Toxicities toward New Therapeutics. Toxins 2021, 13, 417. 10.3390/toxins13060417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Zhang X.; Han C.; Wan G.; Huang X.; Ivan C.; Jiang D.; Rodriguez-Aguayo C.; Lopez-Berestein G.; Rao P. H.; et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 2015, 520, 697–701. 10.1038/nature14418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack M. The Role of Exotoxin A in Pseudomonas Disease and Immunity. Reviews of Infectious Diseases 1983, 5, S979–S984. 10.1093/clinids/5.Supplement_5.S979. [DOI] [PubMed] [Google Scholar]
- LUMOXITI (moxetumomab pasudotox-tdfk) for injection, for intravenous use; U.S. FDA, 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761104s000lbl.pdf (accessed 07-26-2023).
- Holmes R. K. Biology and Molecular Epidemiology of Diphtheria Toxin and the tox Gene. Journal of Infectious Diseases 2000, 181, S156–S167. 10.1086/315554. [DOI] [PubMed] [Google Scholar]
- Bachanova V.; Frankel A. E.; Cao Q.; Lewis D.; Grzywacz B.; Verneris M. R.; Ustun C.; Lazaryan A.; McClune B.; Warlick E. D.; et al. Phase I Study of a Bispecific Ligand-Directed Toxin Targeting CD22 and CD19 (DT2219) for Refractory B-cell Malignancies. Clin. Cancer Res. 2015, 21, 1267–1272. 10.1158/1078-0432.CCR-14-2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandish P. E.; Palmieri A.; Antonenko S.; Beaumont M.; Benso L.; Cancilla M.; Cheng M.; Fayadat-Dilman L.; Feng G.; Figueroa I.; et al. Development of Anti-CD74 Antibody–Drug Conjugates to Target Glucocorticoids to Immune Cells. Bioconjugate Chem. 2018, 29, 2357–2369. 10.1021/acs.bioconjchem.8b00312. [DOI] [PubMed] [Google Scholar]
- Wang R. E.; Liu T.; Wang Y.; Cao Y.; Du J.; Luo X.; Deshmukh V.; Kim C. H.; Lawson B. R.; Tremblay M. S.; et al. An Immunosuppressive Antibody–Drug Conjugate. J. Am. Chem. Soc. 2015, 137, 3229–3232. 10.1021/jacs.5b00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Pearson A. D.; Lim R. K. V.; Rodgers D. T.; Li S.; Parker H. B.; Weglarz M.; Hampton E. N.; Bollong M. J.; Shen J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody–drug Conjugate. Molecular Therapy 2016, 24, 2078–2089. 10.1038/mt.2016.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Efficacy of ABBV-3373 in Participants With Moderate to Severe Rheumatoid Arthritis (RA). ClinicalTrials.gov; NIH, 2021. https://clinicaltrials.gov/study/NCT03823391?tab=history&a=12 (accessed 07-26-2023).
- Thomsen K. L.; Møller H. J.; Graversen J. H.; Magnusson N. E.; Moestrup S. K.; Vilstrup H.; Grønbæk H. Anti-CD163-dexamethasone conjugate inhibits the acute phase response to lipopolysaccharide in rats. World Journal of Hepatology 2016, 8, 726. 10.4254/wjh.v8.i17.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S.; Tan M. W. Antibody-Antibiotic Conjugates: A Novel Therapeutic Platform against Bacterial Infections. Trends in molecular medicine 2017, 23, 135–149. 10.1016/j.molmed.2016.12.008. [DOI] [PubMed] [Google Scholar]
- Walsh S. J.; Bargh J. D.; Dannheim F. M.; Hanby A. R.; Seki H.; Counsell A. J.; Ou X.; Fowler E.; Ashman N.; Takada Y.; et al. Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. 10.1039/D0CS00310G. [DOI] [PubMed] [Google Scholar]
- Borrok M. J.; Jung S. T.; Kang T. H.; Monzingo A. F.; Georgiou G. Revisiting the Role of Glycosylation in the Structure of Human IgG Fc. ACS Chem. Biol. 2012, 7, 1596–1602. 10.1021/cb300130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koniev O.; Wagner A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. 10.1039/C5CS00048C. [DOI] [PubMed] [Google Scholar]
- Dovgan I.; Ursuegui S.; Erb S.; Michel C.; Kolodych S.; Cianférani S.; Wagner A. Acyl Fluorides: Fast, Efficient, and Versatile Lysine-Based Protein Conjugation via Plug-and-Play Strategy. Bioconjugate Chem. 2017, 28, 1452–1457. 10.1021/acs.bioconjchem.7b00141. [DOI] [PubMed] [Google Scholar]
- Moreau M.; Raguin O.; Vrigneaud J.-M.; Collin B.; Bernhard C.; Tizon X.; Boschetti F.; Duchamp O.; Brunotte F.; Denat F. DOTAGA-Trastuzumab. A New Antibody Conjugate Targeting HER2/Neu Antigen for Diagnostic Purposes. Bioconjugate Chemistry 2012, 23, 1181–1188. 10.1021/bc200680x. [DOI] [PubMed] [Google Scholar]
- von Witting E.; Hober S.; Kanje S. Affinity-Based Methods for Site-Specific Conjugation of Antibodies. Bioconjugate Chem. 2021, 32, 1515–1524. 10.1021/acs.bioconjchem.1c00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.; Kim J. H.; Kwon S. J.; Seo J. W.; Park S. H.; Kim J.; Jin J.; Hong J. H.; Kang H. J.; Sharma C.; et al. Site-Selective Antibody–Drug Conjugation by a Proximity-Driven S to N Acyl Transfer Reaction on a Therapeutic Antibody. J. Med. Chem. 2022, 65, 5751–5759. 10.1021/acs.jmedchem.2c00084. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Fujii T.; Stofleth J. T.; Takasugi R.; Takahashi K.; Matsuda Y. Scale-Up Synthesis of Site-Specific Antibody–Drug Conjugates Using AJICAP Second-Generation Technology. Org. Process Res. Dev. 2023, 27, 1136–1143. 10.1021/acs.oprd.3c00111. [DOI] [Google Scholar]
- Sun M. M. C.; Beam K. S.; Cerveny C. G.; Hamblett K. J.; Blackmore R. S.; Torgov M. Y.; Handley F. G. M.; Ihle N. C.; Senter P. D.; Alley S. C. Reduction–Alkylation Strategies for the Modification of Specific Monoclonal Antibody Disulfides. Bioconjugate Chem. 2005, 16, 1282–1290. 10.1021/bc050201y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasco J. M. J. M.; Faustino H.; Trindade A.; Gois P. M. P. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chemistry A European Journal 2019, 25, 43–59. 10.1002/chem.201803174. [DOI] [PubMed] [Google Scholar]
- Shen B.-Q.; Xu K.; Liu L.; Raab H.; Bhakta S.; Kenrick M.; Parsons-Reponte K. L.; Tien J.; Yu S.-F.; Mai E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- Fontaine S. D.; Reid R.; Robinson L.; Ashley G. W.; Santi D. V. Long-Term Stabilization of Maleimide–Thiol Conjugates. Bioconjugate Chem. 2015, 26, 145–152. 10.1021/bc5005262. [DOI] [PubMed] [Google Scholar]
- Lahnsteiner M.; Kastner A.; Mayr J.; Roller A.; Keppler B. K.; Kowol C. R. Improving the Stability of Maleimide–Thiol Conjugation for Drug Targeting. Chemistry A European Journal 2020, 26, 15867–15870. 10.1002/chem.202003951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanjee H. H.; Saebi A.; Buslov I.; Loftis A. R.; Buchwald S. L.; Pentelute B. L. Protein–Protein Cross-Coupling via Palladium–Protein Oxidative Addition Complexes from Cysteine Residues. J. Am. Chem. Soc. 2020, 142, 9124–9129. 10.1021/jacs.0c03143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial M.; Postma A. Disulfide conjugation chemistry: a mixed blessing for therapeutic drug delivery?. Therapeutic Delivery 2017, 8, 359–362. 10.4155/tde-2017-0003. [DOI] [PubMed] [Google Scholar]
- Kasper M.-A.; Glanz M.; Stengl A.; Penkert M.; Klenk S.; Sauer T.; Schumacher D.; Helma J.; Krause E.; Cardoso M. C.; et al. Cysteine-Selective Phosphonamidate Electrophiles for Modular Protein Bioconjugations. Angew. Chem., Int. Ed. 2019, 58, 11625–11630. 10.1002/anie.201814715. [DOI] [PubMed] [Google Scholar]
- Kasper M.-A.; Glanz M.; Oder A.; Schmieder P.; von Kries J. P.; Hackenberger C. P. R. Vinylphosphonites for Staudinger-induced chemoselective peptide cyclization and functionalization. Chemical Science 2019, 10, 6322–6329. 10.1039/C9SC01345H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier R.; Ceballos J.; Guidotti N.; Simonet-Davin R.; Fierz B.; Waser J. ″Doubly Orthogonal″ Labeling of Peptides and Proteins. Chem. 2019, 5, 2243–2263. 10.1016/j.chempr.2019.06.022. [DOI] [Google Scholar]
- Bird M.; Nunes J.; Frigerio M.. Bridged Cysteine Conjugations. In Antibody-Drug Conjugates: Methods and Protocols; Tumey L. N., Ed.; Springer US: New York, 2020; pp 113–129. [DOI] [PubMed] [Google Scholar]
- Hu Q.-Y.; Imase H.. Methods for making conjugates from disulfide-containing proteins. Patent WO2014083505, 2014.
- Schumacher F. F.; Sanchania V. A.; Tolner B.; Wright Z. V. F.; Ryan C. P.; Smith M. E. B.; Ward J. M.; Caddick S.; Kay C. W. M.; Aeppli G.; et al. Homogeneous antibody fragment conjugation by disulfide bridging introduces ‘spinostics’. Sci. Rep. 2013, 3, 1525. 10.1038/srep01525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte N.; Livanos M.; Miranda E.; Morais M.; Yang X.; Rajkumar V. S.; Chester K. A.; Chudasama V.; Baker J. R. Tuning the Hydrolytic Stability of Next Generation Maleimide Cross-Linkers Enables Access to Albumin-Antibody Fragment Conjugates and tri-scFvs. Bioconjugate Chem. 2018, 29, 486–492. 10.1021/acs.bioconjchem.7b00795. [DOI] [PubMed] [Google Scholar]
- Morais M.; Forte N.; Chudasama V.; Baker J. R.. Application of Next-Generation Maleimides (NGMs) to Site-Selective Antibody Conjugation. In Bioconjugation: Methods and Protocols; Massa S., Devoogdt N., Eds.; Springer: New York, 2019; pp 15–24. [DOI] [PubMed] [Google Scholar]
- Lee M. T. W.; Maruani A.; Baker J. R.; Caddick S.; Chudasama V. Next-generation disulfide stapling: reduction and functional re-bridging all in one. Chemical Science 2016, 7, 799–802. 10.1039/C5SC02666K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-J.; Li Y.-Y.; Liu X.-Y.; Lu X.-L.; Cao X.; Jiao B.-H. Marine Antibody–Drug Conjugates: Design Strategies and Research Progress. Marine Drugs 2017, 15, 18. 10.3390/md15010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N.; Kancharla J.; Kaushik S.; Ansari A.; Hossain S.; Goyal R.; Pandey M.; Sivaccumar J.; Hussain S.; Sarkar A.; et al. Development of a facile antibody–drug conjugate platform for increased stability and homogeneity. Chemical Science 2017, 8, 2387–2395. 10.1039/C6SC05149A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh S. J.; Omarjee S.; Dannheim F. M.; Couturier D.-L.; Bexheti D.; Mendil L.; Cronshaw G.; Fewster T.; Gregg C.; Brodie C.; et al. Divinylpyrimidine reagents generate antibody–drug conjugates with excellent in vivo efficacy and tolerability. Chem. Commun. 2022, 58, 1962–1965. 10.1039/D1CC06766D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witus L. S.; Netirojjanakul C.; Palla K. S.; Muehl E. M.; Weng C.-H.; Iavarone A. T.; Francis M. B. Site-Specific Protein Transamination Using N-Methylpyridinium-4-carboxaldehyde. J. Am. Chem. Soc. 2013, 135, 17223–17229. 10.1021/ja408868a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D.; Nilchan N.; Nanna A. R.; Li X.; Cameron M. D.; Roush W. R.; Park H.; Rader C. Site-Selective Antibody Functionalization via Orthogonally Reactive Arginine and Lysine Residues. Cell Chemical Biology 2019, 26, 1229–1239. e1229 10.1016/j.chembiol.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sornay C.; Hessmann S.; Erb S.; Dovgan I.; Ehkirch A.; Botzanowski T.; Cianférani S.; Wagner A.; Chaubet G. Investigating Ugi/Passerini Multicomponent Reactions for the Site-Selective Conjugation of Native Trastuzumab**. Chemistry A European Journal 2020, 26, 13797–13805. 10.1002/chem.202002432. [DOI] [PubMed] [Google Scholar]
- Lu R.-M.; Hwang Y.-C.; Liu I. J.; Lee C.-C.; Tsai H.-Z.; Li H.-J.; Wu H.-C. Development of therapeutic antibodies for the treatment of diseases. Journal of Biomedical Science 2020, 27, 1. 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Vijayasankaran N.; Shen A.; Kiss R.; Amanullah A. Cell culture processes for monoclonal antibody production. mAbs 2010, 2, 466–479. 10.4161/mabs.2.5.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari P.; Zacharias N.; Ohri R.; Sadowsky J.. Site-Specific Conjugation to Cys-Engineered THIOMAB Antibodies. In Antibody-Drug Conjugates: Methods and Protocols; Tumey L. N., Ed.; Springer US: New York, 2020; pp 51–69. [DOI] [PubMed] [Google Scholar]
- Casi G.; Huguenin-Dezot N.; Zuberbühler K.; Scheuermann J.; Neri D. Site-Specific Traceless Coupling of Potent Cytotoxic Drugs to Recombinant Antibodies for Pharmacodelivery. J. Am. Chem. Soc. 2012, 134, 5887–5892. 10.1021/ja211589m. [DOI] [PubMed] [Google Scholar]
- Procopio-Melino R.; Kotch F. W.; Prashad A. S.; Gomes J. M.; Wang W.; Arve B.; Dawdy A.; Chen L.; Sperry J.; Hosselet C.; et al. Cysteine metabolic engineering and selective disulfide reduction produce superior antibody-drug-conjugates. Sci. Rep. 2022, 12, 7262. 10.1038/s41598-022-11344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy G.; Reier J.; Garcia A.; Martin T.; Rice M.; Wang J.; Prophet M.; Christie R.; Dall’Acqua W.; Ahuja S.; et al. Development of a high yielding expression platform for the introduction of non-natural amino acids in protein sequences. mAbs 2020, 12, 1684749. 10.1080/19420862.2019.1684749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stech M.; Kubick S. Cell-Free Synthesis Meets Antibody Production: A Review. Antibodies 2015, 4, 12–33. 10.3390/antib4010012. [DOI] [Google Scholar]
- Evans E. G. B.; Millhauser G. L.. Chapter Nineteen - Genetic Incorporation of the Unnatural Amino Acid p-Acetyl Phenylalanine into Proteins for Site-Directed Spin Labeling. In Methods in Enzymology; Qin P. Z., Warncke K., Eds.; Academic Press, 2015; Vol. 563, pp 503–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axup J. Y.; Bajjuri K. M.; Ritland M.; Hutchins B. M.; Kim C. H.; Kazane S. A.; Halder R.; Forsyth J. S.; Santidrian A. F.; Stafin K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16101–16106. 10.1073/pnas.1211023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson J. W.; Smith A. K.; Wilding K. M.; Bundy B. C.; Knotts T. A. I. V. The Effects of p-Azidophenylalanine Incorporation on Protein Structure and Stability. J. Chem. Inf. Model. 2020, 60, 5117–5125. 10.1021/acs.jcim.0c00725. [DOI] [PubMed] [Google Scholar]
- Ko W.; Jin S.; Lee J.; Kim D.; Park J.; Kim S.; Lee H. S. Efficient and Site-Specific Antibody Labeling by Strain-promoted Azide–Alkyne Cycloaddition. Bulletin of the Korean Chemical Society 2015, 36, 2352–2354. 10.1002/bkcs.10423. [DOI] [Google Scholar]
- Tookmanian E. M.; Fenlon E. E.; Brewer S. H. Synthesis and protein incorporation of azido-modified unnatural amino acids. RSC Adv. 2015, 5, 1274–1281. 10.1039/C4RA14244F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman E. S.; Heibeck T. H.; Gill A.; Li X.; Murray C. J.; Madlansacay M. R.; Tran C.; Uter N. T.; Yin G.; Rivers P. J.; et al. Production of Site-Specific Antibody–Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjugate Chem. 2014, 25, 351–361. 10.1021/bc400490z. [DOI] [PubMed] [Google Scholar]
- Syed J.; Palani S.; Clarke S. T.; Asad Z.; Bottrill A. R.; Jones A. M. E.; Sampath K.; Balasubramanian M. K. Expanding the Zebrafish Genetic Code through Site-Specific Introduction of Azido-lysine, Bicyclononyne-lysine, and Diazirine-lysine. International journal of molecular sciences 2019, 20, 2577. 10.3390/ijms20102577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam T. J.; Wold E.; Wahl A.; Smider V. V. Antibody Conjugates with Unnatural Amino Acids. Mol. Pharmaceutics 2015, 12, 1848–1862. 10.1021/acs.molpharmaceut.5b00082. [DOI] [PubMed] [Google Scholar]
- VanBrunt M. P.; Shanebeck K.; Caldwell Z.; Johnson J.; Thompson P.; Martin T.; Dong H.; Li G.; Xu H.; D’Hooge F.; et al. Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody–Drug Conjugates Using Click Cycloaddition Chemistry. Bioconjugate Chem. 2015, 26, 2249–2260. 10.1021/acs.bioconjchem.5b00359. [DOI] [PubMed] [Google Scholar]
- Kyung Jin L.; Deokhee K.; Hee-Sung P. Site-Specific Labeling of Proteins Using Unnatural Amino Acids. Mol. Cells 2019, 42, 386–396. 10.14348/molcells.2019.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Cheung J. W.; Carpenter T.; Jones S. K.; Luong N. H.; Tran N. C.; Jacobs S. E.; Galbada Liyanage S. A.; Cropp T. A.; Yin J. Enhancing the incorporation of lysine derivatives into proteins with methylester forms of unnatural amino acids. Bioorg. Med. Chem. Lett. 2020, 30, 126876. 10.1016/j.bmcl.2019.126876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oller-Salvia B.; Kym G.; Chin J. W. Rapid and Efficient Generation of Stable Antibody–Drug Conjugates via an Encoded Cyclopropene and an Inverse-Electron-Demand Diels–Alder Reaction. Angew. Chem., Int. Ed. 2018, 57, 2831–2834. 10.1002/anie.201712370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler C.; Sauter P. F.; Wawryszyn M.; Girona G. E.; Gupta K.; Landry J. J. M.; Fritz M. H.-Y.; Radic K.; Hoffmann J.-E.; Chen Z. A.; et al. Genetic code expansion for multiprotein complex engineering. Nat. Methods 2016, 13, 997–1000. 10.1038/nmeth.4032. [DOI] [PubMed] [Google Scholar]
- Li X.; Nelson C. G.; Nair R. R.; Hazlehurst L.; Moroni T.; Martinez-Acedo P.; Nanna A. R.; Hymel D.; Burke T. R. Jr; Rader C. Stable and Potent Selenomab-Drug Conjugates. Cell Chemical Biology 2017, 24, 433–442. 10.1016/j.chembiol.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri P.; Martin M. S.; Lino B. R.; Scheck R. A.; Van Deventer J. A. Dual Noncanonical Amino Acid Incorporation Enabling Chemoselective Protein Modification at Two Distinct Sites in Yeast. Biochemistry 2023, 62, 2098–2114. 10.1021/acs.biochem.2c00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Welborn M.; Zhu T.; Yang N. J.; Santos M. S.; Van Voorhis T.; Pentelute B. L. π-Clamp-mediated cysteine conjugation. Nat. Chem. 2016, 8, 120–128. 10.1038/nchem.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Dai P.; Vinogradov A. A.; Gates Z. P.; Pentelute B. L. Site-Selective Cysteine–Cyclooctyne Conjugation. Angew. Chem., Int. Ed. 2018, 57, 6459–6463. 10.1002/anie.201800860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z.; Zhang X.-N.; Nasertorabi F.; Cheng Q.; Li J.; Katz B. B.; Smbatyan G.; Pei H.; Louie S. G.; Lenz H.-J.; et al. Synthesis of site-specific antibody-drug conjugates by ADP-ribosyl cyclases. Science Advances 2020, 6, eaba6752 10.1126/sciadv.aba6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine E. B.; Alter G. Understanding the role of antibody glycosylation through the lens of severe viral and bacterial diseases. Glycobiology 2020, 30, 241–253. 10.1093/glycob/cwaa018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickgiesser S.; Rieker M.; Mueller-Pompalla D.; Schröter C.; Tonillo J.; Warszawski S.; Raab-Westphal S.; Kühn S.; Knehans T.; Könning D.; et al. Site-Specific Conjugation of Native Antibodies Using Engineered Microbial Transglutaminases. Bioconjugate Chem. 2020, 31, 1070–1076. 10.1021/acs.bioconjchem.0c00061. [DOI] [PubMed] [Google Scholar]
- Ratnayake A. S.; Chang L.-p.; Tumey L. N.; Loganzo F.; Chemler J. A.; Wagenaar M.; Musto S.; Li F.; Janso J. E.; Ballard T. E.; et al. Natural Product Bis-Intercalator Depsipeptides as a New Class of Payloads for Antibody–Drug Conjugates. Bioconjugate Chem. 2019, 30, 200–209. 10.1021/acs.bioconjchem.8b00843. [DOI] [PubMed] [Google Scholar]
- Chio T. I.; Demestichas B. R.; Brems B. M.; Bane S. L.; Tumey L. N. Expanding the Versatility of Microbial Transglutaminase Using α-Effect Nucleophiles as Noncanonical Substrates. Angew. Chem., Int. Ed. 2020, 59, 13814–13820. 10.1002/anie.202001830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S.; Washietl S.; Eisenhaber F. Protein prenyltransferases. Genome Biology 2003, 4, 212. 10.1186/gb-2003-4-4-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-j.; Choi H.-J.; Yun M.; Kang Y.; Jung J.-E.; Ryu Y.; Kim T. Y.; Cha Y.-j.; Cho H.-S.; Min J.-J.; et al. Enzymatic Prenylation and Oxime Ligation for the Synthesis of Stable and Homogeneous Protein–Drug Conjugates for Targeted Therapy. Angew. Chem., Int. Ed. 2015, 54, 12020–12024. 10.1002/anie.201505964. [DOI] [PubMed] [Google Scholar]
- Beerli R. R.; Hell T.; Merkel A. S.; Grawunder U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PloS one 2015, 10, e0131177 10.1371/journal.pone.0131177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmand T. J.; Bousbaine D.; Chan A.; Zhang X.; Liu D. R.; Tam J. P.; Ploegh H. L. One-Pot Dual Labeling of IgG 1 and Preparation of C-to-C Fusion Proteins Through a Combination of Sortase A and Butelase 1. Bioconjugate Chem. 2018, 29, 3245–3249. 10.1021/acs.bioconjchem.8b00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund V.; Piater B.; Fischer F.; Kolmar H.. SpyLigase-Catalyzed Modification of Antibodies. In Enzyme-Mediated Ligation Methods; Nuijens T., Schmidt M., Eds.; Springer: New York, 2019; pp 171–192. [DOI] [PubMed] [Google Scholar]
- Grünewald J.; Klock H. E.; Cellitti S. E.; Bursulaya B.; McMullan D.; Jones D. H.; Chiu H.-P.; Wang X.; Patterson P.; Zhou H.; et al. Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Phosphopantetheinyl Transferases. Bioconjugate Chem. 2015, 26, 2554–2562. 10.1021/acs.bioconjchem.5b00558. [DOI] [PubMed] [Google Scholar]
- York D.; Baker J.; Holder P. G.; Jones L. C.; Drake P. M.; Barfield R. M.; Bleck G. T.; Rabuka D. Generating aldehyde-tagged antibodies with high titers and high formylglycine yields by supplementing culture media with copper(II). BMC Biotechnology 2016, 16, 23. 10.1186/s12896-016-0254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; Kudirka R.; Albers A. E.; Barfield R. M.; de Hart G. W.; Drake P. M.; Jones L. C.; Rabuka D. Hydrazino-Pictet-Spengler Ligation as a Biocompatible Method for the Generation of Stable Protein Conjugates. Bioconjugate Chem. 2013, 24, 846–851. 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- Szijj P. A.; Kostadinova K. A.; Spears R. J.; Chudasama V. Tyrosine bioconjugation – an emergent alternative. Organic & Biomolecular Chemistry 2020, 18, 9018–9028. 10.1039/D0OB01912G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins J. J.; Damen J. A. M.; Wijdeven M. A.; Lelieveldt L. P. W. M.; van Delft F. L.; Albada B. Non-Genetic Generation of Antibody Conjugates Based on Chemoenzymatic Tyrosine Click Chemistry. Bioconjugate Chem. 2021, 32, 2167–2172. 10.1021/acs.bioconjchem.1c00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguzza B. C.; Nichols C. L.; Briggs S. L.; Cullinan G. J.; Johnson D. A.; Starling J. J.; Baker A. L.; Bumol T. F.; Corvalan J. R. F. New antitumor monoclonal antibody-vinca conjugates LY203725 and related compounds: design, preparation, and representative in vivo activity. J. Med. Chem. 1989, 32, 548–555. 10.1021/jm00123a007. [DOI] [PubMed] [Google Scholar]
- Wang W.; Vlasak J.; Li Y.; Pristatsky P.; Fang Y.; Pittman T.; Roman J.; Wang Y.; Prueksaritanont T.; Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Molecular Immunology 2011, 48, 860–866. 10.1016/j.molimm.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Huang W.; Giddens J.; Fan S.-Q.; Toonstra C.; Wang L.-X. Chemoenzymatic Glycoengineering of Intact IgG Antibodies for Gain of Functions. J. Am. Chem. Soc. 2012, 134, 12308–12318. 10.1021/ja3051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons T. B.; Struwe W. B.; Gault J.; Yamamoto K.; Taylor T. A.; Raj R.; Wals K.; Mohammed S.; Robinson C. V.; Benesch J. L. P.; et al. Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed. 2016, 55, 2361–2367. 10.1002/anie.201508723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Ponte J. F.; Yoder N. C.; Laleau R.; Coccia J.; Lanieri L.; Qiu Q.; Wu R.; Hong E.; Bogalhas M.; et al. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. 10.1021/acs.bioconjchem.7b00062. [DOI] [PubMed] [Google Scholar]
- Fang J.; Guo L.; Zhang Y.; Guo Q.; Wang M.; Wang X.. The Target Atlas for Antibody-Drug Conjugates across Solid Cancers. Research Square 2023. 10.21203/rs.3.rs-2884661/v1 [DOI] [PubMed] [Google Scholar]
- Perez H. L.; Cardarelli P. M.; Deshpande S.; Gangwar S.; Schroeder G. M.; Vite G. D.; Borzilleri R. M. Antibody-drug conjugates: current status and future directions. Drug Discov Today 2014, 19, 869–881. 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- von Minckwitz G.; Huang C.-S.; Mano M. S.; Loibl S.; Mamounas E. P.; Untch M.; Wolmark N.; Rastogi P.; Schneeweiss A.; Redondo A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. New England Journal of Medicine 2019, 380, 617–628. 10.1056/NEJMoa1814017. [DOI] [PubMed] [Google Scholar]
- Stepan L. P.; Trueblood E. S.; Hale K.; Babcook J.; Borges L.; Sutherland C. L. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J. Histochem Cytochem 2011, 59, 701–710. 10.1369/0022155411410430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedder T. F.; Tuscano J.; Sato S.; Kehrl J. H. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 1997, 15, 481–504. 10.1146/annurev.immunol.15.1.481. [DOI] [PubMed] [Google Scholar]
- Pfeifer M.; Zheng B.; Erdmann T.; Koeppen H.; McCord R.; Grau M.; Staiger A.; Chai A.; Sandmann T.; Madle H.; et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586. 10.1038/leu.2015.48. [DOI] [PubMed] [Google Scholar]
- Staudacher A. H.; Brown M. P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required?. Br. J. Cancer 2017, 117, 1736–1742. 10.1038/bjc.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohl W. R. Current progress in innovative engineered antibodies. Protein Cell 2018, 9, 86–120. 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumura R.; Manabe S.; Takashima H.; Koga Y.; Yasunaga M.; Matsumura Y. Influence of the dissociation rate constant on the intra-tumor distribution of antibody-drug conjugate against tissue factor. Journal of controlled release: official journal of the Controlled Release Society 2018, 284, 49–56. 10.1016/j.jconrel.2018.06.016. [DOI] [PubMed] [Google Scholar]
- Singh A. P.; Guo L.; Verma A.; Wong G. G.; Thurber G. M.; Shah D. K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: a Quantitative Investigation. Aaps j 2020, 22, 28. 10.1208/s12248-019-0387-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun Y. V.; Audette C. A.; Ye Y.; Xie H.; Ruberti M. F.; Phinney S. J.; Leece B. A.; Chittenden T.; Blättler W. A.; Goldmacher V. S. Antibody-Drug Conjugates Designed to Eradicate Tumors with Homogeneous and Heterogeneous Expression of the Target Antigen. Cancer research 2006, 66, 3214–3221. 10.1158/0008-5472.CAN-05-3973. [DOI] [PubMed] [Google Scholar]
- Khera E.; Cilliers C.; Smith M. D.; Ganno M. L.; Lai K. C.; Keating T. A.; Kopp A.; Nessler I.; Abu-Yousif A. O.; Thurber G. M. Quantifying ADC bystander payload penetration with cellular resolution using pharmacodynamic mapping. Neoplasia 2021, 23, 210–221. 10.1016/j.neo.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalouni C.; Doll S. Fate of Antibody-Drug Conjugates in Cancer Cells. Journal of Experimental & Clinical Cancer Research 2018, 37, 20. 10.1186/s13046-017-0667-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun Y. V.; Goldmacher V. S. Cell killing by antibody–drug conjugates. Cancer letters 2007, 255, 232–240. 10.1016/j.canlet.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Hamblett K. J.; Jacob A. P.; Gurgel J. L.; Tometsko M. E.; Rock B. M.; Patel S. K.; Milburn R. R.; Siu S.; Ragan S. P.; Rock D. A.; et al. SLC46A3 Is Required to Transport Catabolites of Noncleavable Antibody Maytansine Conjugates from the Lysosome to the Cytoplasm. Cancer research 2015, 75, 5329–5340. 10.1158/0008-5472.CAN-15-1610. [DOI] [PubMed] [Google Scholar]
- Casi G.; Neri D. Noninternalizing Targeted Cytotoxics for Cancer Therapy. Mol. Pharmaceutics 2015, 12, 1880–1884. 10.1021/mp500798y. [DOI] [PubMed] [Google Scholar]
- Giugliano F.; Corti C.; Tarantino P.; Michelini F.; Curigliano G. Bystander effect of antibody-drug conjugates: fact or fiction?. Curr. Oncol Rep 2022, 24, 809–817. 10.1007/s11912-022-01266-4. [DOI] [PubMed] [Google Scholar]
- Strohl W. R. Current progress in innovative engineered antibodies. Protein & Cell 2018, 9, 86–120. 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damelin M.; Zhong W.; Myers J.; Sapra P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3494–3507. 10.1007/s11095-015-1624-3. [DOI] [PubMed] [Google Scholar]
- Yu S.; Lim A.; Tremblay M. S.. Next Horizons: ADCs Beyond Oncology. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin M., Ed.; Springer International Publishing: Cham, 2018; pp 321–347. [Google Scholar]
- FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment; U.S. FDA, 2023. https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed 08-04-2023).
- FDA Converts Novel Alzheimer’s Disease Treatment to Traditional Approval; U.S. FDA, 2023. https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval. (accessed 08-04-2023).
- Nicolò E.; Giugliano F.; Ascione L.; Tarantino P.; Corti C.; Tolaney S. M.; Cristofanilli M.; Curigliano G. Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treat. Rev. 2022, 106, 102395. 10.1016/j.ctrv.2022.102395. [DOI] [PubMed] [Google Scholar]
- Fighting Viruses: What Is Passive Immunization?; Caltech, 2023. https://scienceexchange.caltech.edu/topics/covid-19-coronavirus-sars-cov-2/passive-immunization#:~:text=Passive%20immunization%2C%20or%20passive%20immunotherapy,short%2Dterm%20protection%20against%20infection. (accessed 06-14-2023).
- Nagasawa D. T.; Fong C.; Yew A.; Spasic M.; Garcia H. M.; Kruse C. A.; Yang I. Passive immunotherapeutic strategies for the treatment of malignant gliomas. Neurosurg Clin N Am. 2012, 23, 481–495. 10.1016/j.nec.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolting B. Linker technologies for antibody-drug conjugates. Methods in molecular biology (Clifton, N.J.) 2013, 1045, 71–100. 10.1007/978-1-62703-541-5_5. [DOI] [PubMed] [Google Scholar]
- Pitiot A.; Heuzé-Vourc’h N.; Sécher T. Alternative Routes of Administration for Therapeutic Antibodies—State of the Art. Antibodies 2022, 11, 56. 10.3390/antib11030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner B.; Richter W.; Schmidt J. Subcutaneous Administration of Biotherapeutics: An Overview of Current Challenges and Opportunities. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy 2018, 32, 425–440. 10.1007/s40259-018-0295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznar M. A.; Tinari N.; Rullán A. J.; Sánchez-Paulete A. R.; Rodriguez-Ruiz M. E.; Melero I. Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally. J. Immunol. 2017, 198, 31–39. 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- Conilh L.; Sadilkova L.; Viricel W.; Dumontet C. Payload diversification: a key step in the development of antibody–drug conjugates. Journal of Hematology & Oncology 2023, 16, 3. 10.1186/s13045-022-01397-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson A. D.; McPherson M. J.; Waegell W.; Goess C. A.; Stoffel R. H.; Li X.; Zhou J.; Wang Z.; Yu Y.; Hernandez A. Jr; et al. Design and Development of Glucocorticoid Receptor Modulators as Immunology Antibody–Drug Conjugate Payloads. J. Med. Chem. 2022, 65, 4500–4533. 10.1021/acs.jmedchem.1c02099. [DOI] [PubMed] [Google Scholar]
- Keam S. J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. 10.1007/s40265-020-01281-4. [DOI] [PubMed] [Google Scholar]
- Syed Y. Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. 10.1007/s40265-020-01337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TROP-2: A widely expressed cell surface protein. mNSCLC; Daiihi Sankyo, Inc. and AstraZeneca, 2023. https://trop2spotlight.com/trop2-expression?utm_source=google&utm_medium=cpc&utm_term=antibody%20drug%20conjugate&utm_campaign=CNHCPDUBNSCLCDATDSIATreatmentADC&gclid=CjwKCAjwp6CkBhB_EiwAlQVyxYfIYpnniWBhG1LqOTnr6DCgmG6KOdT8e7q7vzrNIt6gwHKImI3FvRoCyH0QAvD_BwE&gclsrc=aw.ds (accessed 06-12-2023).
- Wong J. L.; Rosenberg J. E. Targeting nectin-4 by antibody-drug conjugates for the treatment of urothelial carcinoma. Expert Opin Biol. Ther 2021, 21, 863–873. 10.1080/14712598.2021.1929168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Song Y.; Tian W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. 10.1186/s13045-020-00876-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R.; Hammond S. A.; Oberst M.; Wilkinson R. W. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. Journal for ImmunoTherapy of Cancer 2014, 2, 1–10. 10.1186/s40425-014-0029-x.24829758 [DOI] [Google Scholar]
- Collins D. M.; Bossenmaier B.; Kollmorgen G.; Niederfellner G. Acquired Resistance to Antibody-Drug Conjugates. Cancers (Basel) 2019, 11, 394. 10.3390/cancers11030394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loganzo F.; Sung M.; Gerber H.-P. Mechanisms of Resistance to Antibody–Drug Conjugates. Molecular Cancer Therapeutics 2016, 15, 2825–2834. 10.1158/1535-7163.MCT-16-0408. [DOI] [PubMed] [Google Scholar]
- Hunter F. W.; Barker H. R.; Lipert B.; Rothé F.; Gebhart G.; Piccart-Gebhart M. J.; Sotiriou C.; Jamieson S. M. F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. 10.1038/s41416-019-0635-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos-Luci C.; García-Alonso S.; Díaz-Rodríguez E.; Nadal-Serrano M.; Arribas J.; Ocaña A.; Pandiella A. Resistance to the Antibody–Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer research 2017, 77, 4639–4651. 10.1158/0008-5472.CAN-16-3127. [DOI] [PubMed] [Google Scholar]
- Cancer; WHO, 2022. https://www.who.int/news-room/fact-sheets/detail/cancer (accessed 07-12-2023).
- Conventional treatment; National Cancer Institute, 2023. https://www.cancer.gov/publications/dictionaries/cancer-terms/def/conventional-treatment (accessed 07-12-2023).
- Hansen L. A.; Hughes T. E. Altretamine. DICP 1991, 25, 146–152. 10.1177/106002809102500209. [DOI] [PubMed] [Google Scholar]
- Toschi L.; Finocchiaro G.; Bartolini S.; Gioia V.; Cappuzzo F. Role of gemcitabine in cancer therapy. Future Oncology 2005, 1, 7–17. 10.1517/14796694.1.1.7. [DOI] [PubMed] [Google Scholar]
- Brulikova L.; Hlavac J.; Hradil P. DNA Interstrand Cross-Linking Agents and their Chemotherapeutic Potential. Curr. Med. Chem. 2012, 19, 364–385. 10.2174/092986712803414295. [DOI] [PubMed] [Google Scholar]
- Goswami S.; Ghosh R.; Prasanthan P.; Kishore N. Mode of interaction of altretamine with calf thymus DNA: biophysical insights. J. Biomol. Struct. Dyn. 2023, 41, 3728–3740. 10.1080/07391102.2022.2054472. [DOI] [PubMed] [Google Scholar]
- Mini E.; Nobili S.; Caciagli B.; Landini I.; Mazzei T. Cellular pharmacology of gemcitabine. Annals of Oncology 2006, 17, v7–v12. 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- Nurgali K.; Jagoe R. T.; Abalo R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae?. Frontiers in Pharmacology 2018, 9, 245. 10.3389/fphar.2018.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfarouk K. O.; Stock C.-M.; Taylor S.; Walsh M.; Muddathir A. K.; Verduzco D.; Bashir A. H. H.; Mohammed O. Y.; Elhassan G. O.; Harguindey S.; et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell International 2015, 15, 71. 10.1186/s12935-015-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y. L.; Sapra P.; Bolton J.; Chua J. X.; Durrant L. G.; Stern P. L. Combination Treatment with an Antibody–Drug Conjugate (A1mcMMAF) Targeting the Oncofetal Glycoprotein 5T4 and Carboplatin Improves Survival in a Xenograft Model of Ovarian Cancer. Targeted Oncology 2019, 14, 465–477. 10.1007/s11523-019-00650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oflazoglu E.; Kissler K. M.; Sievers E. L.; Grewal I. S.; Gerber H.-P. Combination of the anti-CD30-auristatin-E antibody-drug conjugate (SGN-35) with chemotherapy improves antitumour activity in Hodgkin lymphoma. Br. J. Hamaetol. 2008, 142, 69–73. 10.1111/j.1365-2141.2008.07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purwanto I.; Utomo B. P.; Ghozali A. Complete Remission of Relapsed Hodgkin’s Lymphoma following Brentuximab Vedotin and Gemcitabine Combination Therapy with Severe Hypotension as Possible Treatment-Related Adverse Event: A Case Report. Case Reports in Oncology 2020, 13, 341–346. 10.1159/000505830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voruz S.; de Leval L.; Cairoli A. Successful salvage therapy for refractory primary cutaneous gamma-delta T-cell lymphoma with a combination of brentuximab vedotin and gemcitabine. Experimental Hematology & Oncology 2021, 10, 32. 10.1186/s40164-021-00225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournilhac O.; Hacini M.; Bouabdallah K.; Laribi K.; Maerevoet M.; Ysebaert L.; Guidez S.; Le Gouill S.; André M.; Dupuis J.; et al. Addition of Brentuximab Vedotin to Gemcitabine in Relapsed or Refractory T-Cell Lymphoma: Results of a Lysa Multicenter, Phase II Study. "the TOTAL Trial". Blood 2020, 136, 15–16. 10.1182/blood-2020-139224. [DOI] [Google Scholar]
- Kan S.; Koido S.; Okamoto M.; Hayashi K.; Ito M.; Kamata Y.; Komita H.; Nagasaki E.; Homma S. Up-regulation of HER2 by gemcitabine enhances the antitumor effect of combined gemcitabine and trastuzumab emtansine treatment on pancreatic ductal adenocarcinoma cells. BMC Cancer 2015, 15, 726. 10.1186/s12885-015-1772-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera A. F.; Carlo-Stella C.; Collins G. P.; Maddocks K. J.; Bartlett N. L.; Savage K. J.; Caimi P. F.; Hess B. T.; Zinzani P. L.; Cruz H. G.; et al. Preliminary Results of a Phase 2 Study of Camidanlumab Tesirine (Cami), a Novel Pyrrolobenzodiazepine-Based Antibody-Drug Conjugate, in Patients with Relapsed or Refractory Hodgkin Lymphoma. Blood 2020, 136, 21–23. 10.1182/blood-2020-137451. [DOI] [Google Scholar]
- Jabeen A.; Huang S.; Hartley J. A.; Van Berkel P. H.; Zammarchi F. Combination of Camidanlumab Tesirine, a CD25-Targeted ADC, with Gemcitabine Elicits Synergistic Anti-Tumor Activity in Preclinical Tumor Models. Blood 2020, 136, 31–32. 10.1182/blood-2020-137004. [DOI] [Google Scholar]
- Zheng C.; Zhou D.; Li W.; Duan Y.; Xu M.; Liu J.; Cheng J.; Xiao Y.; Xiao H.; Gan T.; et al. Therapeutic efficacy of a MMAE-based anti-DR5 drug conjugate Oba01 in preclinical models of pancreatic cancer. Cell Death & Disease 2023, 14, 295. 10.1038/s41419-023-05820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willuda J.; Linden L.; Lerchen H.-G.; Kopitz C.; Stelte-Ludwig B.; Pena C.; Lange C.; Golfier S.; Kneip C.; Carrigan P. E.; et al. Preclinical Antitumor Efficacy of BAY 1129980—a Novel Auristatin-Based Anti-C4.4A (LYPD3) Antibody–Drug Conjugate for the Treatment of Non–Small Cell Lung Cancer. Molecular Cancer Therapeutics 2017, 16, 893–904. 10.1158/1535-7163.MCT-16-0474. [DOI] [PubMed] [Google Scholar]
- Bisht A.; Cox M.; Peddaboina C. S.; Crocamo M.; Fandi A.; Rohlff C. Abstract 1069: OBT076, a clinical-stage ADC, displays synergy with oxaliplatin and cisplatin in preclinical gastric cancer models. Cancer research 2022, 82, 1069–1069. 10.1158/1538-7445.AM2022-1069. [DOI] [Google Scholar]
- Javaid F.; Pilotti C.; Camilli C.; Kallenberg D.; Bahou C.; Blackburn J.; Baker J.; Greenwood J.; Moss S. E.; Chudasama V. Leucine-rich alpha-2-glycoprotein 1 (LRG1) as a novel ADC target. RSC Chemical Biology 2021, 2, 1206–1220. 10.1039/D1CB00104C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K. N.; O’Malley D. M.; Vergote I.; Martin L. P.; Gonzalez-Martin A.; Malek K.; Birrer M. J. Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecologic Oncology 2018, 151, 46–52. 10.1016/j.ygyno.2018.07.017. [DOI] [PubMed] [Google Scholar]
- O’Malley D.; Richardson D.; Vergote I. B.; Gilbert L.; Martin L. P.; Mantia-Smaldone G. M.; Castro C.; Provencher D.; Matulonis U. A.; Malek K.; et al. 1028P - Mirvetuximab soravtansine, a folate receptor alpha (FRa)-targeting antibody-drug conjugate (ADC), in combination with carboplatin and bevacizumab: Initial results from a phase Ib study in patients (pts) with ovarian cancer. Annals of Oncology 2019, 30, v419–v420. 10.1093/annonc/mdz250.036. [DOI] [Google Scholar]
- Quanz M.; Hagemann U. B.; Zitzmann-Kolbe S.; Stelte-Ludwig B.; Golfier S.; Elbi C.; Mumberg D.; Ziegelbauer K.; Schatz C. A. Anetumab ravtansine inhibits tumor growth and shows additive effect in combination with targeted agents and chemotherapy in mesothelin-expressing human ovarian cancer models. Oncotarget 2018, 9 (75), 34103. 10.18632/oncotarget.26135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- I-SPY TRIAL: Neoadjuvant and Personalized Adaptive Novel Agents to Treat Breast Cancer (I-SPY). ClinicalTrials.gov; NIH, 2023. https://clinicaltrials.gov/study/NCT01042379 (accessed 07-12-2023).
- Overview of ADC-Based Combination Therapies; Biopharma PEG, 2023. https://www.biochempeg.com/article/330.html (accessed 05-03-2023).
- Cai X.; Zhan H.; Ye Y.; Yang J.; Zhang M.; Li J.; Zhuang Y. Current Progress and Future Perspectives of Immune Checkpoint in Cancer and Infectious Diseases. Frontiers in Genetics 2021, 12, 785153. 10.3389/fgene.2021.785153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.; Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Research 2020, 30, 660–669. 10.1038/s41422-020-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassannia H.; Ghasemi Chaleshtari M.; Atyabi F.; Nosouhian M.; Masjedi A.; Hojjat-Farsangi M.; Namdar A.; Azizi G.; Mohammadi H.; Ghalamfarsa G.; et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology 2020, 159, 75–87. 10.1111/imm.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zheng J.. Functions of Immune Checkpoint Molecules Beyond Immune Evasion. In Regulation of Cancer Immune Checkpoints: Molecular and Cellular Mechanisms and Therapy; Xu J., Ed.; Springer: Singapore, 2020; pp 201–226. [DOI] [PubMed] [Google Scholar]
- Dong H.; Strome S. E.; Salomao D. R.; Tamura H.; Hirano F.; Flies D. B.; Roche P. C.; Lu J.; Zhu G.; Tamada K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nature Medicine 2002, 8, 793–800. 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Sharpe A. H.; Pauken K. E. The diverse functions of the PD1 inhibitory pathway. Nature Reviews Immunology 2018, 18, 153–167. 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- Leach D. R.; Krummel M. F.; Allison J. P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (New York, N.Y.) 1996, 271, 1734–1736. 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Ning Z.; Liu K.; Xiong H. Roles of BTLA in Immunity and Immune Disorders. Frontiers in Immunology 2021, 12, 654960. 10.3389/fimmu.2021.654960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruhashi T.; Sugiura D.; Okazaki I.-m.; Okazaki T. LAG-3: from molecular functions to clinical applications. Journal for ImmunoTherapy of Cancer 2020, 8, e001014 10.1136/jitc-2020-001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chocarro L.; Blanco E.; Zuazo M.; Arasanz H.; Bocanegra A.; Fernández-Rubio L.; Morente P.; Fernández-Hinojal G.; Echaide M.; Garnica M.; et al. Understanding LAG-3 Signaling. International journal of molecular sciences 2021, 22, 5282. 10.3390/ijms22105282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M.; So T.; Duan W.; Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological Reviews 2009, 229, 173–191. 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby J.; Griffiths J.; Tews I.; Cragg M. S. OX40: Structure and function – What questions remain?. Molecular Immunology 2017, 83, 13–22. 10.1016/j.molimm.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Kim A. M. J.; Nemeth M. R.; Lim S.-O. 4–1BB: A promising target for cancer immunotherapy. Frontiers in Oncology 2022, 12, 968360. 10.3389/fonc.2022.968360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraman M.; Faroudi M.; Allen N. L.; Kmiecik K.; Gliddon D.; Seal C.; Koers A.; Wydro M. M.; Batey S.; Winnewisser J.; et al. FS118, a Bispecific Antibody Targeting LAG-3 and PD-L1, Enhances T-Cell Activation Resulting in Potent Antitumor Activity. Clin. Cancer Res. 2020, 26, 3333–3344. 10.1158/1078-0432.CCR-19-3548. [DOI] [PubMed] [Google Scholar]
- Lichtenegger F. S.; Rothe M.; Schnorfeil F. M.; Deiser K.; Krupka C.; Augsberger C.; Schlüter M.; Neitz J.; Subklewe M. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Frontiers in Immunology 2018, 9, 385. 10.3389/fimmu.2018.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl L.; den Boer A. T.; Schoenberger S. P.; van der Voort E. I. H.; Schumacher T. N. M.; Melief C. J. M.; Offringa R.; Toes R. E. M. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nature Medicine 1999, 5, 774–779. 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- Bartkowiak T.; Curran M. A. 4–1BB Agonists: Multi-Potent Potentiators of Tumor Immunity. Frontiers in Oncology 2015, 5, 117. 10.3389/fonc.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D.-T.; Bac N. D.; Nguyen K.-H.; Tien N. L. B.; Thanh V. V.; Nga V. T.; Ngoc V. T. N.; Anh Dao D. T.; Hoan L. N.; Hung N. P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. International journal of molecular sciences 2019, 20, 1822. 10.3390/ijms20081822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheide R. H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. 10.1016/j.ccell.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole R. M. Pembrolizumab: First Global Approval. Drugs 2014, 74, 1973–1981. 10.1007/s40265-014-0314-5. [DOI] [PubMed] [Google Scholar]
- Markham A.; Duggan S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. 10.1007/s40265-018-1012-5. [DOI] [PubMed] [Google Scholar]
- Mullard A. New checkpoint inhibitors ride the immunotherapy tsunami. Nat. Rev. Drug Discovery 2013, 12, 489–492. 10.1038/nrd4066. [DOI] [PubMed] [Google Scholar]
- Keam S. J. Tremelimumab: First Approval. Drugs 2023, 83, 93–102. 10.1007/s40265-022-01827-8. [DOI] [PubMed] [Google Scholar]
- Lee J. J.; Powderly J. D.; Patel M. R.; Brody J.; Hamilton E. P.; Infante J. R.; Falchook G. S.; Wang H.; Adams L.; Gong L.; et al. Phase 1 trial of CA-170, a novel oral small molecule dual inhibitor of immune checkpoints PD-1 and VISTA, in patients (pts) with advanced solid tumor or lymphomas. Journal of Clinical Oncology 2017, 35, TPS3099–TPS3099. 10.1200/JCO.2017.35.15_suppl.TPS3099. [DOI] [Google Scholar]
- Schöffski P.; Tan D. S. W.; Martín M.; Ochoa-de-Olza M.; Sarantopoulos J.; Carvajal R. D.; Kyi C.; Esaki T.; Prawira A.; Akerley W.; et al. Phase I/II study of the LAG-3 inhibitor ieramilimab (LAG525) ± anti-PD-1 spartalizumab (PDR001) in patients with advanced malignancies. Journal for ImmunoTherapy of Cancer 2022, 10, e003776 10.1136/jitc-2021-003776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safety and Efficacy of LAG525 Single Agent and in Combination With PDR001 in Patients With Advanced Malignancies. ClinicalTrials.gov; NIH, 2022. https://clinicaltrials.gov/study/NCT02460224 (accessed 07-12-2023).
- Brunner A. M.; Esteve J.; Porkka K.; Knapper S.; Vey N.; Scholl S.; Garcia-Manero G.; Wermke M.; Janssen J.; Traer E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and High-Risk Myelodysplastic Syndrome (HR-MDS): Updated Results from a Phase 1b Study. Blood 2020, 136, 1–2. 10.1182/blood-2020-136855.32430499 [DOI] [Google Scholar]
- Zeidan A. M.; Giagounidis A.; Sekeres M. A.; Xiao Z.; Sanz G. F.; Hoef M. V.; Ma F.; Hertle S.; Santini V. STIMULUS-MDS2 design and rationale: a phase III trial with the anti-TIM-3 sabatolimab (MBG453) + azacitidine in higher risk MDS and CMML-2. Future Oncology 2023, 19, 631–642. 10.2217/fon-2022-1237. [DOI] [PubMed] [Google Scholar]
- Study of Efficacy and Safety of MBG453 in Combination With Azacitidine in Subjects With Intermediate, High or Very High Risk Myelodysplastic Syndrome (MDS) as Per IPSS-R, or Chronic Myelomonocytic Leukemia-2 (CMML-2) (STIMULUS-MDS2). ClinicalTrials.gov; NIH, 2022. 3. https://classic.clinicaltrials.gov/ct2/show/NCT04266301 (accessed 07-12-2023).
- Pai-Scherf L.; Blumenthal G. M.; Li H.; Subramaniam S.; Mishra-Kalyani P. S.; He K.; Zhao H.; Yu J.; Paciga M.; Goldberg K. B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. 10.1634/theoncologist.2017-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J.; Mandala M.; Del Vecchio M.; Gogas H. J.; Arance A. M.; Cowey C. L.; Dalle S.; Schenker M.; Chiarion-Sileni V.; Marquez-Rodas I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. New England Journal of Medicine 2017, 377, 1824–1835. 10.1056/NEJMoa1709030. [DOI] [PubMed] [Google Scholar]
- Eggermont A. M. M.; Blank C. U.; Mandala M.; Long G. V.; Atkinson V.; Dalle S.; Haydon A.; Lichinitser M.; Khattak A.; Carlino M. S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. New England Journal of Medicine 2018, 378, 1789–1801. 10.1056/NEJMoa1802357. [DOI] [PubMed] [Google Scholar]
- Robert C.; Ribas A.; Hamid O.; Daud A.; Wolchok J. D.; Joshua A. M.; Hwu W.-J.; Weber J. S.; Gangadhar T. C.; Joseph R. W.; et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. Journal of Clinical Oncology 2018, 36, 1668–1674. 10.1200/JCO.2017.75.6270. [DOI] [PubMed] [Google Scholar]
- Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbandi A.; Chiu F.-Y.; Ungerleider N. A.; Kvadas R.; Mheidly Z.; Sun M. J. S.; Tian D.; Waizman D. A.; Anderson A. Y.; Machado H. L.; et al. Breast cancer cells survive chemotherapy by activating targetable immune-modulatory programs characterized by PD-L1 or CD80. Nature Cancer 2022, 3, 1513–1533. 10.1038/s43018-022-00466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins R. W.; Barbie D. A.; Flaherty K. T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh R. J.; Sundar R.; Lim J. S. J. Immune checkpoint inhibitor combinations—current and emerging strategies. Br. J. Cancer 2023, 128, 1415–1417. 10.1038/s41416-023-02181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafaei S.; Zekiy A. O.; Khanamir R. A.; Zaman B. A.; Ghayourvahdat A.; Azimizonuzi H.; Zamani M. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell International 2022, 22, 2. 10.1186/s12935-021-02407-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khair D. O.; Bax H. J.; Mele S.; Crescioli S.; Pellizzari G.; Khiabany A.; Nakamura M.; Harris R. J.; French E.; Hoffmann R. M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Frontiers in Immunology 2019, 10, 453. 10.3389/fimmu.2019.00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Kumar A. B.; Finnes H.; Markovic S. N.; Park S.; Dronca R. S.; Dong H. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Frontiers in Immunology 2018, 9, 1739. 10.3389/fimmu.2018.01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolò E.; Giugliano F.; Ascione L.; Tarantino P.; Corti C.; Tolaney S. M.; Cristofanilli M.; Curigliano G. Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treatment Reviews 2022, 106, 102395. 10.1016/j.ctrv.2022.102395. [DOI] [PubMed] [Google Scholar]
- Salifu I.; Singh N.; Berraondo M.; Remon J.; Salifu S.; Severson E.; Quintana A.; Peiró S.; Ramkissoon S.; Vidal L.; et al. Antibody-drug conjugates, immune-checkpoint inhibitors, and their combination in advanced non-small cell lung cancer. Cancer Treatment and Research Communications 2023, 36, 100713. 10.1016/j.ctarc.2023.100713. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Ma J.; Chen T.; Yang Y. Case report: The remarkable response of pembrolizumab combined with RC48 in the third-line treatment of metastatic urothelial carcinoma. Frontiers in Immunology 2022, 13, 978266. 10.3389/fimmu.2022.978266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Heijden M. S.; Gupta S.; Galsky M. D.; Derleth C. L.; Lee S.; Kataria R. S.; Powles T. Study EV-302: A two-arm, open-label, randomized controlled phase 3 study of enfortumab vedotin in combination with pembrolizumab versus chemotherapy in previously untreated advanced urothelial carcinoma (aUC) (trial in progress). Journal of Clinical Oncology 2022, 40, TPS589–TPS589. 10.1200/JCO.2022.40.6_suppl.TPS589. [DOI] [Google Scholar]
- Levy B. P.; Reck M.; Yang J. C.-H.; Cappuzzo F.; Rawat S.; Xie J.; Basak P.; Felip E. Datopotamab deruxtecan (Dato-DXd) plus pembrolizumab in treatment-naive advanced/metastatic (adv/met) non–small cell lung cancer (NSCLC) with PD-L1 ≥ 50% and without actionable genomic alterations. Journal of Clinical Oncology 2022, 40, TPS3162–TPS3162. 10.1200/JCO.2022.40.16_suppl.TPS3162. [DOI] [Google Scholar]
- Santoro A.; Moskowitz A. J.; Ferrari S.; Carlo-Stella C.; Fanale M. A.; Francis S.; Sacchi M.; Savage K. J. Nivolumab Combined with Brentuximab Vedotin for Relapsed/Refractory Mediastinal Gray Zone Lymphoma: Primary Efficacy and Safety Analysis of the Phase 2 CheckMate 436 Study. Blood 2020, 136, 44–45. 10.1182/blood-2020-137653. [DOI] [Google Scholar]
- Advani R. H.; Moskowitz A. J.; Bartlett N. L.; Vose J. M.; Ramchandren R.; Feldman T. A.; LaCasce A. S.; Christian B. A.; Ansell S. M.; Moskowitz C. H.; et al. Brentuximab vedotin in combination with nivolumab in relapsed or refractory Hodgkin lymphoma: 3-year study results. Blood 2021, 138, 427–438. 10.1182/blood.2020009178. [DOI] [PubMed] [Google Scholar]
- Camidge D. R.; Barlesi F.; Goldman J. W.; Morgensztern D.; Heist R.; Vokes E.; Angevin E.; Hong D. S.; Rybkin I. I.; Barve M.; et al. A Phase 1b Study of Telisotuzumab Vedotin in Combination With Nivolumab in Patients With NSCLC. JTO Clinical and Research Reports 2022, 3, 100262. 10.1016/j.jtocrr.2021.100262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottey S.; Clarke J.; Aung K.; Machiels J.-P.; Markman B.; Heinhuis K. M.; Millward M.; Lolkema M.; Patel S. P.; de Souza P.; et al. Phase I/IIa Trial of BMS-986148, an Anti-mesothelin Antibody–drug Conjugate, Alone or in Combination with Nivolumab in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2022, 28, 95–105. 10.1158/1078-0432.CCR-21-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy G.; Alexander M.; Camidge D. R. 83TiP A phase II study of mecbotamab vedotin (BA3011), a CAB-AXL-ADC, alone and in combination with nivolumab in adult patients with metastatic NSCLC who had prior disease progression on or are intolerant to a PD-1/L1, EGFR, or ALK inhibitor. Journal of Thoracic Oncology 2023, 18, S88. 10.1016/S1556-0864(23)00337-4. [DOI] [Google Scholar]
- Santoro A.; Moskowitz A. J.; Ferrari S.; Carlo-Stella C.; Lisano J.; Francis S.; Wen R.; Akyol A.; Savage K. J. Nivolumab combined with brentuximab vedotin for relapsed/refractory mediastinal gray zone lymphoma. Blood 2023, 141, 2780–2783. 10.1182/blood.2022017951. [DOI] [PubMed] [Google Scholar]
- Emens L. A.; Esteva F. J.; Beresford M.; Saura C.; De Laurentiis M.; Kim S.-B.; Im S.-A.; Wang Y.; Salgado R.; Mani A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. Lancet Oncology 2020, 21, 1283–1295. 10.1016/S1470-2045(20)30465-4. [DOI] [PubMed] [Google Scholar]
- Hamilton E. P.; Kaklamani V.; Falkson C.; Vidal G. A.; Ward P. J.; Patre M.; Chui S. Y.; Rotmensch J.; Gupta K.; Molinero L.; et al. Impact of Anti-HER2 Treatments Combined With Atezolizumab on the Tumor Immune Microenvironment in Early or Metastatic Breast Cancer: Results From a Phase Ib Study. Clinical Breast Cancer 2021, 21, 539–551. 10.1016/j.clbc.2021.04.011. [DOI] [PubMed] [Google Scholar]
- Moskowitz C. H.; Bastos-Oreiro M.; Ungar D.; Dautaj I.; Kalac M. Safety and Anti-Tumor Activity Study of Loncastuximab Tesirine and Durvalumab in Diffuse Large B-Cell, Mantle Cell, or Follicular Lymphoma. Blood 2019, 134, 2807–2807. 10.1182/blood-2019-128036. [DOI] [Google Scholar]
- Schmid P.; Jung K. H.; Wysocki P. J.; Jassem J.; Ma C. X.; Fernandes R.; Huisden R.; Stewart R.; Vukovic P.; Tablante Nunes A.; et al. 166MO Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): Initial results from BEGONIA, a phase Ib/II study. Annals of Oncology 2022, 33, S199. 10.1016/j.annonc.2022.03.185. [DOI] [Google Scholar]
- Rosenberg J. E.; Flaig T. W.; Friedlander T. W.; Milowsky M. I.; Srinivas S.; Petrylak D. P.; Merchan J. R.; Bilen M. A.; Carret A.-S.; Yuan N.; et al. Study EV-103: Preliminary durability results of enfortumab vedotin plus pembrolizumab for locally advanced or metastatic urothelial carcinoma. Journal of Clinical Oncology 2020, 38, 441–441. 10.1200/JCO.2020.38.6_suppl.441. [DOI] [Google Scholar]
- Zhou L.; Xu H.; Yan X.; Chi Z.; Cui C.; Si L.; Tang B.; Mao L.; Lian B.; Wang X.; et al. RC48-ADC combined with toripalimab, an anti-PD-1 monoclonal antibody (Ab), in patients with locally advanced or metastatic urothelial carcinoma (UC): Preliminary results of a phase Ib/II study. Journal of Clinical Oncology 2021, 39, 4534–4534. 10.1200/JCO.2021.39.15_suppl.4534. [DOI] [Google Scholar]
- Zhou L.; Xu H.; Li S.; Yan X.; Li J.; Wu X.; Chi Z.; Si L.; Cui C.; Kong Y.; et al. Study RC48-C014: Preliminary results of RC48-ADC combined with toripalimab in patients with locally advanced or metastatic urothelial carcinoma. Journal of Clinical Oncology 2022, 40, 515–515. 10.1200/JCO.2022.40.6_suppl.515. [DOI] [Google Scholar]
- Sheng X.; Zhou L.; He Z.; Guo H.; Yan X.; Li S.; Xu H.; Li J.; Chi Z.; Si L.; et al. Preliminary results of a phase Ib/II combination study of RC48-ADC, a novel humanized anti-HER2 antibody-drug conjugate (ADC) with toripalimab, a humanized IgG4 mAb against programmed death-1 (PD-1) in patients with locally advanced or metastatic urothelial carcinoma. Journal of Clinical Oncology 2022, 40, 4518–4518. 10.1200/JCO.2022.40.16_suppl.4518. [DOI] [Google Scholar]
- Zhou L.; Yang K.; Zhang S.; Xu H.; Yan X.; Li S.; Li J.; Cui C.; Chi Z.; Si L.; et al. Disitamab vedotin, a novel humanized anti-HER2 antibody-drug conjugate (ADC), combined with toripalimab in patients with locally advanced or metastatic urothelial carcinoma: An open-label phase 1b/2 study. Journal of Clinical Oncology 2023, 41, 4566–4566. 10.1200/JCO.2023.41.16_suppl.4566. [DOI] [Google Scholar]
- Chen M.; Yao K.; Cao M.; Liu H.; Xue C.; Qin T.; Meng L.; Zheng Z.; Qin Z.; Zhou F.; et al. HER2-targeting antibody–drug conjugate RC48 alone or in combination with immunotherapy for locally advanced or metastatic urothelial carcinoma: a multicenter, real-world study. Cancer Immunology, Immunotherapy 2023, 72, 2309–2318. 10.1007/s00262-023-03419-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodyak N. D.; Bergstrom D. A.; Hyer M.; Lowinger T. B.; Protopopova M.; Veiby O. P.; Yurkovetskiy A. V.; Zhang Q. X.. Combination therapies of HER2-targeted antibody-drug conjugates. Patent WO2018160538, 2018.
- Kulke M.; Hechler T.; Pahl A.; Mueller C.; Werner-Simon S.. Composition comprising a combination of immune checkpoint inhibitor and antibody-amatoxin conjugate for use in cancer therapy. US Patent US20220133902, 2022.
- Fang J.; Jiang J.; Li S.; Su X.. Use of antibody-drug conjugate in combination with immune checkpoint inhibitor in treatment of urothelial cancer. Patent WO2022242692, 2022.
- Iwata T.; Ishii C.; Wada T.; Ishida S.; Kamai Y.. Combination of antibody-drug conjugate and immune checkpoint inhibitor. Patent WO2018110515, 2018.
- Waks A. G.; Keenan T.; Li T.; Tayob N.; Wulf G. M.; Richardson E. T.; Mittendorf E. A.; Overmoyer B.; Krop I. E.; Winer E. P.; et al. A phase Ib study of pembrolizumab (pembro) plus trastuzumab emtansine (T-DM1) for metastatic HER2+ breast cancer (MBC). Journal of Clinical Oncology 2020, 38, 1046–1046. 10.1200/JCO.2020.38.15_suppl.1046. [DOI] [Google Scholar]
- Hamilton E.; Shapiro C. L.; Petrylak D.; Boni V.; Martin M.; Conte G. D.; Cortes J.; Agarwal L.; Arkenau H.-T.; Tan A. R.; et al. Abstract PD3–07: Trastuzumab deruxtecan (T-DXd; DS-8201) with nivolumab in patients with HER2-expressing, advanced breast cancer: A 2-part, phase 1b, multicenter, open-label study. Cancer research 2021, 81, PD3-07. 10.1158/1538-7445.SABCS20-PD3-07. [DOI] [Google Scholar]
- Schmid P.; Im S.-A.; Armstrong A.; Park Y. H.; Chung W.-P.; Nowecki Z.; Lord S.; Wysocki P. J.; Lu Y.-S.; Dry H.; et al. BEGONIA: Phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)—Initial results from arm 1, d+paclitaxel (P), and arm 6, d+trastuzumab deruxtecan (T-DXd). Journal of Clinical Oncology 2021, 39, 1023–1023. 10.1200/JCO.2021.39.15_suppl.1023. [DOI] [Google Scholar]
- Galsky M. D.; Conte G. D.; Foti S.; Yu E. Y.; Machiels J.-P. H.; Doger B.; Necchi A.; Braud F. G. D.; Hamilton E. P.; Hennequin A.; et al. Primary analysis from DS8201-A-U105: A phase 1b, two-part, open-label study of trastuzumab deruxtecan (T-DXd) with nivolumab (nivo) in patients (pts) with HER2-expressing urothelial carcinoma (UC). Journal of Clinical Oncology 2022, 40, 438–438. 10.1200/JCO.2022.40.6_suppl.438.34890214 [DOI] [Google Scholar]
- Han H. S.; Alemany C. A.; Brown-Glaberman U. A.; Pluard T. J.; Sinha R.; Sterrenberg D.; Albain K. S.; Basho R. K.; Biggs D.; Boni V.; et al. SGNLVA-002: Single-arm, open label phase Ib/II study of ladiratuzumab vedotin (LV) in combination with pembrolizumab for first-line treatment of patients with unresectable locally advanced or metastatic triple-negative breast cancer. Journal of Clinical Oncology 2019, 37, TPS1110–TPS1110. 10.1200/JCO.2019.37.15_suppl.TPS1110. [DOI] [Google Scholar]
- Friedlander T. W.; Milowsky M. I.; Bilen M. A.; Srinivas S.; McKay R. R.; Flaig T. W.; Hoimes C. J.; Balar A. V.; Henry E.; Petrylak D. P.; et al. Study EV-103: Update on durability results and long term outcome of enfortumab vedotin + pembrolizumab in first line locally advanced or metastatic urothelial carcinoma (la/mUC). Journal of Clinical Oncology 2021, 39, 4528–4528. 10.1200/JCO.2021.39.15_suppl.4528. [DOI] [Google Scholar]
- Okajima D.; Yasuda S.; Maejima T.; Karibe T.; Sakurai K.; Aida T.; Toki T.; Yamaguchi J.; Kitamura M.; Kamei R.; et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody–drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Molecular Cancer Therapeutics 2021, 20, 2329–2340. 10.1158/1535-7163.MCT-21-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy B.; Paz-Ares L.; Rixe O.; Su W. C.; Yang T. Y.; Tolcher A.; Lou Y.; Zenke Y.; Savvides P.; Felip E.; et al. MA13.07 TROPION-Lung02: Initial Results for Datopotamab Deruxtecan Plus Pembrolizumab and Platinum Chemotherapy in Advanced NSCLC. Journal of Thoracic Oncology 2022, 17, S91. 10.1016/j.jtho.2022.07.152. [DOI] [Google Scholar]
- An Anti-CD276 Antibody–Drug Conjugate Kills Tumor Cells and Vasculature. Cancer Discovery 2017, 7, 550–550. 10.1158/2159-8290.CD-RW2017-078 [DOI] [Google Scholar]
- Xiao D.; Luo L.; Li J.; Wang Z.; Liu L.; Xie F.; Feng J.; Zhou X. Development of bifunctional anti-PD-L1 antibody MMAE conjugate with cytotoxicity and immunostimulation. Bioorganic Chemistry 2021, 116, 105366. 10.1016/j.bioorg.2021.105366. [DOI] [PubMed] [Google Scholar]
- Doi T.; Patel M.; Falchook G. S.; Koyama T.; Friedman C. F.; Piha-Paul S.; Gutierrez M. E.; Abdul-Karim R.; Awad M.; Adkins D. R.; et al. 453O DS-7300 (B7-H3 DXd antibody-drug conjugate [ADC]) shows durable antitumor activity in advanced solid tumors: Extended follow-up of a phase I/II study. Annals of Oncology 2022, 33, S744–S745. 10.1016/j.annonc.2022.07.582. [DOI] [Google Scholar]
- He L.; Wang L.; Wang Z.; Li T.; Chen H.; Zhang Y.; Hu Z.; Dimitrov D. S.; Du J.; Liao X. Immune Modulating Antibody–Drug Conjugate (IM-ADC) for Cancer Immunotherapy. J. Med. Chem. 2021, 64, 15716–15726. 10.1021/acs.jmedchem.1c00961. [DOI] [PubMed] [Google Scholar]
- Solinas G.; Germano G.; Mantovani A.; Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal of Leukocyte Biology 2009, 86, 1065–1073. 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- Sica A.; Allavena P.; Mantovani A. Cancer related inflammation: The macrophage connection. Cancer Letters 2008, 267, 204–215. 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- Szebeni G. J.; Vizler C.; Kitajka K.; Puskas L. G. Inflammation and Cancer: Extra- and Intracellular Determinants of Tumor-Associated Macrophages as Tumor Promoters. Mediators of Inflammation 2017, 2017, 9294018. 10.1155/2017/9294018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Liu R.; Su X.; Pan Y.; Han X.; Shao C.; Shi Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Molecular Cancer 2019, 18, 177. 10.1186/s12943-019-1102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oflazoglu E.; Stone I. J.; Gordon K. A.; Grewal I. S.; van Rooijen N.; Law C.-L.; Gerber H.-P. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 2007, 110, 4370–4372. 10.1182/blood-2007-06-097014. [DOI] [PubMed] [Google Scholar]
- Oflazoglu E.; Stone I. J.; Brown L.; Gordon K. A.; van Rooijen N.; Jonas M.; Law C. L.; Grewal I. S.; Gerber H. P. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br. J. Cancer 2009, 100, 113–117. 10.1038/sj.bjc.6604812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Ulrich M.; Jonas M.; Stone I. J.; Linares G.; Zhang X.; Westendorf L.; Benjamin D. R.; Law C.-L. Tumor-Associated Macrophages Can Contribute to Antitumor Activity through FcγR-Mediated Processing of Antibody–Drug Conjugates. Molecular Cancer Therapeutics 2017, 16, 1347–1354. 10.1158/1535-7163.MCT-17-0019. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Yu Y.; Wang X.; Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Frontiers in Immunology 2020, 11, 583084. 10.3389/fimmu.2020.583084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaynes J. M.; Sable R.; Ronzetti M.; Bautista W.; Knotts Z.; Abisoye-Ogunniyan A.; Li D.; Calvo R.; Dashnyam M.; Singh A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Science Translational Medicine 2020, 12, eaax6337 10.1126/scitranslmed.aax6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequential treatment; National Cancer Institute, 2023. https://www.cancer.gov/publications/dictionaries/cancer-terms/def/sequential-treatment (accessed 07-25-2023).
- Kwon M.; Jung H.; Nam G.-H.; Kim I.-S. The right Timing, right combination, right sequence, and right delivery for Cancer immunotherapy. J. Controlled Release 2021, 331, 321–334. 10.1016/j.jconrel.2021.01.009. [DOI] [PubMed] [Google Scholar]
- Messenheimer D. J.; Jensen S. M.; Afentoulis M. E.; Wegmann K. W.; Feng Z.; Friedman D. J.; Gough M. J.; Urba W. J.; Fox B. A. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin. Cancer Res. 2017, 23, 6165–6177. 10.1158/1078-0432.CCR-16-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles D.; Minckwitz G.; Seidman A. D. Combination Versus Sequential Single-Agent Therapy in Metastatic Breast Cancer. Oncologist 2002, 7, 13–19. 10.1634/theoncologist.2002-0013. [DOI] [PubMed] [Google Scholar]
- Escudier B.; Goupil M. G.; Massard C.; Fizazi K. Sequential therapy in renal cell carcinoma. Cancer 2009, 115, 2321–2326. 10.1002/cncr.24241. [DOI] [PubMed] [Google Scholar]
- Molife C.; Hess L. M.; Cui Z. L.; Li X. I.; Beyrer J.; Mahoui M.; Oton A. B. Sequential therapy with ramucirumab and/or checkpoint inhibitors for non-small-cell lung cancer in routine practice. Future Oncology 2019, 15, 2915–2931. 10.2217/fon-2018-0876. [DOI] [PubMed] [Google Scholar]
- Wahl A. F.; Donaldson K. L.; Mixan B. J.; Trail P. A.; Siegall C. B. Selective tumor sensitization to taxanes with the mAb-drug conjugate cBR96-doxorubicin. Int. J. Cancer 2001, 93, 590–600. 10.1002/ijc.1364. [DOI] [PubMed] [Google Scholar]
- Liu R.; Wang R. E.; Wang F. Antibody-drug conjugates for non-oncological indications. Expert Opinion on Biological Therapy 2016, 16, 591–593. 10.1517/14712598.2016.1161753. [DOI] [PubMed] [Google Scholar]
- McPherson M. J.; Hobson A. D. Pushing the Envelope: Advancement of ADCs Outside of Oncology. Methods in molecular biology (Clifton, N.J.) 2020, 2078, 23–36. 10.1007/978-1-4939-9929-3_2. [DOI] [PubMed] [Google Scholar]
- Gingrich J.How the Next Generation Antibody Drug Conjugates Expands Beyond Cytotoxic Payloads for Cancer Therapy. Journal of Antibody-Drug Conjufates 2020. 10.14229/jadc.2020.04.07.001 [DOI] [Google Scholar]
- Theocharopoulos C.; Lialios P. P.; Samarkos M.; Gogas H.; Ziogas D. C. Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond. Vaccines (Basel) 2021, 9, 1111. 10.3390/vaccines9101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal L. B.; Bule P.; Khan W.; Chella N. An Overview of the Development and Preclinical Evaluation of Antibody–Drug Conjugates for Non-Oncological Applications. Pharmaceutics 2023, 15, 1807. 10.3390/pharmaceutics15071807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aminov R. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Frontiers in Microbiology 2010, 1, 134. 10.3389/fmicb.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig S. G.; Pillow T. H.. Discovery and Development of the First Antibody–Antibiotic Conjugate Linker-Drug. In Complete Accounts of Integrated Drug Discovery and Development: Recent Examples from the Pharmaceutical Industry Vol. 2; American Chemical Society, 2019; Vol. 1332, pp 85–105. [Google Scholar]
- Maxson T.; Mitchell D. A. Targeted treatment for bacterial infections: prospects for pathogen-specific antibiotics coupled with rapid diagnostics. Tetrahedron 2016, 72, 3609–3624. 10.1016/j.tet.2015.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaco M.; Castanho M. A. R. B.; Neves V. The Use of Antibody-Antibiotic Conjugates to Fight Bacterial Infections. Frontiers in Microbiology 2022, 13, 835677. 10.3389/fmicb.2022.835677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg N. J.; Katavolos P.; Achilles Poon K.; Zhong S.; Ljumanovic N.; Kamath A.; Cai H.; Carrasco-Triguero M.; Halpern W. Nonclinical toxicology development of a novel antibody antibiotic conjugate for treating invasive Staphylococcus Aureus infections. Toxicol. Appl. Pharmacol. 2022, 435, 115811. 10.1016/j.taap.2021.115811. [DOI] [PubMed] [Google Scholar]
- Motley M. P.; Banerjee K.; Fries B. C. Monoclonal antibody-based therapies for bacterial infections. Curr. Opin Infect Dis 2019, 32, 210–216. 10.1097/QCO.0000000000000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvilum A.; Johansen M. I.; Glud L. N.; Ivarsen D. M.; Khamas A. B.; Carmali S.; Mhatre S. S.; Søgaard A. B.; Faddy E.; Vor L. d.; et al. Antibody-drug conjugates to treat bacterial biofilms. bioRxiv 2023, 10.1101/2023.01.16.524127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan B. W.; Chowdhury N.; Wood T. K. Combatting bacterial infections by killing persister cells with mitomycin C. Environmental Microbiology 2015, 17, 4406–4414. 10.1111/1462-2920.12873. [DOI] [PubMed] [Google Scholar]
- Verweij J.; Pinedo H. M. Mitomycin C: mechanism of action, usefulness and limitations. Anticancer Drugs 1990, 1, 5–13. 10.1097/00001813-199010000-00002. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Jayaraman N.; Chatterji D. Small-Molecule Inhibition of Bacterial Biofilm. ACS Omega 2020, 5, 3108–3115. 10.1021/acsomega.9b03695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graversen J. H.; Svendsen P.; Dagnæs-Hansen F.; Dal J.; Anton G.; Etzerodt A.; Petersen M. D.; Christensen P. A.; Møller H. J.; Moestrup S. K. Targeting the Hemoglobin Scavenger receptor CD163 in Macrophages Highly Increases the Anti-inflammatory Potency of Dexamethasone. Molecular Therapy 2012, 20, 1550–1558. 10.1038/mt.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graversen J. H.; Moestrup S. K. Drug Trafficking into Macrophages via the Endocytotic Receptor CD163. Membranes (Basel) 2015, 5, 228–252. 10.3390/membranes5020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller L. N. O.; Knudsen A. R.; Andersen K. J.; Nyengaard J. R.; Hamilton-Dutoit S.; Okholm Møller E. M.; Svendsen P.; Møller H. J.; Moestrup S. K.; Graversen J. H.; et al. Anti-CD163-dexamethasone protects against apoptosis after ischemia/reperfusion injuries in the rat liver. Ann. Med. Surg (Lond) 2015, 4, 331–337. 10.1016/j.amsu.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen P.; Graversen J. H.; Etzerodt A.; Hager H.; Røge R.; Grønbæk H.; Christensen E. I.; Møller H. J.; Vilstrup H.; Moestrup S. K. Antibody-Directed Glucocorticoid Targeting to CD163 in M2-type Macrophages Attenuates Fructose-Induced Liver Inflammatory Changes. Mol. Ther Methods Clin Dev 2017, 4, 50–61. 10.1016/j.omtm.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel B.; McPherson M.; Hernandez A.; Goess C.; Mathieu S.; Waegell W.; Bryant S.; Hobson A.; Ruzek M.; Pang Y.; et al. POS0365 ANTI-TNF GLUCOCORTICOID RECEPTOR MODULATOR ANTIBODY DRUG CONJUGATE FOR THE TREATMENT OF AUTOIMMUNE DISEASES. Annals of the Rheumatic Diseases 2021, 80, 412–413. 10.1136/annrheumdis-2021-eular.2213. [DOI] [Google Scholar]
- Deora A.; Hegde S.; Lee J.; Choi C.-H.; Chang Q.; Lee C.; Eaton L.; Tang H.; Wang D.; Lee D.; et al. Transmembrane TNF-dependent uptake of anti-TNF antibodies. mAbs 2017, 9, 680–695. 10.1080/19420862.2017.1304869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcpherson M. J.; Hobson A. D.; Hayes M. E.; MARVIN C. C.; Schmidt D.; Waegell W.; GOESS C.; OH J. Z.; HERNANDEZ A. JR; Randolph J. T.. Glucocorticoid receptor agonist and immunoconjugates thereof. Patent WO2017210471, 2017.
- Everts M.; Kok R. J.; Ásgeirsdóttir S. A.; Melgert B. N.; Moolenaar T. J. M.; Koning G. A.; van Luyn M. J. A.; Meijer D. K. F.; Molema G. Selective Intracellular Delivery of Dexamethasone into Activated Endothelial Cells Using an E-Selectin-Directed Immunoconjugate. J. Immunol. 2002, 168, 883–889. 10.4049/jimmunol.168.2.883. [DOI] [PubMed] [Google Scholar]
- Kern J. C.; Dooney D.; Zhang R.; Liang L.; Brandish P. E.; Cheng M.; Feng G.; Beck A.; Bresson D.; Firdos J.; et al. Novel Phosphate Modified Cathepsin B Linkers: Improving Aqueous Solubility and Enhancing Payload Scope of ADCs. Bioconjugate Chem. 2016, 27, 2081–2088. 10.1021/acs.bioconjchem.6b00337. [DOI] [PubMed] [Google Scholar]
- Han A.; Olson W.; Andrew M. J.. STEROIDS AND PROTEIN-CONJUGATES THEREOF. Patent WO2018089373, 2018.
- Sugo T.; Terada M.; Oikawa T.; Miyata K.; Nishimura S.; Kenjo E.; Ogasawara-Shimizu M.; Makita Y.; Imaichi S.; Murata S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. Journal of controlled release: official journal of the Controlled Release Society 2016, 237, 1–13. 10.1016/j.jconrel.2016.06.036. [DOI] [PubMed] [Google Scholar]
- Ibtehaj N.; Huda R. High-dose BAFF receptor specific mAb-siRNA conjugate generates Fas-expressing B cells in lymph nodes and high-affinity serum autoantibody in a myasthenia mouse model. Clinical Immunology 2017, 176, 122–130. 10.1016/j.clim.2017.01.005. [DOI] [PubMed] [Google Scholar]
- Lehar S. M.; Pillow T.; Xu M.; Staben L.; Kajihara K. K.; Vandlen R.; DePalatis L.; Raab H.; Hazenbos W. L.; Hiroshi Morisaki J.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- Peck M.; Rothenberg M. E.; Deng R.; Lewin-Koh N.; She G.; Kamath A. V.; Carrasco-Triguero M.; Saad O.; Castro A.; Teufel L.; et al. A Phase 1, Randomized, Single-Ascending-Dose Study To Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S, an Anti-Staphylococcus aureus Thiomab Antibody-Antibiotic Conjugate, in Healthy Volunteers. Antimicrob. Agents Chemother. 2019, 63 (6), e02588-18. 10.1128/AAC.02588-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim R. K. V.; Yu S.; Cheng B.; Li S.; Kim N.-J.; Cao Y.; Chi V.; Kim J. Y.; Chatterjee A. K.; Schultz P. G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody–Drug Conjugate. Bioconjugate Chem. 2015, 26, 2216–2222. 10.1021/acs.bioconjchem.5b00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Bhang S. H.; Lee J. H.; Kim H.; Hahn S. K. Tocilizumab–Alendronate Conjugate for Treatment of Rheumatoid Arthritis. Bioconjugate Chem. 2017, 28, 1084–1092. 10.1021/acs.bioconjchem.7b00008. [DOI] [PubMed] [Google Scholar]
- Chen Y.-B.; Perales M.-A.; Li S.; Kempner M.; Reynolds C.; Brown J.; Efebera Y. A.; Devine S. M.; El-Jawahri A.; McAfee S. L.; et al. Phase 1 multicenter trial of brentuximab vedotin for steroid-refractory acute graft-versus-host disease. Blood 2017, 129, 3256–3261. 10.1182/blood-2017-03-772210. [DOI] [PubMed] [Google Scholar]
- Fernandez-Codina A.; Nevskaya T.; Pope J. OP0172 BRENTUXIMAB VEDONTIN FOR SKIN INVOLVEMENT IN REFRACTORY DIFFUSE CUTANEOUS SYSTEMIC SCLEROSIS, INTERIM RESULTS OF A PHASE IIB OPEN-LABEL TRIAL. Annals of the Rheumatic Diseases 2021, 80, 103–104. 10.1136/annrheumdis-2021-eular.2115.33115761 [DOI] [Google Scholar]
- Zhao P.; Zhang N.; An Z. Engineering antibody and protein therapeutics to cross the blood–brain barrier. Antibody Therapeutics 2022, 5, 311–331. 10.1093/abt/tbac028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q.; Liu J.; Liang J.; Liu X.; Li W.; Liu Z.; Ding Z.; Tuo D. Towards improvements for penetrating the blood–brain barrier—recent progress from a material and pharmaceutical perspective. Cells 2018, 7, 24. 10.3390/cells7040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks W. A. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discovery 2016, 15, 275–292. 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- PitchBook; PitchBook, 2023. https://www.pitchbook.com (accessed 07-25-2023).
- Study to Evaluate Adverse Events and Change in Disease Activity in Participants Between 18 to 75 Years of Age Treated With Subcutaneous (SC) Injections of ABBV-154 for Moderately to Severely Active Rheumatoid Arthritis (RA). ClinicalTrials.gov; NIH, 2023. https://clinicaltrials.gov/study/NCT04888585 (accessed 07-14-2023).
- Brentuximab Vedotin for Systemic Sclerosis (BRAVOS). ClinicalTrials.gov; NIH, 2023. https://clinicaltrials.gov/study/NCT03222492 (accessed 07-14-2023).
- Review and Formulation Analysis of 14 Antibody Drug Conjugates (ADCs) Approved by FDA Up To 2022; BOC Sciences, 2023. https://adc.bocsci.com/resource/review-and-formulation-analysis-of-14-antibody-drug-conjugates-adcs-approved-by-fda-up-to-2022.html?hsa_acc=8252592073&hsa_cam=&hsa_grp=&hsa_ad=2348537486689944079&hsa_src=&hsa_tgt=&hsa_kw=&hsa_mt=b&hsa_net=adwords&hsa_ver=3&gclid=CjwKCAjwuqiiBhBtEiwATgvixH3agxenwZ1R8ImxVsNMFJMrUdf-qfsDZ_d9XAbo_G5PQmwRRS1i-hoCkg0QAvD_BwE (accessed 05-01-2023).
- FDA Approved Antibody-Drug Conjugates (ADCs) Up To 2023; Biopharma PEG, 2019. https://www.biochempeg.com/article/74.html (accessed 05-01-2023).
- Antibody-drug conjugates (ADCs) list Approved by FDA (2000–2023); AxisPharm, 2023. https://axispharm.com/antibody-drug-conjugatesadcs-list-approved-by-fda2000-2022/ (accessed 05-01-2023).
- The potential of antibody-drug conjugates (ADCs) to provide breakthroughs in cancer therapy. News Medical Life Sciences; AZoNetwork, 2023. https://www.news-medical.net/whitepaper/20230425/The-potential-of-Antibody-Drug-Conjugates-%28ADCs%29-to-provide-breakthroughs-in-cancer-therapy.aspx (accessed 05-04-2023).
- Tarantino P.; Carmagnani Pestana R.; Corti C.; Modi S.; Bardia A.; Tolaney S. M.; Cortes J.; Soria J.-C.; Curigliano G. Antibody–drug conjugates: Smart chemotherapy delivery across tumor histologies. CA: A Cancer Journal for Clinicians 2022, 72, 165–182. 10.3322/caac.21705. [DOI] [PubMed] [Google Scholar]
- Tridente G.; Tridente G. Gemtuzumab. Adverse Events with Biomedicines: Prevention Through Understanding 2014, 211–217. 10.1007/978-88-470-5313-7_21. [DOI] [Google Scholar]
- DeLeve L.Chapter 30—Cancer Chemotherapy. Drug-Induced Liver Disease, 3rd ed.; Kaplowitz N., DeLeve L. D., Eds., 2013; pp 541–567. [Google Scholar]
- Nicolaou K. C. Chemistry and biology of the calicheamicins. Chemistry & Biology 1994, 1, xxvi–xxvii. 10.1016/1074-5521(94)90030-2. [DOI] [Google Scholar]
- Bross P. F.; Beitz J.; Chen G.; Chen X. H.; Duffy E.; Kieffer L.; Roy S.; Sridhara R.; Rahman A.; Williams G. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clinical cancer research 2001, 7, 1490–1496. [PubMed] [Google Scholar]
- Petersdorf S. H.; Kopecky K. J.; Slovak M.; Willman C.; Nevill T.; Brandwein J.; Larson R. A.; Erba H. P.; Stiff P. J.; Stuart R. K. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood, The Journal of the American Society of Hematology 2013, 121, 4854–4860. 10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles F. J.; Kantarjian H. M.; Kornblau S. M.; Thomas D. A.; Garcia-Manero G.; Waddelow T. A.; David C. L.; Phan A. T.; Colburn D. E.; Rashid A. Mylotarg(gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer: Interdisciplinary International Journal of the American Cancer Society 2001, 92, 406–413. . [DOI] [PubMed] [Google Scholar]
- Selby C.; Yacko L. R.; Glode A. E. Gemtuzumab ozogamicin: back again. Journal of the Advanced Practitioner in Oncology 2019, 10, 68. [PMC free article] [PubMed] [Google Scholar]
- Gbadamosi M.; Meshinchi S.; Lamba J. K. Gemtuzumab ozogamicin for treatment of newly diagnosed CD33-positive acute myeloid leukemia. Future Oncology 2018, 14, 3199–3213. 10.2217/fon-2018-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younes A.; Gopal A. K.; Smith S. E.; Ansell S. M.; Rosenblatt J. D.; Savage K. J.; Ramchandren R.; Bartlett N. L.; Cheson B. D.; De Vos S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. Journal of clinical oncology 2012, 30, 2183. 10.1200/JCO.2011.38.0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pro B.; Advani R.; Brice P.; Bartlett N. L.; Rosenblatt J. D.; Illidge T.; Matous J.; Ramchandren R.; Fanale M.; Connors J. M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J. Clin Oncol 2012, 30, 2190–2196. 10.1200/JCO.2011.38.0402. [DOI] [PubMed] [Google Scholar]
- Doronina S. O.; Toki B. E.; Torgov M. Y.; Mendelsohn B. A.; Cerveny C. G.; Chace D. F.; DeBlanc R. L.; Gearing R. P.; Bovee T. D.; Siegall C. B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nature biotechnology 2003, 21, 778–784. 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- Moskowitz C. H.; Nademanee A.; Masszi T.; Agura E.; Holowiecki J.; Abidi M. H.; Chen A. I.; Stiff P.; Gianni A. M.; Carella A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. 10.1016/S0140-6736(15)60165-9. [DOI] [PubMed] [Google Scholar]
- Prince H. M.; Kim Y. H.; Horwitz S. M.; Dummer R.; Scarisbrick J.; Quaglino P.; Zinzani P. L.; Wolter P.; Sanches J. A.; Ortiz-Romero P. L.; et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet 2017, 390, 555–566. 10.1016/S0140-6736(17)31266-7. [DOI] [PubMed] [Google Scholar]
- Shah N. N.; Stevenson M. S.; Yuan C. M.; Richards K.; Delbrook C.; Kreitman R. J.; Pastan I.; Wayne A. S. Characterization of CD22 expression in acute lymphoblastic leukemia. Pediatr Blood Cancer 2015, 62, 964–969. 10.1002/pbc.25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza F.; Maffini E.; Rondoni M.; Massari E.; Faini A. C.; Malavasi F. CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers (Basel) 2020, 12, 303. 10.3390/cancers12020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H. M.; DeAngelo D. J.; Stelljes M.; Liedtke M.; Stock W.; Gökbuget N.; O’Brien S. M.; Jabbour E.; Wang T.; Liang White J.; et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer 2019, 125, 2474–2487. 10.1002/cncr.32116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiJoseph J. F.; Armellino D. C.; Boghaert E. R.; Khandke K.; Dougher M. M.; Sridharan L.; Kunz A.; Hamann P. R.; Gorovits B.; Udata C.; et al. Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. 10.1182/blood-2003-07-2466. [DOI] [PubMed] [Google Scholar]
- Kantarjian H. M.; DeAngelo D. J.; Stelljes M.; Martinelli G.; Liedtke M.; Stock W.; Gökbuget N.; O’Brien S.; Wang K.; Wang T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. New England Journal of Medicine 2016, 375, 740–753. 10.1056/NEJMoa1509277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A.; Kjeldsberg C. R. Hairy cell leukemia: a review. Medicine 1978, 57, 477–499. 10.1097/00005792-197811000-00001. [DOI] [PubMed] [Google Scholar]
- Cordone I.; Annino L.; Masi S.; Pescarmona E.; Rahimi S.; Ferrari A.; Giubilei E.; Pignoloni P.; Faraggiana T.; Mandelli F. Diagnostic relevance of peripheral blood immunocytochemistry in hairy cell leukaemia. Journal of clinical pathology 1995, 48, 955–960. 10.1136/jcp.48.10.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babusíková O.; Tomova A.; Kusenda J.; Gyarfas J. Flow cytometry of peripheral blood and bone marrow cells from patients with hairy cell leukemia: phenotype of hairy cells, lymphocyte subsets and detection of minimal residual disease after treatment. Neoplasma 2001, 48, 350–357. [PubMed] [Google Scholar]
- Urquhart L. FDA new drug approvals in Q3 2018. Nat. Rev. Drug Discov 2018, 17, 779. 10.1038/nrd.2018.194. [DOI] [PubMed] [Google Scholar]
- Deeks E. D. Polatuzumab vedotin: first global approval. Drugs 2019, 79, 1467–1475. 10.1007/s40265-019-01175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.; Fuji R. N.; Elkins K.; Yu S.-F.; Fuh F. K.; Chuh J.; Tan C.; Hongo J.-A.; Raab H.; Kozak K. R.; et al. In vivo effects of targeting CD79b with antibodies and antibody-drug conjugates. Molecular cancer therapeutics 2009, 8, 2937–2946. 10.1158/1535-7163.MCT-09-0369. [DOI] [PubMed] [Google Scholar]
- Pfeifer M.; Zheng B.; Erdmann T.; Koeppen H.; McCord R.; Grau M.; Staiger A.; Chai A.; Sandmann T.; Madle H.; et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586. 10.1038/leu.2015.48. [DOI] [PubMed] [Google Scholar]
- Urquhart L. FDA new drug approvals in Q2 2019. Nature reviews. Drug discovery 2019, 18, 575. 10.1038/d41573-019-00121-9. [DOI] [PubMed] [Google Scholar]
- Seckinger A.; Delgado J. A.; Moser S.; Moreno L.; Neuber B.; Grab A.; Lipp S.; Merino J.; Prosper F.; Emde M.; et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer cell 2017, 31, 396–410. 10.1016/j.ccell.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Lonial S.; Lee H. C.; Badros A.; Trudel S.; Nooka A. K.; Chari A.; Abdallah A.-O.; Callander N.; Lendvai N.; Sborov D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. lancet oncology 2020, 21, 207–221. 10.1016/S1470-2045(19)30788-0. [DOI] [PubMed] [Google Scholar]
- Jain N.; Stock W.; Zeidan A.; Atallah E.; McCloskey J.; Heffner L.; Tomlinson B.; Bhatnagar B.; Feingold J.; Ungar D.; et al. Loncastuximab tesirine, an anti-CD19 antibody-drug conjugate, in relapsed/refractory B-cell acute lymphoblastic leukemia. Blood advances 2020, 4, 449–457. 10.1182/bloodadvances.2019000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammarchi F.; Corbett S.; Adams L.; Tyrer P. C.; Kiakos K.; Janghra N.; Marafioti T.; Britten C. E.; Havenith C. E.; Chivers S. ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood, The Journal of the American Society of Hematology 2018, 131, 1094–1105. 10.1182/blood-2017-10-813493. [DOI] [PubMed] [Google Scholar]
- Hartley J. A. The development of pyrrolobenzodiazepines as antitumour agents. Expert opinion on investigational drugs 2011, 20, 733–744. 10.1517/13543784.2011.573477. [DOI] [PubMed] [Google Scholar]
- Caimi P. F.; Ai W.; Alderuccio J. P.; Ardeshna K. M.; Hamadani M.; Hess B.; Kahl B. S.; Radford J.; Solh M.; Stathis A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncology 2021, 22, 790–800. 10.1016/S1470-2045(21)00139-X. [DOI] [PubMed] [Google Scholar]
- Erickson H. K.; Park P. U.; Widdison W. C.; Kovtun Y. V.; Garrett L. M.; Hoffman K.; Lutz R. J.; Goldmacher V. S.; Blättler W. A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer research 2006, 66, 4426–4433. 10.1158/0008-5472.CAN-05-4489. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya B.; Wolff J. Maytansine binding to the vinblastine sites of tubulin. FEBS letters 1977, 75, 159–162. 10.1016/0014-5793(77)80075-6. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Trastuzumab emtansine: a review of its use in patients with HER2-positive advanced breast cancer previously treated with trastuzumab-based therapy. Drugs 2014, 74, 675–686. 10.1007/s40265-014-0201-0. [DOI] [PubMed] [Google Scholar]
- Wedam S.; Fashoyin-Aje L.; Gao X.; Bloomquist E.; Tang S.; Sridhara R.; Goldberg K. B.; King-Kallimanis B. L.; Theoret M. R.; Ibrahim A.; et al. FDA Approval Summary: Ado-Trastuzumab Emtansine for the Adjuvant Treatment of HER2-positive Early Breast CancerFDA Approval: Ado-Trastuzumab Emtansine. Clin. Cancer Res. 2020, 26, 4180–4185. 10.1158/1078-0432.CCR-19-3980. [DOI] [PubMed] [Google Scholar]
- Chang E.; Weinstock C.; Zhang L.; Charlab R.; Dorff S. E.; Gong Y.; Hsu V.; Li F.; Ricks T. K.; Song P.; et al. FDA Approval Summary: Enfortumab Vedotin for Locally Advanced or Metastatic Urothelial CarcinomaFDA Approval Summary: Enfortumab Vedotin. Clinical cancer research 2021, 27, 922–927. 10.1158/1078-0432.CCR-20-2275. [DOI] [PubMed] [Google Scholar]
- Challita-Eid P. M.; Satpayev D.; Yang P.; An Z.; Morrison K.; Shostak Y.; Raitano A.; Nadell R.; Liu W.; Lortie D. R.; et al. Enfortumab vedotin antibody–drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer modelsADC cancer therapeutic targeting nectin-4. Cancer research 2016, 76, 3003–3013. 10.1158/0008-5472.CAN-15-1313. [DOI] [PubMed] [Google Scholar]
- Powles T.; Rosenberg J. E.; Sonpavde G.; Loriot Y.; Duran I.; Lee J.-L.; Matsubara N.; Vulsteke C.; Wu C.; Campbell M. S.. Primary results of EV-301: A phase III trial of enfortumab vedotin versus chemotherapy in patients with previously treated locally advanced or metastatic urothelial carcinoma; American Society of Clinical Oncology, 2021. [DOI] [PubMed] [Google Scholar]
- Petrylak D. P.; Rosenberg J. E.; Duran I.; Loriot Y.; Sonpavde G.; Wu C.; Gartner E. M.; Melhem-Bertrandt A.; Powles T.. EV-301: Phase III study to evaluate enfortumab vedotin (EV) versus chemotherapy in patients with previously treated locally advanced or metastatic urothelial cancer (la/mUC); American Society of Clinical Oncology, 2019. [Google Scholar]
- Yu E. Y.; Petrylak D. P.; O’Donnell P. H.; Lee J.-L.; van der Heijden M. S.; Loriot Y.; Stein M. N.; Necchi A.; Kojima T.; Harrison M. R.; et al. Enfortumab vedotin after PD-1 or PD-L1 inhibitors in cisplatin-ineligible patients with advanced urothelial carcinoma (EV-201): a multicentre, single-arm, phase 2 trial. Lancet Oncology 2021, 22, 872–882. 10.1016/S1470-2045(21)00094-2. [DOI] [PubMed] [Google Scholar]
- Shitara K.; Baba E.; Fujitani K.; Oki E.; Fujii S.; Yamaguchi K. Discovery and development of trastuzumab deruxtecan and safety management for patients with HER2-positive gastric cancer. Gastric Cancer 2021, 24, 780–789. 10.1007/s10120-021-01196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meddahi A.; Lemdjabar H.; Caruelle J.-P.; Barritault D.; Hornebeck W. FGF protection and inhibition of human neutrophil elastase by carboxymethyl benzylamide sulfonate dextran derivatives. Int. J. Biol. Macromol. 1996, 18, 141–145. 10.1016/0141-8130(95)01074-2. [DOI] [PubMed] [Google Scholar]
- Modi S.; Saura C.; Yamashita T.; Park Y. H.; Kim S.-B.; Tamura K.; Andre F.; Iwata H.; Ito Y.; Tsurutani J.; et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. New England Journal of Medicine 2020, 382, 610–621. 10.1056/NEJMoa1914510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S.; Saura C.; Yamashita T.; Park Y. H.; Kim S.-B.; Tamura K.; Andre F.; Iwata H.; Ito Y.; Tsurutani J.; et al. Abstract PD3–06: Updated results from DESTINY-breast01, a phase 2 trial of trastuzumab deruxtecan (T-DXd) in HER2 positive metastatic breast cancer. Cancer research 2021, 81, PD3-06. 10.1158/1538-7445.SABCS20-PD3-06. [DOI] [Google Scholar]
- Janjigian Y.; Viglianti N.; Liu F.; Mendoza-Naranjo A.; Puvvada S. 1500TiP A phase Ib/II, multicenter, open-label, dose-escalation and dose-expansion study evaluating trastuzumab deruxtecan (T-DXd; DS-8201) monotherapy and combinations in patients with HER2-overexpressing gastric cancer (DESTINY-Gastric03). Annals of Oncology 2020, 31, S930–S931. 10.1016/j.annonc.2020.08.2006. [DOI] [Google Scholar]
- Lipinski M.; Parks D. R.; Rouse R. V.; Herzenberg L. A. Human trophoblast cell-surface antigens defined by monoclonal antibodies. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 5147–5150. 10.1073/pnas.78.8.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapani E.; Sacchetti A.; Corda D.; Alberti S. Human TROP-2 is a tumor-associated calcium signal transducer. Int. J. Cancer 1998, 76, 671–676. . [DOI] [PubMed] [Google Scholar]
- Wang J.; Day R.; Dong Y.; Weintraub S. J.; Michel L. Identification of Trop-2 as an oncogene and an attractive therapeutic target in colon cancers. Mol. Cancer Ther 2008, 7, 280–285. 10.1158/1535-7163.MCT-07-2003. [DOI] [PubMed] [Google Scholar]
- Kawato Y.; Aonuma M.; Hirota Y.; Kuga H.; Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer research 1991, 51, 4187–4191. [PubMed] [Google Scholar]
- Perrone E.; Manara P.; Lopez S.; Bellone S.; Bonazzoli E.; Manzano A.; Zammataro L.; Bianchi A.; Zeybek B.; Buza N.; et al. Sacituzumab govitecan, an antibody-drug conjugate targeting trophoblast cell-surface antigen 2, shows cytotoxic activity against poorly differentiated endometrial adenocarcinomas in vitro and in vivo. Mol. Oncol 2020, 14, 645–656. 10.1002/1878-0261.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahota S.; Vahdat L. T. Sacituzumab govitecan: an antibody-drug conjugate. Expert Opin Biol. Ther 2017, 17, 1027–1031. 10.1080/14712598.2017.1331214. [DOI] [PubMed] [Google Scholar]
- Bardia A.; Hurvitz S. A.; Tolaney S. M.; Loirat D.; Punie K.; Oliveira M.; Brufsky A.; Sardesai S. D.; Kalinsky K.; Zelnak A. B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. New England journal of medicine 2021, 384, 1529–1541. 10.1056/NEJMoa2028485. [DOI] [PubMed] [Google Scholar]
- Bardia A.; Tolaney S. M.; Loirat D.; Punie K.; Oliveira M.; Rugo H. S.; Brufsky A.; Kalinsky K.; Cortés J.; O’Shaughnessy J.; et al. LBA17 ASCENT: A randomized phase III study of sacituzumab govitecan (SG) vs treatment of physician’s choice (TPC) in patients (pts) with previously treated metastatic triple-negative breast cancer (mTNBC). Annals of Oncology 2020, 31, S1149–S1150. 10.1016/j.annonc.2020.08.2245. [DOI] [Google Scholar]
- O’Shaughnessy J.; Punie K.; Oliveira M.; Lynce F.; Tolaney S. M.; Dalenc F.; Sharma P.; Tsai M. L.; Bardia A.; Cortes J.; et al. Assessment of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) cohort by agent in the phase 3 ASCENT study of patients (pts) with metastatic triple-negative breast cancer (mTNBC). Journal of Clinical Oncology 2021, 39, 1077–1077. 10.1200/JCO.2021.39.15_suppl.1077. [DOI] [Google Scholar]
- Li J.; Wang R.; Gao J. Novel anticancer drugs approved in 2020. Drug Discov Ther 2021, 15, 44–47. 10.5582/ddt.2021.01013. [DOI] [PubMed] [Google Scholar]
- Kaplon H.; Reichert J. M. Antibodies to watch in 2021. MAbs 2021, 13, 1860476. 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura N.; Sento S.; Yoshizawa Y.; Sasabe E.; Kudo Y.; Yamamoto T. Current Trends and Future Prospects of Molecular Targeted Therapy in Head and Neck Squamous Cell Carcinoma. International journal of molecular sciences 2021, 22, 240. 10.3390/ijms22010240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognetti D. M.; Johnson J. M.; Curry J. M.; Mott F.; Kochuparambil S. T.; McDonald D.; Fidler M. J.; Stenson K.; Vasan N. R.; Razaq M.; et al. Results of a phase 2a, multicenter, open-label, study of RM-1929 photoimmunotherapy (PIT) in patients with locoregional, recurrent head and neck squamous cell carcinoma (rHNSCC). Journal of Clinical Oncology 2019, 37, 6014–6014. 10.1200/JCO.2019.37.15_suppl.6014. [DOI] [Google Scholar]
- Jiang J.; Li S.; Shan X.; Wang L.; Ma J.; Huang M.; Dong L.; Chen F. Preclinical safety profile of disitamab vedotin: a novel anti-HER2 antibody conjugated with MMAE. Toxicol. Lett. 2020, 324, 30–37. 10.1016/j.toxlet.2019.12.027. [DOI] [PubMed] [Google Scholar]
- Peng Z.; Liu T.; Wei J.; Wang A.; He Y.; Yang L.; Zhang X.; Fan N.-F.; Luo S.; Gong J.; et al. A phase II study of efficacy and safety of RC48-ADC in patients with locally advanced or metastatic HER2-overexpressing gastric or gastroesophageal junction cancers. Journal of Clinical Oncology 2020, 38, 4560–4560. 10.1200/JCO.2020.38.15_suppl.4560. [DOI] [Google Scholar]
- Sheng X.; He Z.; Han W.; Zhou A.-P.; Luo H.; Shi Y.; Hu C.; Liu Z.; Guo H.; Yao X.; et al. An open-label, single-arm, multicenter, phase II study of RC48-ADC to evaluate the efficacy and safety of subjects with HER2 overexpressing locally advanced or metastatic urothelial cancer (RC48-C009). Journal of Clinical Oncology 2021, 39, 4584–4584. 10.1200/JCO.2021.39.15_suppl.4584. [DOI] [Google Scholar]
- Alley S. C.; Harris J. R.; Cao A.; Heuvel E. G.-v. d.; Velayudhan J.; Satijn D.; Verploegen S.; Dominguez T.; Breij E. C. Abstract 221: Tisotumab vedotin induces anti-tumor activity through MMAE-mediated, Fc-mediated, and Fab-mediated effector functions in vitro. Cancer research 2019, 79, 221–221. 10.1158/1538-7445.AM2019-221. [DOI] [Google Scholar]
- Liu Y.; Jiang P.; Capkova K.; Xue D.; Ye L.; Sinha S. C.; Mackman N.; Janda K. D.; Liu C. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer research 2011, 71, 6492–6502. 10.1158/0008-5472.CAN-11-1145. [DOI] [PubMed] [Google Scholar]
- Coleman R. L.; Lorusso D.; Gennigens C.; González-Martín A.; Randall L.; Cibula D.; Lund B.; Woelber L.; Pignata S.; Forget F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2021, 22, 609–619. 10.1016/S1470-2045(21)00056-5. [DOI] [PubMed] [Google Scholar]
- Ab O.; Whiteman K. R.; Bartle L. M.; Sun X.; Singh R.; Tavares D.; LaBelle A.; Payne G.; Lutz R. J.; Pinkas J.; et al. IMGN853, a Folate Receptor-α (FRα)–Targeting Antibody–Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRα-Expressing TumorsAnti-FRα Maytansinoid ADC for FRα-Positive Tumors. Molecular cancer therapeutics 2015, 14, 1605–1613. 10.1158/1535-7163.MCT-14-1095. [DOI] [PubMed] [Google Scholar]
- FDA grants accelerated approval to mirvetuximab soravtansine-gynx for FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or peritoneal cancer; U.S. FDA, 2022. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-mirvetuximab-soravtansine-gynx-fra-positive-platinum-resistant (accessed 06-15-2023).
- Hoimes C. J.; Flaig T. W.; Milowsky M. I.; Friedlander T. W.; Bilen M. A.; Gupta S.; Srinivas S.; Merchan J. R.; McKay R. R.; Petrylak D. P.; et al. Enfortumab Vedotin Plus Pembrolizumab in Previously Untreated Advanced Urothelial Cancer. Journal of Clinical Oncology 2023, 41, 22–31. 10.1200/JCO.22.01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killmurray C.Datopotamab Deruxtecan/Pembrolizumab Combo Show Anti-tumor Activity in Advanced NSCLC. Targeted Oncology; MJH Life Sciences, 2023. https://www.targetedonc.com/view/datopotamab-deruxtecan-pembrolizumab-combo-show-anti-tumor-activity-in-advanced-nsclc (accessed 09-22-2023).
- Goto Y.; Su W.-C.; Levy B. P.; Rixe O.; Yang T.-Y.; Tolcher A. W.; Lou Y.; Zenke Y.; Savvides P.; Felip E.; et al. TROPION-Lung02: Datopotamab deruxtecan (Dato-DXd) plus pembrolizumab (pembro) with or without platinum chemotherapy (Pt-CT) in advanced non-small cell lung cancer (aNSCLC). Journal of Clinical Oncology 2023, 41, 9004–9004. 10.1200/JCO.2023.41.16_suppl.9004. [DOI] [Google Scholar]
- McNulty R.First-Line Sacituzumab Govitecan Plus Pembrolizumab Shows Promising Early Activity in NSCLC. AJMC; MJH Life Sciences, 2023. https://www.ajmc.com/view/first-line-sacituzumab-govitecan-plus-pembrolizumab-shows-promising-early-activity-in-nsclc (accessed 09-22-2023).
- Gilead’s Phase 2 EVOKE-02 Study of Trodelvy® (sacituzumab govitecan-hziy) in Combination With KEYTRUDA® (pembrolizumab) Demonstrates Promising Clinical Activity in First-Line Metastatic Non-Small Cell Lung Cancer; Gilead, 2023. https://www.gilead.com/news-and-press/press-room/press-releases/2023/9/gileads-phase-2-evoke02-study-of-trodelvy-sacituzumab-govitecanhziy-in-combination-with-keytruda-pembrolizumab-demonstrates-promising-clinica (accessed 09-22-2023).
- Colombo R.; Rich J. R. The therapeutic window of antibody drug conjugates: A dogma in need of revision. Cancer Cell 2022, 40, 1255–1263. 10.1016/j.ccell.2022.09.016. [DOI] [PubMed] [Google Scholar]
- Lowe D.The Mysteries of Antibody-Drug Conjugates. Science; AAAS, 2022. https://www.science.org/content/blog-post/mysteries-antibody-drug-conjugates (accessed 06-29-2023).
- Criscitiello C.; Morganti S.; Curigliano G. Antibody–drug conjugates in solid tumors: a look into novel targets. Journal of Hematology & Oncology 2021, 14, 20. 10.1186/s13045-021-01035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makawita S.; Meric-Bernstam F. Antibody-Drug Conjugates: Patient and Treatment Selection. American Society of Clinical Oncology Educational Book 2020, 105–114. 10.1200/EDBK_280775. [DOI] [PubMed] [Google Scholar]
- Tolcher A. W. Antibody drug conjugates: The dos and don’ts in clinical development. Pharmacol Ther 2022, 240, 108235. 10.1016/j.pharmthera.2022.108235. [DOI] [PubMed] [Google Scholar]
- Ventana signs companion diagnostic agreement with international biotech for antibody drug conjugate program. Fierce Pharma; Questex LLC, 2014. https://www.fiercepharma.com/pharma/ventana-signs-companion-diagnostic-agreement-international-biotech-for-antibody-drug (accessed 05-03-2023).
- Companion Diagnostics; Novodiax, Inc., 2021. https://www.novodiax.com/companion-diagnostics/ (accessed 05-03-2023).
- Guo Y.; Wang Y.; Peng X.; Coukos G.; Li C. Abstract NT-099: NOVEL ANTIBODY DRUG CONJUGATE (ADC) AND COMPANION DIAGNOSTICS AGAINST CD248+ CANCER. Clin. Cancer Res. 2019, 25, NT-099-NT-099. 10.1158/1557-3265.OVCASYMP18-NT-099. [DOI] [Google Scholar]
- Bao G.; Tang M.; Zhao J.; Zhu X. Nanobody: a promising toolkit for molecular imaging and disease therapy. EJNMMI Research 2021, 11, 6. 10.1186/s13550-021-00750-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W.; Ding C.; Zheng D.; Ma X.; Yi L.; Tong X.; Wu C.; Xue C.; Yu Y.; Zhou Q. Nanobody Conjugates for Targeted Cancer Therapy and Imaging. Technol. Cancer Res. Treat 2021, 20, 153303382110101. 10.1177/15330338211010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Mo Y.; Tang M.; Shen J.; Qi Y.; Zhao W.; Huang Y.; Xu Y.; Qian C. Bispecific Antibodies: From Research to Clinical Application. Frontiers in Immunology 2021, 12, 626616. 10.3389/fimmu.2021.626616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y.; Nam S. M.; Moon A. Antibody-drug conjugates and bispecific antibodies targeting cancers: applications of click chemistry. Arch Pharm. Res. 2023, 46, 131–148. 10.1007/s12272-023-01433-6. [DOI] [PubMed] [Google Scholar]
- Shim H. Bispecific Antibodies and Antibody-Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. 10.3390/biom10030360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Galisteo A.; Compte M.; Álvarez-Vallina L.; Sanz L. When three is not a crowd: trispecific antibodies for enhanced cancer immunotherapy. Theranostics 2023, 13, 1028–1041. 10.7150/thno.81494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.