

Abstract
Aims
Left ventricular remodelling subsequent to myocardial infarction (MI) constitutes a pivotal underlying cause of heart failure. Intervention with the nontoxic endogenous aryl hydrocarbon receptor (AHR) agonist 2‐(1′H‐indole‐3′‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester (ITE) in the acute phase of MI has been shown to ameliorate cardiac function, but its role in the chronic phase remains obscured. This study explores the beneficial role of ITE in delaying the progression of heart failure in the chronic phase of MI.
Methods and results
MI rats established by ligating the left anterior descending coronary artery were treated with the indicated concentration of the AHR agonist ITE or vehicle alone. Echocardiography was performed to determine cardiac structure and function; myocardial morphology and fibrosis were observed by haematoxylin and eosin and Masson's trichrome staining; serum biochemical indices, BNP, and inflammatory cytokine levels were detected by enzyme‐linked immunosorbent assay; F4/80+iNOS+M1 macrophages and F4/80+CD206+M2 macrophages were detected by immunofluorescence; the terminal deoxynucleotidyl transferase‐mediated dUTP nick end labelling assay was used to detect the apoptosis of cardiomyocytes; ultrastructural changes in myocardial tissue were observed by transmission electron microscopy; and Cyp1a1, Akt, P‐Akt, p70S6K, P‐p70S6K, Bcl‐2, Bax, caspase‐3, and cleaved caspase‐3 protein levels were determined via Western blotting. We found that therapy with the AHR agonist ITE rescued cardiac remodelling and dysfunction in rats with MI and attenuated myocardial fibrosis, inflammation, and mitochondrial damage. Further studies confirmed that ITE dose‐dependently improved myocardial cell apoptosis after MI, as demonstrated by reduced levels of the apoptosis‐related proteins cleaved caspase‐3 and Bax but increased expression levels of Bcl‐2. These effects were attributed to ITE‐induced activation of AHR receptors, leading to the down‐regulation of Akt and p70S6K phosphorylation.
Conclusions
The AHR agonist ITE alleviates cardiomyocyte apoptosis through the Akt/p70S6K signalling pathway, thereby rescuing left ventricular adverse remodelling and cardiac dysfunction after MI.
Keywords: AHR agonist, Myocardial infarction, Ventricular remodelling, Apoptosis, Akt/p70S6K
Introduction
Myocardial infarction (MI), a disease that seriously affects human lifespan and quality of life, is currently a main cause of death. Clinical application of various revascularization strategies, such as early coronary reperfusion therapy, can help save dying myocardium and significantly reduce mortality. 1 , 2 However, pathological myocardial remodelling and unavoidable heart failure after MI have become public health problems. 1 Therefore, improving cardiac remodelling has long been the focus of an effective strategy to improve patient outcomes after MI.
Due to the non‐regeneration of myocytes, myocardial tissue in the infarct and peri‐infarct areas undergoes apoptosis and necrosis after MI due to ischaemia, hypoxia, and inflammatory reactions. 3 The massive loss of cardiomyocytes alters the electrical conduction system of the myocardium and causes systolic and diastolic dysfunction, leading to heart failure and arrhythmias. 4 Apoptosis is required for the orderly clearance of cells in organisms. In contrast, necrosis is the premature death of cells caused by external disease, injury, or insufficient blood supply. As necrotic cells die, an inflammatory response is triggered, causing collateral damage to surrounding cells. Normally, apoptosis removes cells without triggering adverse immune and inflammatory responses. However, when apoptosis is dysregulated, cell death contributes to disease onset and progression. Apoptosis has been widely demonstrated in hypertrophic cardiomyopathy, ischaemic cardiomyopathy, myocarditis, and dilated cardiomyopathy. 5 Myocardial ischaemia can cause apoptosis through a variety of pathways. 6 Studies have shown that cardiomyocyte apoptosis plays a role in different types of cardiomyopathy and heart failure and that apoptotic cardiomyocyte death often accompanies irreversible congestive heart failure. 7 Inhibition of cardiomyocyte apoptosis has been shown to delay the development of cardiac dilation and systolic dysfunction and improve heart failure.
The aryl hydrocarbon receptor (AHR) belongs to a subfamily of the basic helix–loop–helix/Per‐ARNT‐Sim (bHLH/PAS) family and is a transcription factor that regulates adaptive metabolism and environmental responses. 8 Currently, an increasing number of studies have found that the activation of AHR by endogenous AHR ligands can regulate various biological processes, such as the immune response, growth factor signalling, cell cycle proliferation, differentiation, arrest, and apoptosis. 9 AHR has been shown to play an important role in the regulation of the pathophysiological process of cardiac development 10 and cardiovascular and neurological diseases. 11 Activation of AHR protects cardiomyocytes from apoptosis induced by antitumor‐related drugs. 12 Nontoxic endogenous AHR ligand 2‐(1′H‐indole‐3′‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester (ITE) therapy in the acute phase of MI can improve cardiac function by activating AHR, 13 but the effects of ITE in the chronic phase of MI are unknown. Therefore, we speculate that AHR activation by ITE could halt the development of heart failure in the chronic stage of MI.
Methods
Induction of the rat myocardial infarction model and treatment with ITE
This animal experiment was approved by the Laboratory Animal Ethics Committee of Fujian Medical University. Eight‐week‐old male Sprague–Dawley rats (body weight of 200 ± 10 g) were purchased from Shanghai Slack Company. The MI model was established by permanent ligation of the left anterior descending (LAD) coronary artery under ventilator support. 14 Rats that survived after surgery were verified by cardiac ultrasound after 1 week and randomly divided into the MI group (MI, n = 8), MI with 1.6 mg/kg/day ITE treatment group (MI + ITE1.6, n = 8), and MI with 8 mg/kg/day ITE treatment group (MI + ITE8.0, n = 8). The high and low doses of ITE used in this study were described in the literature. 15 In addition, the control group (control, n = 8), sham group (sham, n = 8), control + ITE1.6 group (n = 8), and control + ITE8.0 group (n = 8) were set up; dimethyl sulfoxide (DMSO) (Sigma‐Aldrich, St. Louis, MO, USA), PEG300, Tween 80 (VICMED Company), and normal saline were added to ITE (Eisai Company) at a ratio of 1:3.5:1:9.5, mixed well, and administered by intraperitoneal injection once a day.
Echocardiography
At 7 days after MI and 28 days after MI, GE Vivid E9 Doppler echocardiography with a 12S probe (frequency of 4–12 MHz, equipped with an EchoPAC 113.1.3 workstation) was used for echocardiography under anaesthesia with 2% sodium pentobarbital. Left ventricular (LV) volumes and ejection fraction (EF) were assessed with the biplane Simpson's method. The LV M‐mode images at the level of the papillary muscle were recorded from the short‐axis view. Pulsed‐wave Doppler blood flow images of the apical four‐chamber view at the mitral level and tissue Doppler images of the lateral and septal mitral annulus were recorded. Heart rate (HR) was recorded by synchronized electrocardiography. Images were quantified and analysed by the EchoPAC 113.1.3 image analysis system (GE Healthcare). Operators for all experiments were blinded to group allocation during all analytical procedures.
Analysis of blood serum
Blood samples were collected from the abdominal aorta, centrifuged to isolate serum, and stored at −80°C until assay. Glucose (GLU), blood urea nitrogen (BUN), serum creatinine (SCr), serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatine kinase (CK), and CK isoenzyme (CK‐MB) were determined by the colorimetric method utilizing an automatic biochemical analyser (Roche, Shanghai, China). Circulating BNP (SEA541RA, Cloud‐Clone Corp., China), tumour necrosis factor‐α (TNF‐α) (KE20018, Proteintech Group, China), interleukin‐1β (IL‐1β) (KE20005, Proteintech Group, China), interleukin‐17 (IL‐17) (SEA063RA, Cloud‐Clone Corp., China), and interleukin‐10 (IL‐10) (SEA056RA, Cloud‐Clone Corp., China) were measured by rat ultrasensitive enzyme‐linked immunosorbent assay (ELISA) kit according to the manufacturer's instructions.
Histology and immunofluorescence
Global cardiac mass index and LV mass index were calculated as whole cardiac mass/body weight and left cardiac mass/body weight. For histological analysis, the mid‐papillary slice of the left ventricle was fixed in 4% formaldehyde and paraffin embedded. Haematoxylin and eosin (HE) staining and Masson's trichrome staining were performed to evaluate the infarct size and the extent of interstitial fibrosis. To quantify cardiomyocyte area and fibrotic percentage, heart section images were obtained using an optical microscope and then analysed using ImageJ Version 1.8.0 (National Institutes of Health, USA). Three visual fields were randomly selected for each heart tissue section, and their average values were taken as data for each rat. For immunofluorescence staining, paraffin‐embedded cardiac tissue sections were deparaffinized and rehydrated. After blocking, the sections were stained overnight at 4°C with one of the following antibodies: F4/80 (sc‐377009, Santa Cruz Biotechnology, USA), inducible nitric oxide synthase (iNOS) (YT3169, ImmunoWay, USA), and CD206 (24595S, Cell Signaling Technology, UK), and then separately reacted with the corresponding Alexa Fluor 488‐conjugated anti‐IgG (ab150077, Abcam, USA) and Alexa Fluor 594‐conjugated anti‐IgG (ab150116, Abcam, USA) secondary antibodies. Staining was visualized using a Zeiss 510 confocal microscope (Carl Zeiss). Image analysis was conducted using Image‐Pro Plus.
TUNEL fluorescence staining
After dewaxing the paraffin section, the tissue was treated with proteinase K working solution in the DeadEnd™ Fluorometric TUNEL (terminal deoxynucleotidyl transferase‐mediated dUTP nick end labelling) System kit (Promega, China) for 15–30 min at 21–37°C. After rinsing, equilibration buffer was added dropwise and incubated at room temperature for 5–10 min. Recombinant terminal deoxynucleotidyl transferase (rTdT) incubation buffer was added dropwise and incubated at 37°C for 1 h in the dark, and then 2× saline sodium citrate (SSC) was added and incubated at room temperature for 15 min to terminate the reaction. DAPI‐containing fluorescence quencher was added and the samples were covered with a coverslip; a Zeiss LSM 780 laser was used to scan a total of observation and filming under a focusing microscope, randomly selected five fields of view, the apoptotic cells stained with TUNEL were regarded as positive cells, and the percentage of apoptosis in each field of view was counted by Image‐Pro Plus 6.0 software as cell apoptosis index (AI).
Western blotting
Protein samples (25 μg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), followed by transfer to polyvinylidene difluoride (PVDF) membranes. Next, the samples were examined by immunoblotting with primary antibodies against serine/threonine protein kinase (Akt) (1:1000, 9272S, Cell Signaling Technology, UK), phosphorylated Akt (1:1000, 9271S, Cell Signaling Technology, UK), 70 kDa ribosomal protein S6 kinase (p70S6K) (1:1000, 9202, Cell Signaling Technology, UK), phosphorylated p70S6K (1:1000, 9204, Cell Signaling Technology, UK), caspase‐3 (1:1000, 9662, Cell Signaling Technology, UK), cleaved caspase‐3 (1:1000, 9661, Cell Signaling Technology, UK), Bcl‐2 (1:1000, ab194583, Abcam, USA), Bax (1:1000, ab32503, Abcam, USA), and β‐actin (1:1000, 20536‐1‐AP, Proteintech Group, China) at 4°C overnight. Then, horseradish peroxidase (HRP)‐conjugated secondary antibody (1:3000, GB23303, Servicebio, China) was incubated for 1 h at room temperature and visualized by using an enhanced chemiluminescence (ECL) detection kit (Meilunbio, China).
Transmission electron microscopy
Fresh frozen tissues were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, and transmission electron microscopy (TEM) fixation buffer (Electron Microscopy Sciences, Hatfield, USA) at 4°C. The samples were subsequently post‐fixed in 2% aqueous osmium tetroxide, dehydrated, embedded, cut into 100 nm ultrathin sections with a Leica UC6 ultramicrotome, stained with uranyl acetate and lead citrate for 5–15 min, washed with distilled water, and observed under a Philips EM208 transmission electron microscope photograph.
Statistical analysis
All data were analysed by the SPSS 13.0 software system. Data are expressed as mean ± SD. The Kolmogorov–Smirnov test, Shapiro–Wilk test, Q–Q Plot, and histograms were used to test the normality of the data distribution. ANOVA was employed for the comparison of means among multiple groups. A P < 0.05 was considered a significant difference.
Results
Induction of the myocardial infarction model in rats and design for ITE therapy
We generated a rat MI model by performing LAD coronary artery ligation surgery. The significant elevation of the ST segment in electrocardiogram (ECG) denoted successful LAD ligation (Supporting Information, Figure S1 ). In the present study, we designed a preclinical trial in which rats with similar levels of cardiac dysfunction after MI were randomized to receive either vehicle or ITE therapy (Figure 1 A ). All rats enrolled the study (N = 82) underwent baseline echocardiography and were then randomly assigned to four groups: MI surgery (N = 50), sham (N = 8), control (N = 8), control + ITE1.6 (N = 8), and control + ITE8.0 (N = 8). Twenty MI rats died during or 1 week after surgery, leaving 30 surviving MI rats. The survival rate of rats 1 week after MI was consistent with that of other similar studies. 16 Echocardiograms at 1 week after surgery were used to assess MI severity. Of the remaining 30 MI rats, 6 were excluded with LVEF outside the range of 40–50% because they either already developed severe heart failure or did not exhibit signs of heart dysfunction. The remaining 24 rats with LVEF ranging from 40% to 50% entered the prospective, randomized trial in which rats were assigned to either ITE (N = 8 for 1.6 mg/kg/day; N = 8 for 8 mg/kg/day) or vehicle (N = 8) treatment. ITE was delivered daily by intraperitoneal injection at 1 week after MI. Following ITE treatment for 4 weeks, rats underwent echocardiography (Figure 1 B ). As shown in Table 1 , 7 days after MI surgery, compared with the sham group, echocardiographic parameters including LV end‐diastolic diameter (LVDd), LV end‐systolic diameter (LVDs), and the ratio of mitral valve flow in early diastole and peak annulus motion velocity (E/e′) were significantly increased in the MI group (P < 0.05), whereas the ventricular septal end‐diastolic thickness (IVSd), LVEF, and LV short‐axis fractional shortening (FS) were significantly decreased (P < 0.05) (Table 1 ). These results indicate successful induction of the MI model.
Figure 1.
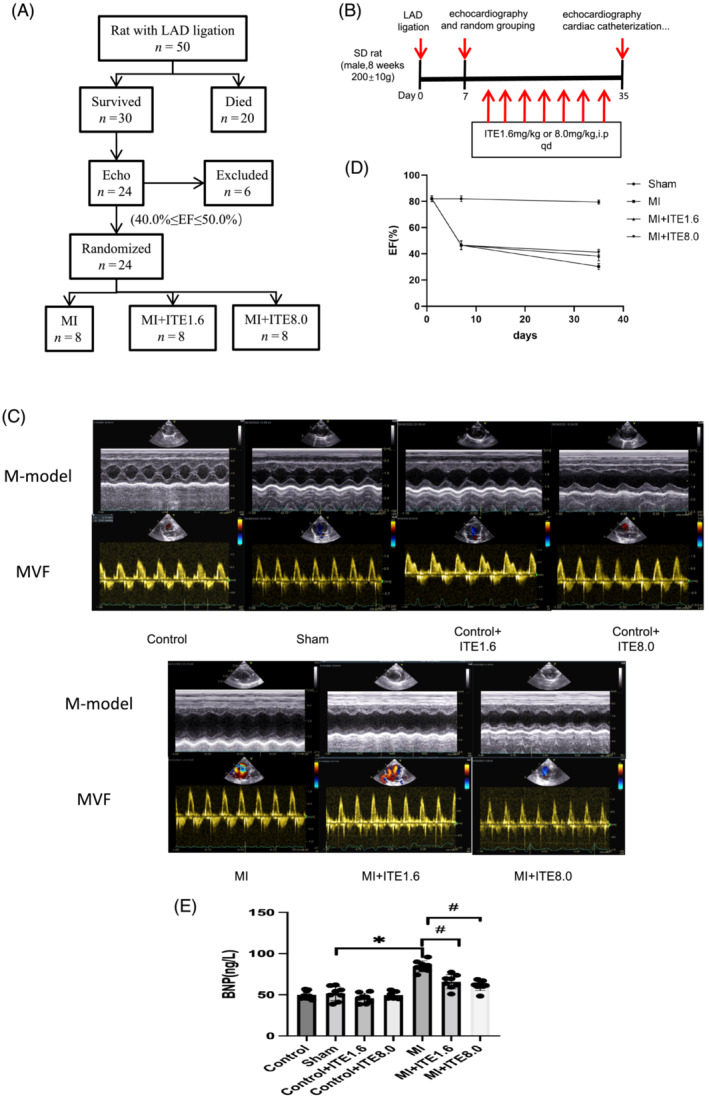
ITE ameliorates cardiac dysfunction and heart failure in MI rats. (A) Flow chart of inclusion criteria based on echocardiographic ejection fraction (EF) used to obtain a cohort of MI mice with similar levels of cardiac dysfunction that were then randomized to receive vehicle or ITE. (B) Schematic diagram describing the animal experiments. (C) Representative M‐mode echocardiograms and mitral valve inflow (MVF) from control, sham, control + ITE1.6, control + ITE8.0, MI, MI + ITE1.6, and MI + ITE8.0. (D) The EF timeline shows the changes in EF at different time points in sham, MI, MI + ITE1.6, and MI + ITE8.0 rats. (E) Serum BNP levels measured by ELISA. N = 6–8 per group. Data are presented as mean ± SD. *P < 0.05 vs. sham group. # P < 0.05 vs. MI group.
Table 1.
Changes of cardiac structure and function in rats 1 week after MI
|
Control n = 8 |
Sham n = 8 |
MI‐1W n = 24 |
|
|---|---|---|---|
| HR (b.p.m.) | 476.36 ± 4.26 | 492.34 ± 6.22 | 481.22 ± 6.23 |
| IVSd (mm) | 1.63 ± 0.02 | 1.60 ± 0.07 | 1.49 ± 0.28* |
| LVDd (mm) | 6.37 ± 0.84 | 6.32 ± 0.72 | 8.47 ± 0.84* |
| LVDs (mm) | 3.48 ± 0.32 | 3.32 ± 0.27 | 6.42 ± 0.68* |
| LVPWd (mm) | 171 ± 0.09 | 1.63 ± 0.07 | 1.55 ± 0.13 |
| EF (%) | 80.68 ± 0.81 | 82 ± 2.27 | 46.57 ± 3.38* |
| FS (%) | 44.30 ± 0.83 | 42.58 ± 0.72 | 19.31 ± 1.09* |
| E/e′ | 11.50 ± 1.63 | 12.37 ± 1.73 | 19.39 ± 2.14* |
E/e′, the ratio of mitral valve flow in early diastole and peak annulus motion velocity; EF, ejection fraction; FS, left ventricular short‐axis shortening; HR, heart rate; IVSd, ventricular septal end‐diastolic thickness; LVDd, left ventricular end‐diastolic diameter; LVDs, left ventricular end‐systolic diameter; LVPWd, left ventricular posterior wall end‐diastolic thickness; MI, myocardial infarction.
Data are presented as mean ± SD.
p < 0.05 vs. sham group.
ITE therapy halts the development of heart failure in myocardial infarction rats
To evaluate the effects of ITE on rat cardiac structure and function, we examined rat heart function by echocardiography after ITE intervention. After 4 weeks of ITE treatment, LV dilatation was attenuated in both the MI + ITE1.6 and MI + ITE8.0 groups, as evidenced by a sharp decrease in LVDs and E/e′ (P < 0.05) and an increase in EF and FS (P < 0.05), but there was no significant difference in LVDd (Table 2 and Figure 1 C ). The EF timeline shows the changes in EF at different time points in the sham, MI, MI + ITE1.6, and MI + ITE8.0 groups (Figure 1 D ). Next, we detected the serum BNP level of the rats and found similar results: the serum BNP level of the MI rats with ITE treatment was significantly decreased compared with that of the MI group (P < 0.05) (Figure 1 E ), but ITE treatment alone or sham surgery did not affect BNP levels. Collectively, our data illustrate the beneficial effects of ITE on cardiac structure and function after MI.
Table 2.
Effects of ITE treatment on cardiac structure and function in MI rats
| Control | Sham | Control + ITE1.6 | Control + ITE8.0 | MI | MI + ITE1.6 | MI + ITE8.0 | |
|---|---|---|---|---|---|---|---|
| HR (b.p.m.) | 488.63 ± 5.08 | 492.34 ± 6.22 | 485.02 ± 6.13 | 478.64 ± 8.22 | 488.44 ± 5.86 | 487.28 ± 6.33 | 491.02 ± 6.12 |
| IVSd (mm) | 1.73 ± 0.04 | 1.70 ± 0.07 | 1.78 ± 0.08 | 1.72 ± 0.07 | 1.35 ± 0.23* | 1.33 ± 0.10* | 1.32 ± 0.24* |
| LVDd (mm) | 6.64 ± 0.84 | 6.32 ± 0.72 | 6.79 ± 0.66 | 6.49 ± 0.74 | 10.81 ± 1.07* | 9.38 ± 0.82* | 9.41 ± 0.67* |
| LVDs (mm) | 3.71 ± 0.49 | 3.24 ± 0.58 | 3.97 ± 0.42 | 3.49 ± 0.66 | 8.05 ± 0.45* | 7.34 ± 0.39* , # | 7.13 ± 0.41* , # |
| LVPWd (mm) | 1.68 ± 0.08 | 1.66 ± 0.07 | 1.67 ± 0.08 | 1.66 ± 0.06 | 1.28 ± 0.13* | 1.32 ± 0.09* | 1.33 ± 0.16* , # |
| EF (%) | 80.71 ± 0.81 | 79.51 ± 1.54 | 78.86 ± 1.36 | 77.26 ± 2.33 | 30.29 ± 2.33* | 38.24 ± 3.51* , # | 41.17 ± 2.20* , # |
| FS (%) | 44.12 ± 0.72 | 45.04 ± 0.66 | 43.24 ± 0.88 | 44.64 ± 0.73 | 12.35 ± 2.34* | 16.10 ± 1.03* , # | 17.11 ± 1.10* , # |
| E/e′ | 10.69 ± 1.85 | 11.59 ± 1.24 | 11.05 ± 2.13 | 10.74 ± 2.13 | 32.73 ± 3.73* | 25.80 ± 2.92* , # | 26.65 ± 2.43* , # |
E/e′, the ratio of mitral valve flow in early diastole and peak annulus motion velocity; EF, ejection fraction; FS, left ventricular short‐axis shortening; ITE, 2‐(1′H‐indole‐3′‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; IVSd, ventricular septal end‐diastolic thickness; LVDd, left ventricular end‐diastolic diameter; LVDs, left ventricular end‐systolic diameter; LVPWd, left ventricular posterior wall end‐diastolic thickness; MI, myocardial infarction.
n = 8 per group. Dataare presented as mean ± SD.
p < 0.05 vs. sham group.
p < 0.05 vs. MI group.
ITE suppresses cardiac remodelling and fibrosis inmyocardial infarction rats
To further elucidate the mechanism by which ITE improves cardiac structure and function after MI, we performed histological analysis of rat hearts. First, morphological analysis demonstrated that the global cardiac mass index and LV mass index were lower in the MI + ITE1.6 and MI + ITE8.0 groups than in the MI group (P < 0.05) (Figure 2 A ), suggesting attenuation of cardiac hypertrophy and cardiac remodelling.
Figure 2.
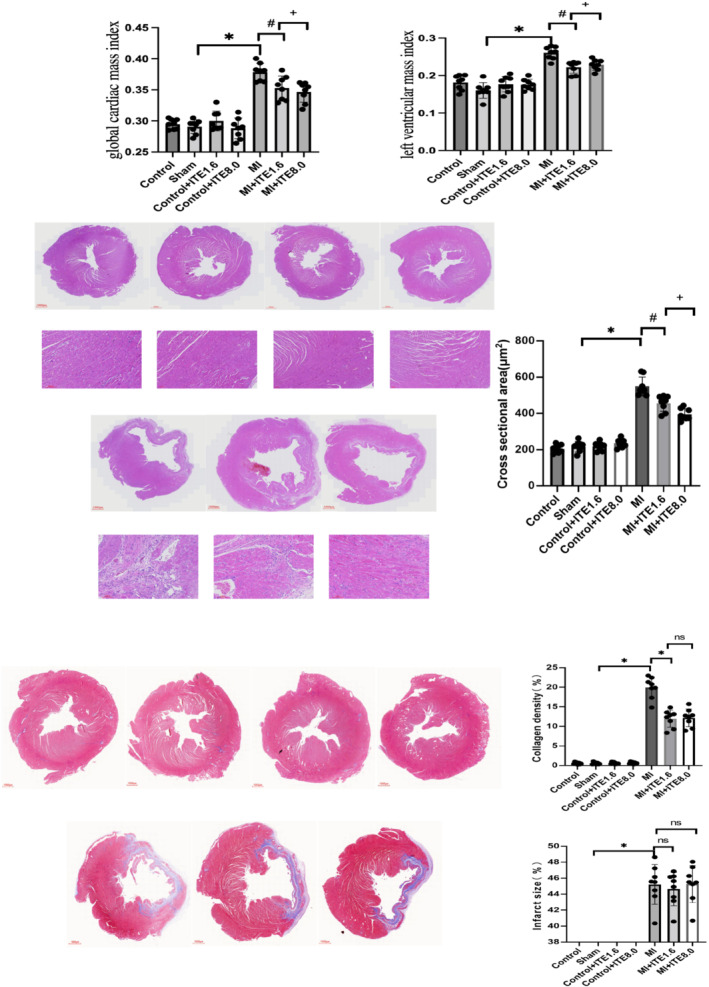
ITE suppresses cardiac remodelling and fibrosis in MI rats. (A) Global cardiac mass index and left ventricular mass index calculated as whole cardiac mass/body weight and left cardiac mass/body weight, respectively. N = 8 per group. Data are presented as mean ± SD. *P < 0.05 vs. sham group. # P < 0.05 vs. MI group. (B) Representative HE staining obtained from control, sham, control + ITE1.6, control + ITE8.0, MI, MI + ITE1.6, and MI + ITE8.0. (C) Masson's trichrome staining of transverse heart sections at the papillary level to detect the collagen density and infarct size; collagen density is presented as a percentage of collagen‐occupying area relative to total LV area; infarct size was calculated as total infarct circumference divided by total LV circumference; n = 8 per group. Data are presented as mean ± SD. *P < 0.05 vs. sham group. # P < 0.05 vs. MI group.
Next, we observed in HE staining that compared to MI, there was the swelling of myocardial cells at the edge of MI in the MI + ITE1.6 and MI + ITE8.0 groups (Figure 2 B ). We further assessed infarct size and collagen deposition in the infarct area using Masson's trichrome staining. After 4 weeks of ITE intervention, Masson's trichrome staining at the papillary muscle level showed that ITE treatment did not influence the infarct size. However, the collagen density in the MI + ITE1.6 and MI + ITE8.0 groups was significantly decreased compared with that in the MI group (P < 0.05) (Figure 2 C ), while there was no significant difference among the control, control + ITE1.6, control + ITE8.0, and sham groups. Collectively, our findings suggest that ITE may halt cardiac remodelling and dysfunction by alleviating cardiomyocyte apoptosis and fibrosis.
ITE attenuates cardiomyocyte apoptosis after myocardial infarction
Previous studies 4 have shown that massive apoptosis of cardiomyocytes is an important cause of cardiac dysfunction. Therefore, a TUNEL assay was performed to detect apoptosis in each group. In comparison with the sham group, a significant decrease in TUNEL‐positive cells was observed in the border area of the hearts in the MI + ITE1.6 and MI + ITE8.0 groups (P < 0.05), and the myocardial AI in the ITE treatment groups showed a dose‐dependent decrease (P < 0.05) (Figure 3 A ). Several studies 17 have confirmed that Bcl‐2 and Bax are anti‐apoptotic proteins and pro‐apoptotic proteins, respectively, and caspase‐3 is an apoptosis marker protein. The ratio of Bcl‐2 to Bax and the expression level of caspase‐hydrolyzed fragments can reflect the apoptosis state of cells. Immunoblot analysis revealed significantly decreased expression of cleaved caspase‐3 but increased expression of Bcl‐2/Bax in myocardium lysates harvested from peri‐infarct areas of hearts in the MI + ITE1.6 and MI + ITE8.0 groups compared with that from the MI group, and such changes presented ITE concentration dependence (P < 0.05). ITE treatment alone or sham surgery did not affect the expression of Bcl‐2/Bax or cleaved caspase‐3 (Figure 3 B and Supporting Information, Figure S2 ). Our results suggest that the AHR agonist ITE attenuates cardiomyocyte apoptosis after MI.
Figure 3.
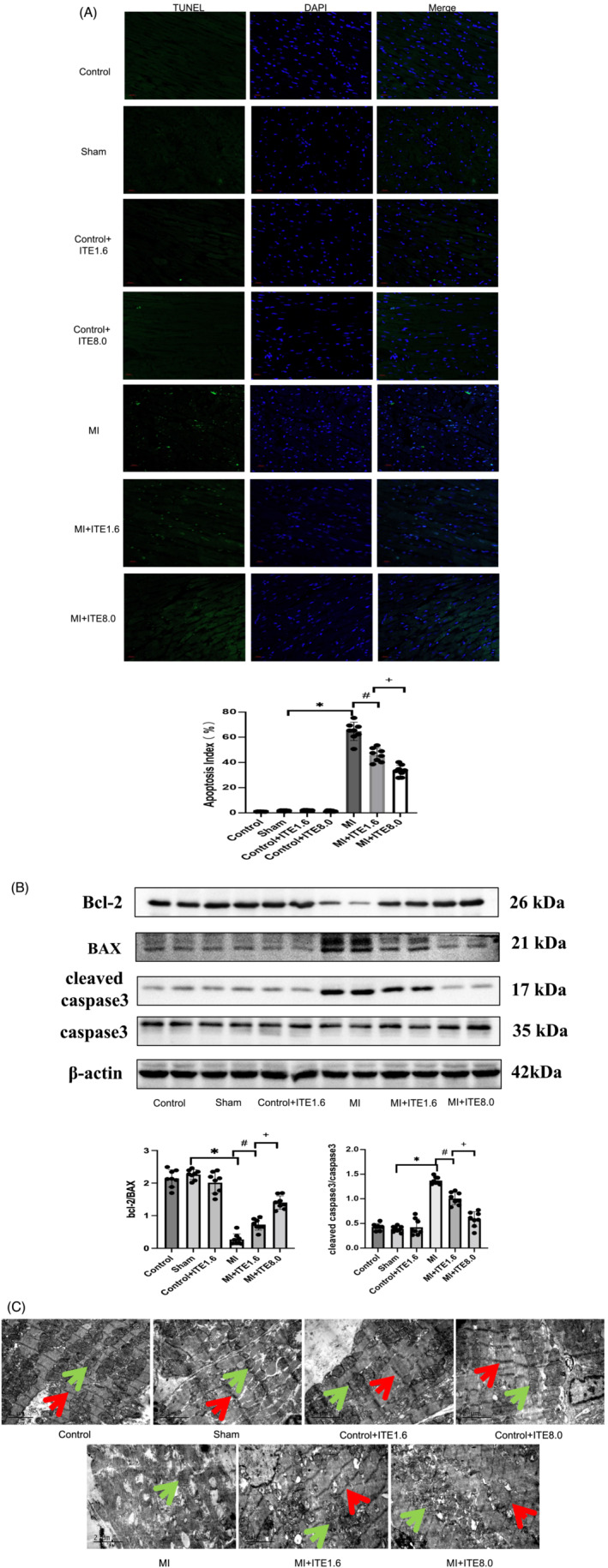
ITE attenuates cardiomyocyte apoptosis after MI. (A) Representative images obtained from border areas of MI‐operated hearts from control, sham, control + ITE1.6, control + ITE8.0, MI, MI + ITE1.6, and MI + ITE8.0 are shown. DAPI staining denotes nuclei; myocardial apoptosis index measured as the positive cell divided by total cell. (B) Expression of apoptosis‐related proteins including Bcl‐2, Bax, caspase‐3, and cleaved caspase‐3 in myocardial tissue lysates harvested from peri‐infarct areas of control, sham, control + ITE1.6, MI, MI + ITE1.6, and MI + ITE8.0 (n = 6 per group); the results were compared using two‐way ANOVA. Data are presented as mean ± SD. (C) Representative ultrastructure of cardiomyocytes from peri‐infarct regions obtained from the control, sham, control + ITE1.6, MI, MI + ITE1.6, and MI + ITE8.0 groups; red arrows point to sarcomeres, and green arrows point to mitochondria. n = 8 per group. Data are presented as mean ± SD. *P < 0.05 vs. sham group. # P < 0.05 vs. MI group.
In addition, we also used TEM to observe the ultrastructure of cardiomyocytes. In the MI group, myofilament arrangement was disorganized and sparse with a large number of breaks, mitochondria were visibly swollen, cristae structure was destroyed, vacuole‐like changes were observed, and lysosomes were increased. Compared with the MI group, the MI + ITE1.6 and MI + ITE8.0 groups had a more orderly arrangement of myofilaments, clearer mitochondrial crista structure, fewer vacuole‐like changes, and a small number of autophagic lysosomes (Figure 3 C ).
The effect of the AHR agonist on post‐infarction inflammation
The inflammatory response after MI is also an important factor affecting cardiac remodelling and the development of heart failure. As shown in Figure 3 A , the serum TNF‐α, IL‐1β, and IL‐17 levels in the MI rats treated with ITE were significantly decreased compared with those in the MI group (P < 0.05) (Figure 4 A ), and up‐regulation of serum IL‐10 was observed in MI rats treated with ITE, but ITE treatment alone or sham surgery did not affect these levels. M1‐type macrophages are usually considered proinflammatory macrophages, while M2‐type macrophages are anti‐inflammatory macrophages. 18 Previous studies 13 have shown that ITE can inhibit inflammation by affecting macrophage polarization. Consistent with this, the F4/80+iNOS+M1 macrophage population in infarcted heart tissue of the MI rats with ITE treatment was far lower than that in the MI group (Figure 4 B ), whereas the F4/80+CD206+M2 macrophage population was increased in MI rats with administration of ITE (Figure 4 C ).
Figure 4.
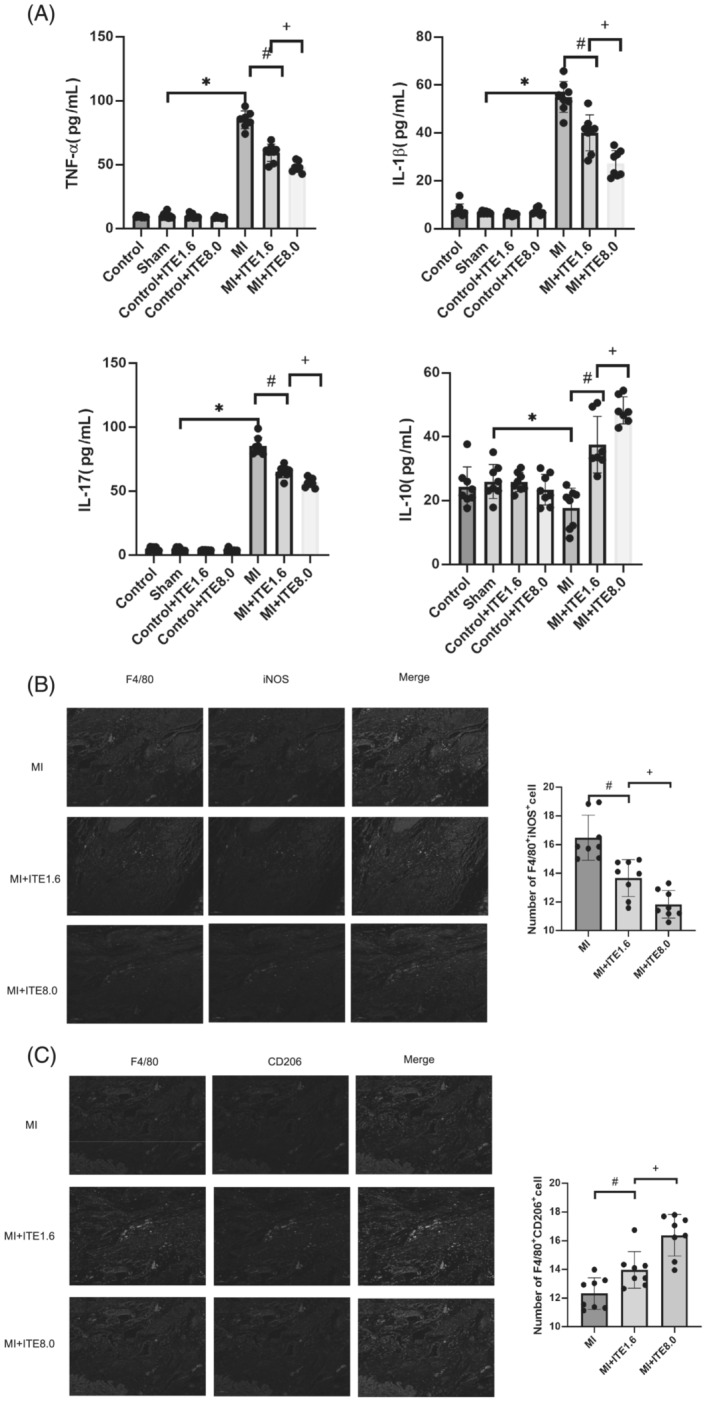
The effect of the AHR agonist on post‐infarction inflammation. (A) Serum TNF‐α, IL‐1β, IL‐17, and IL‐10 levels measured by ELISA. N = 6–8 per group. Data are presented as mean ± SD. *P < 0.05 vs. sham group. # P < 0.05 vs. MI group. + P < 0.05 vs. MI group. (B) Representative confocal IF images of M1 macrophages obtained from infarcted areas of MI‐operated hearts from MI, MI + ITE1.6, and MI + ITE8.0 are shown. Sections were stained for F4/80 (green) and iNOS (red) (left). The number of F4/80+iNOS+ cells was counted (right). N = 6–8 per group. Scale bars = 50 μm. Data are presented as mean ± SD. # P < 0.05 vs. MI group. + P < 0.05 vs. MI group. (C) Representative confocal IF images of M2 macrophages obtained from infarcted areas of MI‐operated hearts from MI, MI + ITE1.6, and MI + ITE8.0 are shown. Sections were stained for F4/80 (green) and CD206 (red) (left). The number of F4/80+CD206+ cells was counted (right). N = 6–8 per group. Scale bars = 50 μm. Data are presented as mean ± SD. # P < 0.05 vs. MI group. + P < 0.05 vs. MI group.
The AHR agonist regulates the activation of Akt and p70S6K
To confirm ITE‐induced AHR activation in MI hearts, we examined the protein expression of cytochrome P450 1A1 (Cyp1a1), which has been used as a biomarker for AHR activation. The MI group showed decreased Cyp1a1 expression compared with the sham and control groups, while the MI + ITE1.6 and MI + ITE8.0 groups showed a gradient increase in Cyp1a1 levels compared with the MI group (P < 0.05). ITE treatment alone or sham surgery did not affect the expression of Cyp1a1 (Figure 5 A and Supporting Information, Figure S3A ).
Figure 5.
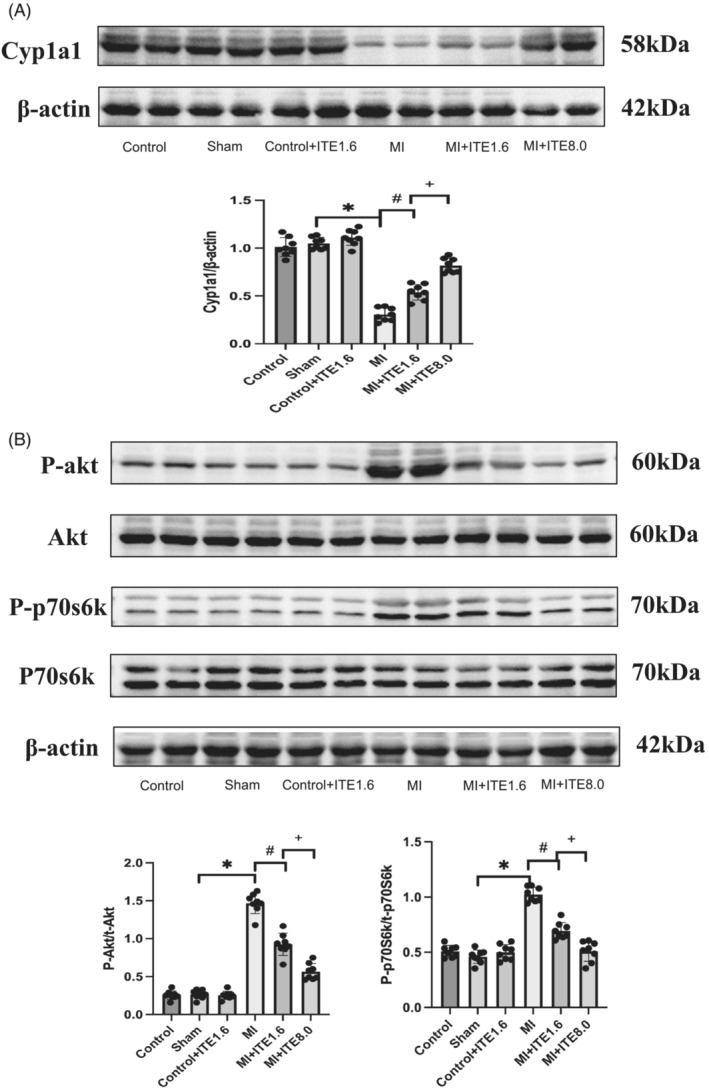
The AHR agonist regulates the activation of Akt and p70S6K. (A) Expression of the AHR activation biomarker protein Cyp1a1 in myocardial tissue lysates harvested from peri‐infarct areas of the control, sham, control + ITE1.6, MI, MI + ITE1.6, and MI + ITE8.0 groups (n = 6 per group); the results were compared using two‐way ANOVA. Data are presented as mean ± SD. (B) Expression of Akt/p70S6K signalling pathway proteins, including total Akt (t‐Akt), phosphorylated Akt (P‐Akt), total p70S6K (t‐p70S6K), and phosphorylated p70S6K (P‐p70S6K), in myocardial tissue lysates harvested from peri‐infarct areas of control, sham, control + ITE1.6, MI, MI + ITE1.6, and MI + ITE8.0 (n = 6 per group); the results were compared using two‐way ANOVA. Data are presented as mean ± SD.
Akt, a key molecule in the PI3K/Akt signalling pathway, is the centre of multiple intracellular signal transduction pathway networks and is involved in a variety of cellular processes, including cell survival, GLU metabolism, and cellular protein synthesis in cardiomyocytes. 7 p70S6K is one of the main signalling molecules downstream of Akt/mTOR signalling, and some studies 19 have shown that down‐regulation of p70S6K can inhibit apoptosis. We detected proteins related to the Akt/p70S6K signalling pathway, and the results showed that compared with the control and sham groups, the expression levels of phosphorylated Akt and p70S6K in myocardial tissue harvested from the peri‐infarct area of the MI group were significantly increased (P < 0.05), while there was a dose‐dependent decrease in P‐Akt and phosphorylated p70S6K (P‐p70S6K) levels in the MI + ITE1.6 and MI + ITE8.0 groups compared with the MI group (P < 0.05). ITE treatment alone or sham surgery did not affect the expression of P‐Akt and P‐p70S6K. There was no significant difference in the levels of total Akt and p70S6K among the groups (Figure 5 B and Supporting Information, Figure S3B ), while administration of ITE alone did not affect Akt and p70S6K phosphorylation. The results suggested that ITE may reduce the apoptosis of cardiomyocytes by activating the AHR receptor to down‐regulate the phosphorylation level of the Akt/p70S6K signalling pathway.
Effects of ITE on biochemical indicators in rats
Finally, the liver and renal functions as well as other biochemical indicators of the rats in each group were tested using blood serum collected from the abdominal aorta. Our results showed that the triglyceride (TG) and very‐low‐density lipoprotein (VLDL) levels were significantly lower in the MI group than in the sham group (P < 0.05), but there was no significant difference among the MI, MI + ITE1.6, and MI + ITE8.0 groups (Table 3 ). In addition, there was no significant difference in GLU, total serum cholesterol (TC), low‐density lipoprotein (LDL), or high‐density lipoprotein (HDL) among the groups, and we did not observe any liver, skeletal muscle, or kidney toxicity related to ITE treatment (Table 3 ).
Table 3.
Effects of ITE on biochemical indicators in rats
| Control | Sham | Control + ITE1.6 | Control + ITE8.0 | MI | MI + ITE1.6 | MI + ITE8.0 | |
|---|---|---|---|---|---|---|---|
| GLU (mM) | 15.05 ± 2.66 | 16.52 ± 2.44 | 15.29 ± 4.05 | 14.37 ± 5.24 | 14.31 ± 2.02 | 17.23 ± 3.62 | 18.62 ± 3.67 |
| ALT (U/L) | 44.71 ± 9.57 | 42.38 ± 7.94 | 45.88 ± 11.01 | 48.78 ± 9.57 | 41.29 ± 9.64 | 34.75 ± 4.77 | 40.13 ± 13.95 |
| AST (U/L) | 148.86 ± 71.78 | 232.37 ± 81.53 | 237.75 ± 107.97 | 263.84 ± 96.57 | 224.57 ± 77.95 | 191.13 ± 75.97 | 138.5 ± 53.21 |
| BUN (mM) | 7.66 ± 3.79 | 6.58 ± 1.21 | 6.91 ± 1.84 | 6.28 ± 2.36 | 6.84 ± 1.01 | 6.00 ± 0.76 | 6.53 ± 1.13 |
| SCr (μM) | 33.71 ± 5.94 | 32.58 ± 5.34 | 30.00 ± 4.33 | 34.27 ± 3.36 | 34.00 ± 6.19 | 31.25 ± 6.14 | 39.50 ± 10.49 |
| CK (U/L) | 1460.43 ± 322.32 | 1394 ± 320.57 | 1470.88 ± 351.33 | 1592.68 ± 310.84 | 1520.57 ± 586.15 | 1831.50 ± 832.07 | 1266.25 ± 399.58 |
| CK‐MB (U/L) | 1209.29 ± 256.33 | 1304 ± 205.83 | 1172.63 ± 318.14 | 1142.67 ± 196.99 | 1526.14 ± 320.40 | 1186.63 ± 312.93 | 1232.88 ± 280.66 |
| UA (μM) | 34.57 ± 8.87 | 39.63 ± 13.21 | 29.69 ± 11.50 | 34.52 ± 10.56 | 39.08 ± 10.31 | 40.30 ± 20.03 | 43.66 ± 19.91 |
| TC (mM) | 1.44 ± 0.29 | 1.68 ± 0.31 | 1.78 ± 0.35 | 1.66 ± 0.39 | 1.50 ± 0.34 | 1.41 ± 0.30 | 1.38 ± 0.20 |
| TG (mM) | 0.75 ± 0.49 | 0.77 ± 0.34 | 0.83 ± 0.29 | 0.68 ± 0.44 | 0.40 ± 0.12* | 0.31 ± 0.18* | 0.47 ± 0.22 |
| LDL (mM) | 0.31 ± 0.09 | 0.32 ± 0.09 | 0.35 ± 0.08 | 0.33 ± 0.08 | 0.33 ± 0.09 | 0.32 ± 0.10 | 0.33 ± 0.07 |
| HDL (mM) | 0.78 ± 0.17 | 0.77 ± 0.15 | 0.83 ± 0.18 | 081 ± 0.22 | 0.70 ± 0.12 | 0.71 ± 0.15 | 0.75 ± 0.12 |
| VLDL (mM) | 0.34 ± 0.22 | 0.36 ± 0.14 | 0.37 ± 0.13 | 0.41 ± 0.18 | 0.18 ± 0.06* | 0.14 ± 0.08* | 0.21 ± 0.10* |
ALT, serum alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CK, creatine kinase; CK‐MB, creatine kinase myocardial band; GLU, glucose; HDL, high‐density lipoprotein; ITE, 2‐(1′H‐indole‐3′‐carbonyl)‐thiazole‐4‐carboxylic acid methyl ester; LDL, low‐density lipoprotein; MI, myocardial infarction; SCr, serum creatinine; TC total serum cholesterol; TG, triglyceride; UA, uric acid; VLDL, very‐low‐density lipoprotein.
n = 7–8 per group. Data are presented as mean ± SD.
p < 0.05 vs. sham group.
Discussion
In the present study, we investigated whether intervention with the AHR agonist ITE at the chronic stage of MI could halt post‐infarction cardiac remodelling and the development of heart failure. While ITE did not exhibit a significant effect on the progression of ventricular enlargement or infarct size following MI, our findings demonstrate that it effectively delays ventricular remodelling through the attenuation of myocardial hypertrophy, myocardial fibrosis, and myocardial apoptosis. We found that the AHR agonist ITE ameliorated cardiac remodelling in rats with MI and inhibited cardiomyocyte apoptosis by down‐regulating the Akt/p70S6K signalling pathway.
Pathological myocardial remodelling and the inevitable development of heart failure subsequent to MI pose formidable challenges for survivors. To counteract cardiac remodelling, the clinical implementation of angiotensin‐converting enzyme (ACE) inhibitors, beta‐blockers, and aldosterone antagonists has been widespread. 20 Nevertheless, the initial administration of these medications is frequently impeded by hypotension, bradycardia, and other associated side effects. Consequently, it becomes imperative to investigate the underlying mechanisms and novel pharmacological targets implicated in cardiac remodelling, with the ultimate aim of intervening and delaying adverse cardiac remodelling processes.
The myocardium is a terminally differentiated tissue; thus, maintaining cardiomyocyte viability in ischaemic disease is critical. Apoptosis is prevalent in ischaemic heart disease. 21 In the early stages of MI, infiltration of inflammatory cells into the region of damaged myocardium and replacement of apoptotic necrosis of cardiomyocytes by fibroblasts and myofibroblasts are essential to prevent ventricular wall rupture. However, the excessive activation of apoptotic signals leads to the programmed death of a large number of cardiomyocytes around the infarct area, which largely hinders the recovery of cardiac function. Inhibition of cardiomyocyte apoptosis attenuates cardiac dysfunction and remodelling after MI. Bax is a key cytokine that induces apoptosis after MI. 22 Bcl‐2 is an anti‐apoptotic protein that binds to mitochondrial membranes and blocks the release of cytochrome c to inhibit cell death. Bax is considered a pro‐apoptotic molecule that maintains an inactive conformation in normal cells. When stimulated by apoptotic signals, Bax undergoes conformational activation and blocks the anti‐apoptotic effect of Bcl‐2. 23 The ratio of Bcl‐2 and Bax has been used as a marker representing the role of apoptosis. Caspase‐3 is a class of cysteine proteases responsible for the proteolysis of many important proteins associated with apoptosis and is also considered a marker for most types of apoptosis. 24 Chen et al. 22 found that increasing the ratio of Bcl‐2 to Bax in the myocardium and reducing cardiomyocyte apoptosis could improve cardiac function after MI. AHR can protect cells from apoptosis, oxidative stress, and endoplasmic reticulum (ER) stress. 25 Our study confirmed that after intervention with the AHR agonist ITE, the ratio of Bcl‐2/Bax was increased and the level of cleaved caspase‐3 was decreased; the apoptosis of cardiomyocytes during MI was reduced, and the cell survival rate was improved. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) is an exogenous ligand of AHR. Consistent with our study, TCDD‐induced AHR activation can inhibit genotoxic therapy‐induced tumour cell apoptosis in tumour therapy. 26 In acute kidney injury models, 27 activation of AHR can inhibit inflammation and cellular apoptosis to reduce the adverse effects of ischaemia–reperfusion.
Akt is a key molecule in the PI3K/Akt signalling pathway, and some studies 28 have shown that inhibition of the Akt‐related signalling pathway can inhibit cardiomyocyte apoptosis, reduce myocardial fibrosis, and improve MI‐induced cardiac dysfunction. However, when Akt signalling is up‐regulated, it can promote the expression of myocardial fibrosis and related inflammatory factors in MI. 28 Proteomic analysis 29 found that the phosphorylation levels of various proteins changed after AHR was activated by ligands. In both AHR knockout cells 30 and animal 31 models, Akt phosphorylation levels were elevated. One study found 32 that the deletion of AHR can damage the related signalling pathways of the Akt pathway and increase the apoptosis sensitivity of cells. Our present study found that the phosphorylation level of Akt was increased after MI, and reducing the phosphorylation level of Akt by ITE could alleviate cardiomyocyte apoptosis, which was consistent with the above findings. However, some studies 33 have also found that ischaemic preconditioning or drug preconditioning can activate the PI3K/Akt signalling pathway in cardiomyocytes. This can induce a series of subsequent responses, including alleviation of apoptosis, elimination of intracellular reactive oxygen species, inhibition of neutrophil activation and aggregation, and protection of mitochondrial function. This difference may be related to the timing of Akt activation. Shiojima et al. 34 found that short‐term Akt activation promotes cardiac growth, while long‐term Akt activation induces pathological hypertrophy and heart failure.
p70S6K is one of the downstream molecules of the Akt/mTOR pathway. 35 Some studies 27 , 30 have found that AHR can regulate apoptosis through the Akt signalling pathway. Some studies have shown a decrease in pP70 in acute MI, 19 but in chronic MI 36 or chronic heart failure, 37 pP70 is increased, and inhibition can be protective. This study confirmed that the phosphorylation level of p70S6K increased after MI, and ITE intervention reduced the phosphorylation level of p70S6K, which was consistent with the trend of the phosphorylation level of Akt. Therefore, we speculated that ITE may reduce myocardial apoptosis by regulating Akt/p70S6K. Consistent with our experimental results, Lechpammer et al. 38 found that inhibiting the activation of the PI3K/Akt/mTOR signalling pathway can down‐regulate p70S6K phosphorylation and alleviate brain damage caused by ischaemia and hypoxia. Shi et al. 39 found that down‐regulation of the overactivated mTOR/p70S6K pathway during MI can inhibit apoptosis, promote autophagy, reduce the secretion of inflammatory factors, and improve chronic heart failure.
However, there are still limitations in this experiment. AHR is a ligand‐specific receptor, and its function is related to the structure of the ligand. The environmental toxicant TCDD is an exogenous ligand of AHR. Studies have found that long‐term exposure to TCDD to activate AHR can promote hypertension 40 and atherosclerosis 41 and increase the risk of ischaemic heart disease, 42 and endogenous AHR ligands are generally considered nontoxic. In this study, liver, skeletal muscle, or kidney toxicity related to ITE treatment was not observed, and thus, it must be further verified by longer intervention times. Second, whether Akt/p70S6K is a key signalling pathway in the protective effect of ITE on the heart after MI needs further verification, and the effect of ITE needs to be further verified by knocking out AHR in vivo or in vitro. At present, the mechanism by which ITE improves ventricular remodelling and cardiomyocyte apoptosis after MI still needs to be further explored.
In conclusion, the AHR agonist ITE reduces cardiomyocyte apoptosis by down‐regulating the Akt/p70S6K pathway and thus improves ventricular remodelling and cardiac function after MI. The results imply that ITE intervention might be a potential therapeutic approach for treating post‐MI heart failure.
Conflict of interest
None declared.
Funding
This work was supported by the Joint Funds for the Innovation of Science and Technology of Fujian Province (Grant Number 2018Y9088).
Supporting information
Figure S1. Changes of electrocardiogram (ECG) before and after MI surgery in rats. A, Normal ECG before LAD ligation, no ST‐segment elevation. B, ECG after LAD ligation, ST‐segment elevation can be seen.
Figure S2. Expression of apoptosis‐related proteins including bcl‐2, Bax, caspase‐3 and cleaved caspase‐3 in myocardial tissue lysates harvested from peri‐infarct areas of Control and Control+ITE8.0 (n = 8 per group); the results were compared using 2‐way ANOVA. Data are presented as mean±SD.
Figure S3. A, Expression of AhR activation biomarker proteins Cyp1a1 in myocardial tissue lysates harvested from peri‐infarct areas of Control and Control+ITE8.0 (n = 8 per group); the results were compared using 2‐way ANOVA. Data are presented as mean±SD. B, Expression of Akt/p70S6k signaling pathway proteins including t‐Akt, P‐Akt, t‐p70S6k and P‐p70S6k in myocardial tissue lysates harvested from peri‐infarct areas of Control and Control+ITE8.0 (n = 8 per group); the results were compared using 2‐way ANOVA. Data are presented as mean±SD. t‐Akt, total Akt; P‐Akt, phosphorylated Akt; t‐p70S6k, total p70S6k; P‐p70S6k, phosphorylated p70S6k.
Lin, X. , Liu, W. , Chu, Y. , Zhang, H. , Zeng, L. , Lin, Y. , Kang, K. , Peng, F. , Lin, J. , Huang, C. , and Chai, D. (2023) Activation of AHR by ITE improves cardiac remodelling and function in rats after myocardial infarction. ESC Heart Failure, 10: 3622–3636. 10.1002/ehf2.14532.
Contributor Information
Chunkai Huang, Email: chun-kai-huang@outlook.com.
Dajun Chai, Email: dajunchai-fy@fjmu.edu.cn.
References
- 1. Ozaki Y, Hara H, Onuma Y, Katagiri Y, Amano T, Kobayashi Y, et al. CVIT expert consensus document on primary percutaneous coronary intervention (PCI) for acute myocardial infarction (AMI) update 2022. Cardiovasc Interv Ther 2022;37:1‐34. doi: 10.1007/s12928-021-00829-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cuenin L, Lamoureux S, Schaaf M, Bochaton T, Monassier JP, Claeys MJ, et al. Incidence and significance of spontaneous ST segment re‐elevation after reperfused anterior acute myocardial infarction—Relationship with infarct size, adverse remodeling, and events at 1 year. Circ J 2018;82:1379‐1386. doi: 10.1253/circj.CJ-17-0671 [DOI] [PubMed] [Google Scholar]
- 3. Joshi C, Bapat R, Anderson W, Dawson D, Hijazi K, Cherukara G. Detection of periodontal microorganisms in coronary atheromatous plaque specimens of myocardial infarction patients: A systematic review and meta‐analysis. Trends Cardiovasc Med 2021;31:69‐82. doi: 10.1016/j.tcm.2019.12.005 [DOI] [PubMed] [Google Scholar]
- 4. Li L, Fu W, Gong X, Chen Z, Tang L, Yang D, et al. The role of G protein‐coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur Heart J 2021;42:1415‐1430. doi: 10.1093/eurheartj/ehaa878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wei Z, Luo L, Hu S, Tian R, Liu Z. KDM2B overexpression prevents myocardial ischemia‐reperfusion injury in rats through regulating inflammatory response via the TLR4/NF‐κB p65 axis. Exp Ther Med 2022;23:154. doi: 10.3892/etm.2021.11077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 2019;116:2672‐2680. doi: 10.1073/pnas.1821022116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walkowski B, Kleibert M, Majka M, Wojciechowska M. Insight into the role of the PI3K/Akt pathway in ischemic injury and post‐infarct left ventricular remodeling in normal and diabetic heart. Cell 2022;11:1553. doi: 10.3390/cells11091553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nebert DW. Aryl hydrocarbon receptor (AHR): “Pioneer member” of the basic‐helix/loop/helix per‐Arnt‐sim (bHLH/PAS) family of “sensors” of foreign and endogenous signals. Prog Lipid Res 2017;67:38‐57. doi: 10.1016/j.plipres.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kou Z, Dai W. Aryl hydrocarbon receptor: Its roles in physiology. Biochem Pharmacol 2021;185:114428. doi: 10.1016/j.bcp.2021.114428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carreira VS, Fan Y, Kurita H, Wang Q, Ko CI, Naticchioni M, et al. Disruption of Ah receptor signaling during mouse development leads to abnormal cardiac structure and function in the adult. PLoS ONE 2015;10:e0142440. doi: 10.1371/journal.pone.0142440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mallah MA, Mallah MA, Liu Y, Xi H, Wang W, Feng F, et al. Relationship between polycyclic aromatic hydrocarbons and cardiovascular diseases: A systematic review. Front Public Health 2021;9:763706. doi: 10.3389/fpubh.2021.763706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qian C, Yang C, Lu M, Bao J, Shen H, Deng B, et al. Activating AhR alleviates cognitive deficits of Alzheimer's disease model mice by upregulating endogenous Aβ catabolic enzyme neprilysin. Theranostics 2021;11:8797‐8812. doi: 10.7150/thno.61601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seong E, Lee JH, Lim S, Park EH, Kim E, Kim CW, et al. Activation of aryl hydrocarbon receptor by ITE improves cardiac function in mice after myocardial infarction. J Am Heart Assoc 2021;10:e020502. doi: 10.1161/JAHA.120.020502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yurista SR, Sillje HHW, Oberdorf‐Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium–glucose co‐transporter 2 inhibition with empagliflozin improves cardiac function in non‐diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail 2019;21:862‐873. doi: 10.1002/ejhf.1473 [DOI] [PubMed] [Google Scholar]
- 15. Wu Y, Chen X, Zhou Q, He Q, Kang J, Zheng J, et al. ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS ONE 2014;9:e86549. doi: 10.1371/journal.pone.0086549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu Z, Yu L, Li X, Li X. Protective mechanism of trimetazidine in myocardial cells in myocardial infarction rats through ERK signaling pathway. Biomed Res Int 2021;2021:9924549. doi: 10.1155/2021/9924549 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17. Chen Z, Wu J, Li S, Liu C, Ren Y. Inhibition of myocardial cell apoptosis is important mechanism for ginsenoside in the limitation of myocardial ischemia/reperfusion injury. Front Pharmacol 2022;13:806216. doi: 10.3389/fphar.2022.1070736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Z, Zhao L, Zhou X, Meng X, Zhou X. Role of inflammation, immunity, and oxidative stress in hypertension: New insights and potential therapeutic targets. Front Immunol 2022;13:1098725. doi: 10.3389/fimmu.2022.1091188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oeing CU, Jun S, Mishra S, Dunkerly‐Eyring BL, Chen A, Grajeda MI, et al. MTORC1‐regulated metabolism controlled by TSC2 limits cardiac reperfusion injury. Circ Res 2021;128:639‐651. doi: 10.1161/CIRCRESAHA.120.317710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jia S, Liu Y, Yuan J. Evidence in guidelines for treatment of coronary artery disease. Adv Exp Med Biol 2020;1177:37‐73. [DOI] [PubMed] [Google Scholar]
- 21. Chunhacha P, Pinkaew D, Sinthujaroen P, Bowles DE, Fujise K. Fortilin inhibits p53, halts cardiomyocyte apoptosis, and protects the heart against heart failure. Cell Death Dis 2021;7:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen P, Liu J, Ruan H, Zhang M, Wu P, Yimei D, et al. Protective effects of salidroside on cardiac function in mice with myocardial infarction. Sci Rep 2019;9:18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quiles JM, Gustafsson AB. The role of mitochondrial fission in cardiovascular health and disease. Nat Rev Cardiol 2022;19:723‐736. doi: 10.1038/s41569-022-00703-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jalil HM, Ghazi HF. NLRP3 inflammasome gene polymorphisms variably associated with its serum levels in acute myocardial infarction. Pak J Biol Sci 2020;23:612‐618. doi: 10.3923/pjbs.2020.612.618 [DOI] [PubMed] [Google Scholar]
- 25. Guerrina N, Aloufi N, Shi F, Prasade K, Mehrotra C, Traboulsi H, et al. The aryl hydrocarbon receptor reduces LC3II expression and controls endoplasmic reticulum stress. Am J Physiol Lung Cell Mol Physiol 2021;320:L339‐L355. doi: 10.1152/ajplung.00122.2020 [DOI] [PubMed] [Google Scholar]
- 26. Benoit L, Jornod F, Zgheib E, Tomkiewicz C, Koual M, Coustillet T, et al. Adverse outcome pathway from activation of the AhR to breast cancer‐related death. Environ Int 2022;165:107323. doi: 10.1016/j.envint.2022.107323 [DOI] [PubMed] [Google Scholar]
- 27. Tao S, Guo F, Ren Q, Liu J, Wei T, Li L, et al. Activation of aryl hydrocarbon receptor by 6‐formylindolo[3,2‐b]carbazole alleviated acute kidney injury by repressing inflammation and apoptosis. J Cell Mol Med 2021;25:1035‐1047. doi: 10.1111/jcmm.16168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao X, Ren Y, Ren H, Wu Y, Liu X, Chen H, et al. The mechanism of myocardial fibrosis is ameliorated by myocardial infarction‐associated transcript through the PI3K/Akt signaling pathway to relieve heart failure. J Int Med Res 2021;49:3000605211031433. doi: 10.1177/03000605211031433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grosskopf H, Walter K, Karkossa I, von Bergen M, Schubert K. Non‐genomic AhR‐signaling modulates the immune response in endotoxin‐activated macrophages after activation by the environmental stressor BaP. Front Immunol 2021;12:620270. doi: 10.3389/fimmu.2021.620270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi F, Aloufi N, Traboulsi H, Trempe JF, Eidelman DH, Baglole CJ. Endogenous regulation of the Akt pathway by the aryl hydrocarbon receptor (AhR) in lung fibroblasts. Sci Rep 2021;11:23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moreno‐Marin N, Merino JM, Alvarez‐Barrientos A, Patel DP, Takahashi S, Gonzalez‐Sancho JM, et al. Aryl hydrocarbon receptor promotes liver polyploidization and inhibits PI3K, ERK, and Wnt/β‐catenin signaling. iScience 2018;4:44‐63. doi: 10.1016/j.isci.2018.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yi T, Wang J, Zhu K, Tang Y, Huang S, Shui X, et al. Aryl hydrocarbon receptor: A new player of pathogenesis and therapy in cardiovascular diseases. Biomed Res Int 2018;2018:6058784. doi: 10.1155/2018/6058784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang T, Zhang L, Ding M, Li M. Protective effect of vasicine against myocardial infarction in rats via modulation of oxidative stress, inflammation, and the PI3K/Akt pathway. Drug Des Devel Ther 2019;13:3773‐3784. doi: 10.2147/DDDT.S220396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 2005;115:2108‐2118. doi: 10.1172/JCI24682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong BS, Wu WT, Su JH, Goan YG, Wu YJ. Flaccidoxide induces apoptosis through down‐regulation of PI3K/AKT/mTOR/p70S6K signaling in human bladder cancer cells. Anticancer Res 2021;41:6123‐6133. doi: 10.21873/anticanres.15432 [DOI] [PubMed] [Google Scholar]
- 36. Fan Y, Cheng Y, Li Y, Chen B, Wang Z, Wei T, et al. Phosphoproteomic analysis of neonatal regenerative myocardium revealed important roles of checkpoint kinase 1 via activating mammalian target of rapamycin C1/ribosomal protein S6 kinase b‐1 pathway. Circulation 2020;141:1554‐1569. doi: 10.1161/CIRCULATIONAHA.119.040747 [DOI] [PubMed] [Google Scholar]
- 37. Oeing CU, Nakamura T, Pan S, Mishra S, Dunkerly‐Eyring BL, Kokkonen‐Simon KM, et al. PKG1α cysteine‐42 redox state controls mTORC1 activation in pathological cardiac hypertrophy. Circ Res 2020;127:522‐533. doi: 10.1161/CIRCRESAHA.119.315714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lechpammer M, Tran YP, Wintermark P, Martinez‐Cerdeno V, Krishnan VV, Ahmed W, et al. Upregulation of cystathione β‐synthase and p70S6K/S6 in neonatal hypoxic ischemic brain injury. Brain Pathol 2017;27:449‐458. doi: 10.1111/bpa.12421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shi B, Huang Y, Ni J, Chen J, Wei J, Gao H, et al. Qi Dan Li Xin pill improves chronic heart failure by regulating mTOR/p70S6k‐mediated autophagy and inhibiting apoptosis. Sci Rep 2020;10:6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kopf PG, Huwe JK, Walker MK. Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin are associated with increased superoxide. Cardiovasc Toxicol 2008;8:181‐193. doi: 10.1007/s12012-008-9027-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shan Q, Qu F, Chen N. 2,3,7,8‐Tetrachlorodibenzo‐p‐dioxin (TCDD) and polychlorinated biphenyl coexposure alters the expression profile of microRNAs in the liver associated with atherosclerosis. Biomed Res Int 2020;2020:2652756. doi: 10.1155/2020/2652756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collins JJ, Bodner KM, Aylward LL, Bender TJ, Anteau S, Wilken M, et al. Mortality risk among workers with exposure to dioxins. Occup Med (Lond) 2016;66:706‐712. doi: 10.1093/occmed/kqw167 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Changes of electrocardiogram (ECG) before and after MI surgery in rats. A, Normal ECG before LAD ligation, no ST‐segment elevation. B, ECG after LAD ligation, ST‐segment elevation can be seen.
Figure S2. Expression of apoptosis‐related proteins including bcl‐2, Bax, caspase‐3 and cleaved caspase‐3 in myocardial tissue lysates harvested from peri‐infarct areas of Control and Control+ITE8.0 (n = 8 per group); the results were compared using 2‐way ANOVA. Data are presented as mean±SD.
Figure S3. A, Expression of AhR activation biomarker proteins Cyp1a1 in myocardial tissue lysates harvested from peri‐infarct areas of Control and Control+ITE8.0 (n = 8 per group); the results were compared using 2‐way ANOVA. Data are presented as mean±SD. B, Expression of Akt/p70S6k signaling pathway proteins including t‐Akt, P‐Akt, t‐p70S6k and P‐p70S6k in myocardial tissue lysates harvested from peri‐infarct areas of Control and Control+ITE8.0 (n = 8 per group); the results were compared using 2‐way ANOVA. Data are presented as mean±SD. t‐Akt, total Akt; P‐Akt, phosphorylated Akt; t‐p70S6k, total p70S6k; P‐p70S6k, phosphorylated p70S6k.