

Abstract
Aneuploidy, the presence of chromosome gains or losses, is a hallmark of cancer. Here, we describe KaryoCreate (Karyotype CRISPR Engineered Aneuploidy Technology), a system that enables generation of chromosome-specific aneuploidies by co-expression of an sgRNA targeting chromosome-specific CENPA-binding ɑ-satellite repeats together with dCas9 fused to mutant KNL1. We designed unique and highly specific sgRNAs for 19 of the 24 chromosomes. Expression of these constructs leads to missegregation and induction of gains or losses of the targeted chromosome in cellular progeny, with an average efficiency of 8% for gains and 12% for losses (up to 20%) validated across 10 chromosomes. Using KaryoCreate in colon epithelial cells, we show that chromosome 18q loss, frequent in gastrointestinal cancers, promotes resistance to TGFβ, likely due to synergistic hemizygous deletion of multiple genes. Altogether, we describe an innovative technology to create and study chromosome missegregation and aneuploidy in the context of cancer and beyond.
Keywords: aneuploidy, centromere; chromosome gains and losses; kinetochore; DNA damage; mitosis; chromosome missegregation
Graphical Abstract
INTRODUCTION
Aneuploidy, i.e. chromosomal gains or losses, is rare in normal tissues1–3 as it causes cellular stress phenotypes4,5. Despite its detrimental effect, aneuploidy is common in cancer, where specific chromosomes tend to be gained or lost more frequently than others2–6. We and others have proposed that recurrent patterns of aneuploidy are selected for in cancer to maximize oncogene dosage and minimize tumor-suppressor gene dosage4,7.
A challenge in studying aneuploidy is the lack of straightforward methods to generate cell models with a specific chromosome added or removed. Common methods to induce aneuploidy utilize chemical inhibition of mitotic proteins, e.g. MPS1, resulting in random chromosome missegregation8,9. Microcell-mediated chromosome transfer induces chromosome gains but this method is quite complicated10,11. Centromere inactivation of the Y chromosome can induce its missegregation12,13. Newer strategies to induce chromosome losses involve using CRISPR/Cas9 to eliminate all or part of chromosomes5,14,15. Other recently described methods use non-centromeric repeats to induce specific losses or, more rarely, gains of chromosomes 1 and 916,17.
Human centromeres consist of repetitive α-satellite DNA hierarchically organized in megabase-long arrays called higher-order repeats (HOR), a subset of which bind CENPA, a histone H3 variant critical to kinetochore function18–21. In humans, HORs are generally specific to individual chromosomes: 15 autosomes and the 2 sex chromosomes have unique centromeric arrays19 and the rest can be grouped in two families based on centromere similarity (chromosomes 1, 5, 19 and chromosomes 13, 14, 21, 22). CENPA-bound centromeric sequences direct the kinetochore assembly which enables microtubule binding to mitotic chromosomes22. The KMN network (KNL1/MIS12 complex/NDC80 complex) is important in modulating kinetochore-microtubule attachments23. In mitosis, each sister kinetochore must be attached to opposite spindle poles to allow their equal and correct segregation24. Properly attached chromatids experience an inter-kinetochore mechanical tension required to satisfy the spindle assembly checkpoint (SAC) and allow progression into anaphase24,25. SAC activation triggers the activity of Aurora B kinase, which destabilizes kinetochore-microtubule attachments by phosphorylating different targets including NDC80 and KNL126,27. Aurora B activity is counteracted by the action of PP1 phosphatase, recruited to the kinetochores through KNL128. The balance between kinase and phosphatase activities determines the fate of the kinetochore-microtubule attachment and the timing of the metaphase-to-anaphase transition.
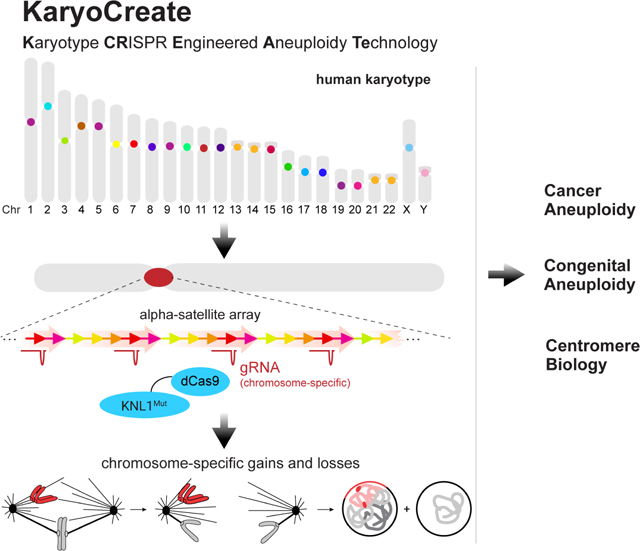
Here we describe KaryoCreate, a CRISPR/Cas9-based technology that uses gRNAs targeting chromosome-specific human centromeric repeats to direct a mutant KNL1/dCas9 construct that interferes with normal mitotic functions, generating chromosome-specific aneuploidy. Using this method, we obtain cell models of highly recurrent aneuploidies in human gastro-intestinal cancers and present data supporting tumor-associated phenotypes occurring after chromosome 18q loss in colorectal cells.
RESULTS
Computational prediction of sgRNAs targeting chromosome-specific α-satellite centromeric repeats
To design chromosome-specific centromeric sgRNAs, we primarily relied on the genome assembly from the Telomere-to-Telomere (T2T) consortium29. For centromeres resolved in previous assemblies, we confirmed the sgRNA predictions from T2T using the hg38 reference genome30, to reduce the risk of bias associated with a single assembly31,32. To maximize the likelihood of interfering with chromosome segregation, we focused our design on centromeric HORs found to bind to CENPA in chromatin immunoprecipitation (ChIP) experiments (defined as “Live”, or HOR_L, by the T2T)21,33. For any given chromosome, the ideal sgRNA has 1) high on-target specificity (i.e. does not bind to centromeres on other chromosomes or to other genomic locations), 2) high number of binding sites on the repetitive HOR_L and 3) high efficiency in tethering dCas9 to the DNA. For each chromosome, we started by identifying all possible Cas9 sgRNAs targeting its HOR_L. We performed this analysis for all 24 human chromosomes (Tables S1, S2).
Next, we determined two parameters that define the specificity and efficiency of each sgRNA (both percentages, with 100% the best score): a chromosome specificity score, defined as the ratio of the number of binding sites on the target centromere to the total number of binding sites across all centromeres, and a centromere specificity score, defined as the ratio of the number of binding sites in centromeric regions to the number of sites across the whole genome. We also predicted the efficiency of each sgRNA based on GC content34, sgRNA activity from published studies35,36 (see Methods), and total number of binding sites to the specific centromere (Fig. 1A).
Figure 1.

Prediction and validation of chromosome-specific sgRNAs targeting human α-satellite centromeric sequences
(A) Schematic representation of the computational prediction of chromosome-specific centromeric sgRNAs based on specificity score and predicted efficiency.
(B) Idiogram of human karyotype reporting the number of sgRNAs predicted with specificity ≥99% and validated by imaging for each chromosome.
(C) Left: Proliferation assay of centromeric sgRNAs in hCECs expressing Cas9 or empty vector (EV). sgRNAα-β refers to a sgRNA specific for chromosome α where β is the sgRNA serial number. Percentage of live cells relative to EV determined 7 days after transduction by cell counting. Mean and S.D. (standard deviation) are from triplicates; p-values are from Wilcoxon test comparing each condition to NC (*=p<0.05); conditions with significant p-values are in red. Imaging validation is also indicated (see (D).) Right: Western blot showing Cas9 expression.
(D) Top: Imaging validation of centromere targeting in hCEC clones (containing 3 copies of chr7 or chr13) expressing 3xmScarlet-dCas9 and the indicated sgRNAs. Representative images of interphase are shown (percentages of cells displaying the expected number of foci are in Table S1). Scale bars: 5 𝝻M. Bottom: Low-pass WGS confirming specific aneuploidies in the two clones.
(E) Imaging of hCECs (trisomic for chr7) expressing sgRNA7–1 or sgRNA18–4 showing colocalization of 3xmScarlet-dCas9 foci (red) and chromosome 7 or 18 centromeric FISH probes (green); FISH protocol was used after PFA fixation. Colocalization is quantified at right (mean and S.D. from triplicates).
(F)Validation of additional sgRNAs as in (D).
Using thresholds of 99% for both chromosome and centromere specificity scores, a GC content ≥40%, a minimum of 400 sgRNA binding sites, sgRNA activity35,36 >0.1, and representation in hg38, we designed at least one sgRNA for 19 of the 24 human chromosomes (all except 21, 22, Y; Fig. 1B; Table S1), with 1590 binding sites per chromosome on average. Increasing the chromosome specificity score from 99% to 100% resulted in at least one sgRNA for 16 chromosomes.
Experimental validation of sgRNAs targeting α-satellite centromeric repeats on 15 human chromosomes
To assess the activity of the predicted sgRNAs, we co-expressed selected sgRNAs with Cas9 and monitored cell proliferation, since the presence of several double-strand breaks at the centromere is likely to decrease cell viability37. We used hTERT TP53−/− human colonic epithelial cells (hCECs)38 and hTERT TP53 WT retinal pigment epithelial cells (RPEs) expressing p21 (CDKN1A) and RB (RB1) shRNAs39. We transduced Cas9-expressing RPEs and hCECs with a lentiviral vector expressing either a centromeric or a negative control sgRNA (sgNC) that does not target the human genome40. Hereafter we refer to each centromeric sgRNA as sgChrα-β, where α is the specific targeted chromosome and β is the serial number of the designed sgRNA.
We first tested 3 sgRNAs predicted for chromosomes 7 and 13, and 4 for chromosome 18. Compared to sgNC, hCECs and RPEs expressing sgChr7–1, sgChr7–3, sgChr13–3, or sgChr18–4 exhibited at least 50% reduction in proliferation, while the other sgRNAs did not result in significant differences (Fig. 1C; Fig. S1A). We selected the sgRNAs exhibiting the greatest reduction in proliferation for additional testing.
To confirm that the sgRNAs targeted the intended centromeres, we designed a dCas9–0based imaging system comprising three mScarlet fluorescent molecules fused to the N-terminus of endonuclease-dead Cas9 (3xmScarlet-dCas9). To achieve consistently high expression, we FACS-sorted 3xmScarlet-dCas9-transduced hCECs for strong fluorescent signal. hCECs co-expressing 3xmScarlet-dCas9 and sgChr7–1, sgChr13–3, or sgChr18–4 (but not sgNC) showed bright nuclear foci (Fig. 1D). Notably, the sgRNAs that did not cause a decrease in proliferation in the presence of Cas9 failed to form foci (Fig. 1C and data not shown).
To further confirm the chromosome specificity of the sgRNAs, we used two independent approaches. We first utilized hCEC clones with aneuploidies previously identified through whole-genome sequencing (WGS)-based copy number analysis to verify whether the observed number of foci was consistent with the expected DNA copy number. We found that hCEC clones carrying three copies of chromosome 7 or 13 each showed three foci when transduced with sgChr7–1 or sgChr13–3, respectively (Fig. 1D; Fig. S1B). As anticipated, transduction with sgRNAs targeting chromosomes present in two copies led to the formation of two foci per nucleus (Fig. 1D). Next, we confirmed that the 3xmScarlet-dCas9 foci localized at specific centromeres by fluorescence in-situ hybridization (FISH) using centromeric probes. We confirmed colocalization of FISH signals for both chromosomes 7 (sgChr7–1) and 18 (sgChr18–4) with mScarlet foci (Fig. 1E). Altogether, these experiments indicate that the computationally predicted sgRNAs can recruit dCas9 to the expected specific centromere.
We tested 75 additional sgRNAs in hCECs and confirmed the formation of the expected number of foci for 24 sgRNAs targeting 15 different chromosomes (2, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 16, 18, 19, X; Fig. 1F, Fig. S1C, Table S1). We also confirmed 4 sgRNAs in RPEs (Fig. S1D).
Altogether, we designed and validated 24 chromosome-specific sgRNAs targeting the centromeres of 15 different human chromosomes. Interestingly, the predicted sgRNA efficiency evaluated using a previously published algorithm36 did not correlate with the ability of sgRNAs to form foci (r=0.2; p=0.5; Fig. S1E, left). Instead, for the sgRNAs that formed foci, there was a significant correlation between the intensity of the signal of the foci and the number of binding sites at the centromeres predicted based on the CHM13 genome reference (r=0.65, p=0.03; Fig. S1E, right).
Centromeric targeting of KNL1Mut-dCas9 induces modest mitotic delay and chromosome missegregation
To induce chromosome missegregation, we built and tested four dCas9 fusion proteins predicted to disrupt kinetochore-microtubule attachments (Fig. 2A, Fig. S2A). KNL1S24A;S60A-dCas9 and KNL1RVSF/AAAA-dCas9 utilize the KNL1 N-terminal portion (amino acid (aa) 1–86)28,41 and contain mutations with opposing effects in disrupting the cross-regulation between Aurora B and PP1 (Fig. S2A). KNL1S24A;S60A is predicted to be always bound to PP1 as its mutated residues cannot be phosphorylated by Aurora B41 (Fig. S2A); KNL1RVSF/AAAA contains a mutation affecting the RVSF motif (aa 58–61) preventing it from interacting with PP1 and recruiting it to the centromere28 (Fig. S2A). NDC80-CH1-dCas9 and NDC80-CH2-dCas9 were designed to render the interaction between kinetochores and microtubules hyperstable and refractory to Aurora B destabilization. These constructs contain one (NDC80-CH1) or two (NDC80-CH2) CH domains (aa 1–207), the region of NDC80 responsible for binding microtubules. CH domains normally contain 6 residues whose phosphorylation by Aurora B inhibits the interaction with microtubules; our constructs have all 6 residues mutated, preventing Aurora-B-mediated regulation42 (Fig. S2A).
Figure 2.

KNL1Mut-dCas9 targeted to centromeres induces modest mitotic delay and chromosome missegregation
(A) Left: Maps of KNL1RVSF/AAAA-dCas9 and dCas9-KNL1RVSF/AAAA constructs. Right: Western blot showing the expression of the indicated constructs in hCECs.
(B) Top: Time-lapse imaging of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and the indicated sgRNA. Cells were analyzed for time spent in mitosis and for lagging chromosomes (quantified in C and D), and representative images are shown (see Movies S1 for time-lapse series). Bottom: Analysis performed in H2B-GFP hCECs co-expressing 3xmScarlet-KNL1Mut-dCas9 and sgChr7–1, indicating specific chromosome missegregation.
(C) Quantification of mitotic duration (time spent between metaphase and anaphase onset) of cells in (B) (mean and S.D. from triplicates; ≥25 dividing cells analyzed per condition).
(D) Quantification as in (C) reporting % of mitoses showing lagging chromosomes.
(E) Immunofluorescence (IF) analysis of mitotic HCT116 cells expressing KNL1Mut-dCas9 and sgChr7–1 or sgChr18–4 or sgNC stained as indicated. White arrows point to misaligned chromosomes.
(F) Quantification of chromosome congression defects in (E) (mean and S.D. from triplicates).
(G) Analysis of micronuclei in hCECs expressing KNL1Mut-dCas9 and sgChr7–1, sgChr18–4, or sgNC. The percentage of cells with micronuclei relative to EV was determined 7 days after transduction (mean and S.D. from triplicates; ≥50 cells per condition).
(H) Representative images and quantification of chr-18-containing micronuclei in cells treated as in (G), from triplicate experiments.
Western blot analysis showed that KNL1RVSF/AAAA-dCas9 and KNL1S24A;S60A-dCas9 expression levels were higher than those of NDC80-CH1-dCas9 and NDC80-CH2-dCas9 (Fig. S2B). For the KNL1 constructs, the N-terminal fusions were generally more stable than the C-terminal fusions (Fig. S2C, Fig. 2A). Given their higher protein expression and greater efficiency in inducing chromosome gains and losses compared to the other constructs (see next section), we focused on the KNL1 constructs, particularly KNL1RVSF/AAAA-dCas9, hereafter referred to as KNL1Mut-dCas9.
To confirm centromeric localization of the fusion protein, we transduced hCECs expressing a fluorescently tagged version of KNL1Mut-dCas9 (3xmScarlet-KNL1Mut-dCas9) with centromeric sgRNAs, as described above. We observed the expected number of foci in the presence of sgChr7–1 and sgChr18–4 (Fig. S2D), indicating that fusing KNL1Mut with dCas9 does not alter the ability of dCas9 to be recruited to centromeres. Next, using live-cell imaging, we examined the effect of KNL1Mut-dCas9 on mitosis duration and chromosome segregation. hCECs constitutively expressing GFP-tagged histone H2B were transduced with KNL1Mut-dCas9 or empty vector (EV) and with sgChr7–1, sgChr18–4, or sgNC. Cells expressing KNL1Mut-dCas9 and either sgChr7–1 or sgChr18–4 progressed more slowly through mitosis than cells transduced with EV and either sgChr7–1 or sgChr18–4 (Fig. 2C): the average time spent in the metaphase-to-anaphase transition increased from 6 minutes to 9 or 10 minutes in the sgChr7–1 or sgChr18–4 condition, respectively (Fig. 2B, 2C). Nonetheless, cells transduced with sgChr7–1 or sgChr18–4 did not arrest in metaphase and completed mitosis, and their proliferation rate was only slightly and non significantly lower than that of cells transduced with sgNC (Fig. S2E). The number of cell divisions with lagging chromosomes increased from <5% to 15% between EV+sgChr7–1 and KNL1Mut-dCas9+sgChr7–1 and from 7% to 23% between EV+sgChr18–4 and KNL1Mut-dCas9+sgChr18–4 (Fig. 2B, upper panel, 2D). Furthermore, live-cell imaging of cells expressing 3xmScarlet-KNL1Mut-dCas9 and sgChr7–1, where mScarlet marks chromosome 7 as in Fig. S2D (polyclonal population), showed that about 80% of the lagging chromosomes observed during mitosis had red foci, consistent with chromosome-specific missegregation (Fig. 2B, lower panel; Movies S1). Note that in this experiment sgNC could not be used as a control as it did not cause foci formation.
To corroborate these data in a different cell line, we performed a similar experiment in the HCT116 (TP53 WT) colon cancer cell line, transducing them with KNL1Mut-dCas9 and either sgNC, sgChr7–1, or sgChr18–4. Immunofluorescence for ɑ-tubulin to visualize the mitotic spindle, CREST serum to visualize the centromeres, and DAPI to assess chromosome alignment showed that the percentage of mitoses with misaligned chromosomes increased from 12% in the sgNC samples to 32% and 35% in the sgChr7–1 and sgChr18–4 conditions, respectively (Fig. 2E, 2F).
Finally, we scored the fraction of KNL1Mut-dCas9-expressing hCECs containing micronuclei (a well-known consequence of missegregation43) 7–9 days after transduction with sgRNAs. The percentage of cells showing micronuclei increased from <2.5% for sgNC to 9% for sgChr7–1 and 14% for sgChr18–4 (Fig. 2G). Furthermore, FISH using a chr18 centromeric probe on cells co-expressing KNL1Mut-dCas9 and sgChr18–4 showed that 85% of micronuclei had a FISH signal (Fig. 2H). We also confirmed this result for chromosomes 7 and 13 (Fig. S2F).
Altogether, these data indicate that tethering KNL1Mut-dCas9 to the centromeres through chromosome-specific sgRNAs can induce chromosome misalignment, lagging chromosomes, modest mitotic delay, and formation of micronuclei containing the targeted chromosome without substantially affecting the rate of cell division.
KaryoCreate allows induction of chromosome-specific gains and losses in human cells
Having designed and validated chromosome-specific sgRNAs and dCas9-based constructs to induce chromosome missegregation, we next tested the capability of this system, designated “KaryoCreate” for Karyotype CRISPR Engineered Aneuploidy Technology, to generate specific aneuploidies in human cell lines (Fig. 3A). We reasoned that transient targeting of the dCas9-based construct to the centromere would be ideal to generate chromosome gains and losses and allow isolation of stable aneuploid lines.
Figure 3.

KNL1Mut-dCas9 is recruited to human centromeres and allows induction of chromosome-specific gains and losses
(A) KaryoCreate conceptualization: Chromosome specificity of human α-satellite centromeric sequences makes it possible to induce missegregation of a specific chromosome while leaving the others unaffected.
(B) Western blot showing the expression of KaryoCreate constructs in hCECs, either through transient transfection with a constitutive promoter (pHAGE-CMV) or through infection with a doxycycline (Doxy)-inducible promoter (pIND20).
(C) KaryoCreate experimental plan with transient KNL1Mut-dCas9 expression and (transient or constitutive) sgRNA expression; cells are harvested after 7–9 days for validation by FISH and can then be plated to create single-cell clones.
(D) Representative FISH images using probes specific for chr7 or chr18 on hCECs showing gains and losses after KaryoCreate with the indicated sgRNAs.
(E) Quantification of the experiment shown in (D) for chr7 (left) or chr18 (right); see also Table S2 for automated image quantification. Mean and S.D. from triplicates.
(F) Representative metaphase spreads from hCECs treated as in (D) and analyzed by FISH using probes specific for chr7 and chr18 as indicated.
(G) Quantification of FISH signals from (F) (mean and S.D. from triplicates).
We first designed a system based on doxycycline-inducible expression of KNL1Mut-dCas9 (constructed in the pIND20 vector44) and constitutive sgRNA expression (pLentiGuide-Puro-FE, Fig. 3B, 3C; see Methods). We tested KaryoCreate in hCECs co-transduced with pIND20-KNL1Mut-dCas9 or pIND20-GFP (control) and with sgNC, sgChr7–1, or sgChr18–4. Cells were treated with doxycycline for 7–9 days, and analyzed by FISH. As expected, 95% of control cells (GFP with sgNC) showed two copies of chromosomes 7 and 18 (Fig. 3D, 3E). This percentage did not significantly change in cells expressing KNL1Mut-dCas9 and sgNC, indicating that in the absence of a centromere-specific sgRNA, KNL1Mut-dCas9 does not induce chromosome missegregation (Fig. 3D, 3E; see Table S2 for automated quantification). Compared to sgNC, sgChr7–1 expression in hCECs transduced with KNL1Mut-dCas9 significantly increased the percentages of cells showing chromosome loss, i.e. <2 copies (from 3% to 16%; p=0.01), or gain, i.e. >2 copies (from 2.8% to 12.5%; p=0.03), of chromosome 7, but not loss or gain of chromosome 18 (3% versus 3.2%). We next tested sgChr18–4, finding significant increases in loss (from 2% to 17.5%; p=0.01) and gain (from 2.5% to 14%; p=0.02) of chromosome 18 but not chromosome 7 (Fig. 3D, 3E; see Table S2 for automated quantification). Furthermore, we obtained comparable results when we restricted the FISH analysis to metaphase spreads as opposed to nuclei (Fig. 3F, 3G).
We also developed two additional KaryoCreate systems: one based on transient co-transfection of KNL1Mut-dCas9 driven by a constitutive promoter (pHAGE vector) and an sgRNA-expressing vector (pLentiGuide-Puro-FE) and another based on a degrader approach whereby KNL1Mut-dCas9 is fused to an FKBP-based degradation domain45 and is stabilized only after treatment with the small molecule Shield-1 (see Methods). Overall, the three methods gave similar results (Fig. S3A).
We next analyzed the frequency of aneuploidy induced by other constructs generated for KaryoCreate (NDC80-CH1-dCas9 and NDC80-CH2-dCas9, described above; see Fig. S2A–S2C), finding that the other fusion proteins induced aneuploidy with similar or lower efficiency than KNL1Mut-dCas9 (KNL1RVSF/AAAA-dCas9; Fig. S3B). KNL1S24A;S60A-dCas9 produced similar levels of induced aneuploidy to KNL1Mut-dCas9 (KNL1RVSF/AAAA-dCas9), while NDC80-CH1-dCas9 and NDC80-CH2-dCas9 showed lower but appreciable efficiency (see Fig. S2B). Notably, after normalization for the corresponding expression level (shown in Fig. S2B), KNL1S24A;S60A-dCas9 induced a higher absolute level of aneuploidy than KNL1RVSF/AAAA-dCas9, while NDC80-CH1-dCas9 and NDC80-CH2-dCas9 showed the highest induction of aneuploidy (Fig. S3B, see Discussion). Finally, we measured aneuploidy induced by expression of dCas9 (with sgRNAs), finding this to be approximately 30% of the level induced by KNL1RVSF/AAAA-dCas9 (Fig. S3B). Interestingly, about 90% of the aneuploidy events induced by dCas9 were losses and 10% were gains, whereas for KNL1RVSF/AAAA-dCas9 and especially KNL1S24A;S60A-dCas9, 55–65% were losses (Fig. S3C). This indicates that just the recruitment of dCas9 to centromeres at least partially inhibits its normal function, leading mainly to chromosome losses, and that the simultaneous expression of mutant forms of KNL1 (especially KNL1S24A;S60A-dCas9) has a significant additive effect on aneuploidy induction that is biased toward chromosome gains (see Discussion).
We next set out to evaluate which parameters and conditions affect KaryoCreate’s efficiency, focusing on KNL1Mut-dCas9 due to its higher absolute level of aneuploidy induction compared to other constructs (see Discussion). Higher levels of KNL1Mut-dCas9 expression induced greater aneuploidy: a 3-fold increase in KNL1Mut-dCas9 expression led to a 2-fold increase in gains or losses (Fig. S3D). Next, combining multiple sgRNAs targeting the same chromosome (sgChr7–1 + sgChr7–3 or sgChr9–3 + sgChr9–5) did not increase the percentage of aneuploid cells over that due to individual sgRNAs, despite the increase in predicted binding sites achieved by combining the sgRNAs (Fig. S3E, 3F). We also tested whether FACS sorting, based on a cell surface marker encoded on the target chromosome, could increase the percentage of cells with gains or losses. We sorted cells transduced with KNL1Mut-dCas9 and sgChr7–1 based on high (top 15%) or low (bottom 15%) expression of EPHB4, a gene on chromosome 7 encoding a cell surface ephrin receptor. The percentage of cells with chromosome 7 gain increased from 12% to 26% from unsorted to high-EPHB4 cells (Fig. S3G), and the percentage of cells with chromosome 7 loss increased from 8% to 16% from unsorted to low-EPHB4 cells. Finally, a time-course experiment showed that sustained KaryoCreate activity increased aneuploidy progressively after 1, 2, or 3 cell cycles (2, 4, and 6 days after doxycycline; Fig. S3H). Altogether, the results indicate that KaryoCreate can induce chromosome-specific aneuploidy.
KaryoCreate allows induction of arm-level and chromosome-level gains and losses across human chromosomes
FISH analyses showed that targeting chromosome 7 does not affect chromosome 18 and vice versa, but did not rule out erroneous targeting of other chromosomes. To extend our analysis of KaryoCreate’s specificity across all chromosomes, we performed high-throughput single-cell RNA sequencing (scRNA-seq) to estimate genome-wide DNA copy number profiles across thousands of cells46–48. To infer copy number, we use the mean expression of genes across each chromosome or arm as a proxy for DNA copy number and then estimated the percentage of gains and losses for each arm by comparing the DNA copy number distribution of each experimental sample to that of the control population (e.g. sgNC or untreated cells). To prove the ability to infer arm-level copy number through scRNA-seq, we compared scRNA-seq and bulk shallow WGS results for hCEC cell lines with specific gains and losses. Analysis of a trisomic chromosome 7 clone showed that the percentage of cells with chromosome 7 gain was 91% by FISH and 80% by scRNA-seq. Similarly, analysis of the more complex karyotype (+chr7, −chr18, +19p) showed that the percentage of cells with chromosome 7 gain was 88% by FISH and 76% by scRNA-seq, and that for chromosome 18 loss was 87% by FISH and 81% by scRNA-seq (Fig. S4A, S4B). Notably, scRNA-seq slightly underestimated aneuploidy, especially gains, likely because a change from 2 to 3 copies represents an increase in DNA and RNA of 33%, while loss of 1 copy from 2 copies corresponds to a decrease of 50%. Overall, the patterns of aneuploidy inferred by scRNA-seq recapitulated those revealed by bulk WGS, confirming the validity of scRNA-seq for analyzing genome-wide gains and losses in single cells.
We performed scRNA-seq on diploid hCECs 7 days after KaryoCreate for chromosome 7 (sgChr7–1), chromosome 18 (sgChr18–4), and sgNC to estimate the frequency of induced aneuploidy (Fig. 4; pIND20 vector, expression level intermediate compared to those in Fig. S3D). For each sample, we estimated arm-level gains or losses for most chromosomes, except those with few (<20) genes detected on the p arm. First, we confirmed that the expression of KNL1Mut-dCas9 with the sgNC construct did not significantly induce aneuploidy compared to that in cells treated with the EV control (Fig. 4, Fig. S4C), as it led to very low percentages of gains and losses across chromosomes, averaging 0.9% for gains and 1.2% for losses. We confirmed the induction of chromosome-specific gains or losses after KaryoCreate, consistent with our FISH experiments (Fig. 3D, 3E). For example, scRNA-seq showed 10% gains and 17% losses for chromosome 18 (sgChr18–4) (Fig. 4, Table S3) and 9% and 11% gains and losses for chromosome 7 (sgChr7–1), respectively (Fig. 4, Table S3). Most importantly, scRNA-seq confirmed that KaryoCreate-induced aneuploidy was highly specific, with an average background level of nonspecific aneuploidy of 1% (Fig. 4, Table S3). Notably, the gains (0.9%) and losses (1.2%) observed in the sgNC sample across chromosomes are about 3 times lower than those observed by DNA FISH (3% for both gains and losses) (Fig. 3E), again suggesting that scRNA-seq underestimates aneuploidy, and especially gains, compared to FISH (Table S3).
Figure 4.

KaryoCreate induces both arm-level and chromosome-level gains and losses across different human chromosomes
Heatmap depicting arm-level copy numbers inferred from scRNA-seq analysis in KaryoCreate experiments using the indicated sgRNAs. scRNA-seq was used to quantify the presence of chromosome- or arm-level gains or losses using a modified version of CopyKat (see Methods). Rows represent individual cells, columns represent chromosomes, and colors represent gains in red and losses in blue. ‘Higher expression of KNL1Mut-dCas9’ indicates that the cells were transduced with a larger amount of the construct (as in Fig. S3D). See also Table S3 for quantification of arm- and chromosome-level events.
We further tested KaryoCreate using sgRNAs targeting additional chromosomes, including 6, 8, 9, 12, 16, and X, that were previously confirmed to induce foci with mScarlet-dCas9 (Fig. 4; see also Fig. 1 and Fig. S1). We performed KaryoCreate with the diploid hCECs expressing KNL1Mut-dCas9 (pIND20) and analyzed the cells through scRNA-seq 7 days after doxycycline induction. In all cases, cells expressing the chromosome-specific sgRNAs showed more gains and losses of the targeted chromosome than those expressing sgNC. The chromosome-specific gains and losses differed among the chromosomes and ranged between 5% and 12% for gains (average across 10 chromosomes: 8%) and between 7% and 17% for losses (average across 10 chromosomes: 12%) (Fig. 4, Table S3). Importantly, gains or losses of the non-targeted chromosomes never exceeded those in the sgNC control.
In agreement with our previous findings (Fig. S3D), the expression levels of the KNL1Mut-dCas9 construct correlated with the efficiency of KaryoCreate: a 3-fold increase in KNL1Mut-dCas9 expression (Fig. S3D) resulted in a 40–50% increase in both gains (from 9% to 16%) and losses (from 11% to 22%) (Fig. 4, compare sgChr7–1 and sgChr7–1 with high KNL1Mut-dCas9 expression). Furthermore, we successfully utilized KaryoCreate for inducing multiple chromosomal gains or losses in the same cells, by transducing cells simultaneously with multiple sgRNAs targeting different chromosomes (sgChr7–1 + sgChr18–4; 8% of cells had changes in both chromosomes 7 and 18 (Fig. S4F) or by utilizing a single sgRNA targeting multiple chromosomes (e.g. sgRNA 13–5 which targets both chromosomes 13 and 21 in hCEC; Fig. 4, Table S3). Finally, we obtained similar results using KaryoCreate in TP53 WT RPEs (Fig. S4D), suggesting that the method can be applied to different cell lines and in cells with an intact TP53 pathway.
Throughout the scRNA-seq analysis, we noted that in addition to whole-chromosome gains and losses, KaryoCreate also induced arm-level events, in which only one chromosomal arm (p or q) is gained or lost. Across the chromosomes tested, approximately 60% of aneuploidy events involved chromosome arms and 40% affected whole chromosomes (Fig. S4E). On average, there were 28% whole-chromosome losses, 17% whole-chromosome gains, 32% arm-level gains, and 23% arm-level losses (Fig. S4E, Table S3). Consistent with arm-level aneuploidy, we observed a modest increase in centromeric foci detected with the DNA damage marker γH2AX after expression of KNL1Mut-dCas9 and sgChr7–1 or sgChr18–4 (but not sgNC) for 10 days in HCT116 cells, in both interphase nuclei and mitotic cells; the average γH2AX signal intensity per cell, normalized to DAPI, also increased (Fig. S4G–S4H and data not shown). In a time-course experiment, γH2AX signal had increased after 4 days of doxycycline treatment (approximately two cell cycles) but not after 2 days (approximately one cell cycle) (Fig. S4I). Notably, the ratio between arm-level and chromosome-level events also increased significantly after 4 (and 6) compared to 2 days of doxycycline treatment (Fig. S3H), indicating that DNA damage signal increases over prolonged binding of KNL1Mut-dCas9 to the centromere and proportionally to arm-level events (see Discussion).
Altogether these data show that KaryoCreate can generate chromosomal gains and losses across individual chromosomes as well as combinations of the human autosomes and sex chromosomes.
18q loss in colon cells promotes resistance to TGFβ signaling likely due to haploinsufficiency of multiple genes
We used KaryoCreate to model 18q loss and chromosome 7 gain, aneuploidy events frequently found in colorectal cancer. Chromosome 18q is lost in about 62% of colorectal cancer (TCGA Dataset;49, Fig. 5A), and patients with 18q loss (N=136) show poorer survival than those without (N=86) (p=0.04, log-rank test, Fig. 5B). Chromosome 7 gain is present in 50% of patients (Fig. 5A).
Figure 5.

Loss of 18q in colon cancer cells promotes resistance to TGFβ signaling
(A) Frequency of copy number alteration in colorectal cancer (TCGA) indicated as percentage of patients with gain or loss for each chromosome.
(B) Kaplan-Meier survival analysis for colorectal cancer patients (TCGA) displaying or not displaying 18q loss (N=number).
(C) Left: Shallow WGS analysis of single-cell-derived clones obtained by KaryoCreate using sgNC or sgChr18–4 performed on diploid hCECs to identify arm-level gains and losses. Each row represents a single clone. Right: Plots of copy number alterations from WGS of two representative clones treated with sgChr18–4.
(D) Bulk RNA-seq showing differential expression analysis between clone 14 (18q loss) and clone 13 (diploid) using DESeq2 and GSEA (performed using the Hallmark gene sets); the top 7 pathways depleted in clone 14 are shown, including TGFβ signaling as the top depleted one.
(E) Effects of TGFβ (20 ng/ml) on clone 13 and 14 growth monitored for 9 days. Cells were counted every 3 days in quadruplicates. p-value is from Wilcoxon test comparing the difference in cell number between treated and untreated clone 14 cultures versus the same difference calculated for clone 13 cultures.
(F) Top 10 predicted tumor-suppressor genes (TSG) on 18q and their genomic locations. TSG were predicted based on the correlation between DNA and RNA levels, survival analysis, and TUSON-based q-value for the prediction of TSGs4 (see Methods).
(G) Western blot analysis for SMAD2, SMAD4, and GAPDH (as control) in clones 13 and 14. Quantification of SMAD2/SMAD4 levels after normalization against GAPDH.
To model these events, we performed KaryoCreate on hCECs using sgChr7–1, sgChr18–4, or sgNC as above (see also Methods). About 20 single-cell-derived clones were derived for each condition and their copy number profiles evaluated by WGS. After KaryoCreate, cells were seeded at low density and allowed to grow into colonies for 3–4 weeks, a longer time than in the experiments above (Fig. 4), during which cells likely experienced selective pressure for the ability to grow as single colonies (Fig. S5A).
Compared to clones derived from the sgNC control population, clones derived from sgChr7–1 showed an increase from 0% in sgNC to 22% in chr7 gains but no losses (0 for both conditions) (Fig. S5B). Clones derived from sgChr18–4 showed an increase from 0% in sgNC to 30% in chr18 loss losses but not gains (0 for both conditions) (Fig. 5C). This recapitulates the recurrent patterns observed in human tumors, where chromosome 18 is frequently lost but virtually never gained (2%), whereas chromosome 7 is frequently gained and almost never lost (0.3%). We did not observe aneuploidy of chromosomes not targeted by KaryoCreate except for 10q gain, which was present in ~20% of clones for all conditions, including sgNC, and was likely present in the initial population. Next, to test whether KaryoCreate clones can be stably propagated, we cultured a chromosome 7 trisomic clone (sgChr7–1 clone 23) for several weeks; we confirmed chromosome 7 gain by FISH and WGS analysis before and after 25 population doublings (Fig. S5C). We obtained similar results for sgChr18–4 clone 14 (data not shown).
Given the association of chromosome 18q loss with poor survival (Fig. 5B), we characterized the phenotypes of clones with or without this loss, starting from two clones derived from the KaryoCreate hCECs with sgChr18–4: one disomic control (clone 13) and one with 18q loss (clone 14). We performed bulk RNA sequencing analyses of each clone and conducted differential expression analysis using DESeq250. Gene-set enrichment analysis (GSEA) for cancer hallmarks showed that the top pathway downregulated in clone 14 compared to clone 13 was TGFβ signaling (enrichment score=−0.59; q-value=0.006), followed by cholesterol homeostasis, myogenesis, and bile acid metabolism (Fig. 5D, Table S4). TGFβ (transforming growth factor beta) normally inhibits the proliferation of colon epithelial cells by promoting their differentiation; its inhibition through intestine niche factors such as Noggin is essential for the proliferation and expansion of colon epithelial cells51. We tested the effect of TGFβ activation in our clones through an in vitro cell proliferation assay in which we cultured clones 13 and 14 in the presence of TGFβ (20 ng/ml) for 10 days. At day 9, TGFβ treatment had reduced cell growth by about 45% for the control clone 13 but <10% for clone 14 (Fig. 5E; p=0.02). Altogether, these data suggest that 18q deletion leads to decreased response to the growth-inhibitory signals derived from TGFβ treatment. We obtained similar results with an independent pair of different clones, clone 10 (diploid) and clone 5 (lacking chromosome 18) (Fig. S5E).
Chromosome 18q harbors the tumor-suppressor gene SMAD4 (located on 18q21.2), encoding a transcription factor critical for mediating response to TGFβ signaling52,53. In colorectal cancer, SMAD4 can be inactivated through point mutation (29% of patients)54 or genomic loss (62% of patients); in 96% of cases of genomic loss, the deletion encompasses the entire chromosome arm. A previous study suggested that mutations may occur before chromosomal instability54. Independently of the timing of SMAD4 mutations versus 18q loss, it is unknown whether the decreased survival in 18q loss patients (Fig. 5B) is a consequence of the complete loss of SMAD4 (due to co-occurring point mutation in the other allele) or is independent of SMAD4 mutation and possibly due to simultaneous loss of several tumor-suppressor genes on 18q, as previously suggested55. To distinguish between these possibilities, we assessed the contribution of 18q loss to patient survival after excluding patients with point mutations in SMAD4: if 18q loss serves to abolish SMAD4 function through deletion of the wild-type allele when one copy of SMAD4 carries a point mutation, we would predict that 18q loss would lose its association with patient survival after patients with SMAD4 mutations are excluded. Interestingly, 18q loss remained a significant predictor of survival after SMAD4-mutated patients were removed, indicating that decreased survival could be a consequence of the deletion of several tumor-suppressor genes on 18q (Fig. S5D, p-value of 0.006, lower than in the analysis including all patients, see Fig. 5B).
To systematically predict tumor-suppressor genes located on 18q, we developed a score using three computational parameters based on the TCGA dataset: 1. correlation between DNA and RNA level of each gene across patients56; 2. association of expression level of each gene with patients’ survival; 3. TUSON-based prediction of the likelihood for a gene to behave as a tumor-suppressor gene based on its pattern of point mutations4 (see Methods). The top ten predicted genes were SMAD2, ADNP2, MBD1, ATP8B1, WDR7, MBD2, DYM, SMAD4, ZBTB7C, and LMAN1 (Fig. 5F, Table S4). SMAD2, a paralogue of SMAD4 located on 18q21.1, is also a transcription factor acting downstream of TGFβ signaling51,57. Thus, concomitant decreases in gene dosage of both SMAD4 and SMAD2 could synergistically mediate the unresponsiveness of cells to TGFβ signaling.
We tested the role of decreased dosage of SMAD2 and SMAD4 proteins in our clone containing 18q loss. We confirmed by both RNA-seq and Western blotting a decrease in both SMAD2 and SMAD4 in clone 14 compared to control clone 13 (Fig. 5G, Table S4; SMAD4 log2FC: −0.78, p<0.0001; SMAD2 log2FC: −0.75, p<0.0001). Furthermore, overexpression of SMAD2 and SMAD4 in clone 14 decreased proliferation rate after TGFβ treatment to a level similar to clone 13 (Fig. S5E, S5F). To further test whether the increased resistance to TGFβ treatment after 18q loss was due to the synergistic effects of decreases in both SMAD2 and SMAD4 (as opposed to SMAD4 only), we derived hCECs with a ~50% decrease in SMAD4 protein level by CRISPR interference (Fig. S5G, S5H). In proliferation assays, cells with 18q loss (clone 14) were more resistant to TGFβ treatment than hCECs with decreased SMAD4 levels (Fig. S5G,H), indicating that 18q loss has a greater effect than a ~50% decrease in SMAD4 expression.
Altogether, our computational and experimental data suggest that chromosome 18q loss, one of the most frequent events in gastro-intestinal cancers, is associated with poor survival and promotes resistance to TGFβ signaling, likely because of the synergistic effect of simultaneous deletion of haploinsufficient genes.
DISCUSSION
Chromosome-specific centromeric sgRNAs
KaryoCreate relies on the design of sgRNAs targeting chromosome-specific α-satellite DNA. Among 75 tested, we validated 24 sgRNAs specific for 16 different chromosomes (Fig. 1, Fig. S1, Table S1). Since centromere sequences vary across the human population, we designed sgRNAs using two genome assemblies (CHM13 and GRCh38) and tested them in different cell lines (hCECs, RPEs, and HCT116), increasing their likelihood of targeting conserved regions.
Our study showcases the design and use of sgRNAs to target human centromeres, yet for some chromosomes we were unable to design or validate specific sgRNAs. Depending on the chromosome, this was due to centromeric sequences sharing high similarity across specific chromosome groups (i.e. acrocentric), to the low GC content of centromeric sequences likely decreasing the gRNA activity, or to a relative paucity of predicted binding sites (e.g. D21Z1, D15Z3, and D3Z1 in the CHM13 assembly have relatively small active centromere regions)21,58. Furthermore, the efficiency of centromeric sgRNAs is not accurately predicted using algorithms for non-centromeric regions35 (Fig. S1E). Moreover, using more than one sgRNA simultaneously did not improve aneuploidy induction (Fig. S3E, S3F). Because of the repetitive nature of centromeres, any pair of sgRNAs is predicted to bind multiple times and relatively close together, potentially inducing competition or interference among KNL1Mut-dCas9 molecules.
Comparison of KaryoCreate with similar technologies
Other strategies have been recently described to induce chromosome-specific aneuploidy targeting non-centromeric repeats and have been successful for chromosome 1 using a sub-telomeric repeat and chromosome 9 using a pericentromeric repeat16,17. Tovini et al. used dCas9 fused to the kinetochore-nucleating domain of CENPT to form an ectopic kinetochore. Truong et al. tethered a plant kinesin to pull the chromatids towards one pole of the mitotic spindle, potentially generating a pseudo-dicentric chromosome, as suggested by the fact that most aneuploidies observed were of part of the targeted chromosome (chromosome 9). Both methods will be especially useful to dissect the spindle assembly checkpoint, the fate of dicentric chromosomes, and the biophysical properties of chromosomal behavior at the metaphasic plate. KaryoCreate is distinct in that it relies on endogenous centromeric sequences to allow the generation of nearly any karyotype of interest. We found that cells progressed normally through the cell cycle with an expected brief delay in metaphase, likely due to attempts at correcting merotelic attachments59,60. Also, in contrast to existing technologies, KaryoCreate can induce specific aneuploidies across several chromosomes or combinations thereof (Table S3). Finally, KaryoCreate enables induction of aneuploidy not only in TP53−/− cells but also in TP53 WT cells such as HCT116 cells (Fig. 2E) and RPEs (Fig. S4D).
Targeting mutant kinetochore proteins to centromeric α-satellites to engineer chromosome-specific aneuploidy
Tethering of chimeric dCas9 with mutant forms of KNL1 or NDC80 to human centromeres induces chromosome- and arm-level gains and losses (Fig. S3B). Our work and other studies suggest that dCas9 itself may induce low-frequency aneuploidy, possibly due to tethering of a bulky protein to the centromeric repeats16,17,42. Remarkably, the expression of chimeric mutants of kinetochore proteins at centromeric regions induces about 3 times as many aneuploidy events compared to dCas9 alone, reasonably due to the disruption of their proper kinetochore functions (Fig. S3B). Future studies will be necessary to clarify this point and may be instrumental in further improving KaryoCreate efficiency. For example, we noted that different mutants show different efficiency of aneuploidy induction relative to their expression level (Fig. S2B, S3B). NDC80 mutants induced aneuploidy efficiently relative to their low expression level, suggesting a higher degree of kinetochore disruption compared to KNL1 fusion (Fig. S2B, S3B). Of the two chimeras containing KNL1 mutants, we predicted that KNL1S24A;S60A-dCas9 would result in a more efficient induction of chromosome gains and losses than KNL1RVSF/AAAA-dCas9, owing to a more efficient inhibition of Aurora-B-mediated error correction through recruitment of PP128,41. Although this was not the case in terms of absolute level of aneuploidy, KNL1S24A;S60A-dCas9 efficiency was higher when normalized for protein expression level (Fig. S3B).
Induction of arm-level gains and losses
About 55% of the aneuploidy generated by KaryoCreate are arm-level events. In addition, we observed more losses (60%) than gains (40%) for both chromosome and arm events. Our findings are consistent with another recent study of similar methods for aneuploidy induction that also found a high level of chromosome losses and predominantly arm-level events17. Our data reveal a small fraction of centromeres positive for γH2AX upon aneuploidy induction with KaryoCreate (Fig. S4G–S4I), especially upon prolonged centromere recruitment of KNL1Mut-dCas9 and proportionally to the ratio between arm-level and chromosome-level events (Fig. S3H). The mere recruitment of a bulky protein to the centromere may influence centromere function, as our data on the effect of dCas9 alone suggest (Fig. S3B)18,31,61–63. When recruited to the highly repetitive centromeric regions, dCas9 may influence chromosome segregation through impaired replication or transcription affecting chromatin, transcripts, and R-loops and, in turn, centromere function62–66.
Chromosome-specific aneuploidy as a driver of cancer hallmarks
We used KaryoCreate to induce missegregation of chromosomes 7 and 18, two of the chromosomes most frequently aneuploid in colorectal tumors. Among the single-cell-derived clones, chromosome 7 tended to be gained and chromosome 18 tended to be lost (Fig. 5C, Fig. S5B), indicating that the selective pressure acting during tumor evolution to shape recurrent patterns of aneuploidy may also act in vitro4,7. In our analyses, 18q loss was a strong predictor of poor survival, consistent with previous studies67,68; in addition the association of 18q loss with survival was independent of SMAD4 point mutations. We showed that chr18q loss can promote resistance to TGFβ signaling in colon cells. While SMAD4 is a frequently mutated tumor-suppressor gene54 on chr18q, the TGFβ resistance phenotype determined by 18q loss may be due not solely to its loss but to the cumulative effect of losing multiple tumor suppressors on the arm. In fact, ~50% reduction in SMAD4 alone was not sufficient to recapitulate resistance to TGFβ signaling seen after 18q loss, and dosage increases in both SMAD4 and SMAD2 could rescue TGFβ resistance in 18q loss cells (Fig. 5E, Fig. S5E–S5H). Thus, chromosome 18 loss may drive TGFβ resistance through hemizygous deletion of (at least) two haploinsufficient genes acting in the same pathway.
Previous studies have proposed that a single cancer-driver gene may confer the strong phenotypic effect of whole-chromosome gain or loss69,70. Other studies, including previous work on chromosome 18, have proposed that the selective advantage of aneuploidy is instead conferred by the cumulative effect of gene dosages of multiple genes4,6,55,71. Our experimental data support this latter hypothesis. Altogether, these data suggest that 18q loss may drive tumor phenotypes in colorectal cancer through the cumulative loss of several tumor-suppressor genes located on the chromosome arm.
Limitations of the study
These data reveal KaryoCreate as a powerful resource to foster our understanding of chromosome missegregation and aneuploidy in several fields of biomedicine, including genetics, centromere biology, and cancer. However, in its current design, KaryoCreate cannot target all human chromosomes, and its efficacy depends on the ability to transfect or infect target cells. Finally, KaryoCreate induces more losses than gains and more arm-level events than whole-chromosome events. Because missegregation likely depends on several mechanisms, further studies will be necessary to clarify and improve the method for wider applications.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to the lead contact, Teresa Davoli (Teresa.Davoli@nyulangone.org, t.davoli@gmail.com).
Materials availability
The plasmids generated in this study are available upon request.
Data and code availability
Single-cell RNA sequencing and bulk RNA sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Whole-genome sequencing data have been deposited at SRA and are publicly available as of the date of publication. Accession number is listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Cas9 antibody | Abcam | Cat#Ab191468 |
| GAPDH antibody | Santa Cruz Biotechnology | Cat#Sc-47724 |
| B-Actin antibody | Cell Signaling Technology | Cat#8844 |
| Goat Anti-Mouse IgG H&L (HRP) | Abcam | Cat#Ab205719 |
| Anti-centromere protein antibody | Antibodies Incorporated | SKU 15-234 |
| Anti-a-Tubulin antibody | Sigma-Aldrich | T9026 |
| CyTM3 AffiniPure Donkey Anti-Mouse IgG | Jackson ImmunoResearch | 715-165-150; RRID: AB_2340813 |
| Alexa Fluor® 647 AffiniPure F(ab’)₂ Fragment Donkey Anti-Human |
Jackson ImmunoResearch | 709-606-149; RRID: AB_2340581 |
| TotalSeq-B0256 anti-human Hashtag 1 Antibody | BioLegend | Cat#394631 |
| TotalSeq-B0256 anti-human Hashtag 2 Antibody | BioLegend | Cat#394633 |
| TotalSeq-B0256 anti-human Hashtag 3 Antibody | BioLegend | Cat#394635 |
| TotalSeq-B0256 anti-human Hashtag 4 Antibody | BioLegend | Cat#394637 |
| TotalSeq-B0256 anti-human Hashtag 5 Antibody | BioLegend | Cat#394649 |
| TotalSeq-B0256 anti-human Hashtag 6 Antibody | BioLegend | Cat#394641 |
| TotalSeq-B0256 anti-human Hashtag 7 Antibody | BioLegend | Cat#394643 |
| TotalSeq-B0256 anti-human Hashtag 8 Antibody | BioLegend | Cat#394645 |
| TotalSeq-B0256 anti-human Hashtag 9 Antibody | BioLegend | Cat#394647 |
| TotalSeq-B0256 anti-human Hashtag 10 Antibody | BioLegend | Cat#394649 |
| Anti-yH2A.X Antibody | Sigma-Aldrich | SKU 05-636 |
| Alexa Fluor® 647 AffiniPuro Goat Anti-Human | Jackson ImmunoResearch | 109-605-044; RRID: AB_2337885 |
| Alexa Fluor® 488 AffiniPuro Donkey Anti-Rabbit | Jackson ImmunoResearch | 711-545-152; RRID: AB_2313584 |
| Anti-Smad2 antibody | Abcam | Cat#Ab40855 |
| Smad4 Antibody | Santa Cruz Biotechnology | Cat#Sc-7966 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Pierce FITC Conjugated Avidin | ThermoFisher Scientific | Cat#21221 |
| Anti-Digoxigenin-Rhodamine, Fab fragments | Sigma-Aldrich | Cat#112077509 1 |
| RO-3306 | Sigma-Aldrich | Cat#SML0569; CAS: 872573-93-8 |
| MG-132 | Tocris | Cat#1748; CAS: 133407-82-6 |
| Colcemid | Roche | Cat#102958920 01 |
| Doxycycline | Sigma | Cat#D5207 |
| Shield-1 | CheminPharma | CIP-S1-0.5nM |
| Lipofectamine 3000 Transfection Reagent | ThermoFisher Scientific | Cat#L3000075 |
| DAPI | Sigma-Aldrich | Cat#MBD0015; CAS:28718-90-3 |
| ProLong Glass Antifade Moutant | ThermoFisher Scientific | Cat#P36980 |
| Critical commercial assays | ||
| Gateway LR Clonase II Enzyme mix | ThermoFisher Scientific | Cat#11791020 |
| Chromosome 7 Control Probe | Empire Genomics | Cat#CHR07-10GR |
| Chromosome 18 Control Probe | Empire Genomics | Cat#CHR18-10-GR |
| NEBNext dsDNA Fragmentase | New England Biolabs | Cat#M0348 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7645L |
| Qubit 2.0 fluorometer | Invitrogen | Cat#Q32866 |
| Qubit dsDNA HS kit | Invitrogen | Cat#Q32854 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| 2100 Bioanalyzer system | Agilent | Cat#G2939BA |
| TruSeq Stranded Total RNA Library Prep Gold | Illumina | Cat#20020598 |
| Agilent 2200 TapeStation System | Agilent | G2964AA |
| Agilent High Sensitivity D1000 ScreenTape | Agilent | 5067-5584 |
| NovaSeq 6000 SP Reagent Kit v1.5 (100 cycles) | Illumina | Cat#20028401 |
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| Chromium Single Cell B Chip Kit | 10X Genomics | PN-1000073 |
| Chromium i7 Multiplex Kit | 10X Genomics | Pn-120262 |
| Experimental models: Cell lines | ||
| hCEC hTERT | Ly et al.38 | PMC:3071083 |
| hCEC hTERT TP53−/− | This paper | N/A |
| HCT116 | ATCC | CCL-247 |
| RPE hTERT | ATCC | CRL-4000 |
| RPE hTERT p21/Rb shRNA | Maciejowski et al.39 | PMID:26687355 |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| gNC: ACGGAGGCTAAGCGTCGCAA | Sanjana et al.40 | N/A |
| Chromosome-specific gRNAs, see table S1 | This paper | N/A |
| SMAD4 CRISPRi gRNA: GGCAGCGGCGACGACGACCA |
Gilbert et al.76 | N/A |
| Recombinant DNA | ||
| plentiGuide-Puro | Chen et al.72 | Addgene #52963 |
| pLentiGuide-Puro-FE | This paper | N/A |
| pX330-U6-Chimeric_BB_CBh-hSpCas9 | Cong et al.74 | Addgene #42230 |
| Chromosome 7 BAC | BACPAC Genomics | RP11-22N19 |
| Chromosome 13 BAC | BACPAC Genomics | RP11-76N11 |
| Chromosome 18 BAC | BACPAC Genomics | RP11-787K12 |
| H2B-GFP plasmid | Titia de Lange lab | pCLRNX-H2B-GFP |
| pHAGE-3xmScarlet-dCas9 | This paper | N/A |
| pHAGE-KNL1Mut-dCas9 | This paper | N/A |
| pIND20-KNL1Mut-dCas9 | This paper | N/A |
| pIND20-GFP | This paper | N/A |
| pHAGE-DD-KNL1Mut-dCas9 | This paper | N/A |
| pHAGE-KNL1S24A;S60A-dCas9 | This paper | N/A |
| pHAGE-dCas9 | This paper | N/A |
| pHAGE-NDC80CH1-dCas9 | This paper | N/A |
| pHAGE-NDC80CH2-dCas9 | This paper | N/A |
| pInducer20 | Meerbrey et al.44 | Addgene #44012 |
| Software and algorithms | ||
| Photoshop v21.2.3 | Adobe | https://www.adobe.com |
| FIJI/ImageJ2 version 2.3.0/1.53f | Schindelin et al.75 | https://imagej.nih.gov/ij/download.html |
| Python 3.7 | Python Software Foundation | https://www.python.org/downloads/ |
| Scikit-image | Van Der Walt et al.77 | https://scikiti-mage.org |
| BWA-mem v0.7.17 | Li et al.78 | https://github.com/lh3/bwa/releases/tag/v0.7.17 |
| Genome Analysis Toolkit v4.1.7.0 | Van der Auwera, 202079 | https://gatk.broadinstitute.org/hc/en-us |
| CopywriteR v1.18.0 | Kuilman et al.80 | https://github.com/PeeperLab/CopywriteR |
| Seq-N-Slide | Dolgalev, 2022.81 | https://github.com/igordot/sns |
| Trimmomatic | Bolger et al.82 | https://github.com/timflutre/trimmomatic |
| STAR | Dobin et al.83 | https://github.com/alexdobin/STAR |
| featureCounts | Liao et al.84 | https://github.com/byee4/featureCounts |
| DESeq2 | Love et al.50 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA pre-ranked | Subramanian et al.85 | https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html |
| CellRanger v6.1 | 10X Genomics | https://support.10xgenomics.com/single-cell-geneexpression/software/overview/welcome |
| Seurat v4.0.3 | Hao et al.86 | https://github.com/satijalab/seurat |
| CopyKat v1.0.5 | Gao et al.46 | https://github.com/navinlabcode/copykat |
| ComplexHeatmap v2.8 | Gu et al.87 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Other | ||
| Code for automated FISH foci counting | This paper | https://doi.org/10.6084/m9.figshare.21843393 |
| Code for single-cell RNA-seq analysis | This paper | https://doi.org/10.6084/m9.figshare.21843393 |
| Glass-bottom microwell dishes | MatTek | Cat# P35G-1.5-14-C |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen Invitrogen |
Cat#NP0007 |
| NuPAGE 4 to 12% Bis-Tris Mini Protein Gels | Cat#NP0322BO X |
|
| Trans-Blot Turbo Mini 0.45 uM LF PVDF Transfer Kit | Bio-Rad | Cat#1704274 |
| Human Cot-1 DNA | ThermoFisher Scientific | Cat#15279011 |
| UltraPure Herring Sperm DNA Solution | ThermoFisher Scientific | Cat#15634017 |
| Proteinase K | Qiagen | Cat#19131 |
| Pierce ECL Western Blotting Substrate | Thermo Scientific | Cat#32209 |
| Sera-Mag Select beads | Cytiva | Cat#29343052 |
| Single-cell and bulk RNA sequencing data | This paper |
GSE217326; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217326 |
| Whole genome sequencing data | This paper | PRJNA899849;https://dataview.ncbi.nlm.nih.gov/object/PRJNA899849 |
| T2T Chm13v2.0.fa.gz assembly | Nurk et al.29 | GCA_00991475 5.4 https://github.com/marbl/CHM13 |
| GRChg38 reference assembly | Schneider et al.30 | GCA_00000140 5.28 https://hgdownload.soe.ucsc.edu/downloads.html |
| TCGA data | NA | https://www.cancer.gov/aboutnci/organization/ccg/research/structuralgenomics/tcga/using-tcga/tools |
| TUSON data | Davoli et al.4 | NA |
All original analysis code has been deposited to GitHub and is available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cells were grown at 37°C with 5% CO2 levels. hTERT TP53 −/−−/− human colonic epithelial cells (hCECs)38 were cultured in a 4:1 mix of DMEM:Medium 199, supplemented with 2% FBS, 5 ng/mL EGF, 1 μg/mL hydrocortisone, 10 μg/mL insulin, 2 μg/mL transferrin, 5 nM sodium selenite, pen-strep, and L-glutamine. hTERT retinal pigment epithelial cells (RPEs)39 either WT (Fig. S4D) or expressing p21 (CDKN1A) and RB (RB1) shRNAs (Fig. S1D), and human colorectal carcinoma-116 cells (HCT116s) were incubated in DMEM, supplemented with 10% FBS, pen-strep, and L-glutamine. For longterm storage, cells were cryopreserved at -−80°C in 70% medium (according to cell line), 20% FBS, 10% DMSO. TP53 was knocked- out in hCECs by transfection with a Cas9-containing plasmid (Addgene #42230) and plLentiGuide-Puro expressing the following sgRNA: GCATGGGCGGCATGAACCGG. Clones were derived and tested for the expression of TP53.
METHODS DETAILS
Cloning of KaryoCreate Constructs
Cas9 and dCas9 without ATG and without stop codon (for N-terminal and C-terminal tagging respectively) were cloned into D-TOPO vector (Thermo #K240020). Cloning of KNL1RVSF/AAAA-dCas9 was achieved by inserting KNL1 PCR product (aa1–86, amplified from Addgene plasmid #4522528) into XhoI-digested pENTR-dCas9 (no ATG) using Gibson assembly. The GGSGGGS linker was added between KNL1 and dCas9. Cloning of KNL1S24A;S60A-dCas9 was achieved starting from KNL1RVSF/AAAA-dCas9 and inserting the appropriate mutations using Gibson assembly. Cloning of NDC80-CH1-dCas9 was achieved by Gibson assembly of NDC80 aa1–207 (generously provided by Dr. Jennifer DeLuca) with BamHI-digested pENTR dCas9 (ATG). Cloning of NDC80-CH2-dCas9 was achieved in a similar way except that 2 CH domains were cloned in tandem separated by a linker (see also Fig. S2A).
To generate an inducible KNL1Mut-dCas9 construct, the FKBP12 degradation domain (DD, Banaszynski 200645) was first amplified from Degron-KI-donor backbone (Addgene #65483) and inserted at the N-terminus of the fusion protein sequence in pENTRKNL1RVSF/AAAA-dCas9 using Gibson cloning. Gateway LR cloning was then used to yield the expression vector, pHAGE-DD-KNL1RVSF/AAAA-dCas9.
pHAGE-3xmScarlet-dCas9 was generated by first assembling three mScarlets in series and inserting them into the BsaI-digested pAV10 vector by Golden Gate cloning. The assembled 3xmScarlet was then inserted into XhoI-digested pENTR-dCas9 using Gibson cloning to form pENTR-3xmScarlet-dCas9.
All pENTR vectors were cloned into specific pDEST vectors by LR reaction (Thermo #11791020) following the manufacturer’s instructions. pDEST vectors used in this study were pHAGE (blast resistance, CMV promoter) or pINDUCER20 (or pIND20, neomycin resistance, doxycycline inducible promoter)44.
Cloning of sgRNAs
We modified the scaffold sequence of pLentiGuide-Puro (Addgene #52963) by Gibson assembly to contain the A-U flip (F) and hairpin extension (E) described by Chen et al72. for improved sgRNA-dCas9 assembly, obtaining pLentiGuide-Puro-FE. sgRNAs were designed and cloned into this pLentiGuide-Puro-FE vector according to the Zhang Lab General Cloning Protocol73 (also https://www.addgene.org/crispr/zhang/) (see also Table S1 for sgRNA sequences). To be suitable for cloning into BbsI-digested vectors, sense oligos were designed with a CACC 5’ overhang and antisense oligos were designed with an AAAC 5’ overhang. The sense and antisense oligos were annealed, phosphorylated, and ligated into either BbsI-digested pLentiGuide-Puro-FE for KaryoCreate and imaging purposes or pX330-U6-Chimeric_BB-CBh-hSpCas974 (Addgene #42230) for CRISPR/Cas9 editing applications. Sequences were confirmed by Sanger sequencing.
Lentivirus production and nucleofection
For transduction of cells, lentivirus was generated as follows: 1 million 293T cells were seeded in a 6-well plate 24 hours before transfection. The cells were transfected with a mixture of gene transfer plasmid (2 μg) and packaging plasmids including 0.6 μg ENV (VSV-G; addgene #8454), 1 μg Packaging (pMDLg/pRRE; addgene #12251), and 0.5 μg pRSV-REV (addgene #12253) along with CaCl2 and 2× HBS or using Lipofectamine 3000 (Thermo #L3000075).The medium was changed 6 hours later and virus was collected 48 hours after transfection by filtering the medium through a 0.45-μm filter. Polybrene (1:1000) was added to filtered medium before infection.
Nucleofection of hCECs was carried out using the Amaxa Nucleofector II (Lonza), using the program optimized for the HCT116 cell line. Approximately 1 million cells suspended in 100 μL of electroporation buffer (80% 125 mM Na2HPO4.∙7H2O), 12.5 nM KCl, 20% 55 mM MgCl2) were subjected to electroporation in the presence of a vector and then immediately returned to normal medium.
KaryoCreate Experiments
We used three main ways to perform KaryoCreate experiments in this paper. The main difference between these methods is the way KNL1Mut-dCas9 and the sgRNA are expressed in the cell.
Methods to express KNL1Mut-dCas9:
KNL1Mut-dCas9 is expressed from a doxycycline-inducible promoter (pIND20-KNL1Mut-dCas9) through a viral vector constitutively integrated in the genome of the target cell. Cells are treated with doxycycline (1 μg/ul) for 7–9 days.
KNL1Mut-dCas9 is expressed from a constitutive promoter (pHAGE-KNL1Mut-dCas9; CMV promoter) through transient transfection.
KNL1Mut-dCas9 is expressed through a viral vector constitutively integrated in the genome of the target cell; the expression level of KNL1Mut-dCas9 is regulated through a degron (pHAGE-DD-KNL1Mut-dCas9; see above)
For the sgRNA, expression is mediated by pLentiGuide-Puro-FE vector through infection or transient transfection. In this paper, otherwise specified, the sgRNA was introduced through infection. For a comparison of the three different methods, see Figure S3A.
Western blot analysis
Cells were harvested by trypsinization, lysed in 2× NuPAGE LDS buffer (Thermo #NP0007) at 106 cells in 100 μl of buffer. DNA was sheared using a 28 1/2-gauge insulin syringe and lysate was denatured by heating at 80°C for 10 min. Lysate equivalent to 105 cells was resolved by SDS/PAGE using a NuPAGE 4–12% Bis-Tris mini gel and transferred to a PVDF membrane (Bio-Rad #1704274). The membrane was then blocked in 5% milk in TBS with 0.1% Tween-20 (TBS-T) for 1 hour at room temperature. Afterward, the membrane was probed with Cas9 (Abcam #ab191468, 1:1000 dilution) and GAPDH (Santa Cruz #sc-47724, 1:10,000 or 1:100,000 dilution) or β-actin (Cell Signaling Technology #8844) primary antibodies and incubated in 1% milk in TBS at 4°C overnight. For SMAD2 and SMAD4 western blots, Abcam Ab40855 and Santa Cruz Biotechnology #Sc-7966 were used.
Subsequently, the membrane was washed three times with TBS-T and incubated with HRP-anti-Mouse secondary Ab (Abcam #ab205719, 1:1000 dilution) in 1% milk/TBS for 1 hour at room temperature. Signals were detected using an ECL system using 1:1 detection solution (Thermo Scientific #32209) after three 10-min washes in TBS-T. Images were acquired using a BIORAD transilluminator.
Fluorescence in situ hybridization (FISH)
For the analyses confirming centromeric localization of 3xmScarlet-dCas9 and localization of specific chromosomes within micronuclei, FISH was performed using an Empire Genomics chromosome 7 control probe (CHR07–10-GR) or chromosome 18 control probe (CHR18–10-GR) on PFA-fixed cells according to the manufacturer’s manual hybridization protocol.
FISH analysis was carried out on interphase nuclei and metaphase spreads prepared as follows: Cells at 70% confluence were harvested by trypsinization (after 3- to 4-hour treatment with 100 ng/mL colcemid (Roche #10295892001) for metaphase spreads), washed with PBS, suspended in 0.075 M KCl at 37°C, and fixed in methanol-acetic acid (3:1) at 4°C. Fixed cells were dropped onto glass slides and then allowed to air dry overnight.
The slides were next incubated with RNase solution (20 μg RNase A in 2× SSC ) for one hour at 37°C in a dark moist chamber. Denaturing was performed using a 70% formamide solution (in 2× SSC) for 3 min at 80°C prior to hybridization. Biotinylated/digoxigeninated probes were obtained by nick translation from BAC DNA (RP11–22N19 for chromosome 7, RP11–76N11 for chromosome 13, and RP11–787K12 for chromosome 18 from the BACPAC Resource Center). 200 ng of each labeled probe, together with 8 μg Human Cot-I DNA (Thermo #15279011) and 3 μg Herring Sperm DNA (Thermo #15634017) were precipitated for 1 hour at −20°C in 1/10 volume of 3 M sodium acetate and 3 volumes of ethanol. The pelleted probe was washed with 70% ethanol, air dried, and resuspended in hybridization solution (50% deionized formamide, 10× dextran sulfate, 2× SSC). The hybridization solution containing the probes was then denatured at 80°C for 10 min and then incubated at 37°C for 20 min to allow annealing of the Cot-I competitor DNA. The sealed hybridized slides were then incubated at 37°C in a dark moist chamber overnight. The following day, slides were washed in 1× SSC at 60°C (3 times, 5 min each) and incubated with a blocking solution (BSA, 2× SSC, 0.1% Tween-20) for 1 hour at 37°C in a moist chamber. Following blocking, the slides were incubated with detection solution containing BSA , 2× SSC , 0.1% Tween-20, and FITC-Avidin conjugated (Thermo #21221), and 10 μl Rhodamine-Anti-Digoxigenin (Sigma #11207750910) to detect the biotin and digoxigenin signals. Finally, slides were washed 3 times (5 min each) with 4× SSC and 0.1% Tween-20 solution at 42°C and then mounted with DAPI to stain DNA (Vector Laboratories #H-1200–10).
Images were acquired using an Invitrogen™ Evos™ M700 imaging system or Nikon TI Eclipse. The number of fluorescent signals was counted in 100 intact nuclei per slide. Adobe Photoshop was used to count the signals and correct the images.
Live-cell imaging
Cells were plated on 35-mm glass-bottom microwell dishes (MatTek P35G-1.5–14-C) 1 day prior to imaging. Imaging was performed at 37°C and 5% CO2 using an Andor Yokogawa CSU-X confocal spinning disc on a Nikon TI Eclipse microscope. Samples were exposed to 488-nm (30-ms) and 561-nm (100-ms) lasers and fluorescence was recorded with a sCMOS Prime95B camera (Photometrics). A 100× objective was used to acquire images at 0.9-μm steps (total range size=9 μm) every 1 or 3 min as indicated in the figure legends. Image analysis was performed using ImageJ and formatting (cropping, contrast adjustment, labeling) was performed in Adobe Photoshop.
Chromosome misalignment staining
HCT116 cells were plated onto coverslips coated with 5 μg/ml fibronectin (Sigma-Aldrich) at 60–70% confluence and synchronized with 7.5 μM RO-3306 (Sigma-Aldrich) for 16 hours at 37°C. Cells were released from RO-3306 for 40 min and then treated with 10 uM MG-132 (Tocris) for 90 min at 37°C. Cells were then fixed with 4% paraformaldehyde for 12 min at room temperature and blocked in 5% BSA for 30 min. Samples were stained with the following antibodies for 90 min at room temperature: anti-ɑ-Tubulin (Sigma-Aldrich #T9026, 1:1500 dilution) and anti-centromeric antibody (Antibodies Incorporated SKU 15-234, 1:100 dilution). CyTM3 AffiniPure (Jackson ImmunResearch #715-165-150) and Alexa 647-labeled (Jackson ImmunoResearch #709-606-149) secondary antibodies were used 1:400 for 45 min at room temperature. Coverslips were mounted using Mowiol. Cells were imaged using a Leica SP5 confocal microscope with a magnification objective of 63×. FIJI software was used for image analysis.
Low-pass whole-genome sequencing
Genomic DNA was extracted from trypsinized cells using 0.3 μg/μL Proteinase K (Qiagen #19131) in 10 mM Tris, pH 8.0, for 1 hour at 55°C and then heat inactivated at 70°C for 10 min. DNA was digested using NEBNext® dsDNA Fragmentase® (NEB #M0348S) for 25 min at 37°C and then subjected to magnetic DNA bead cleanup with Sera-Mag Select Beads (Cytiva #293430452), 2:1 bead/lysate ratio by volume. DNA libraries with an average library size of 320 bp were created using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (NEB #E7645L) according to the manufacturer’s instructions. Quantification was performed using a Qubit 2.0 fluorometer (Invitrogen #Q32866) and the Qubit dsDNA HS kit (Invitrogen #Q32854). Libraries were sequenced on an Illumina NextSeq 500 at a target depth of 4 million reads in either paired-end mode (2 × 36 cycles) or single-end mode (1 × 75 cycles).
RNA bulk sequencing
Clones were plated in 6-well plates 1 day before collection. On the day of collection, cells were checked for confluency within 70–90% and normal morphology. Cells were washed twice with PBS and stored at −80°C immediately. RNA was purified for bulk sequencing using the Qiagen RNeasy Mini Kit (Qiagen #74106). RNA concentration and integrity were assessed using a 2100 BioAnalyzer (Agilent #G2939BA). Sequencing libraries were constructed using the TruSeq Stranded Total RNA Library Prep Gold (Illumina #20020598) with an input of 250 ng and 13 cycles final amplification. Final libraries were quantified using High Sensitivity D1000 ScreenTape (Agilent #5067-5584) on a 2200 TapeStation (Agilent #G2964AA) and Qubit 1× dsDNA HS Assay Kit (Invitrogen #Q32854). Samples were pooled equimolar with sequencing performed on an Illumina NovaSeq6000 SP 100 Cycle Flow Cell v1.5 as Paired-end 50 reads.
Clone derivation
hCECs were transduced with pHAGE-DD-KNL1Mut-dCas9 and a sgRNA vector and DD-KNL1Mut-dCas9 was stabilized with 100 nM Shield-1 (CheminPharma #CIP-S1, 0.5 nM) for 9 days. Three days after Shield-1 treatment, 20–500 cells were plated per 15-cm plate and were incubated in normal culture conditions until colonies were visible (~2–3 weeks). Colonies were then picked by applying wax cylinders to the area surrounding each clone, trypsinizing the cells, and moving them to separate wells in 48-well plates for further expansion.
Single-cell RNA sequencing scRNA-seq libraries were prepared using the 10× Chromium Single-Cell 3’ v3 Gene Expression kit according to the manufacturer’s instructions, including the manufacturer’s protocol for cell surface protein (hashtag antibody) feature barcoding. Up to 10 TotalSeqB hashtag antibodies (BioLegend) were used for multiplexing samples in each sequencing run.
Immunofluorescence for centromeric damage
Cells were grown on poly-L-lysine coverslips, fixed in PFA (Sigma-Aldrich 8187081000) 2% in 1× PBS, and washed three times in 1× PBS. Fixed cells were permeabilized with 1× PBS and 0.2% Triton (Sigma-Aldrich X100, 500 ml) for 5 min at room temperature and washed again before being blocked with PBS-0.1% Tween 20 (Sigma-Aldrich P1379, 500 ml) plus 5% BSA for 10 min. Cells were then incubated with primary antibodies, γH2AX (Sigma-Aldrich 05–636) diluted 1:200 and CREST (Antibodies Incorporated 15-234-0001). After 45 min, cells were washed three times with 1× PBS and 0.1% Tween 20 and then incubated with the secondary antibodies anti-Mouse Alexa-488 (Jackson ImmunoResearch 711-545-152) and anti-Human Alexa 647 (Jackson ImmunoResearch 109-605-044). After 30 min, cells were washed twice with 1× PBS and 0.1% Tween 20 and once with 1× PBS with DAPI (Sigma-Aldrich 28718-90-3) diluted 1:750 from a 0.5 mg/ml stock. After 5 min, cells were washed one last time with 1× PBS and mounted using ProLong Glass Antifade Mountant (Thermo Scientific P36980). Images were acquired using a Thunder Leica fluorescent microscope at a 100× magnification and with a 0.2 μm z-stack and then processed using FIJI-ImageJ 75 to obtain a maximum projection.
Quantification of centromeric damage
For each cell, the number of γH2AX and CREST colocalizing foci was scored using maximum projection images.
Quantification of the fluorescent mean intensity signal
FIJI software was used to select the area of each cell and measure the signal mean intensity of the maximum projection images.
Overexpression or downregulation of SMAD2 and SMAD4
To overexpress human SMAD2 and SMAD4, cDNA for each gene was cloned into pHAGE vectors. CRISPRi (CRISPR-inhibition) was used to downregulate SMAD4 expression by transducing dCas9 into the cells using a pHAGE-dCas9 vector together with a CRISPR-interference sgRNA (GGCAGCGGCGACGACGACCA) from Gilbert et al76 cloned into pLentiGuide-Puro-FE .
QUANTIFICATION AND STATISTICAL ANALYSIS
Replicates, statistical analyses and scale bars
For each experiment we report in the figure legends the sample size and whether triplicates or duplicates were performed. Unless otherwise specified, triplicates or duplicates were biological, not technical. Unless otherwise specified,; p-values are from the Wilcoxon test. If not otherwise specified,; at least 50 nuclei or cells were analyzed in the FISH or IF experiments. Also, if not otherwise specified the scale bars in the FISH and IF images represent 5 μM.
Computational sgRNA prediction
The CHM13 centromeric sequences and whole-genome reference were downloaded from the T2T Consortium (https://github.com/marbl/CHM13)29 and the hg38 reference genome from the UCSC genome browser. For the CHM13 centromeric sequences, the HOR region with the classification “Live” or “HOR_L” was selected. For each HOR_L region, all possible SpCas9 sgRNA sites with a pattern comprising 20 nucleotides followed by NGG as PAM were searched. For each possible sgRNA, the numbers of binding sites in the centromeric HOR_L regions of each chromosome and in the whole genome were counted. The number of sgRNA binding sites was also determined using the hg38 reference. The GC content for each sgRNA was also determined.
For each sgRNA, two scores were determined: the chromosome specificity score, defined as the ratio between the number of binding sites on the centromere (HOR_L) of the target chromosome (chromosome that we intend to target) and the total number of sites across all centromeres (HOR_L) (given as a fraction or as a percentage after multiplication by 100), and the centromere specificity score, defined as the ratio between the number of binding sites on the centromere (HOR_L) of the target chromosome and the number of binding sites across the whole genome (given as a fraction or as a percentage after multiplication by 100).
The sgRNA efficiency was evaluated based on 3 parameters: 1) GC content, 2) total number of binding sites in the centromere of the target chromosome, and 3) sgRNA activity predicted from previous studies by Doench et al35,36. With that method, the sgRNA activity is calculated based on 72 genetic features36, which include the presence of certain nucleotides at specific positions along the sgRNA and the GC content. For a particular guide ,the model weights for the features will be and the intercept will be int. The activity is then given via logistic regression as:
Predicted sgRNA activity falls into the range , with as the worst score and as the best score. Since CHM13 is a female-derived (XX) cell line, all binding sites for chromosome Y were evaluated based on hg38. Predicted sgRNAs are listed in Table S1.
Automated image quantification of FISH foci
In addition to manual counting of FISH foci (shown in Fig. 3 and Fig. S3), an automated image quantification was also performed (Table S2). FISH counts were calculated automatically using an in-house-developed python script, available publicly at https://github.com/davolilab/FISH-counting. Individual nuclei were segmented by applying an automatic threshold to the DAPI channel after smoothing and contrast enhancement. Thresholded objects were filtered for area and solidity to remove erroneously segmented regions. For probe detection within segmented nuclei, a white tophat filter was applied to remove small spurious regions, and then the “blob_log” function from scikit-image package77 was utilized to identify and count fluorescent spots. Since it was observed that some FISH probes were incorrectly doubly counted, a distance cutoff was applied so that spots within a set (minimal) distance count as one spot. Then, the probe numbers were aggregated and the percentages for different spot counts were calculated. The script was run under a python 3.7 environment; for more details, see the github repository.
Quantification of foci intensity
The regions corresponding to the FISH foci were determined by the threshold function of Fiji. Then, the average intensity of each determined region was calculated as the representative of the brightness of the focus by Fiji (used in Fig S1E).
Low-pass whole-genome sequencing analysis
Low-pass (~0.1–0.5×) whole-genome sequencing reads of cells were aligned to reference human genome hg38 by using BWA-mem (v0.7.17; https://github.com/lh3/bwa/releases/tag/v0.7.17)78, and duplicates were removed using GATK (Genome Analysis Toolkit, v4.1.7.0) (https://gatk.broadinstitute.org/hc/en-us)79 with default parameters to generate analysis-ready BAM files. BAM files were processed by the R Package CopywriteR (v1.18.0; https://github.com/PeeperLab/CopywriteR)80 to call the arm-level copy numbers.
Bulk RNA-seq analysis pipeline
RNA sequencing reads were processed, quality controlled, aligned, and quantified using the Seq-N-Slide software(https://github.com/igordot/sns)81. In brief, total RNA sequencing reads were trimmed using Trimmomatic (https://github.com/timflutre/trimmomatic)82 and mapped to the GENCODE human genome hg38 by STAR(https://github.com/alexdobin/STAR)83. featureCounts(https://github.com/byee4/featureCounts)84 was used to quantify reads and generate a genes-sample counts matrix. Differential gene expression (DGE) analysis was completed with DESeq2 in R (https://bioconductor.org/packages/release/bioc/html/DESeq2.html)50. Gene ranks from DGE were used for pathway analysis using the GSEA preranked utility (https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html)85. Further plotting and statistical analyses were completed in R.
Single-cell RNA sequencing data pre-processing
The CellRanger v6.1 pipeline (10X Genomics) was used to process single-cell RNA sequencing data. CellRanger count was used to align sequences and generate gene expression matrices. Sequences were aligned to the pre-built GRCh38-2020-A human reference for CellRanger. Gene expression matrices were generated with each column representing a cell barcode and each row representing a gene or hashtag oligo sequences (HTO).
To identify the sample of origin for each cell barcode, the HTO count data from each 10X Chromium experiment were demultiplexed using the Seurat v4.0.3 package for R v4.1 (https://github.com/satijalab/seurat)86. Cell barcodes that could be confidently assigned to a single sample were kept. Several quality control thresholds were applied uniquely to each dataset on total gene number, total UMI counts, and total HTO counts to remove low-quality cells and potential cell doublets. Cells were also discarded if their proportion of total gene counts that could be attributed to mitochondrial genes exceeded 10%.
Modified CopyKat analysis
A modified version of the CopyKat v1.0.5 (https://github.com/navinlabcode/copykat)46 pipeline for R was used to generate a copy number alteration (SCNA) score for each chromosome arm in each cell. Hashtagged samples from the same cell line in each 10X Chromium dataset were grouped together for analysis. Each such group of samples contained a diploid control sample used to set the SCNA value baseline centered around 0. For each analysis, genes expressed in less than 5% of the cells, HLA genes, and cellcycle genes were excluded. The log-Freeman-Tukey transformation was used to stabilize variance and dlmSmooth() was used to smooth outliers. The diploid control sample for each set was used to calculate a baseline expression level for each gene. This value was subtracted from the samples in the set, centering the control sample expression around 0. Genes expressed in less than 10% of cells were then excluded from further analysis. The original CopyKat pipeline splits the transcriptome into artificial segments based on similar expression, and calculates a SCNA value for each segment. Instead, we generated a SCNA value for each chromosome arm by calculating the mean gene expression for the genes on that arm.
A single SCNA value for the entire chromosome 18 was calculated using genes on both the p and q arms of the chromosome instead of each arm individually, due to its relatively small size. SCNA values for chromosomes 13, 14, 15, 21, and 22 were calculated only using genes on their respective q arms. Gains or losses of a chromosome arm relative to the control sample (diploid) were called based on a threshold calculated from the control sample for each chromosome arm. The threshold is calculated as
where the median is calculated from the SCNA values for each arm in the control sample, and the median absolute deviation (MAD) is calculated by the mad() function from the stats R package. Gains (or losses) are then called for a chromosome arm if its SCNA value is above (or below) the threshold for its sample set.
CopyKat data visualization
Heatmaps were generated using the ComplexHeatmap v2.8 R package87. Each row represents one cell, each column represents a chromosome arm, and each value is the corresponding SCNA score. Column widths were scaled to the number of genes on the arm. For the heatmaps, cells were clustered by row of the chromosome of interest. Bar graphs were generated using the ggplot2 v3.3.5 R package.
Survival analysis
For survival analysis, the disease-free interval (DFI) and related clinical data were downloaded from cBioPortal88. Arm-level copy number was downloaded from TCGA Firehose Legacy (https://gdac.broadinstitute.org). For each patient, purity α, ploidy τ, and integer copy number q(x) data were downloaded from GDC (https://gdc.cancer.gov/about-data/publications/pancanatlas). Before the analysis, the arm-level copy number values R(x) were adjusted using the formula below:
Patients with arm-level log2 ratio less than −0.3 would be regarded as an arm-level loss event to evaluate patients based on the presence or absence of 18q arm loss. A log-rank test between the stratified patients and the Kaplan-Meier method was used to calculate the p-value and plot survival curves. Patients for whom clinical survival information was unavailable were excluded from the analysis. In addition, a Cox proportional hazards (PH) regression model was used to calculate each gene’s hazard ratio (HR) between the top 50% and bottom 50% expression.
Gene rank score analysis
For each gene on chromosome 18, we calculated the DNA-RNA Spearman’s correlation (rho value) from the TCGA-COADREAD dataset. Genes with no or very low frequency of SCNA (−0.02 < DNA log2FC < 0.02 in >70% of the patients) were removed because for those genes very little or no variance at the DNA level is likely to influence the correlation value. The Cox proportional-hazards model was then applied to estimate the association between the expression level of each gene and patients’ survival. The TUSON algorithm for predicting the likelihood for a gene to behave as a tumor-suppressor gene (TSG) based on its pattern of point mutation was from Davoli et al.4 and was applied to the latest available TCGA dataset of point mutations. A gene rank score was generated based on the rank sum of the following three parameters: DNA-RNA correlation, hazard ratio from Cox proportional hazards regression, and q-value from TUSON-based TSG prediction. In other words, for each gene, the (three) rank position values determined based on the three parameters listed above were summed (Table S4A).
Supplementary Material
Figure S1 (related to Fig. 1) Prediction and validation of chromosome-specific sgRNAs targeting human α-satellite centromeric sequences (A) Left: Proliferation assay on RPEs p21/Rb shRNA expressing Cas9 or empty vector (EV) transduced with lentiviral vectors expressing the indicated sgRNAs. The same number of cells were plated in 6-well plates and the percentage of live cells relative to EV was determined 7 days after transduction. Mean and S.D. from triplicates, p-values from Wilcoxon test (*=p<0.05). Imaging validation is also indicated in red. Right: Western blot showing Cas9 expression. (B) Left: Imaging of hCECs (47, +7) expressing 3xmScarlet-dCas9 and sgChr7–1 in the polyclonal population and in a derived clone (clone 8) with high 3xmScarletdCas9 expression. As compared to the polyclonal population, clone 8 contains a higher percentage of cells showing the expected foci. Average frequency of cells displaying foci is shown for the polyclonal and clonal populations (>100 cells counted; in triplicates). Right: Western blot analysis of the expression level of 3xmScarlet-dCas9 in the polyclonal population and clone 8. The percentage of cells showing foci was 45% in the hCEC polyclonal population transduced with 3xmScarlet-dCas9 and increased to 72% in clone 8. (C) Imaging of hCECs expressing 3xmScarlet-dCas9 and the indicated sgRNAs. Representative images of interphase cells are shown; the percentage of cells displaying foci is shown in Table S1. See also Fig. 1F. (D) Imaging of RPEs p21/Rb shRNA expressing 3xmScarlet-dCas9 fusion and the indicated sgRNAs. Representative images of interphase cells are shown. (E) Left: Correlation between the intensity of the signal of the 3xmScarlet-dCas9 foci (measured with ImageJ/Fiji) and the sgRNA activity score(Doench et al., 2016, 2014) of cells treated as in (C). Right: Correlation between the intensity of the signal of the 3xmScarlet-dCas9 foci and the number of predicted sgRNA binding sites on the specific centromere (based on the T2T genome assembly) of cells treated as in (C). Pearson correlation coefficients and corresponding p-values are shown.
Figure S2 (related to Fig. 2) Analysis of KNL1Mut-dCas9 and other fusion proteins targeted to centromeres (A) Maps of the dCas9, KNL1RVSF/AAAA-dCas9, KNL1S24A;S60A-dCas9, NDC80-CH1-dCas9, and NDC80-CH2-dCas9 constructs. The predicted function of each construct is indicated on the right. See text for details. (B) Western blot showing the expression of the indicated constructs in hCECs. (C) Western blot showing the expression of the indicated constructs, in which different mutated segments of KNL1 or NDC80 are fused to the N- or C-terminus of dCas9; see also (A). L: linker with amino acid sequence GGSGGGS. (D) Imaging of hCECs (47, +7) expressing 3xmScarlet-KNL1Mut-dCas9 and transduced with sgChr7–1 or sgChr18–4. (E) Proliferation rate of hCECs transduced with KNL1Mut-dCas9 and with the indicated sgRNAs. Mean and S.D. from triplicates are shown for each time point. (F) FISH imaging and quantification of micronuclei containing chromosome 7 or 13 in hCECs treated with KNL1Mut-dCas9 and the indicated sgRNA (as in Fig. 2G); quantification of micronuclei counts is shown below. Experiments were performed in duplicates, and for each replicate, at least 100 cells were scored.
Figure S3 (related to Fig. 3) Analysis of KNL1Mut-dCas9 and other fusion proteins targeted to centromeres for the induction of chromosome-specific gains and losses (A) KaryoCreate experiment in hCECs comparing the efficiency of different methods for delivering KNL1Mut-dCas9, as quantified by FISH. Methods: (1) transfection of pHAGE-KNL1Mut-dCas9, whose expression of KNL1Mut-dCas9 is driven by the CMV promoter; (2) lentiviral-mediated transduction with pIND20-KNL1Mut-dCas9, whereby the vector is integrated in the genome of the target cells and expression of KNL1Mut-dCas9 is driven by doxycycline treatment (1 μg/ml); (3) lentiviral-mediated transduction with pHAGE-DD-KNL1Mut-dCas9, whereby expression of KNL1Mut-dCas9 is driven by treatment with shield-1 to stabilize the protein. All cells were transduced with sgChr7–1, and FISH quantification of chr7 gains/losses is shown (mean and S.D. from triplicates). (B) KaryoCreate experiment comparing the efficiency of different constructs in inducing chromosome gains and losses. hCECs were transduced with sgChr7–1 and the indicated constructs. FISH quantification for chr7 gains/losses is shown (mean and S.D. from triplicates), along with the aneuploidy level (% of chr7 gains/losses) normalized to the expression level of each construct (as in Fig. S2B). Note that after normalization, the induction of aneuploidy is greatest for NDC80CH2-dCas9 and is higher for KNL1S24A;S60A-dCas9 than for KNL1RVSF/AAAA-dCas9. (C) Left: Western blot analysis of the indicated constructs. Right: KaryoCreate experiment to compare the efficiency of different constructs in inducing chromosome gains and losses. hCECs were transduced with sgChr7–1 and the indicated constructs, and FISH quantification of chr7 gains/losses is shown. (D) Left: Western blot analysis of dCas9 expression in hCECs transduced with KNL1Mut-dCas9 using different amount of virus (about 3 times more virus in the HIGH versus LOW sample, i.e. MOI of 6 for HIGH and 2 for LOW). The corresponding quantification (through ImageJ) is shown below. Right: FISH quantification of chr7 gains/losses in cells expressing KNL1Mut-dCas9 transduced with sgChr7–1 using different amounts of virus at 9–10 days after transduction (mean and S.D. from duplicates). (E) FISH quantification of chr7 gains/losses in hCECs transduced with KNL1Mut-dCas9 and with sgChr7–1 and/or sgChr7–3 (mean and S.D. from duplicates). (F) Single-cell sequencing quantification of chr9 gains/losses in hCECs were transduced with KNL1Mut-dCas9 and with sgNC, sgChr9–3 and/or sgChr9–5 (mean and S.D. from technical duplicates). (G) Left: FACS sorting results for hCECs treated as in (D) using an MOI of 2 after sorting for low or high expression of the cell surface protein EPHB4, encoded by a gene on chr7. Right: FISH quantification of the % of chr7 gains or losses in each condition (N=100 nuclei; mean and S.D. from duplicates). *, p-value<0.05 (Welch two-sample t-test). (H) scRNA-seq analysis of chromosome or arm gains/losses (as in Fig. 4) in hCECs transduced with KNL1Mut-dCas9 (via infection with pIND20-KNL1Mut-dCas9 lentiviral vector) and sgChr7–1. Cells were treated with doxycycline for the indicated number of days to induce construct expression; experiment performed in duplicate.
Figure S4 (related to Fig. 4) Analysis of KaryoCreate across chromosomes and conditions (A) Analysis of hCEC clones with different aneuploidies by bulk WGS (left) and scRNA-seq (right). Arm-level copy number events were inferred from each method (see Methods) and the derived copy number profiles are shown for both methods. See also (B). (B) FISH and scRNA-seq analyses of hCEC clones with chr7 trisomy or more complex karyotypes and the percentage of aneuploid cells was quantified using both methods. Mean values from duplicates are shown. (C) A heatmap depicting gene copy numbers inferred from scRNA-seq analysis following KaryoCreate control experiments. hCECs were transduced either with empty vector or with KNL1Mut-dCas9 together with a negative control sgRNA (sgNC), and scRNA-seq was performed as in (B) to estimate % of gains and losses across chromosomes. (D) A heatmap depicting gene copy numbers inferred from scRNA-seq analysis following KaryoCreate. KaryoCreate for different individual chromosomes (or combination of chromosomes) was performed on RPEs. scRNA-seq was used to estimate the presence of chromosome- or arm-level gains or losses using a modified version of CopyKat. The median expression of genes across each chromosome arm is used to estimate the DNA copy number. The % of gains/losses for each arm (reported below each heatmap) is estimated by comparing the DNA copy number distribution of each experimental sample (chromosome-specific sgRNA) to that of the negative control (sgRNA NC; see also Methods). Heatmap rows represent individual cells, columns represent different chromosomes, and the color represents the copy number change (gain in red and loss in blue). (E) Average proportions (%) of whole-chromosome and arm-level gains/losses. The percentage of the indicated events were calculated as the average among the aneuploid cells generated using KaryoCreate for chromosomes 6, 7, 8, 9, 12, 16, and X )mean values from duplicates). (F) A heatmap depicting chromosome copy numbers inferred from scRNA-seq analysis following KaryoCreate. KaryoCreate was performed on hCECs using two sgRNAs targeting chromosome 7 (sgChr7–1) and 18 (sgChr18–4). scRNA-seq was used to estimate the presence of chromosome- or arm-level gains/losses using a modified version of CopyKat as in (D). Heatmap rows represent individual cells, columns represent different chromosomes, and the color represents the copy number change (gain in red and loss in blue). (G) Immunofluorescence (IF) assay showing DNA damage in HCT116 cells expressing KNL1Mut-dCas9 and sgNC, sgChr7–1, or sgChr18–4. IF was performed for γH2AX (green), CREST (red) to visualize centromeres, and DAPI (blue). Representative images are shown. (H) Quantification of experiment shown in (G). Left: number of DNA damage foci colocalizing with CREST in each cell, quantified and normalized to the total number of CREST foci in the cell. Right: total γH2AX signal per cell, quantified and normalized to the total DAPI signal. p-values are from Wilcoxon test. (I) Left: The total γH2AX signal per cell as determined by IF analysis of hCECs expressing KNL1Mut-dCas9 (pIND20 vector) and sgNC or sgChr7–1 for γH2AX (green) and DAPI (blue), quantified and normalized to the total DAPI signal. Right: Western blot analysis of KNL1Mut-dCas9 expression before or after treatment with doxycycline to induce construct expression. p-values are from Wilcoxon test.
Figure S5 (related to Fig. 5) Dissection of the consequences of 18q loss in colorectal cancer (A) Schematic of experimental plan to apply KaryoCreate across different chromosomes to derive single-cell clones with specific gains or losses. (B) Shallow WGS analysis of single-cell-derived clones obtained by KaryoCreate using sgNC or sgChr7–1 performed on diploid hCECs (as indicated. (C) Representative FISH images and copy number plots from WGS analysis of hCEC sgChr7–1 clone 23 (B) before or after 25 population doublings in culture. (D) Survival analysis (Kaplan-Meier curve) for colorectal cancer patients (TCGA – COADREAD) displaying or not displaying 18q loss, after exclusion of patients with SMAD4 point mutation. (E) Proliferation rates of the indicated hCEC clones 13 and 14 (18q loss) (as in Fig. 5E) after the overexpression of the indicated genes. Mean and S.D. are shown for triplicates; p-values are from Wilcoxon test (*=p<0.05). Proliferation rates for hCEC clones 10 and 5 (18 loss) with and without TGFβ are also shown. (F) Western blot showing SMAD2 and SMAD4 levels in hCEC clone 13 after overexpression of GFP, SMAD2, SMAD4, or SMAD2 + SMAD4. Related to Fig. S5E. (G) Proliferation rates of the indicated hCEC cell lines (clone 14 and hCEC transduced with dCas9 and a SMAD4 or NC sgRNA) when cultured in the presence of TGFβ (20 ng/ml) for 9 days; cells were counted every 3 days in triplicates. p-value is derived from the Wilcoxon test. (H) Western blot showing SMAD4 levels in hCECs transduced with dCas9 and a SMAD4 or NC sgRNA. Related to Fig. S5G.
Table S1. Prediction of sgRNA for each chromosome with CHM13 genome; Related to Figure 1 (see also Methods). The first tab shows sgRNA predictions for 76 selected sgRNAs across all chromosomes except chromosome Y. Columns A-C: chromosome, sgRNA name, and sgRNA sequence. Column D: binding sites (CHM13-specific chromosome centromere HOR_L) representing the number of binding sites on the centromere (HOR_L) of the target chromosome (the chromosome that we intended to target). Column E: binding sites (CHM13-all centromere horL) representing the total number of sites across all centromeres (HOR_L). Column F: chromosome specificity score, defined as the ratio between Columns D and E (given as a fraction, or as a percentage after multiplication by 100). Column G: binding sites (CHM-13 whole genome) representing the number of binding sites on the CHM-13 genome of the target chromosome. Column H: binding sites (HG38 whole genome) representing the number of binding sites on the HG38 genome of the target chromosome. Column I: centromere specificity score, defined as the ratio between the number of binding sites on the centromere (HOR_L) of the target chromosome and the number of binding sites across the whole genome (given as a fraction, or as a percentage after multiplication by 100). Column J: GC content. Column K: activity score, generated based on Doench’s method36. Column L: HOR_L length from the CHM13 genome. Column M: binding sites (CHM13-specific chromosome centromere Not HOR_L) representing the number of binding sites on the centromere (Not HOR_L) of the target chromosome. Column N Validated_by_Imaging, indicating whether the sgRNA can be validated by imaging or not. Column O percentage of cells showing foci (hCEC) related to Figure 1/S1. Columns P-R: binding HOR_L (which HORL region the sgRNA comes from), HOR_L in this chromosome (how many HORL region we have for this specific chromosome), and HOR in this chromosome (how many HOR_L and Not HOR_L region we have for this specific chromosome), respectively. The following tabs contain predictions of sgRNAs for every single chromosome, as indicated.
Table S2. Automated (python-based) quantification of FISH foci after KaryoCreate; Related to Figure 3. This table lists the number of FISH foci quantified using an Automated FISH counting (see Methods) designed to score FISH signals in interphase cells.
Table S3. scRNA-seq-based quantification of arm-level gains and losses after KaryoCreate; Related to Figure 4. Quantification of the frequency of arm-level or chromosome-level gains and losses after KaryoCreate for the indicated chromosomes, based on scRNA-seq (see Methods). Tab S4A refers to Fig. S4A–B and tabs S4B and S4C refer to Fig. 4.
Table S4. Genetic information about the genes on chromosome 18; related to Figs. 5 and S5.
Tab S4A. Genetic information about the genes on chromosome 18.
Tab S4B. Differential expression analysis of hCEC clones comparing clones with or without chromosome 18 loss (Loss vs WT).
Tab S4C. GSEA analysis of hCEC clones comparing clones with or without chromosome 18 loss, Pathways Depleted.
Tab S4D. GSEA analysis of hCEC clones comparing clones with or without chromosome 18 loss, Pathways Enriched.
Supplemental Movie S1. Live-cell imaging showing normal mitosis or lagging chromosome; Related to Figure 2. Clip 1. Live-cell imaging corresponding to Figure 2B, showing a normal mitosis. Shown are time-lapse images taken every 3 min of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and sgChr7–1. Clip 2. Live-cell imaging corresponding to Figure 2B, showing a lagging chromosome and micronucleus. Shown are time-lapse images taken every 3 min of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and sgChr7–1. Clip 3. Live-cell imaging corresponding to Figure 2B, showing a lagging chromosome. Shown are time-lapse images taken every 3 min of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and sgChr18–4. Clip 4. Live-cell imaging corresponding to Figure 2B, showing a lagging chromosome. Shown are time-lapse images taken every minute of hCECs expressing H2B-GFP, 3xmScarlet-KNL1Mut-dCas9, and sgChr7–1.
Highlights.
We designed chromosome specific sgRNAs for targeting 19 of 24 human chromosomes
KaryoCreate facilitates sgRNA-dCas9 mediated recruitment of mutant kinetochore proteins
KaryoCreate enables the creation of human cells with distinct karyotypes
Engineered 18q loss promotes tumor-associated phenotypes in colon epithelial cells
ACKNOWLEDGEMENTS
We acknowledge the NYU Langone Genome Technology Center(P30CA016087) and the NYU Langone High Performance Computing (HPC) Core. We acknowledge all the members of the Davoli lab, Jef Boeke, Liam Holt, Gregory Brittingham, and Glennis Logson for helpful insights and discussion and Rebecca M. Barr for help with editing. We thank Jennifer DeLuca for the NDC80 constructs and Karen Miga for the initial computational validation of centromeric sgRNAs. T.D. was supported by the Cancer Research UK Grand Challenge and the Mark Foundation for Cancer Research (C5470/A27144), R37 R37CA248631, the MRA Young Investigator Award, and T32 GM136542. S.K. and D.F were supported by grant U24CA210972 from the US National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium, and contract S21-167 from Leidos Biomedical Research, respectively. C.T. and S.S., E.D.T. and S.G were supported by the Italian Association for Cancer Research (AIRC-MFAG 2018 ID.21665 and AIRC Start-Up 2020 ID. 25189) and the Rita-Levi Montalcini program from MIUR C.T. and S.S. were supported by Ricerca Finalizzata (GR-2018-12367077) and Fondazione Cariplo. S.M.L. was supported by NIH Grants R01DE026644, P01 CA106451, P50 CA097007, and P30 CA023100.
Footnotes
Declaration of interests
T.D. is on the Scientific Advisory Board (SAB) of io9. S.M.L. is a co-founder of io9 and a member of Biological Dynamics, Inc. SAB. T.D. and N.B are inventors on patent application no. 63/375,181.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Knouse KA, Wu J, Whittaker CA, and Amon A. (2014). Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. U. S. A 111, 13409–13414. 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knouse KA, Davoli T, Elledge SJ, and Amon A. (2017). Aneuploidy in Cancer: Seq-ing Answers to Old Questions. Annu. Rev. Cancer Biol 1, 335–354. 10.1146/annurev-cancerbio-042616-072231. [DOI] [Google Scholar]
- 3.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, and Elledge SJ (2013). Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962. 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. (2018). Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689.e3. 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.William WN, Zhao X, Bianchi JJ, Lin HY, Cheng P, Lee JJ, Carter H, Alexandrov LB, Abraham JP, Spetzler DB, et al. (2021). Immune evasion in HPV- head and neck precancer-cancer transition is driven by an aneuploid switch involving chromosome 9p loss. Proc. Natl. Acad. Sci. U. S. A 118, e2022655118. 10.1073/pnas.2022655118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, Moore DA, Grönroos E, Rowan A, Dewhurst SM, et al. (2020). Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 587, 126–132. 10.1038/s41586-020-2698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santaguida S, Tighe A, D’Alise AM, Taylor SS, and Musacchio A. (2010). Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol 190, 73–87. 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, and Taylor SS (2010). Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol 190, 25–34. 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fournier RE (1981). A general high-efficiency procedure for production of microcell hybrids. Proc. Natl. Acad. Sci. U. S. A 78, 6349–6353. 10.1073/pnas.78.10.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, and Storchova Z. (2012). Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol 8, 608. 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ly P., Teitz LS., Kim DH., Shoshani O., Skaletsky H., Fachinetti D., Page DC., and Cleveland DW. (2017). Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat. Cell Biol 19, 68–75. 10.1038/ncb3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, Flanagan AM, Behjati S, Page DC, Campbell PJ, et al. (2019). Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet 51, 705–715. 10.1038/s41588-019-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rayner E, Durin M-A, Thomas R, Moralli D, O’Cathail SM, Tomlinson I, Green CM, and Lewis A. (2019). CRISPR-Cas9 Causes Chromosomal Instability and Rearrangements in Cancer Cell Lines, Detectable by Cytogenetic Methods. CRISPR J. 2, 406–416. 10.1089/crispr.2019.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuo E, Huo X, Yao X, Hu X, Sun Y, Yin J, He B, Wang X, Shi L, Ping J, et al. (2017). CRISPR/Cas9-mediated targeted chromosome elimination. Genome Biol. 18, 224. 10.1186/s13059-017-1354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tovini L, Johnson SC, Andersen AM, Spierings DCJ, Wardenaar R, Foijer F, and McClelland SE (2022). Inducing Specific Chromosome Mis-Segregation in Human Cells. EMBO J 42: e111559. 10.15252/embj.2022111559 [DOI] [Google Scholar]
- 17.Truong MA, Cané-Gasull P, Vries S.G. de, Nijenhuis W, Wardenaar R, Kapitein LC, Foijer F, and Lens SMA (2022). A motor-based approach to induce chromosome-specific mis-segregations in human cells. EMBO J 42: e111587. 10.15252/embj.2022111587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barra V, and Fachinetti D. (2018). The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun 9, 4340. 10.1038/s41467-018-06545-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayden KE (2012). Human centromere genomics: now it’s personal. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol 20, 621–633. 10.1007/s10577-012-9295-y. [DOI] [PubMed] [Google Scholar]
- 20.Schueler MG, and Sullivan BA (2006). Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet 7, 301–313. 10.1146/annurev.genom.7.080505.115613. [DOI] [PubMed] [Google Scholar]
- 21.Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, Hoyt SJ, Uralsky L, Ryabov FD, Shew CJ, et al. (2022). Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178. 10.1126/science.abl4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Musacchio A, and Desai A. (2017). A Molecular View of Kinetochore Assembly and Function. Biology 6, E5. 10.3390/biology6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheeseman IM (2014). The kinetochore. Cold Spring Harb. Perspect. Biol 6, a015826. 10.1101/cshperspect.a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musacchio A. (2015). The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr. Biol. CB 25, R1002–1018. 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 25.Stern BM., and Murray AW. (2001). Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr. Biol. CB 11, 1462–1467. 10.1016/s0960-9822(01)004511. [DOI] [PubMed] [Google Scholar]
- 26.Liu D, and Lampson MA (2009). Regulation of kinetochore–microtubule attachments by Aurora B kinase. Biochem. Soc. Trans 37. [DOI] [PubMed] [Google Scholar]
- 27.Papini D, Levasseur MD, and Higgins JMG (2021). The Aurora B gradient sustains kinetochore stability in anaphase. Cell Rep. 37, 109818. 10.1016/j.celrep.2021.109818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, and Lampson MA (2010). Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J. Cell Biol 188, 809–820. 10.1083/jcb.201001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. (2022). The complete sequence of a human genome. Science 376, 44–53. 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. (2017). Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864. 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan LL, and Sullivan BA (2020). Genomic and functional variation of human centromeres. Exp. Cell Res 389, 111896. 10.1016/j.yexcr.2020.111896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willard HF (1991). Evolution of alpha satellite. Curr. Opin. Genet. Dev 1, 509–514. 10.1016/s0959-437x(05)80200-x. [DOI] [PubMed] [Google Scholar]
- 33.Uralsky LI, Shepelev VA, Alexandrov AA, Yurov YB, Rogaev EI, and Alexandrov IA (2019). Classification and monomer-by-monomer annotation dataset of suprachromosomal family 1 alpha satellite higher-order repeats in hg38 human genome assembly. Data Brief 24, 103708. 10.1016/j.dib.2019.103708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, and Root DE (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol 32, 1262–1267. 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyers RM., Bryan JG., McFarland JM., Weir BA., Sizemore AE., Xu H., Dharia NV., Montgomery PG., Cowley GS., Pantel S., et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ly P, Eskiocak U, Kim SB, Roig AI, Hight SK, Lulla DR, Zou YS, Batten K, Wright WE, and Shay JW (2011). Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia N. Y. N 13, 348–357. 10.1593/neo.101580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T. (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bajaj R, Bollen M, Peti W, and Page R. (2018). KNL1 Binding to PP1 and Microtubules Is Mutually Exclusive. Structure 26, 1327–1336.e4. 10.1016/j.str.2018.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, and Salmon ED (2006). Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969–982. 10.1016/j.cell.2006.09.047. [DOI] [PubMed] [Google Scholar]
- 43.Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 108, 3665–3670. 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banaszynski LA, Chen L-C, Maynard-Smith LA, Ooi AGL, and Wandless TJ (2006). A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004. 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, Kumar T, Hu M, Sei E, Davis A, et al. (2021). Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol 39, 599–608. 10.1038/s41587-020-00795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.The Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Massagué J, Blain SW, and Lo RS (2000). TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103, 295–309. 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 52.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, et al. (2015). Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47. 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 53.van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodford-Richens KL., Rowan AJ., Gorman P., Halford S., Bicknell DC., Wasan HS., Roylance RR., Bodmer WF., and Tomlinson IPM. (2001). SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc. Natl. Acad. Sci 98, 9719–9723. 10.1073/pnas.171321498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, et al. (1996). Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat. Genet 13, 343–346. 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 56.Cheng P, Zhao X, Katsnelson L, Camacho-Hernandez EM, Mermerian A, Mays JC, Lippman SM, Rosales-Alvarez RE, Moya R, Shwetar J, et al. (2022). Proteogenomic analysis of cancer aneuploidy and normal tissues reveals divergent modes of gene regulation across cellular pathways. eLife 11, e75227. 10.7554/eLife.75227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eppert K, Scherer SW, Ozcelik H, Pirone R, Hoodless P, Kim H, Tsui LC, Bapat B, Gallinger S, Andrulis IL, et al. (1996). MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell 86, 543–552. 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 58.Dumont M, Gamba R, Gestraud P, Klaasen S, Worrall JT, De Vries SG, Boudreau V, Salinas‐Luypaert C, Maddox PS, Lens SM, et al. (2020). Human chromosome‐specific aneuploidy is influenced by DNA ‐dependent centromeric features. EMBO J. 39. 10.15252/embj.2019102924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, and Salmon ED (2001). Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol 153, 517–527. 10.1083/jcb.153.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gregan J, Polakova S, Zhang L, Tolić-Nørrelykke IM, and Cimini D. (2011). Merotelic kinetochore attachment: causes and effects. Trends Cell Biol. 21, 374–381. 10.1016/j.tcb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whinn KS, Kaur G, Lewis JS, Schauer GD, Mueller SH, Jergic S, Maynard H, Gan ZY, Naganbabu M, Bruchez MP, et al. (2019). Nuclease dead Cas9 is a programmable roadblock for DNA replication. Sci. Rep 9, 13292. 10.1038/s41598-019-49837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giunta S, Hervé S, White RR, Wilhelm T, Dumont M, Scelfo A, Gamba R, Wong CK, Rancati G, Smogorzewska A, et al. (2021). CENP-A chromatin prevents replication stress at centromeres to avoid structural aneuploidy. Proc. Natl. Acad. Sci 118, e2015634118. 10.1073/pnas.2015634118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bury L, Moodie B, Ly J, McKay LS, Miga KH, and Cheeseman IM (2020). Alpha-satellite RNA transcripts are repressed by centromere-nucleolus associations. eLife 9, e59770. 10.7554/eLife.59770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McNulty SM, Sullivan LL, and Sullivan BA (2017). Human Centromeres Produce Chromosome-Specific and Array-Specific Alpha Satellite Transcripts that Are Complexed with CENP-A and CENP-C. Dev. Cell 42, 226–240.e6. 10.1016/j.devcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan FL, Marshall OJ, Saffery R, Won Kim B, Earle E, Choo KHA, and Wong LH (2012). Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc. Natl. Acad. Sci 109, 1979–1984. 10.1073/pnas.1108705109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kabeche L, Nguyen HD, Buisson R, and Zou L. (2018). A mitosis-specific and R loop–driven ATR pathway promotes faithful chromosome segregation. Science 359, 108–114. 10.1126/science.aan6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarli L., Bottarelli L., Bader G., Iusco D., Pizzi S., Costi R., DâTMAdda T, Bertolani M, Roncoroni L, and Bordi C. (2004). Association Between Recurrence of Sporadic Colorectal Cancer, High Level of Microsatellite Instability, and Loss of Heterozygosity at Chromosome 18q. Dis. Colon Rectum 47, 1467–1482. 10.1007/s10350-004-0628-6. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka T, Watanabe T, Kazama Y, Tanaka J, Kanazawa T, Kazama S, and Nagawa H. (2006). Chromosome 18q deletion and Smad4 protein inactivation correlate with liver metastasis: a study matched for T- and N- classification. Br. J. Cancer 95, 1562–1567. 10.1038/sj.bjc.6603460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, et al. (2014). Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 156, 1298–1311. 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trakala M, Aggarwal M, Sniffen C, Zasadil L, Carroll A, Ma D, Su XA, Wangsa D, Meyer A, Sieben CJ, et al. (2021). Clonal selection of stable aneuploidies in progenitor cells drives high-prevalence tumorigenesis. Genes Dev. 35, 1079–1092. 10.1101/gad.348341.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, et al. (2012). A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proc. Natl. Acad. Sci. U. S. A 109, 8212–8217. 10.1073/pnas.1206062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li G-W, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell 155, 1479–1491. 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji - an Open Source platform for biological image analysis. Nat. Methods 9, 10.1038/nmeth.2019. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPRMediated Control of Gene Repression and Activation. Cell 159, 647–661. 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van der Walt S, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, Yu T, and scikit-image contributors (2014). scikit-image: image processing in Python. PeerJ 2, e453. 10.7717/peerj.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van der Auwera GA (2020). Genomics in the cloud : using Docker, GATK, and WDL in Terra First edition. (O’Reilly Media; ). [Google Scholar]
- 80.Kuilman T, Velds A, Kemper K, Ranzani M, Bombardelli L, Hoogstraat M, Nevedomskaya E., X G., de Ruiter J., Lolkema MP., et al. (2015). CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 16, 49. 10.1186/s13059-015-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dolgalev Igor (2022). Seq-N-Slide. 10.5281/ZENODO.5550459. [DOI] [Google Scholar]
- 82.Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 85.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gu Z, Eils R, and Schlesner M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 88.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e11. 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (related to Fig. 1) Prediction and validation of chromosome-specific sgRNAs targeting human α-satellite centromeric sequences (A) Left: Proliferation assay on RPEs p21/Rb shRNA expressing Cas9 or empty vector (EV) transduced with lentiviral vectors expressing the indicated sgRNAs. The same number of cells were plated in 6-well plates and the percentage of live cells relative to EV was determined 7 days after transduction. Mean and S.D. from triplicates, p-values from Wilcoxon test (*=p<0.05). Imaging validation is also indicated in red. Right: Western blot showing Cas9 expression. (B) Left: Imaging of hCECs (47, +7) expressing 3xmScarlet-dCas9 and sgChr7–1 in the polyclonal population and in a derived clone (clone 8) with high 3xmScarletdCas9 expression. As compared to the polyclonal population, clone 8 contains a higher percentage of cells showing the expected foci. Average frequency of cells displaying foci is shown for the polyclonal and clonal populations (>100 cells counted; in triplicates). Right: Western blot analysis of the expression level of 3xmScarlet-dCas9 in the polyclonal population and clone 8. The percentage of cells showing foci was 45% in the hCEC polyclonal population transduced with 3xmScarlet-dCas9 and increased to 72% in clone 8. (C) Imaging of hCECs expressing 3xmScarlet-dCas9 and the indicated sgRNAs. Representative images of interphase cells are shown; the percentage of cells displaying foci is shown in Table S1. See also Fig. 1F. (D) Imaging of RPEs p21/Rb shRNA expressing 3xmScarlet-dCas9 fusion and the indicated sgRNAs. Representative images of interphase cells are shown. (E) Left: Correlation between the intensity of the signal of the 3xmScarlet-dCas9 foci (measured with ImageJ/Fiji) and the sgRNA activity score(Doench et al., 2016, 2014) of cells treated as in (C). Right: Correlation between the intensity of the signal of the 3xmScarlet-dCas9 foci and the number of predicted sgRNA binding sites on the specific centromere (based on the T2T genome assembly) of cells treated as in (C). Pearson correlation coefficients and corresponding p-values are shown.
Figure S2 (related to Fig. 2) Analysis of KNL1Mut-dCas9 and other fusion proteins targeted to centromeres (A) Maps of the dCas9, KNL1RVSF/AAAA-dCas9, KNL1S24A;S60A-dCas9, NDC80-CH1-dCas9, and NDC80-CH2-dCas9 constructs. The predicted function of each construct is indicated on the right. See text for details. (B) Western blot showing the expression of the indicated constructs in hCECs. (C) Western blot showing the expression of the indicated constructs, in which different mutated segments of KNL1 or NDC80 are fused to the N- or C-terminus of dCas9; see also (A). L: linker with amino acid sequence GGSGGGS. (D) Imaging of hCECs (47, +7) expressing 3xmScarlet-KNL1Mut-dCas9 and transduced with sgChr7–1 or sgChr18–4. (E) Proliferation rate of hCECs transduced with KNL1Mut-dCas9 and with the indicated sgRNAs. Mean and S.D. from triplicates are shown for each time point. (F) FISH imaging and quantification of micronuclei containing chromosome 7 or 13 in hCECs treated with KNL1Mut-dCas9 and the indicated sgRNA (as in Fig. 2G); quantification of micronuclei counts is shown below. Experiments were performed in duplicates, and for each replicate, at least 100 cells were scored.
Figure S3 (related to Fig. 3) Analysis of KNL1Mut-dCas9 and other fusion proteins targeted to centromeres for the induction of chromosome-specific gains and losses (A) KaryoCreate experiment in hCECs comparing the efficiency of different methods for delivering KNL1Mut-dCas9, as quantified by FISH. Methods: (1) transfection of pHAGE-KNL1Mut-dCas9, whose expression of KNL1Mut-dCas9 is driven by the CMV promoter; (2) lentiviral-mediated transduction with pIND20-KNL1Mut-dCas9, whereby the vector is integrated in the genome of the target cells and expression of KNL1Mut-dCas9 is driven by doxycycline treatment (1 μg/ml); (3) lentiviral-mediated transduction with pHAGE-DD-KNL1Mut-dCas9, whereby expression of KNL1Mut-dCas9 is driven by treatment with shield-1 to stabilize the protein. All cells were transduced with sgChr7–1, and FISH quantification of chr7 gains/losses is shown (mean and S.D. from triplicates). (B) KaryoCreate experiment comparing the efficiency of different constructs in inducing chromosome gains and losses. hCECs were transduced with sgChr7–1 and the indicated constructs. FISH quantification for chr7 gains/losses is shown (mean and S.D. from triplicates), along with the aneuploidy level (% of chr7 gains/losses) normalized to the expression level of each construct (as in Fig. S2B). Note that after normalization, the induction of aneuploidy is greatest for NDC80CH2-dCas9 and is higher for KNL1S24A;S60A-dCas9 than for KNL1RVSF/AAAA-dCas9. (C) Left: Western blot analysis of the indicated constructs. Right: KaryoCreate experiment to compare the efficiency of different constructs in inducing chromosome gains and losses. hCECs were transduced with sgChr7–1 and the indicated constructs, and FISH quantification of chr7 gains/losses is shown. (D) Left: Western blot analysis of dCas9 expression in hCECs transduced with KNL1Mut-dCas9 using different amount of virus (about 3 times more virus in the HIGH versus LOW sample, i.e. MOI of 6 for HIGH and 2 for LOW). The corresponding quantification (through ImageJ) is shown below. Right: FISH quantification of chr7 gains/losses in cells expressing KNL1Mut-dCas9 transduced with sgChr7–1 using different amounts of virus at 9–10 days after transduction (mean and S.D. from duplicates). (E) FISH quantification of chr7 gains/losses in hCECs transduced with KNL1Mut-dCas9 and with sgChr7–1 and/or sgChr7–3 (mean and S.D. from duplicates). (F) Single-cell sequencing quantification of chr9 gains/losses in hCECs were transduced with KNL1Mut-dCas9 and with sgNC, sgChr9–3 and/or sgChr9–5 (mean and S.D. from technical duplicates). (G) Left: FACS sorting results for hCECs treated as in (D) using an MOI of 2 after sorting for low or high expression of the cell surface protein EPHB4, encoded by a gene on chr7. Right: FISH quantification of the % of chr7 gains or losses in each condition (N=100 nuclei; mean and S.D. from duplicates). *, p-value<0.05 (Welch two-sample t-test). (H) scRNA-seq analysis of chromosome or arm gains/losses (as in Fig. 4) in hCECs transduced with KNL1Mut-dCas9 (via infection with pIND20-KNL1Mut-dCas9 lentiviral vector) and sgChr7–1. Cells were treated with doxycycline for the indicated number of days to induce construct expression; experiment performed in duplicate.
Figure S4 (related to Fig. 4) Analysis of KaryoCreate across chromosomes and conditions (A) Analysis of hCEC clones with different aneuploidies by bulk WGS (left) and scRNA-seq (right). Arm-level copy number events were inferred from each method (see Methods) and the derived copy number profiles are shown for both methods. See also (B). (B) FISH and scRNA-seq analyses of hCEC clones with chr7 trisomy or more complex karyotypes and the percentage of aneuploid cells was quantified using both methods. Mean values from duplicates are shown. (C) A heatmap depicting gene copy numbers inferred from scRNA-seq analysis following KaryoCreate control experiments. hCECs were transduced either with empty vector or with KNL1Mut-dCas9 together with a negative control sgRNA (sgNC), and scRNA-seq was performed as in (B) to estimate % of gains and losses across chromosomes. (D) A heatmap depicting gene copy numbers inferred from scRNA-seq analysis following KaryoCreate. KaryoCreate for different individual chromosomes (or combination of chromosomes) was performed on RPEs. scRNA-seq was used to estimate the presence of chromosome- or arm-level gains or losses using a modified version of CopyKat. The median expression of genes across each chromosome arm is used to estimate the DNA copy number. The % of gains/losses for each arm (reported below each heatmap) is estimated by comparing the DNA copy number distribution of each experimental sample (chromosome-specific sgRNA) to that of the negative control (sgRNA NC; see also Methods). Heatmap rows represent individual cells, columns represent different chromosomes, and the color represents the copy number change (gain in red and loss in blue). (E) Average proportions (%) of whole-chromosome and arm-level gains/losses. The percentage of the indicated events were calculated as the average among the aneuploid cells generated using KaryoCreate for chromosomes 6, 7, 8, 9, 12, 16, and X )mean values from duplicates). (F) A heatmap depicting chromosome copy numbers inferred from scRNA-seq analysis following KaryoCreate. KaryoCreate was performed on hCECs using two sgRNAs targeting chromosome 7 (sgChr7–1) and 18 (sgChr18–4). scRNA-seq was used to estimate the presence of chromosome- or arm-level gains/losses using a modified version of CopyKat as in (D). Heatmap rows represent individual cells, columns represent different chromosomes, and the color represents the copy number change (gain in red and loss in blue). (G) Immunofluorescence (IF) assay showing DNA damage in HCT116 cells expressing KNL1Mut-dCas9 and sgNC, sgChr7–1, or sgChr18–4. IF was performed for γH2AX (green), CREST (red) to visualize centromeres, and DAPI (blue). Representative images are shown. (H) Quantification of experiment shown in (G). Left: number of DNA damage foci colocalizing with CREST in each cell, quantified and normalized to the total number of CREST foci in the cell. Right: total γH2AX signal per cell, quantified and normalized to the total DAPI signal. p-values are from Wilcoxon test. (I) Left: The total γH2AX signal per cell as determined by IF analysis of hCECs expressing KNL1Mut-dCas9 (pIND20 vector) and sgNC or sgChr7–1 for γH2AX (green) and DAPI (blue), quantified and normalized to the total DAPI signal. Right: Western blot analysis of KNL1Mut-dCas9 expression before or after treatment with doxycycline to induce construct expression. p-values are from Wilcoxon test.
Figure S5 (related to Fig. 5) Dissection of the consequences of 18q loss in colorectal cancer (A) Schematic of experimental plan to apply KaryoCreate across different chromosomes to derive single-cell clones with specific gains or losses. (B) Shallow WGS analysis of single-cell-derived clones obtained by KaryoCreate using sgNC or sgChr7–1 performed on diploid hCECs (as indicated. (C) Representative FISH images and copy number plots from WGS analysis of hCEC sgChr7–1 clone 23 (B) before or after 25 population doublings in culture. (D) Survival analysis (Kaplan-Meier curve) for colorectal cancer patients (TCGA – COADREAD) displaying or not displaying 18q loss, after exclusion of patients with SMAD4 point mutation. (E) Proliferation rates of the indicated hCEC clones 13 and 14 (18q loss) (as in Fig. 5E) after the overexpression of the indicated genes. Mean and S.D. are shown for triplicates; p-values are from Wilcoxon test (*=p<0.05). Proliferation rates for hCEC clones 10 and 5 (18 loss) with and without TGFβ are also shown. (F) Western blot showing SMAD2 and SMAD4 levels in hCEC clone 13 after overexpression of GFP, SMAD2, SMAD4, or SMAD2 + SMAD4. Related to Fig. S5E. (G) Proliferation rates of the indicated hCEC cell lines (clone 14 and hCEC transduced with dCas9 and a SMAD4 or NC sgRNA) when cultured in the presence of TGFβ (20 ng/ml) for 9 days; cells were counted every 3 days in triplicates. p-value is derived from the Wilcoxon test. (H) Western blot showing SMAD4 levels in hCECs transduced with dCas9 and a SMAD4 or NC sgRNA. Related to Fig. S5G.
Table S1. Prediction of sgRNA for each chromosome with CHM13 genome; Related to Figure 1 (see also Methods). The first tab shows sgRNA predictions for 76 selected sgRNAs across all chromosomes except chromosome Y. Columns A-C: chromosome, sgRNA name, and sgRNA sequence. Column D: binding sites (CHM13-specific chromosome centromere HOR_L) representing the number of binding sites on the centromere (HOR_L) of the target chromosome (the chromosome that we intended to target). Column E: binding sites (CHM13-all centromere horL) representing the total number of sites across all centromeres (HOR_L). Column F: chromosome specificity score, defined as the ratio between Columns D and E (given as a fraction, or as a percentage after multiplication by 100). Column G: binding sites (CHM-13 whole genome) representing the number of binding sites on the CHM-13 genome of the target chromosome. Column H: binding sites (HG38 whole genome) representing the number of binding sites on the HG38 genome of the target chromosome. Column I: centromere specificity score, defined as the ratio between the number of binding sites on the centromere (HOR_L) of the target chromosome and the number of binding sites across the whole genome (given as a fraction, or as a percentage after multiplication by 100). Column J: GC content. Column K: activity score, generated based on Doench’s method36. Column L: HOR_L length from the CHM13 genome. Column M: binding sites (CHM13-specific chromosome centromere Not HOR_L) representing the number of binding sites on the centromere (Not HOR_L) of the target chromosome. Column N Validated_by_Imaging, indicating whether the sgRNA can be validated by imaging or not. Column O percentage of cells showing foci (hCEC) related to Figure 1/S1. Columns P-R: binding HOR_L (which HORL region the sgRNA comes from), HOR_L in this chromosome (how many HORL region we have for this specific chromosome), and HOR in this chromosome (how many HOR_L and Not HOR_L region we have for this specific chromosome), respectively. The following tabs contain predictions of sgRNAs for every single chromosome, as indicated.
Table S2. Automated (python-based) quantification of FISH foci after KaryoCreate; Related to Figure 3. This table lists the number of FISH foci quantified using an Automated FISH counting (see Methods) designed to score FISH signals in interphase cells.
Table S3. scRNA-seq-based quantification of arm-level gains and losses after KaryoCreate; Related to Figure 4. Quantification of the frequency of arm-level or chromosome-level gains and losses after KaryoCreate for the indicated chromosomes, based on scRNA-seq (see Methods). Tab S4A refers to Fig. S4A–B and tabs S4B and S4C refer to Fig. 4.
Table S4. Genetic information about the genes on chromosome 18; related to Figs. 5 and S5.
Tab S4A. Genetic information about the genes on chromosome 18.
Tab S4B. Differential expression analysis of hCEC clones comparing clones with or without chromosome 18 loss (Loss vs WT).
Tab S4C. GSEA analysis of hCEC clones comparing clones with or without chromosome 18 loss, Pathways Depleted.
Tab S4D. GSEA analysis of hCEC clones comparing clones with or without chromosome 18 loss, Pathways Enriched.
Supplemental Movie S1. Live-cell imaging showing normal mitosis or lagging chromosome; Related to Figure 2. Clip 1. Live-cell imaging corresponding to Figure 2B, showing a normal mitosis. Shown are time-lapse images taken every 3 min of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and sgChr7–1. Clip 2. Live-cell imaging corresponding to Figure 2B, showing a lagging chromosome and micronucleus. Shown are time-lapse images taken every 3 min of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and sgChr7–1. Clip 3. Live-cell imaging corresponding to Figure 2B, showing a lagging chromosome. Shown are time-lapse images taken every 3 min of hCECs expressing H2B-GFP, KNL1Mut-dCas9, and sgChr18–4. Clip 4. Live-cell imaging corresponding to Figure 2B, showing a lagging chromosome. Shown are time-lapse images taken every minute of hCECs expressing H2B-GFP, 3xmScarlet-KNL1Mut-dCas9, and sgChr7–1.
Data Availability Statement
Single-cell RNA sequencing and bulk RNA sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession number is listed in the key resources table. Whole-genome sequencing data have been deposited at SRA and are publicly available as of the date of publication. Accession number is listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Cas9 antibody | Abcam | Cat#Ab191468 |
| GAPDH antibody | Santa Cruz Biotechnology | Cat#Sc-47724 |
| B-Actin antibody | Cell Signaling Technology | Cat#8844 |
| Goat Anti-Mouse IgG H&L (HRP) | Abcam | Cat#Ab205719 |
| Anti-centromere protein antibody | Antibodies Incorporated | SKU 15-234 |
| Anti-a-Tubulin antibody | Sigma-Aldrich | T9026 |
| CyTM3 AffiniPure Donkey Anti-Mouse IgG | Jackson ImmunoResearch | 715-165-150; RRID: AB_2340813 |
| Alexa Fluor® 647 AffiniPure F(ab’)₂ Fragment Donkey Anti-Human |
Jackson ImmunoResearch | 709-606-149; RRID: AB_2340581 |
| TotalSeq-B0256 anti-human Hashtag 1 Antibody | BioLegend | Cat#394631 |
| TotalSeq-B0256 anti-human Hashtag 2 Antibody | BioLegend | Cat#394633 |
| TotalSeq-B0256 anti-human Hashtag 3 Antibody | BioLegend | Cat#394635 |
| TotalSeq-B0256 anti-human Hashtag 4 Antibody | BioLegend | Cat#394637 |
| TotalSeq-B0256 anti-human Hashtag 5 Antibody | BioLegend | Cat#394649 |
| TotalSeq-B0256 anti-human Hashtag 6 Antibody | BioLegend | Cat#394641 |
| TotalSeq-B0256 anti-human Hashtag 7 Antibody | BioLegend | Cat#394643 |
| TotalSeq-B0256 anti-human Hashtag 8 Antibody | BioLegend | Cat#394645 |
| TotalSeq-B0256 anti-human Hashtag 9 Antibody | BioLegend | Cat#394647 |
| TotalSeq-B0256 anti-human Hashtag 10 Antibody | BioLegend | Cat#394649 |
| Anti-yH2A.X Antibody | Sigma-Aldrich | SKU 05-636 |
| Alexa Fluor® 647 AffiniPuro Goat Anti-Human | Jackson ImmunoResearch | 109-605-044; RRID: AB_2337885 |
| Alexa Fluor® 488 AffiniPuro Donkey Anti-Rabbit | Jackson ImmunoResearch | 711-545-152; RRID: AB_2313584 |
| Anti-Smad2 antibody | Abcam | Cat#Ab40855 |
| Smad4 Antibody | Santa Cruz Biotechnology | Cat#Sc-7966 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Pierce FITC Conjugated Avidin | ThermoFisher Scientific | Cat#21221 |
| Anti-Digoxigenin-Rhodamine, Fab fragments | Sigma-Aldrich | Cat#112077509 1 |
| RO-3306 | Sigma-Aldrich | Cat#SML0569; CAS: 872573-93-8 |
| MG-132 | Tocris | Cat#1748; CAS: 133407-82-6 |
| Colcemid | Roche | Cat#102958920 01 |
| Doxycycline | Sigma | Cat#D5207 |
| Shield-1 | CheminPharma | CIP-S1-0.5nM |
| Lipofectamine 3000 Transfection Reagent | ThermoFisher Scientific | Cat#L3000075 |
| DAPI | Sigma-Aldrich | Cat#MBD0015; CAS:28718-90-3 |
| ProLong Glass Antifade Moutant | ThermoFisher Scientific | Cat#P36980 |
| Critical commercial assays | ||
| Gateway LR Clonase II Enzyme mix | ThermoFisher Scientific | Cat#11791020 |
| Chromosome 7 Control Probe | Empire Genomics | Cat#CHR07-10GR |
| Chromosome 18 Control Probe | Empire Genomics | Cat#CHR18-10-GR |
| NEBNext dsDNA Fragmentase | New England Biolabs | Cat#M0348 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7645L |
| Qubit 2.0 fluorometer | Invitrogen | Cat#Q32866 |
| Qubit dsDNA HS kit | Invitrogen | Cat#Q32854 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| 2100 Bioanalyzer system | Agilent | Cat#G2939BA |
| TruSeq Stranded Total RNA Library Prep Gold | Illumina | Cat#20020598 |
| Agilent 2200 TapeStation System | Agilent | G2964AA |
| Agilent High Sensitivity D1000 ScreenTape | Agilent | 5067-5584 |
| NovaSeq 6000 SP Reagent Kit v1.5 (100 cycles) | Illumina | Cat#20028401 |
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10X Genomics | PN-1000075 |
| Chromium Single Cell B Chip Kit | 10X Genomics | PN-1000073 |
| Chromium i7 Multiplex Kit | 10X Genomics | Pn-120262 |
| Experimental models: Cell lines | ||
| hCEC hTERT | Ly et al.38 | PMC:3071083 |
| hCEC hTERT TP53−/− | This paper | N/A |
| HCT116 | ATCC | CCL-247 |
| RPE hTERT | ATCC | CRL-4000 |
| RPE hTERT p21/Rb shRNA | Maciejowski et al.39 | PMID:26687355 |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| gNC: ACGGAGGCTAAGCGTCGCAA | Sanjana et al.40 | N/A |
| Chromosome-specific gRNAs, see table S1 | This paper | N/A |
| SMAD4 CRISPRi gRNA: GGCAGCGGCGACGACGACCA |
Gilbert et al.76 | N/A |
| Recombinant DNA | ||
| plentiGuide-Puro | Chen et al.72 | Addgene #52963 |
| pLentiGuide-Puro-FE | This paper | N/A |
| pX330-U6-Chimeric_BB_CBh-hSpCas9 | Cong et al.74 | Addgene #42230 |
| Chromosome 7 BAC | BACPAC Genomics | RP11-22N19 |
| Chromosome 13 BAC | BACPAC Genomics | RP11-76N11 |
| Chromosome 18 BAC | BACPAC Genomics | RP11-787K12 |
| H2B-GFP plasmid | Titia de Lange lab | pCLRNX-H2B-GFP |
| pHAGE-3xmScarlet-dCas9 | This paper | N/A |
| pHAGE-KNL1Mut-dCas9 | This paper | N/A |
| pIND20-KNL1Mut-dCas9 | This paper | N/A |
| pIND20-GFP | This paper | N/A |
| pHAGE-DD-KNL1Mut-dCas9 | This paper | N/A |
| pHAGE-KNL1S24A;S60A-dCas9 | This paper | N/A |
| pHAGE-dCas9 | This paper | N/A |
| pHAGE-NDC80CH1-dCas9 | This paper | N/A |
| pHAGE-NDC80CH2-dCas9 | This paper | N/A |
| pInducer20 | Meerbrey et al.44 | Addgene #44012 |
| Software and algorithms | ||
| Photoshop v21.2.3 | Adobe | https://www.adobe.com |
| FIJI/ImageJ2 version 2.3.0/1.53f | Schindelin et al.75 | https://imagej.nih.gov/ij/download.html |
| Python 3.7 | Python Software Foundation | https://www.python.org/downloads/ |
| Scikit-image | Van Der Walt et al.77 | https://scikiti-mage.org |
| BWA-mem v0.7.17 | Li et al.78 | https://github.com/lh3/bwa/releases/tag/v0.7.17 |
| Genome Analysis Toolkit v4.1.7.0 | Van der Auwera, 202079 | https://gatk.broadinstitute.org/hc/en-us |
| CopywriteR v1.18.0 | Kuilman et al.80 | https://github.com/PeeperLab/CopywriteR |
| Seq-N-Slide | Dolgalev, 2022.81 | https://github.com/igordot/sns |
| Trimmomatic | Bolger et al.82 | https://github.com/timflutre/trimmomatic |
| STAR | Dobin et al.83 | https://github.com/alexdobin/STAR |
| featureCounts | Liao et al.84 | https://github.com/byee4/featureCounts |
| DESeq2 | Love et al.50 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA pre-ranked | Subramanian et al.85 | https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html |
| CellRanger v6.1 | 10X Genomics | https://support.10xgenomics.com/single-cell-geneexpression/software/overview/welcome |
| Seurat v4.0.3 | Hao et al.86 | https://github.com/satijalab/seurat |
| CopyKat v1.0.5 | Gao et al.46 | https://github.com/navinlabcode/copykat |
| ComplexHeatmap v2.8 | Gu et al.87 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Other | ||
| Code for automated FISH foci counting | This paper | https://doi.org/10.6084/m9.figshare.21843393 |
| Code for single-cell RNA-seq analysis | This paper | https://doi.org/10.6084/m9.figshare.21843393 |
| Glass-bottom microwell dishes | MatTek | Cat# P35G-1.5-14-C |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen Invitrogen |
Cat#NP0007 |
| NuPAGE 4 to 12% Bis-Tris Mini Protein Gels | Cat#NP0322BO X |
|
| Trans-Blot Turbo Mini 0.45 uM LF PVDF Transfer Kit | Bio-Rad | Cat#1704274 |
| Human Cot-1 DNA | ThermoFisher Scientific | Cat#15279011 |
| UltraPure Herring Sperm DNA Solution | ThermoFisher Scientific | Cat#15634017 |
| Proteinase K | Qiagen | Cat#19131 |
| Pierce ECL Western Blotting Substrate | Thermo Scientific | Cat#32209 |
| Sera-Mag Select beads | Cytiva | Cat#29343052 |
| Single-cell and bulk RNA sequencing data | This paper |
GSE217326; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE217326 |
| Whole genome sequencing data | This paper | PRJNA899849;https://dataview.ncbi.nlm.nih.gov/object/PRJNA899849 |
| T2T Chm13v2.0.fa.gz assembly | Nurk et al.29 | GCA_00991475 5.4 https://github.com/marbl/CHM13 |
| GRChg38 reference assembly | Schneider et al.30 | GCA_00000140 5.28 https://hgdownload.soe.ucsc.edu/downloads.html |
| TCGA data | NA | https://www.cancer.gov/aboutnci/organization/ccg/research/structuralgenomics/tcga/using-tcga/tools |
| TUSON data | Davoli et al.4 | NA |
All original analysis code has been deposited to GitHub and is available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.