

Significance
CRISPR-Cas RNA-guided nucleases, Cas9 and Cas12, are the primary tools of the new generation of genome editing methods. These CRISPR effector nucleases are thought to have evolved from transposon-encoded RNA-guided nucleases, IscB and TnpB, respectively. We performed a comprehensive evolutionary analysis of TnpB to reveal an immensely diverse set of candidate systems for genome editing. We further showed that Cas12 nucleases evolved from TnpB on numerous, independent occasions. Additionally, TnpB apparently was apparently recruited for diverse other, primarily, regulatory functions, which was accompanied by inactivation of the nuclease. These findings reveal extensive functional and evolutionary flexibility of transposon-encoded proteins and provide many avenues for further exploration of RNA-guided biological systems as well as multiple applications.
Keywords: OMEGA-TnpB, CRISPR-Cas12, evolution, classification, diversity
Abstract
The TnpB proteins are transposon-associated RNA-guided nucleases that are among the most abundant proteins encoded in bacterial and archaeal genomes, but whose functions in the transposon life cycle remain unknown. TnpB appears to be the evolutionary ancestor of Cas12, the effector nuclease of type V CRISPR-Cas systems. We performed a comprehensive census of TnpBs in archaeal and bacterial genomes and constructed a phylogenetic tree on which we mapped various features of these proteins. In multiple branches of the tree, the catalytic site of the TnpB nuclease is rearranged, demonstrating structural and probably biochemical malleability of this enzyme. We identified numerous cases of apparent recruitment of TnpB for other functions of which the most common is the evolution of type V CRISPR-Cas effectors on about 50 independent occasions. In many other cases of more radical exaptation, the catalytic site of the TnpB nuclease is apparently inactivated, suggesting a regulatory function, whereas in others, the activity appears to be retained, indicating that the recruited TnpB functions as a nuclease, for example, as a toxin. These findings demonstrate remarkable evolutionary malleability of the TnpB scaffold and provide extensive opportunities for further exploration of RNA-guided biological systems as well as multiple applications.
Obligate Mobile Element Guided Activity (OMEGA) modules are a recently characterized class of diverse RNA-guided DNA-targeting systems with potential for applications in genome editing, nucleic acid sensing, and beyond (1, 2). OMEGA systems consist of an effector nuclease and an associated ωRNA that guides the effector to a target DNA sequence. All currently known OMEGA systems are associated with transposons of the IS200/IS605 superfamily, most of which are nonautonomous and consist of an OMEGA module alone. However, some of these transposons are autonomous and additionally encode a serine or tyrosine transposase. Three families of OMEGA effectors have been identified—IscB, IsrB, and TnpB—all of which contain an RuvC nuclease domain, but differ with respect to additional domains as well as the structure of their associated ωRNAs (1–8). Although the RNA-guided DNA cleavage by OMEGA systems has been characterized in mechanistic detail, the biological role of these RNA-guided nucleases in the transposon life cycle remains uncertain.
ωRNAs are highly structured RNAs with multiple hairpins and an additional, typically variable region that serves as a guide for target recognition, and, in contrast to CRISPR RNAs (crRNAs), are typically longer (≥~60 bp) (1). In OMEGA systems capable of mobilization, the guide is encoded immediately outside the transposon ends and thus targets DNA sequences closely similar to the sequences adjacent to the transposon’s insertion sites (1, 2). Often, multiple, nearly identical copies of mobilized OMEGA systems can be found in the same genome, allowing for a single OMEGA system to use guides encoded by additional copies of the transposon (1). Moreover, standalone ωRNAs that are evidently mobilized yet lack an associated OMEGA effector or transposase have been identified in the same genomes as effector-containing OMEGA loci (1). These standalone ωRNAs were shown to function in trans with compatible OMEGA effectors (1). Although the biological role of standalone ωRNAs is unknown, they apparently provide for targeting additional sites without requiring duplication of the entire OMEGA system.
IscB and TnpB are distant homologs of Cas9 and Cas12, respectively (3, 9), with which they share the homologous RuvC-like nuclease domain, suggesting that these transposon-encoded nucleases could be the evolutionary ancestors of the type II and type V CRISPR effectors (10). Further work elucidated the evolutionary relationship between IsrB, IscB, and Cas9 in greater detail, demonstrating that the extant Cas9s likely evolved from IscB in a single evolutionary event, but that CRISPR arrays associated with IscBs on multiple, independent occasions, suggesting a general propensity for OMEGA modules to evolve into CRISPR systems (1). In contrast, the Cas12 variants likely evolved from TnpB on multiple, independent occasions (Fig. 1A) (4, 9). Complementing the phylogenomic analyses, the structures of the ternary complexes of IscB and IsrB with ωRNA and the target DNA have been solved, revealing the scaffolding role of ωRNA that in type II CRISPR-Cas systems apparently was taken over by the REC lobe of Cas9 (6–8).
Fig. 1.
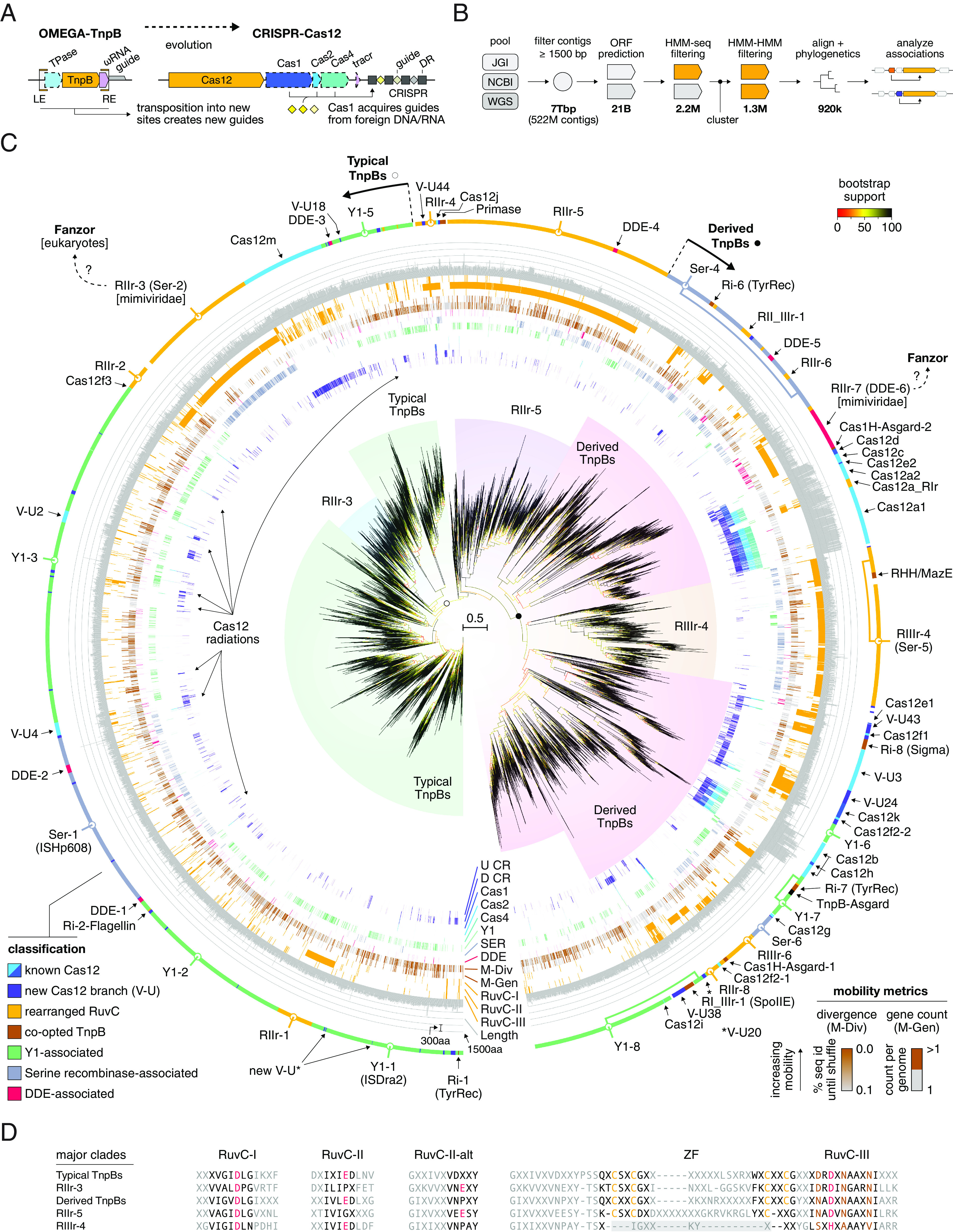
Comprehensive phylogenomic analysis of TnpBs and Cas12s. (A) Overview of differences between OMEGA-TnpB and CRISPR-Cas12. (B) Analysis pipeline used to generate protein sequences for phylogenetic analysis. The first bold number (from left to right) shows the size of the nucleotide database while the second bold numbers and beyond show the number of proteins remaining after various filtering steps. (C) Phylogenetic analysis of 6931 TnpB cluster representatives (at 50% sequence identity) using IQ-Tree2. Bootstrap values are shown as a gradient from red to yellow to black. Major clades are shown around the tree along with their designations. Colored lines around the tree indicate association rates with various elements: upstream CRISPR array (U CR), downstream CRISPR array (D CR), Cas1, Cas2, Cas4, Y1 TnpA (Y1), serine recombinase TnpA (SER). Next, mobility or nonmobility as determined by genome copy counts from complete genomes (when available) are shown in black and brown lines. Then, noncanonical catalytic amino acids for RuvC-I, II, and III are shown as orange lines. Last, trimmed protein lengths are shown as gray bars. The outermost ring contains classifications of TnpBs and Cas12s. Systems with conserved substitutions in the RuvC active site are classified as rearranged and marked accordingly. (D) Alignment of TnpB major clade consensus sequences in the RuvC-I, II, III, and ZF regions.
Notwithstanding the advances in the structural and functional characterization of the OMEGA systems, the detailed evolutionary history of the diverse TnpBs and Cas12s remains to be reconstructed. Here, we present a comprehensive survey of the diversity of TnpBs and Cas12s in bacterial and archaeal genomes and metagenomes along with phylogenetic analysis detailing the relationships between various TnpBs and Cas12 subtypes. We investigate the genomic context of these genes, in particular, derivatives of TnpB with catalytically rearranged RuvC-like nuclease domains and inactivated TnpB derivatives that appear to have been repurposed (exapted) for various functions.
Results
Diversity and Phylogeny of TnpB.
We conducted a comprehensive genomic and metagenomic census of TnpBs and Cas12s from all publicly available prokaryotic genomic sources, followed by a detailed phylogenetic analysis. To this end, TnpBs and Cas12s were identified in NCBI, JGI, and Whole Genome Shotgun (WGS) databases using a permissive hidden Markov model (HMM) sequence search against TnpB and Cas12 profiles (SI Appendix). The extracted set of protein sequences was first made nonredundant by clustering at 90% identity and then clustered at 50% identity (SI Appendix). HMM-HMM alignments were used to further filter sequences containing TnpB RuvC-like domains. We then iteratively aligned and filtered the cluster representatives, producing the final alignment of 6,931 sequences that was used to infer a maximum likelihood phylogenetic tree using IQ-Tree2 (Fig. 1B and SI Appendix). Minor branches were inferred automatically using TreeCluster (11) and were then examined manually and split further if they contained different variants of TnpBs or Cas12s as assessed by analysis of the active site residues, transposase associations, and CRISPR associations. Minor branches were then merged into major branches if they contained the same active site residues, and similar transposon and CRISPR associations. The major branches were then finally merged into five major clades (Fig. 1C) based on their active site residues (Fig. 1D), with each clade including distinct associations. In contrast to CRISPR systems, TnpBs do not have obvious guide boundaries. To better understand the placement of RNAs and guides for TnpBs, we aligned the downstream regions of representative TnpBs from each minor branch and found multiple branches with clear RNA scaffold-guide boundaries (SI Appendix).
To highlight general features of the clades, we developed the following designation scheme: Y1-# (IS200/605 Y1 TnpA-associated TnpBs), Ser-# (Serine Recombinase-associated TnpBs), RIr-#, RIIr-#, RIIIr-# (RuvC-I, II, III catalytically rearranged TnpBs, respectively), and additional designations specific to other branches with distinct properties and associations. The first major clade is the set of typical TnpBs, which contain the Y1-1 and Ser-1 major branches. This branch includes the recently experimentally characterized OMEGA systems from Deinococcus radiodurans (ISDra2) (2) and Ktedonobacter racemifer (1). Also, TnpB from the best-studied IS608 from Helicobacter pylori belongs to this clade (12). These TnpBs are comparatively small proteins (typically, between 300aa and 450aa) that exhibit consistent, albeit infrequent, associations with the respective transposase classes as well as multiple CRISPR associations, largely lacking the CRISPR adaptation module. The TnpBs of this clade contain a DRDXN motif in the RuvC-III region (Fig. 1D). The second major clade consists of derived TnpBs containing a NADXN motif in the RuvC-III region and also included TnpBs with various rearrangements of the catalytic site as well as inactivated derivatives. Three other major clades of TnpBs, RIIr-3 (RuvC-II rearrangement), RIIIr-4 (RuvC-III rearrangement), and RIIr-5 (RuvC-II rearrangement), include distinct catalytic site rearrangements, in the RuvC-II, RuvC-III, and RuvC-II regions, respectively, as described in detail below (Fig. 1D). In addition, RIIIr-4 lacks the C4 zinc finger (ZF) motif that is conserved in the other major clades. The conservation of the catalytic site rearrangements in each of these three major clades suggests that these TnpBs are catalytically active.
In addition to the five major clades, the tree includes numerous branches with different characteristic protein lengths, catalytic site rearrangements and associations with other genes. Some large branches, such as RIIr-7, are associated with transposases not previously known to be associated with TnpB. Several large branches include derived forms of TnpB with both catalytic site rearrangements and associations with distinct transposases and auxiliary genes. These include tyrosine recombinases, DDE transposases (named after the D+D+E catalytic motif), sigma factors, and SpoIIE-like protein phosphatases and also encompass multiple CRISPR connections, including the CRISPR-associated transposon (CAST) Cas12k (for the complete list of analyzed sequences, an expanded view of the tree, and alignments for all major branches, see SI Appendix, Tables S1 and S2.
Taxonomy and Genomic Features of TnpBs and Cas12s.
To identify CRISPR-Cas12 systems derived from TnpB, we examined tree clades that contained at least two clusters of TnpB associated with Cas1 and/or CRISPR (excluding loci in which CRISPR arrays were linked to CRISPR-Cas systems other than type V; SI Appendix). We identified 40 distinct branches of putative Cas12s (V-U6 – V-U45, collectively referred to as V-U* and described in detail in SI Appendix) which are candidates for type V subtypes and variants, along with multiple previously defined Cas12 subtypes that are widely distributed across the tree (Fig. 1C). In general, Cas12s, in contrast to TnpBs, are relatively longer and are found more often in organisms that live at lower temperatures (SI Appendix, Fig. S1). The presence of multiple distinct clades of Cas12s is in accord with previous hypotheses on independent evolution of Cas12 from TnpB on many occasions (4, 9). Some of these Cas12s, such as the greater Cas12f branch, are consistently associated only with CRISPR arrays but not with Cas1, Cas2, or Cas4 (Fig. 1C). We found that in almost all evolutionarily conserved associations between Cas12 and CRISPR, the array is located downstream of the cas12 gene, mimicking the location of the ωRNA downstream of TnpB (Fig. 1C). This arrangement contrasts that of IscB and Cas9, where ωRNA or the CRISPR array are typically located upstream of the genes encoding IscB and Cas9d (subtype II-D), the likely evolutionary intermediate between IscB and typical Cas9s (1). The distinct architectures of the loci encoding these RNA-guided nucleases likely reflect the origin of CRISPR arrays from the respective ωRNAs (1). Of the 40 Cas12-U groups identified in this work, 15 have no members in NCBI genomes, underscoring the importance of metagenome analysis for discovering CRISPR systems. One of the identified systems, V-U24, contains a noncanonical RuvC-I active site and was found to be associated with an HTH domain protein of unknown function (SI Appendix, Fig. S2).
TnpBs are abundant among prokaryotes and are far more common than Cas12s, being found in 63% of archaea and 24% of bacteria, in contrast to 0.4% and 1.4%, respectively, for Cas12 (Fig. 2A). TnpBs are widely spread across diverse archaeal and bacterial lineages and are particularly common in the archaeal TACK superphylum and the bacterial phylum Aquificae (Fig. 2B). Analysis of complete archaeal and bacterial genomes reveals a patchy distribution of TnpB, with many genomes lacking these genes. Certain bacterial phyla, such as Tenericutes, Chlorobi, Chlamydiae, and Planctomycetes are especially TnpB poor (SI Appendix, Table S3).
Fig. 2.
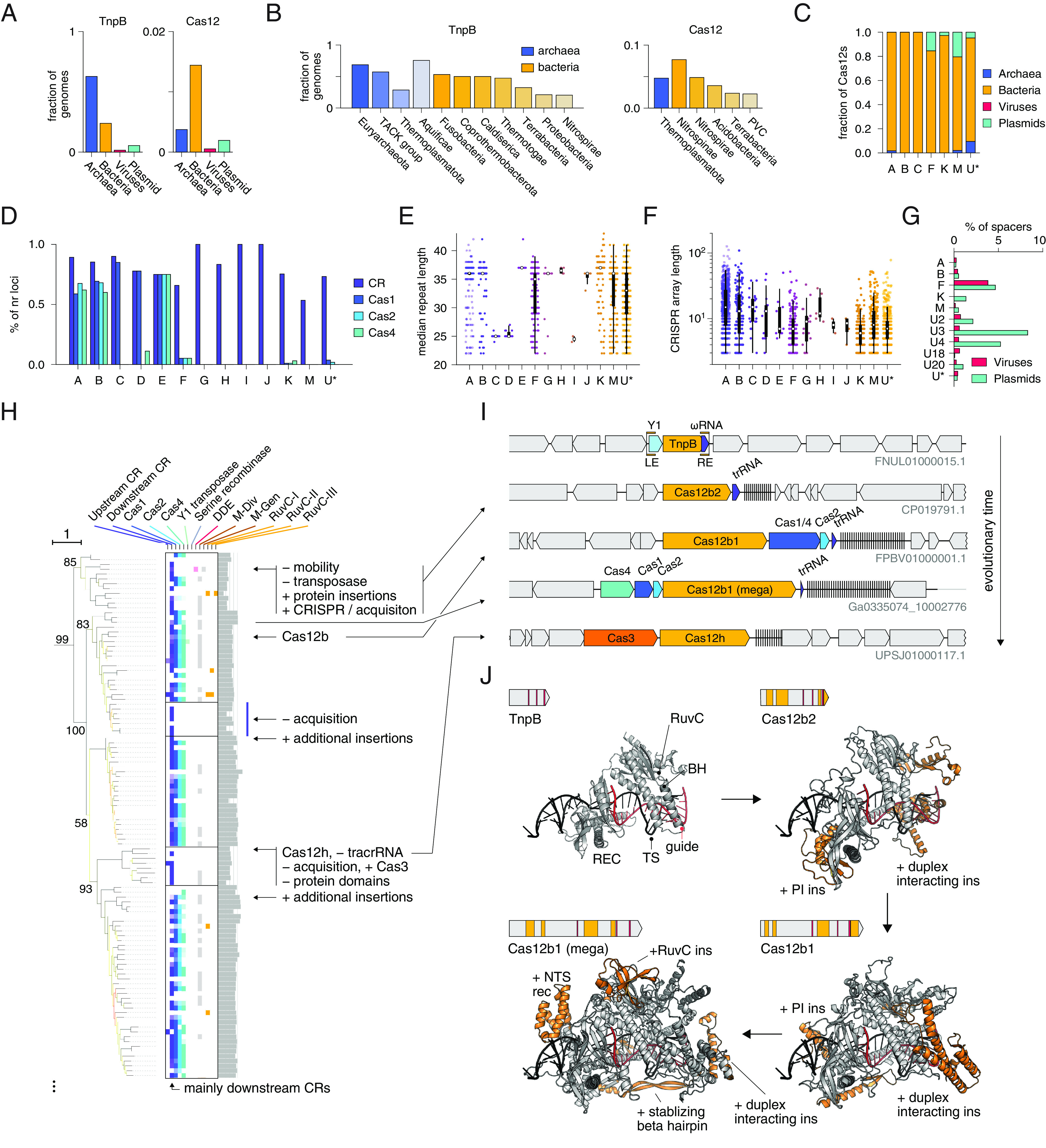
Transposon associations of TnpB and features of the corresponding genomic loci and proteins. (A) Distribution of TnpBs and Cas12 across archaea, bacteria, viruses, and plasmids. (B) Occurrence of TnpB and Cas12 in various phyla sorted by abundance. Phyla with lower abundance of TnpBs and Cas12s are not shown. (C) Distribution of Cas12 subtypes across archaea, bacteria, viruses, and plasmids. (D) Fraction of redundant loci containing CRISPR arrays (CR), Cas1, Cas2, and Cas4 for various Cas12 subtypes. (E) Median repeat length distributions of various Cas12 subtypes. Box and whisker plots shown; median (white circle), 25th and 75th percentiles(thick vertical black line), interquartile range (thin vertical black line). (F) CRISPR array length distributions for various Cas12 subtypes. Box and whisker plots as in (E). (G) Distribution of CRISPR array spacer matches of various Cas12 subtypes against viruses and plasmids. X axis shown as percentage of matches relative to all unique spacers (at 50% sequence identity). (H) Zoom-in of phylogenetic tree from Fig. 1 focusing on Cas12b evolution from TnpB. (I) Inferred evolution of Cas12b from TnpB, as well as potential evolution of Cas12h from Cas12b. Putative Y1-7 branch ancestor shown on top. (J) AlphaFold2 structural prediction of evolutionary stages of Cas12b evolution superimposed upon the Cas12b guide and DNA. The upper left of each Inset includes the effector protein along with RuvC active site positions (red) and insertions relative to recent common ancestor (orange).
Among the Cas12s, subtypes V-A, B, and C are represented almost exclusively in bacterial chromosomes, whereas subtypes V-F, V-K, V-M, and the diverse V-U* variants are often present on plasmids, and V-U* variants are additionally found in various archaea (Fig. 2C). The type V subtypes also vary in the content of other cas genes (Fig. 2D). The V-A, V-B, and V-E subtypes are stably associated with cas1, cas2, and cas4, whereas V-C and V-D only associate with cas1. Cas12s became associated with the CRISPR adaptation machinery (Cas1, Cas2, and/or Cas4) on at least 12 independent occasions (Fig. 1C). The subtypes V-G, V-H, V-I, V-J, V-K, V-M, and most of the V-U* variants are associated with CRISPR arrays, but not with any other cas genes. Thus, type V CRISPR-Cas systems evolved from OMEGA-TnpBs on numerous, independent occasions, giving rise to Cas12s that became linked to CRISPR arrays, either via de novo evolution of an array by serial duplication of ωRNA segments or as a result of insertion of a transposon near a preexisting array. Subsequent evolution proceeded along parallel lines in different type V subtypes and involved gradual increase of the effector size by accretion of protein domains enhancing the interaction with the guide RNA and the target, as well as capture of adaptation modules, on multiple, independent occasions.
Most type V subtypes have characteristic CRISPR DR lengths (Fig. 2E), with the exception of V-M and V-F, which is a highly heterogeneous group encompassing multiple tree branches. In contrast, CRISPR array lengths are highly variable within each Cas12 subtype (Fig. 2F). Typically, the derived V-A and V-B systems contain much longer CRISPR arrays than subtypes V-F, V-M, and many of the V-U* that appear to have relatively recently evolved from OMEGA-TnpB (Fig. 2F). Notably, subtype V-K loci contain the shortest arrays among all type V subtypes, consistent with its role as an RNA-guided target selector for associated Tn7 transposons (13).
CRISPR arrays in different Cas12 subtypes contain spacers targeting viruses and plasmids at different frequencies, suggesting different biological roles (Fig. 2G). In particular, the spacers in V-K-associated arrays target plasmids, but not viruses, consistent with the CAST inserting into plasmids via the RNA-guided route. However, the majority of the previously characterized Type V subtypes target viruses, consistent with their role in adaptive immunity (14). The spacers in V-F arrays target a mix of phages and plasmids, whereas the spacers of V-U18 identified here primarily target viruses. For the rest of the identified V-U* variants, there were too few spacer matches to make conclusions on targeting specificity (SI Appendix, Table S4).
Evolution of Cas12s from TnpBs.
Most of the known Cas12s belong to the Derived TnpB major clade, with the exception of Cas12m, Cas12j, and Cas12f3 (Fig. 1C). The same does not hold, however, for the predicted V-U systems (V-U6 - V-U45): Only 13 out of 40 belong to this clade. In many cases, known or predicted Cas12s group with potentially mobile TnpB clades or with each other (Fig. 1C).
Although phylogenetic artifacts cannot be completely ruled out due to the substantial sequence divergence, some of the relationships among Cas12s and TnpBs are strongly supported. In particular, we confirmed several previously reported affinities, such as those of Cas12c with Cas12d (15) as well as Cas12a1-Cas12a2 and Cas12b1-Cas12b2 (16). The latter case is of special interest because we now can trace the origin of Cas12b1 from the ancestral Cas12b2 and reconstruct the overall evolution within this clade in greater detail (Fig. 2 H and I). Although we cannot confidently identify the direct TnpB ancestors of the Cas12b1/Cas12b2 clade, it appears to emerge from a TnpB branch most often associated with Y1 TnpA (e.g., Y1-6 and Y1-7 on the Fig. 1C) (SI Appendix, Additional Files 2 and 5 and Table S1). The Cas12b2 branch that includes experimentally uncharacterized Type V-B2 effectors is the deepest in the Cas12b1/Cas12b2 clade (Fig. 2H). This placement is consistent with the smaller size of Cas12b2 proteins (~700 to 800 aa) compared to the other Cas12bs. Furthermore, some of these predicted type V-B2 loci also encode the adaptation module consisting of a Cas1-Cas4 fusion and Cas2. This fused cas1-cas4 gene so far was found only in type V-B systems (15). Cas12b2s are currently found mostly in Planctomycetota genomes. By comparing the AlphaFold2 model of a putative ancestral TnpB and the previously solved structure of Cas12b1 (17), we identified multiple insertions of alpha-helical subdomains that accreted en route from TnpB to Cas12b1 and likely interact with the guide:target heteroduplex and the PAM (Fig. 2J). Another clade in the same subtree (Fig. 2H) consists of large, ~1,500 aa in size, Cas12b1 proteins which acquired additional domains that might enhance the interaction with the guide RNA:target DNA duplex (Fig. 2J). Notably, the loci in this clade encode a distinct adaptation module (Fig. 2I). There are several additional, sporadic expansions of the protein size along this subtree, with the largest Cas12b protein reaching 1,888 aa (Fig. 2H). Another subclade includes proteins similar to previously reported Cas12h (18). Although a weak similarity between Cas12h with Cas12b was identified before (18), our tree reconstruction suggests that an ancestor of Cas12h secondarily lost some of the previously acquired subdomains because Cas12h family sequences are typically shorter (~900 aa) than Cas12b1 (~1,200 aa), and also lost the adaptation module. We further observed that the Cas12h variant likely gained association with the Cas3 effector characteristic of type I CRISPR systems, potentially leading to an alternative mode of function for this variant. The Cas3 protein in this system contains an active HD domain as well as a complete, apparently active helicase domain, and Cas12 is also predicted to be an active nuclease. Overall, these findings suggest that evolution of Cas12 proceeds in different directions including both expansion and secondary reduction of the effector protein, yielding broad size variation, and gain/loss of CRISPR-Cas modules.
Catalytic Site Rearrangements and Inactivation in TnpBs and Cas12s.
In two major clades and 8 large branches, the RuvC-II motif of TnpB (or Cas12) is altered, resulting in a rearranged catalytic site (Fig. 1C). In the largest of these groups (RIIr-5), the catalytic glutamate of the RuvC-II motif is replaced with glycine (Fig. 1D). AlphaFold2 predictions of the structures of these TnpBs suggest that the loss of this catalytic residue is compensated by an alternative glutamate which is located in a protruding loop that faces toward the catalytic site and is involved in similar hydrogen bonding interactions with the RuvC-I aspartate as the canonical RuvC-II glutamate (Fig. 3). This architecture of the putative altered catalytic site is similar to that in the RuvX Holliday junction resolvase family, in which the catalytic glutamate is located in β-5 as opposed to β-4 of the RNaseH fold in the canonical RuvC (19, 20).
Fig. 3.
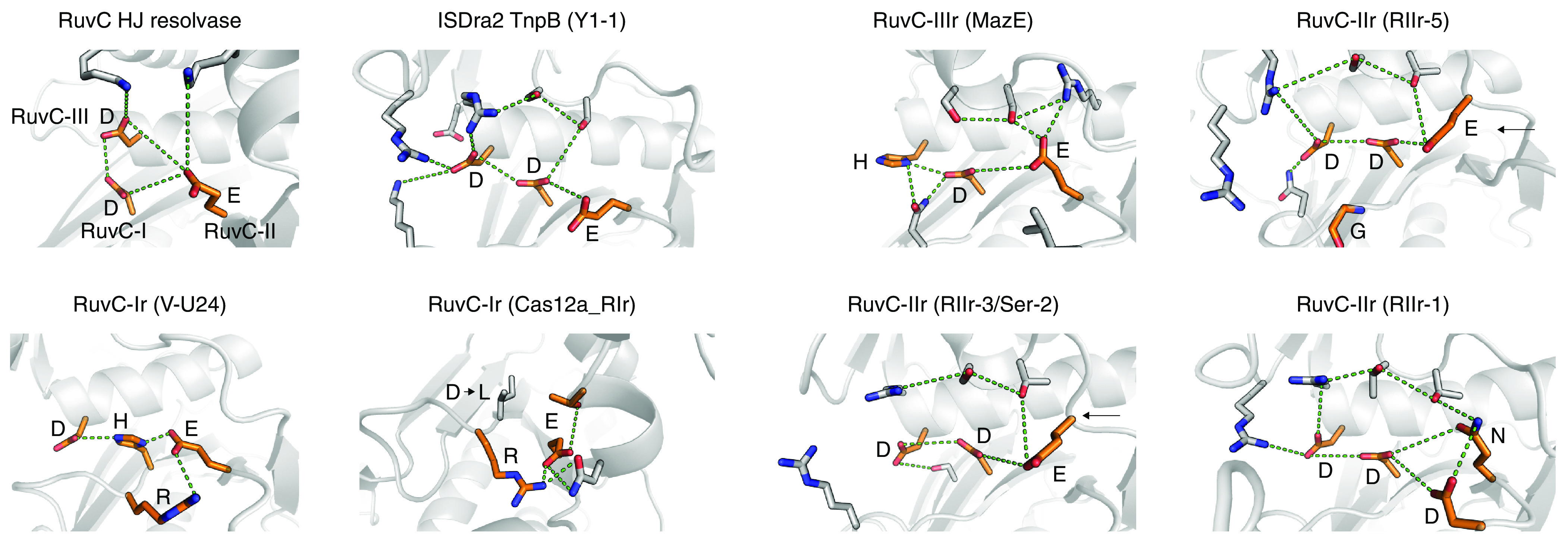
Catalytic site rearrangements of TnpB. Structural comparisons of various catalytic site rearrangements in TnpBs and Cas12s using AlphaFold2 predictions. RuvC HJ resolvase is from PDB: 1HJR.
Additionally, in six large branches, the RuvC-III catalytic motif is altered (Figs. 1C and 3). In the largest of these (RIIIr-4), including TnpBs associated with an RHH/MazE-like antitoxin, the catalytic aspartate of the RuvC-III is mutated to histidine (Figs. 1D and 3). The conservation of specific alterations of the catalytic sites in these branches of the TnpB tree implies that many groups of TnpBs with amino acid replacements in the catalytic site retain the nuclease activity, potentially with altered substrate specificities, as observed with alternative catalytic arrangements across the diversity of the RNaseH-fold enzymes (19). Additionally, apparent complete inactivation of TnpBs and Cas12s was observed, where all 3 catalytic residues are mutated. Large branches of Cas12s with altered catalytic sites are observed in Cas12a, Cas12k, and Cas12f subtypes, as well as various V-U* subtypes such as V-U24, V-U43, and V-U44. Large branches of inactivated TnpBs include Ri-7 and Ri-8, where consistent associations with other proteins were detected. Subtype V-M presents a notable pattern of gradual inactivation of the catalytic site (Fig. 1C and SI Appendix, Fig. S3A), and it has been shown that at least some Cas12m variants suppress plasmid replication by downregulation of essential genes, without cleaving the target (21).
The catalytic site rearrangements are nonuniformly distributed across the TnpB tree (SI Appendix, Fig. S3A). In most Y1-associated groups, about 15% of the TnpBs carry rearranging or inactivating catalytic amino acid replacements, whereas among the different groups of serine recombinase-associated TnpBs, the fraction of those with catalytic site alterations is much more variable. Rearrangements of the catalytic site of TnpB furthermore show a nonrandom distribution across bacterial and archaeal taxa (SI Appendix, Fig. S3B). Several rearrangements are found nearly exclusively in archaea, such as those in the Ser-6, MazE, RIIIr-6, RIIr-6, and RIIr-8 branches. Furthermore, some branches with rearranged catalytic site are specifically represented in viruses (RIIr-4, Y1-5, and RIIIr-4) or plasmids (Y1-1, Y1-5, DDE-5, and others) which likely disseminate TnpBs including the catalytically rearranged variants among diverse bacteria and archaea.
Most TnpBs and some Cas12s contain a conserved C4 Zinc Finger (ZF) between the RuvC-II and RuvC-III domain. The function of these ZFs is unclear, and they are lost or mutated in some large TnpB branches (SI Appendix, Fig. S3 C and D), suggesting lineage-specific roles, such as protein stabilization or ion concentration-mediated modulation of the enzymatic activity.
Associations between TnpB and Diverse Transposases.
We next analyzed the associations between each TnpB cluster and various genes including transposases and CRISPR spacer acquisition (adaptation) machinery. In contrast to IscB and IsrB, TnpBs are strongly associated with Y1 transposases (IS605 group) and serine recombinases (IS607 group) (Fig. 4) as previously described (22, 23). Notably, however, the associations are not consistent within a clade, and many cases of apparent loss and gain of transposases were observed, indicating that the genomic association between TnpB and Y1/serine recombinases is polyphyletic—in four of the five major clades (all except RIIr-3), TnpBs associate with both Y1 and Serine Recombinase type TnpAs on separate, independent occasions (Fig. 1C). The strong preferential connection between TnpB and Y1/serine recombinases, compared to other transposases, implies that these are functional rather than stochastic associations, that is, these transposases and TnpB function in conjunction in the respective transposon life cycles. Previously, IS200 and IS605 were not known to carry any cargo genes; however, we observed that, on rare occasions, some members of specific large branches, for example, Ser-3, are associated with an additional HTH domain protein and other cargo genes, such as an ATP-dependent RNA helicase or a WhiA/meganuclease-like protein, as well as many proteins with no similarity to known domains (Fig. 4).
Fig. 4.
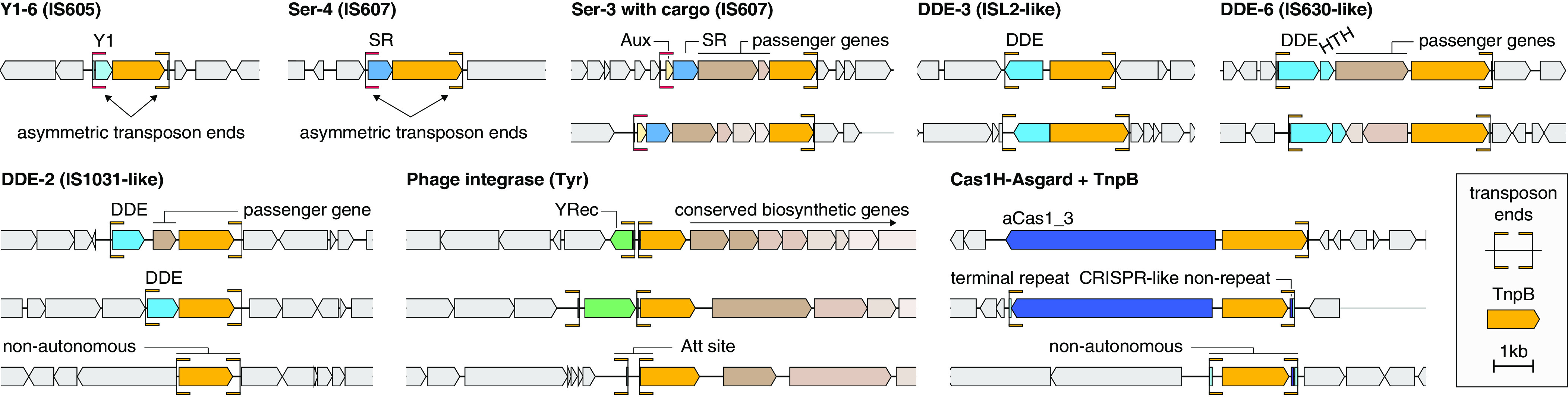
Transposons associated with TnpBs. Genomic loci of TnpBs along with their associated transposons.
On some occasions, the Y1/serine recombinases are replaced with phage tyrosine family integrases, DDE transposases, or Asgard Cas1s, variants of Cas1 (aCas1s) (24) (Fig. 4). In the DDE-6 large branch, the TnpBs are associated with DDE transposases of the IS630 transposon family along with a separate HTH domain protein. In this case, the transposon ends are symmetrical, in contrast to the transposon ends of the IS605 and IS608-like transposons (Fig. 4). We identified several of these IS630-linked TnpBs in viral genomes, largely, in members of the Klosneuvirinae subfamily of the giant virus family Mimiviridae that likely infect various unicellular eukaryotes (25). These IS630-encoded TnpBs might represent the evolutionary link between prokaryotic TnpBs and the previously reported Fanzors, eukaryotic TnpB homologs found in fungi, some unicellular eukaryotes and giant viruses that are also associated with diverse transposons (26) (Fig. 1C).
We observed additional associations between TnpB and phage integrase-like tyrosine recombinases (Fig. 4) that apparently occurred on at least three independent occasions (Fig. 1C) and all coincide with complete inactivation of TnpB. The tyrosine recombinase gene occasionally appears upstream of the TnpB in a seemingly random orientation relative to the TnpB and is itself often surrounded by inverted repeats that are indicative of attachment sites. On some occasions, the tyrosine recombinase is missing but the apparent attachment site is still present upstream of TnpB.
Our analysis places aCas1-associated TnpBs in two distinct Asgard archaea-linked groups (Figs. 1C and 4, Cas1H-Asgard-1 and Cas1H-Asgard-2). The aCas1 is likely to be the transposase of a distinct family of transposons given that the aCas1-TnpB locus is often flanked with repeats (Fig. 1C). Additional analysis of these loci revealed a distinct region downstream of tnpB containing a hypervariable region flanked by a single upstream conserved T nucleotide and a downstream conserved GTTCACTCA motif, which is nearly identical to the 5′ GTTCACTGC motif of a previously studied CRISPR array (SI Appendix, Fig. S4) (27). This hypervariable region could encompass the spacer-like guide of TnpB’s ωRNA, though further work is required to understand the nature of these conserved regions.
To further assess whether these diverse transposase associations were functional, we used sequence conservation to identify putative transposon ends. We found asymmetric terminal repeats surrounding Y1-associated TnpBs, asymmetric ends surrounding serine transposase-associated TnpBs, and inverted terminal repeats for all six DDE transposase-associated TnpBs (Fig. 4 and SI Appendix, Additional File 10). In most examined cases, the TnpB 3′ end was found within 250 bp of the transposon end, suggesting that they employ related mechanisms for obtaining and expressing ωRNA guides. In some cases, we found that solo TnpB is surrounded by transposon ends that are nearly identical to those from systems containing the transposases (SI Appendix, Additional Files 10 and 11), suggesting that some TnpBs are mobilized by transposases in trans. These observations further imply that association of TnpB with diverse transposons is functionally relevant and that the function(s) of TnpB is transferable between different transposons.
Mobility of TnpB.
The evolutionary dynamics of transposon copy number depends on many factors such as the host lifestyle, evolutionary bottlenecks, and stress (28, 29). Generally, transposon families show high genomic flux, that is, they are prone to gain, loss, expansion, and contraction within the same genome (30). Previously, we observed that at least one group of TnpB homologs associated with a CRISPR array (Cas12f) apparently lost mobility as indicated by the absence of (nearly) identical copies of the gene in the same genome (9).
We sought to characterize mobility across the entire diversity of TnpBs and to identify groups of immobilized TnpBs that could have been recruited for alternative roles. To this end, we calculated a mobility metric (M-gen in Fig. 1C) that estimates the presence or absence of multiple copies of nearly identical TnpBs within the same genome for complete genomes (Brief Methods). We compared the mobility indicators for TnpB and 28 other transposons families, including IS3, IS4, IS5, and ISNCY which are also highly abundant in prokaryotic genomes (29). TnpB differed from most other transposons by frequent immobilization, with 89% of the clusters found to be immobile compared to the median of 66% for other transposon families (SI Appendix, Fig. S5A). Furthermore, the distributions of the copy number per genome were similar for IS3, IS4, and IS5 transposons, whereas TnpB is clearly distinct (SI Appendix, Fig. S6). The shapes of all distributions are compatible with evolution under a birth–death model (31), but the TnpB distribution has a steeper slope and, accordingly, lacks large, highly proliferated (>30 copies) clusters that comprise up to 1% of clusters for other transposons. These observations are compatible with the apparent lower mobility of TnpB-containing elements.
We further sought to determine whether the presence of TnpB correlated with the mobility of the corresponding IS elements. We compared different IS architectures and found that IS605 (TnpB and Y1 transposase) is the most mobile one, followed by IS607 (TnpB and serine transposase), IS1341 (coding for a single TnpB), and IS200 (Y1 transposase alone) (SI Appendix, Fig. S5A). Standalone serine transposases showed the lowest recent mobility (SI Appendix, Fig. S5A). Thus, transposons containing TnpBs appear to be more mobile, on average, than their counterparts lacking TnpB (χ2 P-value 2 × 10−128). Furthermore, active TnpB nucleases are more mobile compared with the (predicted) inactive ones (χ2 P-value 2 × 10−5, SI Appendix, Fig. S5A). Last, solo TnpBs are often found within transposon ends related to IS200/605, suggesting that they could be mobilized (32). Indeed, we observed that solo TnpBs are also mobile, with a higher mobility rate than solo Y1 or serine transposases. These observations together suggest that TnpB cooperates with the associated transposases, in cis or in trans, to facilitate the mobility of IS elements and are consistent with the proposed role of TnpB in transposition (1, 2), whereas inactive TnpBs could have acquired different functions.
Next, we explored the taxonomic distribution of various transposon-associated large branches of the TnpB tree in archaea and bacteria. In general, archaea contain a notably greater number of tnpB genes per genome than bacteria, but fewer genes demonstrating recent mobility (SI Appendix, Fig. S5A). These observations seem to suggest that, compared to bacteria, the high abundance of archaeal TnpBs results from a much longer-term accumulation and/or more frequent acquisition by horizontal gene transfer (HGT). Among archaea, high recent TnpB mobility was observed in Methanosarcina and Sulfolobus (SI Appendix, Table S5). Among bacteria, the genera with the highest abundance of (nearly) identical TnpB copies (15 or more from at least one TnpB cluster), which indicates high recent mobility, are Kurthia, Geobacillus, Aeromonas, Helicobacter, Clostridium, Synechococcus, Megamonas, Caldibacillus, and Limosilactobacillus (SI Appendix, Table S5).
We next investigated within-clade differences in TnpB mobility by comparing the average mobility within a given genome (M-gen) across large branches (SI Appendix, Fig. S5B). We found that the fraction of mobile TnpBs was relatively constant across the different large branches of TnpBs associated with Y1 or serine recombinases, with the exception of Y1-6, which showed a lower mobility. The major clades with catalytic site rearrangements (Ser-2, Ser-5, and RIIr-5) all showed similar levels of mobility (SI Appendix, Fig. S5B). We further checked whether the mobility estimated from the sequence divergence before mobilization (M-div in Fig. 1C and SI Appendix) varied across branches and found that DDE-2, DDE-3, DDE-5, and DDE-6 mobilize regularly, suggesting that these are active IS elements (SI Appendix, Fig. S5C). Furthermore, Y1-7 apparently mobilizes often relative to its diversification rate compared to other TnpBs associated with Y1 transposases (SI Appendix, Fig. S5C). We further found that serine recombinase and DDE associated TnpBs mobilize less frequently than Y1 associated TnpBs (SI Appendix, Fig. S5C). Cas12s and tyrosine recombinase-associated TnpBs also were found to be mobile, likely due to HGT and placement in genomic recombination hotspots (SI Appendix, Fig. S5C).
Last, we investigated the potential effect of mobility on the extent of TnpB diversification. We estimated the number of lineages formed per cluster of mobile vs. immobile TnpBs and found that mobile TnpBs are associated with substantially more lineages than immobile ones (P < 1e−4, Kolmogorov–Smirnov test, SI Appendix, Fig. S5D), suggesting that mobilization of IS200/605-like transposons plays a role in TnpB lineage generation and diversification. TnpB could be involved in transposon life cycles through a number of potential mechanisms (SI Appendix, Fig. S5E and Discussion).
Immobilization and Exaptation of TnpB.
Immobilization of transposons, especially, when accompanied by inactivation of transposon-encoded enzymes, suggests the possibility of recruitment for alternative functions, known as exaptation (33–35). To identify potential cases of TnpB exaptation, we examined several low-mobility branches with substantial species diversity (at least, including different species within a genus) as well as systems with neighboring gene associations reported here (Fig. 1C and SI Appendix, Table S1 and Additional Files 5, 8, and 9). Multiple cases of apparent exaptation in diverse contexts were identified (Fig. 5A).
Fig. 5.
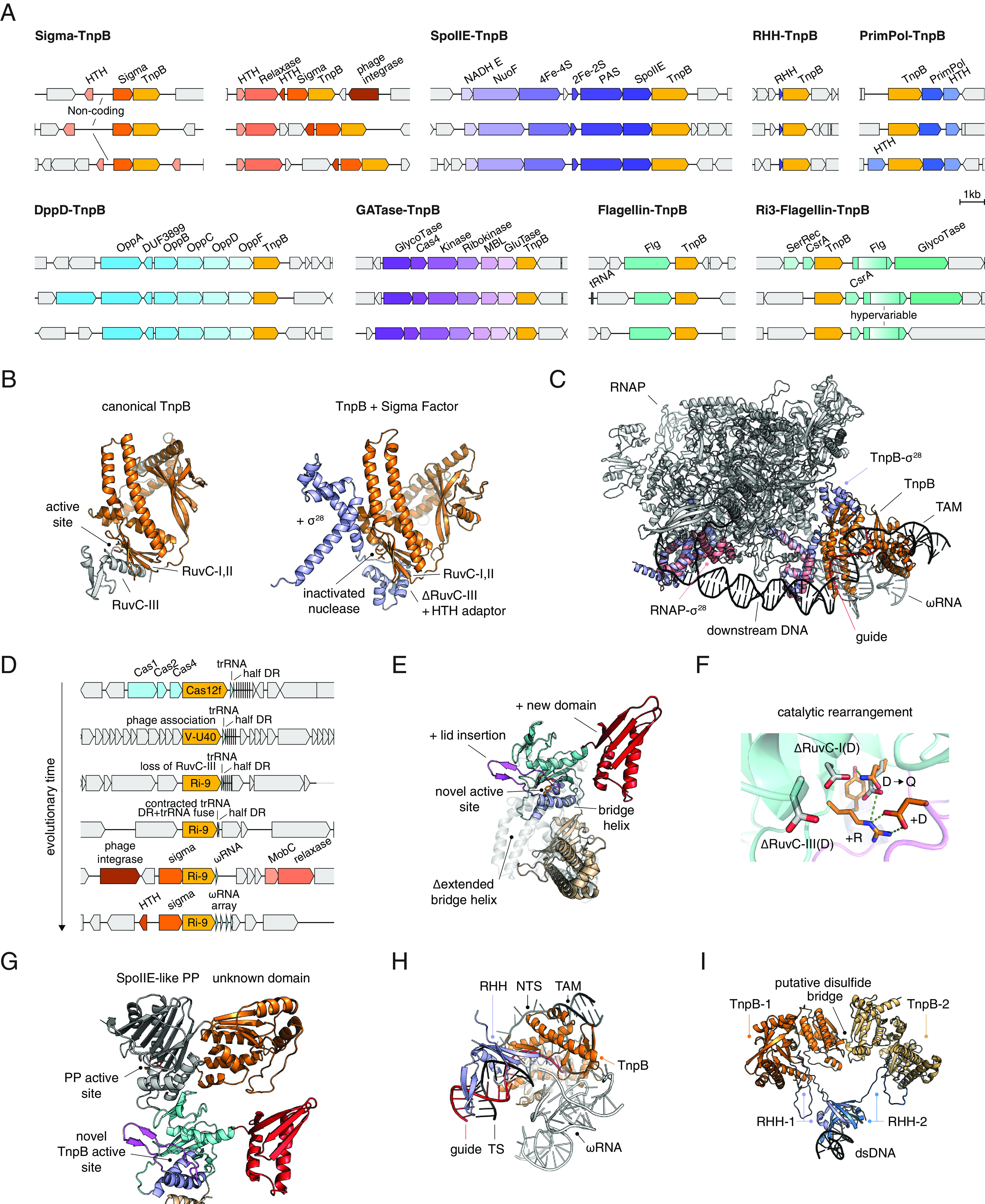
Derived and exapted TnpB systems. (A) Various identified systems with TnpBs recruited for alternative biological functions. (B) Structural comparison of TnpB with the cofolded TnpB + Sigma Factor system. (C) Structural superimposition of TnpB + Sigma Factor with the Sigma28-RNAP complex (PDB: 6PMI). (D) Probable evolutionary scenario of conversion from Cas12f into an inactivated TnpB system associated with RpoE (Sigma), relaxases, MobC, phage integrases. The likely evolution of the associated RNAs is also shown. (E) Structural model of the TnpB from the SpoIIE-TnpB system. A previously unreported lid domain covers the RuvC active site of TnpB. (F) Rearranged catalytic site of the SpoIIE associated TnpB with potentially novel function. (G) Cofolding of SpoIIE with associated TnpB. (H) Structural model of the TnpB from the RHH-TnpB (MazE-TnpB) system. (I) Structural model of the RHH-TnpB complex from the RHH-TnpB system, along with superimposition of the RHH on a related DNA binding protein (PDB: 2MRU).
Sigma-TnpB.
The largest of these exapted TnpB clades includes more than 60 genomes from Flavobacteriales, Chitinophagales, Sphingobacteriales, and Prevotellaceae species in the phylum Bacteroidota (Figs. 1C and 5A and SI Appendix, Tables S1 and S6 and Additional Files 8 and 9). These TnpBs are tightly linked to the rpoE gene which encodes an extracytoplasmic RNA polymerase sigma factor specific for Bacteroidetes that is involved in heat shock response (36, 37) (https://www.ncbi.nlm.nih.gov/genome/annotation_prok/evidence/TIGR02985/). Typically, an Xre family transcriptional regulator is encoded in the vicinity (SI Appendix, Fig. S7A). In some clusters, there is a separate conserved relaxase gene upstream of TnpB, whereas in others, there is a large noncoding region between TnpB and the Xre protein (SI Appendix, Fig. S7A). The nuclease domain in this group of TnpBs is most likely inactive as indicated by the replacement of the catalytic residues in all three catalytic motifs of the RuvC-like nuclease.
The clade of RpoE-associated inactivated TnpBs is lodged within a larger clade in which most other proteins are known or predicted to be active nucleases, largely, of the Cas12f1 and V-U40-43 groups (Fig. 1C), that are associated with divergent CRISPR-like arrays with long direct repeats resembling ωRNA arrays (1). Thus, the RpoE-associated TnpBs appear to have evolved from already immobilized CRISPR effectors (Cas12f1) by inactivation of the nuclease (we denote these inactivated nucleases TnpB despite their apparent origin from Cas12, to emphasize that they are no longer associated with CRISPR-Cas systems). In this case, the CRISPR array likely evolved into a regulatory RNA because the CRISPR array association is lost in this clade (Fig. 1C). We observed the occasional presence of highly irregular ωRNA-like arrays with unusually long repeats (>60 bp) that might reflect an alternative guide capture mechanism (1) (cluster 17474, SI Appendix, Fig. S8 A and B). These repeats have sharp boundaries that in ωRNA separate the scaffold from the guide. In addition to the noncoding ωRNA, there is also a large, variable in size, yet highly conserved noncoding region between the Xre and RpoE genes (SI Appendix, Fig. S7C).
The apparent inactivation of these TnpBs and the association with RpoE suggest that they are involved in gene expression regulation. To investigate this hypothesis further, we built paired AlphaFold models of these TnpBs together with the associated RpoEs and observed that in the complex, the missing ZF motif and RuvC-III motif were replaced by a previously unreported HTH-like domain that appears to mediate the interaction with the RpoE (Fig. 5B). The interface was predicted with low position alignment error (PAE) < 7.5Å, indicating high confidence in the position of the distinct domain of RpoE relative to TnpB. We superimposed the RpoE -TnpB model with the experimentally determined structure of the RNAPol-Sigma28 complex and observed that the orientation of the predicted RpoE-TnpB was compatible with RNA-guided dsDNA binding by TnpB and recruitment of the RNA polymerase by RpoE (Fig. 5C). Therefore, we hypothesize that the RNA-guided DNA-targeting function of TnpB was co-opted as a mechanism for target gene recognition that is alternative to the promoter sequence recognition by RpoE.
By tracing the evolution of the RpoE-linked TnpBs, we delineated the likely evolutionary scenario for this complex system (Fig. 5D and SI Appendix, Fig. S8). The branching order in this clade of the tree (Fig. 1C) implies that Cas12f lost the catalytic activity along with the RuvC-III motif. Subsequently, the CRISPR array was minimized, leaving only a half DR in close proximity to the tracrRNA. The half DR and the tracrRNA then fused, leading to the reemergence of an ωRNA that maintains the same 3′ end of the ωRNA scaffold as the original CRISPR DRs in Cas12f loci. Concomitantly, the newly formed inactivated TnpB gained association with RpoE, MobC, relaxase genes, HTH domains, and phage integrases (Fig. 5D). The entire ωRNA subsequently duplicated to form ωRNA arrays capable of targeting multiple sites.
SpoIIE-TnpB.
Another relatively small clade of nonmobile TnpBs that includes diverse Clostridia (Figs. 1C and 5A) likely followed a similar evolutionary trajectory. This clade is positioned adjacent to a large branch that includes recently described but not yet experimentally characterized type V system (38) denoted here as V-U38 (Fig. 5A). Similarly to the RpoE-associated TnpBs, this tnpB gene is located near a long noncoding region that could encode a ωRNA-like RNA that defines the target of TnpB-based regulation. The tree topology suggests that, as in the case of the RpoE-associated TnpBs, this TnpB was first immobilized, giving rise to a group of active Cas12s, and then, a member of this group lost the nuclease activity and evolved into a family of regulators (Fig. 1C). In this case, TnpB is associated with a SpoIIE-like serine/threonine phosphatase and an iron-only hydrogenase. These TnpBs encompass several notable modifications to the TnpB fold, in particular, a lid insertion that covers the TnpB active site, apparent deletion of the bridge helix that typically interacts with the RNA-DNA duplex and a distinct domain inserted at the C terminus (Fig. 5E; see Fig. 2J for reference). Although the canonical catalytic site of TnpB is inactivated in these proteins, there is a conserved arginine in RuvC-I paired with a conserved aspartate in the lid insert that might confer a distinct biochemical activity (Fig. 5F). Moreover, TnpB in this system appears to form an interaction interface with the SpoIIE-like phosphatase as predicted by paired AlphaFold modeling (Fig. 5G). The SpoIIE-like phosphatase also contains an uncharacterized globular domain that appears to be separate from the phosphatase domain (Fig. 5G). Given that in sporulating bacteria, such as Clostridia, the SpoIIE phosphatase dephosphorylates and hence activates the RpoF sigma factor involved in sporulation (39, 40), the complex of TnpB with the SpoIIE phosphatase is likely involved in transcription regulation similar to TnpB-RpoE.
Flagellin-TnpB.
We identified two clades that feature TnpBs encoded in a putative operon with flagellar hook-associated protein FlgL (Fig. 5A) (41). The association between TnpB and FlgL apparently evolved in these clades independently (SI Appendix, Additional File 5). One clade is specific for Enterobacteriaceae and the operon apparently consists of two genes, flgL and tnpB. In addition to the replacement of the catalytic residues in RuvC-I and RuvC-II, these TnpBs are truncated at the C terminus and thus completely lack the RuvC-III. The second clade, designated Ri3-Flagellin is present in several Oscillospiraceae genomes (Fig. 5A). In this case, the flgL gene contains a hypervariable region in the middle of the protein and is also associated with a gene encoding a CsrA-like RNA binding protein. The long 3′ untranslated regions downstream of tnpB genes in both cases likely encode guide RNAs. As in other cases of TnpB immobilization, a regulatory role of these systems appears likely.
DppD-TnpB.
Another example of potential exaptation of inactivated TnpB was identified in several Enterococcaceae species. In these loci, TnpB is the distal gene in a putative operon encoding components of a peptide ABC type transporter, and the 3’ untranslated region is long enough to accommodate an RNA guide (Fig.5A). Most likely, TnpB in this case also performs regulatory functions. Notably, unlike the Sigma-TnpB and SpoIIE-TnpB cases, in flagellin-TnpB and DppD-TnpB, the immobilized and inactivated TnpBs seem to have evolved directly from mobile, active TnpBs, without a CRISPR effector intermediate (SI Appendix, Table S1).
RHH/MazE-TnpB.
The larger groups of immobilized, apparently exapted tnpB genes encode derived forms predicted to be inactive. Other nonmobile clades encompass TnpBs that are predicted to be active nucleases (SI Appendix, Table S5). In most cases, these tnpB genes are not linked to other genes, with a few notable exceptions. One such exception is an archaeal clade that includes members from several lineages of euryarchaeal and crenarchaeal TnpBs with a rearranged catalytic site (SI Appendix). These tnpB genes are located downstream of a gene encoding a small DNA-binding protein of either the AbrB/MazE or the ribbon-helix-helix (RHH) family (Fig. 5A). The RHH proteins are common antitoxins in type II toxin-antitoxin (TA) systems (42). In most of these cases, there is a long untranslated 3′ region that could encode a guide RNA. Considering the presence of a putative antitoxin and a patchy distribution of this system in archaea, the typical feature of toxin-antitoxin modules, the most plausible hypothesis is that these two genes comprise a TA system in which the TnpB nuclease is a toxin. Using AlphaFold, we cofolded the catalytically rearranged TnpB with the associated RHH protein (Fig. 5H). The model shows that the RHH antitoxin likely interacts with TnpB in such a manner as to prevent the interaction between TnpB and the guide RNA-DNA heteroduplex. Inhibition of the RNA-guided target DNA binding resulting in prevention of the target cleavage likely accounts for the role of the RHH protein as the antitoxin to the TnpB toxin. Analysis of a cofolding model including dimers of both TnpBs and the RHH protein revealed an interface between the TnpBs including a disulfide bridge, which would stabilize the complex, especially, at high temperature, and a possible DNA interacting component that may conditionally regulate transcription (Fig. 5I).
PrimPol-TnpB.
A distinct branch specific for Deinococcus species includes catalytically active TnpBs that form a predicted operon with genes encoding a Primase-Polymerase (PrimPol) (43) and an HTH domain protein (SI Appendix, Table S1). Previous work with CRISPR-associated PrimPols (CAPPs), albeit from a different subfamily, has shown that the DNA polymerase activity of PrimPol contributes to spacer acquisition in cooperation with Cas1 and Cas2 (44). Thus, the PrimPol that is associated with TnpB in Deinococci also might aid guide acquisition.
GATase-TnpB.
Another archaeal clade consists of TnpBs from the Sulfolobaceae. In most of these loci, the tnpB gene is located next to a gene for glutamine amidotransferase (GATase), and there is not enough room for an RNA gene downstream of the TnpB although, as noted previously, an ωRNA gene can overlap with the TnpB CDS (Fig. 5A) (1). However, these TnpBs are unusually small (~280 aa) and appear to consist of the wedge and catalytically active RuvC nuclease domains only, so it is uncertain whether they can bind a guide RNA. The function of these systems remains unclear but is likely to require the nuclease activity of TnpB, possibly, in an RNA-independent manner.
Other nonmobile TnpB systems.
Many other nonmobile TnpBs appear to be fixed in the evolution of some lineages despite the fact that the RuvC domain appears to be active (SI Appendix, Table S5). For example, a nonmobile branch that is specific to Halobacteria (e.g., WP_004214924.1 from Natrialba magadii) includes very short TnpB proteins (~220 aa) that consist of the RuvC domain alone and are unlikely to bind a guide RNA. Some clades show no or low mobility even when TnpBs are associated with transposases (e.g., WP_000978855.1 from Bacillus thuringiensis and WP_157823306.1 from Bifidobacterium longum). Thus, some autonomous TnpB-encoding transposons appear to retain mobility potential for extended periods of evolution without amplification in the genomes, suggesting the possibility of exaptation.
Discussion
Apart from core proteins involved in essential cellular functions, TnpB is one of the most abundant proteins in bacteria and archaea. Until recently, however, these proteins received virtually no attention from researchers. The situation changed dramatically after TnpBs were identified as likely ancestors of type V CRISPR-Cas effectors (1, 2). The demonstration that TnpBs are RNA-guided nucleases solidified the scenario for type V CRISPR origin. In this work, we showed that the association between TnpB and CRISPR evolved independently on a strikingly large number of occasions - more than 50 cases already identified, with many more likely to be discovered. TnpBs are typically encoded in IS200/IS605 transposons, either nonautonomous ones in which tnpB is the only gene or, less commonly, autonomous ones that also encode a transposase.
Comparative analysis of TnpB sequences supplemented by structural modeling demonstrated flexibility of the catalytic scaffold of these nucleases, with the catalytic glutamate of the RuvC-II motif replaced by an alternative glutamate in large TnpB clades. The structural models strongly suggest that this rearrangement produces a distinct configuration of the catalytic site rather than inactivation. The mechanistic and functional consequences of this catalytic site rearrangement, in particular, its effect on the substrate specificity and kinetics of TnpB, remain to be studied experimentally.
The presence of TnpB appears to enhance the mobility of the encompassing transposon (SI Appendix, Fig. S4A) and to help the transposon persist within the population (45, 46). As indicated by recent results, TnpB targeting of dsDNA at sites from which a transposon was excised could potentially initiate homology directed repair with a transposon-containing locus, resulting in transposon restoration in the original site and thus acting as an alternate mechanism of transposon propagation (SI Appendix, Fig. S5E) (45). TnpB also could play a number of other mechanistic roles in transposon maintenance (SI Appendix, Fig. S5E). In particular, TAM-independent ssDNA cleavage by TnpB (1, 2) might help target the transposon locus during replication by producing a stalled replication fork, which is the preferred substrate for transposon excision by IS608 TnpA (12). Second, TnpB might employ TAM-dependent dsDNA targeting (1, 2) to cleave homologous loci where the transposon is not inserted, decreasing the fitness of cells with uninserted sites, possibly, with the aid of the collateral ssDNA cleavage. The association between TnpB and many types of transposases suggests that its function is partially agnostic to the transposition mechanism. The diversity of the associated transposases further implies a degree of modularity of the guide adapter hairpin of the respective ωRNAs, which encompass the transposon ends. This is supported by recent work on engineering Cas12f-CRISPR systems for genome editing, which demonstrated that removing the crRNA:tracrRNA hybrid hairpin from the engineered sgRNA did not inhibit the cleavage activity of Cas12f (47).
The frequent mobilization of transposons containing TnpB (and also IsrB and IscB) results in constant exchange of the guide sequences used by TnpB. This rapid swapping of guide sequences by transposons coupled to the transposon’s life cycle and fitness likely drove the evolution of the ability of TnpB to function with arbitrary guide sequences, rendering it a reprogrammable RNA-guided system.
In this study, we systematically assessed the genomic mobility of TnpBs conjecturing that immobilization is associated with exaptation of TnpB for cellular functions unrelated to transposon activity. The origin of type V CRISPR-Cas systems is a well characterized and arguably the most prominent case of such exaptation (9, 48). Indeed, as RNA-guided nucleases, TnpBs and IscBs appear to be preadapted to evolve into CRISPR effectors. The numerous independent origins of type V CRISPR effectors, Cas12s, from TnpB seem to imply that the CRISPR arrays evolved via duplication of segments of ωRNA although insertion of tnpB near preexisting CRISPR arrays cannot be ruled out. Furthermore, in different, independent lineages of evolving Cas12s, the same evolutionary trend is observed, namely, accretion of additional protein domains resulting in the increased protein size and the formation of the REC lobe. However, the reverse trend, that is, secondary shrinking of the effector protein, is observable as well. Apart from the origin of Cas12, we identified many other cases of apparent TnpB exaptation where TnpB forms an evolutionarily conserved link with another protein(s) although none of these is as common as the formation of type V CRISPR-Cas systems. These instances fall into two categories, one involving apparent inactivation of the nuclease catalytic site and the other one where the catalytic site remains intact. The inactivated TnpBs likely perform regulatory functions that require binding but not cleavage of the target DNA or other proteins to specific sites in the target DNA. The most clear-cut case of a likely regulatory function is the association of TnpB with the sigma factor RpoE, a well-characterized transcription regulator. These exaptations of inactivated TnpBs recapitulate the previously explored cases of Cas12 inactivation, namely, Cas12k that was recruited for RNA-guided transposition by the V-K CAST and Cas12m which inactivates plasmids without cleavage, apparently, via downregulation of transcription (13, 21). The exaptation of catalytically active TnpBs likely resulted in the formation of a distinct toxin–antitoxin module whereas in other cases, the functions of exapted TnpBs and the associated proteins remain obscure. The numerous identified cases of TnpB exaptation fit the “guns for hire” concept whereby the same components are alternatively employed by mobile genetic elements and by cellular organisms, often, for defense functions (49). Overall, the diverse TnpBs and Cas12s described here, along with their exapted variants, comprise an expansive resource of potential tools that could advance genome editing technologies and offer many other applications.
Brief Methods
For the purpose of comprehensive identification of TnpBs and Cas12s, a representative set of TnpB sequences was obtained using the HHblits with eight iterations. The sequences were aligned using mafft, and two contiguous regions were extracted from the alignment: 1) conserved N-terminal domain and RuvC-I and 2) RuvC-II, ZF, and RuvC-III. These aligned regions were converted into two TnpB HMM profiles for HMMER and HHSearch (50–52). Additional Cas12 profiles were obtained (53), covering Cas12a, Cas12b, Cas12c, Cas12d, Cas12e, Cas12g, Cas12h, Cas12i, and V-U1-5. These profiles were employed to search a custom genomic database that was constructed by combining all publicly available, nonembargoed data from JGI, and all publicly available data from NCBI, and NCBI WGS.
For comprehensive phylogenetic analysis of TnpB and Cas12, sequences were clustered using MMSeq2, representative sequences from each cluster were realigned using muscle5, and the phylogenetic tree was constructed using IQTree2 (54–56).
CRISPR arrays were predicted for 10 kb windows around each TnpB and Cas12 gene using PILERCR, CRT, CRISPRDetect and CRISPRFinder (57–60). CRISPR spacer matches were identified by BLASTN search of a Combined Prokaryotic Plasmid and Phage database generated from NCBI plasmid and phage sequences.
For determination of TnpB and Cas12 mobility, the M-div mobility metric was computed as follows. For each 90% sequence identity protein cluster, a maximum of 2,000 loci were sampled, prioritizing loci with larger TnpB to contig edge distances first. Windows of 5 kb were extracted from each side of a TnpB or Cas12 gene, keeping the upstream and downstream windows in separate lists. Only windows with a minimum size of 2,000 were retained for further analysis. For the upstream and downstream windows separately, megablast was used with a word size of 16 to detect sequence similarity. A matrix of e-values was created from the corresponding pairwise megablast searches. The TnpBs from the passing windows were aligned using MAFFT and used to construct a matrix of pairwise protein sequence identity. If e-values were above 1e−5 in the megablast search, the locus was considered rearranged. For all loci pairs considered to be rearranged relative to one another, the corresponding sequence divergence (1 minus sequence identity) for the two TnpBs in the pair as determined by MAFFT was considered the “percentage sequence divergence before mobilization.” For each cluster, the minimum “percentage sequence identity before mobilization” was used as the final M-div metric, with a maximum allowable value of 0.1. Microcluster M-div metrics were aggregated into M-div metrics per cluster (50% cluster) by taking the minimum M-div value of all microclusters in the cluster.
For the analysis of TnpB mobility in complete archaeal and bacterial genomes, 15,913 TnpB/Cas12 family sequences were clustered with MMSeqs2 (55) with 0.8 and 0.98 sequence similarity thresholds, method “cluster,” 0.333 coverage, 0.1 e-value and cluster-mode 2. These clusters were used to estimate TnpB mobility in the genomes with permissive and strict threshold, respectively. TnpB family sequences were defined as mobile if the same or another sequence from the same cluster (separately for 0.8 and 0.98 thresholds) is present in the same genome. The same approach was used to calculate mobility values for other families of mobile elements, using NCBI CDD profiles for transposase identification (SI Appendix).
Protein structure models were constructed using AlphaFold2 implemented under CollabFold (61, 62).
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank G. Faure for helpful discussions. S.A.S., K.S.M., and E.V.K. are supported by the Intramural Research Program of the NIH of the USA (National Library of Medicine). F.Z. is supported by the Howard Hughes Medical Institute, K. Lisa Yang and Hock E. Tan Molecular Therapeutics Center at MIT, Broad Institute Programmable Therapeutics Gift Donors, James and Patricia Poitras, BT Charitable Foundation, David Cheng, and Robert Metcalfe.
Author contributions
H.A.-T., F.Z., and E.V.K. designed research; H.A.-T., S.A.S., K.S.M., Y.I.W., and S.K. performed research; H.A.-T. contributed new reagents/analytic tools; H.A.-T., S.A.S., K.S.M., Y.I.W., S.K., and E.V.K. analyzed data; and H.A.-T., F.Z., and E.V.K. wrote the paper.
Competing interests
F.Z. is a scientific advisor and cofounder of Editas Medicine, Beam Therapeutics, Pairwise Plants, Arbor Biotechnologies, and Aera Therapeutics. F.Z. is also a scientific advisor for Octant.
Footnotes
Reviewers: J.F., Universite Paris-Saclay; and D.G.S., Iowa State University.
Contributor Information
Feng Zhang, Email: zhang@broadinstitute.org.
Eugene V. Koonin, Email: koonin@ncbi.nlm.nih.gov.
Data, Materials, and Software Availability
This paper analyzes publicly available data. Accession numbers for the datasets are listed in supplementary files. Any additional information required to reanalyze the data reported here is available from the lead contact upon request, Han Altae-Tran. All tables and files can be found at Zenodo under the DOI: 10.5281/zenodo.8339301. All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Altae-Tran H., et al. , The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 374, 57–65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karvelis T., et al. , Transposon-associated TnpB is a programmable RNA-guided DNA endonuclease. Nature 599, 692–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapitonov V. V., Makarova K. S., Koonin E. V., ISC, a novel group of bacterial and archaeal DNA transposons that encode Cas9 homologs. J. Bacteriol. 198, 797–807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koonin E. V., Makarova K. S., Origins and evolution of CRISPR-Cas systems. Philos. Trans. R Soc. B Biol. Sci. 374, 20180087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakagawa R., et al. , Cryo-EM structure of the transposon-associated TnpB enzyme. Nature 616, 390–397 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirano S., et al. , Structure of the OMEGA nickase IsrB in complex with ωRNA and target DNA. Nature 610, 575–581 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuler G., Hu C., Ke A., Structural basis for RNA-guided DNA cleavage by IscB-ωRNA and mechanistic comparison with Cas9. Science 376, 1476–1481 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato K., et al. , Structure of the IscB-ωRNA ribonucleoprotein complex, the likely ancestor of CRISPR-Cas9. Nat. Commun. 13, 6719 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shmakov S., et al. , Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 15, 169–182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faure G., et al. , CRISPR-Cas in mobile genetic elements: Counter-defence and beyond. Nat. Rev. Microbiol. 17, 513–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balaban M., Moshiri N., Mai U., Jia X., Mirarab S., TreeCluster: Clustering biological sequences using phylogenetic trees. PLoS One 14, e0221068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavatine L., et al. , Single strand transposition at the host replication fork. Nucleic Acids Res. 44, 7866–7883 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strecker J., et al. , RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marino N. D., et al. , Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 362, 240–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makarova K. S., et al. , Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makarova K. S., Wolf Y. I., Koonin E. V., Classification and nomenclature of CRISPR-Cas systems: Where from here? CRISPR J. 1, 325–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H., Gao P., Rajashankar K. R., Patel D. J., PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell 167, 1814–1828.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan W. X., et al. , Functionally diverse type V CRISPR-Cas systems. Science 363, 88–91 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majorek K. A., et al. , The RNase H-like superfamily: New members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 42, 4160–4179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thakur M., et al. , Novel insights into ATP-stimulated cleavage of branched DNA and RNA substrates through structure-guided studies of the holliday junction resolvase RuvX. J. Mol. Biol. 433, 167014 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Wu W. Y., et al. , The miniature CRISPR-Cas12m effector binds DNA to block transcription. Mol. Cell 82, 4487–4502.e7 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Ton-Hoang B., et al. , Transposition of ISHp608, member of an unusual family of bacterial insertion sequences. EMBO J. 24, 3325–3338 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kersulyte D., Mukhopadhyay A. K., Shirai M., Nakazawa T., Berg D. E., Functional organization and insertion specificity of IS607, a chimeric element of Helicobacter pylori. J. Bacteriol. 182, 5300–5308 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makarova K. S., et al. , Unprecedented diversity of unique CRISPR-Cas-related systems and Cas1 homologs in Asgard Archaea. CRISPR J. 3, 156–163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulz F., et al. , Giant viruses with an expanded complement of translation system components. Science 356, 82–85 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Bao W., Jurka J., Homologues of bacterial TnpB_IS605 are widespread in diverse eukaryotic transposable elements. Mob. DNA 4, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P., et al. , Bioinformatics analyses of Shigella CRISPR structure and spacer classification. World J. Microbiol. Biotechnol. 32, 38 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Siguier P., Gourbeyre E., Chandler M., Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siguier P., Gourbeyre E., Varani A., Ton-Hoang B., Chandler M., Everyman’s guide to bacterial insertion sequences. Microbiol. Spectr. 3, MDNA3-0030–2014 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Puigbò P., Lobkovsky A. E., Kristensen D. M., Wolf Y. I., Koonin E. V., Genomes in turmoil: Quantification of genome dynamics in prokaryote supergenomes. BMC Biol. 12, 66 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karev G. P., Wolf Y. I., Rzhetsky A. Y., Berezovskaya F. S., Koonin E. V., Birth and death of protein domains: A simple model of evolution explains power law behavior. BMC Evol. Biol. 2, 18 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He S., et al. , The IS200/IS605 family and “Peel and Paste” single-strand transposition mechanism. Microbiol. Spectr. 3 (2015), 10.1128/microbiolspec.MDNA3-0039-2014. [DOI] [PubMed] [Google Scholar]
- 33.Frenkel-Pinter M., et al. , Adaptation and exaptation: From small molecules to feathers. J. Mol. Evol. 90, 166–175 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koonin E. V., Dolja V. V., Krupovic M., The logic of virus evolution. Cell Host Microbe 30, 917–929 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Gould S. J., Vrba E. S., Exaptation—a missing term in the science of form. Paleobiology 8, 4–15 (1982). [Google Scholar]
- 36.Mecsas J., Rouviere P. E., Erickson J. W., Donohue T. J., Gross C. A., The activity of sigma E, an Escherichia coli heat-inducible sigma-factor, is modulated by expression of outer membrane proteins. Genes Dev. 7, 2618–2628 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Hiratsu K., Amemura M., Nashimoto H., Shinagawa H., Makino K., The rpoE gene of Escherichia coli, which encodes sigma E, is essential for bacterial growth at high temperature. J. Bacteriol. 177, 2918–2922 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shmakov S. A., et al. , CRISPR arrays away from genes. CRISPR J. 3, 535–549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barák I., Muchová K., Labajová N., Asymmetric cell division during Bacillus subtilis sporulation. Future Microbiol. 14, 353–363 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Diallo M., et al. , Transcriptomic and phenotypic analysis of a spoIIE mutant in Clostridium beijerinckii. Front. Microbiol. 11, 556064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song W. S., Hong H. J., Yoon S.-I., Structural study of the flagellar junction protein FlgL from Legionella pneumophila. Biochem. Biophys. Res. Commun. 529, 513–518 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Makarova K. S., Wolf Y. I., Koonin E. V., Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 41, 4360–4377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo H., et al. , Crystal structure and biochemical studies of the bifunctional DNA primase-polymerase from phage NrS-1. Biochem. Biophys. Res. Commun. 510, 573–579 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Zabrady K., Zabrady M., Kolesar P., Li A. W. H., Doherty A. J., CRISPR-associated primase-polymerases are implicated in prokaryotic CRISPR-Cas adaptation. Nat. Commun. 12, 3690 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meers C., et al. , Transposon-encoded nucleases use guide RNAs to selfishly bias their inheritance. bioRxiv [Preprint] (2023). 10.1101/2023.03.14.532601 (Accessed 2 April 2023). [DOI]
- 46.Pasternak C., et al. , ISDra2 transposition in Deinococcus radiodurans is downregulated by TnpB. Mol. Microbiol. 88, 443–455 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Kim D. Y., et al. , Efficient CRISPR editing with a hypercompact Cas12f1 and engineered guide RNAs delivered by adeno-associated virus. Nat. Biotechnol. 40, 94–102 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koonin E. V., Makarova K. S., Evolutionary plasticity and functional versatility of CRISPR systems. PLoS Biol. 20, e3001481 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koonin E. V., Makarova K. S., Mobile genetic elements and evolution of CRISPR-Cas systems: All the way there and back. Genome Biol. Evol. 9, 2812–2825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinegger M., et al. , HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics 20, 473 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eddy S. R., Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katoh K., Misawa K., Kuma K.-I., Miyata T., MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shmakov S. A., Makarova K. S., Wolf Y. I., Severinov K. V., Koonin E. V., Systematic prediction of genes functionally linked to CRISPR-Cas systems by gene neighborhood analysis. Proc. Natl. Acad. Sci. U.S.A. 115, E5307–E5316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minh B. Q., et al. , IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinegger M., Söding J., MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Edgar R. C., MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edgar R. C., PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8, 18 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bland C., et al. , CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Biswas A., Staals R. H. J., Morales S. E., Fineran P. C., Brown C. M., CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics 17, 356 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Couvin D., et al. , CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirdita M., et al. , ColabFold: Making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
This paper analyzes publicly available data. Accession numbers for the datasets are listed in supplementary files. Any additional information required to reanalyze the data reported here is available from the lead contact upon request, Han Altae-Tran. All tables and files can be found at Zenodo under the DOI: 10.5281/zenodo.8339301. All other data are included in the manuscript and/or SI Appendix.