

Abstract
Recently, a lot of attention has been dedicated to double- or triple-atom catalysts (DACs/TACs) as promising alternatives to platinum-based catalysts for the oxygen reduction reaction (ORR) in fuel cell applications. However, the ORR activity of DACs/TACs is usually theoretically understood or predicted using the single-site association pathway (O2 → OOH* → O* → OH* → H2O) proposed from Pt-based alloy and single-atom catalysts (SACs). Here, we investigate the ORR process on a series of graphene-supported Fe–Co DACs/TACs by means of first-principles calculation and an electrode microkinetic model. We propose that a dual channel for electron acceptance–backdonation on adjacent metal sites of DACs/TACs efficiently promotes O–O bond breakage compared with SACs, which makes ORR switch to proceed through dual-site dissociation pathways (O2 → O* + OH* → 2OH* → OH* → H2O) from the traditional single-site association pathway. Following this revised ORR network, a complete reaction phase diagram of DACs/TACs is established, where the preferential ORR pathways and activity can be described by a three-dimensional volcano plot spanned by the adsorption free energies of ΔG(O*) and ΔG(OH*). Besides, the kinetics preferability of dual-site dissociation pathways is also appropriate for other graphene- or oxide-supported DACs/TACs. The contribution of dual-site dissociation pathways, rather than the traditional single-site association pathway, makes the theoretical ORR activity of DACs/TACs in better agreement with available experiments, rationalizing the superior kinetic behavior of DACs/TACs to that of SACs. This work reveals the origin of ORR pathway switching from SACs to DACs/TACs, which broadens the ideas and lays the theoretical foundation for the rational design of DACs/TACs and may also be heuristic for other reactions catalyzed by DACs/TACs.
Keywords: double-atom catalysts (DACs), triple-atom catalysts (TACs), ORR pathway, dual-site dissociation pathway, DFT calculation
1. Introduction
Atomically dispersed metal catalysts (ADMCs) with well-defined structures, especially for single-atom catalysts (SACs) and multi-atom catalysts (such as double-atom catalysts (DACs), triple-atom catalysts (TACs)), have attracted great interest in heterogeneous catalysis in recent years.1,2 SACs exhibit distinct catalytic properties compared to metal nanoparticles (NPs) due to their uniform active sites and high metal utilization.3−5 However, SACs only provide an isolated adsorption site for catalytic reactions. For catalytic reactions involving the adsorption and coupling of multiple molecules, such as the hydrocarbon hydrogenation reaction and CO2 reduction reaction (CO2RR), it is rather challenging to improve the catalytic performance of SACs.6 Compared with SACs, the multiple active sites of DACs/TACs may render them versatile binding modes toward adsorbates due to their diverse synergistic interactions, thus improving their ability to surpass the intrinsic performance by circumventing the scaling relationship.7,8 Nowadays, DACs/TACs have attracted considerable research interest, since they exhibit remarkable catalytic performances in a variety of reactions, including thermocatalysis,9−11 photocatalysis,12 and electrocatalysis.13−15
The electrochemical oxygen reduction reaction (ORR) has gained widespread attention by virtue of its significance in energy storage and conversion, including metal–air batteries and proton-conducting membrane fuel cells.2,16 Platinum-based nanomaterials have been investigated experimentally and computationally in depth over the past decade because they are effective and durable ORR catalysts,17−21 but they are too expensive for large-scale applications. Nonprecious-metal carbon-based atomically dispersed catalysts with metal–nitrogen coordination sites, especially for Fe-involved or Co-involved composites,22−24 have attracted broad research interest in recent years because of their relatively high ORR activity and low cost.25,26 Many experimental tests and theoretical studies have been devoted to elucidating the electrocatalytic mechanism of ORR. The current understanding of the superior ORR performance of DACs/TACs to SACs is still based on the single-site associative mechanism (O2 → OOH* → O* → OH* → H2O). Only one of the metal sites (M1) in DACs/TACs serves as an active center, while the others are spectators modifying the electronic configuration of M1, thus enhancing the intrinsic activity of the M1 site.23,27 Besides, theoretical or high-throughput screening for DACs/TACs aided by density functional theory (DFT) calculation is similar to that for Pt-based nanomaterials, which is still based on the associative mechanism28 and the activity volcano trend spanned by adsorption free energies of OH*.29−33 However, in contrast to metal particles with continuous zero valence metal sites, the contiguous metal cations of DACs/TACs provide a desired synergistic effect altering intrinsic catalytic ability, which in turn renders the possibility to tune a reaction pathway. Compared with SACs, DACs/TACs possess multiple active sites to bind with the adsorbent with different modes, so that the reaction can proceed through another pathway with kinetics preferability.34−37 In recent experimental work, Wang et al.38 and Liu et al.39 adjusted the atomic distance of single-atom pairs to induce O–O radical breakage without forming redundant *OOH intermediates, which causes the ORR to follow the dissociated ORR pathway. Therefore, it is reasonable to infer that unique charge distribution at the active site of DACs/TACs may contribute to promoting the delocalized electron transfer between the orbital of O2 and DACs/TACs, and accordingly, ORR may possess new reaction pathways40 on DACs/TACs, which accounts for their superior activity to SACs.41−43
Herein, we identify a dual channel for electron acceptance–backdonation between adjacent metal sites of DACs/TACs and the O–O bond, leading to greater activation and easier cleavage of the O–O bond on DAC/TACs than those on SACs. A total of 15 Fe–Co DACs and 20 Fe–Co TACs supported on graphene are established to evaluate the ORR process, demonstrating the universality of preferential pathway switching from the single-site association pathway to the dual-site dissociation pathway, due to O–O bond activation by the dual channel for electron acceptance–backdonation. Following this revised ORR network, a complete reaction phase diagram of DACs/TACs is established. It shows that the reaction pathways, potential determining step, and activity of ORR over DACs/TACs can be described by a three-dimensional volcano plot spanned by the adsorption free energies of ΔG(O*) and ΔG(OH*). Finally, the domination of the dual-site dissociation pathway on ORR can be extended to other carbon- or oxide-supported DACs/TACs. Hence, this work has a deep understanding and rationalizes the superior ORR kinetic behavior of DACs/TACs to SACs in available experiments, which opens up a new way for the rational design of DACs/TACs.
2. Computational Methods
All of the computations were carried out by the spin-polarized DFT method, as implemented in the Vienna Ab initio Simulation Package (VASP) 5.4 code.44,45 The exchange–correlation energy was modeled by the Perdew–Burke–Ernzerhof (PBE) functional.46,47 Among the extensively theoretical computational works, a N-doped graphene monolayer has been regarded as an efficient theoretical model to represent the carbon-based support of ADMCs.38,48−50 In this work, graphene was modeled with a (5 × 5) supercell, and a vacuum slab of 15 Å was inserted in the z direction for surface isolation to prevent the interaction between two neighboring surfaces. The DFT + U method with U – J = 3.0 eV was used to describe the strong correlation of the localized Fe/Co 3d states.51 The magnetic moment of Fe/Co was calculated, allowing the magnitude to change during the optimization. An energy cutoff of 500 eV was adopted for the plane-wave basis. In structural optimizations, the Brillouin zone was sampled by 3 × 3 × 1 k-points using the Monkhorst–Pack scheme, while denser k-points of 6 × 6 × 1 were employed for electronic property computations. The energy and force convergence criteria are 10–5 eV and 0.02 eV/Å, respectively. The adsorption free energy of each adsorbate is defined as follows
| 1 |
where ΔE is the energy change from DFT calculations, ΔEZPE is the zero-point energy change, and ΔS is the entropy change at 298.15 K. The adsorption free energy ΔGOOH*, ΔGO*, and ΔGOH* are relative to the free energy of stoichiometrically appropriate amounts of H2O (l) and H2 (g). The solvent effect (ΔEsolv) was tested by using an implicit solvation model of the VASPsol code.52,53 At the common ORR temperature of 298.15 K, the dielectric constant of water is 78.36 F/m. Referring to relevant calculation references,53,54 the dielectric constant of 80 was approximately set to simulate the aqueous electrolyte. The effective surface tension parameter was assigned to 0 in VASPsol to neglect the cavitation energy contribution. The Debye length is set to 3.0 Å. ΔEsolv refers to the stabilization of the adsorbate because of the interaction with surrounding water.
Activation barriers for all proton–electron transfer steps of intermediates were set at 0.26 eV taken from Pt(111),18 which has been successfully applied in ADMCs.33,55 We calculated the activation barriers of the reaction involved with O–O bond breaking, using the climbing image nudged elastic band (CINEB)56 method.
The reaction free energies at constant potential were determined by combining the capacitor model developed by Chan and Nørskov57,58 and the computational hydrogen electrode (CHE) model proposed by Nørskov et al.59 We first used the capacitor model57,58 to convert the constant-charge simulations to the actual electrochemical constant-potential conditions. The charge transfer (the Bader charge of the surface) and work function of state 1(Φ1) and state 2 (Φ2) were calculated based on the optimized structures from density functional theory (DFT) calculations. All of the calculated charge transfers were referenced to state 2 in each of the elementary steps, and a change of work function was used to build the linear relations. The free energy changes between the two states (i.e., state 1 and state 2) at constant work functions thus are given by
| 2 |
| 3 |
in which G1(Φ1) and G2(Φ2) represent the DFT-calculated free energies of the optimized initial and final states, respectively, Φ1 and Φ2 are the work functions of the optimized initial state and final state relative to the vacuum energy level, and q1 and q2 are the excess surface charges, at states 1 and 2 simulated with constant charge, respectively. Φ(U = 0 V) is the value of Φ at which the horizontal line ΔG = ΔGCHE(U = 0 V) and line ΔG = ΔG(Φ) intersect.
3. Results and Discussion
3.1. Dual Channel for Electron Acceptance–Backdonation on Adjacent Metal Sites to Activate the O–O Bond
Generally, the widely accepted ORR pathway is a single-site associative mechanism (O2 → OOH* → O* → OH* → H2O), which was originally developed from extended metal surfaces and is now also commonly employed in ADMCs. However, the mechanism of O2 reduction can be very different on DACs/TACs compared with extended metal surfaces or SACs. Active centers in DACs/TACs not only optimize the electronic structure of adjacent metal centers through the short/medium-range electron interaction but also provide an additional adsorption site to change the intermediate adsorption. Previous reports have noted the probable existence of side-on adsorption of O2* or OOH*, where each adjacent active site binds to an O atom to form dual-side adsorption on DACs/TACs,60 offering the opportunity to further accelerate multi-intermediate conversion reactions.61,62
The “acceptance–backdonation” mechanism, usually used to explain the adsorption and activation process of small molecules (N2*, CO*, etc.) on transition metal catalysts,63−65 was introduced to understand the activation mechanism of O2 molecules on ADMCs. When adsorbates containing O–O bonds are adsorbed on SACs (Figure 1a), the lone-pair electrons from the σ orbital of the O–O bond transfer to empty d-orbitals of isolated metal atoms, and simultaneously, the partially filled d-electrons of isolated metal atoms transfer electrons to π* orbitals of the O–O bond, thus activating the O–O bond. However, electrons only transfer from one side of the O–O bond upon adsorption on solo metal centers of SACs, namely, a single channel of electron acceptance–backdonation. As shown in Figure 1b, the adjacent metal sites of DACs/TACs could provide an additional adsorption site to form a side-on O2 adsorption mode, so we suggest a “dual channel of electron acceptance–backdonation” mechanism to understand the activation mechanism of the O–O bond on DACs. The lone-pair electrons from the σ orbital of O2 may transfer from both sides of the O–O bond (or dual channels) to empty the d orbital of the adjacent metal sites, and the d-electrons of the metal sites may also donate back part of the electrons to O2 through dual channels. Compared with SACs, the dual channel of electron acceptance–backdonation of DACs/TACs enables more efficient orbital overlap between metal and O atoms, leading to better activation of the O–O bond. When the active centers of DACs/TACs are involved in the interaction with the O–O bond, both M1–O and M2–O bonds have strong binding abilities, so that both M1 and M2 have a large pulling force on the O–O bond, which is conducive to the dissociation of the O–O bond. Due to the “dual channel of electron acceptance–backdonation” mechanism-induced O–O bond activation, dissociation pathways involving O–O breakage during ORR on DACs/TACs should not be overlooked. Here, we further supplement possible ORR routes for DACs/TACs, as shown in Figure 1c, where O2 and OOH intermediates proceed to dissociation on DACs/TACs and generate stable 2OH* and O* + OH* intermediates. The complete reaction pathway with all possible intermediates and electrochemical steps is constructed, where pathway I is a traditional single-site associative pathway and pathways II–IV are dual-site dissociation pathways.
Figure 1.

(a) Schematic diagram of the single channel of electron acceptance–backdonation for the O–O bond activated by SACs. (b) Schematic diagram of the dual channel of electron acceptance–backdonation for the O–O bond activated by DACs/TACs. (c) Schematic depiction of the single-site associative pathway and dual-site dissociation pathways for O2 electrochemical reduction.
There are many experimental reports on N-doped graphene as the supporting material for dual metal sites.66−68 Besides, FeCo DACs have been reported more than once to possess superior ORR activity to Fe SACs or Co SACs in the experiment.23,32,69 Therefore, Fe1Co1–N6 and Fe1–N4 embedded in graphene were chosen as prototypes (Figure 2a), and the “dual channel of electron acceptance–backdonation” mechanism was applied to rationalize the more activation of the O–O bond and higher ORR activity on Fe1Co1-DAC than those on Fe-SAC. Adsorption of a single O2 molecule on Fe1Co1-DAC occurs via a side-on configuration with the formation of Fe–O and Co–O bonds (Figure S1). It is actually more favorable by 0.09 eV than that of the endon configuration. Figures 2a,b and S2a show the activation mechanism of O2* adsorbed on Fe-SAC and Fe1Co1-DAC according to the projected density of state and partial differential charge density projecting to the specific energy level.70,71 When O2* is adsorbed on Fe-SAC, the electrons of the σ orbital of the O–O bond contributed by O 2px orbital transfer to dxz and dyz orbitals of Fe in the energy level from −5 to −7 eV. At the same time, the electrons from the partially filled Fe dz2 orbital flow back into the π* orbitals of the O–O bond in the energy level of 0––2 eV. Fe1Co1-DAC provides an additional adsorption site to form bonds with both ends of O2, forming a dual channel of electron acceptance–backdonation in the corresponding energy level, which could be conducive to the sufficient activation of O2. The O–O bond length is elongated to 1.33 Å on Fe1Co1-DAC, which is longer than that on Fe-SAC (1.30 Å). The O–O bond strength on Fe1Co1-DAC (ICOHP = −6.48 eV) is weaker than that on Fe1-SAC (ICOHP = −6.93 eV). These could be ascribed to the strong hybridization between Fe 3dz2, Co 3dz2, and O 2pz orbitals in O2@Fe1Co1-DAC, which increases delocalized electron transfer between O2 and FeCo sites and promotes dissociation of O2 for ORR. The dual channel of electron acceptance–backdonation is still observed between the O–O bond of OOH* on Fe1Co1-DAC (Figure S2).
Figure 2.

Partial differential charge density projecting to the specific energy level. The projected density of state between the O2 molecule on (a) Fe-SAC and (b) Fe1Co1-DAC. (The blue area represents the loss of electrons, and the yellow area represents the gain of electrons. The Fermi energy is set to 0 eV.) (c) The reaction free energy (at 0.9 V vs RHE) of the competing steps between single-site association and dual-site dissociation mechanisms on Fe1Co1-DAC, according to the constant-charge and constant-potential method. (d) The O–O bond cleavage barrier of O2* along with hydrogenation on Fe-SAC and Fe1Co1-DAC. The associative pathway barrier of O2* + H → OOH* on Fe1Co1-DAC. The free energies of Fe-SAC and Fe1Co1-DAC with no adsorption are set as references for other intermediates along the reaction (G = 0 eV).
To evaluate the thermodynamic and kinetic priority of O–O bond dissociation on DACs/TACs, the reaction free energy of the competing steps between single-site association and dual-site dissociation mechanisms on Fe1Co1-DAC is shown in Figure 2c. O2* is preferable to be dissociated and hydrogenated simultaneously to O* + OH* with a ΔG of −1.46 eV, which is much lower than that of OOH* formation (ΔG = −0.17 eV). The step OOH* → 2OH* (ΔG = −1.64 eV) is more exothermic than the step OOH* → O* (ΔG = −1.39 eV). In addition to constant-potential calculation, the constant-charge calculation also supports the thermodynamic priority of dual-site dissociation mechanisms (Figures 2c and S3). Subsequently, Figures 2d and S4 show that the H-aided O2 dissociation is more facile than O2 direct dissociation of the O–O bond for dual-site dissociation mechanisms. Figure 2d and Table S1 also show that the cleavage barrier of the O–O bond upon O2* + H* into O* + OH* is much lower on Fe1Co1-DAC (0.14 eV) than that on Fe-SAC (0.90 eV), and it is also much lower than the associative pathway barrier of O2* + H* → OOH* on Fe1Co1-DAC (0.62 eV). The reason for the facile cleavage of the O–O bond is attributed to the effective activation of the O–O bond on the Fe–Co dual site. Thus, thanks to the dual channel of electron acceptance–backdonation, the dual-site dissociation mechanism shows thermodynamic and kinetic feasibility on DAC and should not be neglected in the ORR network.
3.2. Contribution of Dual-Site Dissociation Pathways for Enhanced ORR Activity
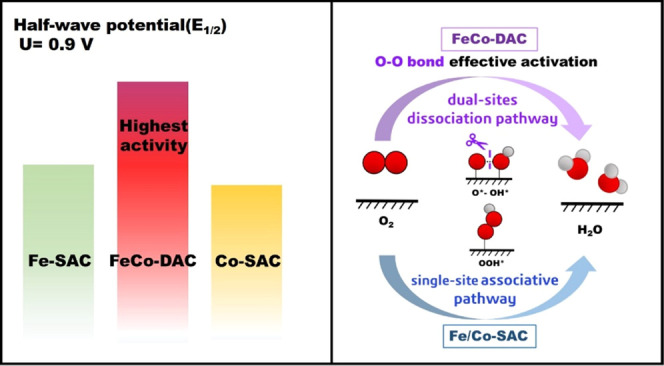
To determine the predominant mechanisms in ORR on DAC/TACs, free energy profiles for Fe1Co1-DAC are shown in Figure 3a. ORR is a four-electron reaction, and the onset potential is limited by the step with the smallest free energy drop, which is also the rate-determining step of ORR. A smaller free energy drop in the rate-determining step indicates a lower onset potential for ORR. Based on thermodynamic analysis, the rate-determining step of ORR on pathways III and IV (dissociation mechanism) is identified to be O* → OH*, and ΔG values are −0.91 and −0.92 eV, respectively, much lower than that of pathway I (association mechanism), where ΔG is only −0.67 eV from O2* to OOH*. Accordingly, the rate-determining steps of pathways III and IV are both more exothermic than that of pathway I, which indicates that the dual-site dissociation mechanism contributes more activity in ORR. Moreover, we perform an electrode microkinetic model to simulate polarization curves (see details in the Supporting Information). For convenience, the rate for electrochemical reduction steps is calculated by assuming an activation energy of 0.26 eV for proton transfers to oxygen-involved intermediates in aqueous solution as previously suggested.18 In Figure 3b, when Fe1Co1-DAC only proceeded through the single-site association mechanism (named Fe1Co1-associate), it shows similar ORR activity to Fe-SAC, which is inconsistent with experimental results.69,72,73 Obviously, when the dual-site dissociation mechanism is considered (named Fe1Co1-complete), there is a remarkable ORR performance improvement on Fe1Co1-DAC. Attributed to the consideration of complete reaction pathways I–VI, the simulated half-wave potential (E1/2) and current density at 0.9 V versus reversible hydrogen electrode (RHE) on Fe1Co1-DAC were superior to those on Fe/Co-SAC, which is in good agreement with that reported in experiments (Figure 3c and Table S2).69 The current density and dominant contribution of the dual-site dissociation pathway were not significantly changed when the calculated barrier of the O–O bond dissociation step rather than 0.26 eV was adopted in the microkinetic model (Figure S4). Thus, the reaction pathway switch to the dual-site dissociation mechanism rationalizes the superior ORR kinetic behavior of Fe1Co1-DAC to Fe/Co-SAC.
Figure 3.

(a) Calculated free energy profiles of reaction pathways I–IV for ORR on Fe1Co1-DAC. (b) Simulated polarization curves of Fe/Co-SAC and Fe1Co1-DAC through the sole single-site association pathway and complete pathways. (c) Comparison of half-wave potential (E1/2, V vs RHE) and current density (mA/cm2) at 0.9 V versus RHE on Fe1Co1-DAC and Fe/Co-SAC between the theoretical microkinetic model and the experiment reference.69 (d) Simulated polarization curves of Fe1Co1-DAC and the contribution from various pathways. (e) The coverage of the intermediate and (f) degree of rate control for each elementary step in the reaction network as a function of output potential on Fe1Co1-DAC. Note: coverage and degree of rate control profiles for only the relevant states are shown in the respective panel for each metal, and those not explicitly shown indicate zero coverage.
To further identify the contribution of the dual-site dissociation pathway in the ORR kinetics, we analyze the contribution of each pathway to the total polarization curve for Fe1Co1-DAC, as shown in Figure 3d. Obviously, pathway I (red) contributes a little current density, while pathways III (pink) and IV (green) contribute much more degree of flux, indicating that the dual-site dissociation pathway is dominant. In addition, Figure 3e–f shows potential-dependent intermediate coverage and degree of rate control for each elementary step (details of degree of rate control are available in the Supporting Information) for Fe1Co1-DAC. The O* + OH* formation from the hydrogenation of O2*, involved in the dual-site dissociation mechanism, is found to be the rate-limiting step, indicating that ORR proceeds dominantly through the dual-site dissociation pathway. Besides, within the working potential range, the coverage of both O* + OH* and 2OH* intermediates belonging to the dual-site dissociation pathway is much larger than that of OOH* involved in the single-site association pathway. It also suggests a predominant contribution of the dual-site dissociation pathway to ORR activity on Fe1Co1-DAC.
3.3. Pathways Switch to the Dual-Site Dissociation Mechanism on DACs/TACs
To further evaluate the feasibility of pathway switch to the dual-site dissociation mechanism on DACs/TACs and its impact on ORR activity, we also employ a thermodynamic and microkinetic model from DFT-derived energetics on other Fe/Co DACs/TACs embedded in N-doped graphene. As shown in Figure 4a, we have constructed a total of 15 Fe–Co DAC (named FexCo(2–x)-y-NC, y = 1–5, x = 0–2) and 20 Fe–Co TAC (named FexCo(3–x)-y-NC, y = 1–5, x = 0–3) models, in which Fe–Co bi-atoms and tri-atoms are embedded in a N-doped graphene sheet with five different types of environmental coordination. We evaluated the thermodynamic and electrochemical stabilities of these 35 models by the antiaggregation energy (Eanti-agg) and dissolution potential (Udiss), which are defined as follows.
| 4 |
| 5 |
where EFe/Co,bulk is the total energy of the metal atom in its most stable bulk structure, EFexCo2–x-NC and EFexCo2–x-1-NC are the total energies of FexCo2–x-NC and the substrate in which an Fe/Co atom has been dissolved, and Udiss0 (UFe0 = −0.447V, UCo0 = −0.28 V) and n are the standard dissolution potential of the bulk metal and the number of electrons transferring involved in the dissolution, respectively. According to our definition, systems with Eanti-agg < 0 eV (for the system containing both Fe and Co atoms, Eanti-agg = max[Eanti-agg-Fe, Eanti-agg-Co] < 0) are considered to be thermodynamically stable, while materials with Udiss ≥ 0 V versus RHE are regarded as electrochemically stable. The values of Eanti-agg and Udiss are listed in Table S3. Figure 4b shows the Eanti-agg of the heteronuclear Fe–Co DACs/TACs and homonuclear Fe/Co DACs/TACs examined in this study. We finally screened out 10 systems that meet the stability criteria, as shown in Figure 4b,c marked in red. Since stable horizontal adsorption of oxygen molecules on DACs/TACs is a necessary prerequisite for the dual channel of electron acceptance–backdonation mechanism, we studied the adsorption of O2 on the 10 DACs/TACs that meet the stability criteria. Among them, only O2 on Co2-2 DAC cannot be horizontally adsorbed, while the other 9 systems can horizontally adsorb O2. In some DACs/TACs, horizontal adsorption of O2 is thermodynamically less favorable than vertical adsorption and is regarded as a high-energy metastable structure. Even though the proportion of O2 horizontal adsorption may be lower than that of O2 vertical adsorption throughout the catalytic reaction, it can dictate catalysis with extraordinary activity arising from its unique dual channel for electron acceptance–backdonation mechanism and accordingly dual-site dissociation pathways. The key role of high-energy metastable structure-enabled catalysis has been demonstrated before.74
Figure 4.

(a) Geometric structures of Fe–Co DACs/TACs with coordination configurations 1–5 (FexCo2–x-1–5-NC/ FexCo3–x-1–5-NC). (b, c) Computed antiaggregation energy and dissolution potential of metal atoms in Fe–Co DACs/TACs, respectively (Fe–Co DACs/TACs with Eanti-agg < 0 eV and Udiss ≥ 0 V vs RHE are marked in red).
Then, we present and discuss the pre-adsorption phase diagram of 10 stable systems (see details in the Supporting Information), which allows us to analyze the most stable pre-adsorption situation of each system under the operating potential.
| 6 |
A spontaneously absorbed different ORR intermediate (OH*, O*, or O2*) with the lowest free energy (G) at a given set of potential (U) and pH = 0 determines the stable form of the surface in those conditions. Each intermediate species along the catalytic path for ORR is characterized by a straight line with a negative slope, which is determined by the number of electrons to be transferred. m is the number of electrons to be transferred (1 for m-G(OH*), 2 for m-G(O*), and 4 for m-G(O2*)). The intermediate corresponding to the lowest line at a common ORR working potential (U = 0.9 V) is regarded as the most stable form in this work. Therefore, the most stable form of 10 stable systems is determined for further study of the reaction mechanism, as shown in Figures S5 and S6. We reveal a potential-dependent structural switching of the active site between planar and non-planar structures during ORR electrocatalysis, which provides important insights, since the axial coordination effect can adjust the electronic structure of the central atom.
Taking Co3-1 TAC as an example to evaluate the priority of dual-site dissociation mechanisms in a complete ORR network induced by the dual channel of electron acceptance–backdonation. It is apparent that the features of the O–O bond activation of Co3-1 TAC agree well with those of Fe1Co1-DAC, namely, a dual channel of electron acceptance–backdonation is formed, which could be conducive to the sufficient activation of O2 (Figure S7a). The O–O bond cleavage barrier of Co3-1 TAC is lower than that of Co-SAC (Figure S7b), which indicates the reasonability of dual-site dissociation mechanisms on Co3-1 TAC. As shown in Figure 5a, the ORR activity of complete reaction pathways (Co3-1 complete, black), primarily contributed by dual-site dissociation pathways, is remarkably improved compared with that derived from the sole association pathway (Co3-1 association, dotted). Therefore, dual-site dissociation pathways rather than the single-site association pathway account for the superior ORR of Co3 to Co1 in the experiment.75 Moreover, 2OH* and OH* are abundant species at the working potential range, accompanied by the hydrogenation of 2OH*(2OH* transfer to OH*) being the second rate-limiting step (Figure S7c,d), which unequivocally indicates that ORR proceeds exclusively through the dual-site dissociation mechanism on Co3-1 TAC. These results above suggested that the reaction pathway switch to the dual-site dissociation pathway rationalizes the superior kinetic behavior of Co3-1 TACs to Co-SAC.
Figure 5.

(a) Simulated polarization curves of Co3-TAC. (b) Comparison of half-wave potential (E1/2, V vs RHE) and current density at 0.9 V versus RHE on Co-SAC and Co3-TAC between the theoretical microkinetic model and the experiment reference.75 (c) Simulated polarization curves of Fe2-2 DAC and (d) the simulated E1/2 value and j at 0.9 V are in good agreement with those reported in experiments.23
The dual-site dissociation mechanism is still observed on Fe2-2 DAC. Within the working potential range, the coverage of the 2OH* intermediate belonging to the dual-site dissociation pathway is much larger than that of OOH* involved in the single-site association pathway (Figure S8). Besides, the ORR activity of the complete reaction pathway (Fe2-2 complete, black), which is mainly contributed by the dual-site dissociation pathway, is significantly higher than that of the single association pathway (Fe2-2 association, dotted), as shown in Figure 5c. Due to the involvement of the dual-site dissociation pathway in the reaction network, the half-wave potential of Fe2-2 increases relative to that of Fe1-SAC, rationalizing the superior kinetic behavior of Fe2-2 DACs to Fe-SAC in the experiment23,61,62 (Figure 5d). Likewise, the consideration of the dual-site dissociation mechanism increases the onset potential or the half-wave potential (Figure S10 and Tables S4 and S5) of the other 8 FeCo DACs/TACs systems compared with the single-site association pathway. Although the barrier of O2 dissociation upon hydrogenation by surface H* on Fe2-2 DAC is not as favorable as Fe1Co1-1 DAC and Co3-1 TAC (Table S1), when the energy barrier of O2 dissociation was brought into the microkinetic model, it was found that the polarization curve changes little and the dual-site dissociation path is still dominant (Figure S5), which indicate that the direct hydrogenation of O2 by surface H* or solvent proton does not affect the conclusion of this study. Dual-site dissociation pathways rather than the single-site association pathway contribute most of the current flux in ORR, which suggests the reliability and feasibility of pathway switch in the ORR network for DACs/TACs. Although the metal active center and environmental coordination configurations of these DACs/TACs systems are different, the ORR mechanisms possess fairly similar features, implying that the presence of adjacent metal sites on DACs/TACs plays a key role in ORR pathway switch to the dual-site dissociation mechanism. We note that the limitation of CHE modeling applied here is to consider the pH effect on the reactivity of catalysts, since the free energy correction of the pH effect is offset when the potentials are regulated to the reversible hydrogen electrode (RHE) scale. The pH effect on the adsorption behavior of intermediates, which leads to the difference in the ORR activity of ADMCs on acidic and alkaline media, will be considered in our future work by using grand canonical ensemble DFT.54,76
3.4. Reaction Phase Diagram and Activity Trend of ORR on DACs/TACs
The theoretical prediction of the catalytic activity trend has been widely focused on due to its significant value in providing a guideline for a catalyst design, where the complete consideration of the reaction pathway is essential. Based on the identified dual-site dissociation pathway and the revised ORR network in Figure 1c, a new reaction phase diagram (RPD) of complete ORR pathways I–VI can be constructed to predict the pathway priority and activity trend in this work among DACs/TACs. The free energies of OOH*, O* + OH*, O*, 2OH*, and OH* on 10FexCo2–x-NC/FexCo3–x-NC systems are calculated and plotted in Figure S11. The DFT adsorption energies, zero-point energies, entropy, and solvation corrections needed to calculate free energies are available in Table S6, and the structure configurations are shown in Figure S12. The scaling relations between ΔG(OH*), ΔG(O*), and ΔG(OOH*) are still applicable in these systems, which are in good agreement with previous reports.28,40,77 Importantly, two new scaling relations are identified for 2OH* and O* + OH* adsorbed on adjacent active sites
| 7 |
| 8 |
The ΔG(OH*) term represents the free energy of a single OH* adsorbed on the active site, and the ΔG(2OH*) term represents the free energy of two OH* adsorbed on adjacent Fe–Co sites. In other words, ΔG(2OH*) is not equal to 2ΔG(OH*) on Fe–Co DACs/TACs with adjacent active sites, as two OH* can bond to different active sites and have an effect on the free energy of each other. In the same way, the free energy of O* + OH* is no longer thermodynamically equivalent to ΔG(OH*) + ΔG(O*). Since the free adsorption energy of each intermediate shows scaling relations with the combination between ΔG(O*) and ΔG(OH*) (Figure S12a), the free energy change of each elementary step along ORR pathways I–VI is relationally correlated with ΔG(O*) and ΔG(OH*), as shown in Figure S12b–e. As the adsorption free energies of ΔG(O*) and ΔG(OH*) show a good linear relationship with each parameter in ORR kinetics, ΔG(O*) and ΔG(OH*) are chosen as independent variables to evaluate the trend of preferential ORR pathways and ORR activity. A more accurate consideration of both catalytic mechanisms and the prediction of the activity trend can be conducted by a collective description of complete reaction pathways, named the reaction phase diagram (RPD) for simplicity in this work. The ORR current density on different catalysts with a given value of the descriptor (ΔG(O*), ΔG(OH*)), jRPD(ΔG(O*), ΔG(OH*)) is determined by the following expression
| 9 |
where all possible reaction pathways (denoted i) are established, considering all forward and backward elementary steps. e is the elementary charge, ρ is the surface density of active sites, and TOFe– is the turnover frequency of electron transfer. All considered pathways are compared to each other to identify one contributing the highest current density to the total ORR. As shown in Figure 6a, a reaction phase diagram at the electrode potential of 0.9 V versus RHE is established, where the predominant reaction pathways can be derived from the RPD at various given O* and OH* free energies. We find in this work that an ideal catalyst possessing high ORR activity is in the range of ΔG(O*) = 2.4–3.0 eV and ΔG(OH*) = 1–1.2 eV. Pathway IV is more favored over Fe1Co1-1, Fe1Co1-2, Fe2-1, Fe2-2, Co2-1, Co2-2, and Co3-1, and pathway III is more preferred on Fe1Co2-1, which suggests that DACs/TACs are more likely to follow the dual-site dissociation mechanism. The comparison between the predicted ORR activity and the DFT-calculated ones shows an excellent agreement (Figure 6b), which also illustrates the prediction accuracy of the three-dimensional volcano plot spanned by the adsorption free energies of ΔG(O*) and ΔG(OH*).
Figure 6.

Contour plot of current density at electrode potentials of 0.9 V versus RHE as a function of ΔG(O*) and ΔG(OH*) accompanied by the reaction pathway phase diagram. (b) The comparison between predicted and computed current densities at 0.9 V versus RHE.
Understanding how the electronic characteristics of the active center affect ΔG(O*) and ΔG(OH*) helps to guide the modification of the DAC/TAC active center and makes them closer to the volcanic summit. Taking the Fe active center in FeCo DACs/TACs as an example, Figure S13a shows the d-electron configurations of iron cations. The iron cations are regarded as +3 valence based on Bader charges analysis in Figure S14. The stable spin configurations of +3 valence iron cations may be in the intermediate-spin state and low-spin state. The intermediate-spin state of Fe3+ enables t2g and eg orbitals to be occupied by one unpaired electron with two and single spin, making the whole d-shell act as selective gates to promote the transfer of local spin currents. The calculated effective magnetic moments of Fe1Co1-1 and Fe2-2 are 3.77 μeff and 2.48 μeff, respectively. We further obtained the number of unpaired d-electrons (n) of Fe3+ ion via the following equation
| 10 |
where the number of unpaired d-electrons (n) of Fe2-2 is about 1.67, which means Fe3+ ions have a low-spin state without eg filling, so that no electron is occupied in the σ* antibonding orbital of Fe2-2, leading to a relatively stronger Fe/OH* interaction (a bond order of 2, Figure S13c). The number of unpaired d-electrons (n) of Fe1Co1-1 is about 3, which indicates an intermediate magnetic moment of the Fe center and the orbital interaction with *OH resulting in a lower bond order of 1.5 (Figure S13b). Since a higher on-site magnetic moment of the Fe center leads to a lower value of bond order, representing a weaker chemical bond strength, a positively linear correlation is reasonably found between ΔG(OH*) and the on-site magnetic moment of the Fe center (Figure S13d), clearly indicating that the spin configurations on the Fe center are the intrinsic factor dominating the ORR activity.
3.5. ORR Pathway Conversion on Other DACs
In addition to FeCo DACs/TACs supported on N-doped graphene, other metal elements or other substrates for ADMCs, such as RuCo-graphene78 and FePt-oxide,79 have also been reported exhibiting high ORR activity. Thus, we investigate the applicability of pathway switch from the single-site associative mechanism to the dual-site dissociation mechanism on the PtFe@Fe2O3(012)79 surface and RuCo@graphene.78 A comprehensive analysis of the calculation results shows that the oxygen molecules can be adsorbed separately on the appropriately distanced Pt and Fe atoms of PtFe@Fe2O3(012), forming a dual channel of electron acceptance–backdonation, which could directly catalyze O–O radical breakage and cause the ORR to follow a fast-kinetic dual-site dissociation mechanism. As shown in Figure S15, the current densities (j) of pathways III (pink −3.39 mA cm–2) and IV (green −5.56 mA cm–2) are closer to the total current density (dotted line −5.97 mA cm–2), indicating that the dual-site dissociation pathway is dominant. Moreover, the dual-site dissociation mechanism brings about better and reasonable onset potentials (Eonset = 1.17 V), which is in better agreement with the one (Eonset = 1.15 V) obtained in experiments, compared to the single-site associative mechanism (Eonset = 0.75 V).79 The features of the O–O bond activation on RuCo@graphene agrees well with that on PtFe@Fe2O3, and the ORR mechanisms possess fairly similar features, implying that the domination of the dual-site dissociation pathway is still applicable in this system.
Since the dual-site dissociation pathway of ORR requires two adsorption sites for oxygen-involved intermediates, we inferred that the oxyphilic property of the metal site might be an important factor in determining the feasibility of the dual-site dissociation pathway. Campbell et al.80 demonstrated that the enthalpy of oxidation formation of metal adsorptive atoms can be used to effectively evaluate the oxygen-affinity ability of metal elements, which has been further validated in recent experiments and calculations.81 Therefore, three metal elements with large differences in the enthalpy of oxidation formation (ΔHmetal-oxide)82 in the fourth, fifth, and sixth rows of the periodic table were selected to form FeM-DACs supported on N-doped graphene (M = Ti, Co, Cu, Y, Zn, Ag, Au, W, and Ta; Figure 7). As shown in Figure 7a, in the case of ΔHmetal-oxide ≥ 0 of M, O2 cannot be horizontally adsorbed in the corresponding FeM-DACs after structural relaxation, that is, they cannot go through the dual channel for electron acceptance–backdonation mechanism. In the case of ΔHmetal-oxide of M < 0, O2 can bind FeM-DACs with side-on adsorption mode. Thanks to the dual channel for electron acceptance–backdonation mechanism, these FeM-DACs allowing O2 side-on adsorption all show the thermodynamic priority of the dual-site dissociation pathway to the single-site association pathway (Figure 7b). Therefore, we come up with a general insight into the ORR mechanism switch from SACs to DACs/TAC that ΔHmetal-oxide of metal elements at the dual adjacent sites of DACs/TACs may be an efficient descriptor to judge the feasibility of the side-on adsorption mode of O2 and the dual-site dissociation pathway.
Figure 7.

(a) Correlation between oxygen adsorption energy (ΔGO2) and the ΔHmetal-oxide of M of FeM-DACs. (b) The reaction free energy of the competing steps between single-site association and dual-site dissociation pathways on FeM-DACs.
Thus, dual-site dissociation pathways involving O–O breakage during ORR should not be overlooked in the future rational design of DACs/TACs. Besides, this work also indicates that the enhancement of a dual channel for electron acceptance–backdonation on DACs/TACs is a promising approach to efficiently promote O–O bond breakage, accordingly improving the ORR activity due to the reaction pathway switch.
4. Conclusions
In summary, we propose the “dual channel for electron acceptance–backdonation” mechanism to rationalize the more activation of the O–O bond on Fe1Co1-DAC than Fe-SAC, which makes ORR switch to proceed through dual-site dissociation pathways from the traditional single-site association pathway. According to the polarization curve calculated by the DFT-based microkinetic model, the ORR activity of complete reaction pathways, primarily contributed by dual-site dissociation pathways, is remarkably improved compared with that derived from the sole association pathway. To further evaluate the feasibility of pathway switch to the dual-site dissociation mechanism on DACs/TACs and its impact on ORR activity, we have constructed a total of 15 Fe–Co DAC and 20 Fe–Co TAC models embedded in N-doped graphene. Although the metal active center and environmental coordination configurations of these DACs/TACs systems are different, the preferential ORR pathway possesses fairly similar features. Following this revised ORR network, a complete reaction phase diagram of DACs/TACs is established, where the preferential ORR pathway and ORR activity can be described by a three-dimensional volcano plot spanned by the adsorption free energies of ΔG(O*) and ΔG(OH*). Besides, the kinetics preferability of dual-site dissociation pathways is also appropriate for other graphene-supported or oxide-supported DACs/TACs. The contribution of dual-site dissociation pathways makes the theoretical ORR activity of DACs/TACs in better agreement with the available experiment than the traditional single-site association pathway, rationalizing the superior kinetic behavior of DACs/TACs to SACs. The enthalpy of oxidation formation (ΔHmetal-oxide) of the metal element at the double adjacent sites of DACs/TAC is found as an effective descriptor to judge the feasibility of the side-on adsorption mode of O2 and the dual-site dissociation pathway. This work reveals the origin of ORR pathway switching from SACs to DACs/TACs and indicates that dual-site dissociation pathways during ORR should not be overlooked in the rational design of DACs/TACs.
Acknowledgments
This work is supported by the National Key Research and Development Program of China (2021YFA1500501) and the National Natural Science Foundation of China (21975016 and 22005025).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00432.
Pathway of the oxygen reduction reaction (ORR); structures and adsorption energies of reactants, intermediates, transition states, and products; comparison of predicted and calculated results about the polarization curves; and analyses related to the O2* adsorption energy, microkinetic models, energy descriptor to regulate reaction pathways I–IV for ORR (PDF)
Author Contributions
§ J.L. and H.X. contributed equally to this work. CRediT: Jin Liu writing-original draft; Haoxiang Xu supervision, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Chen Z. W.; Chen L. X.; Yang C. C.; Jiang Q. Atomic (single, double, and triple atoms) catalysis: frontiers, opportunities, and challenges. J. Mater. Chem. A 2019, 7, 3492–3515. 10.1039/C8TA11416A. [DOI] [Google Scholar]
- Liu M.; Wang L.; Zhao K.; Shi S.; Shao Q.; Zhang L.; Sun X.; Zhao Y.; Zhang J. Atomically dispersed metal catalysts for the oxygen reduction reaction: synthesis, characterization, reaction mechanisms and electrochemical energy applications. Energy Environ. Sci. 2019, 12, 2890–2923. 10.1039/C9EE01722D. [DOI] [Google Scholar]
- Yang X. F.; Wang A.; Qiao B.; Li J.; Liu J.; Zhang T. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. 10.1021/ar300361m. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Ji S.; Chen C.; Peng Q.; Wang D.; Li Y. Single-Atom Catalysts: Synthetic Strategies and Electrochemical Applications. Joule 2018, 2, 1242–1264. 10.1016/j.joule.2018.06.019. [DOI] [Google Scholar]
- Wang J.; Li Z.; Wu Y.; Li Y. Fabrication of Single-Atom Catalysts with Precise Structure and High Metal Loading. Adv. Mater. 2018, 30, 1801649 10.1002/adma.201801649. [DOI] [PubMed] [Google Scholar]
- Jiao J.; Lin R.; Liu S.; Cheong W. C.; Zhang C.; Chen Z.; Pan Y.; Tang J.; Wu K.; Hung S. F.; Chen H. M.; Zheng L.; Lu Q.; Yang X.; Xu B.; Xiao H.; Li J.; Wang D.; Peng Q.; Chen C.; Li Y. Copper atom-pair catalyst anchored on alloy nanowires for selective and efficient electrochemical reduction of CO2. Nat. Chem. 2019, 11, 222–228. 10.1038/s41557-018-0201-x. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Zhang C.; Liu Z.; Chen C.; Li Y. Structural Regulation with Atomic-Level Precision: From Single-Atomic Site to Diatomic and Atomic Interface Catalysis. Matter 2020, 2, 78–110. 10.1016/j.matt.2019.11.014. [DOI] [Google Scholar]
- Zhang J.; Huang Q.-a.; Wang J.; Wang J.; Zhang J.; Zhao Y. Supported dual-atom catalysts: Preparation, characterization, and potential applications. Chin. J. Catal. 2020, 41, 783–798. 10.1016/S1872-2067(20)63536-7. [DOI] [Google Scholar]
- Zhou X.; Han K.; Li K.; Pan J.; Wang X.; Shi W.; Song S.; Zhang H. Dual-Site Single-Atom Catalysts with High Performance for Three-Way Catalysis. Adv. Mater. 2022, 34, 2201859 10.1002/adma.202201859. [DOI] [PubMed] [Google Scholar]
- Yan H.; Lin Y.; Wu H.; Zhang W.; Sun Z.; Cheng H.; Liu W.; Wang C.; Li J.; Huang X.; Yao T.; Yang J.; Wei S.; Lu J. Bottom-up precise synthesis of stable platinum dimers on graphene. Nat. Commun. 2017, 8, 1070 10.1038/s41467-017-01259-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. C.; Ma X. L.; Li Y.; Wang Y. G.; Xiao H.; Li J. Heterogeneous Fe3 single-cluster catalyst for ammonia synthesis via an associative mechanism. Nat. Commun. 2018, 9, 1610 10.1038/s41467-018-03795-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P.; Ding J.; Yao Z.; Pu T.; Zhang P.; Huang Z.; Wang C.; Zhang J.; Zecher-Freeman N.; Zong H.; Yuan D.; Deng S.; Shahbazian-Yassar R.; Wang C. Oxo dicopper anchored on carbon nitride for selective oxidation of methane. Nat. Commun. 2022, 13, 1375 10.1038/s41467-022-28987-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Qian Y.; Li H.; Zhang Z.; Mu Y.; Do D.; Zhou B.; Dong J.; Yan W.; Qin Y.; Fang L.; Feng R.; Zhou J.; Zhang P.; Dong J.; Yu G.; Liu Y.; Zhang X.; Fan X. O-coordinated W-Mo dual-atom catalyst for pH-universal electrocatalytic hydrogen evolution. Sci. Adv. 2020, 6, eaba6586 10.1126/sciadv.aba6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. C.; Xiao H.; Li J. Constructing High-Loading Single-Atom/Cluster Catalysts via an Electrochemical Potential Window Strategy. J. Am. Chem. Soc. 2020, 142, 3375–3383. 10.1021/jacs.9b06808. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Si R.; Liu H.; Chen N.; Wang Q.; Adair K.; Wang Z.; Chen J.; Song Z.; Li J.; Banis M. N.; Li R.; Sham T. K.; Gu M.; Liu L. M.; Botton G. A.; Sun X. Atomic layer deposited Pt-Ru dual-metal dimers and identifying their active sites for hydrogen evolution reaction. Nat. Commun. 2019, 10, 4936 10.1038/s41467-019-12887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Li B.; Hu Y.; Wang S.; Yu C. Highly-dispersed and high-metal-density electrocatalysts on carbon supports for the oxygen reduction reaction: from nanoparticles to atomic-level architectures. Mater. Adv. 2022, 3, 779–809. 10.1039/D1MA00858G. [DOI] [Google Scholar]
- Viswanathan V.; Hansen H. A.; Rossmeisl J.; Norskov J. K. Unifying the 2e(-) and 4e(-) Reduction of Oxygen on Metal Surfaces. J. Phys. Chem. Lett. 2012, 3, 2948–2951. 10.1021/jz301476w. [DOI] [PubMed] [Google Scholar]
- Tripković V.; Skúlason E.; Siahrostami S.; Nørskov J. K.; Rossmeisl J. The oxygen reduction reaction mechanism on Pt(111) from density functional theory calculations. Electrochim. Acta 2010, 55, 7975–7981. 10.1016/j.electacta.2010.02.056. [DOI] [Google Scholar]
- Russell A. E. Electrocatalysis: theory and experiment at the interface. Preface. Faraday Discuss. 2008, 140, 9–10. 10.1039/B814058H. [DOI] [PubMed] [Google Scholar]
- Karlberg G. S.; Rossmeisl J.; Nørskov J. K. Estimations of electric field effects on the oxygen reduction reaction. Phys. Chem. Chem. Phys. 2007, 9, 5158–5161. 10.1039/b705938h. [DOI] [PubMed] [Google Scholar]
- Greeley J.; Nørskov J. K. Combinatorial Density Functional Theory-Based Screening of Surface Alloys for the Oxygen Reduction Reaction. J. Phys. Chem. C 2009, 113, 4932–4939. 10.1021/jp808945y. [DOI] [Google Scholar]
- Xiao M.; Zhang H.; Chen Y.; Zhu J.; Gao L.; Jin Z.; Ge J.; Jiang Z.; Chen S.; Liu C.; Xing W. Identification of binuclear Co2N5 active sites for oxygen reduction reaction with more than one magnitude higher activity than single atom CoN4 site. Nano Energy 2018, 46, 396–403. 10.1016/j.nanoen.2018.02.025. [DOI] [Google Scholar]
- Ye W.; Chen S.; Lin Y.; Yang L.; Chen S.; Zheng X.; Qi Z.; Wang C.; Long R.; Chen M.; Zhu J.; Gao P.; Song L.; Jiang J.; Xiong Y. Precisely Tuning the Number of Fe Atoms in Clusters on N-Doped Carbon toward Acidic Oxygen Reduction Reaction. Chem 2019, 5, 2865–2878. 10.1016/j.chempr.2019.07.020. [DOI] [Google Scholar]
- Bezerra C. W. B.; Zhang L.; Lee K.; Liu H.; Marques A. L. B.; Marques E. P.; Wang H.; Zhang J. A review of Fe–N/C and Co–N/C catalysts for the oxygen reduction reaction. Electrochim. Acta 2008, 53, 4937–4951. 10.1016/j.electacta.2008.02.012. [DOI] [Google Scholar]
- Zhao C. X.; Li B. Q.; Liu J. N.; Zhang Q. Intrinsic Electrocatalytic Activity Regulation of M-N-C Single-Atom Catalysts for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2021, 60, 4448–4463. 10.1002/anie.202003917. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Alonso-Vante N. Nonprecious metal catalysts for the molecular oxygen-reduction reaction. physica status solidi (b) 2008, 245, 1792–1806. 10.1002/pssb.200879537. [DOI] [Google Scholar]
- Jiao L.; Zhu J.; Zhang Y.; Yang W.; Zhou S.; Li A.; Xie C.; Zheng X.; Zhou W.; Yu S. H.; Jiang H. L. Non-Bonding Interaction of Neighboring Fe and Ni Single-Atom Pairs on MOF-Derived N-Doped Carbon for Enhanced CO2 Electroreduction. J. Am. Chem. Soc. 2021, 143, 19417–19424. 10.1021/jacs.1c08050. [DOI] [PubMed] [Google Scholar]
- Kulkarni A.; Siahrostami S.; Patel A.; Norskov J. K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. 10.1021/acs.chemrev.7b00488. [DOI] [PubMed] [Google Scholar]
- Wei B.; Fu Z.; Legut D.; Germann T. C.; Du S.; Zhang H.; Francisco J. S.; Zhang R. Rational Design of Highly Stable and Active MXene-Based Bifunctional ORR/OER Double-Atom Catalysts. Adv. Mater. 2021, 33, 2102595 10.1002/adma.202102595. [DOI] [PubMed] [Google Scholar]
- Xu J.; Elangovan A.; Li J.; Liu B. Graphene-Based Dual-Metal Sites for Oxygen Reduction Reaction: A Theoretical Study. J. Phys. Chem. C 2021, 125, 2334–2344. 10.1021/acs.jpcc.0c10617. [DOI] [Google Scholar]
- Zhang L.; Guo X.; Zhang S.; Huang S. Building up the “Genome” of bi-atom catalysts toward efficient HER/OER/ORR. J. Mater. Chem. A 2022, 10, 11600–11612. 10.1039/D2TA02050E. [DOI] [Google Scholar]
- Zhao X.; Liu X.; Huang B.; Wang P.; Pei Y. Hydroxyl group modification improves the electrocatalytic ORR and OER activity of graphene supported single and bi-metal atomic catalysts (Ni, Co, and Fe). J. Mater. Chem. A 2019, 7, 24583–24593. 10.1039/C9TA08661G. [DOI] [Google Scholar]
- Zhu X.; Yan J.; Gu M.; Liu T.; Dai Y.; Gu Y.; Li Y. Activity Origin and Design Principles for Oxygen Reduction on Dual-Metal-Site Catalysts: A Combined Density Functional Theory and Machine Learning Study. J. Phys. Chem. Lett. 2019, 10, 7760–7766. 10.1021/acs.jpclett.9b03392. [DOI] [PubMed] [Google Scholar]
- Liu C.; Li T.; Dai X.; Zhao J.; He D.; Li G.; Wang B.; Cui X. Catalytic Activity Enhancement on Alcohol Dehydrogenation via Directing Reaction Pathways from Single- to Double-Atom Catalysis. J. Am. Chem. Soc. 2022, 144, 4913–4924. 10.1021/jacs.1c12705. [DOI] [PubMed] [Google Scholar]
- Ren W.; Tan X.; Yang W.; Jia C.; Xu S.; Wang K.; Smith S. C.; Zhao C. Isolated Diatomic Ni-Fe Metal-Nitrogen Sites for Synergistic Electroreduction of CO2. Angew. Chem., Int. Ed. 2019, 58, 6972–6976. 10.1002/anie.201901575. [DOI] [PubMed] [Google Scholar]
- Tian S.; Fu Q.; Chen W.; Feng Q.; Chen Z.; Zhang J.; Cheong W. C.; Yu R.; Gu L.; Dong J.; Luo J.; Chen C.; Peng Q.; Draxl C.; Wang D.; Li Y. Carbon nitride supported Fe2 cluster catalysts with superior performance for alkene epoxidation. Nat. Commun. 2018, 9, 2353 10.1038/s41467-018-04845-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D.-H.; Xu C.-Q.; Wang Y.-G.; Li J. Heterogeneous Single-Cluster Catalysts for Selective Semihydrogenation of Acetylene with Graphdiyne-Supported Triatomic Clusters. J. Phys. Chem. C 2019, 123, 10494–10500. 10.1021/acs.jpcc.9b02029. [DOI] [Google Scholar]
- Xie Y.; Chen X.; Sun K.; Zhang J.; Lai W. H.; Liu H.; Wang G. Direct Oxygen-Oxygen Cleavage through Optimizing Interatomic Distances in Dual Single-atom Electrocatalysts for Efficient Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2023, 62, e202301833 10.1002/anie.202301833. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Su H.; Cheng W.; Li Y.; Jiang J.; Liu M.; Yu F.; Wang W.; Wei S.; Liu Q. Regulating the scaling relationship for high catalytic kinetics and selectivity of the oxygen reduction reaction. Nat. Commun. 2022, 13, 6414 10.1038/s41467-022-34169-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L.; Li S. Unconventional Oxygen Reduction Reaction Mechanism and Scaling Relation on Single-Atom Catalysts. ACS Catal. 2020, 10, 4313–4318. 10.1021/acscatal.0c00815. [DOI] [Google Scholar]
- Li F.; Liu X.; Chen Z. 1 + 1′ ≥ 2: Heteronuclear Biatom Catalyst Outperforms Its Homonuclear Counterparts for CO Oxidation. Small Methods 2019, 3, 1800480 10.1103/PhysRevB.50.17953. [DOI] [Google Scholar]
- Chen Z. W.; Yan J. M.; Jiang Q. Single or Double: Which Is the Altar of Atomic Catalysts for Nitrogen Reduction Reaction?. Small Methods 2019, 3, 1800291 10.1002/smtd.201800291. [DOI] [Google Scholar]
- Han X.; Ling X.; Yu D.; Xie D.; Li L.; Peng S.; Zhong C.; Zhao N.; Deng Y.; Hu W. Atomically Dispersed Binary Co-Ni Sites in Nitrogen-Doped Hollow Carbon Nanocubes for Reversible Oxygen Reduction and Evolution. Adv. Mater. 2019, 31, 1905622 10.1002/adma.201905622. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Rostgaard C. The Projector Augmented-Wave Method. Phys. Rev. B 2009, 62, 11556–11570. [Google Scholar]
- Cui P.; Zhao L.; Long Y.; Dai L.; Hu C. Carbon-Based Electrocatalysts for Acidic Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2023, 62, e202218269 10.1002/anie.202218269. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Shin D.; Kim J.; Lim J. S.; Paidi V. K.; Shin T. J.; Jeong H. Y.; Lee K. S.; Kim H.; Joo S. H. Reversible Ligand Exchange in Atomically Dispersed Catalysts for Modulating the Activity and Selectivity of the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2021, 60, 20528–20534. 10.1002/anie.202108439. [DOI] [PubMed] [Google Scholar]
- Liu F.; Shi L.; Song S.; Ge K.; Zhang X.; Guo Y.; Liu D. Simultaneously Engineering the Coordination Environment and Pore Architecture of Metal-Organic Framework-Derived Single-Atomic Iron Catalysts for Ultraefficient Oxygen Reduction. Small 2021, 17, 2102425 10.1002/smll.202102425. [DOI] [PubMed] [Google Scholar]
- Dudarev S. L.; Botton G. A.; Savrasov S. Y.; Humphreys C. J.; Sutton A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA1U study. Phys. Rev. B 1998, 57, 1505–1509. 10.1103/PhysRevB.57.1505. [DOI] [Google Scholar]
- Mathew K.; Kolluru V. S. C.; Mula S.; Steinmann S. N.; Hennig R. G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 2019, 151, 234101 10.1063/1.5132354. [DOI] [PubMed] [Google Scholar]
- Mathew K.; Sundararaman R.; Letchworth-Weaver K.; Arias T. A.; Hennig R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 2014, 140, 084106 10.1063/1.4865107. [DOI] [PubMed] [Google Scholar]
- Hu X.; Chen S.; Chen L.; Tian Y.; Yao S.; Lu Z.; Zhang X.; Zhou Z. What is the Real Origin of the Activity of Fe-N-C Electrocatalysts in the O(2) Reduction Reaction? Critical Roles of Coordinating Pyrrolic N and Axially Adsorbing Species. J. Am. Chem. Soc. 2022, 144, 18144–18152. 10.1021/jacs.2c08743. [DOI] [PubMed] [Google Scholar]
- Rebarchik M.; Bhandari S.; Kropp T.; Mavrikakis M. How Noninnocent Spectator Species Improve the Oxygen Reduction Activity of Single-Atom Catalysts: Microkinetic Models from First-Principles Calculations. ACS Catal. 2020, 10, 9129–9135. 10.1021/acscatal.0c01642. [DOI] [Google Scholar]
- Henkelman G.; Uberuaga B. P.; Jónsson H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. 10.1063/1.1329672. [DOI] [Google Scholar]
- Chan K.; Norskov J. K. Electrochemical Barriers Made Simple. J. Phys. Chem. Lett. 2015, 6, 2663–2668. 10.1021/acs.jpclett.5b01043. [DOI] [PubMed] [Google Scholar]
- Chan K.; Norskov J. K. Potential Dependence of Electrochemical Barriers from ab Initio Calculations. J. Phys. Chem. Lett. 2016, 7, 1686–1690. 10.1021/acs.jpclett.6b00382. [DOI] [PubMed] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Wang F.; Xie W.; Yang L.; Xie D.; Lin S. Revealing the importance of kinetics in N-coordinated dual-metal sites catalyzed oxygen reduction reaction. J. Catal. 2021, 396, 215–223. 10.1016/j.jcat.2021.02.016. [DOI] [Google Scholar]
- Leng K.; Zhang J.; Wang Y.; Li D.; Bai L.; Shi J.; Li X.; Zheng L.; Bai J.; Qu Y. Interfacial Cladding Engineering Suppresses Atomic Thermal Migration to Fabricate Well-Defined Dual-Atom Electrocatalysts. Adv. Funct. Mater. 2022, 32, 2205637 10.1002/adfm.202205637. [DOI] [Google Scholar]
- Zhang N.; Zhou T.; Ge J.; Lin Y.; Du Z.; Zhong C.; Wang W.; Jiao Q.; Yuan R.; Tian Y.; Chu W.; Wu C.; Xie Y. High-Density Planar-like Fe2N6 Structure Catalyzes Efficient Oxygen Reduction. Matter 2020, 3, 509–521. 10.1016/j.matt.2020.06.026. [DOI] [Google Scholar]
- Yang X.; Shang C.; Zhou S.; Zhao J. MBenes: emerging 2D materials as efficient electrocatalysts for the nitrogen reduction reaction. Nanoscale Horiz. 2020, 5, 1106–1115. 10.1039/D0NH00242A. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhang Z.; Qi S.; Fan Y.; Yang Y.; Li W.; Zhao M. Photo-assisted high performance single atom electrocatalysis of the N2 reduction reaction by a Mo-embedded covalent organic framework. J. Mater. Chem. A 2021, 9, 19949–19957. 10.1039/D1TA02691G. [DOI] [Google Scholar]
- Li L.; Yuan K.; Chen Y. Breaking the Scaling Relationship Limit: From Single-Atom to DualAtom Catalysts. Acc. Mater. Res. 2022, 3, 584–596. 10.1021/accountsmr.1c00264. [DOI] [Google Scholar]
- Zhou X.; Gao J.; Hu Y.; Jin Z.; Hu K.; Reddy K. M.; Yuan Q.; Lin X.; Qiu H.-J. Theoretically Revealed and Experimentally Demonstrated Synergistic Electronic Interaction of CoFe Dual-Metal Sites on N-doped Carbon for Boosting Both Oxygen Reduction and Evolution Reactions. Nano Lett. 2022, 22, 3392–3399. 10.1021/acs.nanolett.2c00658. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Biswas A.; Siddharthan E. E.; Thapa R.; Dey R. S. Strategic Modulation of Target-Specific Isolated Fe,Co Single-Atom Active Sites for Oxygen Electrocatalysis Impacting High Power Zn–Air Battery. ACS Nano 2022, 16, 7890–7903. 10.1021/acsnano.2c00547. [DOI] [PubMed] [Google Scholar]
- Yang W.; Huang H.; Ding X.; Ding Z.; Wu C.; Gates I. D.; Gao Z. Theoretical study on double-atom catalysts supported with graphene for electroreduction of nitrogen into ammonia. Electrochim. Acta 2020, 335, 135667 10.1016/j.electacta.2020.135667. [DOI] [Google Scholar]
- Wang J.; Huang Z.; Liu W.; Chang C.; Tang H.; Li Z.; Chen W.; Jia C.; Yao T.; Wei S.; Wu Y.; Li Y. Design of N-Coordinated Dual-Metal Sites: A Stable and Active Pt-Free Catalyst for Acidic Oxygen Reduction Reaction. J. Am. Chem. Soc. 2017, 139, 17281–17284. 10.1021/jacs.7b10385. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wei Z.; Liu J.; Tan W.; Fang X.; Fang D.; Wang X.; Wang D.; Tang J.; Fan X. Oxidization of Al0.5Ga0.5As(001) surface: The electronic properties. Appl. Surf. Sci. 2018, 436, 460–466. 10.1016/j.apsusc.2017.12.043. [DOI] [Google Scholar]
- Cen W.; Liu Y.; Wu Z.; Wang H.; Weng X. A theoretic insight into the catalytic activity promotion of CeO2 surfaces by Mn doping. Phys. Chem. Chem. Phys. 2012, 14, 5769–5777. 10.1039/c2cp00061j. [DOI] [PubMed] [Google Scholar]
- Xiao M.; Chen Y.; Zhu J.; Zhang H.; Zhao X.; Gao L.; Wang X.; Zhao J.; Ge J.; Jiang Z.; Chen S.; Liu C.; Xing W. Climbing the Apex of the ORR Volcano Plot via Binuclear Site Construction: Electronic and Geometric Engineering. J. Am. Chem. Soc. 2019, 141, 17763–17770. 10.1021/jacs.9b08362. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Ji S.; Wang Y.; Dong J.; Chen W.; Li Z.; Shen R.; Zheng L.; Zhuang Z.; Wang D.; Li Y. Isolated Single Iron Atoms Anchored on N-Doped Porous Carbon as an Efficient Electrocatalyst for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2017, 56, 6937–6941. 10.1002/anie.201702473. [DOI] [PubMed] [Google Scholar]
- Xia Z.; Yin Y.; Li J.; Xiao H. Single-atom catalysis enabled by high-energy metastable structures. Chem. Sci. 2023, 14, 2631–2639. 10.1039/D2SC06962H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Shan H.; Yu H.; Zhong C.; Ge J.; Zhang N.; Chu W.; Yan W.; Xu Q.; Wu H.; Wu C.; Xie Y. Nanopore Confinement of Electrocatalysts Optimizing Triple Transport for an Ultrahigh-Power-Density Zinc-Air Fuel Cell with Robust Stability. Adv. Mater. 2020, 32, 2003251 10.1002/adma.202003251. [DOI] [PubMed] [Google Scholar]
- Liu T.; Wang Y.; Li Y. How pH Affects the Oxygen Reduction Reactivity of Fe–N–C Materials. ACS Catal. 2023, 13, 1717–1725. 10.1021/acscatal.2c05540. [DOI] [Google Scholar]
- Calle-Vallejo F.; Krabbe A.; Garcia-Lastra J. M. How covalence breaks adsorption-energy scaling relations and solvation restores them. Chem. Sci. 2017, 8, 124–130. 10.1039/C6SC02123A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Chun H.; Yang T.-C.; Hong S. J.; Yang C.-M.; Han B.; Lee L. Y. S. Tuning the Site-to-Site Interaction in Ru–M (M = Co, Fe, Ni) Diatomic Electrocatalysts to Climb up the Volcano Plot of Oxygen Electroreduction. ACS Nano 2022, 16, 10657–10666. 10.1021/acsnano.2c02324. [DOI] [PubMed] [Google Scholar]
- Gao R.; Wang J.; Huang Z.-F.; Zhang R.; Wang W.; Pan L.; Zhang J.; Zhu W.; Zhang X.; Shi C.; Lim J.; Zou J.-J. Pt/Fe2O3 with Pt–Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 2021, 6, 614–623. 10.1038/s41560-021-00826-5. [DOI] [Google Scholar]
- Campbell C. T.; Sellers J. R. Anchored metal nanoparticles: effects of support and size on their energy, sintering resistance and reactivity. Faraday Discuss. 2013, 162, 9–30. 10.1039/c3fd00094j. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Yang Z. Interfacial properties of NM/CeO2(111) (NM = noble metal atoms or clusters of Pd, Pt and Rh): a first principles study. J. Phys.: Condens. Matter 2010, 22, 475003 10.1088/0953-8984/22/47/475003. [DOI] [PubMed] [Google Scholar]
- Xu H.; Zhu L.; Nan Y.; Xie Y.; Cheng D. Revisit the Role of Metal Dopants in Enhancing the Selectivity of Ag-Catalyzed Ethylene Epoxidation: Optimizing Oxophilicity of Reaction Site via Cocatalytic Mechanism. ACS Catal. 2021, 11, 3371–3383. 10.1021/acscatal.0c04951. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.