

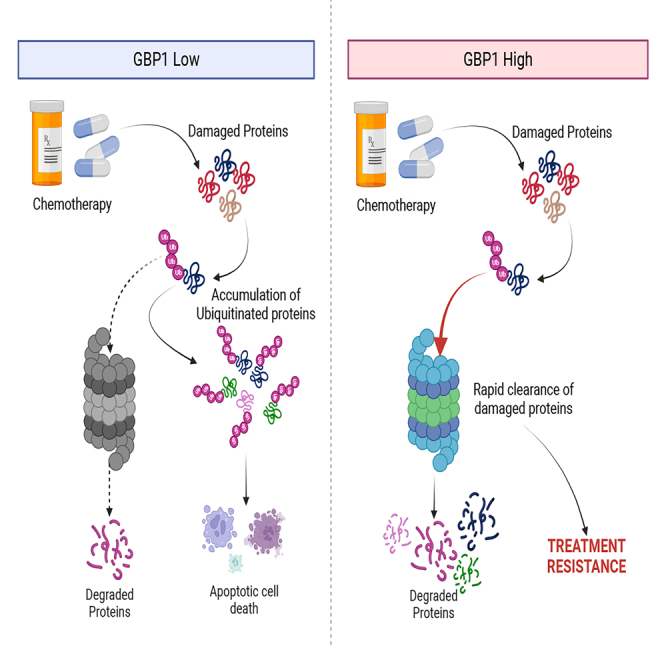
Summary
Guanylate-binding protein 1 (GBP1) is known as an interferon-γ-induced GTPase. Here, we used genetically modified ovarian cancer (OC) cells to study the role of GBP1. The data generated show that GBP1 inhibition constrains the clonogenic potential of cancer cells. In vivo studies revealed that GBP1 overexpression in tumors promotes tumor progression and reduces median survival, whereas GBP1 inhibition delayed tumor progression with longer median survival. We employed proteomics-based thermal stability assay (CETSA) on GBP1 knockdown and overexpressed OC cells to study its molecular functions. CETSA results show that GBP1 interacts with many members of the proteasome. Furthermore, GBP1 inhibition sensitizes OC cells to paclitaxel treatment via accumulated ubiquitinylated proteins where GBP1 inhibition decreases the overall proteasomal activity. In contrast, GBP1-overexpressing cells acquired paclitaxel resistance via boosted cellular proteasomal activity. Overall, these studies expand the role of GBP1 in the activation of proteasomal machinery to acquire chemoresistance.
Subject areas: Pharmacology, Biochemistry, Molecular biology, Cancer, Proteomics
Graphical abstract
Highlights
-
•
GBP1 inhibition delays the ovarian cancer progression
-
•
Overexpression of GBP1 contributes to the development of paclitaxel resistance
-
•
GBP1 modulates the proteasomal activity in ovarian cancer cells
Pharmacology; Biochemistry; Molecular biology; Cancer; Proteomics
Introduction
Ovarian cancer (OC) is the most lethal gynecologic disease in the U.S.1 and more than 80% of patients show relapse after surgery treatment and chemotherapy, as the cells develop resistance to platinum and/or taxane-based drugs.2,3 Drug resistance can arise even before treatment4 and is closely related to the tumor microenvironment.5 Previous studies have identified a large GTPase, guanylate-binding protein 1 (GBP1), that plays a key role in cancer chemoresistance.6,7 GBP1 is an interferon γ (IFNγ)-induced GTPase with critical roles in host defense against microbial and viral infection.8,9 GBP1 is a large two-domain protein where the N-terminal domain consists of GTPase activity and the C-terminal domain is α-helical domain.10 It is part of various biological and cellular functions, including signaling and transport. Studies have shown that GBP1 contributes to the progression of tumors in various cancers such as ovarian, prostate, renal, and lung cancer8,11,.12 Cytokine- and hypoxia-induced overexpression of GBP1 in cancer leads to increased production of class III β-tubulin and recruitment of kinases such as PIM1 into the cytoskeleton, which supports tumor survival and growth.13 GBP1 overexpression has also been associated with resistance to paclitaxel and radiotherapy in ovarian cancer.13,14,15 Chemoresistance is a complex phenomenon and the role of GBP1 in it is not well understood. Here, we systematically studied the molecular biology of GBP1 overexpression and knockdown condition in ovarian cancer cell models. We used a proteomics-based thermal stability assay (CETSA) to understand the mechanism of GBP1 in paclitaxel sensitivity. Our study found that GBP1 in chemoresistance helps to regulate proteasomal degradation of chemotherapy-damped cellular components and that GBP1 inhibition is a promising strategy to improve paclitaxel sensitivity in ovarian cancer.
Results
Inhibition of GBP1 delayed the OC cell proliferation and tumor progression
We used a lentiviral base stable transduction system to achieve GBP1 overexpression (OV) and knockdown (KD) in ovarian cancer (OC) cells. Stably transduced cells were validated for their GBP1 (GBP2b for ID8) expression (Figures 1A–1H) using western blotting. We were able to achieve 2- to 8-fold overexpression (Figures 1A–1D) and 60%–87% knockdown (Figures 1E–1H) in OC cell lines. To confirm the effect of GBP1 modulation on OC cell proliferation and clonogenic potential, we performed the clonogenic assay and found that GBP1 overexpression does not influence the clonogenic potential of OC cells (Figures 1A–1D). In contrast, GBP1 knockdown significantly inhibits the clonogenic potential (Figures 1E–1H). To examine the effect of GBP1 modulation on tumor progression in vivo, we used GBP2b-modulated (Figures 1A and 1E) syngeneic origin ID8 cells in a female C57BL/6 intraperitoneal tumor growth and survival study. We observed that the GBP1 OV cohort rapidly developed ascites with lower median survival (50.5 days) compared to the vehicle control (VC) cohort (61 days) (p = 0.0969) (Figure 2A). At day 40 after intraperitoneal implantation, 6/10 VC mice and 9/10 OV mice had developed ascites (Figure 2B). We did not observe a significant difference in the volume of ascitic fluid or body weight between the VC and OV cohorts (Figure 2B and 2C). On other hand, GBP1 KD significantly (p = 0.0077) improved the median survival (71 days) cohort to the scrambled control (SC) cohort (58.5 days) (Figure 2D). None of the KD cohort mice developed ascites at day 40, whereas 7/10 SC mice developed ascites (Figure 2E). At day 40, no ascitic fluid was observed in the KD cohort (Figure 2E), and ascites-associated weight gain was also not observed (Figure 2F). Overall, these data show that GBP1 inhibition significantly restrains ovarian cancer progression, in vitro and in vivo.
Figure 1.

Inhibition of GBP1 inhibits the clonogenic potency of ovarian cancer cells
(A–H) Human and mouse-origin ovarian cancer (OC) cells were stably transduced with GBP1 (GBP2b in the mouse cell line) overexpression (OV) or vector control (VC) or scrambled control (SC) or GBP1 shRNA (GBP2b in mouse cell line) (KD) using a lentiviral vector and the level of GBP1 expression was confirmed via Western blotting as described in the STAR Methods section. 100–500 OC cells/wells were plated in 12-well tissue culture plates and allowed to grow for 7–10 days. Colonies were stained using crystal violet and confluency was measured. Data are shown as mean ± SD. Significantly different compared with respective controls by Student’s t test.
(A–D) OC cells were either transduced with VC or GBP1-OV lentiviral vector.
(E–H) OC cells were either transduced with SC or GBP1-KD lentiviral vector.
Figure 2.

GBP1 expression modulates tumor progression and ascities formation in the ID8 intraperitoneal ovarian cancer mouse model
1 x 106 ID8 cells stably transduced with GBP2b overexpression (OV) or vector control (VC) or scrambled control (SC) or GBP2b shRNA (KD) were implanted intraperitoneally in 9- to 10-weeks-old female C57BL/6 mice.
(A) Left: representative image of VC and GBP2b-OV mice after 40 days of IP implantation. Right: Kaplan-Meier plot for the survival of VC and OV ID8-implanted mice (n = 10/group).
(B) Left: representative images of SC and GBP2b-KD mice after 40 days of IP implantation. Right: Kaplan-Meier plot for the survival of SC and KD ID8-implanted mice (n = 10/group). Significantly different compared with respective controls (VC or SC) by Log rank (Mantel-Cox) test.
(C and D) Ascites fluid was collected from each mouse after 40 days of IP implantation using an 18G needle. Total fluid volume was measured using 10 mL serological pipette (n = 10/group).
(E and F) The body weight of each mouse was measured before ascites fluid collection. Data are shown as mean ± SD. Significantly different compared with respective controls by Student’s t test.
Cellular thermal shift assay reveals proteasomal interactions of GBP1
The cellular thermal shift assay (CETSA) is based on shifts in protein thermal stability during their different interactions.16 We used CETSA on GBP2b-overexpressed and knockdown ID8 cells with the vehicle and scrambled control, respectively, to identify interactions of GBP1 with other proteins. To get a global overview of protein interactions, we used CETSA followed by mass spectrometry-based proteomic analysis. We normalized the thermal denaturation data for GBP1-OV and KD samples with their respective controls (OV/VC &KD/SC). We examined these data in two ways, (1) gene cluster analysis of proteins either positively or negatively thermal shifted in each group (OV & KD) and (2) identification of proteins which are common in each group with opposite thermal shift. Here, we found that proteins associated with proteasomal degradation were strongly influenced by the modulation of GBP1 levels (Figures 3A and 3B). Thermal stability of proteasomal-associated proteins including proteasome 20S subunit beta (PSMB6), PSMB1, proteasome 20S subunit alpha (PSMA3), and cytochrome c (CYCS) was increased in the GBP1 overexpression condition and decreased in the GBP1 knockdown condition (Figure 3B; Table S1). To identify the interactions of GBP1, we performed a String analysis of combined hits identified in both groups (Figure 3C). We found substantial enrichment of protein clusters associated with the proteasomal protein catabolic process, microtubule-based process, translation, mRNA splicing via spliceosome, and NADP metabolic process (Figure 3C). Gene ontology enrichment analysis for different biological processes suggests that GBP1 knockdown reduces the thermal stability of proteins associated with proteasomal and microtubule-cytoskeleton machinery (Figure 3D; Tables S2 and S4). In contrast, GBP1 overexpression increases the thermal stability of proteasomal machinery (Figure 3E; Tables S3 and S4). To further validate this observation, we performed immunoprecipitation using an anti-GBP1 antibody in human-origin OC cell line-OVCAR8 finding that GBP1 interacts with proteasomal proteins PSMB6, PSMB1, and PSMA3 (Figure 3F).
Figure 3.

GBP1 crosstalk identification
Differential thermal proteome profile of ID8 cells stably transduced with GBP2b overexpression (OV) or vector control (VC) or scrambled control (SC) or GBP2b shRNA (KD). Cells were collected and 106 cells in each PCR tube were incubated at different temperatures (37°C, 41°C, 44°C, 47°C, 50°C, 53°C, 56°C, 59°C, 63°C, or 67°C). Cells were lysed and an equal quantity of soluble protein was labeled with tandem mass tag (TMT), followed by liquid chromatography-tandem mass spectrometry analysis. Thermal stabilities were calculated, and thermal stability shifts in GBP2b presence or absence were calculated.
(A) Dot plot for thermal stability shift of proteins in GBP1 OV and KD groups (normalized with vector/scrambled control).
(B) The melting curves for PSMB1, PSMB6, PSMA3, and CYCS indicate a shift in melting temperature in ID8 cells with GBP2b overexpression or knockdown condition compared to vector/scrambled control.
(C) Significant proteins in common across conditions were graphed in String and showed enrichment for the proteasomal protein catabolic process (gray-filled circles), microtubule-based process (blue-filled circles), translation, NADP metabolic process, and mRNA splicing via spliceosome. The thickness of network edges indicates the strength of data supporting the interaction.
(D and E) Gene ontology enrichment analysis for different biological processes.
(D) Gene ontology of proteins with a negative thermal shift by GBP2b-KD.
(E) Gene ontology of proteins with a positive thermal shift by GBP2b-OV.
(F) Immunoprecipitation with anti-GBP1 or anti-IgG antibodies from whole-cell lysate of OVCAR8 cells, followed by western blot for GBP1, PSMB1, PSMB6, and PSMA3.
GBP1 influences proteasome and tubulin-associated pathways in OC cells
To better understand the broader interactions of GBP1, we performed a global proteomic analysis of GBP2b-modulated ID8 cells. We cultured each cell line at 50%–60% confluency and proteomic analysis was performed on an equal amount of total cell lysates. Gene enrichment analysis suggests that GBP1 overexpression led to the upregulation of eukaryotic translation elongation, selenocysteine synthesis, cellular responses to stress, neutrophil degranulation, regulation of expression of SLITs and ROBOs, RHO GTPases-activated IQGAPs, G2/M transition, HSP90 chaperone cycle for steroid hormone receptors, and loss of proteins required for interphase microtubule organization from the centrosome (Figures 4A and 4B). Moreover, pathways associated with cell cycle checkpoints regulation and apoptosis were downregulated (Figures 4A–4C). These include TP53 regulating metabolic genes, eukaryotic translation initiation, and intrinsic pathway for apoptosis (Figures 4A–4C). GBP1 knockdown led to the enrichment of protein associated with eukaryotic translation initiation and apoptosis (Figures 4D and 4E). In contrast, pathways related to stress response were downregulated including neutrophil degranulation, innate immune system, and cellular response to stress (Figures 4D–4F). GBP1 overexpression and knockdown showed an opposite trend to each other in terms of pathway enrichment. Overall, this global proteome profiling suggests the role of GBP1 in stress response and cell cycle progression.
Figure 4.

Proteome analyses upon GBP1 modulation
Total cell lysates were prepared, and global proteome profiling was performed as described in the STAR Methods section. Gene set enrichment analysis was performed on the proteomics results to determine the enrichment of the Kyoto Encyclopedia of Genes and Genomes pathways upon GBP2b modulation in the ID8 cell line.
(A–C) Enrichment analysis of GBP1-OV ID8 cells compared with VC ID8 cells.
(D–F) Enrichment analysis of GBP1-KD ID8 cells compared with SC ID8 cells.
GBP1 modulates the cellular proteasomal capability and triggers paclitaxel resistance
CETSA and global proteomics analysis suggest that GBP1 may be a key regulator for proteasomal activity and associated chemoresistance. To extend this observation, we probed blots with anti-ubiquitin antibody to detect total ubiquitinated proteins in GBP1-modulated cells. We found that cells with GBP1 overexpression accumulated a lower level of ubiquitinated proteins compared to vector control cells (Figures 5A and 5B), whereas GBP1 knockdown cells accumulated ubiquitinated proteins at the same level as scrambled control (Figures 5A and 5B). To confirm whether these ubiquitination levels are due to modulation of proteasomal activity, we probed proteasomal activity using a proteasome substrate (Succ-LLVY-AMC) utilization-based assay. Results suggest that GBP1-overexpressing cells have significantly higher proteasomal activity compared to VC cells (Figure 5C, left). In contrast, GBP1 KD significantly reduced the proteasomal activity of cells (Figure 5C, right). Based on previous findings of increased proteasomal activity in chemoresistance where increased turnover of chemotherapy-damaged cellular components enables cancer cell survival,17,18,19,20 we hypothesized that GBP1 overexpression-associated elevation of proteasomal activity may enable chemoresistance and its inhibition may sensitize cells to chemotherapy. To test this, we treated GBP1-modulated cells with paclitaxel and its effect on cell proliferation and clonogenic potential. This found that GBP1-overexpressing ID8 and OVCAR8 cells are less responsive to paclitaxel than VC cells (Figures 6A and 6B). GBP1 inhibition in OVCAR8 cells sensitized them to paclitaxel treatment (Figures 6C and 6D). Next, we investigated the effects of GBP1 modulation on paclitaxel-induced proteasomal activity. Western blot analysis of total ubiquitinated proteins found that paclitaxel treatment induces proteasomal activity and that this increase was enhanced by GBP1 overexpression (Figure 6E). In contrast, OVCAR8 cells with GBP1 knockdown accumulated more ubiquitinated proteins than non-treated cells, suggesting a decrease in proteasomal activity (Figure 6E). To analyze the spectrum of relationship between GBP1 and proteasomal activity, we analyzed the GDSC-MGH-Sanger database for pharmacogenomic data of cancer cell lines using CellMinerCDB. Analysis suggests that GBP1 expression positively correlates with the cytotoxic response of proteasomal inhibitors. Cumulative analysis of cell lines from various histologies gave a correlation between GBP1 expression and bortezomib (r = 0.44, p = 2.3e-09) and MG132 (r = 0.29, p = 0.00012) (Figure S1A). We performed clonogenic assay for the cytotoxic effect of proteasome inhibitor (MG132) on transduced OVCAR8 cells and found that GBP1 inhibition sensitizes the OV cells (Figures S1B and S1C). Furthermore, we asked whether pharmacological inhibition of GBP1 using a small-molecule inhibitor could sensitize GBP1-overexpressing cells for paclitaxel treatment. To test this, we used a small-molecule inhibitor of GBP1, SU093 (also called NSC756093) previously developed by our group (Andreoli et al., 2014). SU093 treatment to OC cells inhibits the cellular proteasomal activity compared to vehicle-treated cells (p=<0.0001) (Figure S2A). Immunoblotting analysis suggests that treatment of VC and GBP1-OV OVCAR8 cells with SU093 leads to accumulation of ubiquitinated proteins (Figure S2B). This is similar to our observation in GBP1 KD cells (Figure 5). Moreover, we analyzed the combination of SU093 and paclitaxel using clonogenic assay, which showed that SU093 sensitizes the GBP1-overexpressing OVCAR8 cells for paclitaxel treatment (Figures S2C and S2D). Cumulatively, these results suggest that GBP1 plays a key role in the regulation of cellular proteasomal activity where upregulation of GBP1 in cancer cells may lead to treatment resistance via enhanced proteasomal activity.
Figure 5.

GBP1 modulates proteasomal activity
(A) Immunoblotting of total ubiquitinated proteins in OC cells stably transduced with GBP1 (GBP2b in ID8) overexpression (OV) or vector control (VC) or scrambled control (SC) or GBP1 shRNA (GBP2b in ID8) (KD).
(B) Densitometry analysis of three independent immunoblotting of total ubiquitinated proteins in OC cells stably transduced cells. Data are shown as mean ± SD of triplicate samples. Significant differences compared to respective controls were assessed by Student’s t test.
(C) Proteasomal activity assessment of transduced OC cells. An equal amount of protein from each cell line was analyzed for its proteasomal activity using a proteasome substrate (Succ-LLVY-AMC) utilization-based assay.
Figure 6.

GBP1 influences chemotherapy response
(A–D) 100–300 transduced OC cells/well were plated in 12 well tissue culture plates followed by treatment with paclitaxel (ID8-15 nM and OVCAR8-7.5 nM) after 24 h and allowed to grow for 7–10 days. Colonies were stained using crystal violet and confluency was measured. Data are shown as mean ± SD of triplicate samples. Significant differences compared to respective controls were assessed by Student’s t test.
(E) Immunoblotting of total ubiquitinated proteins in transduced OC cells treated with vehicle (DMSO) (C) or paclitaxel (ID8-15 nM and OVCAR8-7.5 nM) for 24 h.
(F–I) Kaplan-Meier plots (KMplot) for the probability of survival of ovarian cancer patients with GBP1-high (high) and GBP1-low (low) expression (mRNA gene chip data, Affy ID: 202269_x_at). KMplots were plotted using KM-plotter.
(F) KMplot (n = 1435) of ovarian cancer patients with high and low GBP1 expression. The plot includes patients of all the stages (1–4) of ovarian cancer.
(G) KMplot (n = 163) of ovarian cancer patients with disease at stages 1 and 2 with high and low GBP1 expression.
(H) KMplot (n = 73) of ovarian cancer patients with disease at stages 1 and 2 and treated with Taxol with high and low GBP1 expression.
(I) KMplot (n = 94) of ovarian cancer patients with disease at stages 1 and 2 and treated with platinum agents with high and low GBP1 expression.
To further validate this hypothesis, we investigated patient mRNA gene chip data. Overall survival probability comparison using Kaplan-Meier plots (KMplot) between GBP1 low- and GBP1-high ovarian cancer (OV) patient groups (n = 1435) found that the GBP1-high group has significantly lower median survival (18.79 months, p = 1.5e-05) compared to GBP1-low group (26.87 months) (Figure 6F). Additionally, we divided the whole group based on different stages of cancer (stages 1–4). KMplot for the probability of survival of stage 1&2 between GBP1-low and high groups (n = 163) suggested that the GBP1-high group had significantly lower median survival (18.3 months, p = 0.00062) compared to the GBP1-low group (75.63 months) (Figure 6G). We further divided this group (stage 1 + 2) based on the chemotherapy they receive. The patient group who received Taxol treatment (n = 73) was analyzed for their overall survival and again the GBP1-high group had significantly lower median survival (31.43 months, p = 0.002) compared to GBP1-low group (96.79 months) (Figure 6H). The advanced stages group (stages 3 and 4) do not show any significant difference in median survival (Figure S3). A similar trend was observed in the patient group treated with platinum agents (n = 94) where the GBP1-high group had significantly low median survival (31.43 months, p = 0.0019) compared to the GBP1-low group (81.94 months) (Figure 6I). Overall, KMplot analysis for the probability of survival of OV patients supports our finding that GBP1 plays the role in treatment resistance development.
Discussion
Here, we made efforts to understand the GBP1-associated biology and its role in chemoresistance development. GBP1 is majorly known as IFNγ-induced GTPase and plays the role in immune surveillance against pathogenic infection.21 It belongs to the larger GTPase family which includes seven GBPs (GBP1 to 7). Patient archival studies had shown that GBP1 is overexpressed in many cancer types and got enriched in patient populations with treatment resistance.13 The role of GBP1 in cancer and treatment resistance is not entirely clear. Here, we found that GBP1 overexpression does not induce cell proliferation or influence the clonogenic potential of cancer cells, in vitro. Whereas its KD inhibits the clonogenic potential of cancer cells. In vivo experiments using GBP2b-modulated ID8 cells suggest that GBP1 overexpression modernly induces tumor progression, whereas its KD significantly delays tumor progression. Overall, in vitro and in vivo data suggest that GBP1 has a potential role in the regulation of cancer progression.
Mass spectrometry-based proteomics analysis is a powerful tool to dive deeper into the cellular proteome. Mass spectrometry-coupled thermal stability analysis of proteins enabled us to identify the protein-small molecule and protein-protein interactions.16 Here, we have used a unique approach to identify the interactions of GBP1 with other proteins. We have used cells with GBP1 overexpression and knockdown and performed CETSA. Our major goal was to identify protein/s that gain or lose thermal stability in the presence and absence of GBP1. When protein interacts with other molecules, its thermal stability gets changed compared to protein alone. Furthermore, we looked for protein/s commonly present in both the groups (OV/VC and KD/SC) but with opposite thermal behavior. This was to justify the essentiality of GBP1 in the event driven by protein/s interacting with it. We have used mouse ovarian cancer cell line ID8 which is moderately expressing GBP2b (similar to human GBP1) and manipulated its expression. The rationale behind using a mouse cell line was to get consistency analysis in vitro and in vivo. With this strategy, we were able to identify major protein clusters which get influenced by GBP1. This includes proteins associated with proteasomal machinery and microtubules system. Chemotherapies had been developed against both targets and both are associated with therapy resistance. Cancer cells exploit the proteasomal machinery for quick response to stress including therapy-induced stress. Cancer cells tend to have higher proteasomal activity compared to normal cells. We found that GBP1 interacts with proteasome 20S subunits including PSMB1 and PSMB6 and PSMA3. Proteasome 20S subunits are member of proteasomes and are associated with the ATP-dependent degradation of ubiquitinated proteins.22
Cells encounter various stresses including energy stress, heat stress, oxidative stress, hypoxic stress, osmotic stress, and pathogenic stress.23 To overcome these stresses, cells use protein degradation machinery for rapid clearance of damaged protein or to generate raw materials for progression.24 Proteasomal degradation is the key event to maintain cellular homeostasis and is required for cell cycle progression and stress response.25,26 Results from our proteomics experiments suggest that GBP1-overexpressing cells upregulate the proteins and pathways associated with stress response whereas GBP1 knockdown downregulates these pathways. GBP1 overexpression also leads to the downregulation of pathways associated with cell cycle regulations and checkpoints including p53 and CHK1/2. These pathways halt cell cycle progression in response to chemo- and radiotherapy and their inhibition lead the treatment resistance.25 Results suggest the role of GBP1 in resistance development as overexpressing cells accumulated the machinery for better stress response and loosen cellular checkpoints and regulations. Inhibition of proteasome leads to the accumulation of ubiquitinated proteins and ultimately leads to cell death. Based on this, many proteasome inhibitors were developed and used in clinics, including bortezomib.25 CYCS release from mitochondria in response to mitochondrial damage caused by any stress is the key event for the initiation of the apoptotic cell death pathway. Rapid clearance of CYCS from cytosol may help the cells to overcome cellular stress and avoid apoptotic cell death. Clearance of cytosolic CYCS is majorly governed by proteasomal degradation in neuronal and cancer cells.27 Knockdown of GBP1 induces the apoptotic cell death pathway and this is in line with the effect of proteasome inhibitors. CETSA and global proteome profiling pointed out the role of GBP1 in the regulation of proteasome and treatment resistance development. Our results also suggest that GBP1-overexpressing cells show higher proteasomal activity and resistance toward paclitaxel treatment. GBP1 knockdown reverts this with less proteasomal activity and sensitizes toward chemo treatment. TCGA data also support this that ovarian patients with low GBP1 expression overall perform better compared to those with high GBP1 expression.
In summary, this study has shown the potential role of GBP1 in the regulation of proteasomal machinery and chemoresistance development. Combination therapy with GBP1 inhibitor including SU09328 developed by our group may synergize with chemotherapy and help in overcoming resistance. Apart from cancer biology, this knowledge can be useful for diseases associated with neurodegeneration29 and aging30 where lower proteasomal activity causes the problem.
Limitations of the study
This study is limited to genetically overexpressed and knockdown GBP1 cell line conditions and does not include IFNγ-induced GBP1. Also, this study is limited to ovarian cancer cells. It is possible that GBP1 may work differently in other cancer histologies. The effect of GBP1 under various physiological conditions and cancer types will require further investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental models: Cell lines | ||
| ID8 | MilliporeSigma | Cat. No. # SCC145; RRID: CVCL_IU14 |
| OVCAR8 | NCI-DTP repository | RRID: CVCL_1629 |
| SKOV3 | MilliporeSigma | Cat. No. # 91091004; RRID: CVCL_0532 |
| OVCAR3 | NCI-DTP repository | RRID: CVCL_0465 |
| HEK293T | ATCC | Cat. No. # CRL-3216; RRID: CVCL_0063 |
| Recombinant DNA | ||
| mGBP2b overexpression: pLV{Exp}-mCherry:T2A:Puro-EF1A>mGbp2b{NM_010259.2} | VectorBuilder | Vector ID: VB180117-1093kbb |
| mGBP2b shRNA: pLV{shRNA}-EGFP:T2A:Puro-U6>mGbp2b{shRNA# TGGGATTGGCATGTTATAAAC} | VectorBuilder | Vector ID: VB180117-1250gdh |
| hGBP1 overexpression: pLV{Exp}-mCherry:T2A:Puro-EF1A>hGBP1{NM_002053.2} | VectorBuilder | Vector ID: VB170503-1150awy |
| hGBP1 shRNA: pLV{shRNA}-EGFP:T2A:Puro-U6>hGBP1{shRNA# CCAGATGAGTACCTGACATAC} | VectorBuilder | Vector ID: VB170507-1030ruw |
| Vector Control: pLV{Exp}-mCherry:T2A:Puro-EF1A>ORF_Stuffer | VectorBuilder | Vector ID: VB230206-1323fan |
| Scramble shRNA Control: pLV{shRNA}-EGFP:T2A:Puro-U6>Scramble_shRNA {CCTAAGGTTAAGTCGCCCTCG} | VectorBuilder | Vector ID: VB170507-1033npu |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Corning | Cat. No. #10-013-CV |
| RPMI-1640 | Corning | Cat. No. #10-040-CV |
| FBS | Corning | Cat. No. #35-015-CV |
| Antibiotic-Antimycotic solution | Corning | Cat. No. #15240062 |
| Puromycin | InvivoGen | Cat. No. #ant-pr-1 |
| Paclitaxel | Cayman Chemical | Cat. No. # 10461100 |
| DMSO | MP Biomedicals | Cat. No. #196055 |
| SU093 (NSC756093) | Malhotra lab | Andreoli et al., 201428 |
| Antibodies | ||
| GBP1 | Abnova | Cat. No. #H00002633-PW1; RRID: AB_10716038 |
| GBP1 | Novusbio | Cat. No. # NBP2-03972 |
| β-actin | CST | Cat. No. # 4970S; RRID: AB_2223172 |
| Ub | CST | Cat. No. # 43124S; RRID: AB_2799235 |
| PSMB1 | Thermo Scientific | Cat. No. # PA5-49648; RRID: AB_2635102 |
| PSMB6 | Thermo Scientific | Cat. No. # PA1-978; RRID: AB_2172197 |
| PSMA3 | Proteintech | Cat. No. # 11887-I-AP; RRID: AB_2171420 |
| Rabbit IgG | EMD Millipore | Cat. No. # PP64B; RRID: AB_145841 |
| Anti-mouse IgG HRP-linked antibody | CST | Cat. No. #7076; RRID: AB_330924 |
| Anti-rabbit IgG HRP-linked antibody | CST | Cat. No. #7074; RRID: AB_2099233 |
| Critical commercial assays | ||
| M-PER™ lysis solution | Thermo Scientific | Cat. No. #78503 |
| Halt protease and phosphatase inhibitor cocktail | Thermo Scientific | Cat. No. #78440 |
| Tandem Mass Tag (TMT) | Thermo Fisher Scientific | Cat. No. # 90110, Cat. No. #90061 |
| Proteasome Activity Assay kit | Abcam | Cat No. # ab107921 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | Strain #:000664, RRID: IMSR_JAX:000664 |
| Deposited data | ||
| Mass spectrometry proteomics data | PRIDE database |
http://www.ebi.ac.uk/pride Dataset identifier: PXD040444 reviewer_pxd040444@ebi.ac.uk |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/ |
Resource availability
Lead contact
Any requests for resources and reagents or information should be directed to the Lead Contact, Sanjay V Malhotra (malhotsa@ohsu.edu).
Material availability
The materials generated in this study will be distributed upon request. There are restrictions to availability due to a Material Transfer Agreement (MTA).
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. The mass spectrometry proteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD040444. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines
ID8 mouse ovarian surface epithelial cell line (Cat. No. # SCC145) and SKOV3 human Caucasian ovary adenocarcinoma cell line (Cat. No. # 91091004) were purchased from MilliporeSigma, USA and cultured in DMEM media supplemented with 10% FBS and 1% PSA at 5% CO2 and 37°C. OVCAR3 and OVCAR8 cell lines were obtained from the NCI-DTP repository, USA and cultured in RPMI-1640 media supplemented with 10% FBS and 1% PSA at 5% CO2 and 37°C. Each cell line was regularly tested for mycoplasma.
Method details
Drug
Paclitaxel (Cat. No. # 10461100) was purchased from Cayman Chemical, USA, and stock solutions were prepared in DMSO (MP Biomedicals, #196055) and stored at -20°C.
Stable cell line construction
GBP1 (human) or GBP2b (mouse) overexpression and shRNA knockdown vectors were purchased from VectorBuilder, USA. Ovarian cancer cells were transduced with respective lentiviral particles and selected for puromycin resistance followed by cell shorting.
GBP1 (human) or GBP2b (mouse) overexpression and shRNA knockdown vectors were purchased from VectorBuilder, USA. Ovarian cancer cells were transduced with respective lentiviral particles and selected for puromycin resistance followed by cell shorting.
mGBP2b overexpression: pLV{Exp}-mCherry:T2A:Puro-EF1A>mGbp2b{NM_010259.2} (Vector ID: VB180117-1093kbb)
mGBP2b shRNA: pLV{shRNA}-EGFP:T2A:Puro-U6>mGbp2b{shRNA# TGGGATTGGCATGTTATAAAC} (Vector ID: VB180117-1250gdh)
hGBP1 overexpression: pLV{Exp}-mCherry:T2A:Puro-EF1A>hGBP1{NM_002053.2} (Vector ID: VB230206-1323fan)
hGBP1 shRNA: pLV{shRNA}-EGFP:T2A:Puro-U6>hGBP1{shRNA# CCAGATGAGTACCTGACATAC} (Vector ID: VB170507-1030ruw)
Vector Control: pLV{Exp}-mCherry:T2A:Puro-EF1A>ORF_Stuffer (Vector ID: VB010000-9390nka)
Scramble shRNA Control: pLV{shRNA}-EGFP:T2A:Puro-U6>Scramble_shRNA {CCTAAGGTTAAGTCGCCCTCG} (Vector ID: VB170507-1033npu)
Clonogenic assay
200-500 ovarian cancer cells were plated in 12 well plates with 2 mL of respective media and incubated until visible colonies appeared. Cells were washed and fixed with 2% paraformaldehyde followed by staining with 0.5% crystal violet prepared in 2% ethanol. Cells were de-stained with DI water and allowed to dry. Confluency was measured using TECAN Spark multimode plate reader with automated live cell imaging – cell counting and confluence capabilities.
Immunoblotting
Total protein lysates were collected from 60-70% confluent OC cells and lysed using M-PER™ lysis buffer (Thermo Scientific, #78503) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific, #78440). An equal amount (10-20 μg) of proteins was resolved on 4-12%/8-16% gradient SDS-PAGE gel electrophoresis. Proteins were transferred to a PVDF membrane, blocked, and incubated with respective primary antibodies overnight at 4°C. Blots were washed and probed with respective HRP-conjugated secondary antibodies. Blots were developed using Immobilon® Crescendo Western HRP Substrate (Millipore, Germany) and visualized on an iBright™ FL1500 Imaging System (Thermo Scientific, Waltham, MA). The following primary and secondary antibodies were used: GBP1 (IP: Abnova, #H00002633-PW1; IB: 1:2000; Novusbio, # NBP2-03972; 1:2000), β-actin (CST, #4970S; 1:10000), PSMB1 (Thermo Scientific, #PA5-49648, 1:1000), PSMB6 (Thermo Scientific, #PA1-978, 1:1000), PSMA3 (Proteintech #11887-I-AP, 1:1000), Ub (CST, # 43124S, 1:3000), anti-mouse IgG HRP-linked antibody (CST, #7076, 1:5000), and anti-rabbit IgG HRP-linked antibody (CST, #7074, 1:5000). Raw blots are available as supplemental figures (Figures S4–S6).
In vivo studies: ID8 intraperitoneal injection mouse model study
All the animal studies were reviewed and approved by Stanford University’s Institutional Animal Care and Use Committee (APLAC number: 32766) and OHSU’s Institutional Animal Care and Use Committee (IACUC Study number: IP00003247). 9-10 weeks old female C57BL/6J mice were purchased from Jackson Laboratory, USA (Strain #:000664). 1x106 engineered ID8 cells were implanted in each mouse (n=10 mice/group) intraperitonially. Body weight and bally diameter were measured twice a week. Ascitic fluid was collected 40 days after IP implantation (n=10 mice/group).
Cellular thermal shift assay (CETSA)
We adapted the previously described assay by Savitski et al. (2014)31 to study protein interactors of Gbp2b. ID8 engineered cells were cultured in T-175 flasks. At 60% confluency, the cells were washed and lifted using a cell lifter. The cells were then resuspended in PBS and 1x106 cells were transferred to a PCR tube. The CETSA method was applied to ID8-VC, ID8-OV, ID8-SC, and ID8-KD as per the previously described (22). The tubes underwent a temperature cycle of three minutes at different temperatures (37, 41, 44, 47, 50, 53, 56, 59, 63, 67°C) followed by two minutes of room temperature incubation. Next the tubes were rapidly frozen in liquid nitrogen, the cells were lysed with a freeze-thaw cycle, and the soluble and insoluble fractions were separated through centrifugation at 14,000 RPM for 30 minutes at 4°C. An equal amount of soluble fraction from each temperature was then labeled with tandem mass tag (TMT) using the manufacturer’s protocol (TMT10plex™ Isobaric Label Reagent Set, # 90110, Thermo Fisher Scientific, Waltham, MA).
A 3 μL injection of each combined TMT-labeled peptide sample was loaded in a 20 μL sample loop using a Dionex Ultimate Rapid Separation Liquid Chromatography system (Thermo Fisher Scientific) and loaded into a PepMap C18 trap column (Thermo Fisher Scientific) with a flow rate set of 5 μL/min for 10 minutes. A 25 cm long C18 analytical (New Objective) packed in-house with BEH C18, 130 Å, 1.7 μm particle size (Waters) was used to separate TMT-labeled peptides by reversed-phase chromatography. A column heater (MSWIL) was used to heat to column to 60°C. The gradient program consisted of holding mobile phase B (0.1% formic acid in acetonitrile) at 2% for the first 6.0 minutes to trap TMT-labeled peptides, slowly ramped up to 35% over the next 104 minutes followed by an increase to 85% over 5 minutes with a 5-minute hold. The analytical column was re-equilibrated with mobile phase A (0.1 % formic acid in water) at 98% for 10 minutes prior to the next sample injection. Each sample was analyzed in triplicate at a constant flow rate throughout the gradient of 0.3 μL/min. Precursor ions were subject to collision-induced dissociation (CID) on an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific). Top-speed for MS1 was set at 3 seconds with a mass scan range of 375-1600 m/z and a mass resolution of 120,000. The normalized AGC target was to 250 and the precursor fit filter at 50 percent threshold with a 0.7 m/z fit window. The fragmented ions were detected in the ion trap using Turbo scan rate with dynamic exclusion enabled for 60 seconds. The maximum injection time for MS2 was set to 35 ms with an AGC target value set to standard. TMT-labeled peptides were identified by enabling the Real-Time-Search algorithm with search time of 100 ms against the Swiss-Prot database reference to the mouse proteome (17,027 entries, 2019). Carbamidomethyl and TMT6plex on cysteine and lysine residues were added as a fixed modification and methionine oxidation as variable modification for peptide identification. MS3 was performed on identified peptides by enabling synchronous precursor selection (SPS) algorithm with a scan mass range of 400-1600. The total number of SPS precursor ions allowed was set to 10 with an isolation window of 1.2 m/z. Selected precursor ions were subject to higher energy dissociation (HCD) with a collision energy of 65% in the Orbitrap detector. The mass resolution was set to 60,000 and a scan mass range at 100-500 m/z.
Protein levels were quantified using a sum-based bootstrap algorithm that utilized individual spectra-matched peptides and TMT reporter ion intensity, taking into account isotope impurities with Proteome Discoverer 2.4. The protein levels were then compared between the control and Gbp2b-engineered samples, specifically ID8-VC vs. ID8-OV and ID8-SC vs. ID8-KD. To determine the log2 ratio, the lowest temperature was used as a reference and the signal at each temperature was compared to the highest temperature to calculate the percentage of signal loss in the soluble fraction. The final step involved fitting the data to S-curves using a brute-force algorithm in R with the aim of minimizing the difference between the original and Boltzmann-adjusted curves. The melting point differences between the fitted curves (engineered vs. control) with correlations above 0.7, p-values less than 0.01, and consistent thermal shifts in all measurements were considered as potential Gbp2b protein interactors. Finally, we compared the lists of potential protein interactors from both experiments and considered as final candidates those proteins with negative temperature shifts in the Gpb2b KD experiment, proteins destabilized by the absence of Gbp2b, and proteins with positive temperature shifts in the Gbp2b OV experiment, proteins stabilized by the increase of Gbp2b. The output datasets are contained in the Table S1. CETSA_Gbp2b_OV_vs_Gbp2b_KD_Comparison, Table S2. CETSA_Gbp2b_OV, Table S3. CETSA_Gbp2b_KD.
To gain deeper insight into the underlying molecular mechanism of the identified potential protein interactors, we used FunRich32 to conduct a gene enrichment analysis based on Gene Ontology Biological Process. The significance of the mapped categories and their mapped proteins can be found in the Table S4.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE33 partner repository with the dataset identifier PXD040444.
Immunoprecipitation
Cells were lysed using M-PER™ lysis buffer (Thermo Scientific, USA #78503) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific, USA #78440). Precleaned total protein lysates were incubated overnight with anti-GBP1 antibody (1 μg/100 μL, Abnova, #H00002633-PW1) or rabbit IgG (EMD Millipore, USA #PP64B) at 4°C with rocking. Antibody and conjugated proteins were pulled out using Protein A/G magnetic beads (Thermo Scientific, USA #88802). Washed A/G magnetic beads were boiled with 2X Laemmli buffer. Samples were resolved using SDS-PAGE and blots were developed as described above.
Proteasomal activity assay
Each cell was lysed using 1X PBS containing 0.5% NP-40. The proteasomal activity was assayed using a Proteasome Activity Assay kit (Abcam, USA #ab107921) by following the manufacturer's protocol.
Global proteome profiling
Engineered ID8 cells were grown, processed, and TMT-labeled as described above. For proteomic analysis, a 10 μL injection of the combined TMT-labeled peptides were loaded on a PepMap 100 Å C 18 trap column (Thermo Fisher Scientific) using a Dionex Ultimate Rapid Separation Liquid Chromatography system (Thermo Fisher Scientific) at a rate of 5 μl/min for 10 minutes with initial mobile phase A (0.1% formic acid in water) at 98% and mobile phase B (0.1% formic acid in acetonitrile) at 2%. A 25 cm long C18 analytical column (New Objective) packed in house with Magic C18 AQ resin (Michrom Bioresources) was used to separate TMT-labeled peptides by reversed-phase chromatography. A flow rate was set to 0.6 μL/min with gradient parameters as follows; mobile phase B was held at 2% B with a flow rate for the first 10 minutes, increased to 35% B over the next 100 minutes, and rapidly increased to 85% B over 2 minutes with a 7-minute hold time before bringing down mobile phase B to 2 % for column equilibration prior to next sample injection. Each TMT-labeled sample was analyzed in triplicate. A Flex Ion source (Thermo Fisher Scientific) coupled to a LTQ-Orbitrap Elite (Thermo Fisher Scientific) hybrid mass spectrometer was used to ionized TMT-labeled peptides using 1.8 kV. The top 8 most abundant ions per MS1 scan were selected for higher energy dissociation (HCD) in a data-dependent fashion with a collisional energy set to 35 eV. A scan mass resolution of 30,000, FT AGC target of 1e6, and the scan mass range of 400-1800 m/z was set for MS1 precursor ions. For the MS2 scans, the mass range was set at 110-2000 m/z with a mass resolution of 30,000 and the AGC target set at 3e4 with dynamic exclusion enabled for 30 seconds.
The raw data from the LC-MS were analyzed using MaxQuant34 against the Swiss-Prot mouse reference proteome database, which contained 17,027 entries as of 2019. Proteins were identified and quantified based on matched peptides found in each individual MS2 spectrum, using the TMT6plex reporter ion intensities after correcting for isotope impurities. The database search included the digestion of proteins by trypsin with a maximum of two missed cleavages and had a tolerance of 0.5 Da for precursor masses and 10 ppm for fragment masses. The analysis also considered the fixed modification of cysteine carbamidomethylation, lysine and N-terminal TMT6plex modification, as well as variable methionine oxidation. All quantitative information was presented in terms of Z-scores at the protein level, which were calculated by comparing the log2 ratios of the reporter ion intensities to the average of the control cell lines and the proteomics statistical analysis was done using the WSPP model.35 The validity of the null hypothesis was thoroughly examined at the spectrum, peptide, and protein levels by plotting the cumulative distributions. The final statistical comparisons were performed using a Student's t-test. To better understand the molecular mechanism underlying the protein changes detected (p-value < 0.05), we separate the overrepresentation analysis performed according to Reactome Pathway DB into increases ID8 engineered cells minus the corresponding control with positive sign and decreases ID8 engineered cells minus the corresponding control with negative sign. The list of categories and their corresponding mapped proteins could be found in Table S5. Gene_Enrichment analysis_VC vs OV_Increases, Table S6. Gene_Enrichment analysis_VC vs OV_Decreases, Table S7. Gene_Enrichment analysis_SC vs KD_Decreases, Table S8. Gene_Enrichment analysis_SC vs KD_Increases.
Kaplan Meier plot
Kaplan-Meier plots (KMplot) for probability of survival of ovarian cancer patients with GBP1 high (high) and GBP1 low (low) expression (mRNA gene chip data, Affy ID: 202269_x_at). KMplots were plotted using KM-plotter.36
Pharmacogenomic data analysis of cancer cell line using CellMinerCDB
GDSC-MGH-Sanger database for pharmacogenomic data of cancer cell lines was analyzed using CellMinerCDB.37 Cumulative pharmacogenomic data of cell lines (Excluding lung, lymph, skin, and blood cancer cell line) was analyzed for GBP1 mRNA expression and IC50 drug concentration for Bortezomib and MG132 treatment. The dot plot represents the correlation (Y-axis: IC50 value of proteasome inhibitor and X-axis: GBP1 mRNA expression).
Quantification and statistical analysis
Each set of experiments was repeated at least 3 times, with a minimum of 3 replicates. Data was analyzed for its statistical significance using GraphPad Prism 9 software. Each result is represented in Mean ± SD. P-values are denoted using ∗ or numerical values.
Acknowledgments
S.V.M. would like to thank the Knight Cancer Institute, for partial support of this work. D.T. would like to thank the Indo-US Science and Technology Forum (IUSSTF) and the Science and Engineering Research Board (SERB), Department of Sciences & Technology, Government of India, for a SERB Indo-US postdoctoral fellowship.
Author contributions
Conceptualization, D.T. and S.V.M.; methodology, D.T., F.J.G.-M., and A.B.; statistical analysis, D.T. and F.J.G.-M.; investigation, D.T., A.B., F.J.G.-M., and S.J.P.; writing, D.T., F.J.G.-M., A.B., S.J.P., and S.V.M.; supervision, S.J.P. and S.V.M.; all authors read and approved the final manuscript.
Declaration of interests
S.V.M. and D.T. are inventors on the following U.S. Provisional patent application:
Novel N-Hydroxyethyl Didehydroazapodophyllotoxins as GBP1 Inhibitors to Overcome Treatment Resistance in Ovarian Cancer. 2019, WO 2019178091.
Published: October 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108292.
Supplemental information
References
- 1.Sun J., Bogie K.M., Teagno J., Sun Y.H.S., Carter R.R., Cui L., Zhang G.Q. Design and Implementation of a Comprehensive Web-based Survey for Ovarian Cancer Survivorship with an Analysis of Prediagnosis Symptoms via Text Mining. Cancer Inf. 2014;13:113–123. doi: 10.4137/CIN.S14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ledermann J.A., Kristeleit R.S. Optimal treatment for relapsing ovarian cancer. Ann. Oncol. 2010;21 doi: 10.1093/annonc/mdq377. vii218–222. [DOI] [PubMed] [Google Scholar]
- 3.Ushijima K. Treatment for recurrent ovarian cancer-at first relapse. JAMA Oncol. 2010;2010 doi: 10.1155/2010/497429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komarova N.L., Wodarz D. Drug resistance in cancer: principles of emergence and prevention. Proc. Natl. Acad. Sci. USA. 2005;102:9714–9719. doi: 10.1073/pnas.0501870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senthebane D.A., Rowe A., Thomford N.E., Shipanga H., Munro D., Mazeedi M.A.M.A., Almazyadi H.A.M., Kallmeyer K., Dandara C., Pepper M.S., et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017;18 doi: 10.3390/ijms18071586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilla-Moffett D., Barber M.F., Taylor G.A., Coers J. Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J. Mol. Biol. 2016;428:3495–3513. doi: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guenzi E., Töpolt K., Cornali E., Lubeseder-Martellato C., Jörg A., Matzen K., Zietz C., Kremmer E., Nappi F., Schwemmle M., et al. The helical domain of GBP-1 mediates the inhibition of endothelial cell proliferation by inflammatory cytokines. EMBO J. 2001;20:5568–5577. doi: 10.1093/emboj/20.20.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Honkala A.T., Tailor D., Malhotra S.V. Guanylate-Binding Protein 1: An Emerging Target in Inflammation and Cancer. Front. Immunol. 2019;10:3139. doi: 10.3389/fimmu.2019.03139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisch D., Bando H., Clough B., Hornung V., Yamamoto M., Shenoy A.R., Frickel E.M. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 2019;38 doi: 10.15252/embj.2018100926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prakash B., Renault L., Praefcke G.J., Herrmann C., Wittinghofer A. Triphosphate structure of guanylate-binding protein 1 and implications for nucleotide binding and GTPase mechanism. EMBO J. 2000;19:4555–4564. doi: 10.1093/emboj/19.17.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng L., Gou L., Wei T., Zhang J. GBP1 promotes erlotinib resistance via PGK1-activated EMT signaling in non-small cell lung cancer. Int. J. Oncol. 2020;57:858–870. doi: 10.3892/ijo.2020.5086. [DOI] [PubMed] [Google Scholar]
- 12.Zhao J., Li X., Liu L., Cao J., Goscinski M.A., Fan H., Li H., Suo Z. Oncogenic Role of Guanylate Binding Protein 1 in Human Prostate Cancer. Front. Oncol. 2019;9:1494. doi: 10.3389/fonc.2019.01494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Donato M., Mariani M., Petrella L., Martinelli E., Zannoni G.F., Vellone V., Ferrandina G., Shahabi S., Scambia G., Ferlini C. Class III beta-tubulin and the cytoskeletal gateway for drug resistance in ovarian cancer. J. Cell. Physiol. 2012;227:1034–1041. doi: 10.1002/jcp.22813. [DOI] [PubMed] [Google Scholar]
- 14.Duan Z., Foster R., Brakora K.A., Yusuf R.Z., Seiden M.V. GBP1 overexpression is associated with a paclitaxel resistance phenotype. Cancer Chemother. Pharmacol. 2006;57:25–33. doi: 10.1007/s00280-005-0026-3. [DOI] [PubMed] [Google Scholar]
- 15.Fukumoto M., Amanuma T., Kuwahara Y., Shimura T., Suzuki M., Mori S., Kumamoto H., Saito Y., Ohkubo Y., Duan Z., et al. Guanine nucleotide-binding protein 1 is one of the key molecules contributing to cancer cell radioresistance. Cancer Sci. 2014;105:1351–1359. doi: 10.1111/cas.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mateus A., Kurzawa N., Becher I., Sridharan S., Helm D., Stein F., Typas A., Savitski M.M. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 2020;16 doi: 10.15252/msb.20199232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L., Madura K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005;65:5599–5606. doi: 10.1158/0008-5472.CAN-05-0201. [DOI] [PubMed] [Google Scholar]
- 18.Soave C.L., Guerin T., Liu J., Dou Q.P. Targeting the ubiquitin-proteasome system for cancer treatment: discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev. 2017;36:717–736. doi: 10.1007/s10555-017-9705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manasanch E.E., Orlowski R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017;14:417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernández-Vargas H., von Kobbe C., Sánchez-Estévez C., Julián-Tendero M., Palacios J., Moreno-Bueno G. Inhibition of paclitaxel-induced proteasome activation influences paclitaxel cytotoxicity in breast cancer cells in a sequence-dependent manner. Cell Cycle. 2007;6:2662–2668. doi: 10.4161/cc.6.21.4821. [DOI] [PubMed] [Google Scholar]
- 21.Prakash B., Praefcke G.J., Renault L., Wittinghofer A., Herrmann C. Structure of human guanylate-binding protein 1 representing a unique class of GTP-binding proteins. Nature. 2000;403:567–571. doi: 10.1038/35000617. [DOI] [PubMed] [Google Scholar]
- 22.Sahu I., Mali S.M., Sulkshane P., Xu C., Rozenberg A., Morag R., Sahoo M.P., Singh S.K., Ding Z., Wang Y., et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021;12:6173. doi: 10.1038/s41467-021-26427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enenkel C., Kang R.W., Wilfling F., Ernst O.P. Intracellular localization of the proteasome in response to stress conditions. J. Biol. Chem. 2022;298 doi: 10.1016/j.jbc.2022.102083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flick K., Kaiser P. Protein degradation and the stress response. Semin. Cell Dev. Biol. 2012;23:515–522. doi: 10.1016/j.semcdb.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming H., Li B., Jiang J., Qin S., Nice E.C., He W., Lang T., Huang C. Protein degradation: expanding the toolbox to restrain cancer drug resistance. J. Hematol. Oncol. 2023;16:6. doi: 10.1186/s13045-023-01398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanna J., Meides A., Zhang D.P., Finley D. A ubiquitin stress response induces altered proteasome composition. Cell. 2007;129:747–759. doi: 10.1016/j.cell.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 27.Gama V., Swahari V., Schafer J., Kole A.J., Evans A., Huang Y., Cliffe A., Golitz B., Sciaky N., Pei X.H., et al. The E3 ligase PARC mediates the degradation of cytosolic cytochrome c to promote survival in neurons and cancer cells. Sci. Signal. 2014;7:ra67. doi: 10.1126/scisignal.2005309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andreoli M., Persico M., Kumar A., Orteca N., Kumar V., Pepe A., Mahalingam S., Alegria A.E., Petrella L., Sevciunaite L., et al. Identification of the first inhibitor of the GBP1:PIM1 interaction. Implications for the development of a new class of anticancer agents against paclitaxel resistant cancer cells. J. Med. Chem. 2014;57:7916–7932. doi: 10.1021/jm5009902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt M.F., Gan Z.Y., Komander D., Dewson G. Ubiquitin signalling in neurodegeneration: mechanisms and therapeutic opportunities. Cell Death Differ. 2021;28:570–590. doi: 10.1038/s41418-020-00706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frankowska N., Lisowska K., Witkowski J.M. Proteolysis dysfunction in the process of aging and age-related diseases. Front. Aging. 2022;3 doi: 10.3389/fragi.2022.927630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savitski M.M., Reinhard F.B.M., Franken H., Werner T., Savitski M.F., Eberhard D., Martinez Molina D., Jafari R., Dovega R.B., Klaeger S., et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346 doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 32.Fonseka P., Pathan M., Chitti S.V., Kang T., Mathivanan S. FunRich enables enrichment analysis of OMICs datasets. J. Mol. Biol. 2021;433 doi: 10.1016/j.jmb.2020.166747. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 35.Navarro P., Trevisan-Herraz M., Bonzon-Kulichenko E., Núñez E., Martínez-Acedo P., Pérez-Hernández D., Jorge I., Mesa R., Calvo E., Carrascal M., et al. General statistical framework for quantitative proteomics by stable isotope labeling. J. Proteome Res. 2014;13:1234–1247. doi: 10.1021/pr4006958. [DOI] [PubMed] [Google Scholar]
- 36.Lánczky A., Győrffy B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021;23 doi: 10.2196/27633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luna A., Elloumi F., Varma S., Wang Y., Rajapakse V.N., Aladjem M.I., Robert J., Sander C., Pommier Y., Reinhold W.C. CellMiner Cross-Database (CellMinerCDB) version 1.2: Exploration of patient-derived cancer cell line pharmacogenomics. Nucleic Acids Res. 2021;49:D1083–D1093. doi: 10.1093/nar/gkaa968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. The mass spectrometry proteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD040444. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.