

Abstract
The risk of Alzheimer disease (AD) increases with age, family history and informative genetic variants. Sadly, there is still no cure or means of prevention. As in other complex diseases, uncovering genetic causes of AD could identify underlying pathological mechanisms and lead to potential treatments. Rare, autosomal dominant forms of AD occur in middle age as a result of highly penetrant genetic mutations, but the most common form of AD occurs later in life. Large-scale, genome-wide analyses indicate that 70 or more genes or loci contribute to AD. One of the major factors limiting progress is that most genetic data have been obtained from non-Hispanic white individuals in Europe and North America, preventing the development of personalized approaches to AD in individuals of other ethnicities. Fortunately, emerging genetic data from other regions – including Africa, Asia, India and South America – are now providing information on the disease from a broader range of ethnicities. Here, we summarize the current knowledge on AD genetics in populations across the world. We predominantly focus on replicated genetic discoveries but also include studies in ethnic groups where replication might not be feasible. We attempt to identify gaps that need to be addressed to achieve a complete picture of the genetic and molecular factors that drive AD in individuals across the globe.
Introduction
The rapid increase in the prevalence of dementia worldwide is considered by many to be a global emergency1. As we enjoy the benefits of longer lives, we face the blunt reality that dementia affects over 30% of those aged 80 years or older1. Estimations from the WHO suggest that around 55 million people globally have dementia, of whom 40 million are thought to have Alzheimer disease (AD) and over 60% live in low-income and middle-income countries. As the proportion of older people in the population increases in nearly every country, the total number of individuals with AD is projected to rise to approximately 78 million by 2030 and possibly 139 million by 2050. Currently, 6.5 million American individuals are living with AD, which equates to nearly 11% of the population of the USA. By 2050, this number is expected to have doubled to 13.8 million. The risk of late-onset AD increases with age, a family history of the disease and the presence of genes associated with the disease1. With no cure or prevention strategy on the horizon, countries worldwide seek ways to provide humane care for those affected at a cost that is sustainable.
The genetics of Alzheimer disease
Complex genetic disorders, such as AD, are likely to be solved by identifying variants predisposing to disease. These variants and the genes in which they lie can be investigated to determine the mechanisms by which they cause disease. Functional studies can then be performed to yield targets for therapeutic strategies. Over the past four decades, the genetics of AD has been an area of intense interest. According to estimates, 60–80% of the risk of AD is attributable to heritable (genetic) factors2. Despite this relatively high heritability, identifying genes that cause AD has proven to be complex, with both rare and common genetic variants contributing to the disease. The accuracy of identifying a specific genetic cause of dementia in an individual depends on the disease phenotype, age at onset and family history.
Prior to the sequencing of the human genome in 2003, the main research strategy was to focus on the rare, autosomal dominant forms of AD that begin in middle age. Family-based analysis methods (Box 1) were amenable to genetic approaches available at that time and mutations in three genes – APP, PSEN1 and PSEN2 – were discovered between 1985 and 1995 (refs. 3–6). Since then over 300 pathogenic variants have been identified in these three genes, yet at least 25% of the Mendelian forms of AD remain to be defined7–9. Whole-exome sequencing (WES) and whole-genome sequencing (WGS) are now being used to identify additional genes that contribute to these typically highly penetrant forms of the disease. Rare coding variants in PSEN1 and PSEN2 have also been found in multiplex families with late-onset disease10, indicating a genetic continuum between early-onset and late-onset forms. These results suggest that factors other than these variants can affect the age at onset and penetrance of AD11.
Box 1. Types of genetic study.
Family-based studies
Family-based studies have been used for decades to investigate many monogenic disorders. Investigation of multigenerational families facilitated the discovery of presenilin 1 (PSEN1), presenilin 2 (PSEN2) and amyloid precursor protein (APP) in early-onset, autosomal dominant Alzheimer disease (AD). However, families with late-onset AD have also been widely recruited and used in linkage and family-based association studies 209–212. Family-based studies are most feasible when onset of the disorder is at a young age, so that the proband’s parents are still alive213,214. Challenges to this type of analysis include the variable age at onset and penetrance of disease as well as the clinical and genetic heterogeneity of AD, even within a family.
Case–control studies
The case–control design is the most frequent approach used now to identify genetic variants contributing to disease owing to the relative ease in recruiting individuals. This design seeks to identify differences in allele or genotype frequencies between a group of participants with AD and a group of healthy controls. This approach was used to identify the apolipoprotein E association with AD. However, participants in the two groups must be rigorously defined and matched for key factors such as ancestry. Observational studies with a mix of individuals with AD and healthy individuals can also estimate the effect of a variety of genetic and other risk factors on outcomes and can also contribute to our understanding of novel genetic risk factors. Thus, longitudinal, observational studies such as the Alzheimer’s Disease Neuroimaging Initiative can provide a broad assessment to evaluate a wide range of biomarkers and outcomes.
Observational cohort studies
Observational cohort design can also be used to examine the effect of risk factors in a population-based setting in which the number of cases reflects the population incidence rate. The Washington Heights–Inwood Columbia Aging Project is a multi-ethnic cohort consisting of African Americans, non-Hispanic white individuals, and Caribbean Hispanic individuals all recruited from northern Manhattan47. Another well-known longitudinal prospective cohort is the Adult Changes in Thought study that randomly recruited individuals 65 years of age or older, living in Seattle, who were members of Group Health and were cognitively intact at the time of enrolment in Adult Changes in Thought215. A limitation of this approach can be the clinical and genetic heterogeneity within the cohort, which can confound the results.
In 1991, Pericak-Vance and colleagues12 found an association on chromosome 19q that led to discovery of variants in the apolipoprotein E (APOE) gene that increase or decrease the risk of the more typical late-onset form of AD. The APOE gene has three common allelic forms: ε2, ε3 and ε4. The ε4 allele was found to increase the risk of AD, whereas the presence of the ε2 allele was considered to be protective. This study also demonstrated the power of an association study that compares allele and genotype frequencies in two groups: individuals with AD and individuals without disease (case–control design; Box 1).
Over the three decades since the discovery of the association with APOE variants, research has proven that AD is highly heterogeneous with many variants conferring small effects on risk (polygenic)13. The success of the APOE case–control association design and the relative ease of recruiting individuals with and without AD has led to a multitude of studies that use this approach to identify additional genetic risk factors in populations throughout North America, Europe and Asia. Advances in genomic technology have promoted the case–control approach, which now includes genome-wide genotyping of single nucleotide polymorphism (SNPs) as well as WES and WGS. Analyses can focus on association with SNPs or variants or can evaluate the overall evidence of association with a gene, by aggregating the effect of multiple variants within the gene into one test. With the rapid decline in the cost of WGS and the advent of large national and international collaborations, genetic studies now include samples from hundreds of thousands or even a million participants. Over the past 3 years, studies14–17 have identified over 70 different genetic variants associated with AD using this approach (Fig. 1). The majority of these variants were identified in non-Hispanic white individuals mostly from Europe and the USA, and might not generalize to non-white and/or Hispanic individuals. Thus, the existing data are unlikely to fully explain the genetic risks among individuals from Africa, Asia, the Middle East, and Middle and South America. In addition, many of the studies in non-white individuals were small and the findings still need replication to be confirmed.
Fig. 1 |. Genetic loci associated with Alzheimer disease in genome-wide association studies of non-Hispanic white individuals.
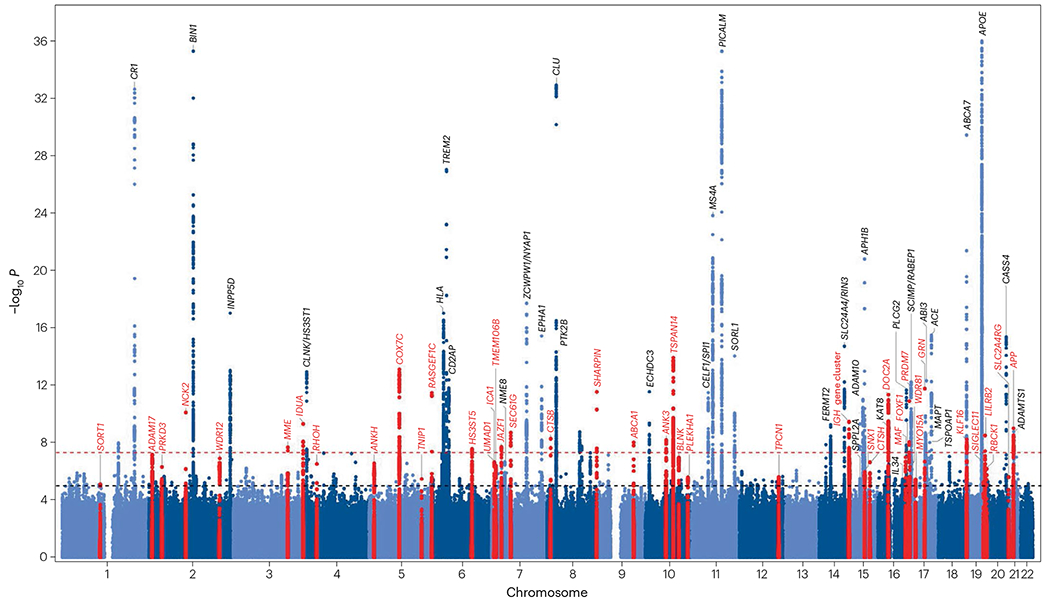
Manhattan plot from a study by Bellenguez and colleagues16 showing the identified genetic loci associated with Alzheimer disease in non-Hispanic white individuals. P values are two-sided raw P values derived from fixed-effect meta-analysis. The threshold for genome-wide significance (P = 5 × 10−8) is indicated by the red dashed line, and the suggestive threshold for genome-wide significance (P = 1 < 10−5) is indicated by the black dashed line. Loci are named for the closest gene to the sentinel variant for each locus. Loci newly identified by Bellenguez et al. are shown in red, whereas loci previously reported are shown in light and dark blue. Adapted from ref. 16, Springer Nature Limited.
Polygenic risk scores provide a quantitative approach to combine the effect of multiple variants and have proven to be an important tool for defining genetic risk in the presence of many different variants with small individual effects. Polygenic risk scores have been evaluated in epidemiological cohorts to estimate the effect of risk factors in a population-based setting in which the number of cases reflects the population incidence rate (Box 1). A polygenic risk score of the AD-associated genetic variants identified in non-Hispanic white individuals is associated with a faster rate of tau-PET accumulation18.
Migration, diversity and ancestry
Human migration and geodemographic events have taken place over thousands of years of human evolutionary history, and have shaped the genetic diversity among current populations. Substantial differences in allele frequencies and the extent to which common alleles are inherited together (linkage disequilibrium) can have profound effects on the associations of genetic variants with traits and disease in different populations. The phylogenetic analyses inferred from global human mtDNA sequences19 almost four decades ago demonstrated that anatomically modern humans originated in Africa and that the population subsequently expanded outwards. The largest split between human populations occurred around 160,000–110,000 years ago in sub-Saharan Africa20,21 (Fig. 2). Over the past 10 years, sequencing of genomes of ancient humans22 and other hominins23 demonstrated that individuals with a greater degree of African ancestry have less linkage disequilibrium than individuals with a smaller degree of African ancestry24,25. This is because, throughout human evolutionary history, the population living in Africa maintained a larger effective size and also had more time for recombination than other populations. By contrast, migration out of Africa within the past 50,000–100,000 years resulted in a distinct genetic pattern of populations living outside Africa showing less genetic diversity with a linear decline of heterozygosity and flattening of the ancestral allele frequency spectrum as a function of geographic distance from Africa and founder event bottlenecks26. These events have resulted in individuals with a higher degree of African ancestry having higher levels of genetic diversity than individuals with a lower degree of African ancestry27. In most genomic studies, individuals are assigned to genetic ancestry categories through principal component analysis, a multivariate approach intended to characterize individuals and populations by ethnobiological origins and relatedness.
Fig. 2 |. Timeline and routes of human migration inferred from genomic data.
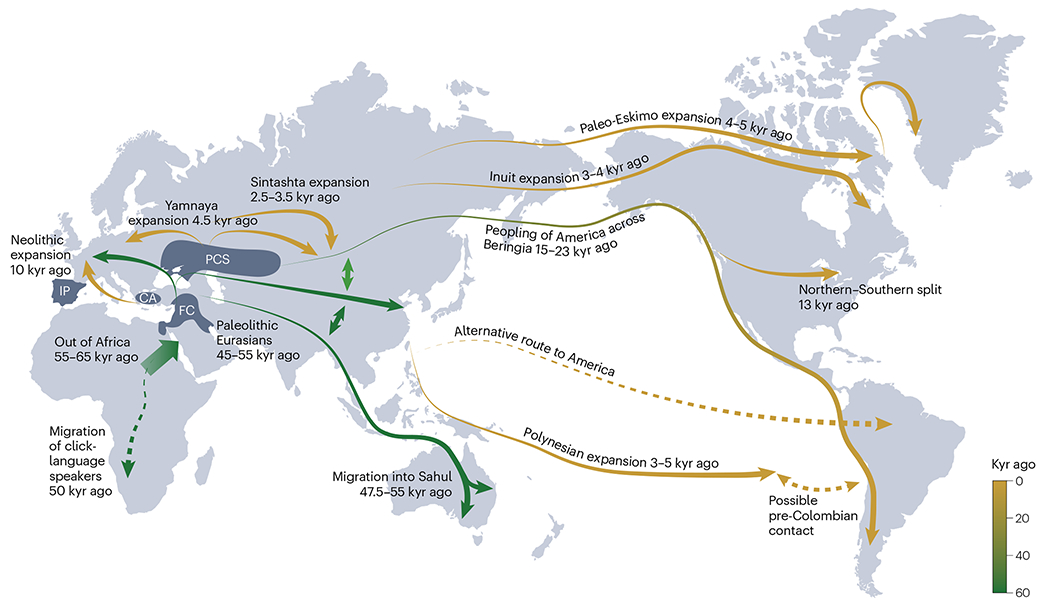
Dashed lines represent routes of migration that remain controversial. CA, Central Anatolia; FC, Fertile Crescent; IP, Iberian Peninsula; kyr, thousand years; PCS, Pontic-Caspian steppe. Adapted from ref. 208, Springer Nature Limited.
These ancestral differences have implications for the assessment of genotypic risk28–30. Although some associations between genetic markers and disease are present across populations – providing important evidence for a shared genetic basis of disease risk – genetic risk prediction models developed from one ancestral group do not perform as well when applied to other ancestral groups31. Common variants can be shared globally but rare variants are usually shared by closely related populations and might be restricted to a single continental group32. The small group of humans that left Africa about 100,000 years ago and populated the rest of the world carried only a subset of the African variations. Thus, populations throughout the world may have genomes that include variants emerging as part of the African migration worldwide. The period in time and the number of individuals migrating would then determine the degree of African ancestry in a given population. However, the subset of human genetic variation left behind in Africa can be studied only in individuals with a high degree of African ancestry33.
African American and Hispanic groups, the largest admixed non-European groups in the USA, account for 12.6% and 16.3%, respectively, of the population34. The first Africans were brought to North America as slaves in the early 17th century and this continued until slavery was abolished in 1865 by the US Congress. The degree of variability in the number of variants is roughly proportional to the degree of recent African ancestry an individual has in their genome35. Thus, for admixed populations, overall genetic disease risk might be determined by multiple genetic variants from a combination of ancestral backgrounds. For admixed groups with African ancestry, it is essential to understand AD risk in indigenous African individuals.
Impact on APOE and other genetic associations with AD.
In most genetic studies conducted in individuals of non-European ancestry, participants self-reported their ancestry, and the researchers performed statistical corrections for ancestral heterogeneity (that is, population substratification owing to genetic ancestral background) or adjustments such as principal components. From studies in individuals of African36–39 and Hispanic ancestry40–44, notable ancestry-related differences have been identified in the genetic architecture of AD. Compared with non-Hispanic white individuals, APOE ε4-associated AD risk is weaker in individuals with African ancestry but stronger in individuals of Japanese ancestry45–47. Preliminary evidence indicates that this variability in risk is driven by the ancestral origin of the DNA at the APOE ε4 locus (for example, local genetic ancestry)48,49. The APOE ε4 amino acid coding sequence does not differ consistently between populations of different ancestry; thus, the source of the variability in genotypic risk associated with APOE ε4 could lie in regulatory regions that affect gene expression. Indeed, in a post-mortem study of individuals with AD, higher levels of APOE expression were observed in the brains of individuals of European ancestry than in those of individuals of African ancestry50.
Genome-wide studies of individuals of African ancestry that used population substratification by principal components have identified many genes, including ABCA7 and ACE, that contain ancestrally specific risk variants36,38,51 (Table 1). In ABCA7, a 44 bp deletion was strongly associated with AD in African Americans52 and also present in Caribbean Hispanic individuals, who are known to have a high proportion of African ancestry (41.8%)53. Functional studies suggest that this ancestry-specific deletion might result in increased production of toxic amyloid-β (Aβ) in neurons and a reduced ability to clear Aβ in microglia52, making it a candidate of interest for development as a therapeutic target. Other rare truncating and splice-altering variants in ABCA7 confer risk in non-Hispanic white individuals54–56.
Table 1 |.
Genetic loci with a P value of ≤10−7 identified in GWAS of AD in African Americans
| SNP | Position (hg38) | Closest gene | Minor allele | MAF | Ref. |
|---|---|---|---|---|---|
| rs115684722 | 1:23,224,0417 | SIPA1L2 | T | 0.003 | 36 |
| rs2633682 | 3:104,690,364 | ALCAM | A | 0.338 | 36 |
| rs168193 | 3:5,260,392 | EDEM1 | G | 0.267 | 36 |
| rs145848414 | 5:174,587,111 | NSG2/MSX2 | A | 0.0416 | 38 |
| rs184179037 | 5:37,483,838 | WDR70 | T | 0.0008 | 36 |
| rs7748513 | 6:41,160,234 | TREM2 a | A | 0.459 | 36 |
| rs112404845 | 7:51,510,325 | COBL | T | 0.01 | 206 |
| rs569584007 | 11:43,145,292 | API5 b | G | 0.0023 | 36 |
| rs115816806 | 11:76,830,796 | ACER3 | G | 0.0083 | 36 |
| rs75739461 | 12:18,318,612 | PIK3C2G | A | 0.0151 | 36 |
| rs9516245 | 13:93,507,547 | GPC6 | C | 0.0189 | 36 |
| rs570487962 | 15:97,449,455 | ARRDC4/IGF1R | C | 0.0008 | 36 |
| rs79537509 | 16:8,238,399 | RBFOX1 a | A | 0.007 | 36 |
| rs115550680 | 19:1,050,421 | ABCA7 a | G | 0.0681 | 38 |
| rs157591 | 19:44,920,677 | APOE a | A | 0.1422 | 38 |
| rs3745495 | 19:50,021,075 | VRK3 c | G | 0.0877 | 36 |
A P value cut-off of ≤10−7 indicates loci suggestive of genome-wide significance. GWAS, genome-wide association study; hg38, Genome Reference Consortium Human Build 38; MAF, minor allele frequency; SNP, single-nucleotide polymorphism.
Locus is also observed in non-Hispanic white individuals.
5 Mb apart from but not in linkage disequilibrium with CELF1/SPI1 locus observed in non-Hispanic white individuals16.
1.7 Mb apart from but not in linkage disequilibrium with CD33 observed in non-Hispanic white individuals17.
Mutations in APP, PSEN1 and PSEN2 have been reported in individuals with early-onset AD from many regions and ancestries, including northern and southern European populations57,58, various Middle Eastern and Arab populations59,60, Caribbean Hispanic populations61–64, Latin American populations65–67, populations from northern and southern Africa68–71, populations from Australia and New Zealand72,73, and a range of Asian populations including those from China, Korea, Taiwan, Japan, India and Malaysia71,74–80. Although some variants are shared across populations, most variants are unique to ancestral groups.
Africa
The Alzheimer’s Disease International report on dementia in sub-Saharan Africa describes the state of dementia in this region and summarizes several trends with respect to the ageing population81. It is noteworthy that of the 46 countries in sub-Saharan Africa, only a small fraction have existing capacity for dementia research and clinical care. Africa comprises mostly low-income to middle-income countries, and the prevalence of AD in the continent is expected to reach approximately 3.5 million by 2030 and 7.6 million by 2050 (ref. 81). Although there are several studies that focus on the epidemiological aspects of AD in Africa, research into the genomics of AD in Africa is in its infancy.
Several candidate gene studies of early-onset AD in Africa identified variants in AD-associated genes previously identified in non-Hispanic white individuals: APP, PSEN1, PSEN2 (refs. 68–70) and TREM2 (ref. 82). The effect of the APOE ε4 allele on AD risk has also been investigated in various groups of individuals with African ancestry83–87. A Moroccan study sequenced exons 16 and 17 of APP in 17 individuals with sporadic AD and eight individuals with familial early-onset AD. This included seven novel frameshift mutations and one novel splice mutation identified in exon 17. A similar targeted study of PSEN1 and PSEN2 in Moroccan individuals with familial and sporadic AD also identified one novel frameshift mutation in PSEN1 and two in PSEN2 (ref. 68). In an investigation of vascular disease-associated polymorphisms in 200 Tunisian individuals with dementia and 300 cognitively healthy Tunisian individuals, polymorphisms in APOE, angiotensin-converting enzyme (ACE) and paraoxonase 1 (PON1) genes were found to be associated with dementia; the association of these variants with AD was not investigated87.
APOE remains the most studied AD gene in individuals of African ancestry. Its known association and substantial contribution to risk in non-Hispanic white individuals88 make it the obvious candidate for global investigation. One of the earliest studies of APOE among African American individuals living in New York City found no association with AD89, but subsequent studies of African American individuals from Indianapolis and Yoruban individuals from Ibadan, Nigeria, found an association between the APOE ε4 allele and incident AD83. However, among the Yoruban individuals, APOE ε4 homozygosity, but not heterozygosity, was a significant risk factor for AD, which indicates a weaker effect of the variant in individuals of African ancestry than in non-Hispanic white individuals. A second study of APOE in Yoruban individuals surmised that a combination of genetic effects (lack of association with APOE) and environmental effects (low cholesterol and lipid levels, and less vascular disease and hypertension, owing to a low-fat diet) might explain the difference84. A similar study in a Nigerian community84 examined the relationship between APOE genotype, cholesterol and AD. Individuals met NINCDS–ADRDA90 criteria for AD, and ICD-10 criteria91 for vascular dementia and other secondary dementias. However, the study lacked autopsy or biomarker information, leaving the possibility of misdiagnosis. Increased levels of cholesterol and LDL were associated with a higher risk of AD, but only in individuals without an APOE ε4 allele. The effect of APOE genotype on AD risk has also been investigated in Kenya but no association between APOE ε4 and AD risk was observed85.
The reports of lower APOE ε4-associated risk of AD in individuals of African ancestry than in non-Hispanic white individuals have triggered additional ancestry-specific studies92,93 and studies to identify genetic variants that might lower APOE ε4 risk on an African ancestry haplotype. A statistically significant interaction between the APOE ε4 allele and the SNP rs10423769 was identified in a discovery dataset from African Americans36,94, and the finding was subsequently replicated by the same investigators in a large Puerto Rican cohort42 and the Yoruban cohort mentioned above95. The rs1042369_A allele was associated with an estimated 70% reduction in the risk of AD for APOE ε4 homozygotes. This African ancestry-specific locus is 2 Mb from the APOE locus and is located in a cluster of pregnancy-specific β1-glycoproteins; it might represent a potential therapeutic target for further investigation.
Latin America
The origin of people currently living in Latin America can be traced to extensive admixture between Native American, European and African populations, and the proportions of different ancestries can vary according to region96–98. This historical population structure in Latin America makes genetic association studies challenging because of the ancestral differences that exist among countries and even regions within Latin America. An estimated 8.5% of adults in Latin America aged 60 years and over have dementia99. The prevalence of dementia in the region is expected to increase by up to 400% by 2040, greater than the increase predicted in Western Europe or North America100.
Mutations in APP, PSEN1 and PSEN2 that cause early-onset AD have been identified in Caribbean Hispanic individuals10,61. In addition, a missense variant in PSEN1 (NM_000021.3: c.1247T>C p.Ile416Thr), originating on an African haplotype, was identified in individuals from a Colombian admixed population101 and a variant at codon 280 in PSEN1 (PSEN1 E280A) was identified in a large Colombian kindred102. A phase II clinical trial (NCT01998841) was then performed in individuals with the latter variant to test the efficacy of a monoclonal antibody (crenezumab) against monomeric and aggregated Aβ1–40 and Aβ1–42 (ref. 103). Although the study did not identify any statistically significant effects of treatment, cognitive, brain imaging and biomarker outcomes showed trends towards favourable outcomes. Compared with non-Hispanic white individuals, among Caribbean Hispanic individuals the effects of APOE ε4 homozygosity on AD risk are blunted and there is no increased risk among heterozygotes43,46.
Genome-wide association studies (GWAS) in Caribbean Hispanic individuals have identified several risk loci near genes known to be associated with AD in non-Hispanic white individuals: PICALM, CLU, BIN1, MS4A gene cluster, CD33, CD2AP, ABCA7 and EPHA1 (refs. 104–106). Subsequent WES or WGS studies in these individuals have also identified rare AD-associated variants in many other genes such as TREM2, SORL1, PINX1, PGL2, ABI3, PTK2B and AKAP9 (refs. 40,44,48,49,107,108). A study using admixture mapping in a large number of Caribbean Hispanic participants to identify AD-associated ancestral blocks for European, African and Native American ancestral components found four such ancestral blocks located on chromosomes 1, 6, 21 and 22. Fine mapping using these ancestral blocks prioritized the GCAT gene on chromosome 22, which was replicated in an independent dataset109 (Fig. 3). Functional studies in human post-mortem tissue and two model systems (Drosophila and zebrafish) suggested that the inflammation-related activity of GCAT is a response to amyloid toxicity, and that reduced GCAT expression exacerbates AD pathology.
Fig. 3 |. Admixture mapping of Alzheimer disease in Caribbean Hispanic individuals.
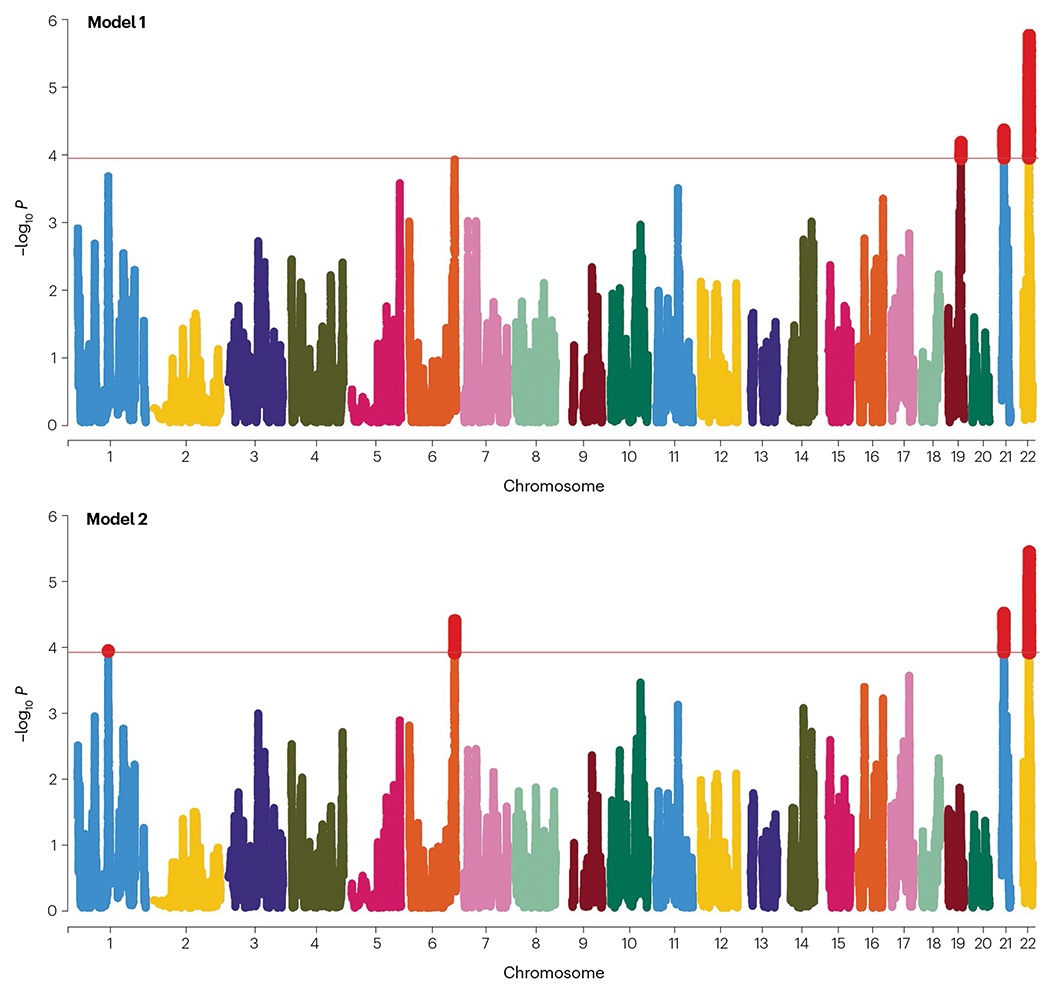
Manhattan plots of analyses conducted in admixMap. Model 1 (upper panel) is adjusted for age, sex, genotype batch, principal components for population stratification, and kinship. Model 2 (lower panel) is in addition adjusted for APOE genotype. The red highlighted parts represent the identified ancestral blocks significant after multiple testing correction. Adapted from ref. 109, Springer Nature Limited.
Evidence of genetic interaction effects has also been observed. Among individuals from Mexico, the presence of the APOE ε4 allele and variants in SORL1 implicated a possible genetic interaction effect that increases AD risk110. A single mutation in the PSEN1 gene (E280A) is the cause of the world’s largest autosomal dominant AD kindred, located in Antioquia, Colombia. The mutation is virtually 100% penetrant for AD. It is noteworthy that a member of this Colombian family escaped cognitive impairment until her seventies. She also carried two copies of the APOE ε3 Christchurch (R136S) mutation11, suggesting that homozygosity for the APOE ε3 allele might be protective.
East Asia
China is likely to have the largest number of individuals with AD in the world. According to a recent meta-analysis, the prevalence of dementia varies from 4.8% in southern China, to 5.2% in central China, 5.5% in northern China and 7.2% in western China; however, not all parts of the country have been surveyed111. East Asia has three major ethnic groups: Han Chinese, Japanese and Korean. As in many other populations, mutations in APP, PSEN1 and PSEN2 have been identified among individuals with familial early-onset AD across all three groups77,112. In addition, candidate gene studies and GWAS have identified AD risk variants in or near several genes that also contain AD risk variants in other ethnic groups113. These genes include APOE, SORL1, SORCS1, TREM2, BIN1, CLU, MS4A4E/MS4A6A, CD33, PICALM, DAPK1, UNC5C, CNTNAP2, SHARPIN and KCNJ15. In Han Chinese individuals, the TREM2 p.H157Y variant reached an odds ratio of 11.01 in a targeted sequencing study114 and an odds ratio of ~3.6 in a meta-analysis of over 7,000 individuals with AD and 7,400 healthy control participants115, indicating the importance of this locus in this population. Additional loci suggested by GWAS or WGS included intergenic regions at SUDS3–SRRM4 and FAM47E–SCARB2 in Japanese cohorts; GCH1, RHOBTB3–GLRX and CHODL in Han Chinese cohorts; and CHD2, CACNA1A and LRIG1 in South Korean cohorts113,116–121 (Table 2).
Table 2 |.
Genetic loci reported with a P value of ≤10−5 by GWAS or WGS studies in East Asian populations
| SNP | Position (hg38) | Closest gene | Minor allele | MAF | Ref. |
|---|---|---|---|---|---|
| Japan | |||||
| rs4598682 | 11:121505242 | SORL1 a | G | 0.192 | 207 |
| rs1992269 | 18:1872316 | ENSG00000266602 | T | 0.018 | 116 |
| rs802571 | 7:146265094 | CNTNAP2 a,b | G | 0.029 | |
| rs11613092 | 12:118455443 | SUDS3 | T | 0.1367 | |
| rs920608 | 4:76217307 | FAM47E/SCARB2 | C | 0.044 | 120 |
| China | |||||
| rs72713460 | 14:54830325 | GCH1 | T | 0.134 | 121 |
| rs2591054 | 15:57320212 | LINC01413 | T | 0.246 | |
| rs73052335 | 19:44916825 | APOC1/APOE | C | 0.092 | |
| rs928771 | 21:38291838 | KCNJ15 | G | 0.154 | 117 |
| rs3777215 | 5:95786296 | RHOBTB3/GLRX | A | 0.168 | |
| rs6859823 | 5:106218683 | ENSG00000252337 | T | 0.369 | |
| rs234434 | 14:97354683 | LINC02325 | G | 0.237 | |
| rs2255835 | 21:18119346 | CHODL | C | 0.321 | |
| South Korea | |||||
| rs1890078 | 10:107218478 | SORCS1 | C | 0.064 | 119 |
| rs12594991 | 15:92973197 | CHD2 | A | 0.141 | |
| rs189753894 | 19:13513675 | CACNA1A | A | 0.359 | |
| rs2280575 | 3:66492439 | LRIG1 | G | 0.059 |
A P value cut-off of ≤10−5 indicates loci suggestive of genome-wide significance. GWAS, genome-wide association study; hg38, Genome Reference Consortium Human Reference37; MAF, minor allele frequency; SNP, single nucleotide polymorphism; WGS, whole-genome sequencing.
Locus is also observed in non-Hispanic white individuals.
Variants in CNTNAP2 have also been reported to be associated with vascular dementia in individuals from Spain174.
Disease-associated loci differ notably among the three ancestral East Asian subgroups (Table 2). Although some of these differences might be explained by limited sample sizes and statistical power, Han Chinese, Japanese and Korean populations have distinct genetic makeups and substantial differences in allele frequencies and linkage disequilibrium patterns, which make the presence of ancestry-specific disease-associated variation likely122. We expect that additional variants and loci will be identified in East Asian populations as the results of additional, larger genetic studies begin to be published. Together, these data further highlight the importance of performing genetic studies in non-European populations to identify more effective, ancestry-informed druggable targets120.
West Asia and the Middle East
Throughout the West Asian and Middle East region, numerous families with early-onset AD and PSEN1 and PSEN2 mutations have been identified123–127. Mendelian forms of AD with novel phenotypes have also been observed. For example, in two individuals from the same family in Turkey, early-onset AD and spastic paraparesis was found to be associated with a heterozygous splicing variant (c.869-1G>A) in PSEN1 (ref. 123). In a family in Israel with a history of cognitive decline or intracerebral haemorrhage across seven generations, affected individuals were found to carry a copy number variant (chr21:27,224,097-27,871,284) that included the APP locus126. A second copy number variant on chromosome 5 co-segregated with the APP duplication in some family members. In this large family, asymptomatic carriers showed cognitive decline in their mid-thirties.
The association between APOE ε4 and AD in West Asian and Middle East populations is robust, although there are regional differences in APOE allele frequencies, specifically a low prevalence of the ε4 allele in some regions128. APOE ε4 has been strongly associated with AD in cohort and clinical studies from Egypt, Iran, Israel, Lebanon, Saudi Arabia and Turkey129–134. In studies of familial and sporadic AD across the West Asian and Middle East region, associations with AD were confirmed for several established candidate genes: TREM2, SORL1, ABCA7, ACE, CD2AP, CLU and EPHA1 (refs. 124,129,134–141). A protective allelic association in PICALM was also reported142. Although there have been few large genome-wide investigations of AD in this region, the identification of common variants in known AD genes confirms the global nature of this disease.
Challenges and future directions
Many of the earlier studies discussed above were limited in size and statistical power to detect more than a few genetic associations. Some smaller studies lacked adjustment for population substratification, which might have affected the findings. Environmental factors such as socioeconomic status or access to health care are also likely to have influenced associations between genetic variants and AD. The ascertainment of larger cohorts consisting of individuals with a range of different ancestries, and the investigation of these cohorts with genome-wide arrays and WGS coupled with adjustment for ancestral background and important environmental factors, will be crucial if we are to fully disentangle the genetic and molecular underpinnings of AD.
New collaborative efforts such as the African Dementia Consortium (AfDC)143 are important milestones towards these goals. The AfDC is a coalition of African dementia researchers that aims to generate a variety of data including clinical, cognitive, epidemiological, socioeconomic, neuroimaging, genomic and biomarker data to characterize and understand AD and related dementias across Africa. The AfDC will work together to define interventions and treatments for dementia across Africa that could also have a worldwide impact. For example, currently only a few African countries have data available on AD prevalence and incidence (Fig. 4). The AfDC will promote research efforts in this area to better characterize the disease across the continent. Genetic studies will also have a key role, with the AfDC collaborating globally to identify new variants for AD, many of which will be unique to African populations. Currently the AfDC includes researchers from nine African countries: Nigeria, Ghana, Benin, Cameroon, Kenya, Uganda, Tanzania, Mozambique and Ethiopia. The AfDC is coordinated from the College of Medicine, University of Ibadan143, and we consider it to be crucial for future success in both research and clinical care in Africa.
Fig. 4 |. Prevalence of dementia in Africa.
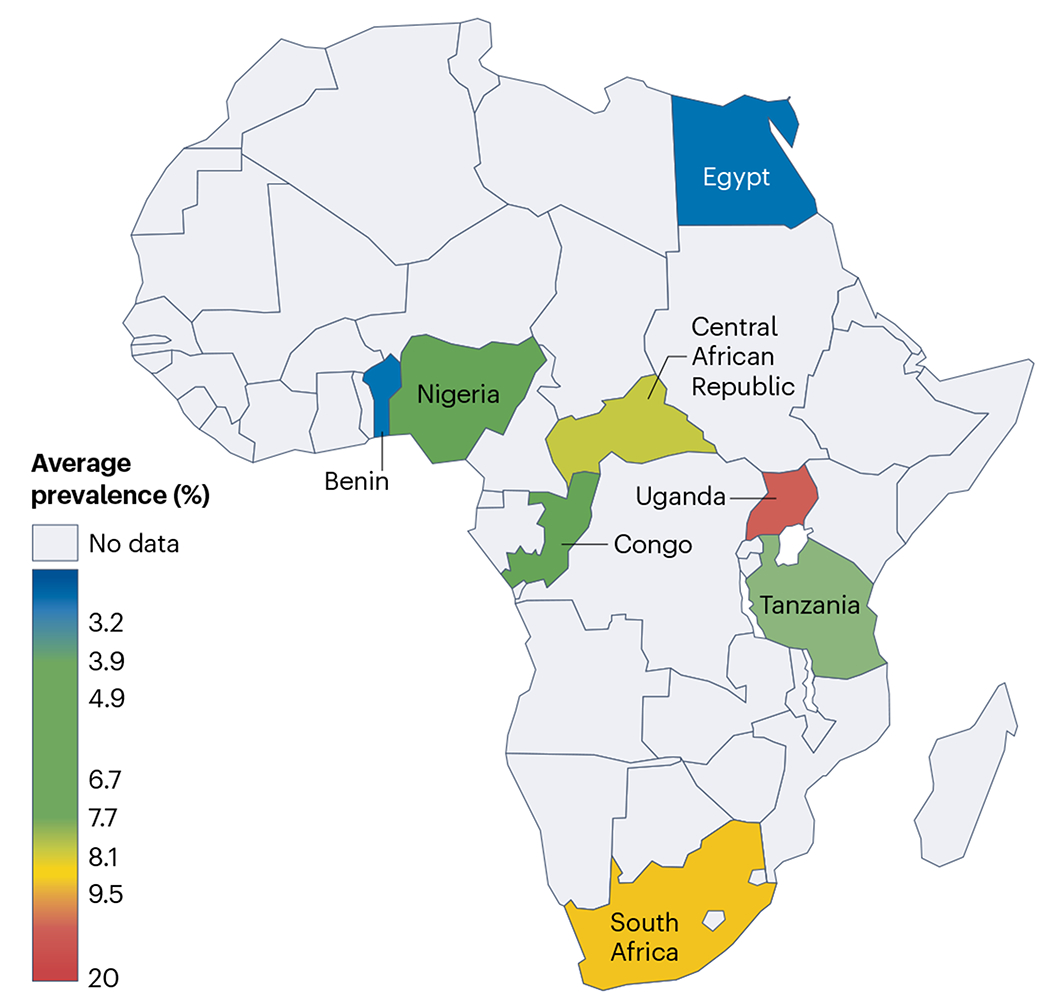
Heat map showing the wide range of dementia prevalence in African countries determined over the past 25 years. Dementia prevalence studies have also been conducted in Senegal and Kenya, but the data are not yet published. Adapted with permission from ref. 143, Wiley.
Several new efforts aim to include participants with a range of different ancestries in studies based in the USA. The largest AD DNA sequencing study in the USA is the Alzheimer’s Disease Sequencing Project, which conducts both WES and WGS studies, and was designed to identify new risk and protective variants that could help to identify putative therapeutic targets. However, like previous genome-wide array studies, the initial large-scale sequencing studies focused primarily on non-Hispanic white individuals, and included a limited number of Hispanic and African American individuals144. The Alzheimer’s Disease Sequencing Project–Follow-Up Study aims to increase the diversity of AD datasets through a specific focus on inclusion of individuals from a range of different ancestral populations145. By the end of 2023, approximately 40,000 whole genomes are expected to have been assembled and released for analysis.
Other efforts to improve the diversity of study populations in AD genetics research include the Research in African-American Alzheimer’s Disease Initiative, which collates whole genomes from multiplex African American families, and the Recruitment and Retention for Alzheimer’s Disease Diversity Genetic Cohorts146, which will collect data from another 4,000 African American individuals and 4,000 Hispanic individuals as well as 5,000 individuals recruited from various sub-Saharan African countries. In addition, Estudio Familiar de Influencia Genetica en Alzheimer is conducting family-based AD genomic studies in Caribbean Hispanic populations, and a WGS study of Korean participants is being performed as part of the Gwangju Alzheimer’s and Related Dementias (GARD) Study. Furthermore, studies are being performed in individuals of native Amerindian ancestry from the southern Peruvian Andes mountains, in Mexican individuals (Mexican Health and Aging Study), and in 2,000 Indian individuals from all parts of India (Longitudinal Aging Study in India). Finally, the Asian Cohort for Alzheimer’s Disease is evaluating approximately 6,000 individuals of Chinese, Korean and Vietnamese ancestry with late-onset AD for genomic analyses. Several of these efforts also involve the acquisition of data on plasma or cerebrospinal fluid (CSF) biomarkers and multiomics. In addition, the ADNI4 study of the Alzheimer’s Disease Neuroimaging Initiative147 aims to enrol 50–60% of its new participants from populations previously under-represented in AD research studies in the USA by implementing improved culturally engaged approaches for recruitment and retention. We expect that these complementary efforts across populations of different ancestries will allow us to further disentangle the genetic contributions of individual ancestries to AD, identify genomic loci that are shared across ethnic groups, and pinpoint loci that are specific to a particular population. This would provide critical information on population-specific AD pathways, potential biomarkers, potential therapeutic targets for drug discovery, and observed health disparities.
Understanding phenotypic heterogeneity
Genetic studies require careful and thorough phenotyping. However, the criteria for the diagnosis of AD have evolved over the past few decades, resulting in somewhat of a moving target. The original diagnostic criteria for AD were created by a panel of experts from the National Institute of Neurological Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINDS-ADRDA) in 1984 (ref. 90). This committee categorized three diagnostic levels: definite AD (neuropathological diagnosis), probable AD (diagnosis without confounding factors), and possible AD (diagnosis with comorbidities). The sensitivity and specificity of the clinical criteria for probable AD compared with post-mortem diagnosis were 81% and 70%, respectively148. These criteria were revised in 2011 (ref. 148) to include pathophysiological changes. At this time, MRI, PET imaging and CSF analyte assays were just starting to be implemented in clinical and research settings, recognizing and reinforcing the observation that the underlying pathological changes related to AD can begin decades prior to the onset of clinical symptoms.
AD can be categorized into early-onset and late-onset forms on the basis of the age at which the first symptoms appeared. However, this classification can be arbitrary149: although the majority of genetic studies use a cut-off age of 65 years, some studies use 60 years of age13,150. Onset before age 65 years accounts for approximately 5–10% of AD13,149, corresponding to ~300,000–700,000 individuals in the USA. Both early-onset and late-onset groups present primarily with memory-predominant phenotypes; however, individuals with early-onset AD can have a more aggressive course of disease with a reduced survival time151. Approximately 25% of individuals with early-onset AD have an atypical clinical presentation characterized by language, visuospatial or executive dysfunction and preserved episodic memory152. Some have considered early-onset AD to represent the purest form of the disease, but this might not be the case. Individuals with the fully penetrant autosomal dominant early-onset form of AD can have an increased burden of white matter hyperintensities, which reflect microvascular pathology, compared with cognitively healthy non-carriers several decades older153. Regardless of the age at onset, extracellular Aβ plaques and intraneuronal neurofibrillary tangles composed of hyperphosphorylated tau protein are consistent at autopsy, and can be accompanied by cerebrovascular disease, Lewy bodies or other aggregated proteins1. TDP43 pathology, hippocampal sclerosis, vascular injury, pronounced brain atrophy154, higher tau burden155 and more widespread tau neuropathology have been associated with early-onset disease156,157. TDP43 deposition might even determine when hippocampal atrophy begins and the rate of neurofibrillary tangle accumulation158.
At brain autopsy, 20% to 30% of individuals with clinically diagnosed AD had cerebrovascular disease or Lewy body pathology, and sometimes both, in addition to plaques and tangles16,159–162 (Fig. 5). The results of neuroimaging and neuropathological studies indicate that cerebrovascular disease co-occurs with primary AD pathology and adds to disease manifestation160,163. Furthermore, a large body of epidemiological cohort studies has implicated a range of highly prevalent vascular risk factors including diabetes, insulin resistance, mid-life obesity and hypertension in dementia risk164. Whether cerebrovascular disease directly causes AD pathology or is a frequent comorbidity that contributes to the clinical phenotype remains an open question. Among individuals with AD, approximately 20% also have α-synuclein pathology, intraneuronal Lewy bodies and Lewy neurites165–167. The presence of Lewy body pathology in AD is associated with earlier symptom onset168, worse cognition169, faster progression170 and parkinsonism171. Individuals with a high tangle load in the limbic system and cerebral neocortex are less likely to express the clinical features typical of dementia with Lewy bodies and tend to be diagnosed as having AD172. MRI studies further suggest that there are four atrophy pattern subtypes in AD: typical, limbic-predominant, hippocampal-sparing, and no atrophy173. The degree of clinical and neuropathological heterogeneity in AD can confound the functional understanding of genetics, making incorporation of additional information to improve diagnosis, such as blood-based biomarkers, critical. For example genetic influences in AD with associated cerebrovascular pathology, Lewy body accumulation or even TDP43 deposits have not been fully explored. Also, incorporating cerebrovascular disease and related risk factors in genetic studies of AD might provide novel information174,175.
Fig. 5 |. Pathology in clinically diagnosed Alzheimer disease.
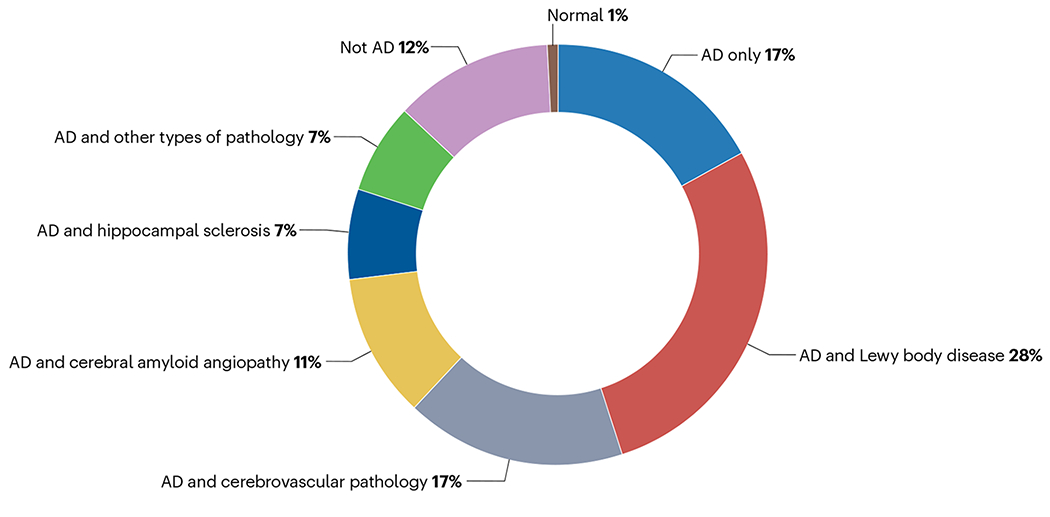
The proportions of other types of pathological changes in individuals with clinically diagnosed Alzheimer disease (AD). The data are from a publication by DeTure and Dickson162, in which they describe the pathological evaluation of 626 individuals from the Mayo Clinic Brain Bank. ‘Not AD’ indicates a completely distinct form of dementia.
Improving diagnostic accuracy
The era of precision medicine has encouraged the development of refinements in the clinical diagnosis of AD, thereby improving phenotype–genotype analyses. The inclusion of MRI, PET and CSF assays in the 2011 research criteria for the diagnosis of AD20 led to a proposed research framework176 that used biomarkers to create classifications that reflect the deposition of Aβ and tau proteins and neurodegeneration – the so-called A/T/N classification177. ‘A’ refers to evidence of Aβ accumulation in the brain provided by PET or low CSF Aβ1–42 concentration. ‘T’ refers to tau pathology indexed with PET or increased tau concentration in CSF. ‘N’ refers to neurodegeneration and would be reflected by regional atrophy on MRI. As discussed in the preceding section, individuals with a clinical diagnosis of AD can have a range of underlying pathological changes, which can begin decades before diagnosis. Therefore, incorporating biomarker assessment into genetic studies might enable more accurate diagnoses and the identification of asymptomatic individuals with causal variants.
PET and CSF biomarkers have excellent sensitivity and specificity for AD and have been compared across countries, which identified variation across laboratories and led to efforts to standardize and harmonize these assays178. These biomarkers have also been used in genetic studies in several highly resourced countries in North America, Europe and Asia (Box 2). Interestingly, APOE ε4 was associated with increased brain Aβ deposition as measured using 11C-labelled Pittsburgh compound B and 18F-florbetapir PET in two relatively small studies in the USA and China179,180. In the study in the USA, SNPs from 20 genes previously associated with AD were analysed and Aβ deposition was found to be associated with variants in ABCA7 and FERMT2. In the largest of these studies to date, RBFOX1, a neuronal RNA binding protein, was found to be associated with brain amyloidosis in a cohort of >4,000 individuals in the USA181.
Box 2. Biomarkers for Alzheimer disease.
Brain MRI
Alzheimer disease (AD) is accompanied by neurodegenerative changes that are detectable with structural MRI years before the clinical diagnosis216. Neurodegeneration presents as patterns of cortical thinning217 and focal atrophy in the medial temporal lobe218. Structural MRI captures cerebral comorbidities in AD, including ischaemic lesions and haemorrhagic lesions.
Amyloid and tau PET imaging
PET is another neuroimaging tool, and the development of molecularly based amyloid and tau tracers has greatly changed the ability of PET imaging to enhance the diagnosis of AD. The need for obtaining and storing radioactive biomarkers, the expense of acquiring the images and the feasibility of imaging large numbers of individuals have prevented the use of PET in individuals with AD in routine clinical settings.
Cerebrospinal fluid biomarkers
Biomarkers in the cerebrospinal fluid (CSF) including amyloid-β (Aβ1–42), total tau (t-tau), and tau phosphorylated at threonine 181 or 217 (p-tau) reflect the underlying pathophysiology of AD and can aid in the differential diagnosis. A recent worldwide multicentre harmonization project generated consensus interpretations of CSF biomarkers in AD219. Some evidence suggests that CSF biomarkers, particularly measures of t-tau and p-tau, can differ between individuals of African ancestry and non-Hispanic white individuals220,221.
Blood-based biomarkers
Technological advances have now made it possible to measure AD biomarkers in plasma222 (see the figure) — single-molecule array-based assays are now available for Aβ1–42, p-tau181 and p-tau217 (ref. 223). Plasma concentrations of neurofilament light chain (NfL), a marker of neuronal injury, are increased in AD and provide a sensitive biomarker for neurodegeneration224,225. Plasma NfL concentrations are associated with neurofibrillary tangle pathology and neurodegeneration measured at autopsy226.
Research into AD biomarkers across racial and ethnic groups has been challenged by small sample sizes, difficulty recruiting participants and selection biases. More work and greater collaboration are required on a global scale.
Most promising blood-based biomarkers for AD by disease stage.
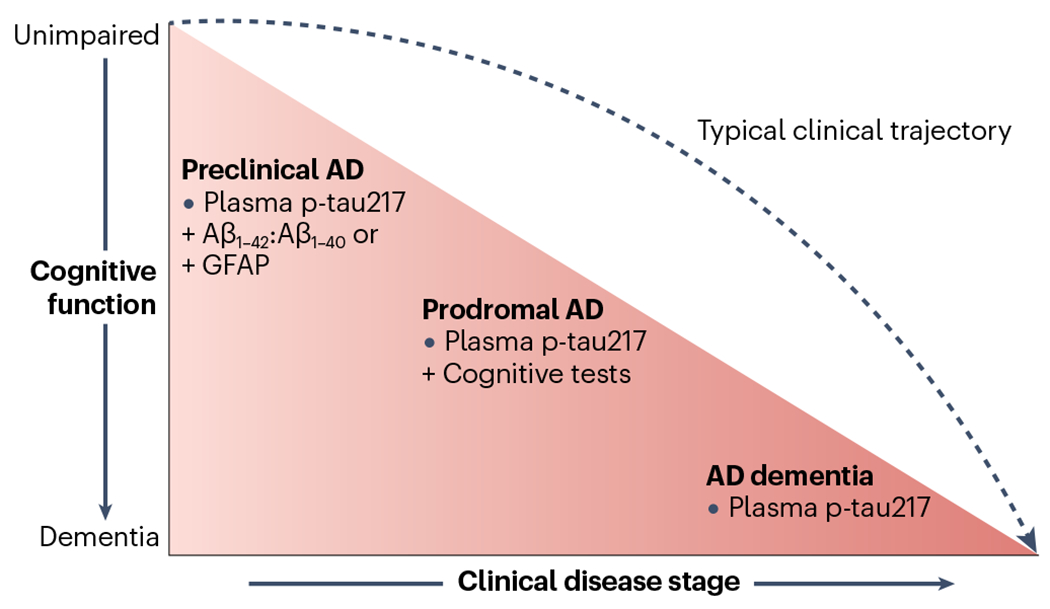
Adapted from ref. 222, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
Several groups have investigated genetic associations with CSF biomarker levels in individuals with AD. CSF concentrations of Aβ1–42, t-tau, and p-tau181 were associated with four variants in the APOE region182, and CSF p-tau concentrations were higher in participants carrying APOE ε4 than in those with other APOE alleles182. In studies with larger sample sizes, additional loci associated with the same CSF biomarkers were uncovered on 3q28 and 6p21.1, near the TREM2 gene cluster183 and 1p32.2 and 6p25 were found to be associated with AD risk, progression and age at onset184. In another study, variants in the MS4A4A gene were associated with increased CSF concentrations of TREM2 and reduced AD risk, indicating that the MS4A gene cluster is a key modulator of soluble TREM2 and AD risk185. Variants in AMTS1, TMEM106B and CPOX have been associated with CSF levels of neurofilament light chain (NfL) in individuals with AD34,186. In a small Chinese cohort, variants in several genes known to be associated with AD were found to also be associated with CSF levels of Aβ1–42, Aβ1–42 to Aβ1–40 ratio, t-tau and p-tau181 (ref. 187).
In addition to the detection of neurodegeneration as part of the A/T/N framework, structural imaging using MRI and diffusion tensor imaging can detect other changes in the brain (such as white matter hyperintensities) that are associated with AD and cognitive dysfunction, but lack sufficient sensitivity or specificity to be considered as biomarkers. There are several studies indicating that these findings can be genetically determined188,189. APOE ε4 and several single nucleotide variants in additional genes previously associated with AD in large genome-wide array studies have been found to be associated with structural changes on MRI190–192. In a Korean cohort, a missense variant in SHARPIN was found to be associated with measures of atrophy as manifested by loss of hippocampal volume and entorhinal cortical thickness, especially among individuals with AD193. Another investigation combined genome-wide array data, information on structural changes from MRI and cognitive measures in individuals with AD, and identified novel loci in or near EHBP1, CEP112, SMOC2 and IL1RAPL1 that were associated with cognitive phenotypes190.
Blood-based biomarkers.
Evidence indicates that recently developed blood-based biomarkers194,195 can strengthen the diagnoses of AD, mild cognitive impairment and cognitive decline, and allow prediction of incident disease. Blood-based biomarkers provide an unparalleled opportunity to incorporate biomarker information in practice worldwide including low-resourced countries where brain imaging and CSF analysis might not be possible. In a genetic association study performed in the UK that included individuals with AD and cognitively healthy controls and was adjusted for age, sex, population stratification and case–control status, the presence of an APOE ε4 allele was associated with levels of all blood-based biomarkers, including Aβ1–40, Aβ1–42, glial fibrillary protein and NfL196 A GWAS from the Alzheimer’s Disease Neuroimaging Initiative found three variants in CD1A that were associated with blood NfL levels in individuals without dementia (mean age 72.9 years)197. In individuals with AD and healthy controls from China, total AD polygenic risk score was associated with plasma concentrations of Aβ1–42, t-tau and NfL, and Aβ1–42 to Aβ1–40 ratio198. Gene-based analyses of the data in this study indicated that ABCA7 and UNC5C, as well as APOE ε4, were the main contributors to the association.
A worldwide approach by the Alzheimer’s Disease Neuroimaging Initiative is a collaborative effort to investigate imaging and biofluid markers for AD across North America, Europe, Argentina, Australia, China,Japan, Korea, Mexico and Taiwan. Collaboration among these developed countries will improve precision in the diagnosis of AD and related disorders. However, biomarker-based studies of populations in low-income countries, which, according to the WHO, includes 58% of people in the world with dementia, remain scarce. Therefore, the inclusion of readily available, blood-based biomarkers in genetic studies would greatly augment phenotype characterization worldwide.
Functional genomics
Investigators have initiated functional studies that use approaches such as epigenomics, transcriptomics, proteomics and metabolomics to better understand the genetic bases of familial and sporadic AD and related disorders (Box 3). Integrated multiomics approaches can be driven by the phenotype, the environment or the genome. The phenotype approach uses multiomics layers to compare individuals with a disease with healthy controls and can define more homogeneous subgroups than clinical diagnosis alone. These subgroups can have unique genetic profiles. The environmental approach uses multiomics analyses to investigate mechanistic links to environmental or medical risk factors such as vascular disease, physical activity, smoking and sleep. The genome approach is a variant-centred or gene-centred integration of harmonized multiomics to determine causality and identify underlying mechanisms.
Box 3. Functional genomics.
Epigenetics
Epigenetics represents a dynamic molecular modification that influences gene expression and can be sensitive to genetic variation, environmental factors and disease state227. Altered DNA methylation is involved in Alzheimer disease (AD) and affects gene expression228. CpG-related single-nucleotide polymorphisms (CGS) alter the sequence of the primary target sites for DNA methylation229 and account for a substantial fraction of allele-specific methylation in the human genome 230,231. More than 80% of CGS have a regulatory role in DNA methylation 232. Epigenome-wide association studies can identify disease-associated methylomic variation.
Transcriptomics
Transcriptomics measures the amounts of transcripts (both mRNA and non-coding RNA (microRNAs, long non-coding RNAs and circular RNAs)) being made, can be used to identify novel genetic associations, and can explain associations between gene expression, disease and genetic variants. Transcriptome sequencing has been used in conjunction with whole-exome and whole-genome sequencing, and multiple types of RNA transcripts have been associated with AD in post-mortem brain 107,233.
Proteomics
Proteomics analyses involve the identification and quantification of proteins present in tissues and biological fluids. Proteins are effectors of biological function, and their levels are not only dependent on corresponding messenger RNA levels but also on host translational control and regulation. Second to genetic mapping, proteomic investigations can validate disease pathways and uncover novel protein networks and mechanisms, such as RNA splicing, development, immunity, membrane transport, lipid metabolism, synaptic function and mitochondrial activity.
Metabolomics
Metabolite levels can be associated with disease or with related underlying biological mechanisms 234, and are influenced by the environment, medications, diet, alcohol and tobacco use, sex235, ethnic group 236–239 and genetic variation 238,240. A comprehensive assessment of exogenous, endogenous and microorganism-derived metabolites can reflect both the environmental exposures and the biological response241. Plasma is a readily available source for metabolomics analysis, and studies using this approach can augment the functional assessment of genetic variants in AD.
Investigating AD-associated genetic variants as quantitative trait loci (QTL) for epigenomic, transcriptomic, proteomic and metabolomic variables can be used to explore how variants in genes perturb pathways leading to AD. Multiomics approaches can also be used to validate variants of uncertain significance. Additionally, associations between genetic variants and exogenous metabolites allow assessment of gene–environment interactions in AD. Non-coding RNAs (ncRNAs) – including long ncRNAs, circular RNAs, small non-coding microRNAs and natural antisense transcripts – have been shown to regulate signalling pathways involved in AD (such as apoptosis, mitochondrial dysfunction and neurotrophic factor depletion in neurons and microglia)199. The role of ncRNAs can be investigated with transcriptomic and epigenetic approaches but to date the specific set of ncRNAs involved in AD has yet to be identified.
The genes that have been consistently associated with AD have shaped approaches to elucidating the causes of AD, and might yet reveal potential targets for therapeutic development; however, the existing multiomics data have some limitations. First, despite the identification of many AD-associated loci, the downstream effects of only a fraction have been studied. Second, the possibility that AD might result from changes in the regulation of gene expression by ncRNAs has not been fully explored200,201. Third, many previous multiomics studies have focused on post-mortem tissues, which generally represent the end stage of AD, so the identified associations could reflect disease progression and not disease risk. Fourth, large multiomics datasets have been collected from non-Hispanic white individuals but few such datasets exist for other ethnic and racial groups, hindering comparisons.
Multiomics approaches offer an opportunity to understand the ‘flow of information’ that underlies disease (Fig. 6). AD is by its very nature a non-linear, genetically driven, pathophysiological disease with high heterogeneity in biological alterations202. Failure of multilevel biological systems underlying AD includes proteostasis (Aβ and tau), synaptic homeostasis, inflammatory and immune responses, lipid and energy metabolism, and oxidative stress. The various ‘omics’ do not exist in isolation, but are part of a poorly understood and highly complex and interdependent system203,204. Therefore, a systems-level understanding of AD, with generation of robust multiomics datasets across diverse ancestries and harmonization of the results to include associated genes, is needed to understand underlying disease mechanisms and to inform our understanding of the biological continuum of this disease.
Fig. 6 |. An integrated multiomics approach.
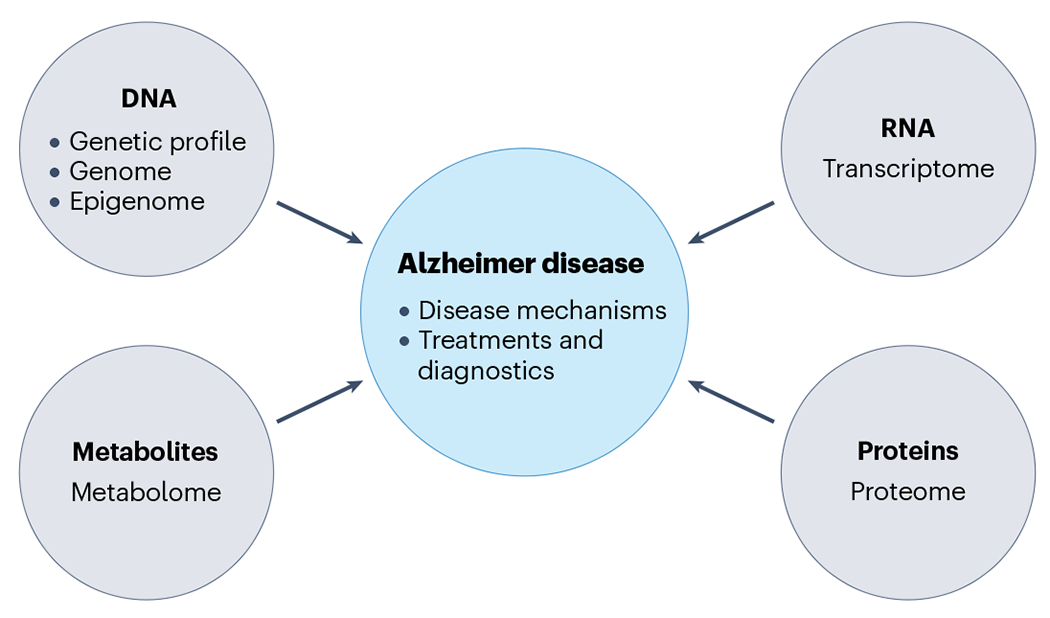
Integrated multiomics approaches can be used to understand how genetic variation leads to disease, to establish which molecular pathways are altered, and to identify new therapeutic targets and diagnostic approaches. Expression quantitative trait loci (eQTLs) would be generated from each omics layer (gene expression from the transcriptome, protein quantitative traits from the proteome, methylation quantitative traits from the metabolome). These multiomics layers would then be integrated into a systems analysis that includes information on the genome and epigenome, with the goal of furthering understanding of disease mechanisms and developing novel treatments and diagnostics.
Conclusions
If we are to fully understand the genetic influences on the common biology of AD, it is crucial that we identify the broadest range of genetic variations that influence AD. Expansion of ongoing efforts to sequence and analyse the genomes in under-sampled areas of the world, ideally combined with acquisition of additional multiomics data and appropriate development of infrastructure, resources, training and ethical guidelines to support this research, will be essential to improve our understanding of global genetic variation profiles and disease. To achieve this aim, a wide range of barriers to research participation will need to be addressed to improve inclusion of under-represented population groups. It will also be essential to address the shortage of biomedical research infrastructure and expertise, expand support for the integration of international collaborative efforts and increase community engagement to broaden the cultural acceptability of biobanking and genomic research. A WGS study of individuals from over 50 ethnolinguistic groups by the Human Health and Heredity in Africa (H3Africa) consortium that was published in 2020 aimed to further characterize African genomic diversity (that is, the region with the greatest level of human genetic variation)205. This effort identified more than three million previously undescribed genetic variants, and refined our understanding of patterns of ancestral admixture, continental migration, gene flow and the response to human disease205, underscoring the scientific imperative for a broader characterization of the global genomic diversity to develop treatments that will be effective for everyone.
To improve precision in genetic studies and the diagnosis of AD in clinical and research settings, it will be crucial to obtain and validate biomarker information in low-income countries across the world. Standardizing and harmonizing a range of phenotypes using imaging and fluid biomarkers would improve the phenotype–genotype analyses of populations across the world. Ultimately, such an endeavour would allow investigators to develop a precision medicine approach, which might differ across ancestral groups. The limited availability of PET facilities and laboratories equipped to analyse biomarkers in some countries underscores the urgent need to develop improved biomarkers that are inexpensive and readily available to all.
Key points.
The genetic variation underlying Alzheimer disease (AD) differs across ethnic groups.
Large-scale genomic studies have identified over 70 genes or genetic loci associated with AD risk, but these data have largely been obtained from populations in Europe and North America, which hinders our understanding of the molecular mechanism(s) underlying the disease in under-represented populations and the development of a personalized therapeutic approach.
Expansion of efforts to sequence and analyse the genomes of people from under-studied areas of the world, combined with acquisition of additional multiomics data and appropriate development of infrastructure, resources, training and ethical guidelines, will be essential to improve our understanding of global genetic variation profiles underlying dementia.
Pathological heterogeneity is the norm in AD and efforts to incorporate this information into genetic studies is underway.
Incorporation of improved biomarkers that can be obtained in low-resource countries will be critical to increase diagnostic accuracy in these efforts.
Acknowledgements
The authors acknowledge support from the National Institute on Aging of the National Institutes of Health in the USA. C.R. (U24AG056270, P30AG066462, U19AG074865, RO1AG064614); M.A.P.-V. (R56AG072547, RO1AG070864, UO1AG057659, UO1AG062943, UO1AG076482, U19AG074865); T.F. (U24AG021886, P30AG072976, U24AG056270), R.M. (U24AG056270, R01AG072474, RF1AG066107, R01AG067501). The authors also thank R. Akinyemi for his help with the review of genetics in Africa and M. Miller for continued encouragement and support of the investigation of AD worldwide.
Glossary
- Kindred
An aggregate of genetically related individuals.
- Principal components
Principal components analysis is a statistical method commonly used in population genetics to identify substructure in the distribution of genetic variation within populations.
- Quantitative trait loci
Regions of DNA each associated with a particular quantitative phenotypic trait.
- Recombination
Genetic recombination is the exchange of genetic material between different individuals which leads to offspring with combinations of traits that differ from those in either parent.
- Variants of uncertain significance
Genetic variants for which association with a specific trait is unclear.
Related links
Alzheimer’s Disease Neuroimaging Initiative: https://adni.loni.usc.edu/
Alzheimer’s Disease Sequencing Project-Follow-Up Study (ADSP-FUS): https://adsp.niagads.org/
Alzheimer’s Disease Sequencing Project-Follow-Up Study: https://www.nia.nih.gov/research/ad-genetics
Asian Cohort for Alzheimer’s Disease: https://acadstudy.org/
Estudio Familiar de Influencia Genetica en Alzheimer (Puerto Rican Alzheimer Disease Initiative; EFIGA): https://dss.niagads.org/cohorts/estudio-familiar-de-influencia-genetica-en-alzheimer-efiga/
Gwangju Alzheimer’s and Related Dementias (GARD) Study: https://dss.niagads.org/cohorts/gwangju-alzheimers-and-related-dementia-gard/
Longitudinal Aging Study in India: https://lasi-india.org/
Mexican Health and Aging Study: https://www.mhasweb.org/Home/index.aspx
Research in African American Alzheimer’s Disease Initiative (REAAADI): https://med.miami.edu/centers-and-institutes/hihg/research-programs/alzheimers-disease-and-related-dementias/research-in-african-american-alzheimer-disease-initiative
WHO dementia fact sheet: https://www.who.int/news-room/fact-sheets/detail/dementia
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 18, 700–789 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Gatz M et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Chartier-Harlin MC et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 353, 844–846 (1991). [DOI] [PubMed] [Google Scholar]
- 4.Goate A et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Sherrington R et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R et al. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum. Mol. Genet 5, 985–988 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Cacace R, Sleegers K & Van Broeckhoven C Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Jarmolowicz AI, Chen HY & Panegyres PK The patterns of inheritance in early-onset dementia: Alzheimer’s disease and frontotemporal dementia. Am. J. Alzheimers Dis. Other Demen 30, 299–306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallon D et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J. Alzheimers Dis 30, 847–856 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Cruchaga C et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One 7, e31039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arboleda-Velasquez JF et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med 25, 1680–1683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pericak-Vance MA et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am. J. Hum. Genet 48, 1034–1050 (1991). [PMC free article] [PubMed] [Google Scholar]
- 13.Wingo TS, Lah JJ, Levey AI & Cutler DJ Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol 69, 59–64 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wightman DP et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet 53, 1276–1282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellenguez C et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet 54, 412–436 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen IE et al. Genome-wide meta-analysis for Alzheimer’s disease cerebrospinal fluid biomarkers. Acta Neuropathol. 144, 821–842 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubinski A et al. Polygenic effect on tau pathology progression in Alzheimer’s disease. Ann. Neurol 10.1002/ana.26588 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Vigilant L, Stoneking M, Harpending H, Hawkes K & Wilson AC African populations and the evolution of human mitochondrial DNA. Science 253, 1503–1507 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Schlebusch CM et al. Genomic variation in seven Khoe-San groups reveals adaptation and complex African history. Science 338, 374–379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gronau I, Hubisz MJ, Gulko B, Danko CG & Siepel A Bayesian inference of ancient human demography from individual genome sequences. Nat. Genet 43, 1031–1034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasmussen M et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463, 757–762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green RE et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramachandran S et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc. Natl Acad. Sci. USA 102, 15942–15947 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakobsson M et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451, 998–1003 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Li JZ et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319, 1100–1104 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg NA et al. Genetic structure of human populations. Science 298, 2381–2385 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Gurdasani D et al. The African Genome Variation Project shapes medical genetics in Africa. Nature 517, 327–332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beltrame MH, Rubel MA & Tishkoff SA Inferences of African evolutionary history from genomic data. Curr. Opin. Genet. Dev 41, 159–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tishkoff SA et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duncan L et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun 10, 3328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rotimi CN & Adeyemo AA From one human genome to a complex tapestry of ancestry. Nature 590, 220–221 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Hong S et al. TMEM106B and CPOX are genetic determinants of cerebrospinal fluid Alzheimer’s disease biomarker levels. Alzheimers Dement. 17, 1628–1640 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Rotimi CN et al. The genomic landscape of African populations in health and disease. Hum. Mol. Genet 26, R225–R236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunkle BW et al. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol. 78, 102–113 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hohman TJ et al. Global and local ancestry in African-Americans: implications for Alzheimer’s disease risk. Alzheimers Dement. 12, 233–243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reitz C et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ε4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bussies PL et al. Use of local genetic ancestry to assess TOMM40-523’ and risk for Alzheimer disease. Neurol. Genet 6, e404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vardarajan BN et al. Whole genome sequencing of Caribbean Hispanic families with late-onset Alzheimer’s disease. Ann. Clin. Transl. Neurol 5, 406–417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vardarajan BN et al. Ultra-rare mutations in SRCAP segregate in Caribbean Hispanic families with Alzheimer disease. Neurol. Genet 3, e178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feliciano-Astacio BE et al. The Puerto Rico Alzheimer Disease Initiative (PRADI): a multisource ascertainment approach. Front. Genet 10, 538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tosto G et al. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann. Clin. Transl. Neurol 2, 810–820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tosto G et al. Association of variants in PINX1 and TREM2 with late-onset Alzheimer disease. JAMA Neurol. 76, 942–948 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farrer LA et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 46.Maestre G et al. Apolipoprotein E and Alzheimer’s disease: ethnic variation in genotypic risks. Ann. Neurol 37, 254–259 (1995). [DOI] [PubMed] [Google Scholar]
- 47.Tang MX et al. The APOE-ε4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA 279, 751–755 (1998). [DOI] [PubMed] [Google Scholar]
- 48.Jonsson T et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368, 107–116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raghavan NS et al. Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer’s disease. Ann. Clin. Transl. Neurol 5, 832–842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griswold AJ et al. Increased APOE ε4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement. 17, 1179–1188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jun GR et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 13, 727–738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cukier HN et al. ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol. Genet 2, e79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bryc K et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl Acad. Sci. USA 107, 786–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kunkle BW et al. Targeted sequencing of ABCA7 identifies splicing, stop-gain and intronic risk variants for Alzheimer disease. Neurosci. Lett 649, 124–129 (2017). [DOI] [PubMed] [Google Scholar]
- 55.De Roeck A et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease. Acta Neuropathol. 134, 475–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cuyvers E et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol. 14, 814–822 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Luukkainen L et al. Mutation analysis of the genes Linked to early onset Alzheimer’s disease and frontotemporal Lobar degeneration. J. Alzheimers Dis 69, 775–782 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Thordardottir S & Graff C Findings from the Swedish study on familial Alzheimer’s disease including the APP Swedish double mutation. J. Alzheimers Dis 64, S491–S496 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Al-Thani HF, Ahmad MN, Younes S & Zayed H Genetic variants associated with Alzheimer disease in the 22 Arab countries: a systematic review. Alzheimer Dis. Assoc. Disord 35, 178–186 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Eryilmaz IE et al. Evaluation of the clinical features accompanied by the gene mutations: the 2 novel PSEN1 variants in a Turkish early-onset Alzheimer disease cohort. Alzheimer Dis. Assoc. Disord 35, 214–222 (2021). [DOI] [PubMed] [Google Scholar]
- 61.Lee JH et al. Disease-related mutations among Caribbean Hispanics with familial dementia. Mol. Genet. Genom. Med 2, 430–437 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Athan ES et al. A founder mutation in presenilin 1 causing early-onset Alzheimer disease in unrelated Caribbean Hispanic families. JAMA 286, 2257–2263 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Bertoli Avella AM et al. A novel presenilin 1 mutation (L174 M) in a Large Cuban family with early onset Alzheimer disease. Neurogenetics 4, 97–104 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Arnold SE et al. Frequency and clinicopathological characteristics of presenilin 1 Gly206Ala mutation in Puerto Rican Hispanics with dementia. J. Alzheimers Dis 33, 1089–1095 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramos C, Aguillon D, Cordano C & Lopera F Genetics of dementia: insights from Latin America. Dement. Neuropsychol 14, 223–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Itzcovich T et al. A novel mutation in PSEN1 (p.T119I) in an Argentine family with early- and late-onset Alzheimer’s disease. Neurobiol. Aging 85, 155.e9–155.e12 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Abdala BB et al. Influence of low frequency PSEN1 variants on familial Alzheimer’s disease risk in Brazil. Neurosci. Lett 653, 341–345 (2017). [DOI] [PubMed] [Google Scholar]
- 68.El Kadmiri N et al. Novel mutations in the amyloid precursor protein gene within Moroccan patients with Alzheimer’s disease. J. Mol. Neurosci 53, 189–195 (2014). [DOI] [PubMed] [Google Scholar]
- 69.El Kadmiri N et al. Novel presenilin mutations within Moroccan patients with early-onset Alzheimer’s disease. Neuroscience 269, 215–222 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Heckmann JM et al. Novel presenilin 1 mutation with profound neurofibrillary pathology in an indigenous Southern African family with early-onset Alzheimer’s disease. Brain 127, 133–142 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Guerreiro RJ et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging 31, 725–731 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joshi P, Gardner M, Lintott C & Anderson T Novel presenilin-1 mutation (Ala275Ser) associated with clinical features of dementia with Lewy bodies. Alzheimer Dis. Assoc. Disord 35, 350–352 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Bechara JA, Brooks WS, Kwok JB, Halliday GM & Schofield PR Familial Alzheimer’s disease in Australia. Alzheimer’s Dement. 16, e040062 (2020). [Google Scholar]
- 74.Syama A et al. Mutation burden profile in familial Alzheimer’s disease cases from India. Neurobiol. Aging 64, 158.e7–158.e13 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Li H et al. The identification of PSEN1 p.Tyr159Ser mutation in a non-canonic early-onset Alzheimer’s disease family. Mol. Cell Neurosci 120, 103715 (2022). [DOI] [PubMed] [Google Scholar]
- 76.Qin Q et al. Gene mutations associated with early onset familial Alzheimer’s disease in China: an overview and current status. Mol. Genet. Genom. Med 8, e1443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bagyinszky E, Youn YC, An SS & Kim S Mutations, associated with early-onset Alzheimer’s disease, discovered in Asian countries. Clin. Interv. Aging 11, 1467–1488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ikeuchi T et al. Mutational analysis in early-onset familial dementia in the Japanese population. The role of PSEN1 and MAPT R406W mutations. Dement. Geriatr. Cogn. Disord 26, 43–49 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Hsu JL, Lin CH, Chen PL, Lin KJ & Chen TF Genetic study of young-onset dementia using targeted gene panel sequencing in Taiwan. Am. J. Med. Genet. B Neuropsychiatr. Genet 186, 67–76 (2021). [DOI] [PubMed] [Google Scholar]
- 80.Bagyinszky E, Ch’ng GS, Chan MY, An SSA & Kim S A pathogenic presenilin-1 Val96Phe mutation from a Malaysian family. Brain Sci. 11, 1328 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alzheimer’s Disease International. Dementia in Sub-Saharan Africa: Challenges and Opportunities (Alzheimer’s Disease International, 2017). [Google Scholar]
- 82.Landoulsi Z et al. Genetic analysis of TREM2 variants in Tunisian patients with Alzheimer’s disease. Med. Princ. Pract 27, 317–322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hendrie HC et al. APOE ε4 and the risk for Alzheimer disease and cognitive decline in African Americans and Yoruba. Int. Psychogeriatr 26, 977–985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hall K et al. Cholesterol, APOE genotype, and Alzheimer disease: an epidemiologic study of Nigerian Yoruba. Neurology 66, 223–227 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen CH et al. A comparative study to screen dementia and APOE genotypes in an ageing East African population. Neurobiol. Aging 31, 732–740 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gureje O et al. APOE ε4 is not associated with Alzheimer’s disease in elderly Nigerians. Ann. Neurol 59, 182–185 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haithem H et al. Association between dementia and vascular disease-associated polymorphisms in a Tunisian population. Int. J. Neurosci 128, 32–41 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in Late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 89.Mayeux R et al. The apolipoprotein epsilon 4 allele in patients with Alzheimer’s disease. Ann. Neurol 34, 752–754 (1993). [DOI] [PubMed] [Google Scholar]
- 90.McKhann G et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services task force on Alzheimer’s disease. Neurology 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- 91.World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines (WHO, 1992). [Google Scholar]
- 92.Blue EE, Horimoto A, Mukherjee S, Wijsman EM & Thornton TA Local ancestry at APOE modifies Alzheimer’s disease risk in Caribbean Hispanics. Alzheimers Dement. 15, 1524–1532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rajabli F et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet. 14, e1007791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rajabli F et al. A locus at 19q13.31 significantly reduces the ApoE ε4 risk for Alzheimer’s disease in African ancestry. PLoS Genet. 18, e1009977 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hendrie HC et al. Incidence of dementia and Alzheimer disease in 2 communities: Yoruba residing in Ibadan, Nigeria, and African Americans residing in Indianapolis, Indiana. JAMA 285, 739–747 (2001). [DOI] [PubMed] [Google Scholar]
- 96.Adhikari K, Chacon-Duque JC, Mendoza-Revilla J, Fuentes-Guajardo M & Ruiz-Linares A The genetic diversity of the Americas. Annu. Rev. Genomics Hum. Genet 18, 277–296 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Adhikari K, Mendoza-Revilla J, Chacon-Duque JC, Fuentes-Guajardo M & Ruiz-Linares A Admixture in Latin America. Curr. Opin. Genet. Dev 41, 106–114 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Salzano FM & Bortolini MC The Evolution and Genetics of Latin American Populations (Cambridge Univ. Press, 2001). [Google Scholar]
- 99.Prince M et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63–75.e2 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Ferri CP et al. Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ramirez Aguilar L et al. Genetic origin of a large family with a novel PSEN1 mutation (Ile416Thr). Alzheimers Dement. 15, 709–719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lopera F et al. Clinical features of early-onset Alzheimer disease in a Large kindred with an E280A presenilin-1 mutation. JAMA 277, 793–799 (1997). [PubMed] [Google Scholar]
- 103.Tariot PN et al. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: a study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. 4, 150–160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guerreiro R et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368, 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vardarajan BN et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann. Neurol 78, 487–498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vardarajan BN et al. Inbreeding among Caribbean Hispanics from the Dominican Republic and its effects on risk of Alzheimer disease. Genet. Med 17, 639–643 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.An F et al. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 8, 114065–114071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 108.Sims R et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet 49, 1373–1384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kizil C et al. Admixture mapping of Alzheimer’s disease in Caribbean Hispanics identifies a new locus on 22q13.1. Mol. Psychiatry 27, 2813–2820 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Toral-Rios D et al. SORL1 polymorphisms in Mexican patients with Alzheimer’s disease. Genes 13, 587 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jia L et al. Dementia in China: epidemiology, clinical management, and research advances. Lancet Neurol. 19, 81–92 (2020). [DOI] [PubMed] [Google Scholar]
- 112.Gan CL, Zhang T & Lee TH The genetics of Alzheimer’s disease in the Chinese population. Int. J. Mol. Sci 21, 2381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miyashita A, Kikuchi M, Hara N & Ikeuchi T Genetics of Alzheimer’s disease: an East Asian perspective. J. Hum. Genet 68, 115–124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jiang T et al. A rare coding variant in TREM2 increases risk for Alzheimer’s disease in Han Chinese. Neurobiol. Aging 42, 217 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Jiang T et al. TREM2 p.H157Y variant and the risk of Alzheimer’s disease: a meta-analysis involving 14,510 subjects. Curr. Neurovasc Res 13, 318–320 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Hirano A et al. A genome-wide association study of late-onset Alzheimer’s disease in a Japanese population. Psychiatr. Genet 25, 139–146 (2015). [DOI] [PubMed] [Google Scholar]
- 117.Jia L et al. Prediction of Alzheimer’s disease using multi-variants from a Chinese genome-wide association study. Brain 144, 924–937 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kang S et al. Potential novel genes for late-onset Alzheimer’s disease in East-Asian descent identified by APOE-stratified genome-wide association study. J. Alzheimers Dis 82, 1451–1460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Park JH et al. Novel Alzheimer’s disease risk variants identified based on whole-genome sequencing of APOE ε4 carriers. Transl. Psychiatry 11, 296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shigemizu D et al. Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl. Psychiatry 11, 151 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou X et al. Identification of genetic risk factors in the Chinese population implicates a role of immune system in Alzheimer’s disease pathogenesis. Proc. Natl Acad. Sci. USA 115, 1697–1706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang Y, Lu D, Chung YJ & Xu S Genetic structure, divergence and admixture of Han Chinese, Japanese and Korean populations. Hereditas 155, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dogan M et al. Clinical and molecular findings in a Turkish family who had a (c.869-1G>A) splicing variant in PSEN1 gene with a rare condition: the variant Alzheimer’s disease with spastic paraparesis. Curr. Alzheimer Res 19, 223–235 (2022). [DOI] [PubMed] [Google Scholar]
- 124.El Bitar F et al. Genetic study of Alzheimer’s disease in Saudi population. J. Alzheimers Dis 67, 231–242 (2019). [DOI] [PubMed] [Google Scholar]
- 125.Guven G et al. A novel PSEN2 p.Ser175Phe variant in a family with Alzheimer’s disease. Neurol. Sci 42, 2497–2504 (2021). [DOI] [PubMed] [Google Scholar]
- 126.Kalfon L et al. Familial early-onset Alzheimer’s caused by novel genetic variant and APP duplication: a cross-sectional study. Curr. Alzheimer Res 19, 694–707 (2022). [DOI] [PubMed] [Google Scholar]
- 127.Lohmann E et al. Identification of PSEN1 and PSEN2 gene mutations and variants in Turkish dementia patients. Neurobiol. Aging 33, 1850.e17–1850.e27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bazrgar M et al. Apolipoprotein E polymorphism in Southern Iran: E4 allele in the lowest reported amounts. Mol. Biol. Rep 35, 495–499 (2008). [DOI] [PubMed] [Google Scholar]
- 129.Abdi S et al. Association of Alzheimer’s disease with genetic variants of apolipoprotein E, clusterin, TNF-α, and IL-6 among elderly Saudis. Curr. Pharm. Biotechnol 23, 1893–1902 (2022). [DOI] [PubMed] [Google Scholar]
- 130.Abyadeh M, Djafarian K, Heydarinejad F, Alizadeh S & Shab-Bidar S Association between apolipoprotein E gene polymorphism and Alzheimer’s disease in an Iranian population: a meta-analysis. J. Mol. Neurosci 69, 557–562 (2019). [DOI] [PubMed] [Google Scholar]
- 131.Amer MS et al. Interaction between apolipoprotein E genotyping, serum inflammatory biomarkers and cognitive functions in Egyptian elderly. Egypt. J. Immunol 28, 1–11 (2021). [PubMed] [Google Scholar]
- 132.Durmaz A et al. Genetic factors associated with the predisposition to late onset Alzheimer’s disease. Gene 707, 212–215 (2019). [DOI] [PubMed] [Google Scholar]
- 133.El Shamieh S, Costanian C, Kassir R, Visvkis-Siest S & Bissar-Tadmouri N APOE genotypes in Lebanon: distribution and association with hypercholesterolemia and Alzheimer’s disease. Per Med. 16, 15–23 (2019). [DOI] [PubMed] [Google Scholar]
- 134.Isbir T et al. Interaction between apolipoprotein-E and angiotensin-converting enzyme genotype in Alzheimer’s disease. Am. J. Alzheimers Dis. Other Demen 16, 205–210 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gharesouran J, Rezazadeh M, Khorrami A, Ghojazadeh M & Talebi M Genetic evidence for the involvement of variants at APOE, BIN1, CR1, and PICALM loci in risk of late-onset Alzheimer’s disease and evaluation for interactions with APOE genotypes. J. Mol. Neurosci 54, 780–786 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Kleinberger G et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med 6, 243ra286 (2014). [DOI] [PubMed] [Google Scholar]
- 137.Manzali SB et al. Association of the CD2AP locus with cognitive functioning among middle-aged individuals with a family history of Alzheimer’s disease. Neurobiol. Aging 101, 50–56 (2021). [DOI] [PubMed] [Google Scholar]
- 138.Mehdizadeh E et al. Association of MS4A6A, CD33, and TREM2 gene polymorphisms with the late-onset Alzheimer’s disease. Bioimpacts 9, 219–225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mehrjoo Z et al. Association study of the TREM2 gene and identification of a novel variant in exon 2 in Iranian patients with late-onset Alzheimer’s disease. Med. Princ. Pract 24, 351–354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rezazadeh M et al. Genetic factors affecting late-onset Alzheimer’s disease susceptibility. Neuromolecular Med. 18, 37–49 (2016). [DOI] [PubMed] [Google Scholar]
- 141.Talebi M et al. ABCA7 and EphA1 genes polymorphisms in late-onset Alzheimer’s disease. J. Mol. Neurosci 70, 167–173 (2020). [DOI] [PubMed] [Google Scholar]
- 142.Masri I, Salami A, El Shamieh S & Bissar-Tadmouri N rs3851179G>A in PICALM is protective against Alzheimer’s disease in five different countries surrounding the Mediterranean. Curr. Aging Sci 13, 162–168 (2020). [DOI] [PubMed] [Google Scholar]
- 143.Akinyemi RO et al. Dementia in Africa: current evidence, knowledge gaps, and future directions. Alzheimers Dement. 18, 790–809 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bis JC et al. Whole exome sequencing study identifies novel rare and common Alzheimer’s-associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 25, 1859–1875 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mena PR et al. The Alzheimer’s disease sequencing project-follow up study (ADSP-FUS): increasing ethnic diversity in Alzheimer’s genetics research with the addition of potential new cohorts. Alzheimer’s Dement. 16, e046400 (2020). [Google Scholar]
- 146.John P Hussman Institute to lead international genetic study of Alzheimer’s disease in people of Hispanic and African ancestry. Inventum; https://physician-news.umiamihealth.org/john-p-hussman-institute-to-lead-international-genetic-study-of-alzheimers-disease-in-people-of-hispanic-and-african-ancestry/ (2022). [Google Scholar]
- 147.Weiner MW et al. Increasing participant diversity in AD research: plans for digital screening, blood testing, and a community-engaged approach in the Alzheimer’s Disease Neuroimaging Initiative 4. Alzheimers Dement. 19, 307–317 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.McKhann GM et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ayodele T, Rogaeva E, Kurup JT, Beecham G & Reitz C Early-onset Alzheimer’s disease: what is missing in research? Curr. Neurol. Neurosci. Rep 21, 4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Reitz C, Rogaeva E & Beecham GW Late-onset vs nonmendelian early-onset Alzheimer disease: a distinction without a difference? Neurol. Genet 6, e512 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bateman RJ et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther 3, 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.van der Flier WM, Pijnenburg YA, Fox NC & Scheltens P Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ε4 allele. Lancet Neurol. 10, 280–288 (2011). [DOI] [PubMed] [Google Scholar]
- 153.Lee S et al. White matter hyperintensities are a core feature of Alzheimer’s disease: evidence from the dominantly inherited Alzheimer network. Ann. Neurol 79, 929–939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Moller C et al. Different patterns of gray matter atrophy in early- and late-onset Alzheimer’s disease. Neurobiol. Aging 34, 2014–2022 (2013). [DOI] [PubMed] [Google Scholar]
- 155.Marshall GA, Fairbanks LA, Tekin S, Vinters HV & Cummings JL Early-onset Alzheimer’s disease is associated with greater pathologic burden. J. Geriatr. Psychiatry Neurol 20, 29–33 (2007). [DOI] [PubMed] [Google Scholar]
- 156.Lowe VJ et al. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141, 271–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Scholl M et al. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 140, 2286–2294 (2017). [DOI] [PubMed] [Google Scholar]
- 158.Josephs KA et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: a longitudinal retrospective study. Lancet Neurol. 16, 917–924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Attems J & Jellinger KA The overlap between vascular disease and Alzheimer’s disease – lessons from pathology. BMC Med. 12, 206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kapasi A, DeCarli C & Schneider JA Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 134, 171–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schneider JA, Arvanitakis Z, Bang W & Bennett DA Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204 (2007). [DOI] [PubMed] [Google Scholar]
- 162.DeTure MA & Dickson DW The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 14, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schneider JA, Wilson RS, Bienias JL, Evans DA & Bennett DA Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62, 1148–1155 (2004). [DOI] [PubMed] [Google Scholar]
- 164.Barnes DE & Yaffe K The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 10, 819–828 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Barker WW et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord 16, 203–212 (2002). [DOI] [PubMed] [Google Scholar]
- 166.Galvin JE, Pollack J & Morris JC Clinical phenotype of Parkinson disease dementia. Neurology 67, 1605–1611 (2006). [DOI] [PubMed] [Google Scholar]
- 167.Lippa CF et al. Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol 153, 1365–1370 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chung EJ et al. Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol. 72, 789–796 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Azar M et al. Cognitive tests aid in clinical differentiation of Alzheimer’s disease versus Alzheimer’s disease with Lewy body disease: evidence from a pathological study. Alzheimers Dement. 16, 1173–1181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kraybill ML et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64, 2069–2073 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Forstl H, Burns A, Luthert P, Cairns N & Levy R The Lewy-body variant of Alzheimer’s disease. Clinical and pathological findings. Br. J. Psychiatry 162, 385–392 (1993). [DOI] [PubMed] [Google Scholar]
- 172.Merdes AR et al. Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 60, 1586–1590 (2003). [DOI] [PubMed] [Google Scholar]
- 173.Ferreira D et al. Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: longitudinal trajectories and clinical applications. Sci. Rep 7, 46263 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Moreno-Grau S et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: the GR@ACE project. Alzheimers Dement. 15, 1333–1347 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Lee AJ et al. FMNL2 regulates gliovascular interactions and is associated with vascular risk factors and cerebrovascular pathology in Alzheimer’s disease. Acta Neuropathol. 144, 59–79 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jack CR Jr. et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Jack CR Jr. et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Balogun WG, Zetterberg H, Blennow K & Karikari TK Plasma biomarkers for neurodegenerative disorders: ready for prime time? Curr. Opin. Psychiatry 36, 112–118 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Apostolova LG et al. Associations of the top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol. 75, 328–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shi Z et al. Amyloid PET in dementia syndromes: a Chinese multicenter study. J. Nucl. Med 61, 1814–1819 (2020). [DOI] [PubMed] [Google Scholar]
- 181.Raghavan NS et al. Association between common variants in RBFOX1, an RNA-binding protein, and brain amyloidosis in early and preclinical Alzheimer disease. JAMA Neurol. 77, 1288–1298 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim S et al. Genome-wide association study of CSF biomarkers Aβ1-42, t-tau, and p-ta181p in the ADNI cohort. Neurology 76, 69–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Babapour Mofrad R et al. Sex differences in CSF biomarkers vary by Alzheimer disease stage and APOE ε4 genotype. Neurology 95, e2378–e2388 (2020). [DOI] [PubMed] [Google Scholar]
- 184.Deming Y et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 133, 839–856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Deming Y et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med 11, eaau2291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hong S et al. Genome-wide association study of Alzheimer’s disease CSF biomarkers in the EMIF-AD multimodal biomarker discovery dataset. Transl. Psychiatry 10, 403 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li WW et al. Association of polygenic risk score with age at onset and cerebrospinal fluid biomarkers of Alzheimer’s disease in a Chinese cohort. Neurosci. Bull 36, 696–704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhao B et al. Common genetic variation influencing human white matter microstructure. Science 372, eabf3736 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhao B et al. Large-scale GWAS reveals genetic architecture of brain white matter microstructure and genetic overlap with cognitive and mental health traits (n = 17,706). Mol. Psychiatry 26, 3943–3955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Homann J et al. Genome-wide association study of Alzheimer’s disease brain imaging biomarkers and neuropsychological phenotypes in the European medical information framework for Alzheimer’s disease multimodal biomarker discovery dataset. Front. Aging Neurosci 14, 840651 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Li JQ et al. GWAS-linked loci and neuroimaging measures in Alzheimer’s disease. Mol. Neurobiol 54, 146–153 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Squillario M et al. A telescope GWAS analysis strategy, based on SNPs-genes-pathways ensamble and on multivariate algorithms, to characterize late onset Alzheimer’s disease. Sci. Rep 10, 12063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Park JY et al. A missense variant in SHARPIN mediates Alzheimer’s disease-specific brain damages. Transl. Psychiatry 11, 590 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Janelidze S et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med 26, 379–386 (2020). [DOI] [PubMed] [Google Scholar]
- 195.Thijssen EH et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med 26, 387–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Stevenson-Hoare J et al. Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer’s disease. Brain 10.1093/brain/awac128 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang ZT et al. Genome-wide association study identifies CD1A associated with rate of increase in plasma neurofilament light in non-demented elders. Aging 11, 4521–4535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jiao B et al. Associations of risk genes with onset age and plasma biomarkers of Alzheimer’s disease: a large case-control study in mainland China. Neuropsychopharmacology 47, 1121–1127 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kim J et al. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener 11, 55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Garcia-Aranda M, Serrano A & Redondo M Regulation of clusterin gene expression. Curr. Protein Pept. Sci 19, 612–622 (2018). [DOI] [PubMed] [Google Scholar]
- 201.Kumar P et al. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS One 8, e69807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hampel H et al. Omics sciences for systems biology in Alzheimer’s disease: state-of-the-art of the evidence. Ageing Res. Rev 69, 101346 (2021). [DOI] [PubMed] [Google Scholar]
- 203.Joyce AR & Palsson BO The model organism as a system: integrating ‘omics’ data sets. Nat. Rev. Mol. Cell Biol 7, 198–210 (2006). [DOI] [PubMed] [Google Scholar]
- 204.Ritchie MD, Holzinger ER, Li R, Pendergrass SA & Kim D Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet 16, 85–97 (2015). [DOI] [PubMed] [Google Scholar]
- 205.Choudhury A et al. High-depth African genomes inform human migration and health. Nature 586, 741–748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mez J et al. Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimers Dement. 13, 119–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Miyashita A et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS One 8, e58618 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Nielsen R et al. Tracing the peopling of the world through genomics. Nature 541, 302–310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tosto G et al. Polygenic risk scores in familial Alzheimer disease. Neurology 88, 1180–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.DeMichele-Sweet MAA et al. Genome-wide association identifies the first risk loci for psychosis in Alzheimer disease. Mol. Psychiatry 26, 5797–5811 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kunkle BW et al. Genome-wide linkage analyses of non-Hispanic white families identify novel loci for familial late-onset Alzheimer’s disease. Alzheimers Dement. 12, 2–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Barral S et al. Linkage analyses in Caribbean Hispanic families identify novel loci associated with familial late-onset Alzheimer’s disease. Alzheimers Dement. 11, 1397–1406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lanoiselee HM et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. 14, e1002270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Golan MP et al. Early-onset Alzheimer’s disease with a de novo mutation in the presenilin 1 gene. Exp. Neurol 208, 264–268 (2007). [DOI] [PubMed] [Google Scholar]
- 215.Kukull WA et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch. Neurol 59, 1737–1746 (2002). [DOI] [PubMed] [Google Scholar]
- 216.Frenzel S et al. A biomarker for Alzheimer’s disease based on patterns of regional brain atrophy. Front. Psychiatry 10, 953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dickerson BC et al. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb. Cortex 19, 497–510 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Killiany RJ et al. MRI measures of entorhinal cortex vs hippocampus in preclinical AD. Neurology 58, 1188–1196 (2002). [DOI] [PubMed] [Google Scholar]
- 219.Delaby C et al. Clinical reporting following the quantification of cerebrospinal fluid biomarkers in Alzheimer’s disease: an international overview. Alzheimers Dement. 18, 1868–1879 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Morris JC et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76, 264–273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chaudhry A & Rizig M Comparing fluid biomarkers of Alzheimer’s disease between African American or Black African and white groups: a systematic review and metaanalysis. J. Neurol. Sci 421, 117270 (2021). [DOI] [PubMed] [Google Scholar]
- 222.Leuzy A et al. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med 14, e14408 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zetterberg H et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS One 6, e28263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gaiottino J et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 8, e75091 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Khalil M et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol 14, 577–589 (2018). [DOI] [PubMed] [Google Scholar]
- 226.Ashton NJ et al. Increased plasma neurofilament light chain concentration correlates with severity of post-mortem neurofibrillary tangle pathology and neurodegeneration. Acta Neuropathol. Commun 7, 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Feinberg AP Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440 (2007). [DOI] [PubMed] [Google Scholar]
- 228.Fransquet PD & Ryan J Micro RNA as a potential blood-based epigenetic biomarker for Alzheimer’s disease. Clin. Biochem 58, 5–14 (2018). [DOI] [PubMed] [Google Scholar]
- 229.Lister R et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gertz J et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 7, e1002228 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Shoemaker R, Deng J, Wang W & Zhang K Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 20, 883–889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhi D et al. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 8, 802–806 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Dehghani R, Rahmani F & Rezaei N MicroRNA in Alzheimer’s disease revisited: implications for major neuropathological mechanisms. Rev. Neurosci 29, 161–182 (2018). [DOI] [PubMed] [Google Scholar]
- 234.Newgard CB Metabolomics and metabolic diseases: where do we stand? Cell Metab. 25, 43–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mittelstrass K et al. Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genet. 7, e1002215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Shah SH et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia 55, 321–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Shah SH, Kraus WE & Newgard CB Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation 126, 1110–1120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Vardarajan B et al. Differences in plasma metabolites related to Alzheimer’s disease, APOE epsilon4 status, and ethnicity. Alzheimers Dement. 6, e12025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wang TJ et al. Metabolite profiles and the risk of developing diabetes. Nat. Med 17, 448–453 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Heath L et al. Manifestations of Alzheimer’s disease genetic risk in the blood are evident in a multiomic analysis in healthy adults aged 18 to 90. Sci. Rep 12, 6117 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Walker DI et al. High-resolution metabolomics of occupational exposure to trichloroethylene. Int. J. Epidemiol 45, 1517–1527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]