

Abstract
Three‐related cats were evaluated for a history of short‐strided gait and temporary recumbency after startle. Neurological examination, electromyography (EMG), muscle biopsies, and a chloride voltage‐gated channel 1 (CLCN1) molecular study were performed. Clinically, all 3 cats presented myotonia with warm‐up phenomenon and myotonic discharges during EMG examination. Muscle biopsies showed normal muscle architecture and variation in the diameter of myofiber size with the presence of numerous hypertrophic fibers. The molecular study revealed a missense variant (c.991G>C, p.Ala331Pro) in exon 9 of the CLCN1 gene, responsible for the first chloride channel extracellular loop. This mutation was screened in 104 control phenotypically normal unrelated cats, and all were wildtype. The alanine at this position is conserved in ClC‐1 (chloride channel protein 1) in different species, and 2 mutations at this amino acid position are associated with human myotonia. This is the third CLCN1 mutation described in the literature associated with hereditary myotonia in cats and the first in domestic animals located in an extracellular muscle ClC‐1 loop.
Keywords: electromyography, feline, neurology, neurophysiology, physiology, physiology‐skeletal muscle
Abbreviations
- AST
aspartate aminotransferase
- CBC
complete blood count
- cDNA
ribonucleic acid
- CIC1
chloride channel protein 1
- CK
creatine kinase
- CLCN1
chloride voltage‐gated channel
- DNA
deoxyribonucleic acid
- EDTA
ethylenediamine tetraacetic acid
- EMG
electromyography
- H&E
hematoxylin and eosin stain
- HM
hereditary myotonia
- mRNA
messenger ribonucleic acid (mRNA)
- PCR
polymerase chain reaction
- RNA
ribonucleic acid
- SNP
single nucleotide polymorphisms
1. INTRODUCTION
Hereditary nondystrophic myotonia associated with chloride voltage‐gated channel 1 (CLCN1) gene mutations occurs in goats, 1 dogs, 2 , 3 , 4 , 5 , 6 horses, 7 buffalos, 8 sheep, 9 and pigs. 10 Myotonia occurs in cats, 11 , 12 , 13 and a mutation associated with this disease in this species is identified. 14 , 15 Here, we describe a clinical and molecular study in 3 cats with hereditary myotonia.
2. CASE REPORT
All procedures were approved by the Institutional Animal Care and Use Committee (0089/2021—CEUA) of São Paulo State University (UNESP). All methods were performed in accordance with the guidelines and regulations of CEUA. Authorization from the owner of the cats was obtained for the genetic study and images used in this manuscript.
3. HISTORY AND CLINICAL EXAMINATION
Three young mixed‐breed cats from the same litter were presented for clinical evaluation with the complaint of stiff gait and recumbency after startle. During the first examination, they were 4 months old: 2 intact males (Cat 1 and Cat 2) and 1 female (Cat 3). The clinical manifestations had been noticed 1 month earlier by the owners who also reported abnormal vocalization patterns and difficulty in cleaning the caudal region of the body (genital area and pelvic limbs). Occurrence of vomiting, regurgitation and difficulty swallowing were denied, as were learning difficulties.
The 3 cats could vocalize only weakly (sometimes just miming meowing), and Cat 2 occasionally kept his tongue exposed and developed transient respiratory stridor during handling. Physical examinations of the 3 cats revealed enlargement of the cervical and proximal appendicular muscles and resistance to opening and then closing the mouth (Video S1). After we forced the cat's mouth open, each presented a delay to close it that became shorter as the forced attempts to open the mouth were repeated (warm‐up effect present). These clinical changes were severe in Cat 1, moderate in Cat 2 and mild in Cat 3. Cat 1 and Cat 2 had moderate periodontal disease, gingivitis, and halitosis. Cat 1 also had a broken upper canine. During neurological examination, the 3 cats exhibited normal consciousness and behavior, but they were reluctant to move. They had short steps and stiff gait. All locomotion abnormalities presented phenotypically varying intensities and became less evident with muscle warm‐up. When stimulated to jump or exposed to a sudden auditory stimulus, the cats contracted the muscles and then fell into lateral recumbency (startle response), a position from which they only came out seconds later (Video S2). There was also a warm‐up effect for these tests. The presence of prolonged blepharospasm and protrusion of the third eyelid in response to digital stimulation of the palpebral fissure were observed (Video S3). No orthopedic abnormalities were identified, and the remaining findings from the physical and neurological examinations, including assessments of posture, postural reaction, spinal reflexes, cranial nerves, and sensation, were all within normal ranges. The cats were re‐examined when they were 6 and 11 months old, and all the signs remained stable. The cats were reluctant to climb or descend obstacles. Interestingly, the cats showed an exaggerated reaction to stimulation in the dorsal region of the lumbar region, resulting in nibbling of the paws or surfaces that were supported (Video S4).
Given the observed muscular hypertrophy, improvement of muscle stiffness with exercise (demonstrating a “warm‐up” phenomenon), startle response, and absence of other neurological signs, the presence of a skeletal muscle channelopathy was suspected. Specifically, hereditary myotonia associated with CLCN1 abnormality emerged as a highly plausible explanation, particularly in light of the absence of episodic weakness or muscle atrophy. To confirm the diagnosis of hereditary myotonia in these feline cases and to rule out other potential muscular abnormalities (such as myopathy presenting inclusions, nemaline myopathy, or glycogen storage disease), a comprehensive evaluation was conducted, which involved complete blood count (CBC), serum biochemical profile, electromyography, muscle biopsies, and molecular evaluation of CLCN1.
CBC and serum biochemical profile (BS‐200E‐Mindray, Shenzen, China) results (performed when cats were 11 months old), including creatine kinase (CK) activity (mean 376 IU/L), were within normal limits, 16 except for a slight increase in AST. At the time of submission of this report, the 3 cats were 2 years old and had shown no change in their clinical condition. The kittens' mother was 15 years old when she gave birth to the litter, and there is no clear information about the father, as the mother has access to the street and neighbor's cats.
Electromyography (EMG) examinations were performed in all 3 cats in the standing position and after sedation. The proximal muscles of the pelvic and thoracic limbs and tongue were evaluated using concentric needle electrodes with electroneuromyography equipment (Neuromap ENMG/PE, Neurotec, Minas Gerais, Brazil). The tracing of the waveform, frequency, amplitude, and sound produced were documented. All 3 cats presented myotonic discharges and characteristic sounds (Figure 1; Audio S1) during EMG examination.
FIGURE 1.
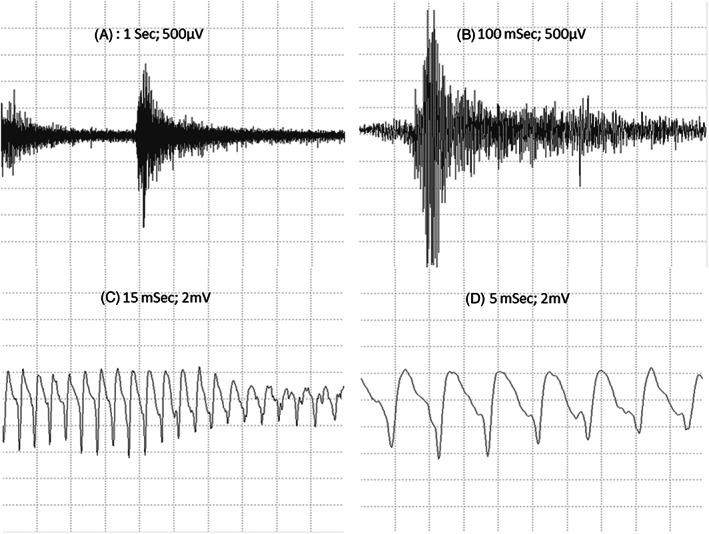
(A and B) Myotonic discharges (increasing and decreasing amplitude and frequency). (C and D) Sustained run of positive waves (initial sharp positivity followed by a slow negative component) at rest.
Immediately after the EMG examination and during anesthesia, biceps femoris muscle biopsy sections of Cat 1 and Cat 2 were obtained for histopathological evaluation (fixed in 10% buffered formalin, embedded in wax and stained with hematoxylin and eosin) and molecular analysis (snap frozen in liquid nitrogen). A sample from 1 control animal (with no previous history of neuromuscular abnormalities was also obtained postmortem for molecular study). Muscle biopsies showed a regular muscle architecture and variation in the diameter of myofiber size (ranging from 40 to 125 μm) with the presence of numerous hypertrophic fibers. There were no histological evidence of inflammation, necrosis, fibrosis central nucleation, sarcoplasmic inclusions or splitting fibers (Figure 2).
FIGURE 2.
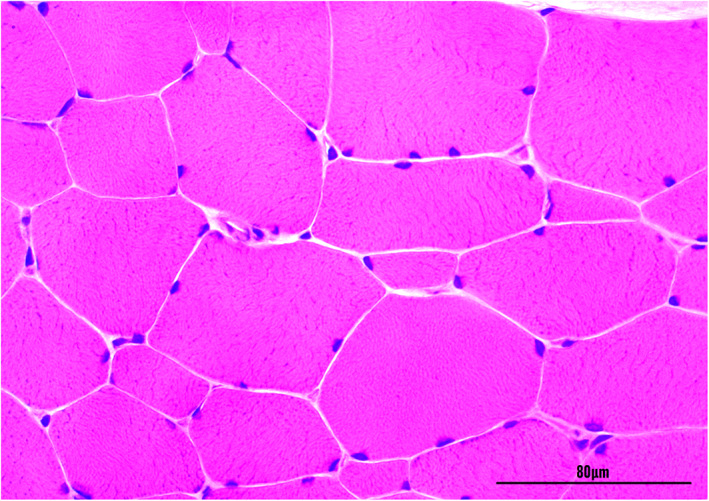
Transverse section of the biceps femoris muscle of cat 1 with hereditary myotonia showing variation in the muscle fiber diameter and hypertrophy. Hematoxylin and eosin stain (×400). Magnification bar = 80 μm.
Blood samples were obtained from all 3 affected animals and stored in ethylenemiamine tetraacetic acid (EDTA) Vacutainer blood collection tubes (Becton Dickinson, São Paulo, São Paulo). These samples were used for CLCN1 exon/intron boundary evaluations and to obtain exon sequences. Genomic DNA was extracted from whole blood using ReliaPrep Blood gDNA Miniprep System (Promega, CA, USA) following the manufacturer's recommendations. Intronic primers previously described 14 and other primers were designed (PrimerQuest software—Integrated DNA Technologies, Iowa, USA) to amplify the 23 exons of the Felis catus CLCN1 gene (RefSeq 101 088 564) (Table S1). The PCR (25 μL) contained 2.5 μL of template DNA (about 125 ng), 0.3 μM of each primer, 12.5 μL of PCR Master Mix (Promega, CA, USA), and 8.5 μL of nuclease‐free water.
To characterize the full coding sequence of the cat CLCN1 gene, total RNA was obtained from muscle samples (≈30 mg) with SV Total RNA Isolation System (Promega, CA,) following the manufacturer's instructions. The RNA was treated with RQ1 RNase‐Free DNase (Promega, CA, USA). First‐strand cDNA synthesis was performed with 800 ng of total RNA per 60 μL of reaction volume using an ImProm‐II Reverse Transcription System (Promega) and random primers following the manufacturer's instructions. To obtain the feline chlorine channel coding gene sequence, sets of primers were designed (PrimerQuest software—Integrated DNA Technologies) based on the CLCN1 mRNA sequence (NM_001305027.1) (Table S2). All amplicons were analyzed by 1.5% agarose gel electrophoresis, purified, and subjected to Sanger sequencing (Table S1). The obtained sequences and electropherograms were analyzed (Geneious v.2019.1.3; Biomatters, Auckland, New Zealand) and compared with the wildtype feline CLCN1 gene sequence.
When analyzing the sequences of 23 CLCN1 exons obtained from the 3 affected cats with the Felis catus CLCN1 gene sequence (Genbank, NM01305027.1), we identified 4 variants. All variants were homozygous in the affected cats. Two of them were synonymous variants, c.1182T>G and c.1650G>T, and are located in exons 11 and 15, respectively. The other 2 were missense variants. The c.1855G>A Ala619Thr variant resulting in the substitution of alanine for threonine does not seem relevant, as these modifications are present in cats (NP_001291956.1) and in other species (Southern elephant seal KAF3819440.1 [Mirounga leonina], polar bear XP_040499815.1 [Ursus maritimus], ferret XP_004772118.1 [Mustela putorius furo], dog NP_001003124.1 [Canis lupus familiaris]). Whereas, the variant c.991G>C p.Ala331Pro in exon 9 (Figure 3) of the CLCN1 gene results in an amino acid replacement of alanine with proline (located in the first extracellular loop). The alanine in this position is conserved in ClC‐1 in different species (Figure 4).
FIGURE 3.
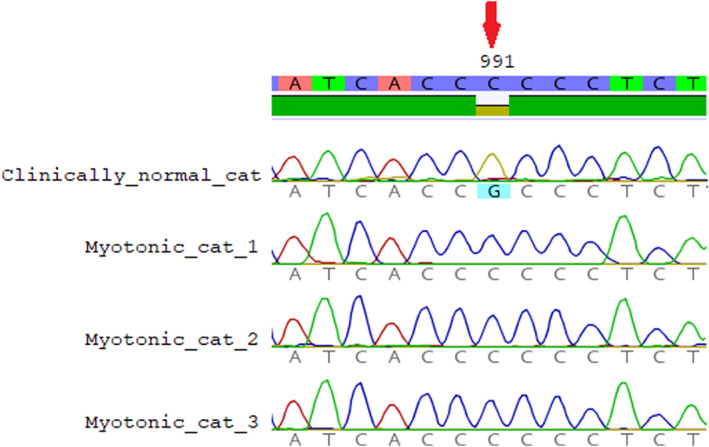
Partial electropherograms from the CLCN1 gene from clinically normal cats presenting the wildtype genotype (G) and 3 myotonic cats (Cats 1, 2, and 3) presenting the homozygous genotype (C). (Red arrow) SNP (c.991G>C). (Geneious v.2019.1.3; Biomatters, Auckland, New Zealand).
FIGURE 4.
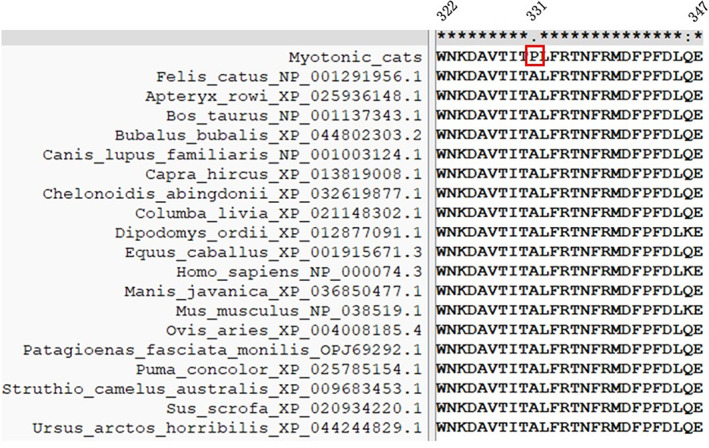
Alignment of CLC‐1 protein reference sequences of 19 species between positions 322 and 347 (upper panel). Highlighted in red is the residue substitution identified in myotonic cats (p.Ala331Pro).
For variants that resulted in a change to the amino acid sequence, PROVEAN (http://provean.jcvi.org/index.php) was used to determine whether the sequence alteration is expected to significantly affect the protein's function. PROVEAN showed a deleterious score of −4.99 (scores of −2.5 or less are predicted to be deleterious) for p.Ala331Pro. For evaluation of protein stability, MUpro: Prediction of Protein Stability Changes for Single Site Mutations from Sequences (http://mupro.proteomics.ics.uci.edu/) was used, and a delta G = −0.96474552 was obtained, indicating decreased stability. Untrained predictors of protein stability changes upon mutation (https://folding.biofold.org/ddgun/method.html) also indicated decreased stability. Using SAAFEC‐SEQ, an online application for calculating folding free energy changes in proteins caused by missense mutations, it was observed that variant p.Ala331Pro destabilizes the protein. The same was observed using PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), which indicates that this substitution probably damages the protein function, with a maximum score.
The SNP c.991G>C (p.Ala331Pro) was screened in 104 control phenotypically normal unrelated cats (DNA bank from the clinical veterinary molecular biology laboratory) using primers designed (PrimerQuest software—Integrated DNA Technologies, I) for amplification of the entire exon 9 (Gandolfi_seq_9). The PCR, Sanger sequencing and electropherogram procedures were the same as those previously described. All 104 cats were wildtype for the p.Ala331Pro variant.
4. DISCUSSION
Among the muscular abnormalities, ion channelopathies, particularly chloride channel disorders resulting in hereditary myotonia, were placed at the top of the differential diagnosis list. This was based on signs of muscular hypertrophy associated with warm‐up and startle response. Other signs in these 3 cats such as abnormal gait, collapse, dysphonia, and difficult swallowing, are associated with myopathy with oval inclusions, 17 nemaline myopathy, 18 glycogen storage disease, 19 and myopathy with tubulin reactive inclusions 20 but were not confirmed during muscle histological evaluation and usually presented muscular weakness and atrophy that were not observed in the cats described here. Abnormalities (such as replacement of muscle fibers by adipose and connective tissue) resulting from muscular dystrophy (Dystrophin‐deficient myopathy) were also subsequently excluded through biopsy and there was no need to perform dystrophin immunohistochemistry.
The observed clinical signs of myotonia, associated with the warm‐up effect and EMG findings, were consistent with the diagnosis of hereditary myotonia, possibly associated with CLCN1 mutation. Hereditary myotonia was phenotypically characterized in 2 young siblings domestic cats 21 and 4 kittens. 11 Subsequently, genetic characterization of the disease was performed in 5 affected cats in Canada 14 and in a 10‐month‐old castrated male domestic longhair cat in North Carolina. 15 The clinical signs in cats in our study are similar to the 4 previous reports in cats. 11 , 14 , 15 , 21
The main clinical signs already described in myotonic cats include stiffness, stilted gait, stiffness reduction during exercise, muscle hypertrophy, blepharospasm, and dysphonia, 11 , 12 , 14 , 15 all of which were observed in our study with different intensities, characterizing a phenotypic variation that is described in cases of myotonia. Tongue exposure and transient respiratory stridor were found in 1 of the 3 cats in this study (Cat 2) in the same way as in previous studies, 14 but open‐mouth breathing was not observed in the cats in our study. Moderate periodontal disease, gingivitis, and halitosis were found in 2 of the 3 cats, as already observed in other myotonic cats. 14 Protrusion of the third eyelid is a previously unrecognized clinical sign in cats with hereditary myotonia. Resistance to opening and then closing the mouth is less described, but it has also been found. 11 We noted the exaggerated reaction to stimulation in the dorsal region of the lumbar region, which resulted in nibbling of the paws or surfaces that supported the cats, in this study, and this has not been previously described.
The EMG findings were consistent with myotonic discharges as in other 2 reports. 14 , 15 Serum CK activity was within normal limits, as described in most cats with hereditary myotonia (only 1 animal in a study showed a slight increase 14 ).
The muscle evaluation in this study showed variation of myofiber diameter, hypertrophy of the muscle fibers and no evidence of other abnormalities. 22 Overall, histological changes in HM are nonspecific, with occasional degeneration of individual myofibers and moderate diffuse myofiber hypertrophy already described in myotonic cats. 11 , 12 , 14 , 21
As in this study, all other mutations related to HM in domestic animals are autosomal recessive. The variant observed in this study is different from those observed in other domestic animals with myotonia (Figure 5), such as the first description of the mutation in cats located in the CBS domain 14 and a recent mutation described in the first transmembrane domain, 15 all of them are present in the ClC‐1 protein. It is interesting to note that a mutation in the extracellular loop was detected for the first time in domestic animals. Three large extracellular loops link helices I‐J (amino acids 322‐347 based on UniProt), helices L‐M (amino acids 427‐457) and helices N‐O (amino acids 499‐521). 23 As seen in several species of mammals, this region is highly conserved, with 100% similarity. As expected from their location, mutations on extracellular loops are typically less drastic than mutations within helices 23 ; however, some mutations have already been described in this loop and associated with hereditary myotonia in humans (p.V327I 24 ; I329T 25 ; A331T 26 ; A331S 27 ; F337C 23 ). Two mutations have been reported for residue R338*, one nonsense 28 and one missense (R338Q). 25 Interestingly, in this same position in humans, 26 the functional consequences of a novel disease‐causing mutation that predicts the substitution of alanine by threonine at position 331 (A331T) by whole‐cell patch‐clamp recording of recombinant mutant channels has been reported. A331T hClC‐1 channels exhibit a novel slow gate that is activated during membrane hyperpolarization and closes at positive potentials. The A331T mutation causes an unprecedented alteration in ClC‐1 gating and reveals novel processes defining transitions between open and closed states in ClC chloride channels. Similarly, A331S causes slow gate activation and may contribute to congenital myotonia, with recessive inheritance in humans. 27 Although we did not perform patch clamp studies, the mutation in these cats is located in a very conserved region in the I‐J extracellular loop; the 2 previous mutations at the same site described in humans and no clinically normal cat presenting this variant indicates that this mutation is likely the cause.
FIGURE 5.
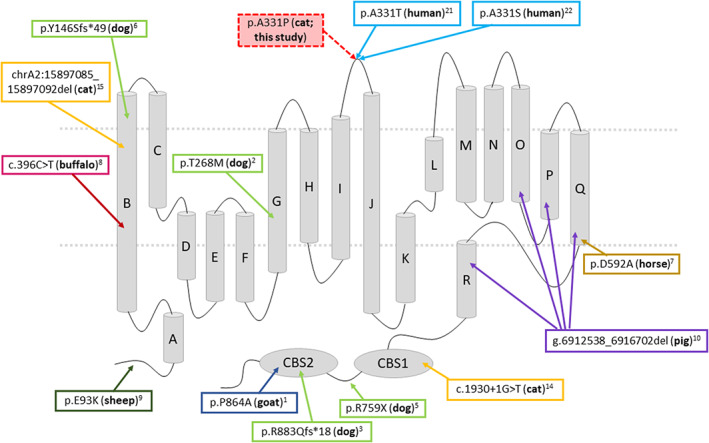
Membrane topology model of the skeletal muscle chloride channel monomer ClC‐1, representing the location of all mutations described in domestic animals associated with hereditary myotonia, the variant detected in this study and close mutations described in humans.
Limitations of this case report include the absence of parental DNA for mutation evaluation, as well as the lack of western blot to confirm protein predicted decreased stability or immunohistochemistry techniques to stain for type 1 and type 2 fibers and localize the voltage‐dependent chloride channel, which could have provided additional information for these cases. Furthermore, because of the owner's decision not to pursue treatment for these 3 cats, primarily influenced by economic and daily management factors, pharmacological intervention was not administered.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Approved by the IACUC (0089/2021‐CEUA) of São Paulo State University (UNESP).
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Table S1: Nucleotide sequences of primers used for specific gene amplification and for conventional PCR studies of CLCN1 gene.
Table S2: cDNA Primers for PCR*.
Video S1: Decreased range of jaw motion, resistance when opening and then closing the mouth and third eyelid protrusion.
Video S2: Stiff gait and recumbency after stimulation
Video S3: Dysphonia and protrusion of the third eyelid
Video S4: Paw nibbling reflex in response to dorsal lumbar region stimulation.
Audio S1: Myotonic discharges characteristic "dive bomber" sound
ACKNOWLEDGMENT
No funding was received for this study.
Corrêa S, Basso RM, Cerri FM, et al. Hereditary myotonia in cats associated with a new homozygous missense variant p.Ala331Pro in the muscle chloride channel ClC‐1 . J Vet Intern Med. 2023;37(6):2498‐2503. doi: 10.1111/jvim.16837
Contributor Information
Fabricio Moreira Cerri, Email: f.cerri@unesp.br.
Alexandre Secorun Borges, Email: alexandre.s.borges@unesp.br.
REFERENCES
- 1. Beck CL, Fahlke C, George AL Jr. Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proc Natl Acad Sci USA. 1996;93:11248‐11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rhodes TH, Vite CH, Giger U, Patterson DF, Fahlke C, George AL. A missense mutation in canine ClC‐1 causes recessive myotonia congenita in the dog. FEBS Lett. 1999;456:54‐58. [DOI] [PubMed] [Google Scholar]
- 3. Finnigan DF, Hanna WB, Poma R, et al. A novel mutation of the CLCN1 gene associated with myotonia hereditaria in an Australian cattle dog. J Vet Intern Med. 2007;21:458‐463. [DOI] [PubMed] [Google Scholar]
- 4. Bhalerao DP, Rajpurohit Y, Vite CH, Giger U. Detection of a genetic mutation for myotonia congenita among Miniature Schnauzers and identification of a common carrier ancestor. Am J Vet Res. 2002;63:1443‐1447. [DOI] [PubMed] [Google Scholar]
- 5. Quitt PR, Hytönen MK, Matiasek K, Rosati M, Fischer A, Lohi H. Myotonia congenita in a Labrador Retriever with truncated CLCN1 . Neuromuscul Disord. 2018;28:597‐605. [DOI] [PubMed] [Google Scholar]
- 6. Rodrigues DJ, Damasceno AD, de Araújo CE, et al. Hereditary myotonia in American Bulldog associated with a novel frameshift mutation in the CLCN1 gene. Neuromuscul Disord. 2020;30:991‐998. [DOI] [PubMed] [Google Scholar]
- 7. Wijnberg ID, Owczarek‐Lipska M, Sacchetto R, et al. A missense mutation in the skeletal muscle chloride channel 1 (CLCN1) as candidate causal mutation for congenital myotonia in a New Forest pony. Neuromuscul Disord. 2012;22:361‐367. [DOI] [PubMed] [Google Scholar]
- 8. Borges AS, Barbosa JD, Resende LA, et al. Clinical and molecular study of a new form of hereditary myotonia in Murrah water buffalo. Neuromuscul Disord. 2013;23:206‐213. [DOI] [PubMed] [Google Scholar]
- 9. Monteagudo LV, Tejedor MT, Ramos JJ, Lacasta D, Ferrer LM. Ovine congenital myotonia associated with a mutation in the muscle chloride channel gene. Vet J. 2015;204:128‐129. [DOI] [PubMed] [Google Scholar]
- 10. Araújo CE, Oliveira CM, Barbosa JD, et al. A large intragenic deletion in the CLCN1 gene causes Hereditary Myotonia in pigs. Sci Rep. 2019;9:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hickford FH, Jones BR, Gething MA, Pack R, Alley MR. Congenital myotonia in related kittens. J Small Anim Pract. 1998;39:281‐285. [DOI] [PubMed] [Google Scholar]
- 12. Toll J, Cooper B, Altschul M. Congenital myotonia in 2 domestic cats. J Vet Intern Med. 1998;12:116‐119. [DOI] [PubMed] [Google Scholar]
- 13. Gaschen F, Jaggy A, Jones B. Congenital diseases of feline muscle and neuromuscular junction. J Feline Med Surg. 2004;6:355‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gandolfi B, Daniel RJ, O'Brien DP, et al. A novel mutation in CLCN1 associated with feline myotonia congenita. PloS One. 2014;9:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woelfel C, Meurs K, Friedenberg S, DeBruyne N, Olby NJ. A novel mutation of the CLCN1 gene in a cat with myotonia congenita: diagnosis and treatment. J Vet Intern Med. 2022;36:1454‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levy JK, Crawford PC, Werner LL. Effect of age on reference intervals of serum biochemical values in kittens. J Am Vet Med Assoc. 2006;228:1033‐1037. [DOI] [PubMed] [Google Scholar]
- 17. Gougeon E, Larcher T, Ledevin M, McGrotty Y, Méheust P. Myopathy with oval inclusions in a domestic shorthair cat. JFMS Open Rep. 2022;8(1):20551169221081418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cooper BJ, De Lahunta A, Gallagher EA, Valentine BA. Nemaline myopathy of cats. Muscle Nerve. 1986;9(7):618‐625. [DOI] [PubMed] [Google Scholar]
- 19. Fyfe JC. Glycogen storage disease in cats. J Am Vet Med Assoc. 1995;206(3):286. [PubMed] [Google Scholar]
- 20. Shelton GD, Sturges BK, Lyons LA, et al. Myopathy with tubulin‐reactive inclusions in two cats. Acta Neuropathol. 2007;114(5):537‐542. [DOI] [PubMed] [Google Scholar]
- 21. Toll J, Cooper B. Feline congenital myotonia. J Small Anim Pract. 1998;39:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Braund KG, Amling KA, Mehta JR, Steiss JE, Scholz C. Histochemical and morphometric study of fiber types in ten skeletal muscles of healthy young adult cats. Am J Vet Res. 1995;56(3):349‐357. [PubMed] [Google Scholar]
- 23. Jehasse K, Jacquerie K, de Froidmont A, et al. Functional analysis of the F337C mutation in the CLCN1 gene associated with dominant myotonia congenita reveals an alteration of the macroscopic conductance and voltage dependence. Mol Genet Genomic Med. 2021;9:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horga A, Rayan DLR, Matthews E, et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. 2013;80:1472‐1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J, Bendahhou S, Sanguinetti MC, Ptacek LJ. Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology. 2000;54:937‐942. [DOI] [PubMed] [Google Scholar]
- 26. Warnstedt M, Sun C, Poser B, et al. The myotonia congenita mutation A331T confers a novel hyperpolarization‐activated gate to the muscle chloride channel ClC‐1. J Neurosci. 2002;22:7462‐7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suetterlin K, Matthews E, Sud R. Translating genetic and functional data into clinical practice: a series of 223 families with myotonia. Brain. 2022;145:607‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brugnoni R, Kapetis D, Imbrici P, et al. A large cohort of myotonia congenita probands: novel mutations and a high‐frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet. 2013;58:581‐587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Nucleotide sequences of primers used for specific gene amplification and for conventional PCR studies of CLCN1 gene.
Table S2: cDNA Primers for PCR*.
Video S1: Decreased range of jaw motion, resistance when opening and then closing the mouth and third eyelid protrusion.
Video S2: Stiff gait and recumbency after stimulation
Video S3: Dysphonia and protrusion of the third eyelid
Video S4: Paw nibbling reflex in response to dorsal lumbar region stimulation.
Audio S1: Myotonic discharges characteristic "dive bomber" sound