

Summary
The C-C chemokine receptor type 5 (CCR5) expressed on immune cells supports inflammatory responses by directing cells to the inflammation site. CCR5 is also a major coreceptor for macrophage tropic human immunodeficiency viruses (R5-HIV-1) and its variants can confer protection from HIV infection, making it an ideal candidate to target for therapy. We developed a stepwise protocol that differentiates induced pluripotent stem cells (iPSCs) from individuals homozygous for the CCR5Δ32 variant and healthy volunteers into myeloid lineage induced monocytes (iMono) and macrophages (iMac). By characterizing iMono and iMac against their primary counterparts, we demonstrated that CCR5Δ32 homozygous cells are endowed with similar pluripotent potential for self-renewal and differentiation as iPSC lines generated from non-variant individuals while also showing resistance to HIV infection. In conclusion, these cells are a platform to investigate CCR5 pathophysiology in HIV-positive and negative individuals and to help develop novel therapies.
Subject areas: Molecular biology, Immunology, Stem cells research
Graphical abstract
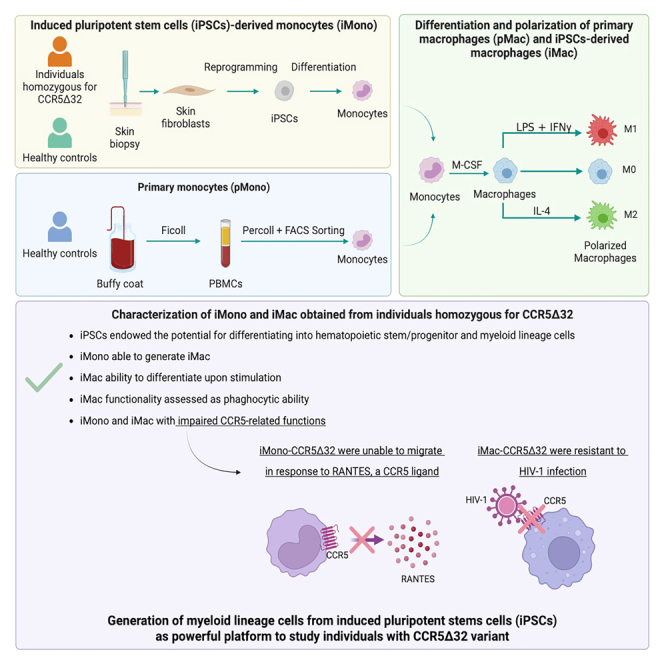
Highlights
-
•
iPSCs derived from CCR5Δ32 subjects can differentiate into myeloid lineage cells
-
•
iMono derived from CCR5Δ32 subjects were unable to migrate in response to RANTES
-
•
iMac derived from CCR5Δ32 subjects were resistant to HIV-1 infection
-
•
CCR5Δ32 subject iMono/iMac are a new platform to investigate CCR5 pathophysiology
Molecular biology; Immunology; Stem cells research
Introduction
The C-C chemokine receptor type 5 (CCR5) is a 7-transmembrane protein member of the G protein–coupled receptor family. It is expressed on the cell surface of monocytes and dendritic cells and on activated T cells, macrophages, and natural killer (NK) cells in both lymphoid and nonlymphoid tissues.1 CCR5 is involved in the inflammatory responses by directing cells to sites of inflammation. Indeed, CCR5 regulates intracellular trafficking and protective cellular and humoral responses by interacting with its natural ligands: Regulated upon Activation, Normal T cell Expressed and Presumably Secreted (RANTES, CCL5), macrophage inflammatory protein-1 alpha (MIP1α, CCL3), MIP1β (CCL4), and monocyte chemotactic protein-2 (MCP-2, CCL8).2,3 In addition, CCR5 is best known as a major coreceptor for macrophage tropic human immunodeficiency viruses (R5-HIV-1 virus).4 In 1996, the CCR5 c.554_585del p.(Ser185fs) (CCR5Δ32) variant was discovered, a 32-base-pair deletion that introduces a frameshift and a premature stop codon into the CCR5 receptor locus, resulting in a nonfunctional receptor.5,6,7 This genetic variant confers protection from HIV infection.5,6,7 Specifically, individuals homozygous for CCR5Δ32 are resistant to certain HIV-1 infection as they do not express functional CCR5 receptors, whereas individuals heterozygous for CCR5Δ32 have a slower progression of HIV infection and development of acquired immunodeficiency syndrome (AIDS) as they are characterized by a reduced CCR5 expression due to the dimerization of mutant and wild-type receptors that interferes with CCR5 transport to the cell surface.5,7
Starting from the identification of the CCR5Δ32 variant and the observation of its role in preventing HIV infection, CCR5 has been proposed as therapeutic target for HIV treatment.8,9 The proof of concept for CCR5 targeting is represented by the so-called Berlin patient who, in 2009, was reported to be cured of HIV with the transplantation of hematopoietic stem cells (HSCs) of a donor homozygous for CCR5Δ32 variant.10,11 Recently, Gupta et al. described another case in which a single allogeneic HSC transplantation with homozygous CCR5Δ32 donor cells was sufficient to achieve HIV-1 remission,12 and a recent study following a 53-year old male with long term HIV-1 cure.13 Among the strategies proposed to target CCR5, its elimination by gene editing represents the most radical approach.8,9 Indeed, the functional knock-out of CCR5 expression in patient cells could allow autologous transplant thus avoiding Graft versus Host Disease (GvHD) and overcoming the issues related to the identification of an HLA-matched donor being also homozygous for CCR5Δ32. Recently, successful disruption of CCR5 gene function has been reported in human pluripotent stem cells (PSCs) using zinc finger nucleases (ZFNs)14 and a combination of transcription activator-like effector nucleases (TALENs) and PiggyBac technology or clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein-9 (Cas9) gene-editing systems combined with PiggyBac technology.15 Kang and colleagues reported the successful targeting of CCR5 in GFP-marked human induced pluripotent stem cells (iPSCs) using CRISPR/Cas9 with single and dual guide RNAs (gRNAs) demonstrating that the dual gRNA approach significantly increased the frequency of biallelic CCR5 gene editing without compromising specificity.16
The use of gene therapy techniques to reproduce the naturally occurring CCR5Δ32 deletion is supported by the fact that individuals homozygous for CCR5Δ32 variant do not show any obvious clinical symptoms because of the redundancy of chemokine interaction with their putative ligands. However, new evidence show that this system is not always able to fully compensate the absence or reduced expression of a given component. Indeed, CCR5Δ32 deletion beside being associated with protection from HIV-1 infection, it was reported also to be deleterious in other situations by increasing risk of susceptibility as well as the probability to develop severe outcomes in several diseases.8,17,18
For these reasons, developing a platform that allow a more detailed characterization of immune cells in individuals homozygous for CCR5Δ32 variant is crucial to extend the knowledge on CCR5 by disclosing the nonredundant roles of this molecules and investigating the effect of CCR5Δ32 deletion on immune cell functionality.17,18 Regarding monocytes and macrophages, their characterization is often limited by the fact that when studied as primary cells, mainly derived from peripheral blood mononuclear cells (PBMCs), they do not self-renew, and their availability is limited in number. These issues can now be overcome by iPSC technology. Indeed, several studies demonstrated that human iPSCs could be a potent source of monocytes and macrophages and that iPSCs can provide an unlimited source of subject genotype-specific cells.19,20
Our group recently generated iPSC lines from individuals homozygous for CCR5Δ32 variant, demonstrating that they are endowed with a similar pluripotent potential for self-renewal and differentiation as iPSC lines generated from non-variant individuals.21 Therefore, in this study we took advantage of iPSC technology to extensively characterize monocytes/macrophages in individuals homozygous for CCR5Δ32 variant.
Results
In vitro differentiation of induced pluripotent stem cells in hematopoietic linage cells
Three individuals carrying the homozygous CCR5Δ32 variant were included in our study recruited from the Clinseq Project.22 Individuals with the homozygous genotype were called and the first three identified individuals who were willing to consent for this additional research were recruited and enrolled in this study.
To evaluate the differentiation ability toward the hematopoietic lineage, iPSCs were cultured in feeder-free and defined induction conditions in the presence of an optimized cocktail of human cytokines (BMP4, VEGF, bFGF, SCF, TPO, FLT-3L, and GM-CSF) to generate iHSPCs. With this protocol, iPSCs were prone to commit to mesoderm lineage and efficiently generate hematopoietic cells (Figure 1). After 3 days of culture, the adherent mesoderm precursor cells partially become floating and gradually increase in number, as they gradually differentiate. Cells in suspension exhibited morphological and phenotypical features reminiscent of primitive human iHSPCs (Figure 2A). At day 10–12 of the differentiation process, flow cytometric characterization showed that the majority of cells obtained were CD34+/CD45+, consistent with an iHSPC immune-phenotype (Figure 2B). In particular, CD34+ cells peaked on day 10 (frequency of CD34+ cells: 51% ± 4.5), and declined thereafter, consistent with a precise timing of appearance of hematopoietic cells in the system. Thus, up to 4–6 x106 human CD34+ cells were generated at day 10 from an initial culture of 0.05x106 iPSCs. Interestingly, the majority of CD34+ cells derived from this procedure hold a CD38− phenotype, consistent with an immature HSPCs population (Figure S1). Of note, the number of CD34+ cells decreased along with the increment of CD45+ cells which peaked at day 19, indicating the occurrence of cell differentiation into mature hematopoietic linage cells (Figure 2B). To further validate the hematopoietic differentiation ability of HSPCs induced from iPSCs, in CFU assay we observed that cells in suspension generated a large number of erythroid colonies (BFU-E) at an early time frame of induction peaking at day 7 and decreasing thereafter. However, the number of myeloid (CFU-GM), and mixed (CFU-Mix) colonies gradually increased during the induction (Figure 2C). The erythroid and myeloid nature of BFU-E and CFU-GM colonies was confirmed by the expression of the glycophorin and CD33 marker in these colonies, respectively (Figure S2). The obtained cells were highly proliferative and displayed functional potential of HSPCs in vitro. Overall, these results demonstrate the efficiency of this short term in vitro system optimized for the induction of hematopoietic lineage cells from iPSCs. Efficient induction of hematopoietic lineage cells, which retain hematopoietic cell potential in defined condition, provides an opportunity to obtain patient-specific cells for iPSC therapy and a useful model for the study of the mechanisms of diseases and drug screening.
Figure 1.

Generation in fully defined condition of hematopoietic lineage cells in suspension from normal iPSCs
Schematic representation of iPSC differentiation toward hematopoietic cells and then further maturation of myeloid lineage cells/monocytes and their following differentiation in macrophages (scale bar: 100 μm).
Figure 2.

Characterization of hematopoietic lineage cells derived from iPSCs
(A) Representative photographs showing the morphological changes observed during the differentiation of iPSCs toward iHSPCs and myeloid lineage cells. One photo for each time point is shown (scale bar: 100 μm).
(B) Frequency of CD34+ and CD45+ cells (upper row) and cumulative CD34+ and CD45+ cell yield (lower row) was assessed at different time points during iPSCs differentiation toward hematopoietic lineage cells.
(C) Proliferation ability and differentiation potential of iHSPCs were assessed in vitro by Colony-forming units (CFUs) assay. The number of Burst-forming unit-erythroid (BFU-E), granulocyte-macrophage progenitor cells (CFU-GM), and multipotential granulocyte, erythroid, macrophage and megakaryocyte progenitor cells (CFU-GEMM, or CFU-Mix) was evaluated at different time points during iPSC differentiation. Data are expressed as number of colonies (x104)/mL. Data are presented as mean ± SD (n = 6). Statistical significance was calculated using ANOVA. See also Figures S1 and S2.
Validation of induced pluripotent stem cells derived monocytes
Once iHSPCs were successfully differentiated from iPSCs, they were further induced to commit to the myeloid lineage using appropriate culture conditions. Specifically, iHSPCs cells were incubated with SCF, TPO, FLT-3L, GM-CSF, and M-CSF, to induce their differentiation in myeloid linage cells and monocytes. The iPSC progressive differentiation toward induced iHSPCs and finally to monocytes is shown by the increased numbers of iHSPCs and iMono, as results of iPSCs progressive differentiation (Figure 3A). A similar cell yield of iMono derived from iPSC was obtained from healthy donors (iMono-Con) and three homozygous CCR5Δ32 individuals (iMono-CCR5Δ32) (Figure 3A). iHSPCs differentiation toward monocyte lineage was further confirmed by FACS analysis as shown by the acquisition of CD45 pan marker of hematopoietic cells, together with the expression of myeloid lineage markers, such as CD11c and CD14 (Figure 3B). As shown in Figure 3B, the expression levels of CD45, CD11c, and CD14 were comparable among primary monocytes (pMono) obtained from healthy donors and iPSC-derived monocytes obtained from healthy donors (iMono-Con) or homozygous CCR5Δ32 individuals (iMono-CCR5Δ32).
Figure 3.

Validation of iPSC-derived monocytes (iMono)
(A) Total number of iHSPCs and iMono generated during the differentiation of iPSCs of healthy controls (light gray bar, n = 6) and CCR5Δ32 individuals (dark gray bar, n = 6). Data are expressed as number of cells (x106).
(B) Surface expression of CD45, CD11c and CD14 in primary monocytes (pMono, white bar, n = 6) and iMono generated during the differentiation of iPSCs of controls (iMono-Con, light gray bar, n = 6) and CCR5Δ32 individuals (iMono-CCR5Δ32, dark gray bar, n = 6). Data are expressed as frequency of positive cells (upper row) and Mean Fluorescence Intensity (MFI) (lower row). Data are presented as mean ± SD. Statistical significance was calculated using ANOVA.
Transcriptome analysis shows similarities between primary and induced pluripotent stem cell-derived monocytes and macrophages
Once we successfully differentiated in vitro iMono, we assessed whether they were phenotypically and functionally similar to their primary counterpart by assessing their ability to give rise to functional macrophages when their differentiation and polarization was induced in vitro. Specifically, we investigated the similarity of transcriptome profile between human primary and iPSC-derived monocytes and macrophages, performing an RNA-seq analysis on samples obtained from healthy controls (Table S1). Looking at the genome-wide overview, iMono and pMono, as well as iMac and pMac, clustered together, while iPSCs formed a separate cluster in the principal components analysis (PCA) and multidimensional scaling (MDS) plot (Figures 4A and 4B). The first principal component (PC1) explained 38% of the variance and clearly segregated iPSCs from monocyte and macrophage samples (Figure 4A) showing how iMono and iMac are transcriptionally much more similar to their primary counterpart than to undifferentiated iPSCs. Such observation was further confirmed by hierarchical clustering (Figure 4B). The second PC (PC2) separated M1 polarized macrophages from M0 and M2 macrophages and monocytes. To better understand transcriptional similarity or changes at the gene level we used a two-factor linear model and draw scatterplots of gene expression levels comparing iMono and iMac with their primary counterpart and with iPSCs. As shown by scatterplots in Figure 4C, iMono displayed a higher similarity (r = 0.51) with pMono compared with iPSCs (r = 0.15). In addition, the similarity was even higher when iMac and pMac were compared (r = 0.78 at M0, r = 0.95 at M1, r = 0.86 at M2) (Figure 4C).
Figure 4.

Similarity of transcriptome between primary and iPSC-derived monocytes and macrophages
Analyses were performed based on RNA-seq data for both primary (p) and iMono and iMacrophages (M0, M1, and M2 according to sample polarization) samples.
(A) PCA analyses based on transcriptome for all healthy control RNA-seq samples.
(B) Hierarchical clustering based on transcriptome among all healthy control samples.
(C) Scatterplot of gene expression levels between primary monocytes and macrophages and iPSC-derived monocytes and macrophages. Comparison between iPSC-derived monocytes and macrophages and iPSCs were also shown as a control. Genes that are highly expressed in iPSC-derived samples are shown in red and genes that are highly expressed in primary monocytes and macrophages or iPSCs are shown in blue.
(D) Heatmap of genes associated with macrophages functions. Columns represent genes, and rows represent samples. See also Table S1.
Further, we showed that iMono and iMac displayed similar expression patterns with pMono and pMac when genes related to macrophage functions were considered (Figure 4D). In particular, M1 iMac showed similar gene expression pattern with M1 pMac for genes encoding for protein associated with M1 polarization, such as CXCL9, CXCL10, CCL5, and CD80. All of them were highly expressed in both M1 iMac and M1 pMac, but lowly expressed in both M0 and M2 either iMac or pMac. In addition, M0 and M2 iMac clustered with M0 and M2 pMac in line with the known similar immune phenotype shared by the two macrophage subtypes (Figure 4D).19
Induced macrophages of healthy controls and C-C chemokine receptor type 5Δ32 individuals are transcriptionally similar
The data shown so far demonstrated that the protocol we optimized is a useful tool to differentiate iPSCs in vitro in a high number of hematopoietic cell precursors that can be efficiently committed into the myeloid cell lineage, such as monocytes, carrying a transcriptomic profile that resemble the primary monocytes.
We validated the same protocol to generate iMono starting from iPSCs obtained from homozygous CCR5Δ32 individuals as this could represent a useful in vitro system to generate hematopoietic cells homozygous for the CCR5Δ32 variant for therapeutic purpose, as the genotype confers resistance to HIV-infection.
A second RNA-seq analysis was performed, and the transcriptome profile of iPSC-derived samples generated from 3 individuals homozygous for CCR5Δ32 was compared with the transcriptome profile of samples obtained from healthy controls (Table S1). PCA revealed striking similarity between samples obtained from homozygous CCR5Δ32 individuals and healthy controls (Figure 5A). Furthermore, no significant differences were observed in the expression of genes encoding for macrophage-polarization surface markers (Figure 5B) and cytokines (Figure 5C) between iPSC-derived samples obtained from homozygous CCR5Δ32 individuals and healthy controls. Among 28,514 detected genes, less than 1,000 genes were differentially expressed (p-value <0.01, Foldchange>2) in cells derived from homozygous CCR5Δ32 individuals compared with healthy control cells in all the three macrophage subtypes (M0, M1, M2 iMac). Among the differentially expressed genes, only 56 were differentially expressed in M2 iMac (Figure 5D). Gene ontology (GO) analysis indicated that the genes that were upregulated in M0 iMac generated from homozygous CCR5Δ32 individuals (iMac-CCR5Δ32) compared with those generated from healthy donors (iMac-con) participate in extracellular matrix organization whereas the genes upregulated in M1 iMac-CCR5Δ32 compared with M1 iMac-con encoded for proteins involved in protein localization to plasma membrane, cell chemotaxis, response to thyroid hormone stimulus, positive regulation of cell migration and cell movement (Figure 5E). Interestingly, we also found a preferential up-regulation of NLRP2 gene in iMac generated from homozygous CCR5Δ32 individuals regardless their polarization (Figure 5F). NLRP2 has been described as an important regulator of immune response, acting as inhibitor of NF-κB signaling, involved in the inflammasome signaling regulation in macrophages.23
Figure 5.

Comparison of iPSC-derived macrophages between individuals homozygous for the CCR5Δ32 variant and healthy controls
Analysis was performed based on RNA-seq data for iPSC-derived monocytes (iMono) and macrophages (iMac; M0, M1, and M2 according to sample polarization) generated from individuals homozygous for the CCR5Δ32 variant (CCR5Δ32) and healthy control samples (ctrl).
(A) PCA analysis based on transcriptome for all iPSC-derived macrophages RNA-seq samples.
(B) Heatmap of macrophages surface makers.
(C) Heatmap of macrophages cytokines. Columns represent genes, and rows represent samples.
(D) Number of differential genes are shown between homozygous CCR5Δ32 individuals and controls at three induced macrophage subtypes.
(E) GO analysis performed on genes up-regulated in homozygous CCR5Δ32 individuals compared to controls in iM0 and iM1 subtypes.
(F) Shared up-regulated genes in homozygous CCR5Δ32 individuals among three induced macrophage subtypes are shown. See also Table S1.
Macrophages derived from induced pluripotent stem cells of healthy controls and C-C chemokine receptor type 5Δ32 individuals show similar immune-phenotype
Once we demonstrated at the transcriptional level that iMono were able to differentiate in polarized iMac similar to pMac, we further investigated whether such observation was confirmed at protein level. As confirmed by confocal microscopy, pan-macrophage marker CD68 was highly expressed by both iMac-Con and pMac regardless of their polarization status (Figure 6A). Moreover, also iMac-CCR5Δ32 expressed CD68 similarly to iMac-Con and pMac (Figure 6A).
Figure 6.

Immunophenotype of primary and iPSC-derived macrophages
(A) Representative photographs of immunofluorescence CD68 staining observed in primary macrophages (pMac, first row, n = 3), iPSC-derived macrophages obtained from healthy controls (iMac-Con, second row, n = 3) and iMac obtained from individuals homozygous for CCR5Δ32 variant (iMac-CCR5Δ32, third row, n = 4) polarized in M0, M1, and M2 macrophages (First, second and third column, respectively). Cells were stained for CD68 (red) and with DAPI for nuclear staining (blue) (scale bar: 20 μm).
(B) Immunophenotype of pMac (n = 4), iMac-Con (n = 3) and iMac-CCR5Δ32 (n = 4) polarized in M0, M1, and M2 macrophages was evaluated by flow cytometry. Data are expressed as MFI. Data shown as Log2(Fold change) with all samples normalized on M0 pMac.
(C and D) Primary and iPSC-derived macrophage secretome profile. Expression levels of IL12p40 as M1 polarization marker (C) and Eotaxin-2/CCL24 as M2 polarization marker (D) were analyzed in primary (white bar; n = 4) and iPSC-derived M0, M1, and M2 macrophages obtained from both controls (light gray bar; n = 4) and CCR5Δ32 individuals (dark gray bar; n = 4). Data are expressed as pg/105 cells and are presented as mean ± SEM. Statistical significance was calculated using ANOVA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. See also Figure S3.
In addition, in order to confirm the macrophage polarization of iMac obtained from healthy controls and homozygous CCR5Δ32 individuals, the surface expression levels of typical pan-macrophage and M1 and M2 specific markers were investigated by FACS analysis. As shown in Figure 6B, iMac-Con was able to respond to in vitro stimulation polarizing toward M1 and M2 macrophages similarly to pMac. Moreover, we observed that also iMac-CCR5Δ32 were able to similarly polarize in M1 and M2 macrophages. In particular, upon M1 polarization, M1 markers - namely CD80, CD86, CD215 and CCR7 – were highly expressed by M1 iMac and pMac whereas the expression levels of M2 markers – namely CD163, CD200R, CD204, CD206 and CD209 – were higher in M0 macrophages and in macrophages that were polarized with stimulus that promote M2 polarization compared to M1-polarized macrophages confirming what we observed at transcript level by RNA-seq (Figure 6B).
Finally, to further confirm the ability of iMono to give rise to properly polarized macrophages, the secretome profile of polarized pMac and iMac was assessed by Luminex technology by measuring in the culture medium the levels of cytokines typically produced by M1 and M2 macrophages. As shown in Figures 6C and 6D and in Figure S3, iMac-Con and iMac-CCR5Δ32 successfully polarized in M1 and M2 macrophages as pMac. In fact, only M1 typical cytokines and chemokines were released at high levels by M1 pMac and iMac generated from either healthy donors or individuals homozygous for CCR5Δ32 variant. In M0 and M2 pMac and iMac, M1 cytokine production was absent or limited to small amount. On the contrary, M2 typical cytokines and chemokines were released in higher amount by M2 polarized pMac and iMac generated from either healthy donors and individuals homozygous for CCR5Δ32 variant. In addition, the secretion of the vast majority of the analyzed pro-inflammatory cytokines and chemokines was significantly higher in M1 iMac-Con than M1 pMac and, in accordance with previous findings,19 also the anti-inflammatory cytokine IL-10, was highly secreted by M1 macrophages and its expression was significantly higher in iMac-Con than pMac. Moreover, despite iMac-CCR5Δ32 properly polarized in M1 macrophages, they produced significantly lower levels of M1 cytokines and chemokines (TNFa, IL1-b, IL-12p40, IL-23, IL-6, IFN-b, CCL2, CCL3, CCL4, CCL20, CXCL2, CXCL11) compared to M1 iMac-Con (Figures 6C, 6D, and S3).
iMac of C-C chemokine receptor type 5Δ32 individuals are endowed with phagocytic activity but their C-C chemokine receptor type 5-related functions are impaired
Once we demonstrated that polarized iMac derived from healthy controls and homozygous CCR5Δ32 individuals acquire a similar phenotype along the macrophages subtype polarization, we proceeded with the assessment of their functionality.
In particular, macrophage phagocytic activity of iMac and pMac was evaluated by using pHrodo Green Zymosan A Bioparticles. As shown in Figure 7A, FACS analysis showed that iMac-Con were endowed with a phagocytic activity similar to pMac. In addition, iMac-CCR5Δ32 showed comparable engulfment of zymosan particles as that of iMac-Con. Regarding macrophage polarization, in all analyzed groups M1 macrophages had a reduced phagocytic ability compared to M0 and M2 macrophages. This observation is consistent with previous work that reported that LPS treatment inhibits phagocytosis in human macrophages.24 Similar results were obtained when the engulfment of zymosan particles was evaluated in confocal microscopy (Figure 7B).
Figure 7.

Functionality of primary and iPSC-derived macrophages
Phagocytic ability of M0, M1 and M2 primary macrophages (pMac, white bar, n = 4) and iPSC-derived macrophages (iMac) obtained from both controls (iMac-Con, light gray bar; n = 3) and individuals homozygous for CCR5Δ32 variant (iMac-CCR5Δ32, dark gray bar; n = 4) was evaluated by using pHrodo Green Zymosan A Bioparticles and was assessed by flow cytometry and confocal microscopy.
(A) Macrophage phagocytic ability assessed by flow cytometry was shown as frequency of Zymosan+ cells.
(B) Representative photographs showing phagocytized zymosan particles (in green) in primary macrophages (pMac, first row, n = 3) and iMac obtained from healthy controls (iMac-Con, second row, n = 3) and iMac obtained from homozygous CCR5Δ32 individuals (iMac-CCR5Δ32, third row, n = 4) polarized in M0, M1, and M2 macrophages (First, second and third column, respectively). Nuclei were stained with DAPI (blue). Scale bar: 20 μm.
(C) The chemotactic ability of primary monocytes (pMono, white circle; n = 3) and iPSC-derived monocytes (iMono) obtained from healthy controls (iMono-Con, light gray square; n = 3) and from homozygous CCR5Δ32 individuals (iMono-CCR5Δ32, dark gray square; n = 3) to migrate in response to increasing the concentration of RANTES was assessed in vitro by a cell migration assay. The number of migrated cells was evaluated at different time points. ∗ iMono-Con vs. iMono-CCR5Δ32; # pMono-Con vs. iMono-CCR5Δ32.
(D and E) Macrophage infectability with HIV was assessed in M1 and M2 polarized primary and iPSC-derived macrophages obtained from healthy controls (iMac-Con) and from homozygous CCR5Δ32 individuals (iMac-CCR5Δ32) by evaluating through qPCR the presence of (D) specific HIV-1 DNA Gag and (E) specific HIV-1 DNA LTR RU5. Amplification of genomic GAPDH as a reference gene was used to control the amount of DNA in each sample. Data shown as mean ± SEM. Statistical significance was calculated using ANOVA. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Since the CCR5Δ32 variant generates a nonfunctional receptor, we assessed the functional relevance of iMono and iMac obtained from individuals homozygous for CCR5Δ32 variant.5,6,7
The first CCR5-dependent function tested was the monocyte ability to migrate in response to RANTES, a natural ligand for CCR5. As shown in Figure 7C, iMono-Con were able to migrate in response to the chemo-attractive factor RANTES similarly to pMono. In particular, both iMono-Con and pMono ability to migrate in response to RANTES was dose and time dependent as progressively increased with the increase in RANTES concentration and in the incubation time. On the contrary, iMono-CCR5Δ32 were unable to migrate in response to RANTES in all evaluated time points and with all tested RANTES concentrations.
As CCR5 acts as coreceptor for HIV-1, we also challenged iMac and pMac with HIV-1 and assessed their infectability by evaluating the presence of specific HIV-1 DNA. As shown in Figure 7D, regardless macrophage polarization, HIV-1 DNA Gag was detectable in both iMac-Con and pMac demonstrating that iMac-Con are similar to pMac and can be infected with HIV-1. Nevertheless, HIV-1 DNA was undetectable in iMac-CCR5Δ32 exposed to HIV-1 demonstrating that the resistance to HIV-1 infection typically associated with the CCR5Δ32 variant was maintained in iMac generated from these individuals. Similar results were obtained when the expression of LTR RU5 was assessed (Figure 7E).
Discussion
The CCR5Δ32 variant, a deletion that led to a non-functional CCR5 receptor, gained growing interest in scientific community as individuals homozygous for the CCR5Δ32 variant are warranted by a natural barrier to HIV-1 infection. On this basis, CCR5 antagonism and gene-edited knockout of the receptor are currently under study as possible therapeutic strategies for several diseases.8
It is critical to recognize that the reduced or absent CCR5 expression on cell surface has been shown to have both positive and negative implications.17,18 In particular, in accordance with the role of CCR5 in mediating immune cell migration to inflammation sites, CCR5Δ32 is associated with increased susceptibility and fatal outcome in some viral infections such as West Nile virus infection and influenza infection.17,18 In addition, a non-redundant role of CCR5 was also described in atherosclerosis development.18 These observations support the idea that the lack of CCR5 appeared to be tolerated and compensated by the redundancy of chemokine system in the context of generalist inflammatory responses, whereas in specific pathological conditions the alternative chemokine/chemokine–receptor interactions that occur can trigger deleterious effects as they generate responses that may be distinct from the CCR5-mediated inflammatory response.18 For these reasons, a deeper knowledge on the immune profile of individuals homozygous for CCR5Δ32 is required to guide the development of therapies based on CCR5 blocking and or deletion. This will allow monitoring whether the CCR5 deletion based on gene-editing techniques generates effects - as could be possible - that are different from the ones that occurred in individuals carrying in homozygous the natural deletion CCR5Δ32.
In this context, very limited sets of tools for the characterization of monocyte/macrophage compartment of individuals homozygous for CCR5Δ32 are currently available. Therefore, taking advantage of the fact that recently we successfully generated hiPSC lines from dermal fibroblasts of individuals homozygous for CCR5Δ32 variant,21 in the present study we characterized iPSC-derived monocytes/macrophages obtained from individuals homozygous for CCR5Δ32 deletion.
The protocol used was demonstrated to be an efficient method for the induction of hematopoietic lineage cells from iPSCs as it allowed to obtain a high yield in iHSPCs. In particular, the yield of CD34+ cells obtained with this protocol was ∼500-1,000 times higher than the yields reported in published protocols starting from embryoid bodies or coculture system with stromal cells.25 Moreover, the obtained cells were highly proliferative and displayed functional potential of HSPCs in vitro. Efficient induction of hematopoietic lineage cells, which retain hematopoietic cell potential in defined condition, provides an opportunity to obtain patient-specific cells for iPSC therapy and a useful model for the study of the mechanisms of diseases and drug screening.
Once obtained, iHSPCs generated from iPSCs of healthy controls and individuals homozygous for CCR5Δ32 were further differentiated into monocytes. Our results not only showed that iMono were similar to pMono but also demonstrated that the efficiency of iMono generation was similar in healthy controls and individuals homozygous for CCR5Δ32. Notably, we demonstrated that, similarly to their primary counterparts, iMono were able to mature and differentiate in polarized macrophages when properly stimulated in vitro. Such observation was confirmed either at transcriptional level by RNA-seq and at protein level with different techniques. Our results confirmed a previous study in which iPSC-derived monocytes/macrophages obtained from healthy donors were compared with primary ones.19 In fact, in accordance with Cao and colleagues, we also observed that M1 cytokine production was increased in iMac compared to pMac. Nevertheless, our study extends the knowledge to iMono/iMac obtained from individuals homozygous for CCR5Δ32 demonstrating that iMono obtained from CCR5Δ32 individuals were able to successfully differentiate into polarized macrophages similarly to iMono obtained from healthy controls as confirmed by the evaluation of their transcriptional profile by RNA-seq. This observation was further confirmed when their immunophenotype was assessed at the protein level by evaluating the expression of monocyte/macrophage markers and M1 and M2 macrophages specific markers. Notably, the evaluation of the secretome profile revealed that iMac-CCR5Δ32 despite being able to properly polarize in M1 macrophages released significantly lower amount of M1 cytokines and chemokines compared to M1 iMac obtained from healthy donors. Little is known about macrophage functionality in CCR5Δ32 individuals. Nevertheless, our results are in accordance with a previous study that described in a murine model of acute and chronic renal allograft rejection that CCR5 deficiency in Ccr5−/− mice induces alternative macrophage activation and improves long term renal allograft outcome.26 The reduced ability to produce M1 cytokines and chemokine shown by iMac-CCR5Δ32 is particularly interesting as it is among the few genes that were differentially expressed by iMac-CCR5Δ32 compared to iMac-Con. We identified the gene encoding for NLRP2 as significantly upregulated in iMac-CCR5Δ32 regardless their polarization. NLRP2 is member of the NLRP (pyrin domain-containing NLR proteins) subfamily and is involved in the regulation of the inflammatory responses by acting an inhibitor of the nuclear factor kB (NF-kB) signaling pathway.23 The role of NLRP2 in modulating inflammatory response was recently investigated in a mouse model of hepatic steatosis.27 In particular, the Authors showed that high fat diet feeding-induced inflammation was significantly accelerated by the loss of NLRP2, as evidenced by the increased expression of pro-inflammatory cytokines and activation of NF-kB pathway.27 As NF-κB is a key transcription factor of M1 macrophages and is required for the induction of a large number of inflammatory genes, including those encoding TNF-α, IL-1β, IL-6, IL-12p40 and cyclooxygenase-2,28 the increased expression of NLRP2 observed in macrophages of CCR5Δ32 individuals could explain at least in part the impaired production of M1 cytokines and chemokines.
By performing a phagocytosis assay with zymosan microparticles, we demonstrated in healthy controls that iMac were functionally similar to pMac. Our results are in line with the results obtained in a previous study where the phagocytosis ability of primary and iPSC-derived macrophages was assessed by using E. coli.19 In addition, we showed also that iMac-CCR5Δ32 were endowed with a phagocytic ability similar to iMac-Con thus demonstrating the proper functionality of iMac obtained from individuals homozygous for CCR5Δ32. Moreover, investigating properties related to CCR5 functionality we demonstrated that the impairment in CCR5-related functions was maintained in iPSC-derived cells obtained from homozygous CCR5Δ32 individuals. In particular, we showed that, unlike monocytes/macrophages obtained from healthy donors, iMono-CCR5Δ32 were unable to migrate in response to RANTES and that iMac-CCR5Δ32 were resistant to HIV infection. Such observations are particularly interesting as support the use of iPSC-derived monocytes/macrophages to study more in detail the monocyte/macrophage compartment in homozygous CCR5Δ32 individuals. This strategy could enable overcoming the limitations related to both the fact that primary monocytes and macrophages do not self-renew, and their availability is limited in number and the rarity of the homozygous CCR5Δ32 genotype in the general population. Concerning this last point, the CCR5Δ32 allele frequency is also variable across human populations, presenting the highest frequencies in European populations – declining from Northern (i.e., > 15% in Norway, Latvia, and Estonia) to Southeastern Eurasia (ranging from 5 to 10%) – and being extremely rare or absent in Asia and African native populations.29,30
In conclusion, we characterized iPSC-derived monocytes/macrophages obtained from individuals homozygous for CCR5Δ32. Despite being performed on a limited number of iPSC lines due to the low frequency of CCR5Δ32 allele in the general population, altogether the results of the present study support the use of iPSC-derived cells as a valuable tool to study monocyte/macrophage compartment in individuals homozygous for CCR5Δ32 Through extensive in vitro characterization, our results demonstrated that functional monocytes/macrophages can be successfully derived from iPSCs obtained from CCR5Δ32 individuals and that iPSC-derived monocytes/macrophages obtained from individuals homozygous for CCR5Δ32 were resistant to CCR5-tropic virus challenge and unable to migrate in response to RANTES, a natural ligand of CCR5 thus confirming that the CCR5 impairment associated with CCR5Δ32 deletion is maintained in iPSC-derived cells. Finally, the impaired M1 cytokine production possibly due to an increased expression of NLRP2 that we observed in M1 macrophages of CCR5Δ32 individuals represents an attribute that deserves to be investigated more in detail to shed light on the possible role of CCR5Δ32 in the functionality of monocytes/macrophage compartment.
Limitations of the study
The main drawback of this study is the small population size used in this study, which is limited by the relatively low frequency of homozygous CCR5Δ32 individuals. In conjunction with this, the differentiation procedures are very complex and the generation of the numbers of isogenic controls required is often cost prohibitive so healthy volunteer control cells were used instead.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC-conjugated mouse monoclonal antibody anti CD3 (Clone: HIT3A) | BioLegend | Cat# 300306; RRID: AB_314041 |
| APC-conjugated mouse monoclonal antibody anti CD16 (Clone: VEP13) | Miltenyi Biotec | Cat#130-091-246; RRID: AB_244305 |
| PE-conjugated Mouse monoclonal antibody anti-human CD34 (Clone: 561) | Biolegend | Cat# 343606; RRID: AB_1732033 |
| FITC-conjugated Mouse monoclonal antibody anti-human CD33 (Clone: HIM3-4) | Biolegend | Cat# 303304; RRID: AB_314344 |
| BV510-conjugated Mouse monoclonal antibody anti-human CD34 (Clone: 581) | Biolegend | Cat# 343528; RRID: AB_2563856 |
| PE-Cy7-conjugated Mouse monoclonal antibody anti-human CD38 (Clone: HB-7) | Biolegend | Cat# 303516; RRID: AB_1279235 |
| APC-conjugated Mouse monoclonal antibody anti-human CD45 (Clone: H130) | Biolegend | Cat# 304012; RRID: AB_314399 |
| PE-conjugated Mouse monoclonal antibody anti-human CD235 (Glycophorin) (Clone: HIR2) | Biolegend | Cat# 306604; RRID: AB_314622 |
| FITC-conjugated Mouse monoclonal antibody anti-human CD14 (Clone: GCD14) | Biolegend | Cat# 325604; RRID: AB_830677 |
| APC-conjugated Mouse monoclonal antibody anti-human CD16 (Clone: 3G8) | Biolegend | Cat# 302012; RRID: AB_314212 |
| PE Cy5-conjugated Mouse monoclonal antibody anti-human CD11b (Clone: IRCF44) | Biolegend | Cat# 301308; RRID: AB_314160 |
| BV421-conjugated Rat monoclonal antibody anti-human CD115 (Clone: 9-4D2-1E4) | BD | Cat#565347; RRID: AB_2739200 |
| BV570-conjugated Mouse monoclonal antibody anti-human CD14 (Clone: M5E2) | Biolegend | Cat# 301831; RRID: AB_10897803 |
| AF700-conjugated Mouse monoclonal antibody anti-human CD16 (Clone: 3G8) | Biolegend | Cat# 302026; RRID: AB_2278418 |
| PE-conjugated Mouse monoclonal antibody anti-human CD209 (Clone: DCN46) | BD | Cat# 551265; RRID: AB_394123 |
| FITC-conjugated Mouse monoclonal antibody anti-human CD80 (Clone: L307-4) | BD | Cat# 557226; RRID: AB_396605 |
| FITC-conjugated Mouse monoclonal antibody anti-human CD86 (Clone: 2331) | BD | Cat# 555657; RRID: AB_396012 |
| APC-conjugated Mouse monoclonal antibody anti-human CD215 (Clone: JM7A4) | Biolegend | Cat# 330209; RRID: AB_2561439 |
| APC Cy7-conjugated Mouse monoclonal antibody anti-human HLA-DR (Clone: L243) | BD | Cat# 335831; RRID: AB_2868692 |
| PE-conjugated Mouse monoclonal antibody anti-human CD195 (CCR5) (Clone: 2D7/CCR5) | BD | Cat# 555993; RRID: AB_396279 |
| PE CF594-conjugated Mouse monoclonal antibody anti-human CD197 (CCR7) (Clone: 150503) | BD | Cat# 562381; RRID: AB_11153301 |
| FITC-conjugated Mouse monoclonal antibody anti-human CD206 (Clone: 19.2) | BD | Cat# 551135; RRID: AB_394065 |
| PE-conjugated Mouse monoclonal antibody anti-human CD163 (Clone: GHI/61) | BD | Cat# 556018; RRID: AB_396296 |
| APC-conjugated Mouse monoclonal antibody anti-human CD200R (Clone: OX-108) | Biolegend | Cat# 329307; RRID: AB_2564350 |
| AF700-conjugated Mouse monoclonal antibody anti-human CD204 (Clone: 351615) | R&D | FAB2708N |
| mouse monoclonal antibody anti-human CD68 (Clone: KP1) | Dako | Cat# M0814; RRID: AB_2314148 |
| AF568-conjugated goat anti-mouse antibody | Invitrogen | Cat# A-11004; RRID: AB_2534072 |
| Bacterial and virus strains | ||
| HIVBaL strain | Gift from G. Scarlatti (IRCCS Ospedale San Raffaele) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel, growth factor reduced | BD Biosciences | 354230 |
| Essential 8™ Medium | Gibco | A1517001 |
| Iscove’s modified Dulbecco’s medium (IMDM) | Invitrogen | 12440053 |
| Ham's F-12 Nutrient Mix | Invitrogen | 11765054 |
| GlutaMAX™ supplement | Invitrogen | 35050061 |
| Albucult (recombinant human - rh Albumin) | Novozymes Delta | RF20-005 |
| a-Monothioglycerol (a-MTG) | Sigma-Aldrich | M6145 |
| Protein-free hybridoma mixture II (PFHMII) | Invitrogen | 12040-077 |
| Ascorbic acid 2 phosphate | Sigma-Aldrich | A8960 |
| Penicillin/streptomycin (50 U Pen G/50 mg streptomycin sulfate) | Invitrogen | 15140122 |
| Insulin Transferrin Selenium-X Supplement | Invitrogen | 515000560 |
| recombinant human (rh) vascular endothelial growth factor (rhVEGF) | PeproTech | 100-20 |
| rh Bone morphogenetic protein 4 (rhBMP4) | R&D systems | 314-BP-050 |
| rh Basic fibroblast growth factor (rhbFGF) | PeproTech | 100-18B |
| rh stem cell factor (rhSCF) | STEMCELL Technologies | 02830 |
| rh Flt-3 Ligand (rhFlt-3 L) | STEMCELL Technologies | 02941 |
| rh Thrombopoietin (rhTPO) | STEMCELL Technologies | 02720 |
| rh Granulocyte-Macrophage Colony-Stimulating Factor (rhGM-CSF) | PeproTech | 300-03 |
| MethoCult™ GF H4434 medium | StemCell Technologies | 04434 |
| rh Macrophage Colony-Stimulating Factor (rhM-CSF) | PeproTech | 300-25 |
| lipopolysaccharides (LPS) purified from Escherichia coli (E. coli) 055:B5 | Sigma-Aldrich | L2880-10MG |
| rhIFN-y | PeproTech | 300-02 |
| rhIL-4 | PeproTech | 200-04 |
| Ficoll Paque™ Premium | GE Healthcare Life Sciences | 17544203 |
| Percoll | Sigma-Aldrich | GE17-0891-01 |
| Zombie Aqua™ Fixable Viability Kit | Biolegend | 423102 |
| Accutase | Merck-Millipore | SCR005 |
| Human TruStain FcX™ (Fc Receptor Blocking Solution) | Biolegend | 422301 |
| BD™ CompBeads Anti-Mouse Ig, κ/Negative Control Compensation Particles Set | BD | 552843 |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 |
| Soluble recombinant–human RANTES (rh-RANTES) | PeproTech | 300-06 |
| Lectin from Phaseolus Vulgaris (PHA-L) | Sigma-Aldrich | L4144-5MG |
| Recombinant human IL-2 (rh-IL-2) | Peprotech | 200-02 |
| Critical commercial assays | ||
| RNeasy mini kit | Qiagen | 74106 |
| TruSeq Stranded mRNA Library Preparation Kit | Illumina | 20020595 |
| KAPA Library Quantification Kit for NGS | Kapa | KK4854 |
| HiSeq 3k/4k PE Cluster Kit | Illumina | PE-410-1001 |
| Magnetic bead-based multi-plex screening assays | R&D | Customized Cytokine Luminex Performance Assay |
| pHrodo™ Green Zymosan BioParticles™ Conjugate for Phagocytosis | Invitrogen | P35365 |
| Transwell-24 units with 5.0 μm pore size | Corning | 3421 |
| Deposited data | ||
| Raw data of RNA-Seq experiments | GEO | GEO: GSE141410 |
| Oligonucleotides | ||
| HIV-1-specific strong-stop DNA (LTR RU5) primers: Fw-5’-GGCTAACTAGGGAACCCACTG-3’ Rev-5’- CTGCTAGAGATTTTCCACACTGAC-3’ |
Primm Srl | Custom |
| DNA Gag primers: Fw-5’-ACATCAAGCAGCCATGCAAAT-3’ Rev-5’-ATCTGGCCTGGTGCAATAGG-3’ |
Primm Srl | Custom |
| Amplification of genomic GAPDH | Applied Biosystem | Hs04420697_g1 |
| Software and algorithms | ||
| FlowJo X 10.0.7r2 software | BD | https://www.bdbiosciences.com/en-us/products/software/flowjo-v10-software; RRID: SCR_008520 |
| Bio-Plex Manager Pro 6.1 analysis software | Bio-Rad | 171-STND01; RRID: SCR_014330 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Manfred Boehm (E-mail: boehmm@nhlbi.nih.gov).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Raw data of RNA-Seq experiments were deposited in GEO (Accession number: GSE141410).
-
•
All data that support the findings of this study are available from the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available for the lead contact upon request.
Experimental model and study participant details
Individuals and study approval
iPSCs were generated from fibroblasts derived from skin punch biopsy samples obtained from three individuals with the homozygous CCR5Δ32 variant (two males (ages: 56, 69 years old) and one female (age: 60 years old) and three healthy volunteers (one male (age: 23 years old) and two females (ages: 66, 25 years old). All of iPSC lines derived from three individuals with the homozygous CCR5Δ32 variant showed the same gene variant as their parental fibroblast.21 This study was approved by the NHLBI’s institutional review board, and samples were collected after obtaining informed written consents from all subjects. Primary monocytes were obtained from PBMCs isolated from buffy coats of four healthy volunteers (three males and one female) who signed consent forms in accordance with the Declaration of Helsinki and with clinical protocols approved by the Institutional Review Board of Humanitas Research Hospital (Ethical Committee of 30/01/2007, name of the protocol “Impiego di buffycoat nella ricerca corrente "processamento dei campioni di sangue"). ClinSeq® study was reviewed and approved by the NHGRI IRB.
Method details
Direct differentiation of iPSCs toward monocytes/macrophages in chemically defined conditions
A step-wise protocol was developed to drive iPSC differentiation into mesoderm precursors under feeder-free and chemical defined conditions, followed by their specific lineage commitment and maturation (Figure 1). iPSCs were cultured and maintained as described previously.21 In particular, in the first step, iPSCs were split onto Matrigel-coated plates (Corning) and cultured making sure the sub-colonies were of small size (10∼30 cells per colony). After 24 hours of recovery in Essential 8™ Medium (Gibco), in order to drive cells into mesoderm progenitor differentiation, cells were allowed to grow in chemically defined differentiation medium (MDM) containing Iscove’s modified Dulbecco’s medium (IMDM, Invitrogen) supplemented with Ham's F-12 Nutrient Mix, GlutaMAX™ supplement (Invitrogen) at 1:1 ratio, Albucult (recombinant human - rh Albumin) (5 mg/ml; Novozymes Delta), a-Monothioglycerol (a-MTG) (3.9 ml per 100 ml; Sigma-Aldrich), Protein-free hybridoma mixture II (PFHMII) (5%; Invitrogen), Ascorbic acid 2 phosphate (50 μg/ml; Sigma-Aldrich), Penicillin/streptomycin (50 U Pen G/50 mg streptomycin sulfate; Invitrogen), Insulin Transferrin Selenium-X Supplement (Invitrogen). During the first two days of culture, MDM was supplemented with recombinant human (rh) vascular endothelial growth factor (rhVEGF, 10 ng/ml; PeproTech), rh Bone morphogenetic protein 4 (rhBMP4, 10 ng/ml; R&D systems), and rh Basic fibroblast growth factor (rhbFGF, 10 ng/ml; PeproTech). From day 3 to day 7 (Step 2), additional supplement of hematopoietic stem/progenitor cells (HSPCs) growth factors and cytokines were further added to the culture medium in order to push cells commitment to HSPCs. In particular, the following growth factors and cytokines were used: rh stem cell factor (rhSCF, 50 ng/ml; PeproTech), rh Flt-3 Ligand (rhFlt-3 L, 50 ng/ml; PeproTech), and rh Thrombopoietin (rhTPO, 50 ng/ml; PeproTech). Starting from day 7 until day 10 (Step 3), rh Granulocyte-Macrophage Colony-Stimulating Factor (rhGM-CSF, 100 ng/ml; PeproTech) was supplied in the culture to enhance HSPCs production and proliferation. During step 4 (from day 10 to day 14), the culture medium was changed to RPMI (Invitrogen) with 10% of FBS (Atlanta Biologicals) plus rhFlt-3 ligand, rhTPO, rhSCF, and rhGM-CSF to further promote HSPC proliferation to promote their differentiation into myeloid lineage cells. After 14 days of induction (Step 5), in order to enrich monocytes generation from myeloid lineage cells, the cells in suspension were collected, seeded into regular cell culture dishes and cultured for 5 additional days in RPMI with 10% of FBS supplemented with rhGM-CSF and rh Macrophage Colony-Stimulating Factor (rhM-CSF, 100 ng/ml; PeproTech), the key regulators of monocyte proliferation and differentiation.31 Finally, the monocytes (iMono) obtained were subsequently differentiated and polarized into resting (M0), M1 and M2 macrophages (iMac).31 For the generation of M0, iMono were cultured in RPMI with 10% of FBS supplemented with rhM-CSF for 7 days (100 ng/ml). For M1 polarization, macrophages (M0) were cultured for additional 24 hours in presence of lipopolysaccharides (LPS) purified from Escherichia coli (E. coli) 055:B5 (100 ng/mL; Sigma-Aldrich) and rhIFN-ƴ (20 ng/mL; PeproTech). For M2 polarization, macrophages (M0) were cultured for additional 24 hours in presence of rhIL-4 (20 ng/mL; PeproTech).
Primary monocytes/macrophages isolation and culture
In order to isolate primary monocytes, PBMCs were isolated using Ficoll Paque™ Premium density gradients (GE Healthcare Life Sciences) from healthy donor buffy coats. Starting from PBMCs, monocytes were first enriched by performing a Percoll (Sigma-Aldrich) density gradient and then cells were first stained with a live-dead exclusion dye (Aqua Zombie; Biolegend) for 15 minutes at room temperature (RT) to discriminate dead and viable cells and then were incubated for 20 minutes at 4°C with monoclonal antibodies for surface antigen staining for CD3 (FITC-conjugated mouse monoclonal antibody anti CD3, BioLegend, clone HIT3A) and CD16 (APC-conjugated mouse monoclonal antibody anti CD16, Miltenyi Biotech, clone VEP13).
CD14+ classical monocytes were sorted by negative selection using FACS Aria (Becton Dickinson). In particular, monocytes were identified on the bases of scatter parameters and, within this gate among the viable cells, classical monocytes were identified as CD3-/CD16- cells. Once obtained, monocytes were differentiated and polarized in M0, M1, and M2 macrophages as described above.
Colony-forming units (CFUs) assay
To evaluate in vitro proliferation ability and differentiation potential of iPSC-derived HSPCs (iHSPCs), the CFUs assay has been performed. At every time point, cells in suspension were collected and washed with phosphate-buffered saline (PBS). Consequently, 1x104 cells were resuspended with MethoCult™ GF H4434 medium (StemCell Technologies), dispensed into petri dishes and cultured at 37°C and 5% CO2. The colony number and type were evaluated at day7 and day14 under microscope. In particular, colonies were classified as Burst-forming unit-erythroid (BFU-E), granulocyte-macrophage progenitor cells (CFU-GM), and multipotential granulocyte, erythroid, macrophage and megakaryocyte progenitor cells (CFU-GEMM, or CFU-Mix).
RNA-seq
RNA-seq analysis was performed on samples of iPSCs, primary cells and iPSC-derived cells obtained from both healthy controls and homozygous CCR5Δ32 individuals, as listed in Table S1.
RNA was isolated using TRIzol-RNaesy (RNeasy mini kit, Qiagen) combination method. Total RNA integrity was assessed using automated capillary electrophoresis on a Fragment Analyzer (Roche). For all samples RNA quality indicator (RQI)> 8.0, a total RNA amount of 500 ng was used as input for library preparation using the TruSeq Stranded mRNA Library Preparation Kit (Illumina). Sequencing libraries were quantified by PCR using KAPA Library Quantification Kit for NGS (Kapa) and assessed for size distribution and absence of adapter dimers on a Fragment Analyzer. Sequencing libraries were pooled and clustered using a cBot2 (Illumina) using a HiSeq 3k/4k PE Cluster Kit before sequencing on a HiSeq 3000 platform (Illumina) with run conditions for paired-end reads at 75 bp length and a 7bp indexing read.
Analysis of RNA-seq data
RNA-seq data quality was assessed using FastQC. Reads obtained for each sample were aligned to the human hg19 reference genome using STAR (v2.5.4a).32 PCA analysis and hierarchical analysis for all RNA-seq samples were performed using modules of DESeq2.33 Expression levels for each gene were calculated as read count per million total sequencing reads (CPM) using EdgeR.34 Scatter plots were shown to comparing gene expression either between iPSC-derived cells and primary cells or between iPSC-derived cells and iPSCs, and Pearson correlation coefficients were calculated.35 To draw the heat maps, known macrophage representative genes were selected and expression levels for each gene were transformed to a z-score among different samples, and hierarchical clustering was performed. Significantly differentially expressed genes (p<0.05) between any two cell types among iPSC-derived macrophages between homozygous CCR5Δ32 individuals and healthy controls were identified using DESeq2.33 GO analysis was performed using DAVID (v6.7).36 Venn diagrams of genes with higher expressions relative to healthy controls in homozygous CCR5Δ32 individuals were drawn using R package.
Clinseq®
The ClinSeq® cohort initially included 1,000 individuals22 of overwhelmingly European background and was subsequently increased to 1,500 individuals, with the addition of 500 participants of African or African-American background.37 All individuals underwent exome sequencing and variant analyses.38 The CCR5 c.554_585del (p.Ser185fs) variant was analyzed by a custom query optimized for indels (details on request). Of the 1,499 sequenced individuals (one was removed for sample contamination), we identified 194 heterozygotes and seven homozygotes, for a MAF of .0693, which was in Hardy Weinberg equilibrium and similar to the expected frequency from gnomAD for a cohort of 2/3 European (Non-Finnish) and 1/3 African or African American, which is 0.1073 and 0.019, respectively, with a weighted average MAF of 0.0772. Individuals with the homozygous genotype were called and the first three identified individuals who were willing to consent for this additional research were recruited and enrolled in this substudy. Genotypes from next generation sequencing data were confirmed with PCR and Sanger sequencing.
Cell immunophenotyping
For the flow cytometric immunophenotyping of iHSCs and monocytes, the suspension cells were harvested at designed time points.
For macrophage immunophenotyping, FACS analysis was performed on in vitro differentiated and polarized (M0, M1, and M2) primary macrophages (pMac) and iPSC-derived macrophages (iMac). pMac and iMac were harvested using non-enzymatic dissociation solution (Accutase, Merck-Millipore).
Following washing with PBS to remove culture medium, cells were incubated with Fc Receptor Blocking Solution (Biolegend) for 15 minutes at 4°C, to prevent non-specific binding, and then stained with a live-dead exclusion dye (Aqua Zombie; Biolegend) for 15 minutes at room temperature (RT) to discriminate dead and viable cells. Subsequently, cells were incubated for 20 minutes at 4°C with monoclonal antibodies listed in the key resources table for surface antigen staining. After the staining, cells were washed with PBS, resuspended, and analyzed as followed. iHSCs and monocytes were acquired on a MACS Quant cytometer (Miltenyi Biotec), while macrophages (both pMac and iMac) were acquired within 24 hours using FACS Aria (Becton Dickinson). FACS data were analyzed with FlowJo X 10.0.7r2 software (BD). Data were compensated by using single-stained antibody-capture beads (CompBeads, BD Biosciences). Fluorescence Minus One (FMO) control for each of these molecules were performed.
For the analysis of CD68 expression, macrophages seeded on coverslips in 24 well-plate were fixed with paraformaldehyde (PFA) 4% for 15 minutes, blocked with PBS with Ca2+ and Mg2+ (PBS+/+) + 0.1% Triton-X100 + 10% normal goat serum for 30 minutes and stained with mouse anti-human CD68 antibody (Dako) for 1 hour at RT. Cells were then washed, incubated with the secondary goat anti-mouse AF568-conjugated antibody (Invitrogen) for 1 hour at RT, washed, stained with DAPI (Invitrogen), and analyzed with an Olympus Fluoview FV1000 laser scanning confocal microscope.
Cytokine measurement
Cell supernatants were collected and cytokine concentrations were measured on a Luminex 100 instrument (Luminex Corp.) according to manufacturer’s specifications using customized magnetic bead-based multi-plex screening assays from R&D systems consisting of IL-1β, IL-6, IL-10, IL-12p40, IL-12p70, IL-23, MCP-1/CCL2, MIP-1α/CCL3, MIP-1β/CCL4, RANTES/CCL5, CCL17, CCL18, MIP-3α/CCL20, MDC/CCL22, Eotaxin-2/CCL24, GRO-β/CXCL2, MIG/CXCL9, ITAC/CXCL11, BCA-1/CXCL13, IFN-β, and TNF-α. Analytes were grouped into panels by the manufacturer according to their abundance in each human macrophage subtype. Data were analyzed using Bio-Plex Manager Pro 6.1 analysis software (Bio-Rad) and reported as pg/105 cells.
Phagocytosis assay
Macrophage phagocytic activity was detected using pHrodo Green Zymosan A Bioparticles (Invitrogen). According to the manufacturer’s manual, Zymosan Bioparticles were diluted in PBS+/+ + 20 mM HEPES (pH 7.4) to a final concentration of 200 μg/ml for phagocytosis assessment by flow cytometry and 400 μg/ml for phagocytosis assessment by confocal microscopy.
Phagocytosis assessment by flow cytometry
Macrophages seeded in 24 well-plate were incubated in presence or absence (negative controls) of zymosan bioparticles for 1 hour at 37°C and with zymosan bioparticles on ice (negative controls for phagocytic activity). After incubation, cells were detached with Accutase (Merck-Millipore), fixed with PFA 1% for 30 minutes and acquired on LSR Fortessa flow cytometer (Beckton Dickinson). As read out of the phagocytic activity, the frequency of zymosan positive macrophages was analyzed using FlowJo X 10.0.7r2 software (BD).
Phagocytosis assessment by confocal microscopy
Macrophages seeded on coverslips in 24 well-plate were incubated in presence or absence (negative controls) of zymosan bioparticles for 1 hour at 37°C or with zymosan bioparticles on ice (negative controls for phagocytic activity). After incubation, cells were fixed with PFA 4% for 15 minutes. Cells were then washed stained with DAPI (Invitrogen, USA), and analyzed with an Olympus Fluoview FV1000 laser scanning confocal microscope.
Monocytes migration assay
The chemotactic ability of primary monocytes (pMono) and iPSC-derived monocytes (iMono) to migrate in response to RANTES was assessed in vitro by a cell migration assay. Monocytes were plated in the transwell inserts of Transwell-24 units with 5.0 μm pore size (Corning). In the lower part of the transwell, DMEM medium (Lonza) was added with recombinant–soluble human RANTES (rh-RANTES; R&D) that was used as an attractant chemokine at different concentrations (1, 3, and 10 ng/ml). Cell migration was assessed after 4, 6 and 8 hours of incubation at 37 C°, 5% of CO2 by counting the cells collected on the bottom of the wells.
Virus expansion
HIV-1BaL was expanded on human PBMCs obtained from buffy coats of healthy volunteers. PBMCs were cultured in RPMI 1640 medium, supplemented with 10% FBS, 2mM L-glutamine, 100 U/mL penicillin/streptomycin and activated with Lectin from Phaseolus Vulgaris (PHA-L, 5 μg/ml; Sigma-Aldrich) for 3 days and subsequently with human recombinant IL-2 (rh-IL-2, 200 IU/mL; PeproTech).
Large quantities of HIV-1BAL strain virions were obtained from the supernatants of virus producing PHA-L and IL-2 activated PBMCs collected at the peak of virus replication. HIV-1 p24 Ag concentrations in the culture supernatants were determined by ELISA (Aalto) and used at final concentration of 2 ng/mL of p24 Ag.39
Detection of specific HIV-1 DNA by qPCR
Before incubation with cells, viral stock was treated (1 hour at RT) with 200 U/ml of RNase-free DNase (Roche). pMac and iMac were incubated with the viral stock for 4 to 6 days and collected for HIV-1 detection.40 Before preparation of cell lysate, control (not infected) and HIV-1BAL-infected cells were collected, washed three times with PBS, then treated with 0.05% trypsin at 37°C for 10 minutes to eliminate not internalized virus, and washed again four times with PBS. Cells were lysate in 50 μL of PCR lysis buffer (50mM KCL, 10mM Tris-HCL pH 8.3, 2.5 mM MgCl2, 0.1 mg/mL gelatin, 0.45% Nonidnet P-40 and 0.45% Tween20, all purchased from Sigma) and treated with 100ug/mL di Proteinase K (Sigma). Cell lysates were subjected to qPCR Syber Green analysis by using HIV-1-specific strong-stop DNA (LTR RU5) primers: LTR RU5 Fw-5’-GGCTAACTAGGGAACCCACTG-3’, Rev-5’- CTGCTAGAGATTTTCCACACTGAC-3’ and DNA Gag primers: Gag Fw-5’-ACATCAAGCAGCCATGCAAAT-3’, Rev-5’-ATCTGGCCTGGTGCAATAGG-3’. Amplification of genomic GAPDH as a reference gene was used to control the amount of DNA in each sample (Applied Biosystem).
Quantification and statistical analysis
Data were shown as mean ±SEM or as mean ±SD, as indicated in figure legends. Statistical significance was calculated using ANOVA. Statistical analyses were performed with either Microsoft Excel or Graph Pad 7-Prism statistical package. The critical significance value was set at 0.05. In addition, significance, group numbers and other details are indicated in individual figure legends.
Additional resources
This study was performed under clinical trial NCT01143454.
Acknowledgments
We thank G. Scarlatti for strain HIVBaL used in the study. NHLBI Core Facilities: Flow Cytometry Core, iPSC Core. We also acknowledge Clesson Turner for assistance with retrieving ClinSeq data.
This work was supported by Intramural Research program of the National Heart, Lung, and Blood Institute (NHLBI): the National Institutes of Health grant 1ZIAHL006079-10. ClinSeq was supported by the National Human Genome Research Institute grants 1ZIAHG200359-14 1ZIAHG200387-09.
This research was also funded by Ministero della Salute (PE-2016-02363915) to DM and by Humanitas Research Hospital grants (Intramural Research and Clinical Funding Programs 5x1000) to DM. FO and AC were supported by a competitive fellowship from the Ph.D. program of Experimental Medicine of University of Milan “La Statale”.
Author contributions
The authors confirm contribution to the article as follows: study conception and design: GC, FC, DM, EP, SDB, DY, and MB; data collection: GC, YM, DY, XP, ZY, YL, YH, FC, EP, JM, FO, AC, LGB, and CLD; analysis and interpretation of results: GC, FC, RLH, DY, YM, XP, EP, JM, SDB, LGB, CLD, and MB; draft article preparation: GC, DY, RLH, YM, XP, FC, EP, FO, DM, MB, and LGB. All authors reviewed the results and approved the final version of the article.
Declaration of interests
LGB is a member of the Illumina Medical Ethics Committee and receives research funding from Merck, Inc. All the other authors declare no competing interests. The section of “In vitro differentiation of iPSCs in hematopoietic linage cells” described in manuscript is registered under a patent “Human iPSC-derived vascular-related and hematopoietic cells for therapies and toxicology/drug screenings” (patent number #10385313 and 11072778). GC and MB receive royalty income.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Declaration of AI and AI-assisted technologies in the writing process
No AI or AI assisted technology was used in generating this body of work, nor in the written article.
Published: October 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108331.
Supplemental information
References
- 1.Samson M., Labbe O., Mollereau C., Vassart G., Parmentier M. Molecular Cloning and Functional Expression of a New Human CC-Chemokine Receptor Gene. Biochemistry. 1996;35:3362–3367. doi: 10.1021/bi952950g. [DOI] [PubMed] [Google Scholar]
- 2.Barmania F., Pepper M.S. C-C chemokine receptor type five (CCR5): An emerging target for the control of HIV infection. Appl. Transl. Genom. 2013;2:3–16. doi: 10.1016/j.atg.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin T., Xu X., Hereld D. Chemotaxis, chemokine receptors and human disease. Cytokine. 2008;44:1–8. doi: 10.1016/j.cyto.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connor R.I., Sheridan K.E., Ceradini D., Choe S., Landau N.R. Change in Coreceptor Use Correlates with Disease Progression in HIV-1–Infected Individuals. J. Exp. Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dean M., Carrington M., Winkler C., Huttley G.A., Smith M.W., Allikmets R., Goedert J.J., Buchbinder S.P., Vittinghoff E., Gomperts E., et al. Genetic Restriction of HIV-1 Infection and Progression to AIDS by a Deletion Allele of the CKR5 Structural Gene. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 6.Liu R., Paxton W.A., Choe S., Ceradini D., Martin S.R., Horuk R., MacDonald M.E., Stuhlmann H., Koup R.A., Landau N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 7.Samson M., Libert F., Doranz B.J., Rucker J., Liesnard C., Farber C.-M., Saragosti S., Lapouméroulie C., Cognaux J., Forceille C., et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 8.Vangelista L., Vento S. The Expanding Therapeutic Perspective of CCR5 Blockade. Front. Immunol. 2018;8:1981. doi: 10.3389/fimmu.2017.01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haworth K.G., Peterson C.W., Kiem H.-P. CCR5-edited gene therapies for HIV cure: Closing the door to viral entry. Cytotherapy. 2017;19:1325–1338. doi: 10.1016/j.jcyt.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Hütter G., Nowak D., Mossner M., Ganepola S., Müssig A., Allers K., Schneider T., Hofmann J., Kücherer C., Blau O., et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 11.Allers K., Hütter G., Hofmann J., Loddenkemper C., Rieger K., Thiel E., Schneider T. Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- 12.Gupta R.K., Abdul-Jawad S., McCoy L.E., Mok H.P., Peppa D., Salgado M., Martinez-Picado J., Nijhuis M., Wensing A.M.J., Lee H., et al. HIV-1 remission following CCR5Δ32/Δ32 haematopoietic stem-cell transplantation. Nature. 2019;568:244–248. doi: 10.1038/s41586-019-1027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen B.-E.O., Knops E., Cords L., Lübke N., Salgado M., Busman-Sahay K., Estes J.D., Huyveneers L.E.P., Perdomo-Celis F., Wittner M., et al. In-depth virological and immunological characterization of HIV-1 cure after CCR5Δ32/Δ32 allogeneic hematopoietic stem cell transplantation. Nat. Med. 2023;29:583–587. doi: 10.1038/s41591-023-02213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Y., Nashun B., Zhou T., Qin L., Qin L., Zhao S., Xu J., Esteban M.A., Chen X. Generation of CD34+ Cells from CCR5-Disrupted Human Embryonic and Induced Pluripotent Stem Cells. Hum. Gene Ther. 2012;23:238–242. doi: 10.1089/hum.2011.126. [DOI] [PubMed] [Google Scholar]
- 15.Ye L., Wang J., Beyer A.I., Teque F., Cradick T.J., Qi Z., Chang J.C., Bao G., Muench M.O., Yu J., et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA. 2014;111:9591–9596. doi: 10.1073/pnas.1407473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang H., Minder P., Park M.A., Mesquitta W.T., Torbett B.E., Slukvin I.I. CCR5 Disruption in Induced Pluripotent Stem Cells Using CRISPR/Cas9 Provides Selective Resistance of Immune Cells to CCR5-tropic HIV-1 Virus. Mol. Ther. Nucleic Acids. 2015;4:e268. doi: 10.1038/mtna.2015.42. [DOI] [PubMed] [Google Scholar]
- 17.Ellwanger J.H., Kaminski V.d.L., Chies J.A. What we say and what we mean when we say redundancy and robustness of the chemokine system – how CCR5 challenges these concepts. Immunol. Cell Biol. 2020;98:22–27. doi: 10.1111/imcb.12291. [DOI] [PubMed] [Google Scholar]
- 18.Ellwanger J.H., Kaminski V.L., Chies J.A.B. CCR5 gene editing - Revisiting pros and cons of CCR5 absence. Infect. Genet. Evol. 2019;68:218–220. doi: 10.1016/j.meegid.2018.12.027. [DOI] [PubMed] [Google Scholar]
- 19.Cao X., Yakala G.K., van den Hil F.E., Cochrane A., Mummery C.L., Orlova V.V. Differentiation and Functional Comparison of Monocytes and Macrophages from hiPSCs with Peripheral Blood Derivatives. Stem Cell Rep. 2019;12:1282–1297. doi: 10.1016/j.stemcr.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Xue C., Shah R., Bermingham K., Hinkle C.C., Li W., Rodrigues A., Tabita-Martinez J., Millar J.S., Cuchel M., et al. Functional Analysis and Transcriptomic Profiling of iPSC-Derived Macrophages and Their Application in Modeling Mendelian Disease. Circ. Res. 2015;117:17–28. doi: 10.1161/CIRCRESAHA.117.305860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen G., Jin H., Yu Z., Liu Y., Li Z., Navarengom K., Schwartzbeck R., Dmitrieva N., Cudrici C., Ferrante E.A., et al. Generation of human induced pluripotent stem cells from individuals with a homozygous CCR5Δ32 mutation. Stem Cell Res. 2019;38 doi: 10.1016/j.scr.2019.101481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., et al. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruey J.M., Bruey-Sedano N., Newman R., Chandler S., Stehlik C., Reed J.C. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J. Biol. Chem. 2004;279:51897–51907. doi: 10.1074/jbc.M406741200. [DOI] [PubMed] [Google Scholar]
- 24.Michlewska S., Dransfield I., Megson I.L., Rossi A.G. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-α. Faseb. J. 2009;23:844–854. doi: 10.1096/fj.08-121228. [DOI] [PubMed] [Google Scholar]
- 25.Kardel M.D., Eaves C.J. Modeling human hematopoietic cell development from pluripotent stem cells. Exp. Hematol. 2012;40:601–611. doi: 10.1016/j.exphem.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Dehmel S., Wang S., Schmidt C., Kiss E., Loewe R.P., Chilla S., Schlöndorff D., Gröne H.J., Luckow B. Chemokine receptor Ccr5 deficiency induces alternative macrophage activation and improves long-term renal allograft outcome. Eur. J. Immunol. 2010;40:267–278. doi: 10.1002/eji.200939652. [DOI] [PubMed] [Google Scholar]
- 27.Li C., Liu Q., Xie L. Suppressing NLRP2 expression accelerates hepatic steatosis: A mechanism involving inflammation and oxidative stress. Biochem. Biophys. Res. Commun. 2018;507:22–29. doi: 10.1016/j.bbrc.2018.10.132. [DOI] [PubMed] [Google Scholar]
- 28.Liu T., Zhang L., Joo D., Sun S.-C. NF-κB signaling in inflammation. Signal Transduct. Targeted Ther. 2017;2 doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulmann-Leal B., Ellwanger J.H., Chies J.A.B. CCR5Δ32 in Brazil: Impacts of a European Genetic Variant on a Highly Admixed Population. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.758358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solloch U.V., Lang K., Lange V., Böhme I., Schmidt A.H., Sauter J. Frequencies of gene variant CCR5-Δ32 in 87 countries based on next-generation sequencing of 1.3 million individuals sampled from 3 national DKMS donor centers. Hum. Immunol. 2017;78:710–717. doi: 10.1016/j.humimm.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Mattiola I., Pesant M., Tentorio P.F., Molgora M., Marcenaro E., Lugli E., Locati M., Mavilio D. Priming of Human Resting NK Cells by Autologous M1 Macrophages via the Engagement of IL-1β, IFN-β, and IL-15 Pathways. J. Immunol. 2015;195:2818–2828. doi: 10.4049/jimmunol.1500325. [DOI] [PubMed] [Google Scholar]
- 32.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai B., Lee J.-E., Jang Y., Wang L., Peng W., Ge K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017;45:6388–6403. doi: 10.1093/nar/gkx234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis K.L., Heidlebaugh A.R., Epps S., Han P.K.J., Fishler K.P., Klein W.M.P., Miller I.M., Ng D., Hepler C., Biesecker B.B., Biesecker L.G. Knowledge, motivations, expectations, and traits of an African, African-American, and Afro-Caribbean sequencing cohort and comparisons to the original ClinSeq(®) cohort. Genet. Med. 2019;21:1355–1362. doi: 10.1038/s41436-018-0341-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnston J.J., Lewis K.L., Ng D., Singh L.N., Wynter J., Brewer C., Brooks B.P., Brownell I., Candotti F., Gonsalves S.G., et al. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am. J. Hum. Genet. 2015;96:913–925. doi: 10.1016/j.ajhg.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mikulak J., Teichberg S., Arora S., Kumar D., Yadav A., Salhan D., Pullagura S., Mathieson P.W., Saleem M.A., Singhal P.C. DC-specific ICAM-3-grabbing nonintegrin mediates internalization of HIV-1 into human podocytes. Am. J. Physiol. Ren. Physiol. 2010;299:F664–F673. doi: 10.1152/ajprenal.00629.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mikulak J., Teichberg S., Faust T., Schmidtmayerova H., Singhal P.C. HIV-1 harboring renal tubular epithelial cell interaction with T cells results in T cell trans-infection. Virology. 2009;385:105–114. doi: 10.1016/j.virol.2008.11.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Raw data of RNA-Seq experiments were deposited in GEO (Accession number: GSE141410).
-
•
All data that support the findings of this study are available from the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available for the lead contact upon request.