

SUMMARY
Mechanisms that prevent accidental activation of the PINK1/Parkin mitophagy circuit on healthy mitochondria are poorly understood. On the surface of damaged mitochondria, PINK1 accumulates and acts as the input signal to a positive feedback loop of Parkin recruitment, which in turn promotes mitochondrial degradation via mitophagy. However, PINK1 is also present on healthy mitochondria, where it could errantly recruit Parkin and thereby activate this positive feedback loop. Here, we explore emergent properties of the PINK1/Parkin circuit by quantifying the relationship between mitochondrial PINK1 concentrations and Parkin recruitment dynamics. We find that Parkin is recruited to mitochondria only if PINK1 levels exceed a threshold and then only after a delay that is inversely proportional to PINK1 levels. Furthermore, these two regulatory properties arise from the input-coupled positive feedback topology of the PINK1/Parkin circuit. These results outline an intrinsic mechanism by which the PINK1/Parkin circuit can avoid errant activation on healthy mitochondria.
Graphical Abstract
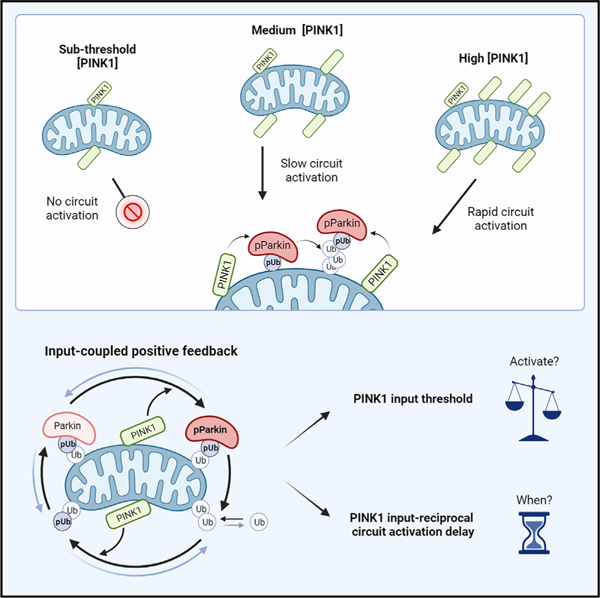
In brief
Waters et al. show that activation of the PINK1/Parkin mitophagy circuit is regulated by a PINK1 input threshold and an input-reciprocal circuit activation delay. The two regulatory behaviors emerge from the positive feedback architecture of the circuit. These findings suggest a mechanism by which cells can avoid errant mitochondrial degradation.
INTRODUCTION
Mitochondrial polarization is critical for mitochondrial function and is a key indicator of mitochondrial health.1–4 Cells maintain a healthy pool of mitochondria by degrading depolarized mitochondria via mitophagy (mitochondrial autophagy).1,5–9 A core molecular circuit mediating the mitophagy decision involves PINK1, a mitochondrial kinase, and Parkin, a cytoplasmic E3 ubiquitin ligase.10 While PINK1 is constitutively recruited to the mitochondrial outer membrane (MOM), polarization-dependent degradation keeps PINK1 levels low on healthy, polarized, mitochondria.6,7 Mitochondrial depolarization prevents this degradation and leads to accumulation of PINK1 on the MOM over time.6,7 Upon PINK1 accumulation, positive feedback in the PINK1/Parkin circuit leads to extensive labeling of the mitochondrial surface with phospho-S65 ubiquitin (pUb) and to pUb-dependent recruitment of downstream mitophagy machinery.10–12
Positive feedback in the PINK1/Parkin circuit occurs through a series of steps (Figure 1A): PINK1 phosphorylates mitochondrial ubiquitin, pUb recruits autoinhibited Parkin to the MOM, PINK1 phosphorylates Parkin’s ubiquitin-like (UBL) domain to activate Parkin, and activated Parkin deposits new ubiquitin on proteins at the MOM.10,13–23 Newly deposited ubiquitin can then be phosphorylated by PINK1, starting new rounds of amplification.10,24 Together, these steps constitute the positive feedback topology of the PINK1/Parkin circuit, where PINK1 provides an input signal, and positive feedback occurs via Parkin’s ubiquitin ligase activity.11
Figure 1. Quantification of input-to-output responses for a minimal PINK1/Parkin synthetic circuit.
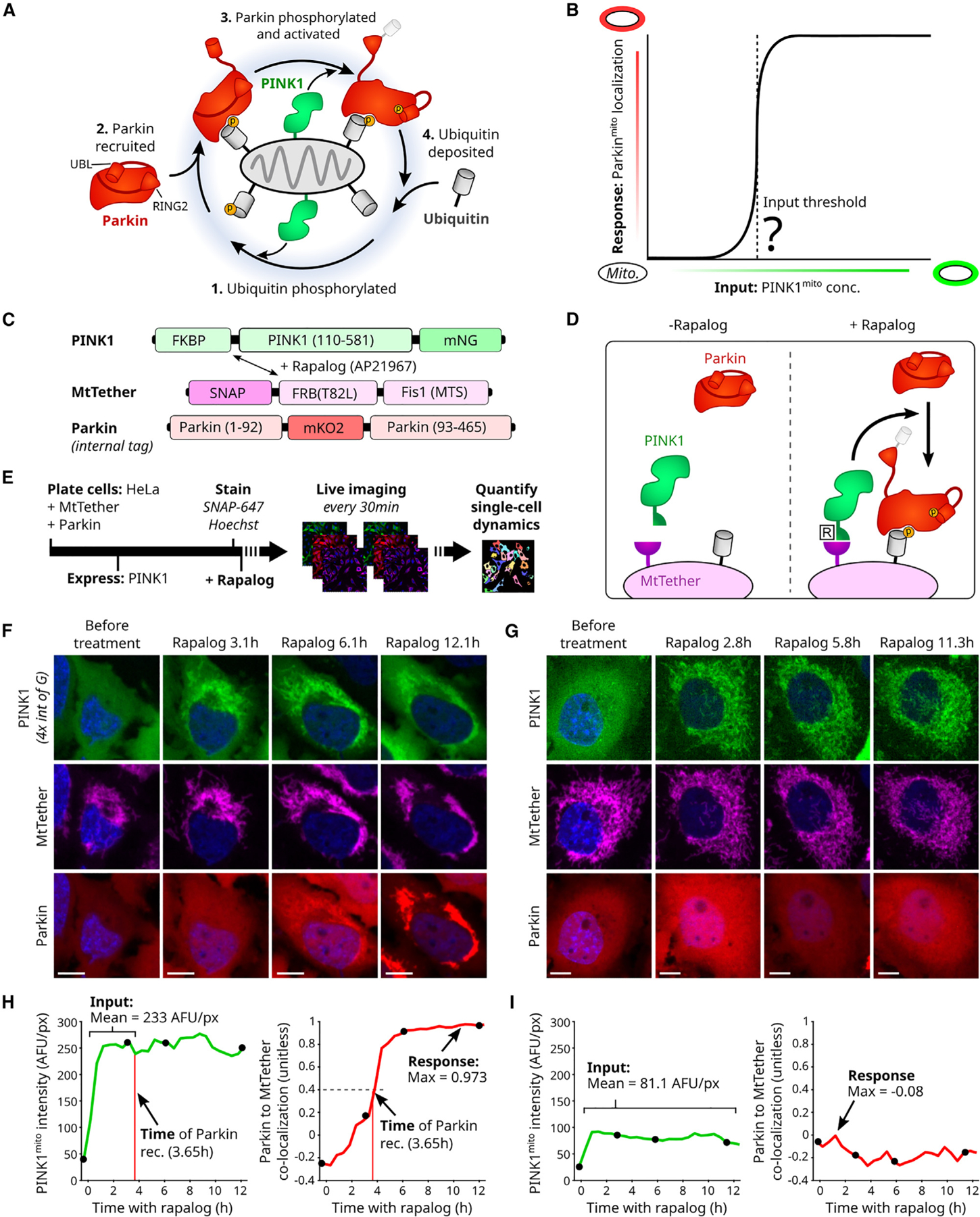
(A) Cartoon of PINK1/Parkin positive feedback loop. Yellow circles: phosphorylation sites. Conformational changes of activated Parkin’s UBL (ubiquitin-like) and catalytic RING2 domains are shown.
(B) Hypothetical input-response relationship (curve) illustrating a PINK1 input threshold for circuit activation (vertical dashed line). Inputs: discrete mitochondrial PINK1 concentrations, held stable over time.
(C and D) PINK1/Parkin synthetic circuit using rapalog-induced recruitment. Mitochondrial targeting sequence (MTS) of PINK1, amino acid 1–109, removed. T82L: mutation required for rapalog binding.
(E) Live-cell imaging approach. Rapalog treatment: 200 nM. SNAP-647: fluorescent SNAP ligand for far-red imaging of MtTether (STAR Methods). Bar length not to scale.
(F and G) Representative time-lapse images of cells with (F) or without (G) Parkinmito recruitment in response to induced recruitment. Scale bars: 10 μm. Relative intensity visualization range noted in (F).
(H and I) Quantification of circuit input (mean concentration), circuit response (max Parkinmito recruitment), and the time of Parkinmito recruitment for cells in (F) and (G). AFU: arbitrary fluorescence units. Co-localization: intensity correlation (Pearson, STAR Methods). Black points: time points in (F) and (G). See also Figures S1 and S2.
While positive feedback in the PINK1/Parkin circuit enables rapid and robust signal amplification on depolarized mitochondria,10,25 healthy mitochondria are somehow able to tolerate constant recruitment and release of PINK16,7 as well as transient depolarization26,27 without triggering runaway positive feedback. A range of theoretical regulatory mechanisms have been proposed for how the PINK1/Parkin circuit could avoid such errant activation.11,13,24,26,28–31 One intriguing hypothesis is that mitochondrial PINK1 concentrations may need to exceed some threshold value to provoke a circuit activation response (Figure 1B).29 However, investigation of such emergent behaviors has been limited by a lack of techniques for holding mitochondrial PINK1 concentrations steady while the corresponding circuit response is measured.26
Here, we investigated whether emergent regulatory behaviors exist in the PINK1/Parkin positive feedback circuit. We used a synthetic circuit approach to quantitatively map the relationship between discrete mitochondrial PINK1 concentrations and Parkin recruitment dynamics. This input-response mapping revealed two regulatory properties: a PINK1 input threshold for circuit activation fate and a PINK1 input-reciprocal delay for circuit activation timing. Further mutational and mathematical evaluation revealed that these two regulatory properties arise intrinsically from the positive feedback topology of the PINK1/Parkin circuit. Together, our results demonstrate a mechanism by which the PINK1/Parkin circuit could avoid unchecked positive feedback activation.
RESULTS
To evaluate emergent behavior of the PINK1/Parkin positive feedback circuit, we adapted a previously described minimal synthetic PINK1/Parkin circuit6,32 for four-color quantitative microscopy. The minimal synthetic circuit was comprised of (1) fluorescently labeled Parkin and (2) fluorescently labeled PINK1 that could be targeted to fluorescently labeled mitochondria using a rapalog (rapamycin analog)-induced recruitment system6,32–34 (Figures 1C and 1D). All components were expressed in HeLa cells, which do not express endogenous Parkin7,35 (STAR Methods). This synthetic circuit approach enabled us to evaluate emergent properties of the circuit by quantifying the relationship between relatively stable circuit inputs (PINK1 concentration on mitochondria) and resulting circuit activation (Parkin localization to mitochondria) in an isolated setting.
Internally tagged Parkin probe
Parkin relocalization from the cytoplasm to phospho-S65 ubiquitin (pUb) on the mitochondria is an established method for measuring PINK1/Parkin circuit activation.6 To track PINK1/Parkin circuit activation, we optimized a fluorescent tagging approach for Parkin. However, traditionally used N-terminal tags are known to perturb Parkin’s conformational stability36 and could affect Parkin’s mitophagy activity. In agreement with this prediction, we found that Parkin, N-terminally tagged with monomeric Kusabira-Orange2 (mKO2), displayed reduced recruitment to and degradation of chemically depolarized mitochondria (Figures S1A–S1H). Upon further investigation, we found that Parkin’s N-terminal NH3+ group appears to engage in a conserved salt bridge (Figures S1I–S1Q)37 that would be disrupted by N-terminal tagging. These observations prompted us to investigate alternative internal tagging locations.
We identified an internal Parkin tagging site, between A92 and G93, that is, (1) within a disordered19,20 and non-conserved linker region (Figure S1D) and (2) distinct from other previously described internal-tagging sites that alter Parkin relocalization dynamics.22 We found that Parkin, internally tagged with mKO2 at A92, displayed untagged-like mitophagy activity (Figures S1E–S1H). Based on these results, we chose to use internally tagged Parkin as both a component of and activation reporter for our minimal synthetic circuit (Figure 1C).
Stable PINK1 recruitment to mitochondria
Traditional mitochondrial depolarization methods cause PINK1 accumulation over time and are therefore not capable of producing stable mitochondrial PINK1 levels.26 To control and quantify PINK1 concentrations on mitochondria, we optimized a previously described rapalog-induced PINK1 recruitment system6,32,38,39 for quantitative microscopy. An FRB/FKBP/rapalog system33,34 was used to control dimerization between mNeonGreen (mNG)-tagged PINK1 and an SNAP-tagged mitochondrial tether (MtTether; Figures 1C, 1D, and S2A; STAR Methods). Upon rapalog treatment, this allows relocalization of PINK1 from the cytoplasm to mitochondria in live cells to be monitored (Figures 1E–1G; STAR Methods). Rapalog-induced PINK1 localization to mitochondria was reasonably stable over time within our measurement precision (Figures 1F–1I and S2B). Notably, PINK1 and MtTether expression was designed to enable a range of mitochondrial PINK1 concentrations across cells in each experiment (Figure S2C; STAR Methods). We verified that PINK1 mediates Parkin recruitment to and degradation of mitochondria (Figure S2D),6,32 that PINK1’s role requires its kinase activity (Figure S2E),32 and that rapalog treatment did not exhibit off-target effects on mTOR signaling in our system (Figure S2F). Consistent with previous reports, activation of the circuit did not cause mitochondrial depolarization.32 This PINK1 and MtTether rapalog-induced recruitment system enabled us to stably direct PINK1 to the mitochondria in our minimal PINK1/Parkin synthetic circuit.
Quantification of synthetic circuit dynamics
To define the behavior of the minimal PINK1/Parkin synthetic circuit, we developed a quantification approach to evaluate the relationship between mitochondrial PINK1 levels and Parkin recruitment at single-cell resolution. Initial observations revealed heterogeneous circuit dynamics where cells with high mitochondrial PINK1 underwent Parkin relocalization to mitochondria (Figure 1F), while cells with low mitochondrial PINK1 did not (Figure 1G). We quantified these circuit dynamics for each cell by tracking mitochondrial PINK1 concentration (via mean PINK1 intensity) and Parkin recruitment (via correlation of Parkin and MtTether intensities) (Figures 2H and 2I; STAR Methods) over time. From these data, we calculated single-cell estimations for (1) the mean concentration of PINK1 on mitochondria after rapalog treatment, (2) the maximal Parkin recruitment response, and (3) the time of Parkin recruitment (Figures 1H and 1I; STAR Methods). This triplet of metrics summarized the behavior of the minimal PINK1/Parkin synthetic circuit.
Figure 2. A PINK1 concentration threshold controls activation of the PINK1/Parkin circuit.
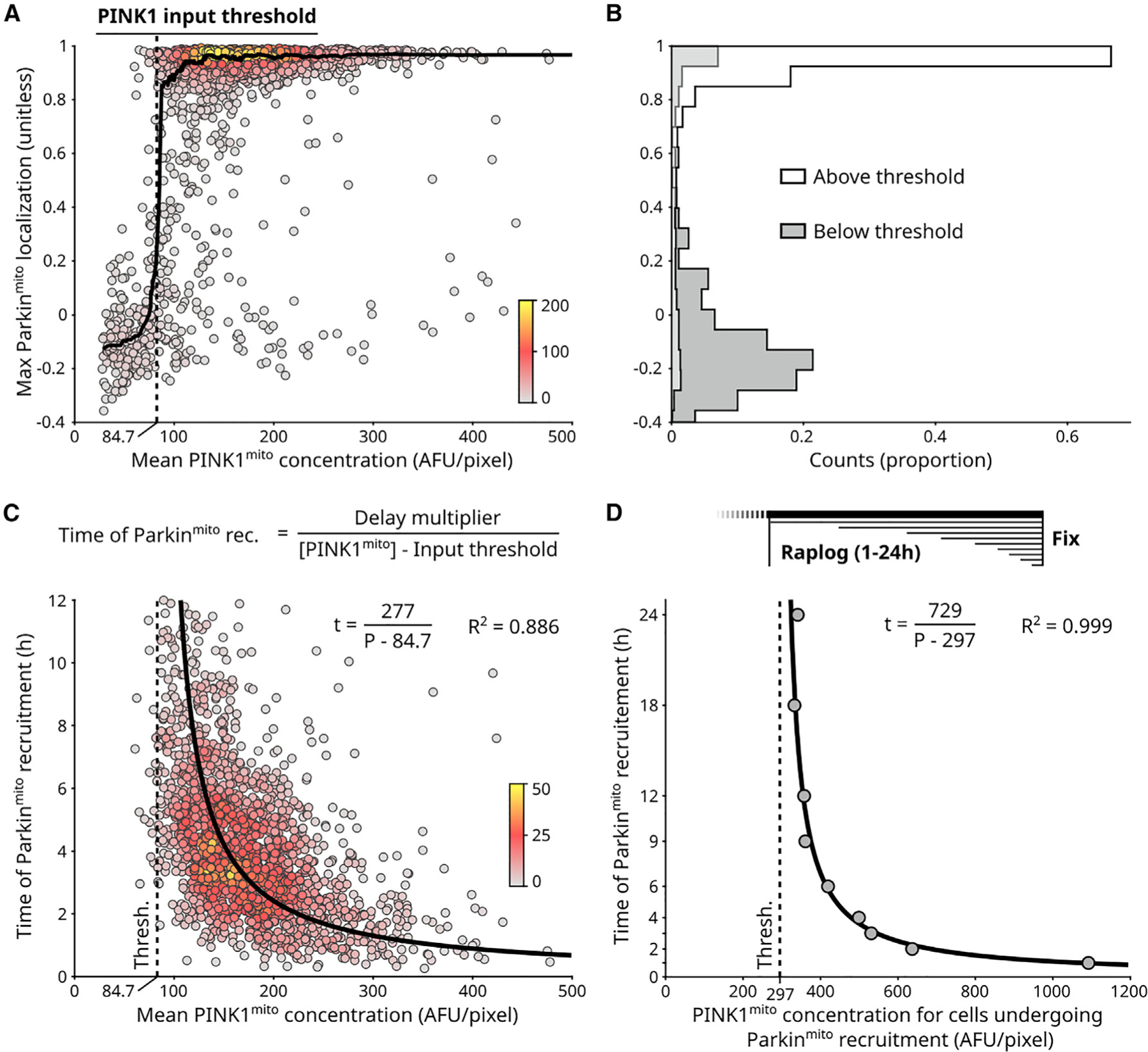
(A) Single-cell measurements for circuit input ( concentration) and circuit response (Parkinmito localization) for n = 1,987 cells (Figures 1H and 1I; STAR Methods). Points: individual cells, colored by local point density (number of nearby points, STAR Methods). Solid line: sliding median. PINK1 input threshold: concentration required for Parkinmito recruitment (STAR Methods).(B) PINK1 input threshold separates cells with and without Parkinmito recruitment.
(C) Timing of Parkinmito recruitment has a reciprocal relationship to . Points: n = 1,676 cells with Parkinmito recruitment from (A) (Figure 1H; STAR Methods), colored by local point density. General equation of reciprocal relationship is shown. Delay multiplier numerator defines hyperbolic relationship. Fitted hyperbolic curve and R2 value are shown. Brackets: concentration.
(D) PINK1 input threshold (dashed line) and timing hyperbola (solid line) quantified using analysis of fixed cells following various durations of rapalog treatment (Figures S2K and S2L; STAR Methods). Points: aggregate quantification from n > 14,000 fixed cells per rapalog treatment duration. See also Figures S2 and S3.
A PINK1 concentration threshold controls PINK1/Parkin circuit activation
To examine emergent behavior of the PINK1/Parkin synthetic circuit, we first evaluated circuit activation fates as a function of PINK1 mitochondrial concentration. Plotting the mean concentration of PINK1 on mitochondria against the associated maximal Parkin recruitment responses of individual cells revealed two distinct response populations (Figure 2A, points). Specifically, cells with higher mitochondrial PINK1 concentrations underwent Parkin recruitment, while cells with lower mitochondrial PINK1 concentrations did not undergo Parkin recruitment (Figure 2A, points). These two populations were distinguished by a sharp inflection point along the PINK1 concentration axis (Figure 2A, sliding median). Because the synthetic circuit in these cells was given time to reach a stable response state, existence of these two populations supports the existence of a PINK1 concentration threshold that mitochondrial PINK1 levels must surpass to detectably recruit Parkin. This ‘‘PINK1 input threshold’’ was quantified as the PINK1 concentration that best separated the two populations (Figures 2A and 2B; STAR Methods). Finally, we checked that cells lacking detectable Parkin recruitment to mitochondria also lacked detectable levels of mitochondrial pUb (Figures S2G and S2H) and that the PINK1 concentration threshold was observed using an alternative Parkin recruitment metric (Figures S2I and S2J). These results suggest that our conclusions are robust with respect to both potential detection limitations and quantification approaches. Taken together, our work shows that the minimal PINK1/Parkin synthetic circuit exhibits a PINK1 concentration threshold for circuit activation.
To further examine emergent behavior of the synthetic circuit, we evaluated circuit activation timing as a function of PINK1 mitochondrial concentration. While a delay for detectable Parkin recruitment is readily observed in depolarization-based assays and can be decreased by overexpressing PINK1,6 the exact relationship of this delay to PINK1 levels is unknown. Plotting the mean concentration of PINK1 on mitochondria against the time of Parkin recruitment of individual cells revealed an inverse relationship that was well fit by a hyperbolic curve (Figure 2C, R2 = 0.886). Specifically, this analysis revealed an ‘‘input-reciprocal delay’’ for Parkin recruitment: doubling the amount by which PINK1 mitochondrial concentration exceeds the PINK1 input threshold will halve the time required for Parkin recruitment. The input-reciprocal delay relationship was described by the numerator of the fit hyperbolic curve, the ‘‘delay multiplier’’ (Figure 2C). Both the input-reciprocal delay behavior as well as the PINK1 input threshold behavior were recapitulated by fixed-cell time series, which had the advantage of allowing quantification from a greater number of cells (Figures 2D, S2K, and S2L). Finally, by leveraging both cell-to-cell heterogeneity in Parkin expression levels (Figures S3A–S3D) and an ultralow strength promoter for Parkin expression (Figures S3E–S3I), we showed that the observed PINK1 input threshold and input-reciprocal delay behaviors were robust across a wide range of Parkin expression levels. Together, the PINK1 input threshold and input-reciprocal Parkin recruitment delay suggest a ‘‘two-factor’’ regulatory paradigm for activation of the PINK1/Parkin circuit where circuit activation is controlled by the amount of PINK1 and the duration of the PINK1 signal.
PINK1 autophosphorylation is not required for PINK1 input threshold behavior
To explore how the observed circuit behaviors could arise, we first evaluated whether PINK1 autophosphorylation is necessary for producing the PINK1-input threshold and/or input-reciprocal delay. PINK1 autophosphorylation at the key S228 locus (Figures 3A and 3B) occurs in trans and increases PINK1’s kinase activity for both Parkin and ubiquitin.28,40,41 We reasoned that such a requirement for two PINK1 molecules to interact could produce a form of threshold or delay behavior. Hence, we made use of PINK1 S228A (non-phosphorylatable) and S228D (phospho-mimetic and non-phosphorylatable) mutants28,40,41 in our minimal PINK1/Parkin synthetic circuit (Figures 3B and S4A).
Figure 3. PINK1 autophosphorylation is not necessary for emergence of PINK1 input threshold or input-reciprocal delay behaviors.
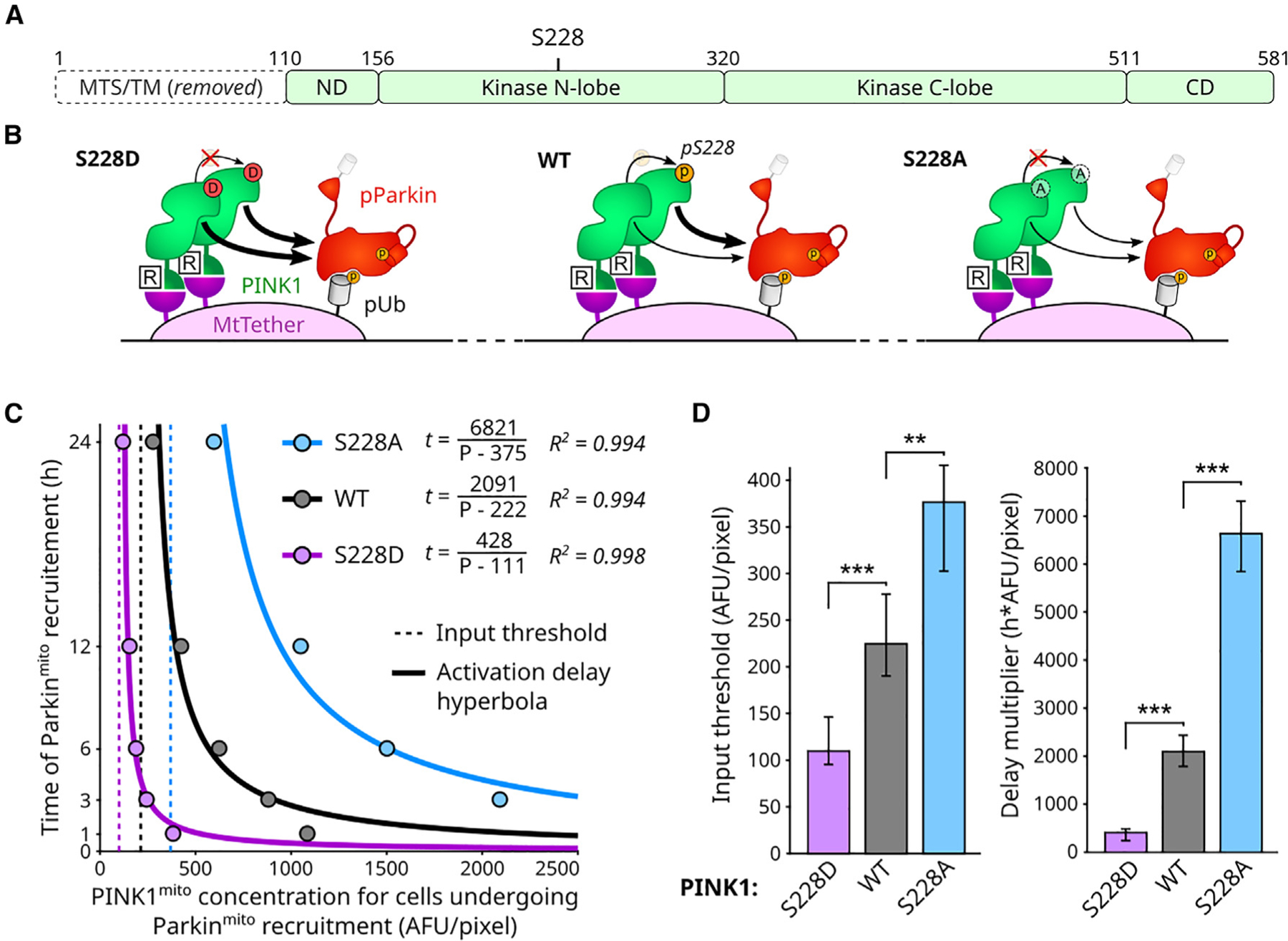
(A) PINK1 domain map. MTS and transmembrane (TM) domain replaced by FKBP domain (Figure 1C). ND: N-terminal domain. CD: C-terminal domain. S228: primary functional PINK1 autophosphorylation site.
(B) Illustration of S228 mutant consequences. S228D: phosphomimicking and non-phosphorylateable. S228A: non-phosphorylateable.
(C) Effects of PINK1 S228 mutations on circuit behavior. Points: aggregate quantification from fixed-cell measurements in Figure S4B.
(D) Statistical analysis of input threshold and delay scaler values for data in (C). Mean, 95% confidence intervals, and statistical significance calculated by bootstrap analysis (B = 10,000; STAR Methods). Bonferroni multiple comparison adjustment. **p < 0.01; ***p < 0.001. See also Figure S4.
We found that the S228A PINK1 mutation caused an increase in both the PINK1 input threshold and delay multiplier, while the S228D mutation caused a decrease in both circuit properties (Figures 3C, 3D, and S4B). S228A PINK1’s retained ability to recruit Parkin (Figure S4B) suggests that autophosphorylation of PINK1 at S228 is not necessary for recruitment of Parkin in our system. The opposing directions of S228A- and S228D-dependent changes (Figures 3C and 3D) support previous observations that PINK1 autophosphorylation at S228 promotes efficient Parkin recruitment.28,40,41 The two mutants define extremes for the range of how autophosphorylation at S228 can affect circuit behavior in our system (Figures 3C and 3D). However, the existence of quantifiable PINK1 input threshold and input-reciprocal delay behaviors at these extremes (Figure 3C) shows that S228 phosphorylation is not necessary for producing these behaviors. Taken together, these data demonstrate that PINK1 S228 autophosphorylation in trans may tune the PINK1 input threshold, and the input-reciprocal delay for Parkin recruitment yet is not necessary for producing these circuit behaviors.
Parkin activation dynamics tune PINK1 input threshold behavior
To further explore how the observed circuit behaviors could arise, we next used previously characterized Parkin mutants (Figure 4A) to rationally perturb Parkin activation dynamics (Figures 4B and S4A). Parkin becomes active and enables positive feedback in the PINK1/Parkin circuit by undergoing a series of dramatic pUb-dependent and PINK1-dependent domain rearrangements (Figure 4B).19–23,42,43 We introduced five Parkin mutations—S65A, K161N, R104A, W403A, or N273K—in our synthetic circuit to alter the kinetics of these domain rearrangements. S65A blocks PINK1’s ability to phosphorylate Parkin’s UBL domain.18 S65A, K161N, and R104A are each expected to decrease Parkin’s catalytic activity by reducing the ability of Parkin’s phosphorylated UBL domain to displace and free Parkin’s catalytic RING2 domain19–21,44 (Figures 4B and S4A). Conversely, W403A is expected to increase Parkin’s catalytic activity by weakening autoinhibitory REP and RING2 interactions and thus allowing Parkin to spend a higher proportion of time in its active conformation16,19,21–23,43,44 (Figures 4B and S4A). (We note that while unphosphorylated W403A Parkin exhibits some baseline catalytic activity16,21,22,44 [Figure 4B, inset], phosphorylation of W403A Parkin by PINK1 is still required to fully activate the mutant.43) Finally, N273K increases Parkin’s affinity for pUb by 10-fold by altering allosteric competition between UBL and pUb binding22,23 (Figures 4B and S4A).
Figure 4. Circuit threshold and delay behaviors are differentially affected by mutations affecting Parkin activation dynamics.
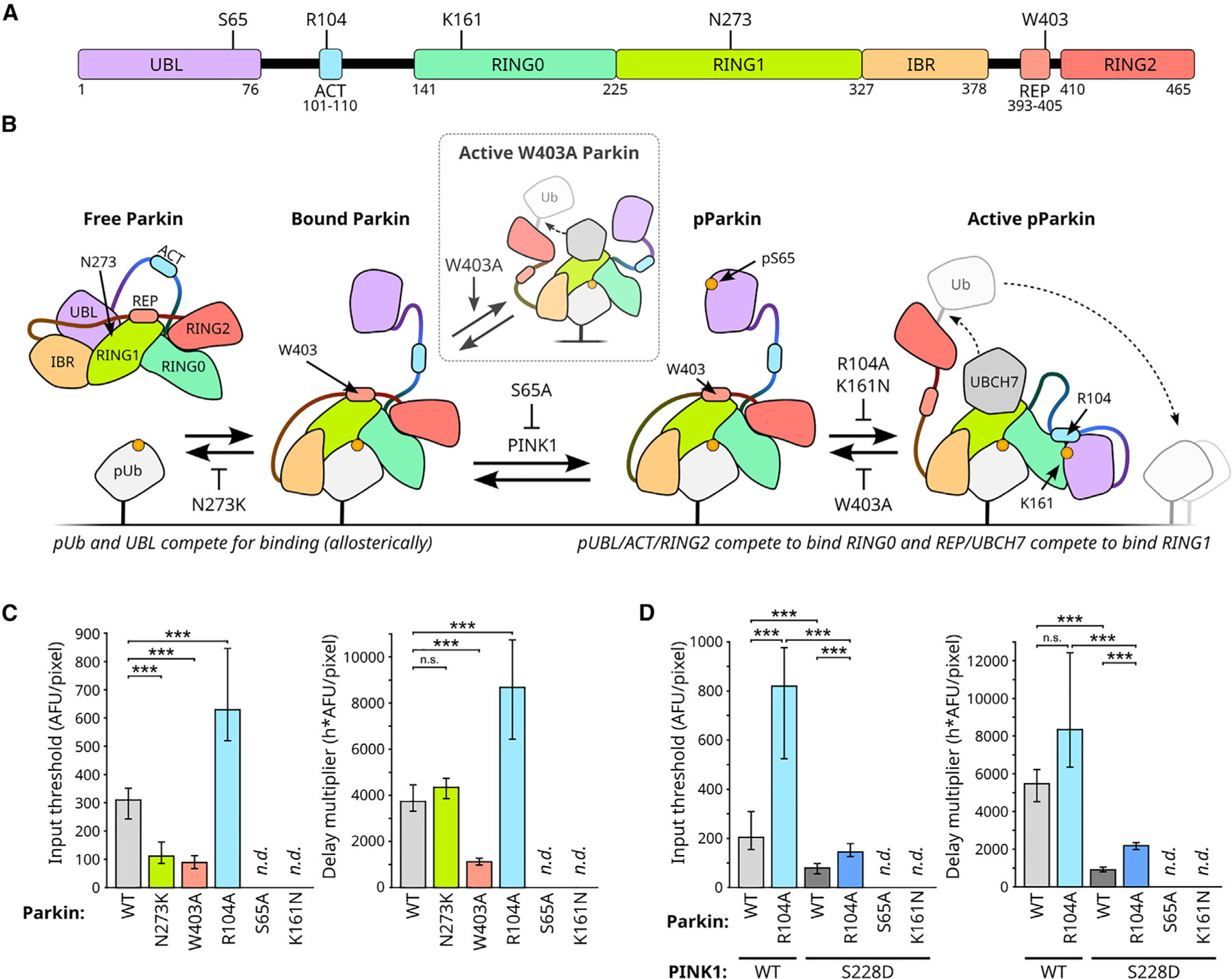
(A) Parkin domain map with location of mutations. UBL: ubiquitin-like. ACT: activating. REP: repressive. Four RING-like domains: RING0, RING1, IBR (in between RING), and RING2.
(B) Effect of selected Parkin mutations on the competition-driven domain rearrangements during parkin activation. UBCH7: an E2 Ub ligase responsible for charging Parkin.23 Inset: unphosphorylated W403A Parkin is partially active. T-bars: repression. For effects in context of full circuit, see Figure S4A.
(C) Effect of Parkin mutations on PINK1 input threshold and reciprocal activation delay. n.d.: not determined due to lack of Parkinmito recruitment. Means, 95% confidence intervals, and statistical significance calculated by bootstrap analysis (B = 10,000; STAR Methods) of fixed cell populations in Figure S4C. Bonferroni multiple comparison adjustment. ***p < 0.001; n.s.: not significant.
(D) S228D PINK1 rescue of R104A Parkin. Representation and analysis as in (C). Single-cell data is shown in Figure S4E. See also Figure S4.
First, S65A and K161N mutations completely blocked Parkin recruitment (Figures 4C, S4C, and S4D). The lack of recruitment of both S65A Parkin and K161N Parkin supports prior observations that positive feedback via parkin activation is critical for activation of the PINK1/Parkin circuit.11,19,22,42 Second, the R104A mutation produced an increased PINK1 input threshold and an increased delay multiplier (Figures 4C, S4C, and S4D), while the activating W403A mutation produced a decrease in both circuit properties (Figures 4C, S4C, and S4D). These opposing effects of the R104A and W403A suggest that Parkin’s activity contributes to generating threshold and delay behaviors in the positive feedback circuit.
Finally, and most interestingly, the N273K mutation decreased the PINK1 input threshold but did not affect the delay multiplier (Figures 4C, S4C, and S4D). This decreased PINK1 input threshold (Figure 4C) suggests that the strength of Parkin’s association with pUb is a critical contributing factor to the PINK1 input threshold behavior (intuitively, increasing Parkin’s affinity for pUb leads to lowered input threshold). The unchanged delay scaler (Figure 4C) demonstrates that the PINK1 input threshold and the input-reciprocal delay can be modulated independently, a property that was not initially obvious. The observation that the delay multiplier was affected by mutations altering Parkin’s activity (W403A and R104A) but not by N273K (Figure 4C) suggests that the timing of circuit activation is primarily influenced by positive feedback in the PINK1/Parkin circuit. Taken together, these results suggest that the PINK1 input threshold is tuned by multiple factors—including Parkin’s binding affinity for pUb and Parkin’s ability to drive positive feedback—while the circuit’s activation delay is dominated by the effect of positive feedback.
To explore the functional interplay between PINK1 activity and Parkin activity, we assessed whether the hyperactive S228D PINK1 mutant could rescue activity defects in the S65A, K161N, or R104A Parkin mutants. The lack of S65A and K161N Parkin recruitment was not rescued by S228D PINK1 (Figure S4E) suggesting a profound activity defect in these Parkin mutants. Conversely, WT and R104A Parkin’s PINK1 input threshold and delay scaler were both decreased for the S228D PINK1 condition with R104A Parkin retaining a higher PINK1 input threshold and delay multiplier compared to WT Parkin (Figures 4D, S4E, and S4F). Thus, PINK1 and Parkin activities cooperate to tune the PINK1 input threshold and delay multiplier. Because each round of positive feedback in the PINK1/Parkin circuit requires contributions by both PINK1 and Parkin (Figure 1A), our results suggest that the identified behaviors arise from the circuit’s positive feedback topology.
Input-coupled positive feedback is sufficient to produce two-factor authentication
To test whether the positive feedback topology of the PINK1/Parkin circuit is sufficient to produce the observed PINK1 input threshold, we investigated mathematical models based on the circuit. A defining feature of the PINK1/Parkin circuit is that it has an ‘‘input-coupled’’ positive feedback topology: the input is continually required for positive feedback, and increased input increases the net rate of feedback. A ‘‘minimal’’ model with this circuit topology is the conversion of a substrate to that requires the input of an enzyme and itself (Figure 5A). Here, abstracts the enzyme PINK1, abstracts the pools of free Ub and free Parkin, and abstracts the pools of pUb and pParkin.
Figure 5. Input threshold and reciprocal activation delay properties arise within a minimal model of input-coupled positive feedback.
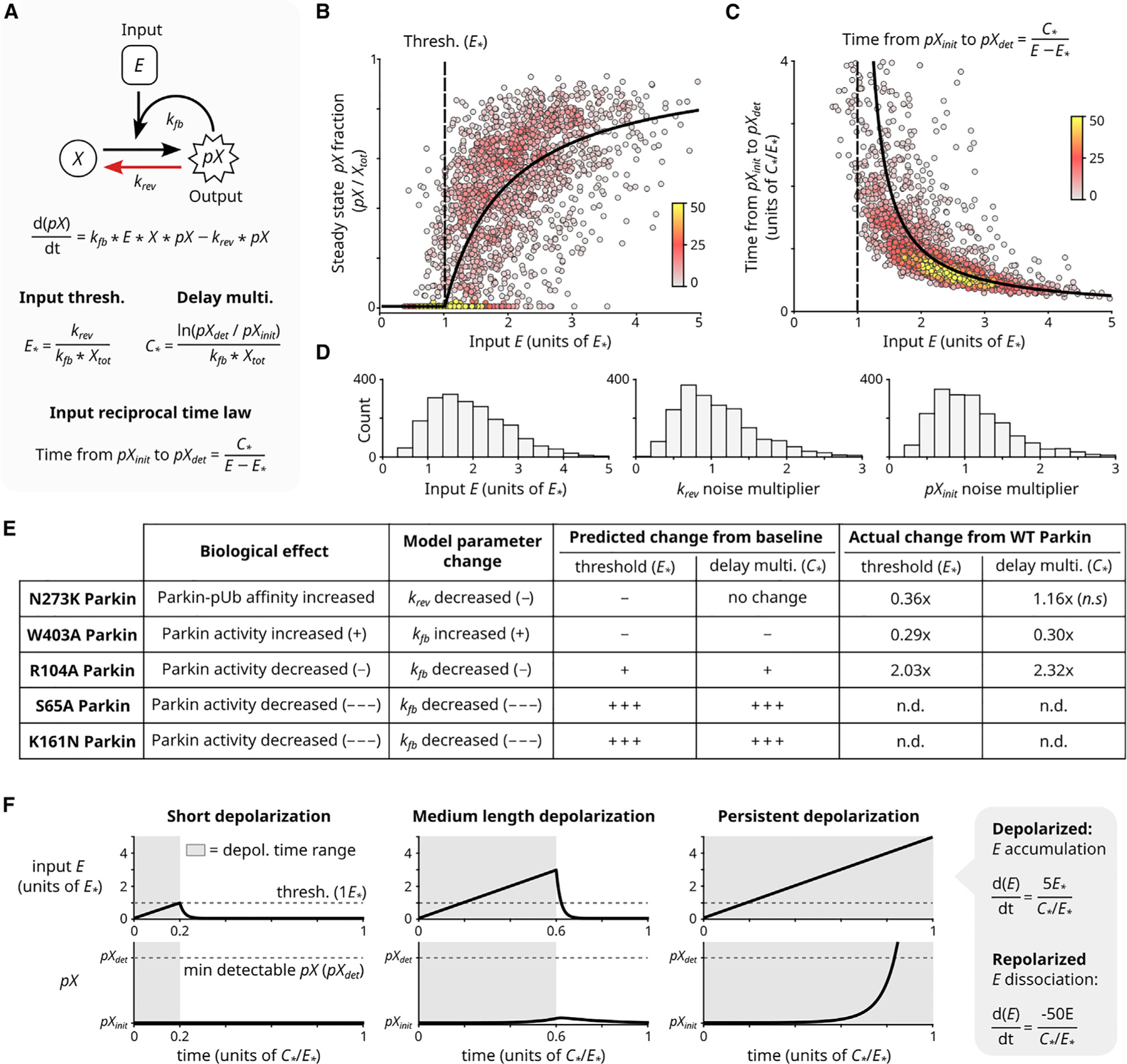
(A) Minimal model of input-coupled positive feedback. : total concentration of (note ). : initial concentration of (note ). : detection concentration of (note ). Parameters and govern input threshold and reciprocal activation delay, respectively. Derivations and model generalization are described in Method S1.
(B–D) System steady-state analysis (B) and relationship between input and time to reach detectable output levels (C). Black curves: algebraic solutions of (A). Points: simulated cell heterogeneity; 2000 cells were simulated, each with a randomly selected value of and randomly selected multiplier for , and . Distributions of randomly selected values are shown in (D) (STAR Methods). Colors: local point density. For analogous experimental data, see Figure S2I and Figure 2C.
(E) The minimal model algebraically predicts experimentally observed effects of Parkin mutations on circuit’s input threshold and reciprocal activation delay behavior (from Figure 4C).
(F) The minimal model can filter out simulated transient depolarization events. Simulated depolarization (and accumulation) for varying lengths of time, followed by repolarization (and dissociation). Rates of accumulation and dissociation are noted (STAR Methods). Short depolarization yields no (i.e., ). Medium depolarization yields negligible (i.e., ). Only sustained depolarization yields detectable (i.e., ). See also Figure S5.
The minimal model can be described by a simple ordinary differential equation and solved analytically (Figure 5A; Method S1). In striking agreement with our experiments, this model displays both a defined input threshold, below which the circuit will not activate, and a hyperbolic law inversely relating input concentration and delay for to reach an arbitrary minimum detection level (e.g., experimentally by our microscope or biologically by downstream mitophagy components) from its initial level (Figures 5B and 5C, analytical curve, and S5A–S5E; Method S1; STAR Methods).
We stress-tested behaviors of the minimal model. First, we found that the input threshold and input-reciprocal delay behaviors emerged from a broad class of generalized models for input-coupled positive feedback, including a detailed model of the PINK1/Parkin circuit with free, bound, and phosphorylated states of both Ub and Parkin (Figures S5F and S5G; Method S1). Second, experimentally observed circuit response heterogeneity could be qualitatively recapitulated by varying single-cell parameters in the minimal mathematical model (Figures 5B–5D, points; STAR Methods). Third, experimentally observed effects of Parkin mutants could be predicted from the analytical expression of threshold and delay (Figure 5E). Finally, we investigated responses of the circuit to transient input fluctuations using numerical simulations (experimentally, the rapalog system is poorly suited to test these properties; dissociation half-life, t1/2 > 2hours). These simulations demonstrated that the emergent circuit threshold and delay behaviors—integrating the level and duration of a PINK1 input—can provide a mitophagy fate decision mechanism to discern between ‘‘false alarms’’ and severe mitochondrial damage (Figure 5F). These simulation results agree with published data showing that circuit activation is readily reversed upon mitochondrial repolarization (Figure 5F, middle).7,26,32 Together these analyses show that the input-coupled positive feedback topology of the PINK1/Parkin pathway is sufficient to generate the regulatory behaviors of input threshold and input-reciprocal circuit activation delay—a conclusion that was not immediately obvious from our initial understanding of the circuit.
DISCUSSION
This study reveals an intrinsic regulatory mechanism by which the PINK1/Parkin positive feedback circuit can avoid errant activation. We used a minimal synthetic circuit approach to directly map circuit inputs (mitochondrial PINK1 concentration) to circuit activation responses (Parkin recruitment) at single-cell resolution. This approach revealed two modes of regulation: (1) that circuit activation can occur only when mitochondrial PINK1 concentrations exceed a threshold value, and (2) that once above this PINK1 input threshold, appreciable circuit activation is delayed by a time inversely proportional to the mitochondrial PINK1 concentration. Furthermore, a combination of mutational and mathematical analyses revealed that these two regulatory behaviors arise from input-coupled positive feedback, a defining feature of the PINK1/Parkin positive feedback circuit topology.
Prior work has proposed mechanisms that could give rise to input threshold and input-reciprocal delay behaviors in the PINK1/Parkin positive feedback circuit.11,13,24,26,28–31 Initial interpretation of the circuit’s topology as a series of feedforward loops led to speculation that a feedforward-based delay could prevent circuit activation by transient PINK1 association.11,24,26 Recent work has further speculated that a relationship between PINK1 levels and the probability to activate this feedforward mechanism could also produce a PINK1 input threshold.29 Here, we propose input-coupled positive feedback as an alternative conceptual framework for understanding the role of positive feedback in the circuit’s topology. Intuitively, input-coupled positive feedback reframes the serialized feedforward loops into a positive feedback format. Thus, our findings that the PINK1 input threshold and input-reciprocal delay exist and that they emerge from the circuit’s topology confirm prior intuition about emergent behaviors of the PINK1/Parkin circuit.
Our mathematical modeling suggests the input threshold and input-reciprocal delay behaviors are intrinsic properties of the PINK1/Parkin circuit’s organization. However, contextual factors could affect whether an input threshold may be experimentally observable for the endogenous circuit. Activation of the endogenous circuit by mitochondrial depolarization is promoted both by secondary depolarization effects (reactive oxygen species formation, ATP depletion)45–47 and by full-length PINK1’s (fl-PINK1) localization to the TOM (translocon of the outer membrane) complex.13,28,32,48 In contrast, the synthetic circuit lacks both sensitizing effects because its activation does not involve mitochondrial depolarization32 and because the PINK fusion is not expected to interact with the TOM complex.32 The higher sensitivity of the endogenous circuit is reflected in the low, nearly undetectable, expression of endogenous PINK1 (Figure S6A). This low expression of endogenous fl-PINK1, coupled with a current lack of techniques for holding fl-PINK1 levels steady on depolarized mitochondria,26 hampers input-response mapping for the endogenous circuit. Evaluation of circuit threshold and delay behaviors in the endogenous context may require the development of new single-molecule analysis or in vitro reconstitution techniques.
PINK1 autophosphorylation in trans at S228 is not required for generating either the PINK1 input threshold or the input-reciprocal delay behaviors in our synthetic circuit. Furthermore, in the synthetic circuit, S228A PINK1 can recruit Parkin, suggesting that S228A PINK1 retains some kinase activity. This conclusion is supported by previous findings that Tribolium castaneum Parkin, with the equivalent S205A mutation, retains some kinase activity for Parkin’s UBL.28 However, previous work has also shown that S228A fl-PINK1 is not capable of recruiting Parkin or generating pUb in depolarization-based assays.28,48 One possible explanation for this discrepancy is that the relatively high expression level of PINK1 in the synthetic circuit could compensate for the activity defect of S228A PINK1. Additionally, it is possible that the focal concentration of fl-PINK1 may be limited by the 2:1 stoichiometry of fl-PINK1’s interaction with the TOM complex on depolarized mitochondria.48,49 The combined kinase activity of just two S228A (or unphosphorylated) fl-PINK1 molecules bound to a single TOM complex might be insufficient to overcome the PINK1 input threshold on a local scale. Thus, PINK1 autophosphorylation could play a role in overcoming the circuit’s PINK1 input threshold on depolarized mitochondria despite not being necessary in the synthetic circuit.
Our results suggest that PINK1 activity and Parkin activation dynamics play a large role in tuning the emergent behaviors of the PINK1/Parkin circuit. Antagonistic enzymes could also contribute to tuning these behaviors. Indeed, we found that pharmacological inhibition of the mitochondrial deubiquitinase USP3011,50–53 slightly decreased both the PINK1 input threshold and delay multiplier (Figures S6B–S6E). The magnitude of the observed effect is consistent with prior work showing that USP30 knockdown only modestly increases parkin recruitment to chemically depolarized mitochondria13 possibly due to USP30’s low activity for pUb.31 However, the presence of a tuning effect suggests that modulation of emergent behaviors by antagonistic factors is at least possible. Other antagonistic factors of interest include cytoplasmic deubiquitinases54 and the putative ubiquitin phosphatase PTEN-L.55 Further exploration of how such enzymatic antagonists tune circuit behaviors may aid in identifying pharmacological approaches for modulating PINK1/Parkin circuit dynamics.
Finally, in addition to producing noise-tolerant circuit activation profiles, input-coupled positive feedback intrinsically provides control over where amplification can occur. For example, because the presence of input is required to maintain positive feedback, PINK1/Parkin circuit activation is not expected to spread to healthy mitochondria where PINK1 is not present at activating levels.3,5,6,56 This contrasts with a more classic form of positive feedback in which the input is decoupled from the feedback rate and for which spatially constrained activation may be lost without additional considerations.57,58 Beyond mitophagy, there are other systems where input-coupled positive feedback may operate to produce robust local responses. In EGF receptor (EGFR) signaling, an EGFR-driven positive feedback loop involving GAB1, PI3K, and PIP3 matches the topology of an input-coupled positive feedback circuit.59,60 A similar positive feedback loop also exists for insulin receptor signaling via IRS, PI3K, and PIP3.60 Taken together, our work motivates future studies into how input-coupled positive feedback may contribute to cellular decisions by balancing signal amplification sensitivity, noise tolerance, and spatial cues.
Limitations of the study
In summary, this study has several limitations. To hold mitochondrial PINK1 levels steady over time, and to facilitate live-cell imaging, a synthetic circuit composed of overexpressed components was used. Further study of the physiological relevance of the PINK1 input threshold and input-reciprocal delay may require the development of novel single-molecule imaging or in vitro reconstitution methodologies. Additionally, the slow dissociation rate of the PINK1-MtTether interaction in the synthetic circuit prevented us from examining the circuit’s response to pulses of mitochondrial PINK1. The use of an optically controlled dimerization system could enable future work to examine circuit responses to transient stimuli and to explore potential hysteresis behaviors of the circuit.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lani F. Wu (Lani.Wu@ucsf.edu).
Materials availability
Plasmids generated in this study will be shared by the lead contact upon request.
Plasmids containing the mNeonGreen domain additionally require an mNeonGreen user license from Allele Biotechnologies.
Data and code availability
Single-cell microscopy quantification data has been deposited at Zenodo and are publicly available as of the date of publication. Sequence maps of plasmids generated for this study have been deposited at Zenodo and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
Original analyis code has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Parkin | Abcam | Cat# ab77924; RRID: AB_1566559 |
| Rabbit monoclonal anti-phospho-Ubiquitin | CST | Cat# 70973; RRID: AB_2799795 |
| Rabbit monoclonal anti-PINK1 | Abcam | Cat# ab216144; RRID: AB_2927726 |
| Rabbit monoclonal anti-pS6(pS235/236) | CST | Cat# 4856; RRID: AB_2181037 |
| Rabbit monoclonal anti-S6 | CST | Cat# 2217; RRID: AB_331355 |
| Rabbit monoclonal anti-GAPDH | CST | Cat# 5174; RRID: AB_10622025 |
| Mouse monoclonal anti-GAPDH | CST | Cat# 97166; RRID: AB_2756824 |
| Rabbit monoclonal anti-TOM20 Alexa Fluor 488 | Abcam | Cat# ab205486; RRID: AB_2943509 |
| Rabbit monoclonal anti-TOM20 Alexa Fluor 647 | Abcam | Cat# ab209606; RRID: AB_2934123 |
|
Biological samples | ||
| Healthy adult human brain tissue lysate (whole) | Novus Biologicals | Cat# NB820–59177 |
|
Chemicals, peptides, and recombinant proteins | ||
| Geneticin (G418 Sulfate) | Life Tech. Corp. | Cat# 10131035 |
| Doxycycline Hydrochloride | Sigma Aldrich | Cat# D3072 |
| SNAP-Cell 647-SiR | NEB | Cat# S9102 |
| Hoechst 33342 | Invitrogen | Cat# H2570 |
| carbonyl cyanide 3-chlorophenylhydrazone (CCCP) | Sigma Aldrich | Cat# C2759 |
| DMSO | Sigma Life Science | Cat# D2650 |
| Rapalog (A/C heterodimerizer; AP21967) | Takara Bio. | Cat# 635055 |
| CMPD39 (USP inhibitor 18) | MedChem Express | Cat# HY-141659 |
| Rapamycin | Sigma Aldrich | Cat# R0395 |
| Halt protease and phosphatase inhibitor cocktail | ThermoFisher | Cat# 78440 |
| Polybrene | EMD Millipore | Cat# TR-1003-G |
|
Critical commercial assays | ||
| Bio-Rad protein assay dye reagent concentrate (Bradford assay kit) | Bio-Rad | Cat# 5000006 |
| Mini-protean TGX 4–20% SDS-PAGE gels | Bio-Rad | Cat# 4561093 |
| Lipofectamine 3000 kit | Invitrogen | Cat# L3000–015 |
| NEBuilder HiFi DNA assembly master mix kit | NEB | Cat# E2621S |
| Q5 Site-directed mutagenesis kit | NEB | Cat# E0554S |
|
Deposited data | ||
| Plasmid maps, single-cell microscopy quantification data, and analysis code | This manuscript | Zenodo: https://doi.org/10.5281/zenodo.8356580 |
|
Experimental models: Cell lines | ||
| Human: HeLa cell line (female) | UCSF cell culture and banking core | Cat# CCLZR030; RRID: CVCL_0030 |
| Human: HEK293T cell line (female) | UCSF cell culture and banking core | Cat# CCLZR076; RRID: CVCL_0063 |
|
Oligonucleotides | ||
| mNeonGreen cDNA | Shaner et al.61 | Allele Biotech Cat# ABP-FP-MNEONSB |
|
Recombinant DNA | ||
| pCMV-Parkin | This manuscript | N/A |
| pCMV-Parkin(E16A) | This manuscript | N/A |
| pCMV-mKO2-Parkin(M1) | This manuscript | N/A |
| pCMV-mKO2-Parkin(M1L) | This manuscript | N/A |
| pCMV-Parkin-A92mKO2 | This manuscript | N/A |
| pCMV-Parkin(C431N)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(S65A)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(R104A)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(K161N)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(N273K)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(W403A)-A92mKO2 | This manuscript | N/A |
| pLv-CMV(trunc)-Parkin-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-SNAP-FRB-FIS1(MTS) | This manuscript | N/A |
| pLv-TetOn-FKBP-PINK1(110–581)-mNeonGreen (Neo) | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(G386A) (110–581)-mNeonGreen | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(S228A) (110–581)-mNeonGreen | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(S228D) (110–581)-mNeonGreen | This manuscript | N/A |
| pCMV-mCherry-Parkin | Narendra et al.5 | Addgene Plasmid #23956 |
| pLL3.7m-Clover-Geminin(1–110)-IRES-mKO2-Cdt(30–120) | Bajar et al.62 | Addgene Plasmid #83841 |
| pLV-EF1a-IRES-Puro | Hayer et al.63 | Addgene Plasmid #85132 |
| pSNAPf | NEB | N9183S |
| pC4-RhE-FRB-Fis1 | Narendra et al.64 | Addgene Plasmid #68056 |
| pC4M-F2E-GFP-FKBP | Narendra et al.64 | Addgene Plasmid #68058 |
| pCMVTNT-PINK1-C-Myc | Beilina et al.65 | Addgene Plasmid #13314 |
| pLv-TetOn-MCS1-P2A-MCS2 (Neo) | Adam Karpf Lab | Addgene Plasmid #89180 |
| pLv-TetOn-FKBP-PINK1 (110–581)-mNG-Neo | This manuscript | N/A |
| pMD2.G | Didier Trono Lab | Addgene Plasmid #12259 |
| pCMV-dR8.91 | NovoPro | Cat# V007548 |
|
Software and algorithms | ||
| MATLAB | https://www.mathworks.com/ | N/A |
| Fiji | Schindelin et al.66 | https://imagej.net/software/fiji/ |
| filt2 MATLAB function | Greene67 | https://www.mathworks.com/matlabcentral/fileexchange/61003-filt2-2d-geospatial-data-filter |
| UCSF ChimeraX software | Pettersen et al.68 | https://www.cgl.ucsf.edu/chimerax/ |
| WebLogo: A sequence Logo generator | Crooks et al.69 | https://weblogo.berkeley.edu/logo.cgi |
| EMBL-EBI Clustal Omega multiple sequence alignment tool | Madeira et al.70 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines and culture conditions
HeLa cells (female, RRID: CVCL_0030) were cultured with a base media consisting of RPMI (Thermo Fisher Scientific, 11875119) with 10% fetal bovine serum (Gemini Bio., #100–106) and 1x antibiotic/antimycotic (Thermo Fisher Scientific, 15240062). As with previous mitophagy studies, HeLa cells were chosen as a cell model for this study, because they lack endogenous Parkin expression.7,35 HeLa cells were obtained from the UCSF Cell and Genome Engineering Core (CCLZR205). Cells were kept at 37C and 5% CO2 during culturing and during live-cell microscopy.
HEK293T cells (female, RRID: CVCL_0030) were cultured for lentivirus generation with a base media consisting of DMEM (Thermo Fisher Scientific, 11965092) with 10% fetal bovine serum and 1x penicillin/streptomycin (Corning, 30–002-CI). HEK293T cells were obtained from the UCSF Cell and Genome Engineering Core (CCLZR076). Cells were cultured at 37C and 5% CO2.
METHOD DETAILS
Plasmid generation
Plasmids for expressing variants of tagged Parkin: untagged Parkin (pCMV-Parkin); Untagged Parkin with E16A mutation (pCMV-Parkin(E16A)); N-terminally tagged wildtype Parkin (pCMV-mKO2-Parkin(M1)); N-terminally tagged Parkin with preserved M1L substitution (pCMV-mKO2-Parkin(M1L)), internally tagged Parkin (pCMV-Parkin-A92mKO2), and internally tagged ligase dead Parkin (pCMV-Parkin(C431N)-A92mKO2). Listed Parkin expression plasmids were derived from the pCMV-mCherry-Parkin (Addgene, #23956)5 which harbors an M1L substitution in Parkin. The mKO2 insert was derived from pLL3.7m-Clover-Geminin(1–110)-IRES-mKO2-Cdt(30–120) (Addgene, #83841).62
The backbone from pLv-EF1a-IRES-Puro (Addgene, #85132)63 was used to create pLv-EF1a-Parkin-A92mKO2 (lentiviral stable expression vector for Parkin), pLv-EF1a-SNAP-FRB-FIS1(MTS) (lentiviral stable expression vector for MtTether-SNAP) and pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen (plasmid used for transient expression of PINK1 by lipofectamine transfection). The SNAP tag insert was derived from pSNAPf (NEB, N9183S). The FRB-Fis1 insert was derived from pC4-RhE-FRB-Fis1 (Addgene, #68056).64 The FKBP insert was derived from pC4M-F2E-GFP-FKBP (Addgene, #68058),64 the PINK1(110–581) insert, lacking the 1–109 mitochondrial targeting region, was derived from pCMVTNT-PINK1-C-Myc (Addgene, #13314)65 and the mNeonGreen insert was obtained as cDNA (Allele Biotech, ABP-FP-MNEONSB).61 The lentiviral doxycycline-inducible PINK1 expression vector, pLv-TetOn-FKBP-PINK1(110–581)-mNeonGreen (Neo), was created using the backbone from pLv-TetOn-MCS1-P2A-MCS2 (Neo) (Addgene, #89180).
Plasmids were assembled using standard cloning methods. Point mutations were added to expression plasmids for variants of tagged Parkin using a NEBuilder HiFi DNA Assembly Master Mix kit (NEB, E2621S). Point mutations were added to pLv-EF1a-Parkin-A92mKO2 and pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen using a Q5 site-directed mutagenesis kit (NEB, E0554S). The truncated CMV, low expression, plasmid for Parkin (pLv-CMV(trunc)-Parkin-A92mKO2) was made by 1) using the NEBuilder HiFi kit to remove the first 408 of 508 bases of the CMV enhancer/promoter region in pCMV-Parkin-A92mKO2, and 2) cloning the resulting partCMV-Parkin-mKO2 insert into the pLV-EF1a-IRES-Puro backbone. The CMV(trunc.) promoter has the sequence ‘CCAAAATCAA CGGGACTTTC CAAAATGTCG TAACAACTCC GCCCCATTGA CGCAAATGGG CGGTAGGCGT GTACGGTGGG AGGTCTATAT AAGCAGAGCT’. All plasmids were grown following transformation into NEB STBL competent E. coli (NEB, C3040H). All inserts were verified via sanger sequencing (Elim Biopharmaceuticals).
Lentiviral transduction of cell lines
Lentivirus was made for pLV-EF1a-SNAP-FRB-FIS1(MTS), pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen, pLv-EF1a-Parkin-A92mKO2, and pLv-EF1a-Parkin-A92mKO2 mutants, using HEK293T cells, pMD2.G (Addgene, 12259), pCMV-dR8.91 (NovoPro, V007548), and a lipofectamine 3000 kit (Invitrogen, L3000–015). Lentivirus was tittered for 10% transduction efficiency with 8ug/mL polybrene (EMD Millipore, TR-1003-G) in growth media.
Triple-positive (MtTether + Parkin + dox-inducible PINK1) stable cells, used in initial synthetic circuit experiments, were made by co-transducing cells with virus made from pLV-EF1a-SNAP-FRB-FIS1(MTS), pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen and pLv-EF1a-Parkin-A92mKO2. Triple positive cells were enriched as follows: TetOn-PINK1-mNG expression cassette was selected for by treatment with 400ug/mL Geniticin (Life Tech. Corp., 10131035), PINK1 expression was induced with 50 ng/mL Doxycycline Hydrochloride (Sigma-Aldrich, D3072), MtTether was stained with 1:2000 SNAP-647 SiR (NEB, s9102) for 30 min, and positive cells were isolated with fluorescence-activated cell sorting (FACS). Following FACS, triple positive cells were allowed to expand and recover in the absence of doxycycline, to eliminate PINK1 expression prior to being frozen for storage.
Double-positive (MtTether + Parkin) HeLa cells, used for PINK1 and Parkin mutant studies, were generated in the same manner but were not transduced with the dox-inducible PINK1 expression cassette. PINK1 was instead expressed in these double-positive HeLa cells by transient transfection of pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen to allow for a greater degree of technical flexibility.
Immunofluorescence
Immunofluorescence and live-cell imaging experiments were performed in 96 well glass bottom, tissue culture treated imaging plates (Thermo Scientific Nunc, 164588). Cells were fixed in PBS (Invitrogen, AM9625) with 4% paraformaldehyde (Electron Microscopy Sciences, 15710) for 20 min at room temperature and were then permeabilized in PBS with 0.5% Triton X-100 (Sigma-Aldrich, 93443) for 20 min. Cells and then washed three times in PBS and blocked in PBS with 0.3% Triton X-100 and 3% Bovine Serum Albumin (BSA) (Fisher bioreagents, BP9703) for 1 h. Blocked cells were then incubated in staining buffer (PBS with 0.3% Triton X-100 and 1% BSA) with primary antibody overnight at 4C and then secondary antibody for 2 h at room temperature when applicable. Cells were stained with fluorescently conjugated primary antibodies for 2 h at room temperature when applicable. When a primary antibody and conjugated antibody of the same host species were used, staining with the conjugated antibody was done separately from and after staining with the secondary antibody to avoid cross-reactivity. Finally, cells were stained with 1:5000 hoechst 33342 (Invitrogen, H3570) in PBS for 10 min, and washed three times in PBS prior to imaging.
Primary antibodies: 1:500 mouse anti-Parkin (Abcam, ab77924); 1:1000 rabbit anti-phospho-Ubiquitin (CST, #70973); 1:1000 rabbit anti-Pink1 (Abcam, ab216144). Secondary antibodies: 1:1000 goat anti-mouse AF (Alexa Fluor) plus 647 (Invitrogen, A32728); 1:1000 goat anti-mouse AF 546 (Invitrogen, A11003); 1:1000 goat anti-rabbit AF plus 647 (Invitrogen, A32728). Conjugated primary antibodies: 1:1000 rabbit anti-TOM20 AF488 (Abcam, ab205486); 1:1000 rabbit anti-TOM20 AF647 (Abcam, ab209606).
Activity assessment of parkin tag variants
HeLa cells were grown in 96 well imaging plates. After 48 h of growth, individual wells were each transfected with 25ng of expression plasmid for one tagged Parkin variant using a Lipofectamine 3000 kit. Media on transfected cells was replaced after 3.5 h and cells were allowed to recover. Wells were treated with either 20uM carbonyl cyanide 3-chlorophenylhydrazone, (CCCP; Sigma Aldrich, C2759) or 2:5000 DMSO (Sigma Life Science, D2650) vehicle control for either 10 h or 30 min with timing such that the treatment ended and cells were fixed 48 h after transfection. Cells were fixed, permeabilized, subjected to immunofluorescence staining for Parkin (mouse anti-Parkin & goat anti-mouse AF647) & TOM20 (rabbit anti-TOM20 AF488), and imaged. Data was collected for three experimental replicates with two plate replicates each. This approach yielded measurements for approximately 60,000 cells per tag variant with DMSO treatment and 150,000 cells per tag variant with CCCP treatment.
Live-cell analysis of synthetic circuit
Triple- or double-positive HeLa cells were seeded in 96 well imaging plates. PINK1 expression was induced in triple-positive cells by treatment with 250 ng/mL doxycycline for 48 h. All growth, stain, wash, and imaging medias used for triple positive cells contained 250 ng/mL doxycycline. PINK1 expression was induced in double-positive cells by transient transfection of 25ng/well of pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen with Lipofectamine 3000. Media on transfected cells was replaced after 3 h and cells were allowed to recover for 48 h.
Prior to imaging, live cells were stained with 1:1000 SNAP-AF647-SiR for 30 min, stained with 1:1000 Hoechst 33342 for 10 min, and washed twice with fresh media for 20 min. Hoechst staining of live HeLa cells was observed to have no effect on HeLa cell viability or growth rate. Cells were imaged in growth media once (~30 min) to capture the pre-treatment state. Cells were then treated with 200nM rapalog (A/C heterodimerizer, previously AP21967) (Takara Bio., 635055) and immediately subjected to live-cell microscopy with images taken every 30 min for 12 h. For live-cell experiments longer than 12 h, imaging frequency was every 45 min. The exact time at which each image was taken was recorded for all experiments.
Synthetic circuit modulation via USP30 inhibitor
Triple positive HeLa cells were seeded, treated with Doxycycline for 48 h, stained, and imaged as with the live-cell imaging of the synthetic circuit described above. A 2-h pretreatment with 1uM CMPD39 (also called USP30 inhibitor 18; MedChem Express, HY-141659) was accomplished by addition to the stain, wash, and imaging medias. Vehicle treatment for CMPD39 was 1:10,000 DMSO.
Fixed-cell analysis of synthetic circuit
Double-positive or triple-positive HeLa cells were seeded as described above. PINK1 expression was induced as described for live-cell experiments above. Individual wells of the 96 well plates were treated with rapalog for treatment durations ranging from 1 h to 24 h. Treatments were started at different times such that all treatments were finished at 72 h following PINK1 expression induction. Cells were then fixed, subjected to immunofluorescence staining with rabbit anti-TOM20 AF647, and imaged.
Assessment of pUbiquitin by immunofluorescence
Double-positive HeLa cells were seeded, transfected to express Pink1, treated with rapalog for 3/6/12/24h, fixed, and permeabilized as described above. Fixed cells were stained with Hoechst and imaged to obtain initial images of Parkin and PINK1 fluorescent protein localization. Fluorescence of mKO2 and mNeonGreen was then bleached using hydrogen peroxide as described previously.71 In short, cells were incubated with a solution of 3% H2O2 with 20mM HCl in PBS for 2 h with exposure to bright light. During this time, the solution was replaced after 1 h. Complete bleaching of mKO2 and mNeonGreen was verified by imaging.
Cells were then subjected to immunofluorescence to detect phospho-Ubiquitin (pUb) and TOM20 using standard techniques as described above. Staining for pUb (rabbit anti-pUb and goat anti-rabbit AF647) was imaged in the Cy5 imaging channel to avoid any possibility of contamination by residual fluorescence from bleached fluorescent proteins. Staining for TOM20 (rabbit anti-Tom20 AF488) was imaged in the FITC channel.
Assessment of endogenous PINK1 expression levels
Parental HeLa cells and double-positive cells were seeded as above. PINK1 was expressed by transient transfection in double-positive, but not in parental, cells. PINK1 transfected cells were treated with rapalog for 3/6/12/24h. Parental untransfected cells were either left untreated or were treated with 20uM CCCP for 3h. Cells were fixed and permeabilized. Fluorescence of mKO2 and mNeonGreen was then bleached as above. Cells were subjected to immunofluorescence to detect PINK1, Parkin, and Tom20. Staining for PINK1 (rabbit anti-PINK1 and goat anti-rabbit AF647) was imaged in the Cy5 imaging channel to avoid any possibility of contamination by residual fluorescence from bleached fluorescent proteins. Staining for TOM20 (rabbit anti-Tom20 AF488) was imaged in the FITC channel. Staining for Parkin (mouse anti-Parkin & goat anti-mouse AF546) was imaged in the TRITC channel.
Microscope imaging parameters
Imaging for assessing activity of Parkin tag variants was performed on a PerkinElmer Operetta microscope, in confocal mode, using a 20× water objective. Experiments assessing the effect of Parkin mutants on the synthetic circuit were imaged on a PerkinElmer Phenix microscope, in confocal mode, using a 40× water objective. All other experiments were imaged on a PerkinElmer Operetta microscope, in confocal mode, using a 40× water objective. The Phenix microscope was used during an extended period when the Operetta microscope was inoperable.
To aid in rough comparison of PINK1 input threshold values across experiments, the imaging parameters for mNeonGreen used in each experiment are listed below. Imaging conditions for fixed cell experiments was changed from 1×1 binning to 2×2 binning after initial experiments to be more comparable with live-cell experiments. Resolution has a small effect on image quantification, meaning that only rough comparison between 1×1 and 2×2 binned experiments is possible, even after accounting for exposure length and intensity binning effects. All illumination light sources were used at 100% power. Imaging parameters are listed for experiments imaged on the Phenix microscope for completeness’ sake only and should not be used to compare results across microscopes.
All live-cell experiments were imaged on an Operetta microscope, with 2×2 pixel binning and a FITC exposure length of 1 s for mNeonGreen fluorescence. Initial fixed-cell circuit behavior quantification experiments as well as the G386A PINK1 mutant assessment were imaged on an Operetta microscope, with 1×1 pixel binning and a FITC exposure length of 10 s for mNeonGreen fluorescence. The fixed-cell PINK1 S228 mutant assessment and pUb IF comparison experiments were imaged on an Operetta microscope, with 2×2 pixel binning and a FITC exposure length of 4 s for mNeonGreen fluorescence. All fixed-cell Parkin mutant assessment experiments were imaged on a Phenix microscope with 2×2 pixel binning and a FITC exposure length of 1 s for mNeonGreen fluorescence.
Western blots
Cell pellets were collected and lysed in RIPA buffer (Sigma, R0278) with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 78440). Lysates were quantified via Bradford assay (BioRad, #5000006). Samples were run on Mini-Protean TGX 4–20% SDS-PAGE gels (Bio-Rad, 4561093). Western blotting was performed using standard approaches and according to antibody manufacturer recommendations. Odyssey blocking buffer (LI-COR Biosciences, 927–50000) diluted 1:1 with 1x TBS (Fisher scientific, BP2471–1) was used for all blocking steps and for antibody staining of S6 and pS6 blots. 3% w/v Milk (LabScientific, M0841) in 1x TBS with 1% Tween 20 (Fisher scientific, BP337–500) was used for antibody staining of Parkin blots. Western blots were imaged using an LI-COR Odyssey CLx infrared imaging system. MTOR signaling assessed via levels of pS6-S235/236, a constitutive downstream target of MTOR signaling.72 Western blot band intensities were quantified in Fiji.66 Rapamycin, (Sigma-Aldrich, R0395). Healthy human brain lysate, whole (Novus Biologicals, NB820–59177). Primary antibodies: 1:1000 rabbit anti-pS6 (Ser235/236; Cell Signaling Technologies, 4856), 1:1000 rabbit anti-S6 (Cell Signaling Technologies, 2217), 1:1000 mouse anti-Parkin (Abcam, ab77924), 1:1000 rabbit anti-GAPDH (Cell Signaling Technologies, 5174), and 1:1000 mouse anti-GAPDH (Cell Signaling Technologies, 97166). Secondary antibodies: 1:5000 goat anti-rabbit IRDye 800CW (LI-COR, 926–32211) and 1:5000 goat anti-mouse IRDye 680LT (LI-COR, 926–68020).
QUANTIFICATION AND STATISTICAL ANALYSIS
Microscopy image processing and cell segmentation
All microscopy image quantification and analyses were done in MATLAB. Images were first subjected to flatfield illumination correction using pre-generated objective-specific flatfield correction estimations. Uniform background fluorescence was then removed for each image. Finally, mild fluorescence crossover from the TRITC channel (mKO2) into the FITC channel (mNeonGreen) was corrected in a pixelwise manner using a multiply and subtract compensation approach. Crossover correction was empirically calibrated for each microscope objective and for each set of exposure times used in this study. Processed images were then subjected to an in-house watershed segmentation pipeline to identify nucleus and cytoplasm boundaries for each cell. A perinuclear region was also identified by removing the outer 25% of pixels from the edge of the cytoplasm region to remove small segmentation errors due to proximity to adjacent cells.
Single-cell quantification for parkin tag variants
For each segmented cell, foreground images were calculated for both the anti-Parkin and anti-TOM20 channels by applying a tophat filter with a radius of 8 pixels to each cell image. Co-localization of anti-Parkin and anti-TOM20 intensity was calculated as the pixel-wise Pearson correlation coefficient of the foreground images for those channels. For each cell, total anti-TOM20 intensity was calculated by identifying a mitochondrial mask (anti-TOM20 foreground image intensity above 2500 AFU) and then calculating the total pixel intensity of the original anti-TOM20 image within that mask.
To compare cells with similar Parkin expression levels, a sliding window approach was used for each construct and each condition. A sliding window was used to identify cells with similar Parkin expression levels, and then the median of a score (colocalization or TOM20 staining intensity) was calculated for the cells in that window. This approach estimated the given score for discrete Parkin expression levels across a range of discrete Parkin expression levels. Window center points were integer Parkin expression values ranging from 3000 to 20000 AFU/pixel. Window size was +/−850 AFU/pixel (representing 1/20 of expression range). Only cells with Parkin expression values between 3000 and 20000 AFU/pixel were used. Next, for each construct, DMSO-treated sliding median values were used to normalize the CCCP-treated sliding median values as described below. When comparing Parkin-TOM20 staining co-localization, the DMSO values at each Parkin expression level were subtracted from the corresponding CCCP-treated values at the corresponding expression level to estimate the change in localization at each parkin expression level. When comparing total cellular anti-TOM20 staining intensity, CCCP-treated values were instead divided by the corresponding DMSO-treated values to estimate the proportion of anti-TOM20 staining remaining at each parkin expression level. Finally, these normalized sliding medians were summarized by taking the mean value across Parkin expression levels. While all constructs were verified to have similar distributions of expression levels, this approach was used to factor out any potential Parkin expression-dependent effects which could theoretically skew the data.
Hypothesis testing was performed using a bootstrap approach. Random sampling with replacement of individual cell datapoints, followed by re-quantification of test metrics, was performed 10,000 times (B = 10,000). Due to all 6 Parkin tag/mutant variants (Untagged, Internal, Nterm-M1, Nterm-M1L, LD, and Untagged-E16A) being assessed in parallel for each CCCP treatment duration, a Bonferroni multiple comparison correction was used (number of comparisons, m = 5). Because the 10h and 30min CCCP treatment durations were separate experiments, they were not considered simultaneous comparisons.
Live-cell quantification for synthetic circuit
Following cell segmentation, foreground images and mitochondrial masks were calculated. Foreground images were calculated for all images by applying a 2D bandpass Gaussian filter (sequential high pass Gaussian with a wavelength of 50 pixels to remove background intensity and low pass Gaussian with wavelength of 3 pixels to reduce pixel noise) using the filt2 MATLAB function.67 A mitochondrial mask was calculated using the MtTether foreground image and a threshold that was calculated on a per-cell basis. This cell-specific threshold was calculated as the median pixel intensity of the original MtTether image raised to the exponent of 0.8 and multiplied by two. This approach was empirically designed to be both effective over a range of MtTether expression levels and at the pixel resolution of the live-cell images.
Next, a variety of cellular phenotypes (features) were quantified for each cell at each timepoint. Intensity co-localization features were calculated as the pixelwise Pearson correlation coefficients between pairs of foreground images. The concentration of PINK1 on mitochondria (shorthand: []) was estimated as the mean mNeonGreen intensity on mitochondria and was calculated as the mean pixelwise foreground intensity in the mitochondrial mask. Co-localization features and mitochondrial PINK1 features were calculated only in the perinuclear region described above to avoid intensity artifacts at cell edges. Expression level features for all channels were calculated as pixel intensity means from the original channel intensity images.
Following feature extraction, single cells were tracked from one timepoint to the next by finding the closest cell, in the next timepoint, both in distance and phenotypic similarity. This was accomplished using a distance metric defined as an empirically selected weighted sum of: the distance between nucleus centroids, the ratio of cell areas, the ratio of nucleus area, and the ratio of marker expression levels between candidate cells.
Only cells that were successfully tracked across all timepoints were used for downstream analyses. Additionally, outlier cells with high or low Parkin or MtTether expression were discarded using a threshold approach that removed approximately 5% of cells in each case. A threshold approach was also used to remove cells with high PINK1 expression levels due to masking of PINK1 recruitment to mitochondria at high expression levels. For the same reason, cells which did not show an increased co-localization between PINK1 and MtTether upon rapalog treatment were also discarded. Finally, cells with PINK1 expression levels that increased 1.5-fold or more over the course of the experiment were discarded.
Circuit inputs and responses from live-cell data
Synthetic circuit input and response estimates were quantified for each successfully tracked cell as follows. First, the maximum Parkinmito localization was calculated as the maximum observed co-localization value between the Parkin and MtTether channels for each cell. The maximum value was used because extended Parkinmito localization eventually caused mitochondrial degradation and an associated decrease in measured co-localization.
Next, the detection time of Parkinmito recruitment was determined as the time at which the co-localization of Parkin and MtTether reached a value of 0.4 for each cell. The value 0.4 was chosen because it is half-way between the values measured for cells with and without Parkinmito recruitment.
Finally, an estimate for the mean concentration over time was calculated. Due to technical noise in the measurement of mean PINK1 intensity on mitochondria, data were smoothed across timepoints using a moving mean with a window size of 5 timepoints for each cell. To avoid including timepoints without recruitment, or with intermediate, recruitment, smoothing did not include the pre-rapalog treatment timepoint or the timepoint immediately following rapalog treatment. The estimate for mean concentration over time was then calculated by taking the mean value of this smoothed data from when recruitment was first detected until either the time when Parkinmito was recruited (max Parkinmito localization >0.4) or the end of the experiment (max Parkinmito localization <0.4).
Global stability over time of PINK1 targeting to mitochondria was quantified by normalizing unsmoothed single-cell traces of mean PINK1 intensity on mitochondria to the time-averaged PINK1 concentration measurement of that cell. The mean and standard deviation of the normalized cell traces were then calculated across cells for each timepoint (Figure S2B).
Live-cell threshold and delay quantification
Synthetic circuit input threshold and reciprocal activation delay hyperbola were calculated from live-cell data as follows. First, a PINK1 input threshold value was calculated by: 1) taking a sliding window over the single-cell concentration measurements (overall window width of 5% of the population size); 2) calculating the percentage of cells within each window exhibiting Parkinmito recruitment (max Parkinmito localization >0.4, or, when applicable, max fraction of Parkin on mitochondria >0.1); and 3) identifying the center point of the first window with at least 50% of cells exhibiting Parkinmito recruitment. This method for calculating the PINK1 input threshold was used because it performed well with the relatively small number of datapoints obtained from live-cell experiments.
Next a hyperbolic curve was fit to the single-cell concentration measurements and the single-cell detection times of Parkinmito recruitment. This hyperbola was fit using a geometric distance, sum of squares, approach. Specifically, the ‘‘delay multiplier’’ in the following function was optimized to obtain a fit: ).
When comparing across conditions, hypothesis testing was performed using a bootstrap approach. Random sampling with replacement of individual cell datapoints, followed by re-quantification of the input threshold and delay multiplier values, was performed 10,000 times. A Bonferroni multiple comparison correction was used.
Single-cell quantification for fixed cells
Single cell quantification for fixed-cell synthetic circuit experiments was performed as follows. Following cell segmentation, foreground images were calculated as for live-cell images above. For experiments imaged with no pixel binning (initial fixed threshold experiment with WT PINK1 and WT Parkin), Gaussian bandpass wavelengths used were 100 pixels and 3 pixels. A Mitochondrial mask was calculated using the anti-TOM20 mitochondrial foreground image and a fixed threshold of 1000 AFU. This approach was empirically determined to perform better for the higher resolution of un-binned images. For 2×2 binned images, (PINK1 and Parkin mutant experiments), the foreground images and mitochondrial mask were calculated exactly as described above for live-cell images. For all experiments, features were calculated the same way as described above for live-cell experiments.
Finally, single-cell data quality control was performed. Mis-segmented cells (small cell areas, small nucleus areas, or high levels of Hoechst staining in the cytoplasm) were discarded. Outlier cells with high or low Parkin expression were discarded using a threshold approach as above. Cells with low anti-TOM20 staining were discarded using a threshold approach to remove cells that had undergone mitochondrial degradation at longer timepoints. Cells with high PINK1 expression levels were discarded as above. Cells with a low co-localization of PINK1 and anti-TOM20 staining (co-localization score <0.6) were discarded to ensure that only cells with verifiable were analyzed.
Fixed-cell threshold and delay quantification
Synthetic circuit input threshold and reciprocal activation delay hyperbola were calculated from fixed-cell data as follows. For each rapalog treatment duration, a variant of Otsu’s thresholding method was used to identify a concentration (mean intensity) value capable of separating the dataset into high and low Parkinmito localization populations. In short, an algorithm searched for a concentration separation value that split the dataset into high and low concentration groups with minimized within-group size-adjusted variance for Parkinmito localization. The separating value estimated the concentration for cells undergoing Parkinmito recruitment at that rapalog treatment duration, denoted here as []. Within this context, the rapalog treatment duration represents time of Parkinmito recruitment, allowing a hyperbolic curve to be fit to the data as with the live-cell experiments. The separating value was not calculated for conditions lacking high and low Parkinmito populations.
Next a hyperbolic curve was fit to the data using a geometric distance, sum of squares, approach. Specifically, the two variables ‘‘input threshold’’ and ‘‘delay multiplier’’ in the following function were concurrently optimized to obtain a fit: ).
When comparing across conditions (e.g., mutants), hypothesis testing was performed using a bootstrap approach. Random sampling of individual cell datapoints with replacement, followed by re-quantification of the input threshold and delay multiplier values, was performed 10,000 times. A Bonferroni multiple comparison correction was used.
Coloring datapoints by local point density
Local point density was calculated by counting the number of nearby points within a radius of 5% of the field of view in each direction (e.g., if the x axis range is [0.200], then the averaging field around a point x is x 10).
Plotting algebraic solutions of minimal math model
The behavior of the minimal model is described by the differential equation:
Properties of the minimal model are illustrated in Figure 5 and S5. For clarity, we make use of normalized units (noted on plot axes). The total amount of is given as . Analysis of model properties, including sources of and constants, is detailed in the Method S1.
In Figure S5B, the forward and backward rates of the model, given by and , respectively, are rewritten as and respectively. The forward and backward rates are plotted for varying values of , as multiples of .
In Figures 5B and S5C, algebraic solutions for the steady state () of as a function of are plotted as and (non-physical) negative values are set to zero.
In Figure S5D, the exponential growth of (over time, ), from an initial value to the value at which it is detected (Method S1), is plotted. The exponential growth function was rewritten as and plotted for varying values of , as multiples of . A value of was used for plotting.
In Figures 5C and S5E, the input reciprocal time law is rewritten as time from to and is plotted (Figures 5A and S5A; Method S1). All analytical curves were plotted with MATLAB.
Heterogeneity simulation for minimal math model
Heterogeneous circuit behaviors were simulated using the minimal model for input-coupled positive feedback as follows. To simulate heterogeneity, we simulated 2000 ‘‘cells’’. For each ‘‘cell’’, values for , , and were sampled as follows.
values were randomly sampled using a gamma distribution (shape parameter: 5.14; scale parameter: 0.38). The gamma distribution was empirically fit to the observed distribution of PINK1 input scores observed in Figure 2A using MATLAB’s ‘‘fitdist’’ function. Noise multiplier values for and for were randomly sampled from log normal distributions (mu: 0; sigma: 0.5).
The detection level, , was set to be , using the unmodified ( with a noise multiplier of 1). Using these parameters, the steady state and time to detectable activation for each ‘‘cell’’ were solved analytically and plotted with MATLAB.
Transient depolarization simulation
Transient depolarization was simulated using the minimal model for input-coupled positive feedback as follows. Values for as a function of time were calculated from the rate equations shown in Figure 5F. Rate of association and dissociation were chosen empirically to allow a time value of 1 (in units of to be roughly interpretable as 1 h. Values of were calculated, as a function of both time and value of , using the exponential growth function described above. A value of was used, as above.
UBL domain structure analysis
PDB accession numbers for structures shown in Figure S1 are as follows: hParkin, PDB: 5c1z; hUbiquitin, PDB: 1f9j; hRAD23B, PDB: 1uel; hNedd8, PDB: 1ndd; hHOIL1, PDB: 2LGY. Visualization and analysis were performed in the UCSF ChimeraX software.68 Hydrogen bonds and salt bridges were predicted using the show hydrogen bonds option in UCSF ChimeraX.
Conservation of ubiquitin-like domains
The amino acid sequence and the metadata for 67594 annotated UBL domains in the Uniprot database were downloaded (April 4, 2019). Analysis performed with MATLAB. Entries with annotation errors were identified and discarded as follows. UBL domain entries with non-standard residues (including “J”, “O”, “U”, “X” or “Z”) or with lengths of less than 65 amino acids were discarded. Domains annotated to start at the protein’s N terminus, but which did not start with methionine were discarded. Entries corresponding to Ubiquitin polymers rather than to UBL domains were identified as starting or ending with tandem glycines. These Ubiquitin entries were discarded to prevent over-representation in the dataset. Finally, any additional UBL domains originating from proteins containing at least one discarded domain were also discarded. This cleanup left 28447 UBL domain entries.
Alignment using traditional methods could not be used successfully due to the large number of sequences and high degree of sequence variability. Instead, the conserved positioning of specific residues in the stereotyped ubiquitin fold were used to align the sequences. Specifically, the following schema was used: hydrophobic residues in the first beta sheet at positions 1, 3 and, 5; a variable-length linker with length ‘‘‘‘ ranging from 7 to 17 residues; hydrophobic residues in the second beta sheet at positions 5+x+1 and 5+x+3; hydrophobic residues in the first alpha helix at positions 5+x+9, 5+x+12, and 5+x+16. These positions correspond to M1, V3, V5, V15, V17, I23, L26 and V30 of Parkin’s UBL domain. Potential alignments were evaluated using scoring system where non-standard hydrophobic residues were allowed but were assigned a penalty score. Furthermore, extreme linker lengths, ‘‘‘‘, were penalized for being too short or too long. Finally, a small tie-breaker bonus was awarded (for choosing between linker lengths) to alignments which contained a positively charged residue at the equivalent position of K27 of Parkin’s UBL. This positively charged residue was used in the alignment because it was present in nearly all the published UBL domain structures which were used as reference when designing the alignment schema. Using this approach, 19623 sequences out of 28447 were successfully aligned for residues surrounding the region of interest, M1 and E16 of Parkin’s UBL.
Finally, aligned sequences were separated into those belonging to N-terminal or internal UBL domains and were submitted to WebLogo369 to create the sequence logos used to visualize residue conservation.
Mammalian parkin sequence alignment
Full protein sequences for mammalian Parkin were downloaded for human (UniProtKB: O60260) pig (UniProtKB: Q2L7G3), dog (UniProtKB: A0A8C0PPD2), rat (UniProtKB: Q9JK66), mouse (UniProtKB: Q9WVS6), and guinea pig (UniProtKB: H0V739). These sequences were aligned using the Clustal Omega multiple sequence alignment tool by EMBL-EBI.70
Supplementary Material
Highlights.
Emergent behaviors arise from the PINK1/Parkin circuit’s input-coupled positive feedback
Parkin recruitment requires mitochondrial PINK1 concentrations to exceed an input threshold
Parkin recruitment time delay is inversely proportional to mitochondrial PINK1 concentration
PINK1 and Parkin mutations tune the input threshold and delay behaviors
ACKNOWLEDGMENTS
We thank the members of the Altschuler-Wu lab for their support, with particular thanks to Capria Rinaldi and Karl Kumbier for technical and analytical suggestions. We thank Jayanta Debnath, Orion Weiner, Matt Jacobson, Richard Youle, and James Ferrell for helpful feedback and perspectives during this study. We thank Wendel Lim for access to his Opera Phenix microscope. We thank the UCSF HDFCCC Laboratory for Cell Analysis Shared Resource Facility for training and assistance with cell sorting on the BD FACS Aria 2. The HDFCCC Laboratory for Cell Analysis Shared Resource Facility is supported through a grant from the National Institute of Health (P30CA082103). Protein structure analyses were performed via UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. ChimeraX development was supported by National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. Graphical abstract was created using BioRender. This work was funded by the National Science Foundation Graduate Research Fellowship Program 2034836 (C.S.W.), a gift from an anonymous donor (C.S.W.), the National Cancer Institute, National Institutes of Healthh R01-CA184984 (L.F.W.), and the Chan Zuckerberg Initiative (L.F.W. and S.J.A.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113260.
DECLARATION OF INTERESTS
S.J.A. and L.F.W. are founders and scientific advisory board members of Nine Square Therapeutics.
REFERENCES
- 1.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27, 433–446. 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Nartiss Y, Steipe B, McQuibban GA, and Kim PK (2012). ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 8, 1462–1476. 10.4161/auto.21211. [DOI] [PubMed] [Google Scholar]
- 3.Yang J-Y, and Yang WY (2011). Spatiotemporally controlled initiation of Parkin-mediated mitophagy within single cells. Autophagy 7, 1230–1238. 10.4161/auto.7.10.16626. [DOI] [PubMed] [Google Scholar]
- 4.Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, et al. (2018). Mitochondrial membrane potential. Anal. Biochem 552, 50–59. 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narendra D, Tanaka A, Suen D-F, and Youle RJ (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol 183, 795–803. 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narendra DP, Jin SM, Tanaka A, Suen D-F, Gautier CA, Shen J, Cookson MR, and Youle RJ (2010). PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol 8, e1000298. 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol 189, 211–221. 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kandul NP, Zhang T, Hay BA, and Guo M (2016). Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat. Commun 7, 13100. 10.1038/ncomms13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suen D-F, Narendra DP, Tanaka A, Manfredi G, and Youle RJ (2010). Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 107, 11835–11840. 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickrell AM, and Youle RJ (2015). The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 85, 257–273. 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ordureau A, Sarraf SA, Duda DM, Heo J-M, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. (2014). Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 56, 360–375. 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ordureau A, Paulo JA, Zhang J, An H, Swatek KN, Cannon JR, Wan Q, Komander D, and Harper JW (2020). Global Landscape and Dynamics of Parkin and USP30-Dependent Ubiquitylomes in iNeurons during Mitophagic Signaling. Mol. Cell 77, 1124–1142.e10. 10.1016/j.molcel.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, and Youle RJ (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol 205, 143–153. 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, and Muqit MMK (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J 460, 127–139. 10.1042/bj20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 17.Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2, 120080. 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, and Hattori N (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep 2, 1002. 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauvé V, Sung G, Soya N, Kozlov G, Blaimschein N, Miotto LS, Trempe J-F, Lukacs GL, and Gehring K (2018). Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol 25, 623–630. 10.1038/s41594-018-0088-7. [DOI] [PubMed] [Google Scholar]
- 20.Gladkova C, Maslen SL, Skehel JM, and Komander D (2018). Mechanism of parkin activation by PINK1. Nature 559, 410–414. 10.1038/s41586-018-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trempe J-F, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, et al. (2013). Structure of Parkin Reveals Mechanisms for Ubiquitin Ligase Activation. Science 340, 1451–1455. 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- 22.Tang MY, Vranas M, Krahn AI, Pundlik S, Trempe J-F, and Fon EA (2017). Structure-guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat. Commun 8, 14697. 10.1038/ncomms14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauvé V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe JF, and Gehring K (2015). A Ubl/ubiquitin switch in the activation of Parkin. Embo J 34, 2492–2505. 10.15252/embj.201592237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riley BE, and Olzmann JA (2015). A Polyubiquitin Chain Reaction: Parkin Recruitment to Damaged Mitochondria. PLoS Genet 11, e1004952. 10.1371/journal.pgen.1004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seirafi M, Kozlov G, and Gehring K (2015). Parkin structure and function. FEBS J 282, 2076–2088. 10.1111/febs.13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowling JL, Skolfield MC, Riley WA, Nolin AP, Wolf LC, and Nelson DE (2019). Temporal integration of mitochondrial stress signals by the PINK1:Parkin pathway. BMC Mol. Cell Biol 20, 33. 10.1186/s12860-019-0220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Reilly CM, Fogarty KE, Drummond RM, Tuft RA, and Walsh JV (2003). Quantitative Analysis of Spontaneous Mitochondrial Depolarizations. Biophys. J 85, 3350–3357. 10.1016/s0006-3495(03)74754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasool S, Soya N, Truong L, Croteau N, Lukacs GL, and Trempe JF (2018). PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 19, e44981. 10.15252/embr.201744981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rusilowicz-Jones EV, Jardine J, Kallinos A, Pinto-Fernandez A, Guenther F, Giurrandino M, Barone FG, McCarron K, Burke CJ, Murad A, et al. (2020). USP30 sets a trigger threshold for PINK1–PARKIN amplification of mitochondrial ubiquitylation. Life Sci. Alliance 3, e202000768. 10.26508/lsa.202000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcassa E, Kallinos A, Jardine J, Rusilowicz-Jones EV, Martinez A, Kuehl S, Islinger M, Clague MJ, and Urbé S (2018). Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep 19, e45595. 10.15252/embr.201745595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gersch M, Gladkova C, Schubert AF, Michel MA, Maslen S, and Komander D (2017). Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat. Struct. Mol. Biol 24, 920–930. 10.1038/nsmb.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lazarou M, Jin SM, Kane LA, and Youle RJ (2012). Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 22, 320–333. 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belshaw PJ, Ho SN, Crabtree GR, and Schreiber SL (1996). Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc. Natl. Acad. Sci. USA 93, 4604–4607. 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, and Crabtree GR (2006). Rapamycin Analogs with Differential Binding Specificity Permit Orthogonal Control of Protein Activity. Chem. Biol 13, 99–107. 10.1016/j.chembiol.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Denison SR, Wang F, Becker NA, Schüle B, Kock N, Phillips LA, Klein C, and Smith DI (2003). Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 22, 8370–8378. 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- 36.Burchell L, Chaugule VK, and Walden H (2012). Small, N-Terminal Tags Activate Parkin E3 Ubiquitin Ligase Activity by Disrupting Its Autoinhibited Conformation. PLoS One 7, e34748. 10.1371/journal.pone.0034748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biswas S, Roy R, Biswas R, and Bagchi A (2020). Structural analysis of the effects of mutations in Ubl domain of PARKIN leading to Parkinson’s disease. Gene 726, 144186. 10.1016/j.gene.2019.144186. [DOI] [PubMed] [Google Scholar]
- 38.Han H, Tan J, Wang R, Wan H, He Y, Yan X, Guo J, Gao Q, Li J, Shang S, et al. (2020). PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep 21, e48686. 10.15252/embr.201948686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pryde KR, Smith HL, Chau K-Y, and Schapira AHV (2016). PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol 213, 163–171. 10.1083/jcb.201509003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun 3, 1016. 10.1038/ncomms2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aerts L, Craessaerts K, De Strooper B, and Morais VA (2015). PINK1 Kinase Catalytic Activity Is Regulated by Phosphorylation on Serines 228 and 402. J. Biol. Chem 290, 2798–2811. 10.1074/jbc.m114.620906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi W, MacDougall EJ, Tang MY, Krahn AI, Gan-Or Z, Trempe J-F, and Fon EA (2019). The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson’s disease. Hum. Mol. Genet 28, 2811–2825. 10.1093/hmg/ddz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stevens MU, Croteau N, Eldeeb MA, Antico O, Zeng ZW, Toth R, Durcan TM, Springer W, Fon EA, Muqit MM, and Trempe JF (2023). Structure-based design and characterization of Parkin-activating mutations. Life Sci. Alliance 6, e202201419. 10.26508/lsa.202201419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sauvé V, Sung G, MacDougall EJ, Kozlov G, Saran A, Fakih R, Fon EA, and Gehring K (2022). Structural basis for feedforward control in the PINK1/Parkin pathway. EMBO J 41, e109460. 10.15252/embj.2021109460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao B, Goh J-Y, Xiao L, Xian H, Lim K-L, and Liou Y-C (2017). Reactive oxygen species trigger Parkin/PINK1 pathway–dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem 292, 16697–16708. 10.1074/jbc.m117.787739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao B, Deng X, Lim GGY, Xie S, Zhou ZD, Lim K-L, and Tan E-K (2017). Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis 8, e3097. 10.1038/cddis.2017.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hung C-M, Lombardo PS, Malik N, Brun SN, Hellberg K, Van Nostrand JL, Garcia D, Baumgart J, Diffenderfer K, Asara JM, and Shaw RJ (2021). AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv 7, eabg4544. 10.1126/sciadv.abg4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rasool S, Veyron S, Soya N, Eldeeb MA, Lukacs GL, Fon EA, and Trempe J-F (2022). Mechanism of PINK1 activation by autophosphorylation and insights into assembly on the TOM complex. Mol. Cell 82, 44–59.e6. 10.1016/j.molcel.2021.11.012. [DOI] [PubMed] [Google Scholar]
- 49.Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, and Matsuda N (2013). A Dimeric PINK1-containing Complex on Depolarized Mitochondria Stimulates Parkin Recruitment. J. Biol. Chem 288, 36372–36384. 10.1074/jbc.m113.509653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rusilowicz-Jones EV, Barone FG, Lopes FM, Stephen E, Mortiboys H, Urbé S, and Clague MJ (2022). Benchmarking a highly selective USP30 inhibitor for enhancement of mitophagy and pexophagy. Life Sci. Alliance 5, e202101287. 10.26508/lsa.202101287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang JR, Martinez A, Lane JD, Mayor U, Clague MJ, and Urbé S (2015). USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep 16, 618–627. 10.15252/embr.201439820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, and Corn JE (2015). USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol 17, 160–169. 10.1038/ncb3097. [DOI] [PubMed] [Google Scholar]
- 53.Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, and Sheng M (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 54.Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, and Vandenberghe W (2014). The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet 23, 5227–5242. 10.1093/hmg/ddu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang L, Cho Y-L, Tang Y, Wang J, Park J-E, Wu Y, Wang C, Tong Y, Chawla R, Zhang J, et al. (2018). PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res 28, 787–802. 10.1038/s41422-018-0056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, Youle AM, Nezich CL, Wu X, Hammer JA, and Youle RJ (2017). Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol 216, 3231–3247. 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jilkine A, Angenent SB, Wu LF, and Altschuler SJ (2011). A Density-Dependent Switch Drives Stochastic Clustering and Polarization of Signaling Molecules. PLoS Comput. Biol 7, e1002271. 10.1371/journal.pcbi.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, and Ferrell JE (2021). C. elegans colony formation as a condensation phenomenon. Nat. Commun 12, 4947. 10.1038/s41467-021-25244-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, and Kholodenko BN (2006). Scaffolding Protein Grb2-associated Binder 1 Sustains Epidermal Growth Factor-induced Mitogenic and Survival Signaling by Multiple Positive Feedback Loops. J. Biol. Chem 281, 19925–19938. 10.1074/jbc.m600482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, Berkhout J, Maiwald T, Kaimachnikov NP, Timmer J, Hoek JB, and Kholodenko BN (2009). Systems-level interactions between insulin–EGF networks amplify mitogenic signaling. Mol. Syst. Biol 5, 256. 10.1038/msb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409. 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bajar BT, Lam AJ, Badiee RK, Oh Y-H, Chu J, Zhou XX, Kim N, Kim BB, Chung M, Yablonovitch AL, et al. (2016). Fluorescent indicators for simultaneous reporting of all four cell cycle phases. Nat. Methods 13, 993–996. 10.1038/nmeth.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hayer A, Shao L, Chung M, Joubert L-M, Yang HW, Tsai F-C, Bisaria A, Betzig E, and Meyer T (2016). Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol 18, 1311–1323. 10.1038/ncb3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narendra D, Kane LA, Hauser DN, Fearnley IM, and Youle RJ (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106. 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, and Cookson MR (2005). Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 102, 5703–5708. 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greene C (2017). Filt2 2D Geospatial Data Filter (MATLAB Central File Exchange) [Google Scholar]
- 68.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: A Sequence Logo Generator. Genome Res 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, Madhusoodanan N, Kolesnikov A, and Lopez R (2022). Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res 50, W276–W279. 10.1093/nar/gkac240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin JR, Fallahi-Sichani M, Chen JY, and Sorger PK (2016). Cyclic Immunofluorescence (CycIF), A Highly Multiplexed Method for Single-cell Imaging. Curr. Protoc. Chem. Biol 8, 251–264. 10.1002/cpch.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chung J, Kuo CJ, Crabtree GR, and Blenis J (1992). Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69, 1227–1236. 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell microscopy quantification data has been deposited at Zenodo and are publicly available as of the date of publication. Sequence maps of plasmids generated for this study have been deposited at Zenodo and are publicly available as of the date of publication. The DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
Original analyis code has been deposited at Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Parkin | Abcam | Cat# ab77924; RRID: AB_1566559 |
| Rabbit monoclonal anti-phospho-Ubiquitin | CST | Cat# 70973; RRID: AB_2799795 |
| Rabbit monoclonal anti-PINK1 | Abcam | Cat# ab216144; RRID: AB_2927726 |
| Rabbit monoclonal anti-pS6(pS235/236) | CST | Cat# 4856; RRID: AB_2181037 |
| Rabbit monoclonal anti-S6 | CST | Cat# 2217; RRID: AB_331355 |
| Rabbit monoclonal anti-GAPDH | CST | Cat# 5174; RRID: AB_10622025 |
| Mouse monoclonal anti-GAPDH | CST | Cat# 97166; RRID: AB_2756824 |
| Rabbit monoclonal anti-TOM20 Alexa Fluor 488 | Abcam | Cat# ab205486; RRID: AB_2943509 |
| Rabbit monoclonal anti-TOM20 Alexa Fluor 647 | Abcam | Cat# ab209606; RRID: AB_2934123 |
|
Biological samples | ||
| Healthy adult human brain tissue lysate (whole) | Novus Biologicals | Cat# NB820–59177 |
|
Chemicals, peptides, and recombinant proteins | ||
| Geneticin (G418 Sulfate) | Life Tech. Corp. | Cat# 10131035 |
| Doxycycline Hydrochloride | Sigma Aldrich | Cat# D3072 |
| SNAP-Cell 647-SiR | NEB | Cat# S9102 |
| Hoechst 33342 | Invitrogen | Cat# H2570 |
| carbonyl cyanide 3-chlorophenylhydrazone (CCCP) | Sigma Aldrich | Cat# C2759 |
| DMSO | Sigma Life Science | Cat# D2650 |
| Rapalog (A/C heterodimerizer; AP21967) | Takara Bio. | Cat# 635055 |
| CMPD39 (USP inhibitor 18) | MedChem Express | Cat# HY-141659 |
| Rapamycin | Sigma Aldrich | Cat# R0395 |
| Halt protease and phosphatase inhibitor cocktail | ThermoFisher | Cat# 78440 |
| Polybrene | EMD Millipore | Cat# TR-1003-G |
|
Critical commercial assays | ||
| Bio-Rad protein assay dye reagent concentrate (Bradford assay kit) | Bio-Rad | Cat# 5000006 |
| Mini-protean TGX 4–20% SDS-PAGE gels | Bio-Rad | Cat# 4561093 |
| Lipofectamine 3000 kit | Invitrogen | Cat# L3000–015 |
| NEBuilder HiFi DNA assembly master mix kit | NEB | Cat# E2621S |
| Q5 Site-directed mutagenesis kit | NEB | Cat# E0554S |
|
Deposited data | ||
| Plasmid maps, single-cell microscopy quantification data, and analysis code | This manuscript | Zenodo: https://doi.org/10.5281/zenodo.8356580 |
|
Experimental models: Cell lines | ||
| Human: HeLa cell line (female) | UCSF cell culture and banking core | Cat# CCLZR030; RRID: CVCL_0030 |
| Human: HEK293T cell line (female) | UCSF cell culture and banking core | Cat# CCLZR076; RRID: CVCL_0063 |
|
Oligonucleotides | ||
| mNeonGreen cDNA | Shaner et al.61 | Allele Biotech Cat# ABP-FP-MNEONSB |
|
Recombinant DNA | ||
| pCMV-Parkin | This manuscript | N/A |
| pCMV-Parkin(E16A) | This manuscript | N/A |
| pCMV-mKO2-Parkin(M1) | This manuscript | N/A |
| pCMV-mKO2-Parkin(M1L) | This manuscript | N/A |
| pCMV-Parkin-A92mKO2 | This manuscript | N/A |
| pCMV-Parkin(C431N)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(S65A)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(R104A)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(K161N)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(N273K)-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-Parkin(W403A)-A92mKO2 | This manuscript | N/A |
| pLv-CMV(trunc)-Parkin-A92mKO2 | This manuscript | N/A |
| pLv-EF1a-SNAP-FRB-FIS1(MTS) | This manuscript | N/A |
| pLv-TetOn-FKBP-PINK1(110–581)-mNeonGreen (Neo) | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(110–581)-mNeonGreen | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(G386A) (110–581)-mNeonGreen | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(S228A) (110–581)-mNeonGreen | This manuscript | N/A |
| pLv-EF1a-FKBP-PINK1(S228D) (110–581)-mNeonGreen | This manuscript | N/A |
| pCMV-mCherry-Parkin | Narendra et al.5 | Addgene Plasmid #23956 |
| pLL3.7m-Clover-Geminin(1–110)-IRES-mKO2-Cdt(30–120) | Bajar et al.62 | Addgene Plasmid #83841 |
| pLV-EF1a-IRES-Puro | Hayer et al.63 | Addgene Plasmid #85132 |
| pSNAPf | NEB | N9183S |
| pC4-RhE-FRB-Fis1 | Narendra et al.64 | Addgene Plasmid #68056 |
| pC4M-F2E-GFP-FKBP | Narendra et al.64 | Addgene Plasmid #68058 |
| pCMVTNT-PINK1-C-Myc | Beilina et al.65 | Addgene Plasmid #13314 |
| pLv-TetOn-MCS1-P2A-MCS2 (Neo) | Adam Karpf Lab | Addgene Plasmid #89180 |
| pLv-TetOn-FKBP-PINK1 (110–581)-mNG-Neo | This manuscript | N/A |
| pMD2.G | Didier Trono Lab | Addgene Plasmid #12259 |
| pCMV-dR8.91 | NovoPro | Cat# V007548 |
|
Software and algorithms | ||
| MATLAB | https://www.mathworks.com/ | N/A |
| Fiji | Schindelin et al.66 | https://imagej.net/software/fiji/ |
| filt2 MATLAB function | Greene67 | https://www.mathworks.com/matlabcentral/fileexchange/61003-filt2-2d-geospatial-data-filter |
| UCSF ChimeraX software | Pettersen et al.68 | https://www.cgl.ucsf.edu/chimerax/ |
| WebLogo: A sequence Logo generator | Crooks et al.69 | https://weblogo.berkeley.edu/logo.cgi |
| EMBL-EBI Clustal Omega multiple sequence alignment tool | Madeira et al.70 | https://www.ebi.ac.uk/Tools/msa/clustalo/ |