

Summary
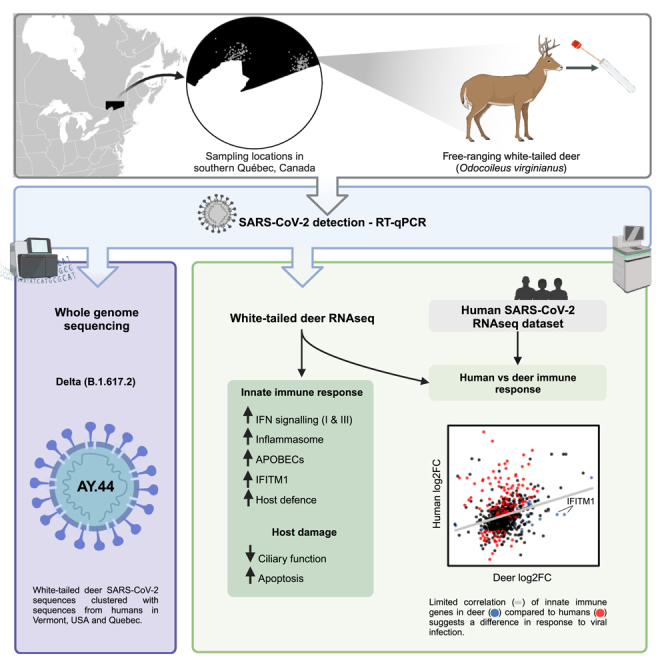
White-tailed deer (WTD) are susceptible to SARS-CoV-2 and represent an important species for surveillance. Samples from WTD (n = 258) collected in November 2021 from Québec, Canada were analyzed for SARS-CoV-2 RNA. We employed viral genomics and host transcriptomics to further characterize infection and investigate host response. We detected Delta SARS-CoV-2 (B.1.617.2) in WTD from the Estrie region; sequences clustered with human sequences from October 2021 from Vermont, USA, which borders this region. Mutations in the S-gene and a deletion in ORF8 were detected. Host expression patterns in SARS-CoV-2 infected WTD were associated with the innate immune response, including signaling pathways related to anti-viral, pro- and anti-inflammatory signaling, and host damage. We found limited correlation between genes associated with innate immune response from human and WTD nasal samples, suggesting differences in responses to SARS-CoV-2 infection. Our findings provide preliminary insights into host response to SARS-CoV-2 infection in naturally infected WTD.
Subject areas: Virology, Wildlife medicine, Wildlife immunology, Genomics, Immunity, Transcriptomics
Graphical abstract
Highlights
-
•
Delta SARS-CoV-2 was detected in white-tailed deer from southern Quebec
-
•
Host expression patterns from naturally infected deer differed from humans
-
•
These deer show increased type I, III interferons, APOBECs and IFITM1 expression
Virology; Wildlife medicine; Wildlife immunology; Genomics; Immunity; Transcriptomics
Introduction
White-tailed deer (Odocoileus virginianus) are considered a priority species for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) surveillance in North America based on existing epidemiological and experimental evidence of SARS-CoV-2 exposure, infection, and transmission.1,2,3,4,5,6,7,8,9,10,11 Multiple focally distributed human to white-tailed deer (WTD) spillover events have been observed, with initial whole genome sequencing (WGS) analyses revealing SARS-CoV-2 lineages in WTD that reflect those contemporaneously circulating in humans.4,5,12 Multiple studies have also documented evidence for onward and sustained deer-to-deer transmission.1,6,10,12,13 This includes the circulation of nearly extinct SARS-CoV-2 lineages (Alpha and Gamma) persisting in WTD in New York and Pennsylvania, USA, with a notable time lapse between the detection of these variants in humans and WTD.1,6 In Ontario, Canada, we previously identified a highly divergent SARS-CoV-2 variant circulating in WTD (B.1.641) with evidence of WTD-to-human transmission;10 this finding supports concerns for the emergence and accumulation of mutations in SARS-CoV-2 while circulating in novel animal hosts.14 This introduces the possibility of further divergent viral evolution and spillback into humans, potentially undermining the effectiveness of medical countermeasures such as antivirals and vaccines.15,16,17
The growing evidence of SARS-CoV-2 circulation among WTD in North America is suggestive of a new non-human maintenance population or reservoir for the virus. Despite this fundamental change to the ecology of SARS-CoV-2, there remains a dearth of knowledge about the course of infection in WTD, the host-immune response, and resultant evolutionary pressures. Experimental data suggest that WTD infected with SARS-CoV-2 exhibit few clinical signs, and pathology is limited.3,7,9 This is in contrast to the spectrum of disease severity observed in humans, underscoring the potential for differing host responses between species. There is a need to integrate data on host-immune response with epidemiological and ecological insights to understand the extent to which WTD host response contributes to their potential role as a maintenance host,18,19 and to discern the implications of species-adapted viruses for human health.
To try to address this we investigated SARS-CoV-2 infection among WTD in southern Québec, Canada as part of a broader pan-Canadian approach to investigate SARS-CoV-2 spillover into wildlife. We then employed viral genomics and host transcriptomics to further characterise SARS-CoV-2 in a novel and ecologically relevant host.
Results
SARS-CoV-2 delta variant of concern detected in white-tailed deer in southern Québec
To discern the prevalence of SARS-CoV-2 in WTD in the region, 258 WTD were sampled in two areas from southern Québec, Canada between November 6–8 2021. The majority of the sampled WTD were adult (92%) and male (79%). We collected 251 nasal swabs and 104 retropharyngeal lymph nodes (RPLNs) and tested for the presence of SARS-CoV-2 ribonucleic acid (RNA) by reverse-transcription polymerase chain reaction (RT-PCR). Longitude and latitude data were obtained for 257 WTD. Chronic wasting disease was not detected in WTD in Quebec in 2021.20
Four nasal swabs were RT-PCR-positive, three of which were confirmed by the Canadian Food Inspection Agency (CFIA) and reported to the World Organization for Animal Health (WOAH) as the first cases of SARS-CoV-2 identified in Canadian wildlife on December 1, 2021 (Table S1).21 Human RNase P was not detected in any of the positive nasal swabs, excluding contamination from human hosts. Of all nasal swabs, 1.6% (4/251; 95% CI 0.5–4.2%) were positive for SARS-CoV-2 RNA (Table 1); no RPLNs were positive; no RPLNs were available from deer with SARS-CoV-2 positive nasal swabs. All positive WTD were adults and three of the four were male. The four SARS-CoV-2-positive deer were harvested through licensed hunting activity in the high deer density region of Estrie (Figure 1).
Table 1.
Results from SARS-CoV-2 RT-PCR testing of nasal swabs and retropharyngeal lymph node tissue from white-tailed deer, in two sampling regions, southern Québec, Canada, November 6-8 2021
| Sampling area | Nasal Swabs |
Retropharyngeal Lymph Nodes |
||
|---|---|---|---|---|
| No. Tested | No. SARS-CoV-2 RNA Positive (%; 95% CI) | No. Testeda | No. SARS-CoV-2 RNA Positive (%; 95% CI)a | |
| Dunham station, Estrie region, High deer density | 150 | 4 (2.7; 0.8–6.9) | NA | NA |
| Browsburg station, Laurentides region, Low deer density | 101 | 0 (0; 0–4.4) | 104 | 0 (0; 0–4.3) |
| Total | 251 | 4 (1.6; 0.5–2.2) | 104 | 0 (0; 0–4.3) |
See also Table S1.
NA, not applicable.
Figure 1.

Locations of white-tailed deer (WTD) and SARS-CoV-2 RT-PCR test results in southern Québec, Canada
Map of southern Québec with locations of SARS-CoV-2 RT-PCR-positive (orange) and -negative (gray) WTD from November 6 - 8 2021 superimposed on (A) a choropleth map of human population density (per km2) by regional county municipalities (thin gray boundaries) and (B) a heatmap of WTD harvest density per 100km2 from 2020 as a proxy for deer population density. Inset shows the location of Québec (outlined) and study region (shaded black) within Canada. See also Figure S1.
Whole genome sequencing for SARS-CoV-2 conducted on three confirmed positive samples generated genome coverage of over 95% with equal to or greater than 10X coverage (mean depth from 1096.8 to 1894.8). On the fourth, higher cycle threshold (Ct) sample, 69.1% genome coverage with at least 10X coverage (mean depth of 116.5) was obtained (Table 2). Sequences were assigned to lineage AY.44, a sublineage of B.1.617.2 (Delta; 0.96–0.99 ambiguity score), while one sample could not be confidently assigned to a Pangolin lineage due to the large number of N bases (31%) in the consensus sequence. Phylogenetic analysis revealed that all sequenced samples clustered together and shared a most recent common ancestor with SARS-CoV-2 sequences from humans in Vermont, USA between 2021 and 10-14 and 2021-10-27 (Figure 2). Phylogenetic analysis of WTD derived SARS-CoV-2 sequences also revealed that all sequenced samples clustered together with WTD-derived SARS-CoV-2 sequences identified in the states of Iowa, Maine, Minnesota, New York, and Pennsylvania (Figure 3).
Table 2.
Read mapping statistics from nf-core/viralrecon analysis for four SARS-CoV-2 positive white-tailed deer nasal swab samples from southern Québec, Canada, November 6-8 2021
| Nasal Swab ID | Genome Coverage (%)a | Mean Coverage Depth | Total Reads | Mapped Reads | 0X positions | <10X positions | Virus name | GISAID Accession No. |
|---|---|---|---|---|---|---|---|---|
| 4055 | 99.6 | 1227.1 | 375796 | 293802 | 121 | 121 | hCoV-19/Canada/QC-WTD-qxic4055/2021 | EPI_ISL_10168587 |
| 4204 | 69.1 | 116.5 | 503532 | 29803 | 3219 | 9245 | QC failure, not uploaded | QC failure, not uploaded |
| 4205 | 95.8 | 1096.8 | 370802 | 252101 | 674 | 1255 | hCoV-19/Canada/QC-WTD-qxic4205/2021 | EPI_ISL_10169675 |
| 4249 | 98.9 | 1894.8 | 583638 | 460000 | 69 | 314 | hCoV-19/Canada/QC-WTD-qxic4249/2021 | EPI_ISL_10170149 |
Genome coverage was calculated as the proportion of Wuhan-Hu-1 (MN908947.3) reference positions with at least 10X read mapping depth.
Figure 2.

Whole-genome phylogenetic tree of four SARS-CoV-2 positive white-tailed deer (WTD) sequences and 17 closely related Canadian and American sequences identified by UShER analysis
IQ-TREE inferred the maximum-likelihood phylogenetic tree with a GTR+F+I + I + R5 substitution model (selected by IQ-TREE’s ModelFinder) from a Nextalign multiple sequence alignment of the 4 WTD, 91 NCBI, 2 GISAID and Wuhan-Hu-1 (MN908947.3) sequences. The tree was manually pruned with BioPython to highlight distinct clades and amino acid mutation patterns. The tree was visualized using the ggtree R library. Amino acid (AA) substitutions and deletions in GISAID and WTD sequences were determined using Nextclade for visualization alongside the tree and other metadata. Some positions within sample 4204 had low or no coverage, however, despite the poor coverage of sample 4204, it still clustered with the other WTD.
Figure 3.

Maximum likelihood phylogenetic tree of the complete SARS-CoV-2 genomes isolated from white-tailed deer (WTD) and mule deer
Phylogeny was reconstructed using IQ-TREE under the GTR+F+I + R4 substitution model. The three high-coverage samples from Québec WTD are labeled on the tree. Branches of the phylogenetic tree are colored by the country of origin and the leaf markers are colored by the state or province of the sequence record origin. The tree is rooted to the outgroup sequence, Wuhan-Hu-1, MN908947.3 (not shown). Ultrafast bootstrap support values are shown at the node of the Québec WTD clade.
Mutations in the S gene and ORF8 detected in white-tailed deer-derived SARS-CoV-2 sequences
We observed multiple mutations in SARS-CoV-2 sequences derived from the four positive WTD samples (Table 3). None of the observed mutations were unique to WTD, however three mutations (open reading frame 1a [ORF1a]:L1853F, M:I82T, and ORF7a:T120I) are speculated to have evolved due to adaptation in WTD; only M:I82T was found in all four sequences.1,22 Other mutations have been previously reported in WTD derived SARS-CoV-2 sequences from other regions (Table 3).
Table 3.
Summary of mutations leading to amino acid changes found in sequences from four SARS-CoV-2 positive white-tailed deer nasal swab samples from southern Québec, Canada, November 6-8 2021
| Gene | Amino Acid Mutation | Nucleotide Mutation | Nasal Swab ID(s) | Max Allele Fraction (%)a | Major Variant?b | Found in other white-tailed deer SARS-CoV-2 sequencesc |
|---|---|---|---|---|---|---|
| orf1ab | ΔM85 | TATG517T | 4204 | 88.9 | Yes | No |
| orf1ab | V1143F | G3692T | 4055; 4205; 4249 | 100 | Yes | No |
| orf1ab | A1306S | G4181T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | Q1784H | G5617T | 4055; 4204; 4205; 4249 | 100 | Yes | No |
| orf1ab | L1853F | C5822T | 4205 | 91.4 | Yes | Yes |
| orf1ab | P2046L | C6402T | 4055; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | H2125Y | C6638T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | S2224F | C6936T | 4249 | 26.1 | No | Yes |
| orf1ab | S2242F | C6990T | 4204; 4205 | 100 | Yes | Yes |
| orf1ab | P2287S | C7124T | 4055; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | A2554V | C7926T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | T2823I | C8733T | 4205 | 100 | Yes | No |
| orf1ab | V2930L | G9053T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | L3116F | C9611T | 4205 | 99.1 | Yes | Yes |
| orf1ab | T3255I | C10029T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | L3606F | G11083T | 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | T3646A | A11201G | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | T4467I | C13665T | 4205 | 98.5 | Yes | Yes |
| orf1ab | I4562M | A13951G | 4249 | 35.9 | No | No |
| orf1ab | N4583K | T14014G | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | H5401Y | C16466T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| orf1ab | I6162T | T18750C | 4055; 4249 | 99.7 | Yes | No |
| orf1ab | T6249I | C19011T | 4205 | 99.7 | Yes | Yes |
| orf1ab | V6265A | T19059C | 4249 | 37.5 | No | No |
| orf1ab | T6436I | C19572T | 4205 | 100 | Yes | Yes |
| orf1ab | T6775I | C20589T | 4055; 4205 | 100 | Yes | Yes |
| S | T19R | C21618G | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| S | T22I | C21627T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| S | A27V | C21642T | 4055; 4249 | 100 | Yes | Yes |
| S | G142D | G21987A | 4055; 4249 | 57.6 | No | Yes |
| S | E156G, ΔFR157-158 | GAGTTCA22028G | 4055; 4205; 4249 | 100 | Yes | Yes |
| S | L452R | T22917G | 4055; 4205; 4249 | 100 | Yes | Yes |
| S | T478K | C22995A | 4055; 4249 | 100 | Yes | Yes |
| S | D614G | A23403G | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| S | P681R | C23604G | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| S | D950N | G24410A | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| S | G1085R | G24815A | 4055; 4204; 4205; 4249 | 100 | Yes | No |
| S | T1117I | C24912T | 4249 | 35.0 | No | Yes |
| ORF3a | T12I | C25427T | 4205 | 99.9 | Yes | Yes |
| ORF3a | S26L | C25469T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| ORF3a | R134H | G25793A | 4055; 4205; 4249 | 100 | Yes | No |
| M | I82T | T26767C | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| ORF7a | T39I | C27509T | 4204; 4205 | 100 | Yes | Yes |
| ORF7a | V82A | T27638C | 4055; 4205; 4249 | 100 | Yes | Yes |
| ORF7a | T120I | C27752T | 4055; 4205; 4249 | 100 | Yes | Yes |
| ORF8 | S43F | C28021T | 4249 | 29.9 | No | Yes |
| ORF8 | L60F, Δ61-66 | TGTGCGTGGATGAGGCTGG28072T | 4205 | 59.3 | No | No |
| ORF8 | L60F | G28073T | 4055; 4249 | 100 | Yes | Yes |
| ORF8 | S67∗, ΔK68 | TCTA28092T | 4205 | 95.6 | Yes | No |
| ORF8 | ΔDF119-120 | AGATTTC28247A | 4055; 4205; 4249 | 87.2 | Yes | Yes |
| ORF8 | F120L | C28253A | 4055; 4249 | 100 | Yes | No |
| N | D63G | A28461G | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| N | R203M | G28881T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| N | G215C | G28916T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
| N | D377Y | G29402T | 4055; 4204; 4205; 4249 | 100 | Yes | Yes |
The maximum allele fraction for each variant observed in all sequenced samples was calculated from the number of alternate allele observations divided by the total number of observations for each variant site.
A variant with an allele fraction of at least 0.75 or 75% was classified as a major variant.
As of 2023-08-28.
Notably, two S gene mutations were observed in the WTD sequences: S:T22I in three samples and S:A27V in one sample. The S:T22I mutation was observed in only one closely related AY.44 sequence from a human from Quebec while S:A27V was not observed in closely related AY.44 sequences in GISAID. The S:T22I mutation has been observed in 16,628 GISAID sequences as of 2023-04-17 from a multitude of lineages. The S:A27V mutation has been observed in 5,889 GISAID sequences as of 2023-04-17. The S:G142D mutation, which is present in 64% of AY.44 sequences in GISAID (171,978/267,019 sequences as of 2023-04-17), is present in two samples as a minor variant (57% and 53% allele fraction, respectively). The S:G142D mutation is prevalent in many Delta sublineage sequences and present in 10,655,444 GISAID sequences as of 2023-04-17. Interestingly, the S:G1085R mutation, which is present in all four sequenced samples and related AY.44 sequences, is only present in 0.1% (279/267,019) in lineage AY.44 and has been identified in 2,574 GISAID sequences as of 2023-04-17.
An inframe deletion leading to a stop codon in the ORF8 was observed at S67/K68 (TCTA to T deletion at nucleotide position 28,092) in sample 4205. Additionally, an ORF8 inframe deletion of 6 amino acids at positions 61–66 and L60F mutation were observed in sample 4205 with an allele fraction of 59% (262/442 observations of TGTGCGTGGATGAGGCTGG to T at nucleotide position 28,072).
As most of the minor alleles were found in sample 4249, it is worth noting that this sample had 15 ambiguous genomic positions with otherwise high completeness (99.1%) and median coverage (1879X). This suggests that more than one SARS-CoV-2 genotype is present within the sequenced sample. This is attributable to either contamination or a mixed infection in the host. Demixing using Freyja v1.3 (https://github.com/andersen-lab/Freyja) revealed the sample was 99.4% AY.44 and 0.1% AY.98. However, no other samples in the same sequencing run contained the 4249-specific variants or were assigned to the AY.98 lineage making contamination less likely. Inter-run contamination is also unlikely since a new flow cell was used for each sequencing run and four different, alternating sets of 96 UD Indices were used to barcode the samples in the library pool to help mitigate index hopping. Moreover, all negative controls met quality control parameters, further suggesting contamination is unlikely. As AY.98 is closely related to AY.44 (differing by only 3 ORF1ab amino acid residues) and is inferred as low abundance, this suggests 4249 more likely represents a real mixed infection of two closely related AY.44 genomes.
Virus isolation
Two of the SARS-CoV-2-positive nasal swabs yielded viable virus when cultured in Vero E6 cells (Figure S2). Resultant sequences from the isolates were comparable to sequences from respective original nasal samples; key mutations found in the original nasal swab sample sequences were also identified in the sequences from the isolates. In addition, we noted several other mutations in the virus upon passaging in Vero E6 cells. Changes noted in 4055 after a single passage included S:R683W and a deletion in ORF 3a, V256-259. The S:R683W mutation has been previously attributed to adaptation in Vero E6 cell lines.23,24 Five low coverage variants were lost in 4249, which may suggest selection for the more abundant genotype.
White-tailed deer nasal microbial community profile an indicator of SARS-CoV-2 infection
The WTD nasal swabs were sequenced by RNAseq, allowing for the taxonomic profiling of the nasal community with Kraken 2 and Bracken (Table S2). SARS-CoV-2 was detected in the reads from WTD samples that tested positive for SARS-CoV-2 (4055 and 4249) (Figure S3A), confirming the RT-PCR results. All swabs were collected and stored in media containing antibiotics and antifungals prior to analysis, which could impact bacterial and fungal profiles. However, the SARS-CoV-2 positive samples clustered together even when excluding SARS-CoV-2 transcriptomic data. SARS-CoV-2-negative WTD samples 4192 and 3719 had a divergent microbial profile compared to other negative samples, most notably for relative increases in Cutibacterium acnes compared to the other WTD nasal swabs (Figure S3A).
SARS-CoV-2 infection elicited antiviral, pro- and anti-inflammatory host response in white-tailed deer
We employed unbiased exploratory transcriptomic analysis to provide insights on WTD host response to SARS-CoV-2 infection. When considering the entire gene expression profile, the WTD samples that tested positive for SARS-CoV-2 (4055 and 4249) clustered apart from the negative samples (Figure S3B). The negative samples further clustered into two groups, with samples 4192 and 3719 clustering away from the rest (Figure S3B). This difference is likely not due to age, sex, or hunting zone since these factors are not unique to the two aforementioned negative samples.
Differential expression analysis identified 316 significant differentially expressed genes (DEGs) (adjusted p < 0.05, >1-fold change), spanning 194 upregulated DEGs and 122 downregulated DEGs (Figures 4A and 4B and Table S3). Top expressed genes included LOC110149600 (an inferred ortholog of human interferon induced transmembrane protein 1 [IFITM1]), LOC110149612 (inferred IFITM1), 2′’-5’-oligoadenylate 2 (OAS2), hematopoietic SH2 domain containing (HSH2D), and apolipoprotein B MRNA editing enzyme catalytic subunit 3H (APOBEC3H) (Table 4 and Figure 4B). Notably, several significant DEGs appear to have duplications of human immune genes, including IFITM1, interferon, alpha-inducible protein 27 like 2A (IFI27L2A), C4a, heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), and XIAP associated factor 1 (XAF1). Upon narrowing down to known SARS-CoV-2 viral attachment and entry factors that have been identified as important for human COVID-19 infection (e.g., angiotensin converting enzyme 2 [ACE2] and transmembrane serine protease 2 [TMPRSS2]), only sialic acid binding Ig like lectin 1 (SIGLEC1) was significantly upregulated in the WTD samples (Figure S4).
Figure 4.

RNAseq analysis of white-tailed deer (WTD) nasopharyngeal swabs comparing uninfected WTD with SARS-CoV-2 infected WTD (4055 and 4249)
(A) Relative expression levels of significant (adjusted p value <0.05) differentially expressed genes with an absolute fold change greater than 3. WTD genes with human orthologs annotated with the Gene Ontology “innate immune response” term indicated underneath.
(B) Volcano plot of the DESeq2 differential gene expression analysis results. Where possible, genes were labeled with their inferred human ortholog gene name. The upper interferon induced transmembrane protein 1 (IFITM1) is associated with LOC110149600 and the lower IFITM1 is associated with LOC110149612 in the WTD genome.
(C) Gene Ontology function enrichment of the significant up and down-differentially expressed genes against a WTD genome background with DAVID. Only terms from biological processes (black dots) and molecular functions (gray dots) are displayed. See also Figures S3 and S4 and Tables S3, S4, S5, S6, and S7.
Table 4.
Top 10 up and down differentially expressed white-tailed deer genes ranked by adjusted p value
| Log2 fold change | Adjusted p value | WTD gene name | Inferred human orthologa |
|---|---|---|---|
| Up | |||
| 5.42 | 1.49x10−13 | interferon-induced transmembrane protein 1-like (LOC110149612) | IFITM1 |
| 5.50 | 1.84x10−11 | interferon alpha-inducible protein 27-like protein 2A (LOC110122860) | |
| 4.84 | 2.75x10−8 | interferon-induced transmembrane protein 1-like (LOC110149600) | IFITM1 |
| 6.34 | 9.71x10−8 | 2′-5′-oligoadenylate synthetase 2 (OAS2) | |
| 3.96 | 9.12x10−6 | pleckstrin homology domain-containing family A member 4-like (LOC110133690) | |
| 3.57 | 1.66x10−5 | hematopoietic SH2 domain containing (HSH2D) | HSH2D |
| 3.44 | 1.66x10−5 | DNA dC- > dU-editing enzyme APOBEC-3H-like (LOC110129971) | APOBEC3H |
| 6.12 | 2.70x10−5 | zonadhesin (ZAN) | ZAN |
| 4.24 | 6.32x10−5 | complement C4-A-like (LOC110139391) | C4B_2 |
| 2.95 | 7.22x10−5 | transporter 1, ATP binding cassette subfamily B member (TAP1) | TAP1 |
| Down | |||
| −25.49 | 5.00x10−7 | heterogeneous nuclear ribonucleoprotein A1-like (LOC110141849) | |
| −22.66 | 2.23x10−5 | cuticlin-2-like (LOC110149771) | |
| −22.56 | 2.31x10−5 | ornithine decarboxylase antizyme 1-like (LOC110136472) | |
| −21.26 | 9.82x10−5 | heterogeneous nuclear ribonucleoprotein A1-like (LOC110143697) | |
| −2.90 | 1.42x10−4 | testis expressed 9 (TEX9) | TEX9 |
| −2.48 | 1.43x10−4 | G protein subunit alpha 14 (GNA14) | GNA11 |
| −3.82 | 2.68x10−4 | janus kinase and microtubule interacting protein 2 (JAKMIP2) | JAKMIP2 |
| −2.82 | 5.32x10−4 | coiled-coil domain containing 181 (CCDC181) | CCDC181 |
| −2.14 | 5.68x10−4 | homer scaffold protein 2 (HOMER2) | HOMER2 |
| −3.04 | 5.68x10−4 | histamine receptor H1 (HRH1) | HRH1 |
See also Table S3. White-tailed deer differential gene expression analysis of SARS-CoV-2 positive samples compared to SARS-CoV-2 negative nasal samples with DESeq2, related to Figure 4 and Table 4, Table S4. White-tailed deer up-regulated differentially expressed genes annotation by the Database for Annotation, Visualization and Integrated Discovery (DAVID), related to Figure 4 and Table 4, Table S5. White-tailed deer up-regulated differentially expressed genes function enrichment compared to a white-tailed deer genome background by the Database for Annotation, Visualization and Integrated Discovery (DAVID), related to Figure 4 and Table 4, Table S6. White-tailed deer down-regulated differentially expressed genes annotation by the Database for Annotation, Visualization and Integrated Discovery (DAVID), related to Figure 4 and Table 4, Table S7. White-tailed deer down-regulated differentially expressed genes function enrichment compared to a white-tailed deer genome background by the Database for Annotation, Visualization and Integrated Discovery (DAVID), related to Figure 4 and Table 4.
If left blank, no human ortholog could be predicted with OrthoFinder.
DEG function enrichment analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Tables S3, S4, S5, and S6) revealed a change in the expression of key host factors in cellular innate immune response (interferon [IFN] signaling, inflammasome, APOBECs), damage (ciliary function and apoptosis), and permissivity (receptors, attachment, and entry factors) in RT-PCR-positive vs. RT-PCR-negative WTD. Significantly enriched Gene Ontology terms among up-regulated DEGs included defense response to virus, apoptotic process, innate immune response, inflammatory response, and chemotaxis, while down-regulated DEGs were involved in microtubule-mediated movement, axonemal dynein complex assembly, and ATP binding (Figure 4C). In humans and mice, orthologs of upregulated WTD DEGs are known to play important roles in type I and type III IFN signaling (e.g., interferon regulatory transcription factor 3 [IRF3], IRF4, IRF5, IRF7, IRF9, interferon lambda 3 [IFNλ3], signal transducer and activator of transcription 2 [STAT2], IFITM1, IFITM2, IFITM3, interferon alpha inducible protein 6 [IFI6], IFI27L2A), complement activation (e.g., C2, C4a), nucleic acid detection (e.g., DExH-box helicase 58 [DHX58]), immune cell recruitment (e.g., colony stimulating factor 1 [CSF1], CC motif chemokine Receptor 1 [CCR1], CXC motif chemokine receptor 2 [CXCR2], RHO family interacting cell polarization regulator 2 [RIPOR2]), apoptosis (e.g., engulfment and cell motility [ELMO], BH3 interacting domain death agonist [BID], XAF1, deoxyribonuclease 1 Like 3 [DNASE1L3]), and host defense (e.g., bone marrow stromal cell antigen 2 [BST2], Z-DNA binding protein 1 [ZBP1], OAS2, adenosine deaminase RNA specific [ADAR]). Orthologous DEGs that were downregulated are associated with microtubules (e.g., dynein cytoplasmic 2 heavy chain 1 [DYNC2H1], dynein light chain roadblock-type 2 [DYNLRB2], dynein axonemal heavy chain 3 [DNAH3], DNAH5, DNAH7) that make up extracellular structures such as cilia, likely reflecting host epithelial damage.
Comparison of white-tailed deer and human host response to SARS-CoV-2 infection
To identify potential differences in SARS-CoV-2 host responses between humans and WTD, we compared the log2 fold change of SARS-CoV-2 positive samples to negative samples from human and WTD nasopharyngeal swabs (Figure 5A and Table S8). The human cohort included 50 SARS-CoV-2 positive patients with a range of disease severity (18 admitted to the intensive care unit [ICU], 16 hospitalized, non-ICU, and 16 outpatients) and 13 SARS-CoV-2 negative individuals.25 The coverage of gene detection in the WTD samples was comparable to that of the human cohort; 67.10% of genes detected ±5.48% (57.93–72.85%) out of a total of 26,670 genes in the WTD reference and the human nasopharyngeal samples had 63.57% of genes detected ±4.53% (54.47–74.43%). When examining all genes associated with innate immune response, there is a weak but statistically significant correlation between WTD and human nasopharyngeal log2 fold change (r = 0.27, p value = 9.49x10−13), with a slightly higher correlation observed when comparing against only outpatients (r = 0.35, p value = 1.45x10−20). For this analysis we only focused on the ∼81% of WTD genes with detected human orthologs and did not include WTD-specific DEGs. We identified several immune-related genes that were significantly up-regulated in WTD and not in the human clinical cohort, including IFNλ1, BST2, and IFITM1 (Figure 5B). In the data from the Butler and colleagues26 human clinical cohort (RNAseq of naso/oropharyngeal swabs collected from 669 patients), some of these genes are significantly differentially expressed in SARS-CoV-2 positive versus negative patients, but still had low log2 fold changes when compared to WTD (0.84, 1.93, and 1.94 respectively), although the expression levels of these genes generally increased when comparing patients with higher viral loads than those with none.26 Key viral entry and attachment factors for human SARS-CoV-2 infection were also examined in this way (Figure S5), with five of the set (ACE2, basigin [BSG], cathepsin V [CTSV], metalloproteinase 2 [MMP2], and SIGLEC1) being significant DEGs in the human nasopharyngeal samples compared to only one (SIGLEC1) in the WTD samples.
Figure 5.

Comparison of the white-tailed deer (WTD) versus human host response to SARS-CoV-2 infection
(A) The WTD expression fold change is plotted here against the human expression fold change from comparable human samples based on the results of an orthology analysis between WTD and human. The genes are labeled with the respective human gene name, if the absolute difference in log2 fold change values between the human and WTD expression profiles is greater than 2.5. Due to duplicated genes in the WTD genome, some points are labeled with the same human gene name. From left to right, cathelicidin antimicrobial peptide (CAMP) genes are associated with LOC110152352, LOC110152353, and LOC110152344 in the WTD genome, interferon omega 1 (IFNω1) genes are associated with LOC110144825 and LOC110144833 and interferon induced transmembrane protein 1 (IFITM1) genes are associated with LOC110149600 and LOC110149612. Points are colored based on significance in their respective differentially gene expression analysis. Genes with very low expression values are not given a p value in the DESeq2 analysis and are not considered significant.
(B) Genes that are significantly differentially expressed in the WTD SARS-CoV-2 infection and have an absolute difference in log2 fold change values between the human and WTD infections greater than 2.5 are plotted here with their corresponding log2 fold change values across different levels of human disease severity. Intensive care unit (ICU), non-ICU, and outpatient are comparisons of human nasopharyngeal swabs between patients from these respective settings, and healthy patients. Genes are labeled with their human ortholog gene name. IFITM1 here corresponds to LOC110149600 in the WTD genome. See also Table S8.
Orthology analysis between human and the texanus subspecies of WTD revealed two interferon induced transmembrane 1 (IFITM1) genes in WTD, compared to a single copy in humans. Both genes were significantly over-expressed in WTD with SARS-CoV-2 infections (log2 fold change of 4.84 and 5.42 with adjusted p values of 2.82x10−8 and 1.53x10−13 for LOC110149600 and LOC110149612, respectively). However, in the human cohort, IFITM1 was not significantly differentially expressed, and log2 fold change values were low. Other WTD genomes (strains 20LAN1187 and brownington 1) were also confirmed to have two IFITM1 genes, with 99% similarity between the sequenced LOC110149600 genes (2–4 nucleotide differences) and 99% similarity between the LOC110149612 genes (6–12 nucleotide differences). LOC110149600 and LOC110149612 are separated on the genome, with an average distance of 26,485 ± 486 base pairs apart. The LOC110149612 transcript has an extended N-terminus compared to LOC110149600 but with respect to their protein sequences, there are only three amino acid substitutions between them: valine/methionine (position 10), leucine/methionine (position 11), and valine/methionine (position 20) for LOC110149612 and LOC110149600, respectively. Near identical IFITM1 sequences were also found in the other two WTD genomes. The identity between the human and WTD IFITM1 proteins was much lower at 66–68%, with differences in the N-terminus including several unique proline substitutions that could impact protein structure. A Fixed Effects Likelihood analysis identified positions 20, 113, and 115 to have evidence of diversifying selection (Table S9), with all three of these positions differing between humans and WTD. A phylogenetic tree based on IFITM1 proteins from boreoeutherian mammals shows multiple instances of IFITM1 gene duplication, all showing the extensive diversification IFITM1 has undergone throughout mammals and the substantial differences between human and WTD IFITM1 (Figure 6).
Figure 6.

Phylogenetic analysis of interferon induced transmembrane protein 1 (IFITM1) in mammals reveals lineage-specific duplications in white-tailed deer and evolutionary diversification
(A) Alignment of IFITM1 from select boreoeutherian mammals. An extended N-terminal start to XP_043307611.1 (Cervus canadensis) and XP_025142924.2 (Bubalus bubalis) has been trimmed from the alignment. Boxed and asterisked positions 20, 113 and 115 were identified as sites of diversifying selection. Alignment is colored with the Clustal color scheme with poorly conserved residues left white using Jalview v2.11.2.6.
(B) A mid-point rooted tree of IFITM1 from select boreoeutherian mammals. Bootstrap values less than 70 were not displayed. See also Table S9.
Discussion
In this report, we detected SARS-CoV-2 in 1.6% (95% CI 0.5–4.2%) of nasal swabs from sampled WTD in the Estrie region of southern Québec; viral sequences were assigned to lineage AY.44, a sublineage of the Delta variant of concern (VOC). Delta was the predominant circulating VOC at the time these animals were sampled. However, several unique mutations observed in the WTD sequences were not observed in the closely related AY.44 sequences from GISAID, supporting sustained deer-deer transmission. The WTD derived sequences from this study were most closely related to SARS-CoV-2 sequences from humans in neighboring Vermont, USA and one sequence from a human in Québec (Figure 2). Although we included SARS-CoV-2 sequences from humans from Québec in the analysis, it is unknown if any of these sequences were from the Estrie region. When compared to other deer derived SARS-CoV-2 sequences, the WTD SARS-CoV-2 sequences from this study were most closely related to sequences from north-eastern USA (Figure 3). While the Estrie region borders Vermont, to our knowledge, there is no evidence of SARS-CoV-2 in WTD or other wildlife species in Vermont reported to date.27
Whole SARS-CoV-2 genome sequences were found to contain two S gene mutations (S:T22I and S:A27V) that were different between WTD SARS-CoV-2 and the most closely related AY.44 sequences from GISAID. These changes are both located in the N terminal domain (NTD) of S1, which harbors antigenic and glycan-binding sites, and may interact with auxiliary receptors.28 Changes at amino acid position 22 and 27 have been noted early in the pandemic, with up to four different amino acid variants at position 27; this plasticity is suggestive of an adaptive evolutionary role.29,30 We have since detected the S:T22I mutation in a highly divergent, WTD-adapted SARS-CoV-2 from Ontario,10 further supporting a relevant role for changes in this region of the S protein. Additionally, in one WTD (4205), two inframe deletions in ORF8 were observed. The ORF8 gene is hypervariable and encodes for a non-essential accessory protein and has been shown to downregulate the major histocompatibility complex class I through autophagic degradation, thus impairing cytotoxic T cell responses during SARS-CoV-2, but not SARS-CoV infection.31 It may also contribute to immune evasion through interferon antagonism.32,33 More recently, ORF8 protein was found to purportedly mimic host interleukin-17 that contributes to severe inflammation in COVID-19.34 Truncation of ORF8 arising from nonsense mutations and deletions have been previously observed in both human and animal derived sarbecoviruses (SARS-CoV and SARS-CoV-2)32,35,36,37 and may be associated with milder disease through enhanced T cell functions. This underscores the potential role of ORF8 in SARS-CoV-2 adaptation.
All RT-PCR-positive WTD were identified in the Estrie region (Table 1; Figure 1). While it is presently unclear how the WTD acquired SARS-CoV-2 infection, there are several notable differences between the sampled regions that may contribute to the spatial heterogeneity of infection: 1) WTD population density and hunter harvest are greater in Estrie (13–15 WTD/km2) compared to the Laurentides (∼1 WTD/km2) (MELCCFP, unpublished data), 2) the sampled regions in Estrie have higher human population density compared to sampled regions in the Laurentides (Figure 1A), and 3) COVID-19 positivity in humans was greater in Estrie (4.2%) during the study period compared to the Laurentides (2.3%).38 Although the spatial clustering of SARS-CoV-2 in WTD has been previously reported in other regions,1,5,6 it is possible that the identification of SARS-CoV-2 in WTD only in the Estrie region could be a result of temporally and geographically limited sampling. More longitudinal surveillance of SARS-CoV-2 in WTD is needed to understand the epidemiology of the virus in this species and how it relates to WTD ecology and transmission dynamics in sympatric humans. These are important considerations for understanding the potential role of WTD as a maintenance population or reservoir for SARS-CoV-2.
Experimental data demonstrate that productive viral replication is limited to the upper respiratory tract with the shedding of infectious virus in nasal secretions of infected WTD.3,9 We successfully isolated viable SARS-CoV-2 from two RT-PCR-positive nasal swabs, demonstrating viral infectivity. This suggests there is a potential risk of contact with infectious SARS-CoV-2 from WTD, and several probable deer-to-human transmission events have been reported to date.10,12 These findings warrant increased awareness of the risks associated with direct human contact with live free-living and captive WTD and their carcasses.39
Work characterizing the WTD host response to SARS-CoV-2 infection is limited. Our study is the first to explore the host response in WTD naturally infected with SARS-CoV-2. Davila and colleagues conducted a comparative transcriptomics analysis of SARS-CoV-2 infected human and WTD primary respiratory tracheal epithelial cells in vitro, finding evidence of differing early innate immune responses.40 Compared to the work by Davila and colleagues,41 there is no overlap in significant DEGs observed in our study. This may be because of the difference in the anatomical site (e.g., nasal cavity vs. trachea) of sampling and differences between in vitro and in vivo systems (e.g., the absence of commensal flora in the former). Additionally, the course of infection for WTD in the present study is unknown and may not be comparable to infection time points for the tracheal cells derived from experimentally infected cells, as well as differences attributable to responses elicited by ancestral SARS-CoV-2 vs. the Delta VOC. Indeed, our data suggest the need for controlled in vivo studies in WTD to map differential host responses upon SARS-CoV-2 infection and delineate the pathological outcomes of infection in this ecologically important mammalian species.
Host expression patterns in SARS-CoV-2 infected WTD were associated with the innate immune response, including signaling pathways related to anti-viral and pro-inflammatory signaling and host damage. There was evidence for type I and III IFN responses (e.g., IRF3, IRF4, IRF5, IRF7, IRF9, INFλ3, STAT2, IFITM1, IFITM2, IFITM3, IFI6, IFI27L2A). The type I IFN response is an important aspect of rapidly controlling viral infection, and dysregulation is associated with severe illness and pathology. Type III IFNs produce a more localized response to infection in comparison to the systemic inflammatory response often induced by type I IFN; type III IFNs also result in prolonged expression of ISGs.42 Upregulation of pro-inflammatory antagonists were also observed. For example, there was increased expression of transcripts for two nuclear factor kappa-light-chain-enhancer of activated B cells (NF-KB) inhibitors (e.g., NFKB inhibitor delta [NFKBID], NFKB inhibitor zeta [NFKBIZ]) which could indicate a mechanism to reduce immunopathology or indicate a switch toward the resolution of inflammation (e.g., tissue repair).43 Additionally, several genes involved in antiviral response and inflammation homeostasis were found to have multiple transcript variants (e.g., IFITM1, XAF1) which may also contribute to protecting the host from inflammation.44,45 Nothing is known about antiviral gene duplication in WTD and their role in antiviral immune response.
When examining all genes associated with innate immune response, there was a weak but statistically significant correlation between WTD and human log2 fold change, with the strongest correlation observed between WTD and outpatients. Previous studies have reported that WTD infected with SARS-CoV-2 do not present with overt signs of infection or pathology; only minor pathological changes associated with rhinitis and loss of cilia in the respiratory epithelium have been observed in experimentally infected WTD.3,7,9 However, the duration of infection for each individual host in our study was unknown, limiting the inferences that can be made regarding the dynamics of innate immune responses. Additionally, it is unclear whether infection with SARS-CoV-2 results in any sublethal effects (e.g., body condition, winter survival, reproduction) despite no overt signs of disease.
There were also DEGs with substantial differences within the innate immune response gene set, notably macrophage receptor with collagenous structure (MARCO), IFNL1, IFITM1, IFITM3, radical s-adenosyl methionine domain containing 2 (RSAD2), BST2, APOBEC3H, low-density lipoprotein receptor-related protein 8 (LRP8), and BPI fold containing family A member 1 (BPIFA1). These genes had a large difference (>2.5) in log2 fold change values between the human and WTD DEGs while also being significantly differentially expressed in the WTD samples. These data indicate that the WTD nasal epithelium expressed genes that encode antiviral proteins, including the IFITM family, in response to SARS-CoV-2 infection. IFITMs are membrane proteins that restrict viral entry for a broad spectrum of enveloped viruses.46,47 In contrast, studies in human cells have shown that SARS-CoV-2 has been shown to hijack IFITMs for efficient cellular entry, a process mediated by specific interactions between the N-terminal region of IFITMs and the viral S protein.45,47 Intriguingly, IFITM1 has undergone a lineage-specific duplication resulting in two copies in WTD and transcripts for both were significantly upregulated in our samples. The amino acid changes between the two WTD IFITM1 are present in other mammalian (e.g., other deer in the Cervus genus and Felinae) lineages and are thus not unique to WTD, but the mutations appear to have evolved independently on several occasions (Figure 6). More importantly, the N- and C-terminal ends of both IFITM1 possess unique differences in WTD compared to human IFITM1. We speculate that these differences might contribute to differences in antiviral properties or efficiency of IFITM1 activity in WTD (and other animals) versus humans, as previously seen with the IFITM3 family in primates,48 and are an important future target for characterizing SARS-CoV-2-WTD molecular interactions.
Surveillance for SARS-CoV-2 in wildlife is ongoing across Canada. Our findings underscore that longitudinal surveillance efforts in WTD in Québec and across Canada are warranted. We provide preliminary insights into unique transcriptional responses in WTD that are naturally infected with SARS-CoV-2. Further work is needed to understand the mechanisms that facilitate virus transmission from humans to deer and among WTD, along with identifying selection pressures within WTD that promote adaptive mutations within the viral genome. Additionally, more longitudinal epidemiological and ecological data is needed to better understand whether WTD truly represent a competent maintenance population or reservoir for SARS-CoV-2. Ongoing coordinated and cross-disciplinary efforts are required to ensure a One Health approach is applied to this critical pandemic challenge by informing evidence-based decision-making for human and animal health.
Limitations of the study
While leveraging annual WTD hunting season resulted in many samples, the present work was conducted over a short time and a relatively small geographic region. Therefore, our study represents a snapshot in time and space. Future work should aim to obtain longitudinal data to investigate maintenance of SARS-CoV-2 in Québec WTD populations and assess spatiotemporal patterns in pathogen ecology. Samples analyzed in this study were derived from harvested WTD and were therefore collected postmortem. Although 98% of samples were collected within 48 h of harvest, it is possible that inhibitors or sample degradation occurred between harvest and sample collection. The low prevalence and available sample material for follow-up analysis resulted in a low sample size of SARS-CoV-2 positive WTD for the transcriptomic analysis. Finally, our study only focuses on free-ranging WTD populations, and we therefore cannot make inferences about SARS-CoV-2 in captive conspecifics.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| White tailed deer nasal swabs | This study | N/A |
| White tailed deer retropharyngeal lymphnodes | This study | N/A |
| Critical commercial assays | ||
| NUCLISENS® EASYMAG® Reagents | bioMérieux | Cat# 280130-280135 |
| Luna Universal one-step rt-qpcr kit | NEB | Cat# E3005L |
| MagMax CORE Nucleic Acid Purification Kit | ThermoFisher Scientific | Cat# A32700 |
| TaqMan Fast Virus 1-step Master Mix | ThermoFisher Scientific | Cat# 4444434 |
| LunaScript RT SuperMix | NEB | Cat# E3010 |
| Q5® High-Fidelity DNA Polymerase | NEB | Cat# M0491 |
| Sample Purification Beads | Illumina | Cat# 20060057 |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Cat# Q33230 |
| Qubit RNA HS | ThermoFisher Scientific | Cat# Q32852 |
| Illumina DNA Prep | Illumina | Cat# 20060059 |
| Illumina DNA/RNA UD Indexes | Illumina | Cat# 20027213, 20027214, 20042666, 20042667 |
| High Sensitivity RNA ScreenTape | Aligent Technologies | Cat# 5067-5579 |
| NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) | NEB | Cat# E7405 |
| NEBNext Ultrea II RNA Library Prep Kit for Illumina | NEB | Cat# E7765 |
| dsDNA High Sensitivity chip | Aligent Technologies | Cat# 5067-4626 |
| Quant-iT dsDNA high-sensitivity | ThermoFisher Scientific | Cat# Q33120 |
| NEBNext Library Quant Kit for Illumina | NEB | Cat# E7630L |
| Deposited data | ||
| White tailed deer SARS-CoV-2 sequences (deposited to GISAID) | This study | EPI_ISL_10169675, EPI_ISL_10170149, EPI_ISL_10168587 |
| White-tailed deer metatranscriptomic sequencing data (deposited to the NCBI Short Read Archive) | This study | BioProject accession no. PRJNA1025888 |
| Experimental models: Cell lines | ||
| Vero E6 cells | ATCC | CRL-1586; RRID: CVCL_0574 |
| Oligonucleotides | ||
| GTTGCAGCCGATCATCAGC | LeBlanc et al.49 | SARS-CoV-2 5’ UTR Forward Primer |
| GACAAGGCTCTCCATCTTACC | LeBlanc et al.49 | SARS-COV-2 5’ UTR Reverse Primer |
| CGGTCACACCCGGACGAAACCTAG | LeBlanc et al.49 | SARS-COV-2 5’ UTR Probe |
| ACAGGTACGTTAATAGTTAATAGCGT | Lu et al.50 | SARS-COV-2 E gene Forward Primer |
| ATATTGCAGCAGTACGCACACA | Lu et al.50 | SARS-COV-2 E gene Reverse Primer |
| ACACTAGCCATCCTTACTGCGCTTCG | Lu et al.50 | SARS-COV-2 E gene Probe |
| TTACAAACATTGGCCGCAAA | Corman et al.51 | SARS-COV-2 N2 gene Forward Primer |
| GCGCGACATTCCGAAGAA | Corman et al.51 | SARS-COV-2 N2 gene Reverse Primer |
| ACAATTTGCCCCCAGCGCTTCAG | Corman et al.51 | SARS-COV-2 N2 gene Probe |
| AGATTTGGACCTGCGAGCG | Lu et al.50 | Human RNAseP Forward Primer |
| AGCGGCTGTCTCCACAAGT | Lu et al.50 | Human RNAseP Reverse Primer |
| TTCTGACCTGAAGGCTCTGCGCG | Lu et al.50 | Human RNAseP Probe |
| ATGCGGCTAATCCCAACCT | Sequences available from Asuragen | Arm-Ent-31 Forward |
| CGTTACGACAGGCCAATCACT | Sequences available from Asuragen | Arm-Ent-31 Reverse |
| CAGGTGGTCACAAAC | Sequences available from Asuragen | Arm-Ent-31 Probe |
| ARTIC primer pools | https://github.com/artic-network/artic-ncov2019/V3 | v3 |
| ARTIC primer pools | https://github.com/artic-network/artic-ncov2019/V4 | v4 |
| Recombinant DNA | ||
| Armored RNA Enterovirus | Asuragen | 42050 |
| Software and algorithms | ||
| nf-core/viralrecon: nf-core/viralrecon Nextflow workflow | https://github.com/nf-core/viralrecon | V2.2 |
| FastQC | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | v0.11.9, v0.11.4; RRID: SCR_104583 |
| Fastp | https://github.com/OpenGene/fastp | v0.20.1, v0.21.0; RRID: SCR_016962 |
| Bowtie2 | https://github.com/BenLangmead/bowtie2 | v2.4.2; RRID: SCR_016368 |
| Mosdepth | https://github.com/brentp/mosdepth/releases | v0.3.1; RRID: SCR_018929 |
| Samtools | https://github.com/samtools/samtools/releases/ | v1.12; RRID: SCR_002105 |
| iVar | https://github.com/andersen-lab/ivar | v1.3.1; RRID: SCR_024045 |
| SnpEff | http://pcingola.github.io/SnpEff/ | v5.0; RRID: SCR_005191 |
| SnpSift | http://pcingola.github.io/SnpEff/ | v4.3t; RRID: SCR_015624 |
| Pangolin | https://github.com/cov-lineages/pangolin/ | v3.1.17 |
| UShER | https://github.com/yatisht/usher/tags | v0.6.2 |
| Nextalign | https://github.com/neherlab/nextalign | v2.13.0 |
| IQ-TREE | https://github.com/iqtree/iqtree2 | v2.2.0.3; RRID: SCR_017254 |
| BioPython | https://github.com/biopython/biopython | v1.79; RRID: SCR_007173 |
| Nexclade | https://github.com/nextstrain/nextclade | v2.13.0 |
| FigTree | http://tree.bio.ed.ac.uk/software/figtree/ | v1.4.4; RRID: SCR_008515 |
| Inkscape | https://inkscape.org/ | v.1.0.2; RRID: SCR_014479 |
| STAR | https://github.com/alexdobin/STAR | v2.5.2b; RRID: SCR_004463 |
| BBMap | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/ | v38.96; RRID: SCR_016965 |
| Kraken 2 | https://github.com/DerrickWood/kraken2 | v2.1.2; RRID: SCR_005484 |
| Bracken | https://github.com/jenniferlu717/Bracken/releases | v2.6.0 |
| R | https://www.r-project.org/ | v4.1.1; RRID: SCR_001905 |
| Salmon | https://github.com/COMBINE-lab/salmon/releases | v1.4; RRID: SCR_017036 |
| DESeq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | v1.34.0; RRID: SCR_015687 |
| OrthoFinder | https://github.com/davidemms/OrthoFinder/releases | v2.5.2; RRID: SCR_017118 |
| MUSCLE | http://www.drive5.com/muscle/ | v3.8.425; RRID: SCR_011812 |
| Stata/SE | http://www.stata.com | 15.1; RRID: SCR_012763 |
| QGIS | http://www.qgis.org | 3.28.2; RRID: SCR_018507 |
| CFIA-NCFAD/Delta-SARS-CoV-2-WTD-QC | https://github.com/CFIA-NCFAD/Delta-SARS-CoV-2-WTD-QC | 1.0.0 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Samira Mubareka (samira.mubareka@sunnybrook.ca).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Sequence data from the three SARS-CoV-2 viruses from white-tailed deer sequenced in this study are deposited in GISAID (https://www.gisaid.org/) under Accession numbers EPI_ISL_10169675 (hCoV-19/Canada/QC-WTD-qxic4205/2021), EPI_ISL_10170149 (hCoV-19/Canada/QC-WTD-qxic4249/2021), and EPI_ISL_10168587 (hCoV-19/Canada/QC-WTD-qxic4055/2021). All white-tailed deer metatranscriptomic sequencing data for this project was deposited into the NCBI Short Read Archive (SRA) under BioProject accession no. PRJNA1025888.
-
•
All codes used in this study is available at the sources referenced in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Ethical statement
Samples were collected post-mortem from white-tailed deer (WTD) harvested by hunters as a part of the regular annual WTD hunting and not for the purpose of this study. As such, no wildlife collection permit was required by the Ministère de l’Environnement, de la Lutte contre les changements climatiques, de la Faune et des Parcs.
Research ethics approval for the previously analyzed human RNA-seq transcriptomics dataset25 was approved by the Sunnybrook Research Ethics Board (Transcriptomic analysis in patients with COVID-19; SUN-5024).
Sample collection and study region
White-tailed deer were sampled during the scheduled hunting season after harvesting by licensed hunters. Samples were collected at two big game registration stations in Dunham and Brownsburg, Québec (Figure S1) when harvested and cleaned carcasses were presented for registration; carcasses were returned to the hunters after sampling. Thus, no clinical or post-mortem examination was possible. No animals were killed for the purpose of this study. The Brownsburg station included collection of retropharyngeal lymph node (RPLN) tissues for Chronic Wasting Disease surveillance conducted by the Ministère de l’Environnement, de la Lutte contre les changements climatiques, de la Faune et des Parcs (MELCCFP) in free-ranging WTD in Québec in early November 2021.52 All samples were collected post-mortem. We collected nasal swabs in 1 mL in-house transport media and retropharyngeal lymph node (RPLN) tissues were collected in dry 2 mL tubes; both sample types were stored at -80°C prior to analysis. Sex, life stage (juvenile or adult) and geographic location of harvest (latitude and longitude) were recorded for each animal. There were 203 males, 53 females, and 2 deer of unknown sex. There were 237 adults, 20 juveniles, and 1 deer of unknown age.
Cells lines
Vero E6 cells, African green monkey cells, from the American Type Culture Collection (ATCC) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1x l-glutamine and penicillin/streptomycin (Pen/Strep; Corning). All cells were incubated at 37C with 5% CO2.
Method details
Biosafety
All work involving live SARS-CoV-2 was performed in the Combined Containment Level 3 Unit (C-CL3 Unit) of the Temerty Faculty of Medicine at the University of Toronto in accordance with institutional biosafety requirements. Personnel conducting the experiment received containment level 3 training.
RT-PCR screening and detection
RNA extractions were performed using 140 μL of nasal swab sample spiked with Armored RNA enterovirus (Asuragen; https://www.asuragen.com) via the Nuclisens EasyMag using Generic Protocol 2.0.1 (bioMérieux Canada Inc., St-Laurent, QC, Canada) according to manufacturer’s instructions. Tissue samples were thawed, weighed, minced with a scalpel, and homogenized in 600 μL of lysis buffer using the Next Advance Bullet Blender (Next Advance, Troy, NY, USA) and a 5 mm stainless steel bead at 5 m/s for 3 minutes. RNA from 30 mg tissue samples was extracted via the Nuclisens EasyMag using Specific Protocol B 2.0.1; RNA was eluted in 50 μL. Reverse-transcription polymerase chain reaction (RT-PCR) was performed using the Luna Universal Probe One-Step RT-qPCR kit (New England BioLabs; https://www.neb.ca). Two gene targets were used for SARS-CoV-2 RNA detection: the 5’ untranslated region (UTR) and the envelope (E) gene.49,51 The cycling conditions were: 1 cycle of denaturation at 60°C for 10 minutes then 95°C for 2 minutes followed by 44 amplification cycles of 95°C for 10 seconds and 60°C for 15 seconds. Quantstudio 3 software (Thermo Fisher Scientific; https://www.thermofisher.com) was used to determine cycle threshold (Ct). All samples were run in duplicate and samples with cycle thresholds (Ct) < 40 for both SARS-CoV-2 targets and armored RNA enterovirus in at least one replicate were considered positive. Positive samples were further analyzed for a human RNase P gene target to rule out potential contamination.50
Original material from positive samples was sent to the Canadian Food Inspection Agency (CFIA) for confirmatory RT-PCR testing and reporting to the World Organization for Animal Health (WOAH).53 Total RNA spiked with Armored RNA enterovirus was extracted using the MagMax CORE Nucleic Acid Purification Kit (ThermoFisher Scientific) and the automated KingFisher Duo Prime magnetic extraction system. The enteroviral armored RNA was used as an exogenous extraction control. Confirmatory RT-qPCR was performed using primers and probe specific for both SARS-CoV-2 E and nucleocapsid (N) genes.50 Master mix for qRT-PCR was prepared using TaqMan Fast Virus 1-step Master Mix (ThermoFisher Scientific) according to manufacturer’s instructions. Reaction conditions were 50°C for 5 minutes, 95°C for 20 seconds, and 40 cycles of 95°C for 3 seconds then 60°C for 30 seconds. Runs were performed by using a 7500 Fast Real-Time PCR System (Thermofisher, ABI). Samples with Ct < 36 for both gene targets were reported to WOAH.
SARS-CoV-2 amplification and sequencing
SARS-CoV-2 whole genome sequencing was performed at Sunnybrook Research Institute (SRI) and analyzed at CFIA. Extracted RNA was reverse transcribed into cDNA; 8 μL RNA was mixed with 8 μL nuclease-free water and 4 μL LunaScript RT SuperMix (New England BioLabs) then incubated at 25°C for 2 minutes followed by an incubation at 55°C for 20 minutes and 95°C for 1 minute before holding at 4°C.
cDNA was amplified using ARTIC v3 primer pools (https://github.com/artic-network/artic-ncov2019). Two separate reactions were prepared by combining 2.5 μL cDNA with 6 μL nuclease-free water, 12.5 μL Q5 Hot Start High-Fidelity 2X Master Mix (New England BioLabs) and 4 μL of 10 μM primer pool 1 or primer pool 2 (Integrated DNA Technologies; https://www.idtdna.com). The PCR cycling conditions included an initial denaturation at 98°C for 30 seconds followed by 35 cycles of 98°C for 15 seconds and 63°C for 5 minutes before holding at 4°C. Both reactions were combined and purified with 1X ratio Sample Purification Beads (Illumina; https://www.illumina.com). The resulting amplicons were quantified using Qubit 1X dsDNA HS Assay Kit (Thermo Fisher Scientific).
Libraries were constructed using Illumina DNA Prep (Illumina) and IDT for Illumina DNA/RNA UD Indexes (Illumina) and sequenced on Illumina MiniSeq using 2x149 paired-end reads. Six negative controls (i.e., two extraction, two RT-PCR, and two library prep controls) are included in each sequencing run and all controls must contain <1% of SARS-CoV-2 genome for the run to pass quality control.
Paired-end Illumina reads for samples RT_PCR-positive samples were analyzed using the nf-core/viralrecon Nextflow workflow (v2.2),54,55,56 which performed the following analysis steps: FastQC (v0.11.9) read quality assessment (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/); fastp (v0.20.1) read quality filtering and trimming;57 read mapping to Wuhan-Hu-1 (GenBank: MN908947.3) SARS-CoV-2 reference sequence with Bowtie2 (v2.4.2);58 read mapping statistics calculation with Mosdepth (v0.3.1)59 and Samtools (v1.12);60,61 ARTIC V3 primer trimming, variant calling and consensus sequence generation with iVar (v1.3.1);62 variant effect analysis and summarization with SnpEff (v5.0)63 and SnpSift (v4.3t),64 respectively; SARS-CoV-2 lineage assignment with Pangolin (v3.1.17).
Phylogenetic analysis was performed with the WTD consensus sequences generated by nf-core/viralrecon and 93 closely related NCBI and GISAID65,66,67 sequences identified by UShER68 using a dataset of 14,323,766 genomes from GISAID, GenBank, COG-UK and CNCB (2023-03-28; https://genome.ucsc.edu/cgi-bin/hgPhyloPlace).69 Multiple sequence alignment (MSA) was performed with Nextalign (v2.13.0)70 of the 4 WTD, 91 NCBI, 2 GISAID and Wuhan-Hu-1 (GenBank: MN908947.3) sequences. A maximum-likelihood tree was inferred using IQ-TREE (v2.2.0.3)71,72 from the Nextalign MSA with Wuhan-Hu-1 reference strain (GenBank: MN908947.3) as the outgroup. The best-fit substitution model was determined by IQ-TREE ModelFinder73 to be GTR+F+I+I+R5. The IQ-TREE phylogenetic tree was pruned with BioPython to highlight distinct clades and amino acid mutation patterns (v1.79)74 for visualization with the R ggtree library (v3.2.0);41 taxa with unique amino acid profiles were maintained with a representative for each unique amino acid profile selected at random. Nexclade CLI (v2.13.0)70 was used to identify amino acid substitutions and deletions in the sequences.
To investigate phylogenetic placement of the SARS-CoV-2 isolated from the Québec WTD relative to other publicly available WTD sequences, we downloaded all complete SARS-CoV-2 genomes isolated from WTD (n = 520) and mule deer (n = 2) available on GISAID (accessed 2023-08-20). Low coverage records including QC-4204 were excluded from the analysis. The Wuhan-Hu-1 (GenBank: MN908947.3) sequence was included as an outgroup. The MSA and maximum likelihood analysis followed the same procedures described for the general phylogenetic analysis (MSA using Nextalign v.2.13.0; maximum likelihood analysis inferred with IQ-TREE v.2.2.0.3). The best-fit substitution model (GTR+F+I+R4) was determined by IQ-TREE ModelFinder using the Bayesian Information Criterion (BIC). Node support was estimated using the ultrafast bootstrap approximation with 1000 replicates.75 Tree visualization was performed using FigTree (http://tree.bio.ed.ac.uk/software/figtree/) and Inkscape v.1.0.2 (https://inkscape.org/).
Virus isolation
Virus isolation was performed on RT-PCR-positive nasal swabs in containment level 3 at the University of Toronto. Vero E6 cells were seeded at a concentration of 3x105 cells/well in a six well-plate. The next day, 250 μL of sample with 16 μg/mL TPCK-treated trypsin (New England BioLabs), 2X penicillin and streptomycin and 2X antibiotic-antimycotic (Wisent; https://www.wisentbioproducts.com/en/) was inoculated onto cells. Plates were returned to a 37°C, 5% CO2 incubator for 1 hour and rocked every 15 minutes. After 1 hour, the inoculum was removed and replaced with DMEM containing 2% FBS, 6 μg/mL TPCK-treated trypsin, 2X penicillin/streptomycin, and 2X antibiotic-antimycotic. Cells were observed daily under a light microscope for cytopathic effect for 5 days post infection. The RT-PCR assay was used to confirm SARS-CoV-2 isolation from supernatant. Comparison of isolate sequences to their original nasal swab sample was conducted for confirmation: cDNA was amplified using ARTIC v4 primer pools (https://github.com/artic-network/artic-ncov2019) and variant calling results were generated using the SIGNAL (SARS-CoV-2 Illumina GeNome Assembly Line) pipeline v1.5.0;76 these were then compared to variant calling data for the original samples using this analysis workflow. Sequencing of the original nasal swab samples was conducted before ARTIC v4 primer pools were available. We subsequently implemented and used ARTIC v4 to improve genome completeness.
RNA-sequencing of white-tailed deer nasal swabs
Sufficient material was available for only two RT-PCR positive nasal swab samples (4055, 4249) for RNA-sequencing (RNA-seq). Eight total RNA samples, including two SARS-CoV-2 RT-PCR-positive and six randomly selected RT-PCR-negative samples, were submitted for RNA sequencing (RNA-seq) at the Donnelly Sequencing Centre at the University of Toronto (http://ccbr.utoronto.ca/donnelly-sequencing-centre). DNase-treated total RNA was quantified using Qubit RNA HS (cat # Q32852, Thermo Fisher Scientific Inc., Waltham, USA) fluorescent chemistry and 5 ng was used to obtain the RNA integrity number (RIN) using the High Sensitivity RNA ScreenTape (cat # 5067-5579, Agilent Technologies Inc., Santa Clara, USA). Lowest RIN was 1.5; median RIN score was 3. RNA-seq libraries were prepared from RNA samples (150ng) using the NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) (NEB Cat# E7405) in conjunction with NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB Cat# E7765). To facilitate the design of WTD-specific ssDNA probes Custom RNA Depletion primers were designed using the tool https://depletiondesign.neb.com/. These were then substituted for the oligos provided in the kit. Ribosomal RNA-depleted libraries had a mean concentration of 15.2ng/uL. 1uL top stock of each purified final library was run on an Agilent Bioanalyzer dsDNA High Sensitivity chip (cat # 5067-4626, Agilent Technologies Inc., Santa Clara, USA). The libraries were quantified using the Quant-iT dsDNA high-sensitivity (cat # Q33120, Thermo Fisher Scientific Inc., Waltham, USA) and were pooled at equimolar ratios after size-adjustment. The final pool was run on an Agilent Bioanalyzer dsDNA High Sensitivity chip and quantified using NEBNext Library Quant Kit for Illumina (cat # E7630L, New England Biolabs, Ipswich, USA).
The quantified pool was hybridized at a final concentration of 320 pM and sequenced paired end 150bp on the Illumina NovaSeq6000 platform using a SP flowcell at a depth of 100M reads per sample.
RNA-seq bioinformatic analysis of white-tailed deer nasal swabs
Raw reads were trimmed with fastp v0.21.0,57 with a front 9 nucleotide trim for R1 and R2 based on a FastQC v0.11.4 quality inspection. To remove the WTD sequences for microbial profiling, we used STAR v2.5.2b77 to create an index of Odocoileus virginianus texanus (GenBank: GCF_002102435.1; also used as a WTD reference in O’Hara et al.,78) with a sjdbOverhang of 100 and align the fastp-processed paired-end reads to this index. WTD-aligned reads were removed with BBMap v38.96 filterbyname.sh.79 The WTD-removed samples were run against the PlusPF pre-built database from May, 17, 2021, consisting of bacteria, archaea, viruses, human, protozoa, and fungi, with Kraken 2 v2.1.2.80 Species abundance was computed with Bracken v2.6.0.81 Distance-based clustering on the species and WTD samples was done with the pheatmap v1.0.12 package in R v4.1.1 and applied to a dot plot displaying the relative abundance of species within the community.
For the differential gene expression analysis, the fastp-process paired-end reads were run in the mapping-based mode against a Odocoileus virginianus texanus (GenBank: GCF_002102435.1) decoy-aware (genome as the decoy sequence) transcriptome using Salmon v1.482 with the validateMappings setting. The quantification files were imported into R (v4.1.1) and a gene mapping file was created with the makeTxDbFromGFF from the DESeq2 v1.34.0 package.83 Transcript quantifications were merged to the gene-level with tximport using the lengthScaledTPM setting. Low count genes with less than 10 counts were pre-filtered out to improve DESeq2 performance as recommended in the DESeq2 vignette. The differential gene expression analysis was done with the DESeq2 function based on the RT-PCR test results as factor levels. Variance-stabilizing transformation (VST) normalized gene expression for significantly differentially expressed genes based on the adjusted p-value and had a log2 fold change greater than 3 were displayed with gene expression scaling (done with the scale function) in a heatmap with the pheatmap package. The plotPCA function from the DESeq2 package was used to plot a PCA of the VST transformed gene expression profiles. Genes identified as significantly (adjusted p-value) up or down-expressed were run on the Database for Annotation, Visualization and Integrated Discovery (DAVID)84 on Oct. 7 2022 to identify functional terms (including Gene Ontology terms) enriched in the significantly up and down-expressed gene set over a WTD genome (GenBank: GCF_002102435.1) background. Notably, these genes and associated processes have been defined using human and mouse studies and as such are used as a surrogate to define likely processes in WTD.
To compare the WTD results with a human cohort, OrthoFinder v2.5.285 was run on a WTD (GenBank: GCF_002102435.1) and human (GRCh38.p13) proteome (GenBank: GCF_000001405.39), with the proteins mapped back to gene names. The log2 fold change values from a previous DESeq2 analysis of RNA-seq human COVID-19 infection gene expression,25 was compared to the WTD DESeq2 results. Briefly, the human RNA-seq transcriptomics dataset is derived from 50 SARS-CoV-2-positive and 13 SARS-CoV-2 negative individuals; nasopharyngeal swab samples were collected from a clinical cohort in the Greater Toronto Area between October 2020 and October 2021.25 The SARS-CoV-2 positive individuals include 16 outpatients, 16 hospitalized (non-ICU) patients, and 18 hospitalized ICU patients.25 The GOBP_INNATE_IMMUNE_RESPONSE human gene set from the Human Molecular Signatures Database (MSigDB) C5: ontology86 was used to subset the genes investigated and the correlation between the human and WTD immune system log2 fold change values was calculated with the cor.test function in R v4.1.1.
IFITM1 comparison within O. virginianus and mammalian IFITM1 phylogenetics
Interferon induced transmembrane protein 1 (IFITM1) duplication in other O. virginianus genomes (GenBank: GCA_023699985.2 and GenBank: GCA_014726795.1) was confirmed via BLASTN searches of those genomes with the two IFITM1 sequences (GenBank: NW_018336621.1:109564-110759 and GenBank: NW_018336621.1:c138855-136683 for LOC110149600 and LOC110149612, respectively) from O. virginianus texanus (GenBank: GCF_002102435.1). Assemblies Genbank: GCA_000191625.1 and GenBank: GCA_000191605.1 were not included as they were extremely small and only fragments of each IFITM1 gene were able to be detected. As for the IFITM1 sequences across mammals, the IFITM1 HomoloGene group 74501, that includes human, chimpanzee, macaque, wolf, and cow IFITM1 sequences was aligned with MUSCLE v3.8.42587 to other similar proteins in Boreoeutheria, including the O. virginianus texanus IFITM1 proteins. A RAxML-pthreads v8.2.1288 tree with the PROTGAMMAAUTO setting and the MRE-based Bootstrapping criterion was generated from the alignment, with JTT likelihood with empirical base frequencies being the best-scoring amino acid model for the tree. A nucleotide alignment including coding sequences from all proteins in the tree, as well as the other O. virginianus IFITM1 coding sequences, was created with MUSCLE v3.8.425 and subsequently trimmed to the best conserved length with codon alignment corrected. A Fixed Effects Likelihood analysis89 on this alignment generated with the Datamonkey Adaptive Evolution Server90 calculated positions with diversifying selection using the default p value threshold of 0.1.
Quantification and statistical analysis
Confidence intervals (CI) for SARS-CoV-2 prevalence of infection were estimated using Stata/SE 15.1 (StataCorp, College Station, Texas, USA; http://www.stata.com) with the Agresti-Coull CI method.91 Diagnostic data were plotted on a map of southern Québec according to the latitude and longitude of each WTD with human population density92 and WTD harvesting density data (MELCCFP, unpublished data). Maps were produced via QGIS 3.28.2 (Quantum GIS Development Team; http://www.qgis.org).
Acknowledgments
We thank the hunters for the deer submissions. Many thanks to the technicians and biologists who assisted with sample collection (Matthew Rokas-Bérubé, Yannicia Fréchette-Hudon, Catherine Greaves, Charles-Étienne Gagnon, Marylou Meyer, Camille Klein, Stéphane Lamoureux, Yannick Bilodeau, François Lebel, Sophie Plante, and Isabelle Laurion), the technicians that assisted with sample analysis (Emily Chien, Mathieu Pinette, Winfield Yim, Melissa Goolia, and Nikki Toledo), and Michelle Nebroski who assisted with sequencing data analysis. We are grateful for the work of D. Bulir in developing the UTR gene target used in the Sunnybrook Research Institute RT-PCR analysis. The graphical abstract for this article was prepared with BioRender.com.
This work was supported by the Public Health Agency of Canada, and Canadian Institutes of Health Research Operating Grant: Emerging COVID-19 Research Gaps and Priorities (466984). J.D.K. is supported by an AMMI Canada/BioMérieux Fellowship in Microbial Diagnostics. Funding and computing resources for F.M were provided by the Shared Hospital Laboratory, Dalhousie University, and the Donald Hill Family. O.L. was supported by the Canadian Safety and Security Program, Laboratories Canada, and Canadian Food Inspection Agency (CFIA) Genomics Development and Research Initiative. A.B. also acknowledges support from Natural Sciences and Engineering Research Council of Canada (NSERC), Canadian Institutes of Health Research (CIHR), and Coronavirus Variants Rapid Response Network (CoVaRR-Net). VIDO receives operational funding for its CL3 facility (InterVac) from the Canada Foundation for Innovation through the Major Science Initiatives. VIDO also receives operational funding from the Government of Saskatchewan through Innovation Saskatchewan and the Saskatchewan Ministry of Agriculture.
Author contributions
Conceptualization: J.D.K., A.M., M.G., J.B., T.B., B.P., and S.M.
Sample collection: J.D.K., A.M., and M.G.
Laboratory analysis: J.D.K, P.A., J.B.S., H.Y.C., K.N., L.Y., L.R.L., and B.P.
Data analysis/Investigation: J.D.K., B.L., P.K., F.M., O.V., and O.L.
Writing – original draft: J.D.K., and B.L.
Writing – review & editing: All authors.
Visualization: J.D.K., B.L., P.K., A.C.D., O.V., and O.L.
Supervision: L.R.L., A.C.D., O.L., B.P., and S.M.
Funding acquisition: A.M., M.G., and S.M.
Co-corresponding authors are justified from One Health and multidisciplinary perspectives: B.P. (animal health perspective and reference laboratory, confirmatory testing and reporting to the World Organization for Animal Health); A.D. (RNAseq computational biology); S.M. (human health perspective and initial virus detection, sequencing, isolation and generation of RNAseq dataset; lead contact).
Declaration of interests
The authors declare no conflicts relevant to this article.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108319.
Contributor Information
Andrew C. Doxey, Email: andrew.doxey@uwaterloo.ca.
Bradley Pickering, Email: bradley.pickering@canada.ca.
Samira Mubareka, Email: samira.mubareka@sunnybrook.ca.
Supplemental information
References
- 1.Caserta L.C., Martins M., Butt S.L., Hollingshead N.A., Covaleda L.M., Ahmed S., Everts M.R.R., Schuler K.L., Diel D.G. White-tailed deer (Odocoileus virginianus) may serve as a wildlife reservoir for nearly extinct SARS-CoV-2 variants of concern. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2215067120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chandler J.C., Bevins S.N., Ellis J.W., Linder T.J., Tell R.M., Jenkins-Moore M., Root J.J., Lenoch J.B., Robbe-Austerman S., DeLiberto T.J., et al. SARS-CoV-2 exposure in wild white-tailed deer (Odocoileus virginianus) Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2114828118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cool K., Gaudreault N.N., Morozov I., Trujillo J.D., Meekins D.A., McDowell C., Carossino M., Bold D., Mitzel D., Kwon T., et al. Infection and transmission of ancestral SARS-CoV-2 and its alpha variant in pregnant white-tailed deer. Emerg. Microbes Infect. 2022;11:95–112. doi: 10.1080/22221751.2021.2012528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hale V.L., Dennis P.M., McBride D.S., Nolting J.M., Madden C., Huey D., Ehrlich M., Grieser J., Winston J., Lombardi D., et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature. 2022;602:481–486. doi: 10.1038/s41586-021-04353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuchipudi S.V., Surendran-Nair M., Ruden R.M., Yon M., Nissly R.H., Vandegrift K.J., Nelli R.K., Li L., Jayarao B.M., Maranas C.D., et al. Multiple spillovers from humans and onward transmission of SARS-CoV-2 in white-tailed deer. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2121644119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marques A.D., Sherrill-Mix S., Everett J.K., Adhikari H., Reddy S., Ellis J.C., Zeliff H., Greening S.S., Cannuscio C.C., Strelau K.M., et al. Multiple Introductions of SARS-CoV-2 Alpha and Delta Variants into White-Tailed Deer in Pennsylvania. mBio. 2022;13:e0210122. doi: 10.1128/mbio.02101-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martins M., Boggiatto P.M., Buckley A., Cassmann E.D., Falkenberg S., Caserta L.C., Fernandes M.H.V., Kanipe C., Lager K., Palmer M.V., Diel D.G. From Deer-to-Deer: SARS-CoV-2 is efficiently transmitted and presents broad tissue tropism and replication sites in white-tailed deer. PLoS Pathog. 2022;18 doi: 10.1371/journal.ppat.1010197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palermo P.M., Orbegozo J., Watts D.M., Morrill J.C. SARS-CoV-2 SARS-CoV-2 neutralizing antibodies in white-tailed deer from Texas. Vector Borne Zoonotic Dis. 2022;22:62–64. doi: 10.1089/vbz.2021.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer M.V., Martins M., Falkenberg S., Buckley A., Caserta L.C., Mitchell P.K., Cassmann E.D., Rollins A., Zylich N.C., Renshaw R.W., et al. Susceptibility of white-tailed deer (Odocoileus virginianus) to SARS-CoV-2. J. Virol. 2021;95 doi: 10.1089/vbz.2021.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickering B., Lung O., Maguire F., Kruczkiewicz P., Kotwa J.D., Buchanan T., Gagnier M., Guthrie J.L., Jardine C.M., Marchand-Austin A., et al. Divergent SARS-CoV-2 variant emerges in white-tailed deer with deer-to-human transmission. Nat. Microbiol. 2022;7:2011–2024. doi: 10.1038/s41564-022-01268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roundy C.M., Nunez C.M., Thomas L.F., Auckland L.D., Tang W., Richison J.J., Green B.R., Hilton C.D., Cherry M.J., Pauvolid-Corrêa A., et al. High seroprevalence of SARS-CoV-2 in white-tailed deer (Odocoileus virginianus) at one of three captive cervid facilities in Texas. Microbiol. Spectr. 2022;10:e0057622. doi: 10.1128/spectrum.00576-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng A., Bevins S., Chandler J., DeLiberto T.J., Ghai R., Lantz K., Lenoch J., Retchless A., Shriner S., Tang C.Y., et al. Transmission of SARS-CoV-2 in free-ranging white-tailed deer in the United States. Nat. Commun. 2023;14:4078. doi: 10.1038/s41467-023-39782-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McBride D.S., Garushyants S.K., Franks J., Magee A.F., Overend S.H., Huey D., Williams A.M., Faith S.A., Kandeil A., Trifkovic S., et al. Accelerated evolution of SARS-CoV-2 in free-ranging white-tailed deer. Res. Sq. 2023 doi: 10.1038/s41467-023-40706-y. rs.3.rs-2574993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan C.C.S., Lam S.D., Richard D., Owen C.J., Berchtold D., Orengo C., Nair M.S., Kuchipudi S.V., Kapur V., van Dorp L., Balloux F. Transmission of SARS-CoV-2 from humans to animals and potential host adaptation. Nat. Commun. 2022;13:2988. doi: 10.1038/s41467-022-30698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashor L., Gagne R.B., Bosco-Lauth A.M., Bowen R.A., Stenglein M., VandeWoude S. SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2105253118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsen H.D., Fonager J., Lomholt F.K., Dalby T., Benedetti G., Kristensen B., Urth T.R., Rasmussen M., Lassaunière R., Rasmussen T.B., et al. Preliminary report of an outbreak of SARS-CoV-2 in mink and mink farmers associated with community spread, Denmark, June to November 2020. Euro Surveill. 2021;26 doi: 10.2807/1560-7917.es.2021.26.5.210009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei C., Shan K.-J., Wang W., Zhang S., Huan Q., Qian W. Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J. Genet. Genom. 2021;48:1111–1121. doi: 10.1016/j.jgg.2021.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker D.J., Banerjee A. Coupling field and laboratory studies of immunity and infection in zoonotic hosts. Lancet. Microbe. 2023;4:E285–E287. doi: 10.1016/S2666-5247(23)00032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandl J.N., Ahmed R., Barreiro L.B., Daszak P., Epstein J.H., Virgin H.W., Feinberg M.B. Reservoir host immune responses to emerging zoonotic viruses. Cell. 2015;160:20–35. doi: 10.1016/S2666-5247(23)00032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ministère de l’Environnement, de la Lutte contre les changements and climatiques, de la Faune et des Parcs . 2021. Chronic Wasting Disease in Cervids Surveillance and Control Operations. [Google Scholar]
- 21.World Organization for Animal Health SARS-CoV-2 in Animals – Situation Report 8. 2021. https://www.oie.int/app/uploads/2022/01/sars-cov-2-situation-report-8.pdf
- 22.Naderi S., Chen P.E., Murall C.L., Poujol R., Kraemer S., Pickering B.S., Sagan S.M., Shapiro B.J. Zooanthroponotic transmission of SARS-CoV-2 and host-specific viral mutations revealed by genome-wide phylogenetic analysis. Elife. 2023;12 doi: 10.7554/elife.83685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y., Liu M.-Q., Luo Y., Jiang R.-D., Si H.-R., Zhu Y., Li B., Shen X.-R., Lin H.-F., Zhao K., et al. Genetic Mutation of SARS-CoV-2 during Consecutive Passages in Permissive Cells. Virol. Sin. 2021;36:1073–1076. doi: 10.1007/2Fs12250-021-00384-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aiewsakun P., Phumiphanjarphak W., Ludowyke N., Purwono P.B., Manopwisedjaroen S., Srisaowakarn C., Ekronarongchai S., Suksatu A., Yuvaniyama J., Thitithanyanont A. Systematic exploration of SARS-CoV-2 adaptation to Vero E6, Vero E6/TMPRSS2, and Calu-3 Cells. Genome Biol. Evol. 2023;15 doi: 10.1093/gbe/evad035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luc J., Lobb B., Hirota J., McGeer A., Mubareka S., Doxey A. Figshare; 2023. Comparative Transcriptomic Analysis of Nasopharyngeal Swabs from Individuals with and without COVID-19. [DOI] [Google Scholar]
- 26.Butler D., Mozsary C., Meydan C., Foox J., Rosiene J., Shaiber A., Danko D., Afshinnekoo E., MacKay M., Sedlazeck F.J., et al. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 2021;12:1660. doi: 10.1038/s41467-021-21361-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Despres H.W., Mills M.G., Schmidt M.M., Gov J., Perez Y., Jindrich M., Crawford A.M.L., Kohl W.T., Rosenblatt E., Kubinski H.C., et al. Surveillance of Vermont wildlife in 2021-2022 reveals no detected SARS-CoV-2 viral RNA. Sci. Rep. 2023;13 doi: 10.1038/s41598-023-39232-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., COVID-19 Genomics UK COG-UK Consortium, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long S.W., Olsen R.J., Christensen P.A., Bernard D.W., Davis J.J., Shukla M., Nguyen M., Saavedra M.O., Yerramilli P., Pruitt L., et al. Molecular architecture of early dissemination and massive second wave of the SARS-CoV-2 virus in a major metropolitan area. mBio. 2020;11:e02707-20. doi: 10.1128/mbio.02707-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saputri D.S., Li S., van Eerden F.J., Rozewicki J., Xu Z., Ismanto H.S., Davila A., Teraguchi S., Katoh K., Standley D.M. Flexible, functional, and familiar: characteristics of SARS-CoV-2 spike protein evolution. Front. Microbiol. 2020;11 doi: 10.3389/2Ffmicb.2020.02112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y., Chen Y., Li Y., Huang F., Luo B., Yuan Y., Xia B., Ma X., Yang T., Yu F., et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2024202118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J.-Y., Liao C.-H., Wang Q., Tan Y.-J., Luo R., Qiu Y., Ge X.-Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286 doi: 10.1016/j.virusres.2020.198074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moriyama M., Lucas C., Monteiro V.S., Iwasaki A. SARS-CoV-2 variants do not evolve to promote further escape from MHC-I recognition. bioRxiv. 2022 doi: 10.1101/2022.05.04.490614. Preprint at. [DOI] [Google Scholar]
- 34.Wu X., Xia T., Shin W.-J., Yu K.-M., Jung W., Herrmann A., Foo S.-S., Chen W., Zhang P., Lee J.-S., et al. Viral mimicry of interleukin-17A by SARS-CoV-2 ORF8. mBio. 2022;13:e0040222. doi: 10.1128/mbio.00402-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fong S.-W., Yeo N.K.-W., Chan Y.-H., Goh Y.S., Amrun S.N., Ang N., Rajapakse M.P., Lum J., Foo S., Lee C.Y.-P., et al. Robust Virus-specific adaptive immunity in COVID-19 patients with SARS-CoV-2 Δ382 variant infection. J. Clin. Immunol. 2022;42:214–229. doi: 10.1007/s10875-021-01142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su Y.C.F., Anderson D.E., Young B.E., Linster M., Zhu F., Jayakumar J., Zhuang Y., Kalimuddin S., Low J.G.H., Tan C.W., et al. Discovery and genomic characterization of a 382-nucleotide deletion in ORF7b and ORF8 during the early evolution of SARS-CoV-2. mBio. 2020;11:e01610-20. doi: 10.1128/mbio.01610-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young B.E., Fong S.-W., Chan Y.-H., Mak T.-M., Ang L.W., Anderson D.E., Lee C.Y.-P., Amrun S.N., Lee B., Goh Y.S., et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. Lancet. 2020;396:603–611. doi: 10.1016/S0140-6736(20)31757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Institut national de and santé publique du Québec (INSPQ) INSPQ; 2021. Données COVID-19 Par Région Sociosanitaire.https://www.inspq.qc.ca/covid-19/donnees/par-region [Google Scholar]
- 39.Public Health Agency of Canada Animals and COVID-19. 2021. https://www.canada.ca/en/public-health/services/diseases/2019-novel-coronavirus-infection/prevention-risks/animals-covid-19.html
- 40.Davila K.M.S., Nelli R.K., Phadke K.S., Ruden R.M., Yongming S., Bellaire B.H., Gimenez-Lirola L.G., Miller L.C. How do deer respiratory epithelial cells weather the initial storm of SARS-CoV-2? bioRxiv. 2023 doi: 10.1101/2023.04.24.538130. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G., Smith D.K., Zhu H., Guan Y., Lam T.T.-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017;8:28–36. doi: 10.1111/2041-210X.12628. [DOI] [Google Scholar]
- 42.Broadbent L., Bamford C.G.G., Lopez Campos G., Manzoor S., Courtney D., Ali A., Touzelet O., McCaughey C., Mills K., Power U.F. An endogenously activated antiviral state restricts SARS-CoV-2 infection in differentiated primary airway epithelial cells. PLoS One. 2022;17 doi: 10.1371/journal.pone.0266412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lieberman N.A.P., Peddu V., Xie H., Shrestha L., Huang M.-L., Mears M.C., Cajimat M.N., Bente D.A., Shi P.-Y., Bovier F., et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han Y., Bai X., Liu S., Zhu J., Zhang F., Xie L., Liu G., Jiang X., Zhang M., Huang Y., et al. XAF1 Protects host against emerging RNA viruses by stabilizing IRF1-dependent antiviral immunity. J. Virol. 2022;96 doi: 10.1128/jvi.00774-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi G., Kenney A.D., Kudryashova E., Zani A., Zhang L., Lai K.K., Hall-Stoodley L., Robinson R.T., Kudryashov D.S., Compton A.A., Yount J.S. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021;40 doi: 10.15252/embj.2020106501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchrieser J., Dufloo J., Hubert M., Monel B., Planas D., Rajah M.M., Planchais C., Porrot F., Guivel-Benhassine F., Van der Werf S., et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020;39 doi: 10.15252/embj.2020106267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prelli Bozzo C., Nchioua R., Volcic M., Koepke L., Krüger J., Schütz D., Heller S., Stürzel C.M., Kmiec D., Conzelmann C., et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021;12:4584. doi: 10.1038/s41467-021-24817-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Compton A.A., Roy N., Porrot F., Billet A., Casartelli N., Yount J.S., Liang C., Schwartz O. Natural mutations in IFITM3 modulate post-translational regulation and toggle antiviral specificity. EMBO Rep. 2016;17:1657–1671. doi: 10.15252/embr.201642771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LeBlanc J.J., Gubbay J.B., Li Y., Needle R., Arneson S.R., Marcino D., Charest H., Desnoyers G., Dust K., Fattouh R., et al. Real-time PCR-based SARS-CoV-2 detection in Canadian laboratories. J. Clin. Virol. 2020;128 doi: 10.1016/j.jcv.2020.104433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu X., Wang L., Sakthivel S.K., Whitaker B., Murray J., Kamili S., Lynch B., Malapati L., Burke S.A., Harcourt J., et al. US CDC Real-time reverse transcription PCR panel for detection of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020;26:1654–1665. doi: 10.3201/eid2608.201246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corman V.M., Landt O., Kaiser M., Molenkamp R., Meijer A., Chu D.K., Bleicker T., Brünink S., Schneider J., Schmidt M.L., et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gagnier M., Laurion I., DeNicola A.J. Control and surveillance operations to prevent chronic wasting disease establishment in free-ranging white-tailed deer in Québec, Canada. Animals. 2020;10:283. doi: 10.3390/2Fani10020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.World Organization for Animal Health Considerations for Sampling, Testing, and Reporting of SARS-CoV-2 in Animals. 2020. https://www.oie.int/app/uploads/2021/03/a-sampling-testing-and-reporting-of-sars-cov-2-in-animals-3-july-2020.pdf
- 54.Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 55.Ewels P.A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M.U., Di Tommaso P., Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 56.Patel H., Varona S., Monzón S., Espinosa-Carrasco J., Heuer M.L., Nf-Core Bot. Underwood A., Gabernet G., Ewels P., MiguelJulia, et al. 2022. Nf-Core/Viralrecon: Nf-Core/viralrecon v2.5 - Manganese Monkey. [DOI] [Google Scholar]
- 57.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinforma. Oxf. Engl. 2018;34:i884–i890. doi: 10.1002/imt2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pedersen B.S., Quinlan A.R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018;34:867–868. doi: 10.1093/bioinformatics/btx699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grubaugh N.D., Gangavarapu K., Quick J., Matteson N.L., De Jesus J.G., Main B.J., Tan A.L., Paul L.M., Brackney D.E., Grewal S., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff. Fly (Austin) 2012;6:80–92. doi: 10.4161/2Ffly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cingolani P., Patel V.M., Coon M., Nguyen T., Land S.J., Ruden D.M., Lu X. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program. Front. Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Elbe S., Buckland-Merrett G. G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khare S., Gurry C., Freitas L., Schultz M.B., Bach G., Diallo A., Akite N., Ho J., Lee R.T., Yeo W., et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021;3:1049–1051. doi: 10.46234/ccdcw2021.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shu Y., McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Euro Surveill. 2017;22 doi: 10.2807/2F1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Turakhia Y., Thornlow B., Hinrichs A.S., De Maio N., Gozashti L., Lanfear R., Haussler D., Corbett-Detig R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021;53:809–816. doi: 10.1038/s41588-021-00862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kruczkiewicz P. 2023. CFIA-NCFAD/Delta-SARS-CoV-2-WTD-QC: 1.0.0 - Zenodo Release. [DOI] [Google Scholar]
- 70.Aksamentov I., Roemer C., Hodcroft E., Neher R. Nextclade: clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021;6:3773. doi: 10.21105/joss.03773. [DOI] [Google Scholar]
- 71.Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M.J.L. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinforma. Oxf. Engl. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minh B.Q., Nguyen M.A.T., von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nasir J.A., Kozak R.A., Aftanas P., Raphenya A.R., Smith K.M., Maguire F., Maan H., Alruwaili M., Banerjee A., Mbareche H., et al. A comparison of whole genome sequencing of SARS-CoV-2 using amplicon-based sequencing, random hexamers, and bait capture. Viruses. 2020;12:895. doi: 10.3390/v12080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O’Hara E., Herbst A., Kommadath A., Aiken J.M., McKenzie D., Goodarzi N., Skinner P., Stothard P. Neural transcriptomic signature of chronic wasting disease in white-tailed deer. BMC Genom. 2022;23:69. doi: 10.1186/s12864-022-08306-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bushnell B. BBMap: A Fast, Accurate, Splice-Aware Aligner. 2014. https://www.osti.gov/biblio/1241166
- 80.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:257. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu J., Breitwieser F.P., Thielen P., Salzberg S.L. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017;3:e104. doi: 10.7717/peerj-cs.104. [DOI] [Google Scholar]
- 82.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., Imamichi T., Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Emms D.M., Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kosakovsky Pond S.L., Frost S.D.W. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 90.Weaver S., Shank S.D., Spielman S.J., Li M., Muse S.V., Kosakovsky Pond S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018;35:773–777. doi: 10.1093/molbev/msx335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agresti A., Coull B.A. Approximate is Better than “Exact” for Interval Estimation of Binomial Proportions. Am. Stat. 1998;52:119–126. doi: 10.2307/2685469. [DOI] [Google Scholar]
- 92.Statistics Canada Population and Dwelling Count Highlight Tables, 2016 Census. 2017. https://www12.statcan.gc.ca/census-recensement/2016/dp-pd/hlt-fst/pd-pl/Table.cfm?Lang=Eng&T=101&S=50&O=A#2016A000224
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequence data from the three SARS-CoV-2 viruses from white-tailed deer sequenced in this study are deposited in GISAID (https://www.gisaid.org/) under Accession numbers EPI_ISL_10169675 (hCoV-19/Canada/QC-WTD-qxic4205/2021), EPI_ISL_10170149 (hCoV-19/Canada/QC-WTD-qxic4249/2021), and EPI_ISL_10168587 (hCoV-19/Canada/QC-WTD-qxic4055/2021). All white-tailed deer metatranscriptomic sequencing data for this project was deposited into the NCBI Short Read Archive (SRA) under BioProject accession no. PRJNA1025888.
-
•
All codes used in this study is available at the sources referenced in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.