

Abstract
Bromine radical (Br•) has been hypothesized to be a key intermediate of bromate formation during ozonation. Once formed, Br• further reacts with ozone to eventually form bromate. However, this reaction competes with the reaction of Br• with dissolved organic matter (DOM), of which reactivity and reaction mechanisms are less studied to date. To fill this gap, this study determined the second-order rate constant (k) of the reactions of selected organic model compounds, a DOM isolate, and monochloramine (NH2Cl) with Br• using γ-radiolysis. The kBr• of all model compounds were high (kBr• > 108 M–1 s–1) and well correlated with quantum-chemically computed free energies of activation, indicating a selectivity of Br• toward electron-rich compounds, governed by electron transfer. The reaction of phenol (a representative DOM moiety) with Br• yielded p-benzoquinone as a major product with a yield of 59% per consumed phenol, suggesting an electron transfer mechanism. Finally, the potential of NH2Cl to quench Br• was tested based on the fast reaction (kBr•, NH2Cl = 4.4 × 109 M–1 s–1, this study), resulting in reduced bromate formation of up to 77% during ozonation of bromide-containing lake water. Overall, our study demonstrated that Br• quenching by NH2Cl can substantially suppress bromate formation, especially in waters containing low DOC concentrations (1–2 mgC/L).
Keywords: bromine radical, bromate, dissolved organic matter, model compounds, reaction kinetics, ozone
Short abstract
The determined reactivity of bromine radicals with dissolved organic matter, surrogates, and monochloramine allowed for assessing bromate mitigation during aqueous ozonation.
1. Introduction
Bromide is ubiquitously present in fresh waters in 10–1000 μg/L1,2 and plays an important role in most oxidative water treatment processes.3 During oxidation processes, bromide is converted to reactive bromine species such as hypobromous acid (HOBr)4 and/or bromine(-containing) radicals (e.g., Br•, Br2•–, BrO•).5−7 HOBr reacts with dissolved organic matter (DOM) to produce potentially harmful brominated disinfection byproducts (Br-DBPs).8,9 Bromine radicals can influence micropollutant abatement5,10−12 and algal toxin degradation.13,14 A special feature of ozonation is the oxidation of bromide to bromate,15 which is a probable human carcinogen with a drinking water standard of 10 μg/L.16,17 Its formation mechanism during ozonation has received considerable research efforts for decades.18 It is formed by a complex interplay between ozone, hydroxyl radical (•OH), and various reactive bromine species,18 characterized by two initial pathways: oxidation of bromide (a) to hypobromite (OBr–) by ozone (eq 1)19,20 or (b) to Br• by •OH (eqs 2 and 3).21,22 The primary products, OBr–/HOBr and Br•, serve as key intermediates by subsequently reacting with ozone or •OH to ultimately form bromate.7,18
| 1 |
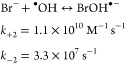 |
2 |
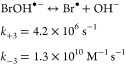 |
3 |
However, the subsequent reactions to bromate can be interrupted by DOM. HOBr reacts partially with residual electron-rich moieties of DOM, which remain after ozone attack, with second-order rate constants (k) ranging from 103 to 107 M–1 s–1.4 Likewise, Br• can react fast with DOM moieties with kBr• of 104 to 108 M–1 s–1.23−25 Recently, kBr• of standard DOM were measured in a range of (0.5–4.2) × 108 Mc–1 s–1.26 During ozonation, the Br• reaction with DOM is in competition to ozone with kBr•, O3 ≈ 1.5 × 108 M–1 s–1.7 A wide range of kBr• has been reported for micropollutants (108 – 1011 M–1 s–1),10 which implies selectivity of Br• to organic compounds. Nevertheless, a systematic investigation on the Br• reactivity with organic moieties including quantitative structure–activity relationship (QSAR) is lacking to date.
Among diverse bromate mitigation strategies, a sequential addition of chlorine and ammonia prior to ozonation has shown good performance.18 Direct addition of monochloramine (NH2Cl) prior to ozonation was also effective to reduce bromate,27,28 but questions remain about the underlying mechanism. NH2Cl was suggested to mitigate bromate by reacting with •OH, HOBr, or bromide.27,28 However, such reactions are either insufficient to account for a substantial mitigation29 or too slow.30
This study aims to understand the role of Br• in bromate formation during ozonation, especially with regard to its reactions with DOM and NH2Cl, with the specific objectives to (1) determine the kBr• of organic model compounds for DOM and establish QSAR, (2) identify products formed during the reaction of phenol (a representative DOM moiety) with Br• and elucidate the underlying reaction mechanisms, and (3) investigate the effect of quenching Br• by NH2Cl and DOM on bromate formation during ozonation of bromide-containing water.
2. Materials and Methods
2.1. Reagents
Details for chemicals and Lake Zurich water composition are provided in Text S1 (Supporting Information). Preparation of DOM and chloride-free NH2Cl stock solutions is described in Text S2 and Text S3, respectively.
2.2. γ-Radiolysis
γ-radiolysis was carried out by a 60Co γ-radiation source (Gammacell 220, Atomic Energy of Canada, Ltd.) with a dose rate of 0.13 kGy/h (yielding a Br• formation rate of 9.7 nM/s) determined by a dosimetry in a formate solution (Text S4).7 Br• was formed by the reaction of 1,2-dibromoethane with e–, according to eqs 4 and 5.23,25,31.32
| 4 |
| 5 |
Samples for determining kBr• of the organic model compounds were prepared as described in Text S5 and Figure S4. Briefly, mixed solutions containing 3.4 μM ibuprofen (as a competitor), 3.4 μM of a model compound, 0.7 mM 1,2-dibromoethane (for generating Br•), 4 mM t-butanol (for scavenging •OH), 50 mM buffer (phosphate for pH 7.1 or borate for pH 10.2), and ∼50 μM dissolved O2 (for scavenging C-centered radicals (Text S6)) were prepared and subjected to γ-radiolysis for 0–20 min. Before and during γ-radiolysis, silver nitrate solution was added for masking bromide which is formed from the reaction of 1,2-dibromomethane with solvated electrons (Text S7). The scavenging rates of the reactive species (e.g., e–, •OH, Br•) in the applied γ-radiolysis condition were estimated based on kinetic information, as shown in Table S10. After a predetermined time point, the sample was taken out from the γ-radiation source and sodium chloride was added to precipitate residual Ag+ as AgCl. Samples for identifying products for the reaction of phenol with Br• were prepared as described in Text S9. Solutions in these vials contained 22 μM phenol, 0.7 mM 1,2-dibromoethane, 40 mM t-butanol, and 50 mM phosphate buffer (pH 7.1).
2.3. Competition Kinetics
kBr• of organic model compounds were determined by competition kinetics with ibuprofen as a competitor based on eq 6 where M and C indicate an organic model compound and a competitor (ibuprofen), respectively. kBr• of ibuprofen was 3.8 × 109 M–1 s–1 (see section 3.1.1).
| 6 |
kBr• for DOM and NH2Cl was determined by measuring the change in ibuprofen concentration over γ-radiolysis time, by applying varying concentration ratios of ibuprofen (1 μM) and DOM (2–15 mgC/L) or NH2Cl (0.01–0.4 mM). Details to derive kBr• under this adapted method are provided in Text S8. All competition kinetics plots are provided in Figures S5 and S6.
2.4. Analyses
High-performance liquid chromatography coupled with a diode array or fluorescence detector (HPLC) and LC coupled with high-resolution tandem mass spectrometry (LC-HRMS/MS) were used for analyzing organic compounds. Reference standards were compared for identified products during the phenol-Br• reaction. For suspected products by LC-HRMS/MS, the confidence level system (level 1 to 5 with level 5 as the lowest level providing only an exact mass)33 was used to categorize the MS results. Ion chromatography coupled with a conductivity detector (IC) was used for analyzing chloride and bromate (Text S10).
2.5. Ozonation
Ozone stock solutions were prepared and standardized as described in Text S11. For ozonation experiments, filtered Lake Zurich water was spiked with 2 μM bromide, 1 mM phosphate buffer (pH 7.6), and 5 μM p-chlorobenzoic acid (pCBA), as mixed concentrations. Additionally, either 10 μM formate, or 4 μM ammonium, or 7 μM NH2Cl, or 15 μM NH2Cl was added to the spiked Lake Zurich water to assess different quenching scenarios. An aliquot of the ozone stock solution (ozone dose 60 μM) was added to the Lake Zurich waters to initiate ozonation. Samples were taken at predetermined reaction times (30 s – 1 h) and analyzed for residual ozone by indigo,34pCBA by HPLC, and bromate by IC. Rct, the ratio of concentrations of •OH and ozone, was determined as described previously35 and used as a control parameter for comparing the different reaction conditions.
2.6. Quantum Chemical Computation
Aqueous-phase free energy (Gaq) of all species were obtained by the sum of electronic energy of a species solvated by explicit water molecules (E0,gas), solvation free energy (ΔGsolv,calc), and gaseous-phase correction for the explicit water molecules (Gcorr,gas) (Text S12). E0,gas was calculated at the level of M06-2X/Aug-cc-pVTZ,36 while ΔGsolv,calc and Gcorr,gas were calculated at the level of M06-2X/Aug-cc-pVDZ with an implicit solvation model (SMD)37 and a continuum solvation method.38 Methods were validated by experimentally determined reduction potentials for halides and aromatic compounds (Text S13). The aqueous free energy of reaction (ΔGreactaq,SET) was determined based on Gaq of reactants and products of the reaction, which was subsequently used for calculating free energy of activation (ΔGactaq) by the Marcus theory39 (Text S14).
3. Results and Discussion
3.1. Reaction Kinetics
3.1.1. Determination of kBr• for Organic Model Compounds
All kBr• for organic model compounds, DOM, and NH2Cl were determined by competition kinetics using ibuprofen as a competitor. kBr• of ibuprofen was determined separately by competition kinetics with 4-iodophenol as a competitor. The reference kBr• of 4-iodophenol was (6.6 ± 0.5) × 109 M–1 s–1 based on the previous values determined by indirect methods using pulse radiolysis or laser flash photolysis.10,23 The obtained kBr• of ibuprofen was (4.6 ± 1.4) × 109 M–1 s–1 (duplicates), higher than the previously reported value of 2.2 × 109 M–1 s–112 by a factor of 2. The final kBr• of ibuprofen used as the reference was (3.8 ± 1.8) × 109 M–1 s–1, an average of our measurements and the reported value. The relative standard deviation in the reference value is relatively large (48%), which systematically affects the results in this study. Table 1 shows that the selected organic model compounds have generally high reactivity toward Br• with kBr• > 108 M–1 s–1. The determined kBr• of anisole, benzoic acid, and phenol agree well to the reported values10 within a factor of <3, which is a typical error range for different kinetic studies. For some of the dissociating compounds, kBr• was measured under two pH conditions (pH 7 and 10) to evaluate the effect of speciation on kBr•. The species with higher electron density (e.g., phenolate and neutral amine) show a 5–20 times higher kBr• than their protonated forms.
Table 1. Measured Apparent Second-Order Rate Constants at Indicated pH for the Reactions of the Selected Organic Model Compounds, Monochloramine, and a DOM Isolate with Br• (kBr•, M–1 s–1 or (mgC/L)−1 s–1 for DOM) and Theoretically Calculated Free Energies of Activation (ΔGactaq,SET, kcal mol–1) for the Reactions of Organic Model Compounds and Monochloramine with Br• by Electron Transfera.
| compounds | group | pKa | pH | measured kBr• (this study) (average ± s.d.) | reported kBr• (previous studies) | σp+e | ΔGactaq,SET (kcal mol–1) |
|---|---|---|---|---|---|---|---|
| benzylamine | 1° amine | 9.3 | 10.0 | (7.4 ± 0.5) × 109 | n.a. | 8.1 | |
| benzylamine | 1° amine | 9.3 | 7.1 | (3.8 ± 0.01) × 108 | n.a. | 20.5 | |
| N,N-dimethylbenzylamine | 3° amine | 8.9 | 10.0 | (1.7 ± 0.2) × 1010 | n.a. | 3.1 | |
| N,N-dimethylbenzylamine | 3° amine | 8.9 | 7.1 | (9.1 ± 5.5) × 108 | n.a. | 20.3 | |
| 4-bromophenol | aromatic | 9.1 | 10.0 | (2.8 ± 0.7) × 1010 | n.a. | –2.15 | 0.1 |
| 4-bromophenol | aromatic | 9.1 | 7.1 | (3.5 ± 0.5) × 109 | n.a. | –0.77 | 8.6 |
| 4-chlorophenol | aromatic | 9.0 | 10.0 | (2.0 ± 1.1) × 1010 | n.a. | –2.19 | 0.1 |
| 4-chlorophenol | aromatic | 9.0 | 7.1 | (3.9 ± 1.3) × 109 | n.a. | –0.81 | 7.9 |
| anisole | aromatic | n.a. | 7.1 | (2.2 ± 0.4) × 109 | 3.3 × 10910 | –0.62 | 6.7 |
| benzeneb | aromatic | n.a. | 7.1 | 3.4 × 108 | n.a. | 0 | 16.5 |
| benzoic acid | aromatic | 4.2 | 7.1 | (2.5 ± 0.4) × 108 | 7.7 × 10810 | –0.02 | 13.7 |
| p-chlorobenzoic acid | aromatic | 4.0 | 7.1 | (3.0 ± 0.4) × 108 | n.a. | 0.09 | 14.9 |
| ibuprofenc | aromatic | 4.4 | 7.1 | (3.8 ± 1.8) × 109 | 2.2 × 10912 | –0.25 | 6.2 |
| naphthalene | aromatic | n.a. | 7.1 | (5.5 ± 0.05) × 109 | n.a. | 5.7 | |
| phenol | aromatic | 10.0 | 7.1 | (3.9 ± 0.6) × 109 | 8.5 × 10910 | –0.92 | 7.5 |
| toluened | aromatic | n.a. | 7.1 | 4.5 × 108 | n.a. | –0.31 | 11.1 |
| 3-phenylpropionic acid | aromatic | 4.7 | 7.1 | (7.3 ± 0.07) × 108 | n.a. | 11.8 | |
| p-benzoquinone | aromatic | n.a. | 7.1 | (4.0 ± 0.5) × 109 | n.a. | 41.2 | |
| sorbic acid | olefin | 4.8 | 7.1 | (5.2 ± 0.1) × 109 | n.a. | 5.0 | |
| trans-cinnamic acid | olefin | 4.5 | 7.1 | (2.1 ± 0.5) × 109 | n.a. | 7.6 | |
| monochloramine | inorganic | 1.4 | 7.1 | (4.4 ± 1.3) × 109 | n.a. | 3.8 | |
| SRFA | DOM | n.a. | 7.1 | (1.7 ± 0.01) × 104 | 2.6 × 104 | n.a. | |
| oxidized SRFA (0.8 gO3/gC) | DOM | n.a. | 7.1 | (1.6 ± 0.01) × 104 | n.a. | n.a. | |
| oxidized SRFA (1.5 gO3/gC) | DOM | n.a. | 7.1 | (2.1 ± 0.3) × 104 | n.a. | n.a. |
Most kBr• were determined in competition with ibuprofen or unless otherwise indicated. kBr• are shown as an average and standard deviation (s.d.) of duplicates except benzene and toluene. The corresponding competition kinetics plots are shown in Figure S5.
Single measurement with a poor linearity of the competition kinetics plot (R2 = 0.67, Figure S5).
kBr• of ibuprofen was determined in competition with 4-iodophenol with kBr• of 4-iodophenol of 6.6 × 109 M–1 s–1.
Single measurement.
3.1.2. QSAR Models of kBr• for Organic Model Compounds with σp+ or ΔGactaq,SET
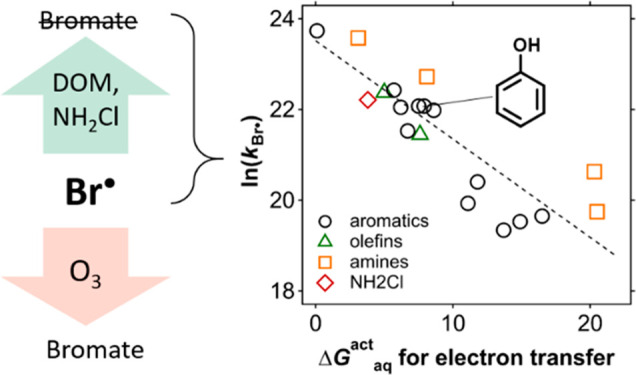
To further validate the selectivity of Br•, a QSAR was assessed for the measured kBr• of the aromatic model compounds based on Hammett constant (specifically σp+).40,41 Quantum-chemically (QC) computed free energies of activation, ΔGactaq,SET, were also tested by assuming a single electron transfer as a reaction mechanism. The QSAR assessment results in a good correlation for both molecular descriptors (R2 = 0.82 for σp+ (Figure S7a) and R2 = 0.92 for ΔGactaq,SET (Figure S7b)) for the aromatic group. Next, the QSAR approach was expanded to the other model compounds beyond aromatic groups and to the aromatic compounds for which Hammett constants are not available, by calculating ΔGactaq,SET. The corresponding results are summarized in Table S8. An overall good QSAR for all model compounds including aromatic compounds (except p-benzoquinone), amines, olefins, and NH2Cl was obtained (R2 = 0.77, Figure 1). Four literature-reported kBr• of phenol, ibuprofen, anisole, and benzoic acid10,12 with our theoretically calculated ΔGactaq,SET values were included in Figure 1 to compare the correlation and they had only an insignificant impact on the overall trend. Because the QSAR was developed based on the ΔGactaq,SET assuming a single electron transfer, it supports a single electron transfer mechanism as the rate-determining step. Only p-benzoquinone appears to undergo a different reaction mechanism based on an exceedingly high ΔGactaq,SET value of 41.2 kcal/mol, significantly higher than for all the other compounds. Instead, p-benzoquinone seems to favor a Br•-addition mechanism to the aromatic ring, according to a lower ΔGactaq,addition of 4.8 kcal/mol.
Figure 1.

Quantitative structure–activity relationship of the measured second-order rate constants for the reactions of all selected model compounds (except p-benzoquinone) with Br• and the computed free energies of activation for electron transfer reactions (see Table 1). The aromatic compounds with reported kBr• in the literature10,12 are labeled as “(ref)” and are also shown for comparison. The literature values were not included in the regression.
3.1.3. kBr• and Calculated ΔGactaq,SET for Ibuprofen
In addition to the simple organic model compounds, micropollutants also likely react by a single electron transfer mechanism, according to the result of ibuprofen agreeing well with the QSAR trend (Figure 1). The ΔGactaq,SET for ibuprofen was calculated as 6.2 kcal/mol, higher than the recently reported value (1.35 kcal/mol),12 despite the use of the same DFT method (with a similar basis set) and the implicit solvation model. The discrepancy may have resulted from the accuracy of solvation energies for Br– and Br•, which is critical to obtain an accurate ΔGactaq,SET. Our calculation method was validated by calculating the one electron reduction potential of Br•/Br– with various DFT and ab initio methods and comparing the result with experimental values as a benchmark (Text S13). Such a validation process was not reported in the previous study.12,26 Another source of uncertainty is the treatment of the solvent reorganization energies in the Marcus theory calculations. Including both outer- and inner-sphere solvent reorganization energies is important because of the potential impacts of both reactants and surrounding water to the overall structure.
3.1.4. kBr• and Calculated ΔGactaq,SET for NH2Cl and Mechanistic Interpretation
NH2Cl reacts fast with Br• with a kBr• of 4.4 × 109 M–1 s–1 (Table 1), higher than the reactivity of NH2Cl with other radical species such as •OH (k•OH,NH2Cl = 5.2 × 108 M–1 s–1 or 5.7 × 108 M–1 s–1).42,43 Previous studies on the reaction of NH2Cl with •OH reported H-atom or Cl-atom abstraction as the main reaction mechanism. However, according to our computation results, for the reaction of NH2Cl with Br•, a single electron transfer shows a clearly lower energy barrier than H-atom or Cl-atom abstraction (ΔGactaq = 3.8, 9.4, and 19.2 kcal/mol, for electron transfer, H-atom abstraction, and Cl-atom abstraction, respectively). The good agreement of the experimental kBr•,NH2Cl with the regression of the QSAR model built upon ΔGactaq,SET (Figure 1) also strongly supports a single electron transfer for the reaction of NH2Cl with Br•. A single electron transfer reaction would form NH2Cl•+ and Br– as products, of which the former can dissociate to NHCl•. NHCl• is also formed in the reaction of NH2Cl with •OH where self-decay or quenching reaction by dissolved oxygen were suggested as follow-up reactions.42
3.1.5. Formation of Transient Adduct from Br• Reactions
Quantum-chemical computations additionally predicted the formation of an energetically stable transient adduct in the reaction coordinate to the oxidized target compound and Br– (Figure S8). The formation of the adduct may not be a rate-determining step, but there is still a reasonable correlation between free energy of adduct formation (ΔGadductaq) and kBr• (Figure S9). In most cases, the most energetically stable adduct among all possible conformers results from the interaction of a bromine atom (Br•) with a benzene ring (Table S7). Exceptions are the neutral forms of amine model compounds (benzylamine and N,N-dimethylbenzylamine) where Br• preferably interacts with the neutral amine-nitrogen over the benzene ring (nitrogen benzylamine-Br adduct ΔGadductaq = −8.4 kcal/mol; adduct on the benzene ring: −1.1 kcal/mol). The preference on the amine-nitrogen was also illustrated by the spin density distribution of the protonated and deprotonated forms of amines (Table S9). For the protonated amines, the spin density is delocalized over the entire structure, indicating less favorable formation of an adduct (ΔGadductaq = −0.4 kcal/mol for the benzylamine-Br adduct). The ΔGadductaq value of p-benzoquinone is higher than for other model compounds with similar kBr• (e.g., ΔGadductaq = 1.2 kcal/mol for p-benzoquinone vs −1.9 kcal/mol for phenol (Table S7)), as observed also for ΔGactaq,SET. The positive ΔGadductaq value of p-benzoquinone indicates unfavorable interaction between Br• and the quinone structure due to the oxidized form of this compound.
3.1.6. Kinetics of Br• Reaction with DOM
The kBr• for the DOM isolate SRFA II was determined as (1.7 ± 0.01) × 104 (mgC/L)−1 s–1 (Table 1), matching to the previously reported value within a factor of < two (kBr• = 2.6 × 104 (mgC/L)−1 s–1).26 The determined value is close to the k•OH of various types of DOM with an average value of (2.2 ± 0.8) × 104 (mgC/L)−1 s–1,44−46 implying a similar DOM scavenging rate for Br• and •OH. kBr• of preozonated SRFA II was also determined to evaluate the effect of DOM oxidation during ozonation. Ozone targets electron-rich moieties of DOM (e.g., phenols),47−49 which are also reactive sites for Br• attack. The extent of preoxidation was controlled by measuring electron donating capacity (EDC), which characterizes antioxidant properties of DOM.47 The oxidation of DOM with specific ozone doses of 0.8 gO3/gC and 1.5 gO3/gC led to a decrease in EDC by (31 ± 5)% and (40 ± 4)%, respectively (Text S2), agreeing with a previous observation.49,50 Nevertheless, kBr• of non- and preozonated DOM remained in a similar range (Table 1), suggesting that DOM oxidation by ozone has a limited effect on its reactivity with Br• and its scavenging rate during ozonation. This is exemplified by p-benzoquinone, a major product of phenol oxidation by ozone,51 for which kBr• is similar as for phenol (Table 1).
3.2. Product Formation and Reaction Mechanisms for the Reactions of Phenol with Br•
3.2.1. Identified Products
Phenols are important DOM moieties and therefore phenol was selected to investigate product formation from the reaction with Br•.47 Its transformation products after reaction with Br• were investigated by LC-HRMS/MS and HPLC. Over a γ-irradiation time of 40 min (theoretically forming 22 μM Br•, according to the dose rate), phenol was degraded by Br• from 21.7 μM to 17.6 μM, suggesting a 1:5 (phenol:Br•) reaction stoichiometry (Figure S10). The reaction led to 2.4 μM p-benzoquinone, 0.5 μM hydroquinone, and 0.2 μM 4-bromophenol, as identified products. The sum of the residual concentrations of phenol and the concentrations of the identified products accounted for 96–98% of the initial phenol concentration. p-Benzoquinone was the major product with a yield of 59% per degraded phenol at 40 min, followed by hydroquinone (14%) and 4-bromophenol (4%) (Figure 2). A small concentration of catechol was also identified at the beginning of the reaction (0.1% at 12 min), but not for >12 min.
Figure 2.
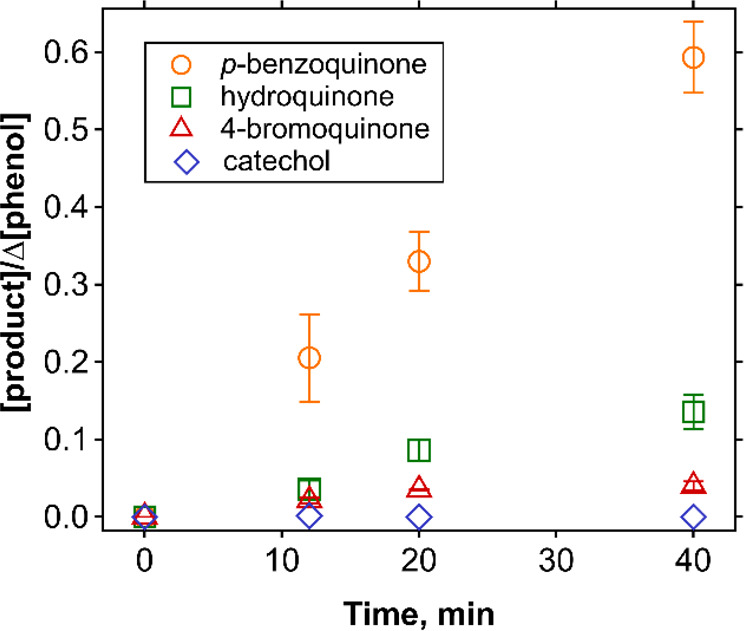
Relative product yields per degraded phenol as a function of the γ-radiolysis time during the reaction of phenol with Br•, for the condition with 22 μM phenol, 0.7 mM 1,2-dibromoethane, 40 mM t-butanol, and 50 mM phosphate buffer (pH 7.1). Concentrations of phenol and the products as a function of time are provided in Figure S10.
3.2.2. Suspected Products
In addition to the identified products with reference standards, some products were detected by nontargeted screenings of LC-HRMS/MS data where peaks with unique retention time and exact mass were extracted by comparing chromatograms of blank samples (containing phenol, no reaction) with those from γ-radiolysis samples (containing phenol, reaction with Br•). Because the mass balance was almost complete by the identified products, they were minor products with yields <4%. Two suspected products featuring molecular formula of C6H3BrO3 and C6H5BrO3 were detected with a confidence level 3 (Text S15). They showed a gradual increase over time during γ-radiolysis experiments with phenol and also p-benzoquinone (Figure S12), suggesting a presence of a quinone structure. For C6H3BrO3, p-benzoquinone substituted by a hydroxy group and a Br was suggested as a possible molecular structure (Scheme 1) based on a comparison of MS spectra with a synthesized isomer (Text S15). The other suspected products consistently detected in the γ-radiolysis sample were: C6H4O3, C12H10O2, C6H6O3, and C6H6O4. C6H4O3 is suspected as hydroxy-benzoquinone or benzoquinone-epoxide (Scheme 1) with a confidence level 3, which could be a precursor of C6H3BrO3 and C6H5BrO3. MS2 spectra of C6H4O3 featured a unique fragmentation pattern (Figure S14) but not specific enough to differentiate the two suspected structures (Figure S15). C12H10O2 is likely to be dihydroxybiphenyl with a confidence level 2, based on the matching MS2 pattern with a spectrum included in the MassBank spectral database (Figure S16).52 C6H6O3 and C6H6O4 are suspected as 6-oxo-2,4-hexydienoic acid and muconic acid, respectively, but without sufficient evidence to confirm these structures (confidence level 4).
Scheme 1. Reaction Pathway for the Reaction of Phenol with Br• Based on the Identified (in squares) and Suspected Products.

The initial step of forming phenoxyl radical is described in detail in Scheme S1.
3.2.3. Phenol-Br• Reaction Mechanism
Based on the identified and suspected products, a reaction pathway of the reaction of phenol with Br• is proposed as in Scheme 1. Phenol is initially transformed by Br• to phenoxyl radical (PhO•), analogously to its reaction with halogen dimer radical anions, X2•– (X = Cl, Br, I).53 The presence of PhO• was supported by the detection of C12H10O2 suspected to be dihydroxybiphenyl, often formed by dimerization of PhO•.53 The formation of PhO• can occur via electron transfer,10,54 H-abstraction,23,25 and Br-addition,25 as depicted in Scheme S1. The QSARs of kBr• determined by this study (Figure 1) supports an electron transfer reaction. After the initial step to PhO•, the reaction pathway further branches to three reactions, resulting in hydroquinone, catechol, or 4-bromophenol, respectively, according to the identified products. Hydroquinone is the major product (>70%), based on the sum of the product yields of hydroquinone and p-benzoquinone (a subsequent oxidation product of hydroquinone). This is likely to further undergo hydroxylation and/or bromination reactions, leading to the suspected products (C6H4O3, C6H5BrO3, and C6H3BrO3). The second pathway is a very minor pathway forming catechol (0.1% yield), a stereoisomer of hydroquinone which was only detected at a short reaction time (12 min). It may be further oxidized to o-benzoquinone, which is difficult to confirm because of its poor stability in aqueous solution.55 The third pathway with 4% yield of 4-bromophenol may imply a possible addition mechanism comparable to the oxidation of phenol by •OH (Scheme S1(3))56 or a radical–radical coupling mechanism of PhO• and Br• confirmed by calculated thermodynamically favorable ΔGreactaq (Text S16). In addition to ring products, ring-opening products such as 6-oxo-2,4-hexydienoic acid and muconic acid are also suspected (Scheme 1). Such short-chain organic acids (up to C6) were identified during oxidation of phenol by •OH57,58 and ozone.59 Dicarbonyl compounds were also reported as ring-opening products from the reaction of phenol with chlorine and •OH,60,61 but they could not be detected by the analytical method applied in this study.
3.3. Mitigation of Bromate Formation by Monochloramine during Ozonation: Role of Br•
As a proven strategy to mitigate bromate during ozonation,18 the chlorine-ammonia pretreatment blocks the initial steps of bromate formation by masking bromide and scavenging •OH (Text S17). As an alternative, a simpler strategy has been applied, which entails the direct addition of NH2Cl before ozonation. It has an advantage over the chlorine-ammonia process by forming less chlorinated DBPs by avoiding free chlorine contact time. Quenching of •OH by NH2Cl is similar to the chlorine-ammonia process. However, an additional benefit such as bromide masking is not expected, because the reaction of NH2Cl with bromide is slow (k = 0.14 M–1 s–1 at pH 7).30 According to our measurement, NH2Cl reacts fast with Br• with kBr• of 4.4 × 109 M–1 s–1 and may therefore be a quencher for this transient species. Additionally, NH2Cl reacts with HOBr with k = 2.8 × 105 M–1 s–1 at pH 7.4,62 This may also add to bromate mitigation, however, only when HOBr significantly builds up during ozonation.
To assess the effect of NH2Cl in quenching Br•, the fraction of Br• reacting with a compound X, f(Br•+X), was calculated based on the measured and reported values for kBr• of NH2Cl, DOM, ozone, and bromide, which are considered the main Br• consumers during ozonation (Text S18). Figure 3 shows the calculated f(Br•+X) in absence or presence of NH2Cl for low dissolved organic carbon (DOC) and Br– levels as in typical Swiss surface waters. Without NH2Cl, most Br• was quenched by DOM (Figure 3a). The low DOC concentration (1 mgC/L) was yet high enough to quench at least 84% of Br• for a range of specific ozone doses of 0.5–1.0 mgO3/mgC, typically applied in drinking water treatment in Switzerland (shaded area in Figure 3). For the same conditions, f(Br•+O3) and f(Br•+Br–) were 8–15% and 0.03%, respectively. The result was markedly changed in the presence of 15 μM NH2Cl where NH2Cl became the major consumer scavenging at least 70% of Br• even for the highest ozone dose of 60 μM. Under these conditions, f(Br•+DOM) was reduced to 20% and more importantly f(Br•+O3) was reduced to 2–4%. This demonstrates, that the Br• + O3 reaction is significantly suppressed by a factor of 4. Conditions with higher DOC and Br– levels simulating wastewater were also assessed and the results are shown in Figure S18. For these conditions, DOM plays an important role even in the presence of 15 μM NH2Cl, quenching 53–57% of Br• for the entire range of ozone concentrations. Therefore, the quenching effect by NH2Cl in the water with high DOC levels is not expected as significant as in the water with low DOC concentrations. Nevertheless, the f(Br•+O3) decreases by a factor of 2, which still mitigates the reaction between Br• and ozone significantly. Br– does not influence the Br• concentration based on the low f(Br•+Br–) of 0.1–0.2%, even with the higher Br– level of 1.3 μM (or 100 μg/L). The importance of DOM relative to the other consumers (ozone and Br–) is additionally illustrated by plotting f(Br•+NH2Cl) as a function of bromide, ozone, or DOC concentration. Figure S19 shows that f(Br•+NH2Cl) remains almost constant throughout the range of bromide and ozone concentrations, whereas it changes as a function of the DOC concentration.
Figure 3.
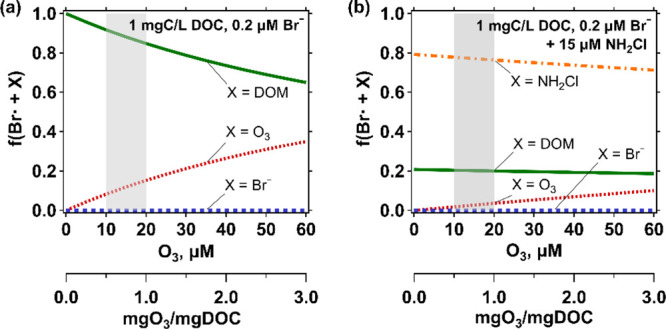
Calculated fractions (see text) of Br• reacting with DOM (green line), ozone (red line), bromide (blue line), or NH2Cl (orange line) as a function of ozone concentration in the (a) absence or (b) presence of NH2Cl. The selected concentrations were 1 mgC/L DOC, 0.2 μM Br– (16 μg/L Br–), and 15 μM NH2Cl. Shaded areas indicate a typical range of ozone doses applied in drinking water treatment in Switzerland (0.5–1.0 mgO3/mgC).
The Br• quenching effect by NH2Cl was further assessed experimentally by measuring bromate formation during ozonation in presence or absence of NH2Cl in a lake water. Figure 4 shows the bromate formation during ozonation of Lake Zurich water containing 1.4 mgC/L DOC, spiked with 2 μM of bromide, with a 60 μM ozone dose at pH 7.6 as a function of the ozone exposure (black four pointed stars). Additional experiments are shown for addition of NH2Cl (green squares (7 μM), red triangles (15 μM)), formate (orange circles), or ammonium (blue diamonds) to Lake Zurich water. Fifteen μM NH2Cl theoretically quenches 66% of Br•, based on the calculated f(Br•+NH2Cl), shown by the red asterisk in Figure S19. Oxidant exposures (ozone and •OH) of the different conditions were assessed by measuring Rct, the ratio of •OH exposure to ozone exposure.35 The corresponding Rct values for each condition are provided in Table 2 and Figure S20a and the ozone decay curves are shown in Figure S20b. To compare bromate formation among the different conditions, slopes of the bromate formation curves were obtained by linear regression of the initial formation (Figure 4b). The reduction in bromate formation of a quenching condition was calculated by comparing the slope of a quenching condition with the slope of a reference condition (Table 2). The addition of 7 μM NH2Cl reduced bromate by 60% compared to the unaltered Lake Zurich water. Doubling the NH2Cl concentration to 15 μM enhanced the reduction of bromate formation to 73%. However, for 15 μM NH2Cl, the Rct increased from 5.6 × 10–9 to 9.9 × 10–9 (Table 2), due to additional consumption of ozone and •OH by NH2Cl (kO3 = 26 M–1 s–1 and k•OH = 5.2 × 108 M–1 s–1 or 5.7 × 108 M–1 s–1).19,42,43 To mimic this change in Rct, another reference condition was tested by using 10 μM formate, which promotes ozone decay by forming O2•– and thereby increases Rct.63 The Rct of the new reference condition with formate was 7.4 × 10–9, very close to the 15 μM NH2Cl condition. Based on this comparison, the bromate mitigation for 15 μM NH2Cl was updated to 77% relative to the formate addition. The bromate formation of the reference condition with formate was augmented by 15% relative to the unaltered Lake Zurich water. This can be explained by the higher Rct in the presence of formate (similar to 15 μM NH2Cl), indicating higher •OH exposure at a given ozone exposure, resulting in higher overall oxidant exposure and bromate. In contrast, an increase in Rct from 7 μM to 15 μM NH2Cl suppressed the bromate formation, in agreement with the Br• quenching effect by NH2Cl.
Figure 4.

(a) Bromate formation as a function of the ozone exposure for five ozonation conditions. Unaltered Lake Zurich water contained 1.4 mgC/L DOC, 2 μM bromide, 1 mM phosphate buffer (pH 7.6), 5 μM pCBA, and a 60 μM ozone dose (black four pointed stars). The other conditions additionally contained 10 μM formate (orange circles), 4 μM ammonium (blue diamonds), 7 μM NH2Cl (green squares), or 15 μM NH2Cl (red triangles), respectively. (b) A close-up for a low range of ozone exposures (dotted rectangle in a). The slopes of the first three data points are provided in Table 2.
Table 2. Conditions of Ozonation Experiments with Lake Zurich Water (1.4 mgC/L DOC, 2.7 mM Alkalinity, 2 μM Bromide (spiked), 0.5 μM pCBA (spiked), 1 mM Phosphate Buffer at pH 7.6 (spiked)) and the Determined Rct (Figure 20a), the Bromate Formation Slopes Expressed As a Function of Ozone Exposure, And the Change in Bromate Formation with Regard to the Reference Conditions.
| no. | condition | comment | Rct | slope (μM/(M s))a | change in bromate formationb (%) | reference condition |
|---|---|---|---|---|---|---|
| 1 | Lake Zurich water (unaltered) | reference for low Rct | 3.7 × 10–9 | 29.5 ± 6.8 | ||
| 2 | Lake Zurich water + 10 μM formate | reference for high Rct | 7.4 × 10–9 | 33.8 ± 3.9 | +15 | 1 |
| 3 | Lake Zurich water + 4 μM NH4+ | NH4+ as HOBr quencher | 4.3 × 10–9 | 18.9 ± 0.8 | –36 | 1 |
| 4 | Lake Zurich water + 7 μM NH2Cl | NH2Cl as HOBr/Br• quencher | 5.6 × 10–9 | 11.8 ± 1.1 | –60 | 1 |
| 5 | Lake Zurich water + 15 μM NH2Cl | NH2Cl as HOBr/Br• quencher | 9.9 × 10–9 | 7.9 ± 0.7 | –73 | 1 |
| –77 | 2 | |||||
| –58 | 3 |
In addition to the NH2Cl experiment, ammonium (NH4+) was tested to further confirm the effect of quenching Br•/HOBr on bromate formation. NH2Cl quenches HOBr as well as Br• (kBr• = 4.4 × 109 M–1 s–1 (Table 1) and kHOBr = 2.8 × 105 M–1 s–1 at pH 7),4,62 NH4+/NH3 only quenches HOBr (kBr• ≪ 9.7 × 107 M–1 s–1, assumed based on k•OH of NH3 (generally more reactive species than NH4+)64 and kHOBr ∼ 106 M–1 s–1 at pH 7–8).4 An NH4+ concentration of 4 μM was selected to obtain a similar HOBr quenching rate as for 15 μM NH2Cl. The condition with 4 μM NH4+ (blue diamonds) leads to a reduction of bromate formation by 36%, mainly due to quenching of HOBr. Therefore, from the 73% bromate mitigation observed for 15 μM NH2Cl, roughly similar contributions can be attributed to the quenching of HOBr and Br•, respectively. The reduction of bromate formation by 15 μM NH2Cl compared to by NH4+ was 58% (by taking the NH4+ condition as reference) and occurred mainly during the initial phase of the ozonation (Figure 4b). During this phase, an enhanced Br• formation is expected from the reaction of bromide with •OH, which are formed in high concentrations during the initial phase of an ozonation.29,65 Therefore, quenching of Br• by NH2Cl during the initial phase slows down bromate formation by reducing the transient Br• concentrations.
3.4. Practical Implications
The good correlation of the experimentally determined kBr• with the molecular descriptors (Hammett constants and the computed ΔGactaq,SET) for diverse functional groups (aromatics, amines, olefins, and NH2Cl) enables a prediction of kBr• for a wider range of compounds. As shown by the case of p-benzoquinone, some compounds react with Br• by different mechanisms than common aromatic rings (e.g., addition instead of electron transfer), which needs further investigation to improve prediction capability for such compounds. The predominant formation of hydroquinone (and subsequently p-benzoquinone) during the phenol-Br• reaction raises concern related to mutagenicity. Their formation is comparable to the phenol reaction with other oxidants (e.g., ozone, •OH) and likely to follow similar subsequent reactions to ring opening compounds. In addition to quinones, brominated phenol was identified during the phenol-Br• reaction but as a minor product only. Accordingly, only a small fraction of Br• will react with DOM to form Br-DBPs, which is likely to be outweighed by other sources of Br-DBP such as bromination of DOM by HOBr. During conventional ozonation, Br• plays a key role in bromate formation by its further reaction with ozone. According to our kinetic result, this reaction can be partially inhibited by the fast reaction of Br• with DOM. For a typical Swiss surface waters with the DOC concentration of ∼1 mgC/L, the majority of Br• is quenched by DOM and the rest reacts with ozone. Despite the generally small fraction, the oxidation of Br• by ozone can lead to a substantial portion of formed bromate, especially during an initial phase of ozonation where •OH exposure is high and Br• becomes an important bromate precursor. This bromate formation pathway involving Br• can be suppressed by NH2Cl, according to our theoretical and experimental assessment. It was demonstrated that the majority of Br• can be quenched by 15 μM NH2Cl for the condition with DOC concentrations on the order of 1–2 mgC/L. Accordingly, bromate reduction of 70–80% was achieved during ozonation of real Lake water even with the excess ozone dose applied in this study (corresponding to ∼2 gO3/gC). About a half of the reduction was related to quenching of Br• and the other was linked to quenching of HOBr. Overall, NH2Cl can interfere in both the •OH (forming Br•) and ozone pathway (forming HOBr), serving as an efficient quenching agent throughout the course of ozonation. Additional benefits of the NH2Cl treatment over the chlorine-ammonia treatment prior to ozonation is mitigating the formation of chlorinated and brominated DBPs, because of a lower reactivity of NH2Cl with DOM and with Br–. The NH2Cl pretreatment would be less efficient for waters with higher DOC concentrations (∼5 mg/L) such as wastewater, but the fraction of Br• oxidized by ozone can be suppressed by a factor of 2, which results in a significant reduction in bromate. If the advanced oxidation processes UV/H2O2 is applied in Br–-containing water, Br– can scavenge •OH to form Br•. However, even for wastewater featuring elevated Br– levels, DOM still quenches about 99% of the •OH (based on 1 μM Br–, 4 mgC/L DOC and k•OH = (2.2 ± 0.8) × 104 (mgC/L)−1 s–1 for DOM44−46 and 1.1 × 109 M–1 s–1 for Br– to Br•).21 Because only a minor fraction of Br• leads to Br-DBPs, small changes are expected in the process performance.
Acknowledgments
This research was funded by the Swiss National Science Foundation (project no. 200021-181975). We thank Paul Scherrer Institute (PSI) for access to the γ-radiolysis facility, Viktor Boutellier at PSI for technical support, Jakob Helbing at Zurich Water Supply for help in collecting Lake Zurich water, the AuA Laboratory at Eawag for analyses of general water quality parameters, Samuel Derrer at Eawag for chemical synthesis, and Elisabeth Muck at Eawag for laboratory support. B.B. and D.M. were partially supported by NSF CHM-1808052. D.M. acknowledges the support by Eawag during his sabbatical.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.2c07694.
Supplementary texts, figures, tables, and scheme to further describe the materials and methods and corroborate results and discussion (PDF)
Author Present Address
‡ Department of Civil and Environmental Engineering, Stanford University, 473 Via Ortega, Stanford, CA 94305, United States
The authors declare no competing financial interest.
Supplementary Material
References
- Soltermann F.; Abegglen C.; Götz C.; von Gunten U. Bromide Sources and Loads in Swiss Surface Waters and Their Relevance for Bromate Formation during Wastewater Ozonation. Environ. Sci. Technol. 2016, 50 (18), 9825–9834. 10.1021/acs.est.6b01142. [DOI] [PubMed] [Google Scholar]
- Flury M.; Papritz A. Bromide in the Natural Environment: Occurrence and Toxicity. Journal of Environmental Quality 1993, 22 (4), 747–758. 10.2134/jeq1993.00472425002200040017x. [DOI] [Google Scholar]
- von Gunten U. Oxidation Processes in Water Treatment: Are We on Track?. Environ. Sci. Technol. 2018, 52 (9), 5062–5075. 10.1021/acs.est.8b00586. [DOI] [PubMed] [Google Scholar]
- Heeb M. B.; Criquet J.; Zimmermann-Steffens S. G.; von Gunten U. Oxidative Treatment of Bromide-Containing Waters: Formation of Bromine and Its Reactions with Inorganic and Organic Compounds — A Critical Review. Water Res. 2014, 48, 15–42. 10.1016/j.watres.2013.08.030. [DOI] [PubMed] [Google Scholar]
- Grebel J. E.; Pignatello J. J.; Mitch W. A. Effect of Halide Ions and Carbonates on Organic Contaminant Degradation by Hydroxyl Radical-Based Advanced Oxidation Processes in Saline Waters. Environ. Sci. Technol. 2010, 44 (17), 6822–6828. 10.1021/es1010225. [DOI] [PubMed] [Google Scholar]
- Lee W.; Lee Y.; Allard S.; Ra J.; Han S.; Lee Y. Mechanistic and Kinetic Understanding of the UV254 Photolysis of Chlorine and Bromine Species in Water and Formation of Oxyhalides. Environ. Sci. Technol. 2020, 54 (18), 11546–11555. 10.1021/acs.est.0c02698. [DOI] [PubMed] [Google Scholar]
- von Gunten U.; Oliveras Y. Advanced Oxidation of Bromide-Containing Waters: Bromate Formation Mechanisms. Environ. Sci. Technol. 1998, 32 (1), 63–70. 10.1021/es970477j. [DOI] [Google Scholar]
- Richardson S. D.; Thruston A. D.; Rav-Acha C.; Groisman L.; Popilevsky I.; Juraev O.; Glezer V.; McKague A. B.; Plewa M. J.; Wagner E. D. Tribromopyrrole, Brominated Acids, and Other Disinfection Byproducts Produced by Disinfection of Drinking Water Rich in Bromide. Environ. Sci. Technol. 2003, 37 (17), 3782–3793. 10.1021/es030339w. [DOI] [PubMed] [Google Scholar]
- Langsa M.; Heitz A.; Joll C. A.; von Gunten U.; Allard S. Mechanistic Aspects of the Formation of Adsorbable Organic Bromine during Chlorination of Bromide-Containing Synthetic Waters. Environ. Sci. Technol. 2017, 51 (9), 5146–5155. 10.1021/acs.est.7b00691. [DOI] [PubMed] [Google Scholar]
- Lei Y.; Lei X.; Yu Y.; Li K.; Li Z.; Cheng S.; Ouyang G.; Yang X. Rate Constants and Mechanisms for Reactions of Bromine Radicals with Trace Organic Contaminants. Environ. Sci. Technol. 2021, 55 (15), 10502–10513. 10.1021/acs.est.1c02313. [DOI] [PubMed] [Google Scholar]
- Cheng S.; Zhang X.; Yang X.; Shang C.; Song W.; Fang J.; Pan Y. The Multiple Role of Bromide Ion in PPCPs Degradation under UV/Chlorine Treatment. Environ. Sci. Technol. 2018, 52 (4), 1806–1816. 10.1021/acs.est.7b03268. [DOI] [PubMed] [Google Scholar]
- Guo K.; Zheng S.; Zhang X.; Zhao L.; Ji S.; Chen C.; Wu Z.; Wang D.; Fang J. Roles of Bromine Radicals and Hydroxyl Radicals in the Degradation of Micropollutants by the UV/Bromine Process. Environ. Sci. Technol. 2020, 54 (10), 6415–6426. 10.1021/acs.est.0c00723. [DOI] [PubMed] [Google Scholar]
- Parker K. M.; Mitch W. A. Halogen Radicals Contribute to Photooxidation in Coastal and Estuarine Waters. Proc. Natl. Acad. Sci. U.S.A. 2016, 113 (21), 5868–5873. 10.1073/pnas.1602595113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker K. M.; Reichwaldt E. S.; Ghadouani A.; Mitch W. A. Halogen Radicals Promote the Photodegradation of Microcystins in Estuarine Systems. Environ. Sci. Technol. 2016, 50 (16), 8505–8513. 10.1021/acs.est.6b01801. [DOI] [PubMed] [Google Scholar]
- von Gunten U.; Hoigne J. Bromate Formation during Ozonization of Bromide-Containing Waters: Interaction of Ozone and Hydroxyl Radical Reactions. Environ. Sci. Technol. 1994, 28 (7), 1234–1242. 10.1021/es00056a009. [DOI] [PubMed] [Google Scholar]
- National Primary Drinking Water Regulations. U.S. EPA. https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed 2022-09-05).
- Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020 on the Quality of Water Intended for Human Consumption (Recast). Official Journal of the European Union 2020, L435, 1–62. [Google Scholar]
- von Sonntag C.; von Gunten U.. Chemistry of Ozone in Water and Wastewater Treatment; IWA Publishing, 2012. [Google Scholar]
- Haag W. R.; Hoigné J. Ozonation of Bromide-Containing Waters: Kinetics of Formation of Hypobromous Acid and Bromate. Environ. Sci. Technol. 1983, 17 (5), 261–267. 10.1021/es00111a004. [DOI] [Google Scholar]
- Liu Q.; Schurter L. M.; Muller C. E.; Aloisio S.; Francisco J. S.; Margerum D. W. Kinetics and Mechanisms of Aqueous Ozone Reactions with Bromide, Sulfite, Hydrogen Sulfite, Iodide, and Nitrite Ions. Inorg. Chem. 2001, 40 (17), 4436–4442. 10.1021/ic000919j. [DOI] [PubMed] [Google Scholar]
- Zehavi D.; Rabani J. Oxidation of Aqueous Bromide Ions by Hydroxyl Radicals. Pulse Radiolytic Investigation. J. Phys. Chem. 1972, 76 (3), 312–319. 10.1021/j100647a006. [DOI] [Google Scholar]
- Kläning U. K.; Wolff T. Laser Flash Photolysis of HCIO, CIO-, HBrO, and BrO- in Aqueous Solution. Reactions of Cl- and Br-Atoms. Berichte der Bunsengesellschaft für physikalische Chemie 1985, 89 (3), 243–245. 10.1002/bbpc.19850890309. [DOI] [Google Scholar]
- Merényi G.; Lind J. Reaction Mechanism of Hydrogen Abstraction by the Bromine Atom in Water. J. Am. Chem. Soc. 1994, 116 (17), 7872–7876. 10.1021/ja00096a050. [DOI] [Google Scholar]
- Scaiano J. C.; Barra M.; Krzywinski M.; Sinta R.; Calabrese G. Laser Flash Photolysis Determination of Absolute Rate Constants for Reactions of Bromine Atoms in Solution. J. Am. Chem. Soc. 1993, 115 (18), 8340–8344. 10.1021/ja00071a048. [DOI] [Google Scholar]
- Guha S. N.; Schoneich C.; Asmus K. D. Free Radical Reductive Degradation of Vic-Dibromoalkanes and Reaction of Bromine Atoms with Polyunsaturated Fatty Acids: Possible Involvement of Br. in the 1,2-Dibromoethane-Induced Lipid-Peroxidation. Arch. Biochem. Biophys. 1993, 305 (1), 132–140. 10.1006/abbi.1993.1402. [DOI] [PubMed] [Google Scholar]
- Lei Y.; Lei X.; Westerhoff P.; Tong X.; Ren J.; Zhou Y.; Cheng S.; Ouyang G.; Yang X. Bromine Radical (Br• and Br2•-) Reactivity with Dissolved Organic Matter and Brominated Organic Byproduct Formation. Environ. Sci. Technol. 2022, 56 (8), 5189–5199. 10.1021/acs.est.2c00549. [DOI] [PubMed] [Google Scholar]
- Pearce R.; Hogard S.; Buehlmann P.; Salazar-Benites G.; Wilson C.; Bott C. Evaluation of Preformed Monochloramine for Bromate Control in Ozonation for Potable Reuse. Water Res. 2022, 211, 118049. 10.1016/j.watres.2022.118049. [DOI] [PubMed] [Google Scholar]
- Ling L.; Deng Z.; Fang J.; Shang C. Bromate Control during Ozonation by Ammonia-Chlorine and Chlorine-Ammonia Pretreatment: Roles of Bromine-Containing Haloamines. Chemical Engineering Journal 2020, 389, 123447. 10.1016/j.cej.2019.123447. [DOI] [Google Scholar]
- Pinkernell U.; von Gunten U. Bromate Minimization during Ozonation: Mechanistic Considerations. Environ. Sci. Technol. 2001, 35 (12), 2525–2531. 10.1021/es001502f. [DOI] [PubMed] [Google Scholar]
- Luh J.; Mariñas B. J. Kinetics of Bromochloramine Formation and Decomposition. Environ. Sci. Technol. 2014, 48 (5), 2843–2852. 10.1021/es4036754. [DOI] [PubMed] [Google Scholar]
- Lal M.; Mahal H. S. Reactions of Alkylbromides with Free Radicals in Aqueous Solutions. International Journal of Radiation Applications and Instrumentation. Part C. Radiation Physics and Chemistry 1992, 40 (1), 23–26. 10.1016/1359-0197(92)90134-2. [DOI] [Google Scholar]
- Lal M.; Mönig J.; Asmus K.-D.; Wardman P. Free Radical Induced Degradation of 1, 2-Dibromoethane. Generation of Free Br+ Atoms. Free Radical Res. Commun. 1986, 1 (4), 235–241. 10.3109/10715768609051633. [DOI] [PubMed] [Google Scholar]
- Schymanski E. L.; Jeon J.; Gulde R.; Fenner K.; Ruff M.; Singer H. P.; Hollender J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48 (4), 2097–2098. 10.1021/es5002105. [DOI] [PubMed] [Google Scholar]
- Bader H.; Hoigné J. Determination of Ozone in Water by the Indigo Method. Water Res. 1981, 15 (4), 449–456. 10.1016/0043-1354(81)90054-3. [DOI] [Google Scholar]
- Elovitz M. S.; von Gunten U. Hydroxyl Radical/Ozone Ratios During Ozonation Processes. I. The Rct Concept. Ozone: Science & Engineering 1999, 21 (3), 239–260. 10.1080/01919519908547239. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120 (1), 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113 (18), 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Bryantsev V. S.; Diallo M. S.; Goddard W. A. III Calculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum Models. J. Phys. Chem. B 2008, 112 (32), 9709–9719. 10.1021/jp802665d. [DOI] [PubMed] [Google Scholar]
- Marcus R. A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15 (1), 155–196. 10.1146/annurev.pc.15.100164.001103. [DOI] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91 (2), 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Lee Y.; von Gunten U. Quantitative Structure-Activity Relationships (QSARs) for the Transformation of Organic Micropollutants during Oxidative Water Treatment. Water Res. 2012, 46 (19), 6177–6195. 10.1016/j.watres.2012.06.006. [DOI] [PubMed] [Google Scholar]
- Poskrebyshev G. A.; Huie R. E.; Neta P. Radiolytic Reactions of Monochloramine in Aqueous Solutions. J. Phys. Chem. A 2003, 107 (38), 7423–7428. 10.1021/jp030198k. [DOI] [Google Scholar]
- Gleason J. M.; McKay G.; Ishida K. P.; Mezyk S. P. Temperature Dependence of Hydroxyl Radical Reactions with Chloramine Species in Aqueous Solution. Chemosphere 2017, 187, 123–129. 10.1016/j.chemosphere.2017.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezonik P. L.; Fulkerson-Brekken J. Nitrate-Induced Photolysis in Natural Waters: Controls on Concentrations of Hydroxyl Radical Photo-Intermediates by Natural Scavenging Agents. Environ. Sci. Technol. 1998, 32 (19), 3004–3010. 10.1021/es9802908. [DOI] [Google Scholar]
- Katsoyiannis I. A.; Canonica S.; von Gunten U. Efficiency and Energy Requirements for the Transformation of Organic Micropollutants by Ozone, O3/H2O2 and UV/H2O2. Water Res. 2011, 45 (13), 3811–3822. 10.1016/j.watres.2011.04.038. [DOI] [PubMed] [Google Scholar]
- Westerhoff P.; Mezyk S. P.; Cooper W. J.; Minakata D. Electron Pulse Radiolysis Determination of Hydroxyl Radical Rate Constants with Suwannee River Fulvic Acid and Other Dissolved Organic Matter Isolates. Environ. Sci. Technol. 2007, 41 (13), 4640–4646. 10.1021/es062529n. [DOI] [PubMed] [Google Scholar]
- Houska J.; Salhi E.; Walpen N.; von Gunten U. Oxidant-Reactive Carbonous Moieties in Dissolved Organic Matter: Selective Quantification by Oxidative Titration Using Chlorine Dioxide and Ozone. Water Res. 2021, 207, 117790. 10.1016/j.watres.2021.117790. [DOI] [PubMed] [Google Scholar]
- Lim S.; Shi J. L.; von Gunten U.; McCurry D. L. Ozonation of Organic Compounds in Water and Wastewater: A Critical Review. Water Res. 2022, 213, 118053. 10.1016/j.watres.2022.118053. [DOI] [PubMed] [Google Scholar]
- Önnby L.; Salhi E.; McKay G.; Rosario-Ortiz F. L.; von Gunten U. Ozone and Chlorine Reactions with Dissolved Organic Matter - Assessment of Oxidant-Reactive Moieties by Optical Measurements and the Electron Donating Capacities. Water Res. 2018, 144, 64–75. 10.1016/j.watres.2018.06.059. [DOI] [PubMed] [Google Scholar]
- Rougé V.; von Gunten U.; Allard S. Efficiency of Pre-Oxidation of Natural Organic Matter for the Mitigation of Disinfection Byproducts: Electron Donating Capacity and UV Absorbance as Surrogate Parameters. Water Res. 2020, 187, 116418. 10.1016/j.watres.2020.116418. [DOI] [PubMed] [Google Scholar]
- Ramseier M. K.; von Gunten U. Mechanisms of Phenol Ozonation—Kinetics of Formation of Primary and Secondary Reaction Products. Ozone: Science & Engineering 2009, 31 (3), 201–215. 10.1080/01919510902740477. [DOI] [Google Scholar]
- Schulze T.; Meier R.; Alygizakis N.; Schymanski E.; Bach E.; Li D. H.; Lauperbe; Raalizadeh; Tanaka S.; Witting M.. MassBank/MassBank-Data: Release Version 2021.12. 10.5281/ZENODO.5775684 [DOI]
- Steenken S.; Neta P.. Transient Phenoxyl Radicals: Formation and Properties in Aqueous Solutions. In The Chemistry of Phenols; John Wiley & Sons, 2003; pp 1107–1152. 10.1002/0470857277.ch16 [DOI] [Google Scholar]
- Zhang K.; Parker K. M. Halogen Radical Oxidants in Natural and Engineered Aquatic Systems. Environ. Sci. Technol. 2018, 52 (17), 9579–9594. 10.1021/acs.est.8b02219. [DOI] [PubMed] [Google Scholar]
- Liao C.-C.; Peddinti R. K. Masked O-Benzoquinones in Organic Synthesis. Acc. Chem. Res. 2002, 35 (10), 856–866. 10.1021/ar000194n. [DOI] [PubMed] [Google Scholar]
- Raghavan N. V.; Steenken S. Electrophilic Reaction of the Hydroxyl Radical with Phenol. Determination of the Distribution of Isomeric Dihydroxycyclohexadienyl Radicals. J. Am. Chem. Soc. 1980, 102 (10), 3495–3499. 10.1021/ja00530a031. [DOI] [Google Scholar]
- Alnaizy R.; Akgerman A. Advanced Oxidation of Phenolic Compounds. Advances in Environmental Research 2000, 4 (3), 233–244. 10.1016/S1093-0191(00)00024-1. [DOI] [Google Scholar]
- Zazo J. A.; Casas J. A.; Mohedano A. F.; Gilarranz M. A.; Rodríguez J. J. Chemical Pathway and Kinetics of Phenol Oxidation by Fenton’s Reagent. Environ. Sci. Technol. 2005, 39 (23), 9295–9302. 10.1021/es050452h. [DOI] [PubMed] [Google Scholar]
- Mvula E.; von Sonntag C. Ozonolysis of Phenols in Aqueous Solution. Org. Biomol. Chem. 2003, 1 (10), 1749–1756. 10.1039/b301824p. [DOI] [PubMed] [Google Scholar]
- Prasse C.; von Gunten U.; Sedlak D. L. Chlorination of Phenols Revisited: Unexpected Formation of α,β-Unsaturated C4-Dicarbonyl Ring Cleavage Products. Environ. Sci. Technol. 2020, 54 (2), 826–834. 10.1021/acs.est.9b04926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasse C.; Ford B.; Nomura D. K.; Sedlak D. L. Unexpected Transformation of Dissolved Phenols to Toxic Dicarbonyls by Hydroxyl Radicals and UV Light. Proc. Natl. Acad. Sci. U.S.A. 2018, 115 (10), 2311–2316. 10.1073/pnas.1715821115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda M.; Margerum D. W. Reactions of Monochloramine with Bromine, Tribromide, Hypobromous Acid and Hypobromite: Formation of Bromochloramines. Inorg. Chem. 1994, 33 (1), 118–123. 10.1021/ic00079a022. [DOI] [Google Scholar]
- Reisz E.; Fischbacher A.; Naumov S.; von Sonntag C.; Schmidt T. C. Hydride Transfer: A Dominating Reaction of Ozone with Tertiary Butanol and Formate Ion in Aqueous Solution. Ozone-Sci. Eng. 2014, 36 (6), 532–539. 10.1080/01919512.2014.891436. [DOI] [Google Scholar]
- Hickel B.; Sehested K. Reaction of Hydroxyl Radicals with Ammonia in Liquid Water at Elevated Temperatures. International Journal of Radiation Applications and Instrumentation. Part C. Radiation Physics and Chemistry 1992, 39 (4), 355–357. 10.1016/1359-0197(92)90244-A. [DOI] [Google Scholar]
- Buffle M.-O.; Schumacher J.; Salhi E.; Jekel M.; von Gunten U. Measurement of the Initial Phase of Ozone Decomposition in Water and Wastewater by Means of a Continuous Quench-Flow System: Application to Disinfection and Pharmaceutical Oxidation. Water Res. 2006, 40 (9), 1884–1894. 10.1016/j.watres.2006.02.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.