

Abstract
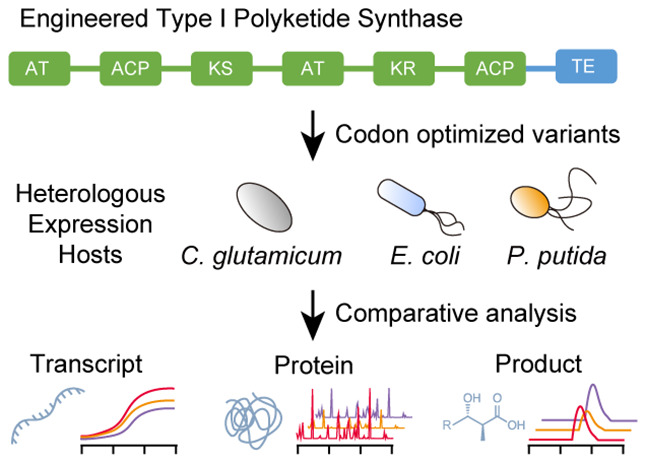
Type I polyketide synthases (T1PKSs) hold enormous potential as a rational production platform for the biosynthesis of specialty chemicals. However, despite great progress in this field, the heterologous expression of PKSs remains a major challenge. One of the first measures to improve heterologous gene expression can be codon optimization. Although controversial, choosing the wrong codon optimization strategy can have detrimental effects on the protein and product levels. In this study, we analyzed 11 different codon variants of an engineered T1PKS and investigated in a systematic approach their influence on heterologous expression in Corynebacterium glutamicum, Escherichia coli, and Pseudomonas putida. Our best performing codon variants exhibited a minimum 50-fold increase in PKS protein levels, which also enabled the production of an unnatural polyketide in each of these hosts. Furthermore, we developed a free online tool (https://basebuddy.lbl.gov) that offers transparent and highly customizable codon optimization with up-to-date codon usage tables. In this work, we not only highlight the significance of codon optimization but also establish the groundwork for the high-throughput assembly and characterization of PKS pathways in alternative hosts.
Keywords: codon optimization, online tool, codon usage, type 1 polyketide synthase, heterologous expression, industrial host
1. Introduction
Type I polyketide synthases (T1PKSs) are a class of natural enzymes primarily found in bacteria and fungi that are responsible for the biosynthesis of secondary metabolites. Over the years, humans have harnessed the therapeutic potential of these metabolites, and numerous indispensable drugs have been discovered.1
The modular architecture of T1PKSs enables the iterative assembly of long carbon chains, while also providing the flexibility for the optional incorporation of functional groups.2 The theoretical design space offered by this modularity has attracted significant research attention, and T1PKSs have been successfully reprogrammed for the production of unnatural polyketides.3−5
However, one of the biggest challenges in PKS engineering is the native host itself. Most of the discovered PKSs originate from the genus Streptomyces, a GC-rich, Gram-positive, and filamentous bacterium. While certain streptomycetes have been highly domesticated and optimized for the production of larger, high value molecules, their efficacy for the production of industrially relevant bulk chemicals is limited.6 To date, little progress has been made in exploring alternative PKS hosts. Besides the required genetic modifications for PKS expression, polyketide titers in non-native hosts are often very low, which can usually be traced back to poor precursor availability or low protein levels.7,8 Common strategies for improving polyketide titers include the supplementation of the media, the exchange of promoters, or codon optimization.4 However, the importance of the latter is often neglected or underestimated.
The host’s codon preference usually has a distinct pattern and can be summarized in codon usage tables.9 The choice of specific codons can impact transcription and translation rates and is also involved in expression control mechanisms. Well-studied examples include the rare Streptomyces codon TTA and the use of alternative start codons such as GTG or TTG.10,11
Codon optimization represents a strategy to address these variations in codon preferences during heterologous gene expression. This approach involves selectively substituting specific codons while preserving the amino acid sequence of the protein. There are three commonly used strategies for this purpose: (i) replacing the original codons with the most frequently used codon of the targeted host, (ii) matching the codon frequency of the targeted host, and (iii) harmonizing the codon frequency of the targeted host with the codon frequency of the native host.12−15 After codon optimization, the final nucleotide sequence will likely be significantly different from the original sequence, and many researchers have to rely on DNA synthesis services to synthesize the codon-optimized gene. Although convenient, the optimization algorithms offered by most synthesis services are not publicly available and their functionality is very limited.16 The recently published open-source codon optimization tool DNA Chisel offers an easy way to apply these aforementioned methods and further customize the resulting nucleotide sequence.15
In this study, we investigated the expression and activity of an engineered T1PKS with different codon variants in the three hosts Corynebacterium glutamicum, Escherichia coli, and Pseudomonas putida. E. coli and C. glutamicum are well-established industrial hosts for the large-scale production of proteins and small molecules, while P. putida has shown enormous potential for the valorization of renewable feedstocks.17−19 By targeting three heterologous hosts and applying the three most common gene optimization methods, we designed 9 codon variants of the engineered PKS (Figure 1). Furthermore, as a conventional approach, we obtained and cloned the native sequences from Streptomyces aureofaciens Tü117 and Saccharopolyspora erythraea NRRL2338, and also tested the effect of two different start codons, GTG and ATG. Due to the large size of PKSs and the metabolic burden of plasmid maintenance, we developed a backbone excision-dependent expression (BEDEX) system to facilitate the cloning process and enable constitutive expression in the heterologous hosts. We further confirmed the universal functionality of BEDEX vectors in our selected hosts and applied the BEDEX system to heterologously express the 11 codon variants of the engineered PKS. To characterize our codon variants in vivo, we measured the PKS protein and transcript levels and demonstrated the production of an unnatural polyketide in each host.
Figure 1.

Engineered polyketide synthase with applied codon optimization strategies and targeted heterologous hosts. The loading module and module 1 originates from the lipomycin polyketide synthase (LipPKS) from Streptomyces aureofaciens Tü117 (green). The thioesterase domain (TE) originates from the erythromycin PKS (EryPKS) from Saccharopolyspora erythraea NRRL2338 (blue). By fusion of these two parts together, the engineered PKS design yields a variety of short-chain 3-hydroxy acids. The gene sequence of the reprogrammed LipPKS was codon optimized using the DNA Chisel algorithms for “Use Best Codon” (yellow), “Match Codon Usage” (orange), and “Harmonize RCA” (red/purple). All algorithms preserve the amino acid sequence of the protein. The “Use Best Codon” method replaces each codon with the most frequently used codon. “Match Codon Usage” matches the codon frequency of the original codon sequence with the codon usage of the targeted host. “Harmonize RCA” applies and matches the codon frequency of the targeted host with the codon usage of the native host. The “Harmonize RCA” algorithm required codon optimization of the LipPKS and EryPKS parts separately. Codon optimizations targeted the three heterologous hosts C. glutamicum, E. coli, and P. putida.
2. Results
2.1. Comparative Analysis on Codon Usage Patterns of Host Organisms
To conduct a comprehensive analysis of the overall codon usage in the target host organisms, we compared the codon usage patterns among three native PKS hosts, our selected heterologous host species, and other well-studied organisms as outgroup references. 16S rRNA sequence-based phylogenetic analysis on these species shows that C. glutamicum is the most closely related species to Streptomyces, and that E. coli and P. putida are more closely related to each other (Figure 2a). Based on the phylogenetic data, it could be concluded that C. glutamicum should be the most suitable heterologous host for genes that are sourced from streptomycetes. However, their codon usage might be vastly different since the GC-content in Streptomyces is about 20% higher than that in C. glutamicum.
Figure 2.

Global analysis of codon usage preferences between different species. (a) 16S rRNA sequence-based phylogenetic analysis of targeted hosts, native polyketide synthase hosts, and outgroup references. Saccharomyces cerevisiae S288C was excluded. (b) Principal component analysis (PCA) of codon usage tables associated with the same species from the phylogenetic analysis. The first two principal components (PCs), accounting for the highest explained variance, were selected and visualized. (c) Comparison of the codon frequency per corresponding amino acid between C. glutamicum, S. aureofaciens, E. coli, and P. putida. The amino acids methionine and tryptophan are encoded by a singular codon and were excluded from this analysis.
Principal component analysis (PCA) was performed on codon usage data for the same set of organisms (Figure 2b). The native PKS hosts S. aureofaciens, S. erythraea, and S. coelicolor are situated in close proximity to each other, whereas the evaluated heterologous hosts C. glutamicum, E. coli, and P. putida are more distant. Notably, despite C. glutamicum belonging to the same phylum, it does not cluster with the other actinobacteria. Furthermore, the significantly higher GC-content of P. putida (>61%) does not seem to lead to increased similarity in codon usage with streptomycetes either. Figure 2c highlights the relative codon frequency per corresponding amino acid (RCF) for C. glutamicum, S. aureofaciens, E. coli, and P. putida. While the RCF in S. aureofaciens has ten distinct maxima (>0.8), P. putida is the only other organism with a similar maximum for the codon TGC. The high GC-content of S. aureofaciens (>72%) most likely leads to these maxima, especially for amino acids with only two codons. In contrast, the proteobacterium E. coli usually prefers codons with lower GC-content but seems to be more tolerable to other codons as well.
All in all, these data might suggest that there is no ideal host of industrial relevance that shows a codon preference similar to that of common native PKS hosts. PCA of codon usage could be a useful tool to assess the success rate of expression of the wild-type (WT) nucleotide sequence in the selected heterologous host. To further evaluate this theory, we performed codon optimization on an engineered PKS from S. aureofaciens Tü117 and compared expression levels with the WT nucleotide sequence in our selected heterologous hosts.
2.2. Codon Optimization of Engineered Lipomycin Polyketide Synthase
The design of our synthetic PKS was based on the work of Yuzawa et al. (2013).21 In short, we removed the first 59 N-terminal amino acids of the lipomycin PKS (LipPKS) and truncated module (M) 1 after the acyl carrier protein (ACP) 1. We then fused the remaining protein to the erythromycin PKS (EryPKS) M6 thioesterase (TE) including the interdomain linker between EryPKS ACP6 and TE6 (Figure 1). The N-terminal truncation leads to improved protein expression, and the TE hydrolyzes the product after the first methylmalonyl-CoA (mmCoA) extension.20 The loading domain of LipPKS is very promiscuous and can utilize isobutyryl-CoA, 2-methylbutyryl-CoA, isovaleryl-CoA and propionyl-CoA.21 The comparably small size and wide range of acceptable acyl-CoAs make this engineered PKS a well-suited candidate for heterologous expression.
While this design has been shown to yield a variety of short-chain 3-hydroxy acids in vivo, its functionality has not yet been evaluated in our chosen heterologous hosts. To increase the probability for functional expression of the engineered PKS, we employed the three codon optimization methods “use best codon” (ubc), “match codon usage” (mcu) and “harmonize relative codon adaptiveness” (hrca) using DNA Chisel, and tested their effect on transcription and translation of the target PKS gene.15 The ubc method corresponds to the previously described codon optimization strategy (i), while mcu and hrca follow the principles of strategies (ii) and (iii), respectively.12−15 Furthermore, we included the WT nucleotide sequence of each part of the PKS and investigated the effect of the two different start codons, GTG and ATG. A full list of the used settings for codon optimization can be found in the Supporting Information (Table S1).
As the applied algorithms are part of a Python-based toolkit with a command line interface (CLI), we developed a user-friendly graphical interface (GUI) to facilitate the use of open-source codon optimization tools. The codon optimization tool is publicly available, requires no prior experience in CLIs and can be reached under https://basebuddy.lbl.gov. In addition to the already implemented codon usage database from Kazusa,9 we also included the latest version of the Codon and Codon Pair Usage Tables (CoCoPUTs) database.22 Compared to the Kazusa database, CoCoPUTs are based on up-to-date sequencing data and also provide codon usage tables for a significantly broader range of organisms.23
2.3. Development of a Backbone Excision-Dependent Expression System
To ensure greater accuracy and consistency of our results in C. glutamicum and P. putida, we utilized serine recombinase-assisted genome engineering (SAGE) to integrate our codon variants into the genome of these hosts.24,25 The strains AG5577 and AG6212 are derived from P. putida and C. glutamicum, respectively, and contain a total of 9 unique attB sites in their genome (Adam Guss, personal communication). These genomic “landing pads” enable highly efficient and precise gene integration via the SAGE system (Figure 3a).
Figure 3.

Extending the SAGE system with backbone excision-dependent expression (BEDEX) vectors. (a) C. glutamicum AG6212 and P. putida AG5577 contain a total of 9 unique attB sites each. Heterologous serine recombinases catalyze the integration of vectors containing the corresponding attP site. Expressing the integrase ΦC31 removes the integrated vector backbone and allows for selection marker recycling. Excising the backbone of BEDEX vectors also removes the repressor LacI. (b) C. glutamicum AG6212, P. putida AG5577, and E. coli BL21 containing BEDEX vector carrying RFP. In E. coli BL21, BEDEX vectors are nonintegrative and maintained by replication. Induction or excision of the BEDEX vector backbone leads to a significant increase in RFP levels (n = 3).
In addition, we sought to further improve the applicability of the SAGE system by combining it with backbone excision-dependent expression (BEDEX) vectors (Figure 3a). The two key elements of BEDEX vectors are the LlacO1 promoter and the excisable lacI gene. In the case of the SAGE system, the integrated plasmid backbone is excised via the transient expression of the ΦC31 integrase. By removing the lacI gene from the host genome, the LlacO1 promoter is no longer repressed and becomes constitutive. To confirm the functionality of the BEDEX vectors in all three host organisms, we used red fluorescent protein (RFP) as a readily quantifiable output (Figure 3b). While the rfp gene was integrated into the genomes of P. putida AG5577 and C. glutamicum AG6212, we relied on plasmid-based expression in E. coli BL21. Due to the presence of the ColE1 origin of replication, the SAGE system vector suite cannot be utilized to engineer E. coli.
In C. glutamicum AG6212, the measured RFP signal was extremely low compared to that of rfp-expressing E. coli and P. putida cells. The C. glutamicum control sample showed a normalized RFP signal of 174 ± 4, which is about 2-fold lower than the uninduced and unexcised integration of the BEDEX vector (data not shown). However, induction or excision of the backbone showed a significant increase in the RFP signal by up to 4-fold.
In P. putida AG5577, the expression was strongly repressed in the presence of lacI. The induction with 200 μM IPTG led to a 44-fold increase in the normalized RFP signal. Excision of the backbone, however, led to a 213-fold increase in signal, and the expression seemed unaffected by the addition of inducer. In E. coli, IPTG-induction achieved a 105-fold change in normalized RFP signal, while maintaining a relatively low signal of 1548 ± 287 in the uninduced state.
These data demonstrate the successful integration of the target gene into the genomes of C. glutamicum and P. putida. In addition, by eliminating lacI from the plasmid backbone, we achieved expression without inducing the LlacO1 promoter.
2.4. Quantification of Heterologous Protein and Transcript
The BEDEX vectors carrying the 11 codon variants of LipPKS were introduced into C. glutamicum AG6212, E. coli BL21, and P. putida AG5577. The LipPKS gene was genomically expressed in C. glutamicum AG6212 and P. putida AG5577, and from plasmid DNA in E. coli BL21. The resulting protein levels of engineered LipPKS are shown in Figure 4a.
Figure 4.

Relative abundance of LipPKS peptides and transcript in heterologous hosts expressing different codon variants of LipPKS. (a) Calculated protein abundance (n = 3) is the relative intensity of the top 3 peptides that correspond to the target protein divided by the intensity of all proteins detected. Hatched bars indicate strains with nonexcisable backbones. Levels of the insolubility marker IbpA are represented by a gray line (n = 3). IbpA is not present in C. glutamicum. (b) Each violin represents the distribution of the relative transcript amount for all target host optimizations with the same optimization strategy (n = 9). Relative transcript was calculated from the Ct value difference between the target transcript and housekeeping gene. The lines within the violins are individual data points. Target host optimization: Cg = C. glutamicum; Ec = E. coli; Pp = P. putida; Optimization strategy: ubc = use best codon; mcu = match codon usage; hrca = harmonize relative codon adaptiveness.
The LipPKS counts in C. glutamicum samples were on average 1 to 2 orders of magnitude lower than in E. coli and P. putida samples. However, if we compare protein abundance instead, values become more similar between organisms. Depending on the host and codon version, the LipPKS abundance ranges from 0 to 3.9 ± 1.5%. Furthermore, compared to the optimized genes, the WT nucleotide sequences performed very poorly: protein levels were either undetectable or extremely low compared to those of the codon optimized variants. However, mutating the original GTG start codon to ATG resulted on average in a 3-fold increase in the peptide counts.
In general, PKS gene codons optimized for a particular organism produced the most PKS when expressed in the target organism. For example, PKS genes that were codon optimized for P. putida (Pp) and were expressed in P. putida achieved a LipPKS abundance of 1.1 ± 0.4% across all three codon optimization methods. When compared to PKS genes that were codon optimized for E. coli (Ec) but expressed in P. putida, LipPKS abundance was as low as 0.2 ± 0.1%. This trend was even more drastic when PKS genes codon optimized for all three organisms were expressed in E. coli: the E. coli codon optimized genes produced on average 22-fold more LipPKS than the genes codon optimized for P. putida and C. glutamicum. A significant outlier of this general observation is the expression of the Cg_hrca codon variant in P. putida. Despite being codon optimized for C. glutamicum, it exhibited the highest peptide abundance in this host. A possible explanation might be the high similarity between the hrca optimized PKS genes (Supplementary Figure S1).
Another noticeable difference was the relative standard deviation between plasmid-based and genomic expression, which can be as high as 121% or as low as 2%, respectively. These data suggest that the expression of LipPKS from a plasmid can lead to greater data variability and might be less reproducible between replicates.
In addition to the LipPKS abundance, we also determined the IbpA levels for E. coli and P. putida samples (Figure 4a). The small heat-shock protein IbpA has been shown to be expressed in the presence of an insoluble protein in the cell. Generally, a high IbpA level is an indicator for misfolded protein and can give a hint about the activity of the heterologously produced protein in these hosts.26 In E. coli, there is a strong Pearson correlation of 0.94 (n = 33) between IbpA and LipPKS levels, whereas no such correlation was observed in P. putida. Interestingly, expressing the WT_ATG construct in P. putida resulted in the highest IbpA signal while producing a relatively low amount of LipPKS (0.09 ± 0.02%). Therefore, the poor similarity in codon usage may have resulted in not only low levels of protein but also insoluble protein.
To date, it is not clear if codon usage affects gene expression on a transcriptional or translational level, especially across different organisms.27,28 To analyze the correlation between transcript and target protein, we measured the LipPKS transcript level by reverse transcription qualitative real-time PCR (RT-qPCR). Transcript levels were calculated from the Ct value difference between the target transcript and housekeeping gene (Supplementary Table S2). The strongest correlation between transcript and protein levels was observed in P. putida (R = 0.67), while the correlation coefficient for the other two hosts was below |0.3| (Supplementary Figure S2). In general, a low transcript amount led to a low protein level, with the exception of codon variants Cg_ubc, Cg_mcu, and Ec_ubc in C. glutamicum. Here, we were not able to detect any transcript, although we detected significant amounts of LipPKS peptides. This was also the case for Ec_mcu and Ec_hrca variants in P. putida. With the exception of the Cg_mcu variant in C. glutamicum, all of these variants were induced at the time of inoculation, which might have affected mRNA levels.
Another noticeable trend was the high transcript amount for the hrca codon optimization, which seemed to be independent of the targeted host (Figure 4b). This trend was especially significant in E. coli where hrca codon optimizations yielded 1266 ± 111% relative transcript, while ubc optimizations resulted in 378 ± 96% relative transcript. Interestingly, for codon variants Pp_hrca and Cg_hrca, the high transcript amount did not lead to high protein levels. These findings support the hypothesis that codon frequency might determine translation rates, as evidenced by the higher LipPKS counts of the target host variants of the hrca optimizations.
2.5. Production of an Unnatural Polyketide by Engineered PKS
The production of the PKS protein itself does not require any genetic modifications of the chosen hosts. However, to demonstrate that our engineered PKS is functional and produces the desired polyketide, we needed to express supplementary pathways. For successful polyketide production in E. coli, the PKS vectors were transformed into the readily available host E. coli K207–3.29 This strain possesses the required mmCoA pathway and phosphopantetheinyl transferase (PPTase) for the supply of the extender substrate and the activation of the ACP domain. Due to the versatile nature of the LipPKS loading module, it exhibits promiscuity toward propionyl-CoA as an alternative starter unit, making the expression of an isobutyryl-CoA pathway unnecessary.21
Given P. putida’s demonstrated broad specificity PPTase and its ability to utilize the isobutyryl-CoA precursor valine, the integration of a heterologous mmCoA pathway is the sole modification required.30,31 As previously reported, the mmCoA mutase and epimerase from Sorangium cellulosum So ce56 is functional in P. putida.32 Therefore, we integrated the unmodified operon under the control of the tac promoter into the MR11 attB site of P. putida AG5577. Unlike the BEDEX vector, the selected vector backbone lacks lacI, which eliminates the need for backbone excision (Figure 5a). The resulting strain was designated as P. putida AG5577mm.
Figure 5.

Production of an unnatural polyketide by engineered heterologous hosts. (a) Serine recombinase-assisted integration of the methylmalonyl-CoA mutase (mut) and epimerase (epi) pathway into P. putida AG5577. mmCoA: methylmalonyl-CoA. (b) Engineered pathway for the production of the LipPKS loading substrate isobutyryl-CoA by the heterologous enzymes Kivd (ketoisovalerate decarboxylase) and CCL4 (2-methylpropanoate–CoA ligase) in C. glutamicum AG6212. Isobutyric and propionic acids were added to the C. glutamicum production medium (green). aldh: endogenous aldehyde dehydrogenase activity. (c) Regression plot of the LipPKS counts and polyketide production levels in the engineered hosts C. glutamicum AG6212cz, E. coli K207–3, and P. putida AG5577mm (n = 3). The calculation of the Pearson correlation included every replicate as a single data point. Target host optimization: Cg = C. glutamicum; Ec = E. coli; Pp = P. putida; Optimization strategy: ubc = use best codon; mcu = match codon usage; hrca = harmonize relative codon adaptiveness.
The third host organism, C. glutamicum, cannot grow on valine as the sole source of carbon, and the existence of an isobutyryl-CoA pathway is unlikely. However, previous studies have confirmed the presence of mmCoA and propionyl-CoA.33,34 Unfortunately, it remains unclear whether there is a type I PKS compatible PPTase in C. glutamicum. To address this, we employed conventional cloning techniques to introduce the PPTase sfp into C. glutamicum AG6212.34 Furthermore, to increase the probability of producing any kind of polyketide, we also integrated the heterologous CoA ligase CCL4 and ketoisovalerate decarboxylase kivd. By relying on endogenous aldehyde dehydrogenase activity, both genes combined could potentially lead to the preferred LipPKS starter unit isobutyryl-CoA.35−37 In addition, we added isobutyric and propionic acids to the production medium to increase precursor availability. Finally, the gene prpDBC2 was removed to facilitate flux through mmCoA by disrupting the methyl citrate cycle (Figure 5b). The final engineered strain was designated as C. glutamicum AG6212cz.
Polyketide production was measured in all three engineered hosts (Figure 5c). The production of the corresponding polyketide was confirmed by an authentic standard (Supplementary Figure S3). In order to synthesize the expected polyketide in vivo, our hosts need to produce one of the required starter units propionyl-CoA, isobutyryl-CoA or 2-methylbutyryl-CoA. However, isobutyryl-CoA is the preferred substrate of the LipPKS loading domain.21 Therefore, in C. glutamicum AG6212cz and P. putida AG5577mm, we chose to focus on the production of (2S,3S)-3-hydroxy-2,4-dimethylpentanoic acid (3H24DMPA). Due to the absence of a branched-chain β-keto acid catabolism, the only possible product in E. coli K207–3 is (2S,3S)-3-hydroxy-2-methylpentanoic acid (3H2MPA). A more detailed reaction of the polyketide synthesis can be found in the Supporting Information (Supplemental Figure S4).
In P. putida and E. coli, the protein and product levels of the codon variants showed Pearson correlation coefficients of 0.80 and 0.81, respectively. The highest product titer for P. putida was achieved by the Cg_mcu codon variant, which was a noticeable outlier when compared to those of other protein and product relations. In E. coli, the highest product titer was achieved by the Ec_mcu codon variant. Interestingly, our in vivo results in P. putida confirm the malonyl-CoA extended product (Supplementary Figure S3), which contradicts the reported and predicted mmCoA specificity of LipPKS AT1.21
Polyketide production in C. glutamicum was very low and barely detectable (Supplementary Figure S3b), despite the significant LipPKS levels. There was also no clear correlation between LipPKS peptides and the polyketide product (R = −0.32). In addition to analyzing LipPKS amounts in this host, we also measured the peptide counts for Sfp, Kivd, and CCL4. However, we were only able to detect peptides that correspond to Sfp and Kivd; CCL4 peptides were not detected (Supplementary Figure S5). Therefore, the strain might be missing the CoA ligase to efficiently activate the starter unit, isobutyric acid.
3. Discussion
In this study, we identified the most suitable codon optimization method for an engineered T1PKS for heterologous expression in C. glutamicum, E. coli, and P. putida. Furthermore, we elucidated the relationship between codon variant, protein level, transcript amount, and product titer.
The Python library DNA Chisel allowed us to do fully customizable and transparent codon optimizations of our target gene. The degree of customization was an important factor to find an appropriate balance between the optimization task and synthesis difficulty. For instance, the implementation of the UniquifyAllKmers constraint can greatly reduce the amount of repetitive sequence, which remains a major hurdle in the chemical synthesis of DNA.38 As a result, the codon optimization efficiency may be constrained by the limitations of current DNA synthesis techniques.
While the Kazusa codon usage database was sufficient in our case, it might become a limiting factor for future investigations of more exotic PKSs or heterologous hosts. The ubc and mcu methods only require the targeted host’s codon usage, whereas the hrca method also requires a codon usage table of the source organism. However, our developed online tool includes the database CoCoPUTs, which greatly improves the codon usage table accuracy and the number of available organisms. During codon optimization, it is often unclear which codon usage database is used, especially by commercial codon optimization tools. This can make a significant difference in the accuracy of the optimized gene. For example, the S. aureofaciens (NCBI: txid1894) codon table from Kazusa contains only 80 CDSs, while the codon table for the same organism on CoCoPUTs contains 37,337 CDSs. The substantial discrepancy is most likely a result of recent advances in genome sequencing and, therefore, more accurate bacterial genotyping.39
Given that our systematic approach amounted to 11 codon variants of a relatively large gene (>7 kb), we sought to avoid relying exclusively on plasmid-based expression systems. Besides well-known issues such as plasmid stability, copy number variations and growth defects, intercompatibility between our different organisms might have become challenging as well.24,40−45 The SAGE system has proven to be an indispensable tool for genetic engineering when reproducibility and throughput are limiting factors. Stable integrations of large genes into the chosen host genomes can be multiplexed, are highly efficient and require minimal knowledge about the targeted host. In combination with BEDEX vectors this system also enables precise control over gene expression and can improve plasmid assembly efficiency by mitigating the cellular burden of strong constitutive promoters or potentially toxic gene products.46 We have also shown that BEDEX vectors are universally functional in our chosen hosts, although measured RFP levels in C. glutamicum were very low. This observation could have multiple reasons, including a noncompatible ribosome binding site (RBS) or promoter.47,48 Nonetheless, the measured differences between the excised and unexcised vector backbones were significant, and PLlacO1 became constitutively active in this host.
In an effort to further evaluate the BEDEX system, we applied it to construct the LipPKS expression strains. As confirmed with RFP, the BEDEX system showed a similar performance during the construction process. However, we encountered some difficulties while excising the backbone of certain codon variants. The presence of highly incompatible codon variants could result in growth inhibition, which then leads to the selection for lacI positive strains. In an attempt to investigate this behavior, we expressed the codon variants Pp_mcu and Ec_mcu from an inducible vector system in P. putida. As previously shown, the Pp_mcu codon optimized variant produced a significant amount of LipPKS peptides and product, whereas the Ec_mcu variant produced only the associated peptides (Figure 4a and 5c). During overexpression of these LipPKS variants, the protein levels for each variant were comparable. However, the levels of the insolubility marker IbpA were significantly higher in P. putida expressing Ec_mcu LipPKS than in the strain expressing Pp_mcu LipPKS (Supplementary Figure S6). Furthermore, we were not able to detect any product formation with the Ec_mcu codon variant. This could be an indicator for the expression of insoluble or misfolded protein by an incompatible codon variant.
The measured differences in LipPKS protein amounts clearly show that codon usage bias is a crucial factor in the heterologous expression of PKSs. Compared to the WT codon version, our best performing codon variants exhibited a minimum 50-fold increase in PKS protein levels, which was also essential to enable detection of the corresponding polyketide. However, we do not fully understand the causes of these differences. In P. putida, our RT-qPCR results might suggest that there is a link between codon usage and transcript level, whereas the E. coli and C. glutamicum data show no such correlation. In the two latter hosts, codon frequency might have a stronger influence on the protein level. Current literature presents evidence for both of these theories.27,28 The prevailing belief is that rare codons interfere with the translation machinery, whereas frequent codons are translated more rapidly.27 Other studies reported that the observed effects are primarily due to variances in transcription efficiency.28,49 In addition to influencing eukaryotic chromatin modifications, it has been hypothesized that nonoptimal gene sequences could facilitate the binding of unknown intragenic transcription factors.49 Furthermore, transcription and translation have no spatial separation in prokaryotes and both processes occur at the same time.50 Consequently, it is challenging to discern transcriptional or translational bottlenecks in these types of organisms.
While our investigation could not provide a satisfying answer to this controversial topic, we still identified an optimal way for the codon optimization of PKSs. According to our data, the host-specific mcu method resulted in the highest protein and product levels, whereas the hrca method gave rise to the highest transcript levels. The choice of the most optimal algorithm is, therefore, dependent on the goal or suspected limitation of the host. The difference in protein production using ATG and GTG start codons could also be confirmed, and our results are consistent with prior studies.51 Utilization of GTG start codons seems to be more prevalent in high GC organisms and the percentage of GTG initiation codons decreases to less than 25% in lower GC organisms (<65%).52 For the heterologous expression of PKSs, the G to A mutation can be easily achieved by site-directed mutagenesis and is a cost-effective way to improve expression levels.
In addition to E. coli, the two other tested hosts have proven to be suitable for the expression of engineered T1PKSs. The activity of the PKS and the production of the expected 3-hydroxy acid product were confirmed in all three hosts. Since we detected sufficient levels of protein in C. glutamicum, it can be assumed that the exceedingly low product levels are a result of the insufficient supply of isobutyryl-CoA. The loading domain of LipPKS has a strong preference for isobutyryl-CoA but does also accept 2-methylbutyryl-CoA, isovaleryl-CoA, and propionyl-CoA.21 However, the catalytic efficiency of utilizing propionyl-CoA is about 4-fold lower than that compared to isobutyryl-CoA. In addition, the employed engineering strategy needs further evaluation of intracellular acyl-CoA concentrations and precursor pathways.
While we successfully identified the most effective strategy for codon optimization of T1PKS, we still lack a complete understanding of the underlying reasons for its success. Such understanding may only come from codon optimizing many different types of genes in many different hosts, a project that would be economically infeasible with current DNA synthesis costs. In addition, it is possible that there are even better methods for codon optimizing genes; indeed, recent developments in artificial intelligence and machine learning could play a crucial role in optimizing heterologous gene sequences in the future.53
4. Methods
4.1. Chemicals, Media, and Culture Conditions
All chemicals were purchased from Sigma-Aldrich (USA) unless otherwise described. The authentic standards for (2S,3S)-3-hydroxy-2,4-dimethylpentanoic acid, 3-hydroxy-4-methylpentanoic acid, (2S,3S)-3-hydroxy-2-methylpentanoic acid, and 3-hydroxy-2,4-dimethylhexanoic acid were synthesized by Enamine (Ukraine).
Precultures of E. coli, C. glutamicum, and P. putida were inoculated from a single colony and grown overnight. For P. putida and E. coli cultures, we used lysogeny broth (LB) medium and temperatures of 30 and 37 °C, respectively. For C. glutamicum cultures, we used brain heart infusion (BHI) medium and a temperature of 30 °C. If applicable, 50 μg/mL kanamycin was added to the medium. The conditions for precultures and main cultures were kept consistent throughout this study unless otherwise described. For main cultures, 2 mL LB or BHI medium was inoculated with 20 μL preculture and grown in a 24-well plate (VWR, USA). Furthermore, no antibiotic was added to cultures of strains that contained a genomically integrated selection marker. The shaking speed was set to 200 rpm. The length and temperature of the cultivation were dependent on the organism and the sample requirements of the analyses. Strains that contained lacI were induced by adding 200 μM IPTG to the medium.
4.2. Plasmids and Strains
Plasmids and strains used in this study can be found in Table 1. All strains and plasmids generated in this work are publicly available through the JBEI registry (https://public-registry.jbei.org/folders/789). The codon optimized gene sequences of LipM1 fused to EryM6-TE were synthesized and cloned into the pBH026 vector backbone by Genscript (USA). The WT nucleotide sequences of LipM1 and EryM6-TE were obtained from S. aureofaciens Tü117 and S. erythraea NRRL2338 genomic DNA, respectively. Gibson primers were designed using the j5 software.54 PCR products and NdeI/XhoI (NEB, USA) digested pAN001 vector DNA were assembled using the Gibson assembly standard protocol. Assembly of pGingerBG-NahR constructs was conducted with PCR amplified vector DNA. Plasmids were isolated with the Qiaprep Spin Miniprep kit (Qiagen, Germany). Primers were synthesized and purchased from IDT (USA). A list of all used primers can be found in the Supporting Information (Table S2).
Table 1. Plasmids and Strains Used in This Study.
| plasmid | description | reference | JBEI part ID |
|---|---|---|---|
| pJH204 | BxB1 integration vector containing BxB1 attP site. Kanamycin selection marker and ColE1 origin flanked by ΦC31 attB and attP site. | (24) | |
| pBH026 | Derivative of BxB1 integration vector pJH204 with promoter LlacO1 repressed by LacI. Kanamycin selection marker, ColE1 origin, and lacI are flanked by ΦC31 attB and attP site. | This work | |
| pBH026 RFP | BxB1 integration vector with rfp gene | This work | JPUB_021284 |
| pK18 | Suicide vector for allelic replacement with KanR, SacB | (55) | |
| pK18 ΔprpDBC2 | Suicide vector for in-frame deletion of prpDBC2 | (34) | |
| pK18 ΔCgl0605::kivd-CCL4 | Suicide vector for replacement of Cgl0605 with kivd from Lactococcus lactis and CCL4 from Humulus lupulus | This work | JPUB_021290 |
| pK18 ΔCgl1016::sfp | Suicide vector for replacement of Cgl1016 with sfp from Bacillus subtilis | (34) | |
| pBH026 LipPKS-Cg_ubc | BxB1 integration vector with ubc codon optimized LipM1+TE gene for expression in C. glutamicum | This work | JPUB_021239 |
| pBH026 LipPKS-Cg_mcu | BxB1 integration vector with mcu codon optimized LipM1+TE gene for expression in C. glutamicum | This work | JPUB_021241 |
| pBH026 LipPKS-Cg_hrca | BxB1 integration vector with hrca codon optimized LipM1+TE gene for expression in C. glutamicum | This work | JPUB_021243 |
| pBH026 LipPKS-Pp_ubc | BxB1 integration vector with ubc codon optimized LipM1+TE gene for expression in P. putida | This work | JPUB_021306 |
| pBH026 LipPKS-Pp_mcu | BxB1 integration vector with mcu codon optimized LipM1+TE gene for expression in P. putida | This work | JPUB_021246 |
| pBH026 LipPKS-Pp_hrca | BxB1 integration vector with hrca codon optimized LipM1+TE gene for expression in P. putida | This work | JPUB_021248 |
| pBH026 LipPKS-Ec_ubc | BxB1 integration vector with ubc codon optimized LipM1+TE gene for expression in E. coli | This work | JPUB_021250 |
| pBH026 LipPKS-Ec_mcu | BxB1 integration vector with mcu codon optimized LipM1+TE gene for expression in E. coli | This work | JPUB_021237 |
| pBH026 LipPKS-Ec_hrca | BxB1 integration vector with hrca codon optimized LipM1+TE gene for expression in E. coli | This work | JPUB_021263 |
| pAN001 | Derivative of pBH026 with additional URA3 marker for yeast assembly | This work | |
| pAN001 LipPKS-WT_ATG | BxB1 integration vector with wildtype LipM1+EryM6-TE nucleotide sequence and ATG start codon | This work | JPUB_021267 |
| pAN001 LipPKS-WT_GTG | BxB1 integration vector with wildtype LipM1+EryM6-TE nucleotide sequence and GTG start codon | This work | JPUB_021265 |
| pJH209 | MR11 integration vector containing MR11 attP site. Kanamycin selection marker and ColE1 origin are flanked by ΦC31 attB and attP site. | (24) | |
| pJH209 Sc_mmCoA | MR11 integration vector with mmCoA pathway from S. cellulosum So56 under the control of Ptac | This work | JPUB_021252 |
| pGW30 | ColE1 vector with apramycin marker expressing ΦC31 integrase under the control of Ptac | (24) | |
| pALC412 | Derivative of pGW30 with optimized expression of ΦC31 integrase for use in C. glutamicum | Adam Guss, personal communication | |
| pGW31 | ColE1 vector with apramycin marker expressing BxB1 integrase under the control of Ptac | (24) | |
| pGW36 | ColE1 vector with apramycin marker expressing MR11 integrase under the control of Ptac | (24) | |
| pGingerBG-NahR | BBR1 vector with gentamicin marker containing the Psal/NahR inducible system | (56) | |
| pGingerBG-NahR LipPKS-Pp_mcu | Derivative of pGingerBG-NahR carrying LipPKS-Pp_mcu | This work | JPUB_021282 |
| pGingerBG-NahR LipPKS-Ec_mcu | Derivative of pGingerBG-NahR carrying LipPKS-Ec_mcu | This work | JPUB_021280 |
| strain | description | reference | JBEI part ID |
|---|---|---|---|
| E. coli XL1 Blue | Agilent | ||
| E. coli BL21(DE3) | NEB | ||
| Carrying replicating vector pBH026 LipPKS-Cg_ubc | This work | ||
| Carrying replicating vector pBH026 LipPKS-Cg_mcu | This work | ||
| Carrying replicating vector pBH026 LipPKS-Cg_hrca | This work | ||
| Carrying replicating vector pBH026 LipPKS-Pp_ubc | This work | ||
| Carrying replicating vector pBH026 LipPKS-Pp_mcu | This work | ||
| Carrying replicating vector pBH026 LipPKS-Pp_hrca | This work | ||
| Carrying replicating vector pBH026 LipPKS-Ec_ubc | This work | ||
| Carrying replicating vector pBH026 LipPKS-Ec_mcu | This work | ||
| Carrying replicating vector pBH026 LipPKS-Ec_hrca | This work | ||
| Carrying replicating vector pAN001 LipPKS-WT_ATG | This work | ||
| Carrying replicating vector pAN001 LipPKS-WT_GTG | This work | ||
| Carrying replicating vector pBH026 RFP | This work | ||
| E. coli K207–3 | Derivative of sfp-expressing E. coli strain BAP1 containing the PCCase pathway from S. coelicolor for the production of mmCoA. Both heterologous pathways are under the control of the T7 promoter. | (29) | |
| Carrying replicating vector pBH026 LipPKS-Cg_ubc | This work | ||
| Carrying replicating vector pBH026 LipPKS-Cg_mcu | This work | ||
| Carrying replicating vector pBH026 LipPKS-Cg_hrca | This work | ||
| Carrying replicating vector pBH026 LipPKS-Pp_ubc | This work | ||
| Carrying replicating vector pBH026 LipPKS-Pp_mcu | This work | ||
| Carrying replicating vector pBH026 LipPKS-Pp_hrca | This work | ||
| Carrying replicating vector pBH026 LipPKS-Ec_ubc | This work | ||
| Carrying replicating vector pBH026 LipPKS-Ec_mcu | This work | ||
| Carrying replicating vector pBH026 LipPKS-Ec_hrca | This work | ||
| Carrying replicating vector pAN001 LipPKS-WT_ATG | This work | ||
| Carrying replicating vector pAN001 LipPKS-WT_GTG | This work | ||
| AG6212 | Derivative of C. glutamicum ATCC13032 with poly attB site replacing Cgl1777–8 | Adam Guss, personal communication | |
| BxB1 integrated pBH026 RFP | This work | JPUB_021285 | |
| BxB1 integrated pBH026 RFP with excised vector backbone | This work | JPUB_021287 | |
| AG6212cz | Derivative of AG6212 with the following modifications: ΔprpDBC2ΔCgl0605::kivd-CCL4ΔCgl1016::sfp | This work | JPUB_021253 |
| BxB1 integrated pBH026 LipPKS-Cg_ubc | This work | JPUB_021254 | |
| BxB1 integrated pBH026 LipPKS-Cg_mcu with excised vector backbone | This work | JPUB_021255 | |
| BxB1 integrated pBH026 LipPKS-Cg_hrca with excised vector backbone | This work | JPUB_021256 | |
| BxB1 integrated pBH026 LipPKS-Pp_ubc with excised vector backbone | This work | JPUB_021257 | |
| BxB1 integrated pBH026 LipPKS-Pp_mcu with excised vector backbone | This work | JPUB_021258 | |
| BxB1 integrated pBH026 LipPKS-Pp_hrca with excised vector backbone | This work | JPUB_021259 | |
| BxB1 integrated pBH026 LipPKS-Ec_ubc | This work | JPUB_021260 | |
| BxB1 integrated pBH026 LipPKS-Ec_mcu with excised vector backbone | This work | JPUB_021261 | |
| BxB1 integrated pBH026 LipPKS-Ec_hrca with excised vector backbone | This work | JPUB_021262 | |
| BxB1 integrated pAN001 LipPKS-WT_ATG with excised vector backbone | This work | JPUB_021267 | |
| BxB1 integrated pAN001 LipPKS-WT_GTG with excised vector backbone | This work | JPUB_021264 | |
| AG5577 | Derivative of P. putida KT2440 with three triple attB sites replacing PP_4740 and PP_2876 and integrated between PP_4217/PP_4218, respectively | Adam Guss, personal communication | |
| BxB1 integrated pBH026 RFP | This work | JPUB_021283 | |
| BxB1 integrated pBH026 RFP with excised vector backbone | This work | JPUB_021286 | |
| BxB1 integrated pBH026 LipPKS-Cg_ubc with excised vector backbone | This work | JPUB_021238 | |
| BxB1 integrated pBH026 LipPKS-Cg_mcu with excised vector backbone | This work | JPUB_021240 | |
| BxB1 integrated pBH026 LipPKS-Cg_hrca with excised vector backbone | This work | JPUB_021242 | |
| BxB1 integrated pBH026 LipPKS-Pp_ubc with excised vector backbone | This work | JPUB_021244 | |
| BxB1 integrated pBH026 LipPKS-Pp_mcu with excised vector backbone | This work | JPUB_021245 | |
| BxB1 integrated pBH026 LipPKS-Pp_hrca with excised vector backbone | This work | JPUB_021247 | |
| BxB1 integrated pBH026 LipPKS-Ec_ubc with excised vector backbone | This work | JPUB_021249 | |
| BxB1 integrated pBH026 LipPKS-Ec_mcu | This work | JPUB_021236 | |
| BxB1 integrated pBH026 LipPKS-Ec_hrca | This work | JPUB_021292 | |
| BxB1 integrated pAN001 LipPKS-WT_ATG with excised vector backbone | This work | JPUB_021294 | |
| BxB1 integrated pAN001 LipPKS-WT_GTG with excised vector backbone | This work | JPUB_021293 | |
| AG5577mm | MR11 integrated pJH209 Sc_mmCoA for the production of mmCoA. | This work | JPUB_021251 |
| BxB1 integrated pBH026 LipPKS-Cg_ubc with excised vector backbone | This work | JPUB_021295 | |
| BxB1 integrated pBH026 LipPKS-Cg_mcu with excised vector backbone | This work | JPUB_021296 | |
| BxB1 integrated pBH026 LipPKS-Cg_hrca with excised vector backbone | This work | JPUB_021297 | |
| BxB1 integrated pBH026 LipPKS-Pp_ubc with excised vector backbone | This work | JPUB_021298 | |
| BxB1 integrated pBH026 LipPKS-Pp_mcu with excised vector backbone | This work | JPUB_021299 | |
| BxB1 integrated pBH026 LipPKS-Pp_hrca with excised vector backbone | This work | JPUB_021300 | |
| BxB1 integrated pBH026 LipPKS-Ec_ubc with excised vector backbone | This work | JPUB_021301 | |
| BxB1 integrated pBH026 LipPKS-Ec_mcu | This work | JPUB_021302 | |
| BxB1 integrated pBH026 LipPKS-Ec_hrca | This work | JPUB_021303 | |
| BxB1 integrated pAN001 LipPKS-WT_ATG with excised vector backbone | This work | JPUB_021305 | |
| BxB1 integrated pAN001 LipPKS-WT_GTG with excised vector backbone | This work | JPUB_021304 |
Heterologous genes were integrated into the host genome or expressed from plasmid DNA. Integrations of pBH026 or pAN001 vectors and backbone excisions were performed using the SAGE system.24 Excision of the backbones of pBH026 or pAN001 using pGW30 or pALC412 leads to the removal of the repressor lacI, the selection marker, and origin of replication. Serine recombinase-assisted integrations into the hosts AG5577 and AG6212 were confirmed by colony PCR (cPCR) using primers that flank the specific attB site in the host genome and primers that anneal to the 5′ and 3′ end of the target gene.
Engineering of C. glutamicum metabolism was performed by first inoculating EPO medium (37 g of BHI powder, 25 g of glycine, 10 mL of Tween, 4 g of isoniazid) with a BHI overnight culture.34 Cells were then grown for 5–6 h until reaching an optical density (OD) of approximately 1 (λ = 600 nm). Following growth, the cells were washed three times with ice-cold 10% glycerol and then resuspended in 1 mL of ice-cold 10% glycerol. From this suspension, 80 μL of competent cells was transferred to ice-cold electroporation cuvettes with a 2 mm gap. Subsequently, 500 ng of plasmid DNA was used for transformation.
After electroporation, cells were resuspended in 1 mL of BHI medium and heat shocked at 46 °C for 5 min. Next, cells were recovered at 30 °C for 1–2 h and cultures were plated on BHI agar supplemented with 25 μg/mL kanamycin and incubated for 2 days. Single colonies were streaked on BHI agar containing 10% sucrose and incubated at 30 °C for 1–2 days to allow for the removal of the selection marker. Finally, individual colonies were picked and verified using cPCR (Table S2).
4.3. Plate Reader Assay for RFP Measurement
Cell cultures were harvested after growing for 24 h at 30 and 37 °C, respectively. After centrifuging 500 μL of sample, the cell pellets were washed with 500 μL of 0.9% NaCl solution. OD and fluorescence measurements were performed in a Biotek Synergy H1M plate reader (BioTek, USA) using a black 96-well plate (Corning, USA) and a 100 μL sample volume. The OD was measured at 600 nm. The fluorescence of RFP was excited at 535 nm, and emission was measured at 620 nm.
4.4. Codon Optimization of Engineered PKS
Codon optimizations were performed by using DNA Chisel and the Kazusa database. The target hosts were E. coli str. K-12 substr. W3110 (NCBI: txid316407), C. glutamicum ATCC13032 (NCBI: txid196627), and P. putida KT2440 (NCBI: txid160488). For the hrca method, the LipPKS part was harmonized with S. aureofaciens (NCBI: txid1894) and the EryPKS part with S. erythraea (NCBI: txid1836). To ensure reproducible results, the Numpy random generator was set to 123. A list of all the applied constraints can be found in the Supporting Information (Table S1).
The website for the GUI based on DNA Chisel (https://basebuddy.lbl.gov) was built using the open-source app framework Streamlit. To view the associated files and scripts, or to run a local version of BaseBuddy, see the Github repository (https://github.com/jbei/basebuddy).
4.5. Proteomics Analysis
Strains for sample preparation were grown at 30 and 37 °C, respectively. After 48 h, 1 mL of the cell culture was collected and consolidated in a 96-well plate. Next, the supernatant was removed by centrifuging the plates at 4000g for 10 min. Cell pellets were then stored at −80 °C until further processing.
Protein was extracted and tryptic peptides were prepared by following established proteomic sample preparation protocols.57 Briefly, cell pellets were resuspended in a Qiagen P2 Lysis Buffer (Qiagen, Germany) to promote cell lysis. Proteins were precipitated with addition of 1 mM NaCl and 4 vol % acetone, followed by two additional washes with 80% acetone in water. The recovered protein pellet was homogenized by pipette mixing with 100 mM ammonium bicarbonate in 20% methanol. Protein concentration was determined by the DC protein assay (BioRad, USA). Protein reduction was accomplished using 5 mM tris 2-(carboxyethyl)phosphine (TCEP) for 30 min at room temperature, and alkylation was performed with 10 mM iodoacetamide (IAM; final concentration) for 30 min at room temperature in the dark. Overnight digestion with trypsin was accomplished with a 1:50 trypsin:total protein ratio. The resulting peptide samples were analyzed on an Agilent 1290 UHPLC system coupled to a Thermo Scientific Orbitrap Exploris 480 mass spectrometer for discovery proteomics.58 Briefly, peptide samples were loaded onto an Ascentis ES-C18 Column (Sigma–Aldrich, USA) and were eluted from the column by using a 10 min gradient from 98% solvent A (0.1% formic acid in H2O) and 2% solvent B (0.1% formic acid in acetonitrile) to 65% solvent A and 35% solvent B. Eluting peptides were introduced to the mass spectrometer operating in positive-ion mode and were measured in data-independent acquisition (DIA) mode with a duty cycle of 3 survey scans from m/z 380 to m/z 985 and 45 MS2 scans with precursor isolation width of 13.5 m/z to cover the mass range. DIA raw data files were analyzed by an integrated software suite DIA-NN.59 The databases used in the DIA-NN search (library-free mode) are the respective microorganisms’ latest Uniprot proteome FASTA sequences plus the protein sequences of the heterologous proteins and common proteomic contaminants. DIA-NN determines mass tolerances automatically based on first pass analysis of the samples with automated determination of optimal mass accuracies. The retention time extraction window was determined individually for all MS runs analyzed via the automated optimization procedure implemented in DIA-NN. Protein inference was enabled, and the quantification strategy was set to Robust LC = High Accuracy. Output main DIA-NN reports were filtered with a global False Discovery Rate (FDR) of 0.01 on both the precursor level and protein group level. The Top3 method, which is the average MS signal response of the three most intense tryptic peptides of each identified protein, was used to plot the quantity of the targeted proteins in the samples.60,61
The generated mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD042749.62 DIA-NN is freely available for download from https://github.com/vdemichev/DiaNN.
4.6. Polyketide Production and Quantification by Liquid Chromatography and Mass Spectrometry
The conditions used for polyketide production were dependent on the production host. C. glutamicum cultures were grown for 96 h, while P. putida cells were cultivated for 48 h. Furthermore, the BHI medium used for C. glutamicum was supplemented with 5 mM isobutyric acid and 5 mM propionate. The LB medium for P. putida contained an additional 20 mM of l-valine.
Polyketide production in E. coli K207–3 followed the same procedure as described by Yuzawa et al. (2017).20 In short, cells were grown at 18 °C for 120 h in LB containing 5 mM propionate, kanamycin and 200 μM IPTG.
After cultivation of each host was completed, 300 μL of the cell culture was harvested and quenched using 300 μL of −80 °C cold methanol. Next, samples were centrifuged at 14,000g for 1 min, and 300 μL of supernatant was transferred into Omega 3K MWCO AcroPrep 96-well filter plates (Pall, USA). After centrifuging for 45 min at 3,000g, the flow-through was collected and used for further analysis.
LC separation of 3-hydroxy acids was conducted using a Kinetex XB-C18 column (100 mm length, 3 mm internal diameter, and 2.6 μm particle size; Phenomenex, USA) with an Agilent 1260 Infinity II LC System (Agilent Technologies, USA) at room temperature. The mobile phase consisted of 0.1% formic acid in water (solvent A) and 0.1% formic acid in methanol (solvent B). The separation of products was achieved with a flow rate of 0.42 mL/min, using the following gradient: 20% to 72.1% B over 6.5 min, 72.1% to 95% B over 1.3 min, and held for 1 min. Subsequently, the flow rate was increased to 0.65 mL/min, and the gradient was 95% to 20% B over 0.2 min, held for 1.2 min.
To identify and quantify the 3-hydroxy acids, the LC system was coupled to an Agilent InfinityLab LC/MSD iQ single quadrupole mass spectrometer (Agilent Technologies, USA), with electrospray ionization (ESI) conducted in negative-ion mode. Identification of 3-hydroxy acids was performed by comparing the mass and retention time to authentic standards.
4.7. Quantitative Reverse Transcription PCR
For RT-qPCR analysis, samples were collected after 24 h by centrifuging 1 mL of cell culture and discarding the supernatant. Total RNA was then extracted using the RNeasy Plus Universal Mini Kit (Qiagen, Germany) following the manufacturer’s instructions. Prior to RNA extraction of C. glutamicum samples, cell pellets were treated with 1 mL of 2 mg/mL lysozyme (Roche, Switzerland) and incubated at 30 °C for 10 min. The RNA extracted from the samples was used as a template for complementary DNA (cDNA) synthesis, which was carried out using the LunaScript RT SuperMix Kit (NEB, USA). Subsequently, qPCR was performed using Luna Universal qPCR Master Mix (NEB, USA). The PCR mixtures were cycled at 95 °C for 1 min (one cycle) followed by 40 cycles at 95 °C for 15 s and 60 °C for 30 s. The amplification profile was monitored on a CFX96 Real-Time PCR Detection System (Bio-Rad, USA). The expression ratio from qPCR was calculated from the Ct value difference between target gene and rpoD for P. putida, E. coli or rpoC for C. glutamicum as housekeeping genes. Primers used for housekeeping genes and target gene quantification can be found in the Supporting Information (Table S2).
4.8. Bioinformatic Analyses
For phylogenetic analysis, 16S rRNA sequences were extracted from the RNAcentral database.63 The sequences were aligned using the CLUSTAL W algorithm, implemented in MEGA 11.64 Subsequently, a phylogenetic tree was constructed by using the maximum-likelihood method with the Tamura-Nei model.
Codon usage data for organisms in part of the Refseq index were obtained from the CoCoPUTS database. For each entry in the database (235,025 entries), the codon usage data was normalized by the total number of respective codons analyzed. Taxonomic classifications corresponding to each Refseq entry were retrieved from the National Center for Biotechnology Information (NCBI) database, providing a hierarchical framework for taxonomic inference. To discern the general similarity of codon usage across organisms, PCA was implemented on the normalized codon tables. The first two principal components, accounting for the highest explained variance, were selected and visualized via a scatter plot.
Acknowledgments
We would like to express our profound appreciation to Adam Guss and his colleagues for generously providing us with the SAGE vector suite, as well as the strains P. putida AG5577 and C. glutamicum AG6212. Their contributions have been instrumental in advancing our research. Additionally, we want to thank Peter Mellinger and Chenyi Li for their assistance during various stages of the cloning process. This research was funded by the DOE Joint BioEnergy Institute (https://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research through contract [DE-AC02-05CH11231] between Lawrence Berkeley National Laboratory and the U.S. Department of Energy, by award EE0008926 from the BioEnergy Technologies Office, Office of Energy Efficiency and Renewable Energy, U.S. Department of Energy, and the Philomathia Foundation. A.A.N. was supported by a National Science Foundation Graduate Research Fellowship, fellow ID [2018253421].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.3c00367.
(Table S1) List of all applied constraints during codon optimization; (Table S2) Primers used in this study; (Figure S1) Principal component analysis of optimized LipPKS sequences; (Figure S2) Regression plot of LipPKS counts and relative transcript for C. glutamicum, E. coli, and P. putida; (Figure S3) LC-MS chromatograms showing the production of unnatural polyketides in C. glutamicum, E. coli, and P. putida; (Figure S4) Polyketide synthesis mechanism with various loading substrates and possible products; (Figure S5) Detection of peptides for supplementary pathways in the C. glutamicum ATCC 13032 derivative AG6212cz; (Figure S6) Expression of the LipPKS codon variants Pp_mcu and Ec_mcu from the inducible vector system pGingerBG-NahR in P. putida (PDF)
Author Contributions
Conceptualization: M.S.; Methodology, M.S.; Investigation: M.S., C.Z., N.L., A.A.N., J.B.R., L.S.K., A.A.V., C.J.P., Y.C.; Writing—Original Draft: M.S.; Writing—Review and Editing: All authors; Resources and supervision: R.W.H., L.M.B., J.D.K.
Author Contributions
¶ N.L. and C.Z. contributed equally.
The authors declare the following competing financial interest(s): J.D.K. has financial interests in Amyris, Ansa Biotechnologies, Apertor Pharma, Berkeley Yeast, Cyklos Materials, Demetrix, Lygos, Napigen, ResVita Bio, and Zero Acre Farms.
Supplementary Material
References
- Weissman K. J. Genetic engineering of modular PKSs: from combinatorial biosynthesis to synthetic biology. Nat. Prod. Rep. 2016, 33, 203–230. 10.1039/C5NP00109A. [DOI] [PubMed] [Google Scholar]
- Donadio S.; Staver M. J.; McAlpine J. B.; Swanson S. J.; Katz L. Modular organization of genes required for complex polyketide biosynthesis. Science 1991, 252, 675–679. 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- Hagen A.; et al. Engineering a polyketide synthase for in vitro production of adipic acid. ACS Synth. Biol. 2016, 5, 21–27. 10.1021/acssynbio.5b00153. [DOI] [PubMed] [Google Scholar]
- Yuzawa S.; Mirsiaghi M.; Jocic R.; Fujii T.; Masson F.; Benites V. T.; Baidoo E. E. K.; Sundstrom E.; Tanjore D.; Pray T. R.; George A.; Davis R. W.; Gladden J. M.; Simmons B. A.; Katz L.; Keasling J. D. Short-chain ketone production by engineered polyketide synthases in Streptomyces albus. Nat. Commun. 2018, 9, 4569. 10.1038/s41467-018-07040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa T.; Fitzgerald B. J.; Keatinge-Clay A. T. Preparative production of an enantiomeric pair by engineered polyketide synthases. Chem. Commun. 2021, 57, 8762–8765. 10.1039/D1CC03073F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B. A.; Khosla C. Biosynthesis of polyketides in heterologous hosts. Microbiol. Mol. Biol. Rev. 2001, 65, 106–118. 10.1128/MMBR.65.1.106-118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B. A.; Admiraal S. J.; Gramajo H.; Cane D. E.; Khosla C. Biosynthesis of Complex Polyketides in a Metabolically Engineered Strain of E. coli. Science (New York, N.Y.) 2001, 291, 1790–1792. 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- Ongley S. E.; Bian X.; Neilan B. A.; Müller R. Recent advances in the heterologous expression of microbial natural product biosynthetic pathways. Nat. Prod. Rep. 2013, 30, 1121–1138. 10.1039/c3np70034h. [DOI] [PubMed] [Google Scholar]
- Nakamura Y.; Gojobori T.; Ikemura T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000, 28, 292. 10.1093/nar/28.1.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskiw B. K.; Bibb M. J.; Chater K. F. The use of a rare codon specifically during development?. Mol. Microbiol. 1991, 5, 2861–2867. 10.1111/j.1365-2958.1991.tb01845.x. [DOI] [PubMed] [Google Scholar]
- Belinky F.; Rogozin I. B.; Koonin E. V. Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 2017, 7, 12422. 10.1038/s41598-017-12619-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P. M.; Li W. H. The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale R. S.; Thompson G. Codon optimization of the gene encoding a domain from human type 1 neurofibromin protein results in a threefold improvement in expression level in Escherichia coli. Protein Expr. Purif. 1998, 12, 185–188. 10.1006/prep.1997.0825. [DOI] [PubMed] [Google Scholar]
- Claassens N. J.; et al. Improving heterologous membrane protein production in Escherichia coli by combining transcriptional tuning and codon usage algorithms. PLoS One 2017, 12, e0184355 10.1371/journal.pone.0184355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulkower V.; Rosser S. DNA Chisel, a versatile sequence optimizer. Bioinformatics 2020, 36, 4508–4509. 10.1093/bioinformatics/btaa558. [DOI] [PubMed] [Google Scholar]
- Fu H.; Liang Y.; Zhong X.; Pan Z.; Huang L.; Zhang H.; Xu Y.; Zhou W.; Liu Z. Codon optimization with deep learning to enhance protein expression. Sci. Rep. 2020, 10, 17617. 10.1038/s41598-020-74091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchert A. J.; Bleem A.; Beckham G. T. RB-TnSeq identifies genetic targets for improved tolerance of Pseudomonas putida towards compounds relevant to lignin conversion. Metab. Eng. 2023, 77, 208–218. 10.1016/j.ymben.2023.04.007. [DOI] [PubMed] [Google Scholar]
- Eggeling L.; Bott M. A giant market and a powerful metabolism: L-lysine provided by Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2015, 99, 3387–3394. 10.1007/s00253-015-6508-2. [DOI] [PubMed] [Google Scholar]
- Rosano G. L.; Ceccarelli E. A. Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 2014, 5, 172. 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzawa S.; et al. Heterologous Gene Expression of N-Terminally Truncated Variants of LipPks1 Suggests a Functionally Critical Structural Motif in the N-terminus of Modular Polyketide Synthase. ACS Chem. Biol. 2017, 12, 2725–2729. 10.1021/acschembio.7b00714. [DOI] [PubMed] [Google Scholar]
- Yuzawa S.; Eng C. H.; Katz L.; Keasling J. D. Broad substrate specificity of the loading didomain of the lipomycin polyketide synthase. Biochemistry 2013, 52, 3791–3793. 10.1021/bi400520t. [DOI] [PubMed] [Google Scholar]
- Athey J.; Alexaki A.; Osipova E.; Rostovtsev A.; Santana-Quintero L. V.; Katneni U.; Simonyan V.; Kimchi-Sarfaty C. A new and updated resource for codon usage tables. BMC Bioinformatics 2017, 18, 391. 10.1186/s12859-017-1793-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb D. D.; Alexaki A.; Katneni U.; Kimchi-Sarfaty C. The Kazusa codon usage database, CoCoPUTs, and the value of up-to-date codon usage statistics. Infect. Genet. Evol. 2019, 73, 266–268. 10.1016/j.meegid.2019.05.010. [DOI] [PubMed] [Google Scholar]
- Elmore J. R.; Dexter G. N.; Baldino H.; Huenemann J. D.; Francis R.; Peabody G. L.; Martinez-Baird J.; Riley L. A.; Simmons T.; Coleman-Derr D.; Guss A. M.; Egbert R. G. High-throughput genetic engineering of nonmodel and undomesticated bacteria via iterative site-specific genome integration. Sci. Adv. 2023, 9, eade1285 10.1126/sciadv.ade1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore J. R.; Furches A.; Wolff G. N.; Gorday K.; Guss A. M. Development of a high efficiency integration system and promoter library for rapid modification of Pseudomonas putida KT2440. Metab. Eng. Commun. 2017, 5, 1–8. 10.1016/j.meteno.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lethanh H.; Neubauer P.; Hoffmann F. The small heat-shock proteins IbpA and IbpB reduce the stress load of recombinant Escherichia coli and delay degradation of inclusion bodies. Microb. Cell Fact. 2005, 4, 6. 10.1186/1475-2859-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen M. A.; Kurland C. G.; Pedersen S. Codon usage determines translation rate in Escherichia coli. J. Mol. Biol. 1989, 207, 365–377. 10.1016/0022-2836(89)90260-X. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Dang Y.; Zhou M.; Li L.; Yu C.-h.; Fu J.; Chen S.; Liu Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E6117–E6125. 10.1073/pnas.1606724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murli S.; Kennedy J.; Dayem L. C.; Carney J. R.; Kealey J. T. Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide B production. J. Ind. Microbiol. Biotechnol. 2003, 30, 500–509. 10.1007/s10295-003-0073-x. [DOI] [PubMed] [Google Scholar]
- Gross F.; Gottschalk D.; Müller R. Posttranslational modification of myxobacterial carrier protein domains in Pseudomonas sp. by an intrinsic phosphopantetheinyl transferase. Appl. Microbiol. Biotechnol. 2005, 68, 66–74. 10.1007/s00253-004-1836-7. [DOI] [PubMed] [Google Scholar]
- Marshall V. D.; Sokatch J. R. Regulation of valine catabolism in Pseudomonas putida. J. Bacteriol. 1972, 110, 1073–1081. 10.1128/jb.110.3.1073-1081.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross F.; et al. Metabolic engineering of Pseudomonas putida for methylmalonyl-CoA biosynthesis to enable complex heterologous secondary metabolite formation. Chem. Biol. 2006, 13, 1253–1264. 10.1016/j.chembiol.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Botella L.; Lindley N. D.; Eggeling L. Formation and metabolism of methylmalonyl coenzyme A in Corynebacterium glutamicum. J. Bacteriol. 2009, 191, 2899–2901. 10.1128/JB.01756-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C.; et al. Improved polyketide production in C. glutamicum by preventing propionate-induced growth inhibition. Nat. Metab. 2023, 5, 1127. 10.1038/s42255-023-00830-x. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Woodruff A. P.; Xiong M.; Zhou J.; Dhande Y. K. A synthetic metabolic pathway for production of the platform chemical isobutyric acid. ChemSusChem 2011, 4, 1068–1070. 10.1002/cssc.201100045. [DOI] [PubMed] [Google Scholar]
- Xu H.; et al. Characterization of the formation of branched short-chain fatty acid:CoAs for bitter acid biosynthesis in hop glandular trichomes. Mol. Plant 2013, 6, 1301–1317. 10.1093/mp/sst004. [DOI] [PubMed] [Google Scholar]
- Lang K.; Zierow J.; Buehler K.; Schmid A. Metabolic engineering of Pseudomonas sp. strain VLB120 as platform biocatalyst for the production of isobutyric acid and other secondary metabolites. Microb. Cell Fact. 2014, 13, 2. 10.1186/1475-2859-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palluk S.; et al. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol. 2018, 36, 645–650. 10.1038/nbt.4173. [DOI] [PubMed] [Google Scholar]
- Muir P.; Li S.; Lou S.; Wang D.; Spakowicz D. J; Salichos L.; Zhang J.; Weinstock G. M.; Isaacs F.; Rozowsky J.; Gerstein M. The real cost of sequencing: scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 53. 10.1186/s13059-016-0917-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeschel S. C.; et al. Novel broad host range shuttle vectors for expression in Escherichia coli, Bacillus subtilis and Pseudomonas putida. J. Biotechnol. 2012, 161, 71–79. 10.1016/j.jbiotec.2012.02.020. [DOI] [PubMed] [Google Scholar]
- Lin-Chao S.; Bremer H. Effect of the bacterial growth rate on replication control of plasmid pBR322 in Escherichia coli. Mol. Gen. Genet. 1986, 203, 143–149. 10.1007/BF00330395. [DOI] [PubMed] [Google Scholar]
- San Millan A.; et al. Integrative analysis of fitness and metabolic effects of plasmids in Pseudomonas aeruginosa PAO1. ISME J. 2018, 12, 3014–3024. 10.1038/s41396-018-0224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Gonzalez A.; Rodriguez-R L. M.; Marston C. K.; Gee J. E.; Gulvik C. A.; Kolton C. B.; Saile E.; Frace M.; Hoffmaster A. R.; Konstantinidis K. T. Genomic Characterization and Copy Number Variation of Bacillus anthracis Plasmids pXO1 and pXO2 in a Historical Collection of 412 Strains. mSystems 2018, 10.1128/mSystems.00065-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J.; et al. Investigation of plasmid-induced growth defect in Pseudomonas putida. J. Biotechnol. 2016, 231, 167–173. 10.1016/j.jbiotec.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Anthony Mason C.; Bailey J. E. Effects of plasmid presence on growth and enzyme activity of Escherichia coli DH5?. Appl. Microbiol. Biotechnol. 1989, 32, 54–60. 10.1007/BF00164823. [DOI] [Google Scholar]
- Silva F.; Queiroz J. A.; Domingues F. C. Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol. Adv. 2012, 30, 691–708. 10.1016/j.biotechadv.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Kang M.-K.; et al. Synthetic biology platform of CoryneBrick vectors for gene expression in Corynebacterium glutamicum and its application to xylose utilization. Appl. Microbiol. Biotechnol. 2014, 98, 5991–6002. 10.1007/s00253-014-5714-7. [DOI] [PubMed] [Google Scholar]
- Li M.; Chen J.; Wang Y.; Liu J.; Huang J.; Chen N.; Zheng P.; Sun J. Efficient Multiplex Gene Repression by CRISPR-dCpf1 in Corynebacterium glutamicum. Front. Bioeng. Biotechnol. 2020, 8, 357. 10.3389/fbioe.2020.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F.; Zhou Z.; Dang Y.; Na H.; Adam C.; Lipzen A.; Ng V.; Grigoriev I. V.; Liu Y. Genome-wide role of codon usage on transcription and identification of potential regulators. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2022590118. 10.1073/pnas.2022590118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster M. W.; Weixlbaumer A. The intricate relationship between transcription and translation. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2106284118. 10.1073/pnas.2106284118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P.; Peterkofsky A.; McKenney K. Translational efficiency of the Escherichia coli adenylate cyclase gene: mutating the UUG initiation codon to GUG or AUG results in increased gene expression. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 5656–5660. 10.1073/pnas.82.17.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas A.; Kropinski A. M. An analysis of initiation codon utilization in the Domain Bacteria - concerns about the quality of bacterial genome annotation. Microbiology (Reading, Engl) 2008, 154, 2559–2661. 10.1099/mic.0.2008/021360-0. [DOI] [PubMed] [Google Scholar]
- Constant D. A.Deep learning-based codon optimization with large-scale synonymous variant datasets enables generalized tunable protein expression. bioRxiv, February 12, 2023. 10.1101/2023.02.11.528149. [DOI]
- Hillson N. J.; Rosengarten R. D.; Keasling J. D. j5 DNA assembly design automation software. ACS Synth. Biol. 2012, 1, 14–21. 10.1021/sb2000116. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; et al. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 1994, 145, 69–73. 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Pearson A. N.; Thompson M. G.; Kirkpatrick L. D.; Ho C.; Vuu K. M.; Waldburger L. M.; Keasling J. D.; Shih P. M. The pGinger Family of Expression Plasmids. Microbiol. Spectr. 2023, 11, e0037323 10.1128/spectrum.00373-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Gin J.; Petzold C. J. Alkaline-SDS cell lysis of microbes with acetone protein precipitation for proteomic sample preparation in 96-well plate format V.1. protocols.io 2023, 10.17504/protocols.io.6qpvr6xjpvmk/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Gin J.; Petzold C. J. Discovery proteomic (DIA) LC-MS/MS data acquisition and analysis v1. protocols.io 2022, 10.17504/protocols.io.e6nvwk1z7vmk/v1. [DOI] [Google Scholar]
- Demichev V.; Messner C. B.; Vernardis S. I.; Lilley K. S.; Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J. C.; Gorenstein M. V.; Li G.-Z.; Vissers J. P. C.; Geromanos S. J. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 2006, 5, 144–156. 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- Ahrné E.; Molzahn L.; Glatter T.; Schmidt A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics 2013, 13, 2567–2578. 10.1002/pmic.201300135. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- et al. RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. 10.1093/nar/gkw1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K.; Stecher G.; Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. 10.1093/molbev/msab120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.