

Abstract
Maternal consumption of a Western-style diet (mWD) during pregnancy alters fatty acid metabolism and reduces insulin sensitivity in fetal skeletal muscle. The long-term impact of these fetal adaptations and the pathways underlying disordered lipid metabolism are incompletely understood. Therefore, we tested whether a mWD chronically fed to lean, insulin-sensitive adult Japanese macaques throughout pregnancy and lactation would impact skeletal muscle oxidative capacity and lipid metabolism in adolescent offspring fed a postweaning (pw) Western-style diet (WD) or control diet (CD). Although body weight was not different, retroperitoneal fat mass and subscapular skinfold thickness were significantly higher in pwWD offspring consistent with elevated fasting insulin and glucose. Maximal complex I (CI)-dependent respiration in muscle was lower in mWD offspring in the presence of fatty acids, suggesting that mWD impacts muscle integration of lipid with nonlipid oxidation. Abundance of all five oxidative phosphorylation complexes and VDAC, but not ETF/ETFDH, were reduced with mWD, partially explaining the lower respiratory capacity with lipids. Muscle triglycerides increased with pwWD; however, the fold increase in lipid saturation, 1,2-diacylglycerides, and C18 ceramide compared between pwCD and pwWD was greatest in mWD offspring. Reductions in CI abundance and VDAC correlated with reduced markers of oxidative stress, suggesting that these reductions may be an early-life adaptation to mWD to mitigate excess reactive oxygen species. Altogether, mWD, independent of maternal obesity or insulin resistance, results in sustained metabolic reprogramming in offspring muscle despite a healthy diet intervention.
Article Highlights
In lean, active adolescent offspring, a postweaning Western-style diet (pwWD) leads to shifts in body fat distribution that are associated with poorer insulin sensitivity.
Fatty acid–linked oxidative metabolism was reduced in skeletal muscles from offspring exposed to maternal Western-style diet (mWD) even when weaned to a healthy control diet for years.
Reduced oxidative phosphorylation complex I–V and VDAC1 abundance partially explain decreased skeletal muscle respiration in mWD offspring.
Prior exposure to mWD results in greater fold increase with pwWD in saturated lipids and bioactive lipid molecules (i.e. ceramide and sphingomyelin) associated with insulin resistance.
Introduction
Intrauterine exposure to maternal obesity, diabetes, or a poor-quality diet (i.e., Westernized high-fat, high-sugar diet [WD]) during development is associated with increased risk for cardiometabolic diseases in youth including insulin resistance, nonalcoholic fatty liver disease, cardiovascular disease, and type 2 diabetes (1,2). Skeletal muscle is a principle regulator of insulin sensitivity and metabolic homeostasis, accounting for the majority of systemic fatty acid oxidation and insulin-mediated glucose disposal (3,4). As such, metabolic dysregulation in muscle, in part due to reductions in mitochondrial mass and function, is a primary contributor to the development and progression of metabolic diseases in adults (5–8). Specifically, skeletal muscle insulin resistance has been associated with reduced mitochondrial abundance, less oxidative phosphorylation (OXPHOS) capacity, blunted lipid oxidation, increased de novo synthesis of bioactive fatty acid species (e.g., diacylglycerides [DGs], ceramides) and/or greater reactive oxygen species (ROS) production in adults (3,9,10) with reduced mitochondrial function observed in overweight children (11). Similar markers of metabolic dysregulation including reduced oxidative capacity, altered gene expression, and development of insulin resistance have also been observed in skeletal muscle of adult offspring exposed to maternal obesity in rodents (12–15). Of concern, metabolic dysregulation is already found in mesenchymal stem cells isolated from umbilical cords of infants born to women with obesity (16). However, few studies have evaluated whether maternal (m)WD in the absence of obesity or insulin resistance induces changes in offspring skeletal muscle.
Previous studies in our established Japanese macaque model of WD-induced maternal obesity identified metabolic dysregulation in multiple tissues of fetal and juvenile offspring (17–23) including reduced insulin sensitivity in skeletal muscle (24) and decreased oxidative metabolism in fetal muscle (18). In this model, we have consistently observed that chronic WD consumption induces obesity and insulin resistance in the majority of, but not all, adult females (25). Leveraging this unique subset of lean and insulin-sensitive dams, we tested whether a WD chronically fed to lean, insulin-sensitive dams during pregnancy and lactation would result in a persistent impairment in offspring skeletal muscle OXPHOS capacity and lipid metabolism. We also evaluated whether weaning offspring exposed to mWD onto a healthy chow diet (postweaning [pw]CD) would attenuate programmed effects at 3 years of age (i.e., early adolescence).
Research Design and Methods
Animals
All animal procedures were conducted under regulatory compliance at the Oregon National Primate Research Center (ONPRC) and Oregon Health & Science University, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. Experiments were designed and reported with reference to the Animals in Research: Reporting In Vivo Experiments (ARRIVE) guidelines (26).
Experimental Design
Adult Japanese macaques were housed in indoor/outdoor pens and fed a CD (15% of calories from fat primarily from soybeans and corn, monkey diet no. 5000; Purina Mills) or WD (37% of calories from fat primarily from corn oil, egg, and animal fat, no. 5LOP, TAD Primate Diet; TestDiet and Purina Mills) ad libitum. Carbohydrate content differed between diets, with sucrose and fructose constituting 19% of WD but only 3% of CD. Females consumed WD for 1–3 years prior to and during the pregnancy from which offspring were studied. Maternal prepregnancy demographics are presented by offspring cohort in Supplementary Table 1. More extensive phenotyping of the adult female macaques has previously been described (27). All births were singleton and delivered vaginally after spontaneous labor. Offspring remained in their home colony until weaning at ∼7–8 months of age, when they were grouped with 6–10 similarly aged juveniles from both maternal diet groups and 1–2 adult females. These new social housing groups were fed either CD or WD.
Offspring from 17 mCD dams and 17 mWD dams were included in this study. The offspring groups include 13 mCD/pwCD (8 female [F], 5 male [M]), 6 mCD/pwWD (2 F, 4 M), 13 mWD/pwCD (5 F, 8 M), and 9 mWD/pwWD (5 F, 4 M). No more than two offspring from the same dam were included in any offspring group per analysis. If two offspring from the same dam were included in the same group, offspring were of the opposite sex. Offspring sex is indicated in figures with use of different symbols.
Offspring Anthropometric Measures
Nonfat mass, fat mass, lean mass, and bone mineral content were measured with DEXA in offspring <1 month prior to necropsy as previously described (23). Offspring body mass, retroperitoneal fat pad mass, crown rump length, and subscapular skin fold thickness were measured at necropsy.
Offspring Intravenous Glucose Tolerance Testing
Intravenous glucose tolerance tests (i.v. GTT) were conducted within 2 months prior to offspring necropsy (at ∼36 months of age) as previously described (23,24). Baseline blood samples were obtained prior to the infusion and at 1, 3, 5, 10, 20, 40, and 60 min after infusion. Glucose was measured immediately with OneTouch Ultra blood glucose monitor (LifeScan), and the remainder of the blood was kept in heparinized tubes on ice for insulin measurement. After centrifugation, samples were stored at −80°C until assayed. Insulin measurements were performed by the Endocrine Technologies Core at ONPRC using a chemiluminescence-based automatic clinical platform (cobas e 411; Roche Diagnostics, Indianapolis, IN). HOMA of insulin resistance (HOMA-IR) was calculated from fasting insulin and glucose values with the following formula: (insulin (µU/L) * glucose (mg/dL))/405.
Offspring Activity Monitoring
Physical activity was continuously monitored in group-housed offspring with Actical accelerometers (Mini Mitter, Bend, OR) affixed to loose-fitting plastic collars (Primate Products, Miami, FL) as previously described (28). These monitors record the total number of activity counts, detected as changes in acceleration in all directions per minute. Activity count data over a 1-month period preceding necropsy are reported as average counts per hour for 24-h and 12-h day and night activities. All data were collected in late spring or early summer to control potential seasonal variability.
Offspring Necropsy Collection
Animals were sacrificed as previously described (28–30) between 37 and 40 months of age. Blood was collected in appropriate tubes for later analysis of insulin, triglycerides, and cholesterol and stored at −80°C for batch analysis by Endocrine Technologies Core at ONPRC. Skeletal muscles including gastrocnemius (gastroc), soleus, vastus lateralis, and rectus femoris were rapidly dissected of fascia and portions were either flash frozen in liquid nitrogen or transferred to a biopsy preservation solution (BIOPS) and shipped overnight for respirometry experiments. Frozen muscle was stored at −80°C until analysis.
Protein Analysis
Frozen gastroc and soleus muscles (50–100 mg) were homogenized mechanically in 0.6 mL buffer (24) with six 2.8-mm ceramic beads (VWR International) in a Bead Ruptor (OMNI International, Kennesaw, GA) at a rate of 6 m/s for 2 × 30 s intervals kept at 4°C with a cryo-cool instrument adaptor. Homogenate was then rotated for 1 h at 4°C on an orbital platform and then centrifuged at 13,000g for 15 min at 4°C. Protein concentration was determined with a BCA kit (Pierce and Thermo Fisher Scientific). Protein abundance was analyzed by capillary immunoassay on a Wes instrument per manufacturer instructions (ProteinSimple, Bio-Techne; San Jose, CA) with 3 µL of 0.25 or 1.25 mg/mL of sample. Antibody concentrations were optimized and multiplexed with target protein abundance quantified with Compass software (ProteinSimple, Bio-Techne) and normalized to a loading control protein. For OXPHOS analysis, abundance of individual complexes was normalized to an internal fluorescent loading control. Equal loading of total protein in soleus and gastroc homogenate was also measured with Stain-Free technology (Bio-Rad Laboratories, Hercules, CA) (Supplementary Figs. 1 and 2). Traditional Western blot was used to measure HADHA and was normalized to GAPDH, as previously described (18). Data were analyzed with Image Lab 5.2 software (Bio-Rad Laboratories). Primary and secondary antibody information can be found in Supplementary Table 2.
Citrate Synthase Activity Assay
Frozen gastroc and soleus muscles (20 mg) were homogenized in 0.7 mL buffer and enzyme activity was measured by spectrophotometry as previously described (18).
Muscle Lipid Analysis
Frozen offspring gastroc (∼50 mg) was dissected of extramuscular adipose tissue and fascia, lyophilized, weighed, and homogenized in 0.9 mL high-performance liquid chromatography–grade water. Homogenate (0.75 mL) plus methyl tert-butyl, as an internal lipid standard, was added to 0.9 mL methanol, and lipid species were serially extracted (18,31,32). Skeletal muscle lipid abundance and composition were analyzed with liquid chromatography–tandem mass spectrometry as previously described (31,32).
Permeabilized Muscle Fiber Bundle Preparation and Respirometry
Mitochondrial respiratory function was measured in permeabilized muscle fiber bundles (PMFBs) with high-resolution respirometry with an Oxygraph-2k system (Oroboros Instruments, Innsbruck, Austria). Muscles fiber bundles (3–5 mg) were dissected from gastroc and soleus and then permeabilized with 30 μg/mL saponin in BIOPS for 30 min, washed, blotted dry, and weighed. All respirometry data were collected at 37°C in a superoxygenated environment (200–400 μmol/L O2), and two titration protocols were run in parallel to measure respiration with lipid and nonlipid substrates as previously described (18). Mitochondrial integrity was confirmed by measurement of respiratory responses to cytochrome c.
Gene Expression
RNA was isolated from frozen gastroc and soleus (25 mg) with Direct-zol RNA MiniPrep kits (Zymo Research, Irvine, CA). cDNA was synthesized from extracted RNA with qScript cDNA Synthesis Kit from Quantabio (Beverly, MA) in a thermal cycler (Eppendorf, Enfield, CT). Expression of target genes were measured with PerfeCTa SYBR Green FastMix (Quantabio) using a CFX384 PCR Detection System (Bio-Rad Laboratories). Gene expression was normalized to the geometric mean of three housekeeping genes, and fold change was calculated with ΔΔCt analysis (33). Primer sequence, efficiencies, genome of origin, and experimental conditions per target/control genes can be found in Supplementary Table 3.
Lipid Peroxidation
Malondialdehyde (MDA) was measured in gastroc (50 mg) with a commercially available kit (no. 700870; Cayman Chemical) according to the manufacturer’s protocol and as previously described (18). Briefly, sample reaction mixture was loaded in duplicate onto a 96-well plate with standards and measured fluorometrically. MDA concentration is expressed relative to protein content.
Protein Carbonylation
Protein carbonyl content in gastroc (∼40 mg) was measured with a commercially available kit (ab126287; Abcam) per the manufacturer’s instructions. Sample homogenate was loaded in duplicate onto a 96-well plate, and data were normalized to total protein content of reaction mixture with the BCA method.
Statistical Analysis
Individual-sample data points with group minimum, maximum, median, and interquartile range are graphed. We calculated sample size using variance from previously published fetal muscle respirometry data, a primary outcome for the current study. For detection of a medium effect size with α = 0.05 and 80% power, this study requires ∼11 offspring (n) for main effects (i.e., m diet or pw diet). Using a factorial design, we tested for interactions of sex by treatment [i.e., m or pw diet] in main outcome measures as previously described [34]) (Supplementary Tables 4 and 5). Sexes were combined when no interactive effect of sex was identified. In subsequent analysis, data were analyzed with a two-way ANOVA for fixed effects of m diet and pw diet, and the interaction (m diet × pw diet). When significant main effects or interactions were identified, a Sidak post hoc analysis was used to test for significance within subgroups. Significant main effects (P < 0.05) are listed above each graph. For post hoc analysis, carets (∧) indicate significant differences between m diets within the same pw diet group, while asterisks (*) indicate significant differences between pw diets within the same m diet group. An unpaired t test was used to compare data sets with only two groups. OXPHOS and VDAC protein abundance were correlated to oxidative stress markers (protein carbonylation and/or lipid peroxidation content) in the following groupings: all offspring, all mCD offspring, and all mWD offspring. All P values for OXPHOS (CI–CV and CI+III) protein abundance and lipid peroxidation were adjusted for multiple comparisons with Bonferroni correction (Supplementary Table 6). All analyses were completed with Prism 9.2 software (GraphPad).
Data and Resource Availability
All data files are available by request.
Results
Adolescent Offspring Physiology Is Altered by mWD and pwWD
At 3 years of age, offspring body weight, nonfat mass, and crown-rump length were not different by group (Table 1). Surprisingly, total fat mass was reduced in offspring consuming pwWD (Table 1). Despite less total fat mass, retroperitoneal fat mass and subscapular skinfold thickness were significantly higher with pwWD, mirroring shifts in body fat distribution typically associated with poorer insulin sensitivity (35). Average daily activity in mWD offspring was significantly greater than mCD, with mWD/pwWD having the highest activity counts (Table 1). There was also an observed increase in activity with pwWD in both groups that did not reach statistical significance (P = 0.08). These data suggest that greater physical activity levels may contribute to reduced total fat mass but do not protect against metabolically unfavorable visceral fat accumulation, especially in mWD offspring.
Table 1.
Adolescent offspring physiological measures and activity at 34 months
| Anthropometrics | CD/CD (n = 13) | CD/WD (n = 6) | WD/CD (n = 13) | WD/WD (n = 9) | m diet | pw diet | m × pw |
|---|---|---|---|---|---|---|---|
| Sex, n | 8 F/5 M | 2 F/4 M | 5 F/8 M | 5 F/4 M | |||
| Body mass (kg) | 6.0 ± 0.2 | 6.1 ± 0.4 | 6.3 ± 0.3 | 6.3 ± 0.3 | ns | ns | ns |
| Nonfat mass (kg) | 4.7 ± 0.2 | 4.8 ± 0.2 | 4.8 ± 0.2 | 5.0 ± 0.2 | ns | ns | ns |
| Fat mass (kg) | 0.94 ± 0.05 | 0.77 ± 0.10 | 0.91 ± 0.05 | 0.78 ± 0.07 | ns | 0.02 | ns |
| Crown rump (cm) | 49 ± 1.2 | 49 ± 1.3 | 49 ± 0.7 | 48 ± 01.4 | ns | ns | ns |
| Subscapular skinfold thickness (cm) | 3.5 ± 0.3 | 3.8 ± 0.3 | 3.1 ± 0.2 | 4.0 ± 0.3a* | ns | 0.03 | ns |
| Retroperitoneal fat mass (g) | 0.73 ± 0.12 | 2.55 ± 0.96a* | 1.25 ± 0.39 | 2.45 ± 0.41a* | ns | 0.001 | ns |
| Activity | CD/CD (n = 10) | CD/WD (n = 6) | WD/CD (n = 12) | WD/WD (n = 9) | m diet | pw diet | m × pw |
|---|---|---|---|---|---|---|---|
| Sex, n | 6 F/4 M | 2 F/4 M | 4 F/8 M | 5 F/4 M | |||
| Daily (24 h) (counts/h) | 353 ± 26 | 370 ± 17 | 393 ± 18 | 453 ± 21∧ | 0.009 | 0.08 | ns |
| Daytime (12 h) (counts/h) | 300 ± 22 | 350 ± 35 | 359 ± 23 | 385 ± 22 | 0.06 | ns | ns |
| Nighttime (12 h) (counts/h) | 57 ± 6 | 60 ± 5 | 66 ± 6 | 66 ± 10 | ns | ns | ns |
| Glucose metabolism | CD/CD (n = 13) | CD/WD (n = 6) | WD/CD (n = 13) | WD/WD (n = 9) | m diet | pw diet | m × pw |
|---|---|---|---|---|---|---|---|
| Fasting glucose (mg/dL) | 56 ± 4 | 59 ± 3 | 55 ± 3 | 55 ± 1 | ns | ns | ns |
| Fasting insulin (µU/mL) | 6.18 ± 1.22 | 18.70 ± 5.4*** | 6.00 ± 1.12 | 11.3 ± 1.8 | 0.08 | 0.0001 | 0.09 |
| Glucose AUC, zero (×103; a.u.) | 10.00 ± 0.35a | 8.46 ± 0.37* | 8.59 ± 0.43 | 8.24 ± 0.38 | ns | 0.03 | ns |
| Insulin AUC, zero (×103; a.u.) | 1.51 ± 0.17a | 2.19 ± 0.33 | 1.70 ± 0.18 | 2.26 ± 0.24 | ns | 0.008 | ns |
| Insulin AUC, baseline (×103; a.u.) | 1.03 ± 0.19a | 1.00 ± 0.17 | 1.44 ± 0.16 | 1.67 ± 0.21 | 0.009 | ns | ns |
| HOMA-IR | 0.7 ± 0.1 | 4.2 ± 1.8** | 0.5 ± 0.1 | 1.4 ± 0.3∧ | 0.05 | 0.0002 | 0.04 |
| Plasma lipids | CD/CD (n = 13) | CD/WD (n = 6) | WD/CD (n = 13) | WD/WD (n = 9) | m diet | pw diet | m × pw |
|---|---|---|---|---|---|---|---|
| Triglycerides (mg/dL) | 42 ± 5 | 53 ± 10 | 46 ± 9 | 33 ± 5 | ns | ns | ns |
| Cholesterol (mg/dL) | 123 ± 6 | 167 ± 9** | 130 ± 7 | 170 ± 9*** | ns | 0.000005 | ns |
| HDL (mg/dL) | 55 ± 2 | 88 ± 5**** | 60 ± 3 | 90 ± 4**** | ns | <0.000001 | ns |
| LDL (mg/dL) | 64 ± 4 | 81 ± 9 | 71 ± 6 | 83 ± 10 | ns | 0.05 | ns |
Body composition, fasting serum values, and activity data are means ± SEM. Glucose AUC was calculated either from zero or from fasting baseline during an i.v. GTT. a.u., arbitrary units; ns, no significant difference. Statistical significance was determined with two-way ANOVA. P values are listed for main effects of m diet, pw diet, and interactions (m × pw). Multiple comparisons following Sidak post hoc analysis are represented by asterisks, bold font highlights significant findings.
P < 0.05,
P < 0.01,
P < 0.001,
P < 0.0001, for significant differences between pw diet within the same m diet group and carets,
P < 0.01, for differences between m diet within same pw diet.
One animal missing from group measure.
Insulin sensitivity and glucose metabolism were impacted by both m and pw diet. Fasting glucose was not different, but fasting insulin was significantly increased by pwWD and to a greater extent in mCD/pwWD offspring (Table 1). During an i.v. GTT, total insulin area under the curve (AUC) was significantly higher in offspring consuming pwWD (main effect, pw diet, P = 0.008). However, when accounting for higher fasting insulin concentrations, we observed a main effect of m diet (P = 0.03) with higher insulin AUCs calculated from baseline in mWD offspring (Table 1), suggesting that mWD may increase peripheral insulin resistance as seen at younger ages (24) requiring greater insulin response. The higher insulin AUC measured may also account for the lower glucose AUC observed in offspring consuming the pwWD. Calculation of HOMA-IR indicates increased insulin resistance with pwWD that is higher in offspring from mCD compared with mWD. Lastly, fasting total cholesterol was significantly increased with pwWD, driven by increases in both HDL and LDL cholesterol (Table 1). There was no difference in offspring fasting triglycerides by m or pw diet.
Persistent Reduction in Muscle Oxidative Capacity in Offspring Exposed to mWD
Skeletal muscle mitochondrial function and/or abundance is strongly associated with systemic insulin sensitivity (8). We previously reported reduced OXPHOS in PMFBs and differentiated myocytes isolated from fetal skeletal muscle exposed to maternal obesity and mWD (18). Here, we examined whether these adaptations persist into adolescence. To address potential muscle-specific differences, we interrogated substrate-specific respiration (i.e., with and without lipids) in the presence of saturating ADP in both the soleus, a highly aerobic muscle with majority type I fibers, and the gastroc, a mixed fiber–type muscle. In the soleus, rates of fatty acid oxidation with palmitoylcarnitine were impacted by m and pw diet (interaction, P = 0.04) such that in the case of mWD fatty acid oxidation was lower for pwCD versus pwWD—with the opposite pattern of response in mCD offspring (Fig. 1A and Supplementary Fig. 3A). However, after the addition of pyruvate (Fig. 1B), respiration was lower with mWD to a similar extent in both pw diet groups (main effect, m diet, P = 0.002). Similarly, maximal CI- and CI+CII-linked respirations were also lower in mWD offspring in the presence of palmitoylcarnitine, with greater reduction in mWD/pwCD offspring (Fig. 1C and D). However, in the absence of palmitoylcarnitine, maximal CI-linked respiration was not different between mWD and mCD (Fig. 1E and Supplementary Fig. 3B), indicating that the presence of long-chain acylcarnitine limited flux of convergent substrate oxidation pathways at CI in mWD groups (Fig. 1C and D). Indeed, when succinate was added (contributing electrons downstream of CI) the maximal CI+II-supported respiration rate in the absence of fatty acids was similar between mCD and mWD offspring and higher in mWD/pwWD than in mWD/pwCD (Fig. 1F). Finally, soleus citrate synthase activity, a marker of mitochondrial content, was not different by group (Fig. 1G), further suggesting that mWD impacts the capacity of skeletal muscle to integrate oxidation of fatty acids with other substrates rather than suppressing total oxidative capacity.
Figure 1.
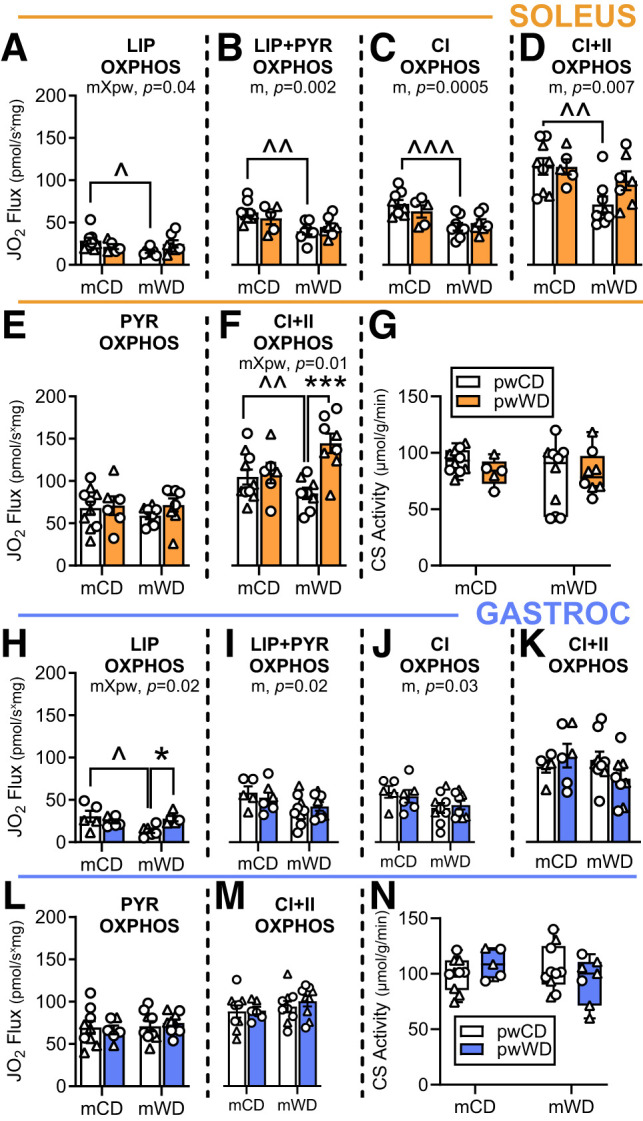
Substrate oxidation in skeletal muscle of adolescent offspring. Average group oxygen flux (JO2) (pmol/s * mg) was measured in PMFB with or without lipid (palmitoylcarnitine [LIP]) and normalized to tissue wet weight in soleus (orange, A–F) or gastroc (blue, H–M). In soleus, rate was measured in the presence of saturating ADP with serial additions of palmitoylcarnitine and malate (A), pyruvate (PYR) (B), glutamate for CI OXPHOS capacity (C), and succinate for maximal CI+CII OXPHOS capacity (D). Respiration rate without lipid was measured in the presence of saturating ADP after titrations of pyruvate and malate (E) and glutamate and succinate CI+CII OXPHOS capacity (F). These measures were repeated, in the same order as above, in the gastroc (blue, H–M). Citrate synthase (CS) activity (µmol/min * g) is shown in soleus (G) and gastroc (N). Respiratory flux and citrate synthase activity were analyzed with two-way ANOVA with Sidak multiple comparisons. P values for significant main effects of m or pw diet are listed above each graph. For post hoc analysis, carets (∧P < 0.05, ∧∧P < 0.01, ∧∧∧P < 0.001) indicate significant differences by m diet within the same pw diet group; asterisks (*P < 0.05, ***P < 0.001) indicate significant differences by pw diet within the same m diet group. Individual data points with group mean and SEM (A–F and H–M) or with the minimum, maximum, and median and interquartile range (G and N) are shown. M offspring are indicated by circles and F offspring by triangles. Sample size for each group by sex: mCD/pwCD, 5–6 F/4 M; mCD/pwWD, 2 F/4 M; mWD/pwCD, 3 F/5–6 M; mWD/pwWD, 4–5 F/4 M.
In the gastroc, fatty acid oxidation capacity was again lower in mWD offspring on the pwCD (Fig. 1H and Supplementary Fig. 3C). Like in the soleus, CI-linked respiration rates in the gastroc supported by subsequent titrations of pyruvate and glutamate were lower in mWD compared with mCD offspring in the presence of palmitoylcarnitine (main effect, m diet [Fig. 1I and J]) but similar across groups after addition of succinate (Fig. 1K), and with all substrates in the absence of palmitoylcarnitine (Fig. 1L and M and Supplementary Fig. 3D). Again, citrate synthase activity was also similar across groups (Fig. 1N). These data indicate that early-life programming by mWD decreases the capacity of muscle to oxidize a combination of metabolic substrates, particularly through CI, when high concentrations of fatty acid are present.
mWD Reduces OXPHOS Enzymes in Exposed Offspring
As a potential mechanism for reduced respiratory capacity in mWD-exposed offspring, we measured the enzymes responsible for mitochondrial OXPHOS. In the soleus, mWD had a main effect to reduce the abundance of subunits in all five OXPHOS complexes while pwWD had a main effect to increase complex abundance. The increase in OXPHOS complexes in the soleus parallels the observed increase in activity levels with pwWD. In pairwise comparisons, CI, CIV, and CV abundance were significantly reduced with mWD compared with mCD in offspring on the pwWD, while CII and CV were significantly reduced with mWD in both offspring pw diet groups (Fig. 2A–G). In contrast, there was no main effect of mWD on expression of electron-transferring flavoprotein (ETF) or ETF dehydrogenase (ETFDH), which coordinate electron transfer from fatty acid β-oxidation to the respiratory chain downstream of CI (Fig. 2F and G). In pairwise comparisons, pwWD increased CIV in mCD offspring and increased CV in both mCD and mWD offspring (Fig. 2D and E). Additionally, there was a main effect of pw diet to increase ETF and ETFDH, with a significant increase in ETF in the mWD group by pairwise comparison (Fig. 2F and G).
Figure 2.

Mitochondrial OXPHOS enzyme abundance in offspring skeletal muscle. Protein abundance of electron transport system and OXPHOS complexes was measured with Simple Western for CI (A), CII (B), CIII (C), and CIV (D), CV (E), ETF (F), and ETFDH (G) in offspring soleus (orange bars). Individual protein abundance was normalized to an internal fluorescent loading control, and CI–CV abundance was normalized to total protein. These measures were repeated in the gastroc (blue, I–P). Representative immunoassay images for soleus (H) and gastroc (P) are shown. Dashed horizontal lines indicate different contrast levels. Data were analyzed with two-way ANOVA for significant main effects of m or pw diet or interactions with Sidak multiple comparisons test. P values for significant main effects of m or pw diet are listed above each graph. For post hoc analysis, carets (∧P < 0.05, ∧∧P < 0.01) indicate significant differences by m diet within the same pw diet group and asterisks (*P < 0.05, **P < 0.01) indicate significant differences by pw diet within the same m diet group. Individual data points for offspring on pwCD or pwWD are shown along with group minimum, maximum, and median and interquartile range. M offspring are indicated by circles and F offspring by triangles. Sample size for each group by sex: mCD/pwCD, 6 F/3 M; mCD/pwWD, 2 F/3 M; mWD/pwCD, 3 F/6 M; mWD/pwWD, 5 F/3 M. For ETF and ETFDH n = mCD/pwCD, 4 F/3-4 M; mCD/pwWD, 2 F/3 M; mWD/pwCD, 3 F/3–4 M; mWD/pwWD, 3–5 F/2–3 M.
Similar to the soleus, there was a main effect of mWD to decrease the expression of all five OXPHOS complex proteins in the gastroc (Fig. 2I–M) but not ETF or ETFDH (Fig. 2N and O). Pairwise comparisons revealed lower expression of CII through CIV in mWD/pwCD groups (Fig. 2I–L) and reduced CV in both pw diet groups (Fig. 2M). In contrast to the soleus, there was no significant main effect of the pw diet on OXPHOS complexes or ETF/ETFDH proteins in the gastroc. Taken together, these data indicate that mWD reduces offspring skeletal muscle expression of respiratory chain complexes but not ETF/ETFDH. This shift may favor a greater flow of electrons from fatty acid β-oxidation to ubiquinone relative to CI and CII in mWD versus mCD offspring, perhaps explaining the partial inhibition of CI- and CI+II-dependent oxidative capacity during convergent palmitoylcarnitine oxidation.
Transcriptional Regulation of OXPHOS in Response to WD
To test whether reductions in OXPHOS proteins in mWD offspring were due to difference in gene transcription, we evaluated the expression of genes that code for subunits of CI–CV from both the mitochondrial and nuclear genomes in soleus and gastroc. In the soleus, COXII (CIV, mitochondrial genome) and UQCRC2 (CIII, nuclear genome) expression was greater with pwWD with post hoc analysis showing a significant increase in UQCRC2 between pw diet in mWD offspring (Fig. 3C and G). Only the nuclear encoded gene for CI, NDUFB8, had a main effect of m diet with a significant decrease in expression in mWD compared with mCD offspring on pwCD (Fig. 3E). In the gastroc, neither mitochondrial nor nuclear encoded OXPHOS genes are significantly impacted by m or pw diets (Supplementary Fig. 4). Although modest, the alterations in gene expression recapitulate patterns observed in the protein abundance of the mitochondrial complexes but do not fully explain either the downregulation of OXPHOS abundance, particularly in the gastroc, nor the subsequent reduction in oxidative capacities in offspring exposed to mWD and pwWD.
Figure 3.

OXPHOS gene expression in offspring soleus. Gene expression of key mitochondrial and nuclear encoded OXPHOS proteins was measured with quantitative PCR in offspring soleus (orange). Mitochondrial encoded genes measured were ND2 for CI (A), CYTB for CIII (B), COXII for CIV (C), and ATP8 for CV (D). Nuclear encoded genes measured were NDUFB8 for CI (E), SDHB for CII (F), UQCRC2 for CIII (G), COX5A for CIV (H), and ATP5F1A for CV (I). Individual gene expression was adjusted to the geometric mean for the expression of housekeeper genes, RPS15, 28S, and RPL13A, and are presented as fold change. Data were analyzed with two-way ANOVA for significant main effects of m or pw diet or interactions with Sidak multiple comparisons test. The P values for significant main effects are listed in each graph. For post hoc analysis, carets (∧∧P < 0.01) indicate significant differences by m diet within the same pw diet group and asterisks (*P < 0.05) indicate significant differences by pw diet within the same m diet group. Individual data points for offspring on pwCD or pwWD are shown along with group minimum, maximum, median, and interquartile range. M offspring are indicated by circles and F offspring by triangles. Sample size for each group by sex: mCD/pwCD, 5 F/3 M; mCD/pwWD, 2 F/3 M; mWD/pwCD, 4 F/3 M; mWD/pwWD, 3 F/3 M.
m and pw Diets Alter Muscle Acylcarnitine Accumulation but Not Lipid Transport or Oxidation Enzyme Abundance
Given the impact of m diet on lipid-associated oxidative capacity in offspring gastroc without reduced ETF or ETFDH abundance (Fig. 2N and O), we next measured upstream regulators of lipid trafficking and β-oxidation. There was no difference in the abundance of two key β-oxidation enzymes, very-long-chain acyl-CoA dehydrogenase (VLCAD) or hydroxyacyl-CoA dehydrogenase α (HADHA) (Fig. 4A and B), by m or pw diet. There was also no difference in the abundance of the inner or outer mitochondrial membrane (OMM) long-chain fatty acid transporters, CPT1β or CPT2, respectively (Fig. 4C and D). Although β-oxidation and lipid transport enzymes were not different, the abundance of intramuscular acylcarnitines was significantly different by pw and m diet, suggesting potential differences in activity and/or lipid transport. Specifically, medium- and long-chain acylcarnitines, C10, C12, C14, C16:1, C18, and C18:1, were much lower with a pwCD in mWD offspring (Supplementary Table 6). Short-chain acylcarnitines, C4 and C6, (main effect, m diet) as well as the unsaturated long-chain acylcarnitines, C18:2 and C18:3 (interactive effect), were also less concentrated with pwCD in mWD compared to mCD offspring.
Figure 4.

Lipid oxidation and signaling enzymes in offspring gastroc. Protein abundance of key regulators of lipid metabolism, including VLCAD (A), HADHA (B), CPT1β (C), and CPT2 (D), upstream signaling kinases, AMPKα2 (E), ACCα/β (F), phosphorylated (p)AMPK (T172) (G), and pACC (S79) (H), and PGC1α (I) was measured by immunoassay in offspring gastroc. Individual protein abundance was adjusted to GAPDH, vinculin, or α-tubulin and ratios were expressed relative to the mCD/pwCD group. J: Representative immunoassay images with loading controls. Dashed horizontal lines indicate different contrast levels. Data were analyzed with two-way ANOVA for significant main effects of m or pw diet or interactions with Sidak multiple comparisons test. P values for significant main effects are listed in each graph. For post hoc analysis, carets (∧P < 0.05, ∧∧P < 0.01) indicate significant differences by m diet within the same pw diet group. Individual data points for offspring on pwCD (open bars) or pwWD (blue bars) are shown along with group minimum, maximum, and median and interquartile range. M offspring are indicated by circles and F offspring by triangles. Sample size for each group by sex: mCD/pwCD, 5–6 F/4 M; mCD/pwWD, 2 F/3 M; mWD/pwCD, 3 F/5–6 M; mWD/pwWD, 5 F/3 M.
AMP-activated protein kinase (AMPK) and acetyl-CoA carboxylase (ACC), key enzymes involved in nutrient sensing and metabolic fuel use, showed a significant decrease in abundance in mWD compared with mCD offspring when consuming pwWD (Fig. 4E and F). Phosphorylation of AMPK and ACC were not different (Fig. 4G and H). Lastly, PGC1α abundance, a master regulator of mitochondrial metabolism and biogenesis, was not different across groups (Fig. 4I).
Prior Exposure to mWD Exacerbates Accumulation of Lipid Metabolites Associated With Muscle Insulin Resistance
Since the shift in the abundance and composition of intramyocellular lipids is associated with muscle insulin resistance (31,36,37), we examined the lipid composition and abundance in offspring gastroc. As expected, the abundance of individual triacylglyceride (TG) species and total TGs (Fig. 5A and B and Supplementary Table 7) increased with pwWD. The composition of the TG pool also changed with pwWD with an increased accumulation of more saturated and shorter-length TGs (Fig. 5A). This shift in TG saturation was exacerbated in mWD offspring with a fivefold greater accumulation with pwWD relative to pwCD animals (Fig. 5C). The abundance of total DGs, including both 1,3-DGs (derived from intramuscular TG lipolysis) and 1,2-DGs (a product of de novo synthesis and phospholipid degradation) (Fig. 5K), was dependent on both the m and pw diet (interaction, P = 0.04). Within mCD offspring, total DGs were lower with pwWD, while the pattern was reversed for mWD offspring (Fig. 5D). However, the percent of 1,2-DGs (Fig. 5E) and the percent of saturated 1,2-DGs (Fig. 5F) was significantly elevated by pwWD in both m groups. Interestingly, the greater difference in the pool of 1,2-DAGs and saturated 1,2-DAGS in mWD offspring was not driven by greater accumulation with pwWD but, rather, was due to reduced content in mWD/pwCD offspring, which have the lowest amount of these intramuscular lipid species (Fig. 5C, E, and F). Total ceramide, saturated ceramide, and total sphingomyelin (SPM) content were not different by m or pw diet (Supplementary Fig. 5A–C). However, saturated SPM, specifically SPM C18:0, and the upstream metabolite ceramide C18:0, a lipid species associated with insulin resistance (37), were most abundant in mWD offspring on pwWD (Fig. 5G–I). The abundance of ceramide C16:0 was not increased with either diet, indicating specificity rather than global enrichment of saturated fatty acids (Fig. 5J). Together, these data suggest that mWD leads to persistent change in lipid handling and metabolism in offspring muscle that is dependent on pw diet; mWD/pwCD offspring have decreased content and mWD/pwWD offspring have equal or higher amounts of saturated TGs and DGs relative to pw-matched controls. The increased accumulation of specific bioactive lipid species—namely, saturated SPM, SPM C18:0, and ceramide C18:0—in response to pwWD may contribute to an earlier decline in skeletal muscle insulin sensitivity (Fig. 5K).
Figure 5.

Measurement of intramuscular lipids in offspring skeletal muscle. Saturated TG species (A), total TG content (B) and saturated (Sat.) TG abundance (C) were measured in gastroc of 3Y offspring. DG abundance (D), percent of 1,2-DGs of total DGs (E), percent of saturated 1,2-DGs out of total 1,2-DGs (F), saturated SPM abundance (G), 18:0 SPM (H), 18:0 ceramide (I), and 16:0 ceramide (J) content were also measured in the same samples. Data were analyzed with two-way ANOVA with Sidak multiple comparisons test. P values for significant effects are listed in each graph. For post hoc analysis, asterisks (*P < 0.05, **P < 0.01, ***P < 0.001) indicate significant differences between pw diets within the same m diet group. Individual data points for offspring with minimum, maximum, and median and interquartile range per group are shown. M offspring are indicated by circles and F offspring by triangles. Sample size for each group by sex: mCD/pwCD, 5 F/3 M; mCD/pwWD, 2 F/3 M; mWD/pwCD, 2 F/6 M; mWD/pwWD, 5 F/3 M. K: An illustration summarizing offspring intramuscular lipid metabolism. TG, triacylglyceride; DG, diacylglyceride; palmCoA, palmitoyl CoA; PC, phosphatidylcholine; SPM, sphingomyelin; beta ox, beta oxidation; LC, long chain; TCA, tricarboxylic acid; Sat., saturated.
Oxidative Stress Linked to CI and VDAC1 Abundance in Muscle From Offspring With Prior Exposure to mWD
Reduced mitochondrial function is often associated with greater oxidative damage and impaired insulin sensitivity (9,38). We previously observed elevated markers of oxidative stress in fetal muscle from offspring of obese dams on mWD (18). Therefore, we assessed markers of oxidative stress in adolescent offspring skeletal muscle. Surprisingly, MDA, a marker of lipid peroxidation, was reduced with mWD (main effect, m diet) in gastroc (Fig. 6A). Similarly, protein carbonylation, a marker of oxidative stress that is not influenced by membrane lipid saturation, was also reduced with pwWD in mWD offspring (Fig. 6B). We next measured the voltage-dependent anion channel (VDAC1/2) abundance, an outer mitochondrial membrane transporter associated with ROS release (39). VDAC1/2 abundance was significantly lower in mWD offspring compared with pw diet–matched mCD offspring (Fig. 6C and D). Indeed, VDAC abundance was approximately fourfold lower in mWD/pwWD offspring relative to mCD/pwCD offspring muscle despite no differences observed in citrate synthase activity (Fig. 1N). VDAC2 was not different, indicating a VDAC1-specific downregulation in mWD offspring (Fig. 6D).
Figure 6.

Intramuscular oxidative stress is lower in mWD offspring. Lipid peroxidation (MDA) (A), protein carbonylation (2,4-dinitrophenylhydrazine [DNPH]) (B), and the protein abundance of VDAC1/2 (C) and VDAC2 (D) were measured in gastroc from 3 year old offspring. Protein abundance data were collected with Simple Western and adjusted to vinculin. Representative immunoassay images are shown (D). Data were analyzed by two-way ANOVA with Sidak multiple comparisons test. P values for significant effects are listed in each graph. For post hoc analysis, carets (∧∧P < 0.01, ∧∧∧∧P < 0.0001) indicate significant differences between m diets within the same pw diet group, while asterisks (*P < 0.05, ****P < .0001) indicate significant differences between pw diets within the same m diet group. M offspring are indicated by circles and F offspring by triangles. Sample size for A–C: mCD/pwCD, 5–6 F/4 M; mCD/pwWD, 2–3 F/3 M; mWD/pwCD, 2–3 F/5–6 M; mWD/pwWD, 4–5 F/2–4 M. Simple regression analysis was run for VDAC1/2 abundance and MDA across all offspring, n = 28 (E), and within m groups (F) or DNPH across all offspring, n = 26 (G), or within m groups (H). Correlations were also run between MDA and CI abundance in all offspring, n = 28 (I), or within m groups (J). Individual data points for offspring are shown with minimum, maximum, and median and interquartile range per group (A–C). Statistically significant correlations are indicated by red lines (E–J) with P value and correlation coefficient (R2) listed. rel., relative.
Correlation analysis was performed for assessment of relationships between markers of oxidative stress and VDAC and OXPHOS abundance in offspring muscle. We found a significant positive linear relationship between VDAC1/2 and MDA (R2 = 0.3, P < 0.002) or protein carbonylation concentration (R2 = 0.3, P < 0.009) across all offspring (Fig. 6E and G) with significant relationships maintained in mWD offspring (Fig. 6F and H). We also found a positive linear relationship between MDA and CI, CIII, and CI+CIII abundance, primary sites for ROS generation, and CV in all offspring (Fig. 6G and Supplementary Table 8). Only CI remained significant in analysis within the mWD offspring (Fig. 6J). These data suggest that reduced OXPHOS content and/or limiting ROS release via VDAC1 may reflect fetal adaptations to elevated ROS in skeletal muscle during development.
Discussion
Future risk for the development of obesity and cardiometabolic disease in youth, including type 2 diabetes, is increased by exposure to maternal obesity and diabetes in utero (40–42). These early exposures may “program” the offspring for metabolic dysfunction; however, the mechanisms and cellular targets mediating these outcomes are not known. Here, we examined the long-term metabolic impact of exposure to mWD during pregnancy and lactation, in the absence of either maternal obesity or insulin resistance, on 3 year old offspring body composition, glucose homeostasis, and skeletal muscle metabolism. We also evaluated the efficacy of a healthy pw diet intervention at ameliorating the effects of early-life exposure to mWD. In summary, offspring exposed to mWD during gestation and lactation weaned to a healthy pw diet had elevated insulin release during i.v. GTT despite a similar body composition compared to mCD offspring and higher physical activity. In skeletal muscle, offspring had significant reductions in oxidative metabolism in the presence of fatty acids concomitant with reduced OXPHOS complex abundance and VDAC1. Further exposure to the pwWD in mWD offspring revealed a greater change in the accumulation of saturated lipids and some bioactive lipid species associated with insulin resistance despite increases in visceral fat accruement similar to those of mCD offspring. The increased accumulation of saturated ceramides in skeletal muscle coupled with reduced oxidative capacity may contribute to worsening systemic insulin sensitivity and partitioning of lipids to adipose stores.
The coordination of mitochondrial oxidation in response to nutrient availability and energy demand (i.e., metabolic flexibility) decreases in parallel with the development of systemic insulin resistance (43,44) and metabolic dysfunction (6). We observed lower oxidative capacity in isolated soleus and gastroc muscle fibers from mWD offspring, regardless of pw diet, when provided a combination of fatty acid and pyruvate. Our data could not be explained by reduced mitochondrial content or decreased abundance of critical lipid trafficking or β-oxidation enzymes or ETF/ETFDH but may be linked to reduced OXPHOS complex abundance. Importantly, skeletal muscle from mWD offspring, including mWD offspring switched to a healthy diet, contained approximately one-half the volume of OXPHOS complexes like CI relative to controls. We propose that the observed loss in maximal CI- and CI+II-linked respiration in the presence of lipids may be due, in part, to greater flow of electrons from fatty acid β-oxidation to ubiquinone via ETF/ETFDH relative to electrons coming from CI and CII in mWD versus mCD offspring. Additionally, patients with CI deficiency have reduced [NAD+]-to-[NADH] ratios that also coincide with impaired β-oxidation (45). As both pathways for lipid and pyruvate oxidation are dependent on an adequate [NAD+]-to-[NADH] ratio to proceed, insufficient CI abundance would shift redox circuitry toward NADH excess and may also explain why flux is most limited when both pathways converge at CI.
Interestingly, with the exception of the CI nuclear gene NDUFB8 in soleus, reduced OXPHOS protein abundance was not related to decreased nuclear or mitochondrial gene expression. Furthermore, in soleus, a more oxidative muscle, pwWD increased the protein abundance of OXPHOS complex, even in the mWD offspring, suggesting an ability to respond to nutrient stresses, albeit starting at a lower baseline level. These baseline differences may be due to posttranscriptional mechanisms related to higher mitochondrial protein turnover and/or less synthesis.
Exposure to the pwWD revealed altered lipid handling in mWD offspring compared with control offspring also on the pwWD. Increased saturated intramyocellular lipid content is a strong predictor of muscle insulin resistance in sedentary adults and children with obesity (37,46,47) and is often associated with impaired lipid oxidation or lipid trafficking in muscle (48). A predominate and unexpected finding across our muscle lipid analysis was a reduction in lipid species accumulation in mWD offspring weaned to pwCD, suggesting reduced uptake or altered fatty acid metabolism. In both soleus and gastroc, reductions in fatty acid oxidation were greatest in this group (i.e., mWD/pwCD) despite no difference in the abundance of key fatty acid oxidation enzymes. However, the activity of CPT1β and/or the abundance of other fatty acid transporters may be responsible for the lower accumulation of fatty acids and fatty acid metabolites in mWD/pwCD muscle. At the molecular level, bioactive lipid metabolites, like 1,2-DG and ceramide, activate signaling cascades that impede insulin signaling transduction (49). The activation of canonical and/or atypical PKCs by elevated 1,2-DG to promote inhibitory phosphorylation of IRS proteins or by ceramide to inhibit Akt activation (50,51) and suppression of insulin signaling is well described in human and animal models of insulin resistance (52). Specifically, in total and saturated 1,2-DGs, there was a greater difference between pw diet treatments in mWD offspring driven by the lower baseline in pwCD muscle. However, whether the magnitude of change in 1,2-DGs (or other lipid species) with pwWD is as important as the total accumulation has not been investigated but may reflect a unique response associated with developmental programming. In contrast to the 1,2-DGs, exposure to mWD led to a greater accumulation of another saturated lipid species, ceramide C18:0, and its downstream metabolite SPM C18:0, in muscle in response to pwWD. Indeed, increased mitochondrial ceramide accumulation has been linked to reduced coenzyme Q levels and reduced electron transport system components and, subsequently, impaired mitochondrial function similar to that seen in skeletal muscle of mWD offspring (53). Importantly, our findings are consistent with an intracellular environment characterized in obesity and type 2 diabetes (31,36,37,54,55).
Increased ROS production and elevated oxidative damage to intracellular molecules are features of mitochondrial dysfunction (56,57). We previously reported increased ROS damage in fetal skeletal muscle, pancreas, and livers from obese mWD dams (17,18,58). Therefore, the absence of elevated markers of lipid peroxidation or protein carbonylation in skeletal muscle of mWD was unanticipated. Indeed, we observed lower levels of oxidative damage in mWD offspring on the pwWD. Lower levels of oxidative damage may result from decreased VDAC1 abundance, which functions in the formation of the mitochondrial permeability transition pore to allow ROS efflux and/or stimulate apoptosis (59). Thus, reduced VDAC may lower oxidative damage by trapping ROS within the mitochondria. Reduced CI and overall lower OXPHOS flux may also act as a countermeasure to ameliorate ROS production (60). Along these lines, we show strong correlations between ROS damage and VDAC1 abundance driven by mWD offspring. We also see a relationship between CI, CI+CIII, and CV and lipid peroxidation. Reduced VDAC1 and OXPHOS abundance may be an adaptation that reprograms ROS handling as a strategy to mediate the excessive oxidative stress previously observed in fetal skeletal muscle, albeit at the cost of mitochondrial health and oxidative efficiency. A higher antioxidant system may also be induced to sequester excess ROS, although this has not been explored here. Further work is required to determine the mechanisms that underlie changes in ROS handling and mitochondrial integrity.
Overall, we show that exposure to an mWD during pregnancy and lactation, even in the absence of maternal obesity and insulin resistance, is sufficient to reprogram offspring lipid handling and impair oxidative metabolism in skeletal muscle, a phenotype typically associated with metabolic disease states or the functional decline in muscle health with aging (1,61). Sex-specific difference in metabolic outcomes including insulin resistance and obesity have been identified in adult offspring exposed to maternal obesity (62). We postulate that these changes observed in cellular metabolism in our peripubertal animals will be exacerbated by future physiological stresses including weight gain, puberty, lack of physical activity, or a chronic pwWD, revealing the elevated and sex-specific risk of cardiometabolic diseases observed in adults from pregnancies complicated by poor maternal nutrition and obesity. Future studies will be aimed at interrogating regulators of cellular quality control processes contributing to downregulated OXPHOS abundance and function with exposure to mWD or obesity.
This article contains supplementary material online at https://doi.org/10.2337/figshare.24147576.
Article Information
Funding. This research was supported by National Institutes of Health (NIH) grants R24 DK090964 (to J.E.F., K.M.A., and P.K.), R01 DK128187 (K.M.A., P.K., and J.E.F.), R01 MH107508 (E.L.S.), and R01 DK089201 (K.M.A.) and by an NIH Center Grant (P51 OD011092) to ONPRC. This work was also supported by the Eugene and Clarissa Evonuk Memorial Graduate Fellowship (K.T.G.). The Endocrine Technologies Core at ONPRC is supported (in part) by NIH grant P51 OD011092 for operation of the ONPRC.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. S.R.W., M.G., E.L.S., K.M.A., P.K., J.E.F., and C.E.M. established and supported the experimental animal model. K.T.G., B.H., B.C.B., T.A.D., S.S., P.K., A.J.C., and C.E.M. collected and analyzed data. K.T.G. and C.E.M. drafted the manuscript. All authors provided critical feedback and reviewed the final manuscript. C.E.M. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 83rd Scientific Sessions of the American Diabetes Association, San Diego, CA, 23–26 June 2023.
Funding Statement
This research was supported by National Institutes of Health (NIH) grants R24 DK090964 (to J.E.F., K.M.A., and P.K.), R01 DK128187 (K.M.A., P.K., and J.E.F.), R01 MH107508 (E.L.S.), and R01 DK089201 (K.M.A.) and by an NIH Center Grant (P51 OD011092) to ONPRC. This work was also supported by the Eugene and Clarissa Evonuk Memorial Graduate Fellowship (K.T.G.). The Endocrine Technologies Core at ONPRC is supported (in part) by NIH grant P51 OD011092 for operation of the ONPRC.
References
- 1. Friedman JE. Developmental programming of obesity and diabetes in mouse, monkey, and man in 2018: where are we headed? Diabetes 2018;67:2137–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wankhade UD, Thakali KM, Shankar K. Persistent influence of maternal obesity on offspring health: mechanisms from animal models and clinical studies. Mol Cell Endocrinol 2016;435:7–19 [DOI] [PubMed] [Google Scholar]
- 3. DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009;32(Suppl. 2):S157–S163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol 1999;277:E1130–E1141 [DOI] [PubMed] [Google Scholar]
- 5. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950 [DOI] [PubMed] [Google Scholar]
- 6. Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell 2014;159:1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 2008;7:45–56 [DOI] [PubMed] [Google Scholar]
- 8. Goodpaster BH. Mitochondrial deficiency is associated with insulin resistance. Diabetes 2013;62:1032–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 2009;119:573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morino K, Petersen KF, Dufour S, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 2005;115:3587–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fleischman A, Kron M, Systrom DM, Hrovat M, Grinspoon SK. Mitochondrial function and insulin resistance in overweight and normal-weight children. J Clin Endocrinol Metab 2009;94:4923–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paulsen ME, Rosario FJ, Wesolowski SR, Powell TL, Jansson T. Normalizing adiponectin levels in obese pregnant mice prevents adverse metabolic outcomes in offspring. FASEB J 2019;33:2899–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saben JL, Boudoures AL, Asghar Z, et al. Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep 2016;16:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelly AC, J Rosario F, Chan J, Cox LA, Powell TL, Jansson T. Transcriptomic responses are sex-dependent in the skeletal muscle and liver in offspring of obese mice. Am J Physiol Endocrinol Metab 2022;323:E336–E353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Latouche C, Heywood SE, Henry SL, et al. Maternal overnutrition programs changes in the expression of skeletal muscle genes that are associated with insulin resistance and defects of oxidative phosphorylation in adult male rat offspring. J Nutr 2014;144:237–244 [DOI] [PubMed] [Google Scholar]
- 16. Boyle KE, Patinkin ZW, Shapiro ALB, et al. Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants. Mol Metab 2017;6:1503–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009;119:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCurdy CE, Schenk S, Hetrick B, et al. Maternal obesity reduces oxidative capacity in fetal skeletal muscle of Japanese macaques. JCI Insight 2016;1:e86612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wesolowski SR, Mulligan CM, Janssen RC, et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Mol Metab 2018;18:25–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thorn SR, Baquero KC, Newsom SA, et al. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes 2014;63:2702–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roberts VHJ, Pound LD, Thorn SR, et al. Beneficial and cautionary outcomes of resveratrol supplementation in pregnant nonhuman primates. FASEB J 2014;28:2466–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ma J, Prince AL, Bader D, et al. High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model. Nat Commun 2014;5:3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elsakr JM, Dunn JC, Tennant K, et al. Maternal Western-style diet affects offspring islet composition and function in a non-human primate model of maternal over-nutrition. Mol Metab 2019;25:73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Campodonico-Burnett W, Hetrick B, Wesolowski SR, et al. Maternal obesity and Western-style diet impair fetal and juvenile offspring skeletal muscle insulin-stimulated glucose transport in nonhuman primates. Diabetes 2020;69:1389–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris RA, Alcott CE, Sullivan EL, et al. Genomic variants associated with resistance to high fat diet induced obesity in a primate model. Sci Rep 2016;6:36123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kilkenny C, Browne W, Cuthill IC, Emerson M; NC3Rs Reporting Guidelines Working Group . Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 2010;160:1577–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elsakr JM, Zhao SK, Ricciardi V, et al. Western-style diet consumption impairs maternal insulin sensitivity and glucose metabolism during pregnancy in a Japanese macaque model. Sci Rep 2021;11:12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sullivan EL, Rivera HM, True CA, et al. Maternal and postnatal high-fat diet consumption programs energy balance and hypothalamic melanocortin signaling in nonhuman primate offspring. Am J Physiol Regul Integr Comp Physiol 2017;313:R169–R179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rivera HM, Kievit P, Kirigiti MA, et al. Maternal high-fat diet and obesity impact palatable food intake and dopamine signaling in nonhuman primate offspring. Obesity (Silver Spring) 2015;23:2157–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Grant WF, Nicol LE, Thorn SR, Grove KL, Friedman JE, Marks DL. Perinatal exposure to a high-fat diet is associated with reduced hepatic sympathetic innervation in one-year old male Japanese macaques. PLoS One 2012;7:e48119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bergman BC, Brozinick JT, Strauss A, et al. Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 2016;59:785–798 [DOI] [PubMed] [Google Scholar]
- 32. Bergman BC, Perreault L, Strauss A, et al. Intramuscular triglyceride synthesis: importance in muscle lipid partitioning in humans. Am J Physiol Endocrinol Metab 2018;314:E152–E164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dymond JS. Explanatory chapter: quantitative PCR. Methods Enzymol 2013;529:279–289 [DOI] [PubMed] [Google Scholar]
- 34. Garcia-Sifuentes Y, Maney DL. Reporting and misreporting of sex differences in the biological sciences. eLife 2021;10:e70817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruiz-Alejos A, Carrillo-Larco RM, Miranda JJ, Gilman RH, Smeeth L, Bernabé-Ortiz A. Skinfold thickness and the incidence of type 2 diabetes mellitus and hypertension: an analysis of the PERU MIGRANT study. Public Health Nutr 2020;23:63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bergman BC, Hunerdosse DM, Kerege A, Playdon MC, Perreault L. Localisation and composition of skeletal muscle diacylglycerol predicts insulin resistance in humans. Diabetologia 2012;55:1140–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perreault L, Newsom SA, Strauss A, et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 2018;3:e96805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Di Meo S, Iossa S, Venditti P. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol 2017;233:R15–R42 [DOI] [PubMed] [Google Scholar]
- 39. Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem 2003;278:5557–5563 [DOI] [PubMed] [Google Scholar]
- 40. Reynolds RM, Allan KM, Raja EA, et al. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ 2013;347:f4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dabelea D, Mayer-Davis EJ, Lamichhane AP, et al. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: the SEARCH Case-Control Study. Diabetes Care 2008;31:1422–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaseva N, Vääräsmäki M, Sundvall J, et al. Gestational diabetes but not prepregnancy overweight predicts for cardiometabolic markers in offspring twenty years later. J Clin Endocrinol Metab 2019;104:2785–2795 [DOI] [PubMed] [Google Scholar]
- 43. Antoun G, McMurray F, Thrush AB, et al. Impaired mitochondrial oxidative phosphorylation and supercomplex assembly in rectus abdominis muscle of diabetic obese individuals. Diabetologia 2015;58:2861–2866 [DOI] [PubMed] [Google Scholar]
- 44. Galgani JE, Moro C, Ravussin E. Metabolic flexibility and insulin resistance. Am J Physiol Endocrinol Metab 2008;295:E1009–E1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Venizelos N, von Döbeln U, Hagenfeldt L. Fatty acid oxidation in fibroblasts from patients with defects in beta-oxidation and in the respiratory chain. J Inherit Metab Dis 1998;21:409–415 [DOI] [PubMed] [Google Scholar]
- 46. Savage DB, Watson L, Carr K, et al. Accumulation of saturated intramyocellular lipid is associated with insulin resistance. J Lipid Res 2019;60:1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weiss R, Dufour S, Taksali SE, et al. Prediabetes in obese youth: a syndrome of impaired glucose tolerance, severe insulin resistance, and altered myocellular and abdominal fat partitioning. Lancet 2003;362:951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab 2000;279:E1039–E1044 [DOI] [PubMed] [Google Scholar]
- 49. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev 2018;98:2133–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol 2003;23:7794–7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bandet CL, Tan-Chen S, Ali-Berrada S, et al. Ceramide analog C2-cer induces a loss in insulin sensitivity in muscle cells through the salvage/recycling pathway. J Biol Chem 2023;299:104815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Szendroedi J, Yoshimura T, Phielix E, et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 2014;111:9597–9602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Diaz-Vegas A, Madsen S, Cooke KC, et al. Mitochondrial electron transport chain, ceramide and coenzyme Q are linked in a pathway that drives insulin resistance in skeletal muscle. 12 March 2023 [preprint]. bioRxiv 2023.03.10.532020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Straczkowski M, Kowalska I, Nikolajuk A, et al. Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 2004;53:1215–1221 [DOI] [PubMed] [Google Scholar]
- 55. Tonks KT, Coster AC, Christopher MJ, et al. Skeletal muscle and plasma lipidomic signatures of insulin resistance and overweight/obesity in humans. Obesity (Silver Spring) 2016;24:908–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab 2012;23:142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A 2001;98:2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nicol LE, Grant WF, Comstock SM, et al. Pancreatic inflammation and increased islet macrophages in insulin-resistant juvenile primates. J Endocrinol 2013;217:207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chaudhuri AD, Choi DC, Kabaria S, Tran A, Junn E. MicroRNA-7 regulates the function of mitochondrial permeability transition pore by targeting VDAC1 expression. J Biol Chem 2016;291:6483–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pryde KR, Taanman JW, Schapira AH. A LON-ClpP proteolytic axis degrades complex I to extinguish ROS production in depolarized mitochondria. Cell Rep 2016;17:2522–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mercken EM, Capri M, Carboneau BA, et al. Conserved and species-specific molecular denominators in mammalian skeletal muscle aging. NPJ Aging Mech Dis 2017;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sandovici I, Fernandez-Twinn DS, Hufnagel A, Constância M, Ozanne SE. Sex differences in the intergenerational inheritance of metabolic traits. Nat Metab 2022;4:507–523 [DOI] [PubMed] [Google Scholar]