

Abstract
Endocrine-resistance remains a major challenge in estrogen receptor α positive (ERα+) breast cancer (BC) treatment and constitutively active somatic mutations in ERα are a common mechanism. There is an urgent need to develop novel drugs with new mode of mechanism to fight endocrine-resistance. Given aberrant ERα activity, we herein report the identification of novel covalent selective estrogen receptor degraders (cSERDs) possessing the advantages of both covalent and degradation strategies. A highly potent cSERD 29c was identified with superior anti-proliferative activity than fulvestrant against a panel of ERα+ breast cancer cell lines including mutant ERα. Crystal structure of ERα‒29c complex alongside intact mass spectrometry revealed that 29c disrupted ERα protein homeostasis through covalent targeting C530 and strong hydrophobic interaction collied on H11, thus enforcing a unique antagonist conformation and driving the ERα degradation. These significant effects of the cSERD on ERα homeostasis, unlike typical ERα degraders that occur directly via long side chains perturbing the morphology of H12, demonstrating a distinct mechanism of action (MoA). In vivo, 29c showed potent antitumor activity in MCF-7 tumor xenograft models and low toxicity. This proof-of-principle study verifies that novel cSERDs offering new opportunities for the development of innovative therapies for endocrine-resistant BC.
Key words: Covalent strategy, Estrogen receptor degraders, Endocrine-resistant breast cancer, X-ray crystallography
Graphical abstract
Novel ERα degrader 29c combining indirect antagonism with covalent strategy, offering new opportunities for the development of innovative therapies for endocrine-resistant breast cancer treatment.

1. Introduction
Breast cancer (BC) is one of the most prevalent malignancies in women globally, with approximately 70% of patients diagnosed as ERα positive (ERα+)1,2. The generally recognized therapeutic strategy is to block ERα signaling for ERα+ BC3,4. Over the past decades, standard-of-care (SOC) endocrine therapies have been developed to antagonize oncogenic ERα function in the clinic5. SOC for BC includes aromatase inhibitors (AIs, e.g., 1a letrozole, 1b exemestane) that block estrogen hormone synthesis and selective estrogen receptor modulators (SERMs, e.g., 2a tamoxifen, 2c raloxifene, 2d lasofoxifene) (Fig. 1A and B) that antagonize ERα signaling pathway6. Although current therapies have shown considerable clinical benefit in the treatment of ERα+ BC, the increased intrinsic and acquired resistance became a major problem7. Statistically, about 25% of patients develop resistance under the pressure of endocrine therapy. In endocrine-resistant settings, up to 90% of breast tumor progression rely on ligand-independent activation of transcription by ERα mutations8. Thus, targeted protein degradation (TPD) therapies capable of removing the proteins, such as selective estrogen receptor degraders (SERDs) or proteolysis targeting chimeras (PROTACs) would be an ideal strategy against the endocrine-resistant BC9, 10, 11, 12. Different from the catalytic mechanism of PROTACs, SERDs achieve antagonism by interfering ERα homeostasis and enforcing programmed degradation13. Fulvestrant (Ful, 3a), the only FDA-approved SERD, presents limited applications due to poor pharmacokinetic (PK) properties14.
Figure 1.

(A, B) Representative AIs and SERMs. (C) Representative SERDs with acidic or basic side chains and (D) SERCAs with electrophilic warheads interaction with C530.
Second-generation SERDs have been developed to overcome drug resistance with improved pharmacokinetic profiles than Ful (Fig. 1C)15,16. The investigation of GDC0810 (3b) was terminated in a phase II due to insufficient clinical benefits. AZD9496 (3c) and LSZ102 (3d) entered phase I clinical trials and was halted due to intolerable side effects or unsatisfying objective response rate (ORR)9. Four oral SERDs: AZD9833 (3f), GDC9545 (3g), SAR439859 (3h) and RAD1901 (3e) are currently under evaluation in phase III clinical trials for advanced BC17. Characteristically, the second-generation SERDs relied on a single mechanism for their drug design strategy, with extended bulky side chains completely displace helix 12 (H12), leading to exposure of the hydrophobic surface, misfolding of ERα proteins, and subsequent proteasomal degradation8. Mutations in the ER gene (ESR1) tend to occur in hot spots that encode amino acids 536, 537 or 538 of ligand-binding domain (LBD), and the most frequent ESR1 mutations are Y537S and D538G18. Therefore, there is a challenge for SERDs design to balance H12 displacement-driven degradation with stabilizing effects of the H12 mutation derived ligand-independent activation19.
Comprehensive structural-biochemical investigations demonstrated that both mutations of ERα ligand binding pocket (LBP) had significantly reduced affinities of ligands and led to incomplete inhibition of target protein cascade with its downstream signaling20. Recent studies have demonstrated the success of covalent inhibitors to block and evade mutational events and maintain potency against mutation targets21. By covalent targeting, high binding affinity and selectivity were obtained22, 23, 24. Based on the process and underlying mechanisms of the ESR1 mutations, mutation-induced conformational change is thought to reduce ligand affinity for ERα25. Compounds H3B-5942 (4a) and H3B-6545 (4b), are the first-reported selective estrogen receptor covalent antagonists (SERCAs)26,27. H3B-6545 was entered into phrase I/II trials (NCT03250676) and the clinical data showed a median response duration of 7.6 months and an objective response rate (ORR) of 17%28. Although 4a‒b showed robust ERα antagonism by covalently targeting cysteine-530 (C530), they were reported no degradation ability to ERα26,27.
Inspired by the covalent strategy that could irreversibly fix wild-type or mutant ERα in the antagonist state, we aim to identify a novel ERα degradation mode that will resolve the challenges of low binding affinity for mutant ERα and balancing H12 displacement-driven degradation with stabilizing effects of the ligand-independent activation of H12 mutation. Previously, our group has developed a novel 7-oxabicyclo[2.2.1]heptane core skeleton-based compound library. Among them, certain compounds exhibited full antagonism and ERα degradation by reposition of helix 11 (H11) within the ERα ligand-binding pocket (LBP), referred as indirect antagonism29, 30, 31, 32. Whereas C530 located at H11 serves as a covalent target of the ERα LBP, we hypothesized that the combination of covalent and degradation strategies can be utilized on the 7-oxabicyclo[2.2.1]heptane core skeleton, thus allowing the covalent warhead to adopt novel degradation associated with noncanonical conformations33. Moreover, the degradation strategy is to address the compensatory increase in targeting protein expression that leads to incomplete inhibition of targeting protein cascade with its downstream signaling20. By covalently enhancing the destabilizing potency of the antagonist against the receptor, it enables the ERα to shift the equilibrium toward an unstable antagonistic conformation in the mutational setting. Additionally, the utilization of covalent strategy can improve the ERα ligand efficiency, thus reducing idiosyngous toxicity (IDT) through smaller and less frequent dosing regimens21.
Given that the ER-LBD is plasticity34 and cysteine reactivity varies among different proteins35,36, the crystal structures of exo-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonic acid phenyl ester (OBHS) or OBH-sulfonamide (OBHSA) complexed with ERα suggested the accessibility to introduce a covalent warhead targeting C530 of H11 (Figure 2, Figure 3). Six most reported cysteine targeting covalent fragments were selected representing Michael-type nucleophilic and substitution nucleophilic warheads21, which allowed to build a focused compounds library with a diverse set of covalent fragments (Fig. 2). Systematic structure–activity relationship (SAR) studies indicated that compound 29c with an OBHSA scaffold had excellent full antagonistic activity and degradability of ERα in Tam-resistant or mutant BC cell lines, as well as potent tumor inhibition in MCF-7 BC xenograft models in vivo. Analysis of the crystal structure of ERα‒29c complex and intact mass spectrometry revealed that 29c disrupted ERα protein homeostasis through covalent targeting C530 and strong hydrophobic interaction clashed on H11, thus enforcing a unique antagonist conformation and driving the ERα degradation. Herein, we report the discovery of novel covalent selective estrogen receptor degraders (cSERDs) for the treatment of ERα+ BC. This study suggests that cSERDs may represent a new option for overcoming clinical endocrine resistance against ERα+ BC.
Figure 2.

Design strategy of novel covalent SERDs.
Figure 3.

Superimposition of crystal structure of OBHS (PDB: 4ZN9, yellow) and OBHSA (PDB: 5KCW, pink) ERα in complexed with ERα LBD. Key residues were shown as sticks (cyan). H-bonds were represented by dotted yellow lines.
2. Results and discussion
2.1. Chemistry
Firstly, we developed a diversity-oriented and functionally synthesis pathway utilizing Diels–Alder reaction to generate a focused set of covalent compounds with the incorporation of distinct structure and chemotype warheads (Scheme 1, Scheme 2, Scheme 3). Next, covalent derivatives based on the OBHS or OBHSA scaffold were obtained through Diels–Alder reaction by a variety of furan derivatives reacted with different warheads (24a‒f) and various sulfonamide dienophiles (27a‒b, 17a‒j), or 3,4-bis(4-hydroxyphenyl) furan (25) and dienophiles with different warheads (13a‒f, 17k) (Scheme 3). Synthesis of key intermediates 11, 23, 25 and 27 have been reported in our previous work30,31,37. For details, the synthesis of various vinyl sulfonates 13a‒f were prepared by the reaction of corresponding acyl chloride with compound 11 under basic reaction conditions (Scheme 1A). Relevant systematic works indicated that the sulfonamide motif and para substitution of the benzenesulfonic group played significant roles in the ERα binding affinities and degradation activity of the OBHSA derivatives38. Thus, the synthesis of Series III mainly focused on further SAR via the N-substitution and phenyl-substitution of dienophiles. The intermediates of sulfonamide dienophiles 17a‒j were prepared according to previous work (Scheme 1B)37. Notably, ortho-substituted phenyl of sulfonamide derivatives could not be accessible using this route, possibly due to the large steric hindrance of methoxy group. Compound 17k was obtained by the reaction of 3,3-dimethylacryloyl chloride under basic conditions.
Scheme 1.

Synthesis of dienophiles 13a‒f and 17a‒k. Reagents and conditions: (a) Boc2O, TEA, DCM, 25 °C, 12 h; (b) TEA, DCM, 25 °C, 2 h; (c) TFA, DCM, 25 °C, 2 h; (d) TEA, THF, 0 °C, 2 h; (e) 15a–e: Trifluoroacetic anhydride, DCM, 25 °C, 3 h; 15h–j: Acetic anhydride, DCM, 25 °C, 3 h; (f) (CH3)2S·BH3, THF, −78 °C, 12 h. (g) BBr3, DCM, −78 °C, 12 h.
Scheme 2.

Synthesis of furan derivatives 24a‒f. Reagents and conditions: (a) TEA, CH3CN, 25 °C, 12 h; (b) NaH, DMSO, 25 °C, 2 h; (c) BBr3, DCM, −20 °C, 12 h; (d) DIBAL-H, THF, −78 °C, 12 h; (e) TEA, THF, 0 °C, 2 h.
Scheme 3.

Synthesis of target compounds 26a‒f, 28a‒f and 29a‒m.
The general synthetic route of compound 23 was described in Scheme 2. The furan derivatives 24a‒f containing the covalent warheads were carefully prepared by the reaction of corresponding acyl chloride under basic reaction conditions at 0 °C.
The target products were obtained via Diels–Alder reaction as shown in Scheme 3. Generally, the final compounds were formed as racemates in thermodynamically favored exo configuration29. In addition, compounds 26a‒f of Series I had no regioisomers due to the symmetrical structure of the furan derivative 25, while compounds 28a‒f of Series II were considered a mixture of regional isomers and, despite our best efforts, they were inseparable. For compounds 29a‒m of Series III, most compounds showed high stereoselectivity in the cyclic addition reaction and trace amounts of regioisomers could be detected by TLC. To investigate the effect of regioisomers on the biological activities, we also isolated the regioisomers 29c′ of the candidate compound 29c by scaling up the reaction to the grams and performed biological evaluation separately. The characterization of the regioisomers 29c and 29c′ was provided in Supporting Information (Supporting Information Fig. S1). In addition, the saturated analog 29k and the unmodified 29l were used as negative controls to verify the effect of pharmacophore group of the “Michael receptor”.
2.2. Evaluation the optimal covalent binding mode and the selectivity of ER isoforms
To initiate structure-based drug design, crystal structure of OBHS (PDB: 4ZN9, yellow) and OBHSA (PDB: 5KCW, pink) in complexed with ERα LBD were superimposed (Fig. 3)39,40. The A-ring phenol groups were highly overlayed and pivotal H-bond interactions with two key residues (E353, R394) were observed. The F ring clashed with His524 in the H11 main chain, indirectly changing the stability of H12 in the agonist conformation. Besides, the tight hydrogen-bonding between S537 in the Y537S mutant and D351 on Helix 3 (H3) was also observed41. The binding modes of OBHS and OBHSA with ERα provides two rational covalent modification sites (Fig. 3). We hoped to launch a set of covalent warheads to these two sites for enhanced covalent degradation. Since cysteine reactivity varies among different proteins, we used a competitive FP assay to quickly screen a diverse set of covalent fragments42. The relative binding affinities (RBA) of all compounds were determined and reported in Table 1. These affinities are discussed as RBA values, where estradiol has an affinity of 100% (Ki values, calculated from RBA values, are also given in Table 1). Different from previous studies, our work was not only to compare the reactivity of warheads, but also to investigate the accessibility of a covalent set of targeted cysteines of ERα. Thus, RBA values could indicate the preliminary information about covalent binding capacity and efficiency of the target compounds to ERα.
Table 1.
Relative binding affinity (RBA) and Ki of all compounds to ERα and ERβa.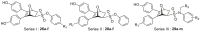
| Entry | Compd. | R1 | R2 | R3 |
Ki (nmol/L) |
RBA (%) |
α/β ratio | ||
|---|---|---|---|---|---|---|---|---|---|
| ERα | ERβ | ERα | ERβ | ||||||
| 1 | 26a | 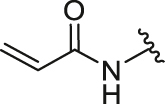 |
/ | / | 0.34 | 1.73 | 8.04 ± 0.87 | 3.86 ± 0.34 | 2.08 |
| 2 | 26b | 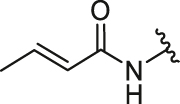 |
/ | / | 0.58 | 16.68 | 4.77 ± 0.16 | 0.40 ± 0.05 | 11.93 |
| 3 | 26c | 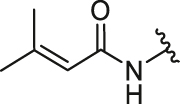 |
/ | / | 0.84 | 5.09 | 3.28 ± 0.17 | 1.31 ± 0.66 | 2.50 |
| 4 | 26d | 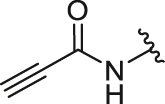 |
/ | / | 3.25 | 13.61 | 0.85 ± 0.08 | 0.49 ± 0.04 | 1.73 |
| 5 | 26e | 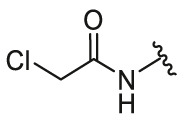 |
/ | / | 2.71 | 12.13 | 1.02 ± 0.13 | 0.55 ± 0.02 | 1.85 |
| 6 | 26f | 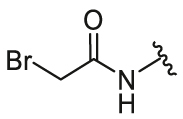 |
/ | / | 0.34 | 1.73 | 8.04 ± 0.37 | 3.86 ± 0.34 | 2.08 |
| 7b | 28a | 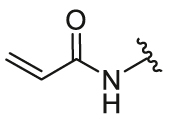 |
/ | / | 0.15 | 5.61 | 18.55 ± 0.34 | 1.19 ± 0.27 | 15.59 |
| 8b | 28b | |
/ | / | 0.2 | 0.64 | 14.08 ± 0.25 | 10.40 ± 0.23 | 1.35 |
| 9b | 28c | |
/ | / | 0.12 | 7.41 | 22.13 ± 0.92 | 0.90 ± 0.25 | 24.59 |
| 10b | 28d | |
/ | / | 0.8 | 7.25 | 3.46 ± 0.02 | 0.92 ± 0.17 | 3.76 |
| 11b | 28e | |
/ | / | 0.29 | 18.53 | 9.44 ± 0.40 | 0.36 ± 0.03 | 26.22 |
| 12b | 28f | |
4-OH | / | 0.35 | 2.03 | 7.93 ± 0.34 | 3.29 ± 0.25 | 2.41 |
| 13 | 29a | |
4-OH | CF3 | 0.14 | 0.99 | 20.03 ± 0.25 | 6.74 ± 0.21 | 2.97 |
| 14 | 29b | |
4-OH | CF3 | 0.14 | 10.59 | 19.82 ± 0.19 | 0.63 ± 0.14 | 31.46 |
| 15 | 29c | |
4-OH | CF3 | 0.09 | 10.93 | 30.44 ± 1.13 | 0.61 ± 0.11 | 49.90 |
| 16c | 29c′ | |
4-OH | CF3 | 0.18 | 3.25 | 15.12 ± 0.31 | 2.05 ± 0.45 | 7.37 |
| 17 | 29d | |
4-Me | CF3 | 0.43 | 4.87 | 6.49 ± 0.21 | 1.37 ± 0.11 | 4.74 |
| 18 | 29e | |
H | CF3 | 0.29 | 23.82 | 9.50 ± 0.09 | 0.28 ± 0.05 | 33.9 |
| 19 | 29f | |
4-OMe | CF3 | 0.19 | 4.07 | 14.48 ± 0.65 | 1.64 ± 0.45 | 8.83 |
| 20 | 29g | |
4-F | CF3 | 0.37 | 6.54 | 7.52 ± 0.29 | 1.02 ± 0.13 | 7.37 |
| 21 | 29h | |
3-OH | CF3 | 0.35 | 15.16 | 7.89 ± 0.11 | 0.44 ± 0.02 | 17.93 |
| 22 | 29i | |
4-OH | CH3 | 0.09 | 1.3 | 29.85 ± 1.09 | 5.12 ± 0.39 | 5.83 |
| 23 | 29j | |
4-OMe | CH3 | 0.63 | 13.08 | 4.36 ± 0.33 | 0.51 ± 0.09 | 8.55 |
| 24 | 29k | 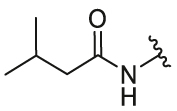 |
4-OH | CF3 | 1.15 | 4.6 | 2.41 ± 0.21 | 1.45 ± 0.12 | 1.66 |
| 25b | 29l | NH2 | 4-OH | CF3 | 1.14 | 2.84 | 2.43 ± 0.27 | 2.35 ± 0.33 | 1.03 |
| 26 | 29m | OH | 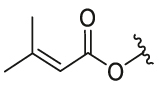 |
CF3 | 0.36 | 8.78 | 7.77 ± 0.43 | 0.76 ± 0.15 | 10.22 |
| 27 | 5 | 0.33 | 2.29 | 8.25 ± 0.67 | 2.91 ± 0.55 | 1.41 | |||
| 28d | 6a | 0.34 | 1.73 | 1.05 ± 0.16 | 0.10 ± 0.02 | 10.5 | |||
Relative binding affinity (RBA) values are the mean ± SD of at least three parallel tests. The RBA values: IC50estradiol/IC50compound × 100 ± the range (RBAestradiol = 100%). A high RBA means that the compound binds well to the ER and a low RBA means that the compound binds poorly to the ER. Ki = (100/RBA) × Kd. For estradiol, the Kd value was 2.76 and 6.67 nmol/L for ERα and ERβ, respectively.
Series II compounds were mixtures of regioisomers.
Compound 29c′ was regioisomer of 29c.
6a was a derivative of OBHSA, X = NCH2CF3, R = 4-OH.
Although RBA values of compounds 26a‒f with covalent warhead on the sulfonate benzene ring of the OBHS skeleton were lower than that of lead compound OBHS (Table 1, entries 1–6 vs 27), compounds 26a‒c (RBAERα = 3.28%–8.04%) showed potential tolerance and accessibility to covalent binding pocket of ERα. Series II was to explore the effects on binding affinity by changing the covalent modification site. The binding affinities of compounds 28a‒c had a sharp improvement compared with Series I (Table 1, entries 7–9 vs 1–3). In addition, binding affinities of compounds 29a‒c were also significantly improved compared with 28a‒c when changing OBHS to OBHSA scaffold. Among them, compound 29c with a warhead of 3,3-dimethylacrylamido possessed the highest ERα binding affinity as 30.44%, while the RBA value of the corresponding regioisomer 29c′ was only 15.12% (Table 1, entries 15 vs 16). To conduct further SAR studies, compounds 29d‒m were obtained by the above synthetic route (Scheme 3B) and RBA values were summarized in Table 1 (entries 17–26). Compounds 29d‒h exhibited significantly decreased RBA values to ERα when the para-hydroxyl group was replaced. Notably, no-covalent compounds 29k and 29l showed significantly decreased the binding affinity of ERα (Table 1, entries 24 and 25 vs 15 and 16).
2.3. Potent antiproliferative activity in ERαWT and ERαMUT breast cancer cell lines
To further investigate reactivity and selectivity of warhead, we evaluated the effects of all target compounds on cell viability of MCF-7 cells. Results expressed as IC50 were summarized in Table 2, and both compounds 5 (OBHS) and 6a (a derivative of OBHSA, X = NCH2CF3, R = 4-OH) were used as the control. In details, compounds 26a‒e of Series I (Table 2, entries 1–5) obtained by introducing a covalent warhead into the sulfonate benzene ring of the OBHS backbone had weak inhibitory activities against MCF-7 cells (IC50 > 0.5 μmol/L). However, in Series II, compounds 28a‒d showed significantly improved inhibitory activity on MCF-7 cells when the warhead was introduced into the phenol ring of the OBHS backbone. Notably, relatively good inhibitory activity was observed for the compound 28c with 3,3-dimethylacrylamido as warhead. Combining with the results of the RBA assay, we could reasonably infer the chemotype and loading position of covalent warhead on the carrier of oxygen-bridged bicycloheptene-like structure. Next, the resulting compounds were investigated by shifting optimized covalent warhead to the OBHSA vectors for a series of exemplary compounds 29a‒m (Series III). Among them, compound 29c (IC50 = 0.056 μmol/L) displayed the best potency and efficacy against MCF-7 cell line, which was 12- and 2.5-fold higher than positive controls 4-hydroxytamoxifen (4-OHT) and fulvestrant (Ful), respectively (Table 2, entries 15 vs 28 and 29). Compounds 29a‒b, 29k and 29l exhibited significantly decreased anti-proliferative potency compared with compound 29c (Table 2, entries 15 vs 13–14, 24–25), suggesting that the covalent warhead fragment of 3,3-dimethylacrylamido was critical for the inhibitory activity of MCF-7 cells. On the other hand, in comparison with compounds 29c′ and 29k, we reconfirmed that the 3,3-dimethylacrylamido was the best covalent warhead and its appropriate modification site of OBHSA vectors was also beneficial for good inhibitory activity. Meanwhile, further structure–activity relationship study indicated that substituents R2 and R3 were essential for their inhibitory activity against MCF-7 cell lines. For instance, when changing the R2 group from 4-OH to 3-OH, 4-F or 4-OMe, the anti-proliferative activity of derivatives 29e‒h decreased (Table 2, entries 18–21). For compounds 29i and 29j, the replacement of CF3 with CH3 group would decrease inhibitory activity on MCF-7 cells (Table 2, entries 15 vs 22 and 19 vs 23).
Table 2.
The cell viability of the target compounds on MCF-7, T-47D, T-47DY537S and T-47DD538G cell lines (IC50, μmol/L)a.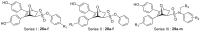
| Entry | Compd. | R1 | R2 | R3 | MCF-7 (μmol/L) | T-47D (μmol/L) | T-47DD538G (μmol/L) | T-47DY537S (μmol/L) |
|---|---|---|---|---|---|---|---|---|
| 1 | 26a | |
/ | / | 7.72 ± 0.64 | >31 | >31 | 27.22 ± 0.73 |
| 2 | 26b | |
/ | / | 9.18 ± 0.95 | 19.38 ± 0.54 | >31 | >31 |
| 3 | 26c | |
/ | / | 3.16 ± 0.63 | 15.84 ± 0.68 | 8.75 ± 0.25 | 12.02 ± 0.40 |
| 4 | 26d | |
/ | / | 1.79 ± 0.40 | 6.91 ± 0.29 | 9.33 ± 0.46 | 8.93 ± 0.25 |
| 5 | 26e | |
/ | / | 3.42 ± 0.32 | 8.20 ± 0.36 | 7.86 ± 0.37 | 8.85 ± 0.61 |
| 6 | 26f | |
/ | / | 0.50 ± 0.30 | 9.11 ± 0.21 | >31 | >31 |
| 7b | 28a | |
/ | / | 5.07 ± 0.71 | >31 | >31 | >31 |
| 8b | 28b | |
/ | / | 0.44 ± 0.11 | 25.58 ± 0.98 | 17.13 ± 0.65 | >31 |
| 9b | 28c | |
/ | / | 0.35 ± 0.02 | 24.92 ± 0.77 | 17.63 ± 0.89 | >31 |
| 10b | 28d | |
/ | / | 0.66 ± 0.12 | 13.35 ± 0.77 | >31 | >31 |
| 11b | 28e | |
/ | / | 1.10 ± 0.14 | 19.72 ± 0.32 | >31 | >31 |
| 12b | 28f | |
4-OH | / | 0.93 ± 0.19 | 8.53 ± 0.12 | >31 | 9.11 ± 0.35 |
| 13 | 29a | |
4-OH | CF3 | 0.18 ± 0.05 | 2.85 ± 0.29 | 5.55 ± 0.24 | >31 |
| 14 | 29b | |
4-OH | CF3 | 0.36 ± 0.04 | 5.12 ± 0.33 | >31 | >31 |
| 15 | 29c | |
4-OH | CF3 | 0.056 ± 0.004 | 0.87 ± 0.15 | 1.91 ± 0.18 | 0.88 ± 0.15 |
| 16c | 29c′ | |
4-OH | CF3 | 0.95 ± 0.11 | 5.52 ± 0.27 | 5.00 ± 0.36 | 4.05 ± 0.27 |
| 17 | 29d | |
4-Me | CF3 | 0.75 ± 0.21 | 8.33 ± 0.22 | >31 | >31 |
| 18 | 29e | |
H | CF3 | 0.66 ± 0.12 | 5.10 ± 0.32 | 5.18 ± 0.31 | 1.79 ± 0.17 |
| 19 | 29f | |
4-OMe | CF3 | 0.094 ± 0.001 | 8.58 ± 0.36 | 8.53 ± 0.47 | >31 |
| 20 | 29g | |
4-F | CF3 | 0.64 ± 0.10 | 5.28 ± 0.23 | 7.82 ± 0.47 | >31 |
| 21 | 29h | |
3-OH | CF3 | 0.98 ± 0.35 | 3.92 ± 0.01 | 2.84 ± 0.14 | 2.99 ± 0.22 |
| 22 | 29i | |
4-OH | CH3 | 0.071 ± 0.027 | 3.49 ± 0.06 | 4.25 ± 0.19 | 1.89 ± 0.17 |
| 23 | 29j | |
4-OMe | CH3 | 0.29 ± 0.03 | 7.92 ± 0.19 | 8.25 ± 0.45 | 12.72 ± 0.53 |
| 24 | 29k | |
4-OH | CF3 | 1.06 ± 0.19 | >31 | >31 | >31 |
| 25 | 29l | NH2 | 4-OH | CF3 | 3.17 ± 0.33 | 8.11 ± 0.37 | >31 | >31 |
| 26 | 29m | OH | |
CF3 | 0.16 ± 0.04 | 7.66 ± 0.60 | 3.68 ± 0.39 | 3.31 ± 0.34 |
| 27d | 6a | 0.25 ± 0.10 | >31 | >31 | >31 | |||
| 28 | 4-OHT | 0.67 ± 0.08 | 1.95 ± 0.20 | 9.13 ± 0.60 | 8.90 ± 0.28 | |||
| 29 | Ful | 0.14 ± 0.05 | 1.76 ± 0.06 | 3.92 ± 0.22 | 2.22 ± 0.17 |
Values are the mean ± SD of at least three parallel tests.
Series II compounds were mixtures of regioisomers.
Compound 29c′ was regioisomer of 29c.
6a was a derivative of OBHSA, X = NCH2CF3, R = 4-OH.
In addition, the toxicity of these compounds in normal breast cell line MCF-10A was investigated. The IC50 values and therapeutic index (TI) were summarized in Supporting Information Table S1. The results indicated that the focused compounds 29c, 29f and 29i displayed nanomolar activity against MCF-7 with IC50 values in the range of 0.056–0.094 μmol/L. Although these compounds showed cytotoxic symptoms, the values of TI were 145.1, 54.89 and 122.82, respectively, which were superior to 4-OHT (TI = 32.09) and inferior to Ful (TI > 357.1). Among them, compound 29c exhibited excellent potency against BC.
Having demonstrated a significant antiproliferative activity of the covalent derivatives in the MCF-7 cell lines, above compounds were also evaluated against another wide-type ERα (ERαWT) T-47D cell line and mutant ERα (ERαMUT) containing T-47DY537S and T-47DD538G cell lines (Table 2). It was found that compounds 29c, 29h and 29i showed better antiproliferation activity than other target compounds. Interestingly, compound 29c exhibited good activity in T-47D, T-47DY537S and T-47DD538G cells, 2- to 2.5-fold higher than Ful (Fig. 4A). The decrease in potency of compounds 29e and 29i further confirmed that R2 = 4-OH and R3 = CF3 group were vital to improved antiproliferative activity.
Figure 4.

(A) Cell viability assays of key compounds and positive controls in ERα+ BC cell lines including T-47D cells, T-47DD538G cells, T-47DY537S cells and LCC-2 cells; (B) Compound 29c induced MCF-7, LCC-2, T-47D, T-47DD538G and T-47DY537S cells apoptosis. Cells were exposed to Ful or 29c for 48 h ∗∗∗P < 0.001, ∗∗P < 0.01 and ∗P < 0.05.
Next, 29c was further characterized in tamoxifen-resistant LCC-2 cells (Table 3). Compound 29c exhibited better anti-proliferation activity against 4-OHT and Ful. In brief, compound 29c is a promising covalent SERD against a panel of ERα+ BC cell lines.
Table 3.
Cell viability assays on tamoxifen-resistant LCC-2 cell lines (IC50, μmol/L)a.
| Entry | Compd. | R1 | R2 | R3 | LCC-2 (μmol/L) |
|---|---|---|---|---|---|
| 1 | 29c | |
4-OH | CF3 | 2.10 ± 0.37 |
| 2 | 29f | |
4-OMe | CF3 | 15.09 ± 1.37 |
| 3 | 29i | |
4-OH | CH3 | 4.07 ± 0.13 |
| 4 | 4-OHT | 22.22 ± 1.93 | |||
| 5 | Ful | 11.04 ± 0.45 |
Values are the mean ± SD of at least three parallel tests.
Given that 29c showed good anti-proliferation activity in the above cell lines, we explored the ability of 29c to induce apoptosis in cell lines by flow cytometry (Supporting Information Fig. S3). Cells were treated with or without 5 μmol/L 29c and 5 μmol/L Ful severed as positive control. As shown in Fig. 4B, 29c significantly induced apoptosis in ERαWT and ERαMUT BC cell lines.
2.4. ER transcription activation assays
Compounds 29c, 29i and 29f were evaluated for ER transcriptional activities in HEK-293T cells. The results are summarized in Table 4, and dose–response curves of representative ligands were shown in Supporting Information Fig. S2. The efficacy of compound 29c to antagonize ERα was quite similar to Ful. When the para-hydroxyl group was replaced into para-methoxy, the antagonistic activity of 29f decreased significantly to ERα, and 29f also showed partial ERα agonistic ability. When changed trifluoroethyl group into ethyl group, 29i decreased the ERα antagonistic activities, while the antagonist ability of ERβ was enhanced.
Table 4.
Transcriptional activity of selected compounds for ERα and ERβa.
| Entry | Compd. | Agonist mode |
Antagonist mode |
||||||
|---|---|---|---|---|---|---|---|---|---|
| ERα |
ERβ |
ERα |
ERβ |
||||||
| EC50 (μmol/L) | Eff (%E2) | EC50 (μmol/L) | Eff (%E2) | IC50 (μmol/L) | Eff (%E2) | IC50 (μmol/L) | Eff (%E2) | ||
| 1 | 29c | – | 5 ± 1 | – | 19 ± 1 | 0.023 | 76 ± 1 | – | 27 ± 2 |
| 2 | 29f | 4.91 | 65 ± 5 | – | 9 ± 2 | 0.21 | 35 ± 2 | – | 13 ± 1 |
| 3 | 29i | – | 15 ± 2 | – | 34 ± 2 | 0.066 | 62 ± 5 | 1.02 | 78 ± 2 |
| 4 | 4-OHT | – | – | – | – | 0.039 | 43 ± 2 | – | 11 ± 3 |
| 5 | Ful | – | – | – | – | 0.013 | 80 ± 2 | 0.22 | 73 ± 1 |
| 6b | 29c | – | 2 ± 1 | – | – | 0.29 | 33 ± 2 | – | – |
| 7b | Ful | – | – | – | – | 0.027 | 71 ± 2 | – | – |
The luciferase activity are standardized in HEK-293T cells (control as 0% and 10 nmol/L E2 as 100%). The values are mean ± SD of at least three independent determinations and the missing parts cannot be accurately determined.
29c and Ful were evaluated for ERα C530S mutant transcriptional activities in HEK-293T cells.
2.5. Covalent SERDs degraded ERαWT and ERαMUT
All compounds were evaluated the degradation ability of ERα on MCF-7 cells. As shown in Fig. 5A, Series I and Series II compounds with OBHS backbone had no significant ERα degradation activity. Therefore, we mainly focused on ERα degradation ability of OBHSA skeleton-based compounds 29c‒m at concentrations of 1 or 5 μmol/L, with Ful and 6a as controls. The immunoblot results showed that compounds 29c, 29i, 29f, 29h, 29j and 29m could be effective in inducing ERα degradation at 1–5 μmol/L. In particular, compounds 29c, 29i and 29f exhibited an efficient ability to degrade ERα at 1 μmol/L. The degradation ability of 29c was more potent than other compounds at the same concentrations. The degradation activity was significantly affected by the compounds 29e, 29g and 29h, in which the para hydroxy group of the sulfonamide was substituted. Compounds 29a‒b, regioisomer 29c′ and saturated analogue of 29k showed impaired degradation ability.
Figure 5.

(A) Immunoblot analysis for ERα protein on the MCF-7 cells treated with indicated compounds at 5 μmol/L for 24 h. (B) Immunoblot analysis of ERα protein on the MCF-7 cells treated with indicated compounds at 1 and 5 μmol/L. (C) Immunoblot analysis of ERα protein treated with gradient concentrations Ful or 29c in the indicated BC cell lines. DC50 and Dmax values were quantified from two independent experiments. All immunoblot was treated with β-actin as the loading control.
In order to further characterize the effect of compound 29c for ERα degradation, we examined the ERα degradation of compound 29c at 0.5, 1 and 5 μmol/L concentrations with 3, 6, 12 or 24 h treatment to determine the time course of its degradation in ERαWT and ERαMUT cell lines. Immunoblot analysis showed that ERα protein of MCF-7 cells was completely degraded by compound 29c at 5 μmol/L concentration over 24 h and LCC-2, T-47D, T-47DY537S and T-47DD538G cell lines completely degraded by compound 29c at 5 μmol/L concentration over 12 h (Supporting Information Fig. S4). Meanwhile, the degradation by 29c in a wide range of concentrations to determine the DC50 (concentration causing 50% ERα degradation) values in BC cell lines. As shown in Fig. 5C, 29c effectively induced ERα degradation in ERαWT and ERαMUT cell lines and DC50 values were ranged from 0.01 to 0.05 μmol/L, where 29c had lower DC50 values than Ful in MCF-7 and T-47D cell lines. Overall, the maximum degradation (Dmax) capacity of 29c was slightly higher than that of Ful at the endpoint of the assay.
The possible mechanism of action of compound 29c mediated ERα degradation had been investigated (Fig. 6A) in ERαWT and ERαMUT cell lines. Compound 29c degraded ERα at 5 μmol/L, whereas 10 μmol/L of the proteasome inhibitor MG-132 had no effect on ERα degradation. When treated with 10 μmol/L MG-132 and 5 μmol/L compound 29c, the latter lost its ability to degrade ERα. Besides, we investigated the 29c induced ubiquitination state of ERα through immunoprecipitant on experiments in the HEK-293T cells and identified K48-specific Ub-binding ERα proteins (Fig. 6B). These results suggested that degradation of ERα by 29c is mediated by the proteasome.
Figure 6.

Further study of degradation of ERα protein by 29c. (A) Intervention experiments of 29c for ERαWT and ERαMUT in the indicated BC cell lines. (B) ERα ubiquitination assay. (C) Immunoblot analysis of ERβ protein treated with 29c. (D) Immunoblot analysis of ERα C530S mutant protein treated with 29c.
Next, we also constructed the C530S plasmid bearing a single point mutation (C530 → S530) that then transfected into HEK-293T cells. Meanwhile, we examined the antagonistic activity and ERα degradation ability of 29c in HEK-293TC530S cells, respectively. We found each of these abilities to be decreased in ERα C530S mutations. Combining the antiproliferative activity data and degradation profiles of the non-covalent derivative 29k, those results demonstrated that C530 played a critical role in covalently targeting ERα with 29c, while the non-covalent compound Ful was unaffected by this mutation (Table 4, entry 6 vs 1 and 7, Fig. 6D). The selective degradation of ERα by 29c was also examined by transfection of ERβ plasmid in HEK-293T cells, and immunoblot analysis showed that 29c (5 μmol/L) had no degradation capacity for ERβ (Fig. 6C). Finally, we performed a proteomic analysis to examine the level of global protein regulation by 29c. The signaling pathways associated with endocrine resistance and breast cancer were significantly enriched through the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Supporting Information Fig. S8).
2.6. Crystal structure and mass spectrometry analysis of 29c in complex with ERα
To further clarify the detailed structural mechanism of 29c, an in-house X-ray crystal structure was determined by soaking 29c into ERα (Y537S/C381S/C417S) crystal (Fig. 7A) at resolution of 2.25 Å (PDB ID 7YMK, Supporting Information Table S2). Y537S mutation occurs frequently in endocrine-resistant BC. The structure analysis offers additional benefit of elucidating how 29c behaves in a clinically relevant setting. While compounds were evaluated as racemates in the present study, high resolution X-ray structure shows only one chiral species with (1S,2R,4S)-configuration bound to ERα LBD. This demonstrates high stereospecificity of ERα in selecting a single enantiomer from a mixture for binding and activation. The crystal structure of the preferred (1S,2R,4S)-enantiomer showed even more dramatic effect on ERα, with H11-12 loop region and H12 (528–547) could not be modeled due to very poor electron density, demonstrating that H12 was highly disordered (Supporting Information Fig. S7). Biochemical and structural evidence indicates that the disorder or displacement of H12 is necessary for the binding of corepressors to generate a greater antagonism of proliferation18,41,43.
Figure 7.

(A) The detailed complex (1S,2R,4S)-29c (orange) with ERα LBD (gray) interaction networks were shown in stereo view. The residues were presented as sticks (green). H-bonds were represented by yellow dotted lines. The 2Fo‒Fc electron density map is contoured at 1σ. H11‒12 loop region and H12 (528–547) could not be modeled due to very poor electron density. (B, C) Crystal structures of E2‒ERα (PDB: 3UUD, cyan) or H3B-5942‒ERα (PDB: 6CHW, purple) were superposed with the 29c‒ERα structure (PDB: 7YMK), with the ligand shown as lines, distances (Å) between H524 or L525 were indicated by dotted green lines.
Moreover, to confirm that C530 is the modification site, intact mass spectrometry analysis of substitutions was performed. Given that there are three cystines (residues 417, 447, and 530) inside the ligand binding pocket of ERα, the control variable mutant ERαY537S and ERαY537S C381S C417S were analyzed. As expected, covalent adducts of ERα containing C530 were found to be detected (Supporting Information Fig. S6). These data confirm that 29c was a small molecule targeting ERα that specifically and covalently bound to residue C530.
The more details on the crystal structure showed that 29c adopts the desired antagonist conformation in the mutant context. The nitrogen of amino group of 29c showed an H-bonding to D351 in H3, which would disrupt the H-bonding between D351 and S537 that contributes to the constitutive activity. The internal H-bond between the phenol of F ring and oxygen of amine group the caused 29c to fold into a unique spatial conformation which helped orient the hydrophobic motif including trifluoroethyl group and F ring towards H11. Crystal complexes of 29c‒ERα superposed with the E2‒ERα structure were shown in Fig. 7B. The trifluoroethyl group strongly clashed with the H11 main chain agonist positioning of H524 and enforcing a 5.8 and 2.7 Å shift on H524 and L525 respectively. H524 and L525 on H11 were pushed to flip outwards, exposing hydrophobic patches that disturbs ERα protein homeostasis40. When superposed with covalent antagonist H3B-5942 (Fig. 7B), it was shown that H524 and L525 were shifted 5.8 and 2.7 Å, respectively. These results suggested that H11 dislocation and H12 distortion of 29c might be important to generate both ER antagonist and SERD profile.
2.7. Pharmacokinetic (PK) studies of 29c
To evaluate the PK profiles of 29c in vivo, 29c was dosed to female Balb/c mice. Compound 29c was administered via intravenous (iv, 2 mg/kg), intraperitoneal (ip, 4 mg/kg) or oral (po, 20 mg/kg). The PK data (Table 5) showed that the half-life (t1/2) values of 29c with ip and po administration were 6.38 and 5.81 h, respectively, and 29c possessed a favorable drug exposure (Cmax (iv): 1.42 μg/mL; Cmax (ip): 0.352 μg/mL) in mice. Next, compound 29c would be involved in pharmacodynamic studies in mice by ip administration due to insufficient oral bioavailability.
Table 5.
In vivo pharmacokinetic parameters of 29c in female micea.
| Route | Dose (mg/kg) | t1/2 (h) | Tmax (h) | Cmax (μg/mL) | AUC (h·μg/mL) | CL (mL/min/kg) | Vss (L/kg) |
|---|---|---|---|---|---|---|---|
| iv | 2 | 2.77 | 0.083 | 1.42 | 1.21 | 27.67 | 4.5 |
| ip | 4 | 6.38 | 0.167 | 0.352 | 0.96 | – | – |
| po | 20 | 5.81 | 0.5 | 0.153 | 0.36 | – | – |
Three plasma samples at each time point. t1/2, half-life time; Tmax, the maximum concentration of time point; Cmax, compound maximum concentration in plasma samples; AUC, area under the concentration curve in plasma samples; CL, plasma clearance rate; Vss, steady state volume of distribution; iv, intravenous administration; ip, intraperitoneal; po, oral administration.
2.8. In vivo breast cancer model
Based on PK profiles of 29c, MCF-7 tumor xenograft models in Balb/c nude mice were used to evaluate the antitumor efficacy of 29c in vivo by intraperitoneal injection. As showed in Fig. 8, the 29c-treated group at ip dose of 2 or 4 mg/kg had a significant tumor growth inhibition (TGI) activity. On Day 32, 29c treatment at a dose of 4 mg/kg almost completely inhibited tumor growth (TGI = 92%), whereas TGI of tamoxifen at dose of 4 mg/kg and Ful at 2 mg/kg were 87% and 79%, respectively. There was no statistically significant difference in tumor weight between 29c and Ful treated control group at a dose of 2 mg/kg (Fig. 8B and D). The tumor proliferation marker protein Ki-67 staining and immunohistochemical (IHC) analysis indicated that 29c significantly reduced tumor cell proliferation (Fig. 8E and Fig. S5A). Besides, 29c did not apparently reduce mice body weight (Fig. 8C). Hematoxylin and eosin (H&E) analysis revealed that intraperitoneal administration of the compound 29c did not show significant changes in the main visceral organs (Fig. 8E). These results indicated that 29c possessed good safety and low toxicity to normal body tissues at efficacious doses. Additionally, ERα IHC analysis and Western-blot analysis also suggested that the high dose at 4 mg/kg of 29c could significantly degrade ERα in MCF-7 xenograft tumors (Fig. 8F and Fig. S5B).
Figure 8.

In vivo study in MCF-7 tumor BALB/c nude mice xenograft models by intraperitoneal injection once every 2 days. (A) Changes in tumor volume of mice were measured every 2 days. (B) Average weight of the dissected tumors in each group. (C) Body weight of mice were measured every 2 days (∗P < 0.05). (D) Representative photos of the dissected tumors on Day 33. (E) Xenografted tumor tissues were stained (scale bar: 100 μm) with Ki-67, ERα and H&E staining (scale bar: 50 μm) of the heart, liver, spleen, and kidney from nude mice in the indicated groups after treatment. (F) Immunoblot analysis for detecting the expression of ERα in MCF-7 xenograft tumors.
3. Conclusions
Endocrine-resistance remains a major challenge in ERα+ breast cancer. There is an urgent need to develop novel drug with new mode to overcome acquired resistance. In this work, we developed a novel series of covalent selective estrogen receptor degraders (cSERDs) possessing the advantages of both covalent and degradation strategies. Herein, a highly potent cSERD 29c is identified with superior anti-proliferative activity than Ful against ERα+ BC cell lines of both wild and mutant types. Crystal structure and intact mass spectra of ERα‒29c complex revealed that 29c disrupted ERα protein homeostasis via covalently targeting C530 and strong hydrophobic interaction collided on H11, thus enforcing a unique antagonist conformation and driving the ERα degradation. These significant effects on ERα homeostasis do not occur directly via long side chains perturbing the morphology of H12 demonstrating a mechanism of action (MoA) distinct from typical ERα degraders. In vivo, 29c has been shown to have potent antitumor activity in MCF-7 tumor xenograft models and low toxicity to normal body tissues. Overall, both in vitro and in vivo, compound 29c holds promise for breast cancer treatment. This proof-of-principle study verifies that the development of novel ERα degraders combining indirect antagonism with covalent strategy, offering new opportunities for the development of innovative therapies for endocrine-resistant breast cancer treatment.
4. Experimental
4.1. General information
All reagents and solvents were available from commercial vendors. 1H NMR and 13C NMR spectra with tetramethylsilane (TMS) internal standard were recorded on a Bruker Advance 400 MHz spectrometer. The chemical shifts were expressed in δ values (ppm). The monitoring of all reactions was thin-layer chromatography (TLC). Purifications were performed by column chromatography (200–300 mesh silica gel). All melting points were tested on RY-1G Tianjin Instruments. The mass raw data was acquired from Thermo Fisher Scientific and analyzed by Xcalibur™. HPLC (Shimadzu) conditions and results were shown in Supporting Information.
4.2. General procedure for target compounds 26a‒g, 28a‒d and 29a‒j
In a round-bottom flask, the furan derivatives (0.6 mmol) and dienophile derivatives (0.72 mmol) were added in THF (1 mL). The mixture was stirred at 90 °C for 12 h under Ar balloon. The solvent was removed and the resulting residue was purified by column chromatography (DCM:MeOH, 50:1–20:1).
4-Acrylamidophenyl-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (26a). 55% yield as yellow solid, mp 83–85 °C. 1H NMR (400 MHz, acetone-d6) δ 9.59 (s, 1H), 8.82 (d, J = 3.2 Hz, 1H), 8.75 (d, J = 3.0 Hz, 1H), 7.75 (d, J = 9.0 Hz, 2H), 7.25–7.19 (m, 6H), 6.81 (dd, J = 13.4, 8.6 Hz, 4H), 6.47–6.31 (m, 2H), 5.72 (dd, J = 9.7, 2.3 Hz, 1H), 5.64–5.62 (m, 1H), 5.43 (d, J = 4.3 Hz, 1H), 3.77 (dd, J = 8.3, 4.5 Hz, 1H), 2.40 (dt, J = 12.0, 4.4 Hz, 1H), 2.27 (dd, J = 12.1, 8.4 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ 163.4, 162.32, 157.7, 157.5, 145.0, 141.3, 138.2, 136.9, 131.6, 129.2, 128.5, 126.7, 124.0, 123.2, 122.6, 120.6, 115.7, 115.5, 84.4, 82.7, 54.1, 19.8. HRMS (ESI) calcd. for C27H23NO7S [M + Na]+ 528.1087, found 528.1088.
4-((E)-But-2-enamido)phenyl-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (26b). 81% yield as yellow solid, mp 86–89 °C. 1H NMR (400 MHz, acetone-d6) δ 9.41 (s, 1H), 8.78 (s, 2H), 7.75 (d, J = 9.0 Hz, 2H), 7.30–7.17 (m, 6H), 6.95–6.79 (m, 5H), 6.14 (dd, J = 15.0, 1.9 Hz, 1H), 5.65 (s, 1H), 5.44 (d, J = 4.3 Hz, 1H), 3.78 (dd, J = 8.3, 4.4 Hz, 1H), 2.42 (dt, J = 12.0, 4.4 Hz, 1H), 2.28 (dd, J = 12.1, 8.4 Hz, 1H), 1.89–1.85 (m, 3H). 13C NMR (100 MHz, acetone-d6) δ 163.8, 157.6, 157.4, 144.9, 141.3, 140.6, 138.4, 137.0, 129.2, 128.6, 125.7, 124.1, 123.3, 122.6, 120.4, 115.7, 115.5, 84.4, 82.8, 60.5, 30.6, 16.9. HRMS (ESI) calcd. for C28H25NO7S [M + Na]+ 542.1244, found 542.1246.
4-(3-Methylbut-2-enamido)phenyl-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (26c). 85% yield as yellow solid, mp 84–86 °C. 1H NMR (400 MHz, CD3OD) δ 7.54 (d, J = 9.0 Hz, 2H), 7.18–7.06 (m, 6H), 6.79–6.65 m, 4H), 5.83 (s, 1H), 5.59 (d, J = 0.9 Hz, 1H), 5.34 (dd, J = 4.4, 1.3 Hz, 1H), 3.67 (dd, J = 8.4, 4.4 Hz, 1H), 2.41–2.34 (m, 1H), 2.19–2.11 (m, 4H), 1.89–1.85 (m, 3H). 13C NMR (100 MHz, CD3OD) δ 166.2, 157.4, 157.3, 153.3, 144.9, 141.1, 137.9, 136.6, 129.0, 128.4, 123.7, 123.0, 122.3, 120.8, 118.2, 115.4, 115.2, 84.4, 82.8, 60.2, 53.5, 30.2, 26.2, 18.9. HRMS (ESI) calcd. for C29H27NO7S [M + Na]+ 556.1400, found 556.1399.
4-Propiolamidophenyl-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate(26d). 65% yield as yellow solid, mp 88–90 °C. 1H NMR (400 MHz, acetone-d6) δ 10.04 (s, 1H), 8.83–8.71 (m, 2H), 7.70 (d, J = 9.0 Hz, 2H), 7.26–7.21 (m, 6H), 6.84–6.78 (m, 4H), 5.64–5.62 (m, 1H), 5.43 (d, J = 4.3 Hz, 1H), 3.82–3.75 (m, 2H), 2.44–2.38 (m, 1H), 2.27 (dd, J = 12.1, 8.3 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ 157.6, 157.5, 149.7, 145.6, 141.3, 137.3, 137.0, 129.2, 128.6, 124.1, 123.3, 122.8, 120.9, 115.7, 115.5, 84.3, 82.8, 77.7, 75.0, 60.6, 30.6. HRMS (ESI) calcd. for C27H21NO7S [M + Na]+ 526.0931, found 526.0935.
4-(2-Chloroacetamido)phenyl-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (26e). 56% yield as yellow solid, mp 99–101 °C. 1H NMR (400 MHz, acetone-d6) δ 9.60 (s, 1H), 8.78–8.61 (m, 2H), 7.69 (d, J = 9.0 Hz, 2H), 7.27–7.18 (m, 6H), 6.85–6.74 (m, 4H), 5.63 (s, 1H), 5.43 (d, J = 4.3 Hz, 1H), 4.24 (s, 2H), 3.77 (dd, J = 8.3, 4.5 Hz, 1H), 2.44–2.37 (m, 1H), 2.30–2.23 (m, 1H). 13C NMR (100 MHz, acetone-d6) δ 164.8, 157.6, 157.5, 145.4, 141.3, 137.5, 137.0, 129.2, 128.6, 124.0, 123.3, 122.7, 120.8, 115.7, 115.5, 84.4, 82.8, 60.6, 43.2, 30.6. HRMS (ESI) calcd. for C26H22ClNO7S [M + Na]+ 550.0698, found 550.0699.
4-(2-Bromoacetamido)phenyl-5,6-bis(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (26f). 60% yield as yellow solid, mp 102–104 °C. 1H NMR (400 MHz, acetone-d6) δ 9.75 (s, 1H), 8.79–8.62 (m, 2H), 7.68 (d, J = 9.0 Hz, 2H), 7.28–7.15 (m, 6H), 6.86–6.74 (m, 4H), 5.64–5.61 (m, 1H), 5.43 (d, J = 4.2 Hz, 1H), 4.03 (s, 2H), 3.77 (dd, J = 8.3, 4.5 Hz, 1H), 2.43–2.35 (m, 1H), 2.30–2.22 (m, 1H). 13C NMR (100 MHz, acetone-d6) δ 164.8, 157.6, 157.4, 145.4, 141.3, 137.7, 137.0, 129.4, 129.2, 128.6, 124.1, 123.3, 122.7, 120.5, 115.7, 115.5, 84.4, 82.8, 60.6, 30.6. HRMS (ESI) calcd. for C26H22BrNO7S [M + Na]+ 594.0193, found 594.0196.
Phenyl-5-(4-acrylamidophenyl)-6-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (28a, mixture of 3:1 isomers). 52% yield as yellow solid. 1H NMR (400 MHz, CD3OD) δ 7.64–7.55 (m, 2H), 7.41–7.07 (m, 10H), 6.80–6.71 (m, 2H), 6.48–6.33 (m, 2H), 5.80–5.72 (m, 1H), 5.66–5.56 (m, 1H), 5.40–5.27 (m, 1H), 3.81–3.63 (m, 1H), 2.58–2.35 (m, 1H), 2.28–2.05 (m, 1H). 13C NMR (100 MHz, CD3OD) δ 164.7, 157.7, 149.3, 143.0, 138.2, 136.1, 131.0, 129.7, 128.6, 127.9, 127.0, 123.2, 121.9, 120.2, 115.3, 84.3, 82.8, 60.4, 29.3. HRMS (ESI) calcd. for C27H23NO6S [M + Na]+ 512.1138, found 512.1133.
Phenyl-5-(4-((E)-but-2-enamido)phenyl)-6-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (28b, mixture of 5:1 isomers). 59% yield as yellow solid. 1H NMR (400 MHz, CD3OD) δ 7.59–7.45 (m, 2H), 7.36–6.95 (m, 10H), 6.93–6.81 (m, 1H), 6.74–6.61 (m, 2H), 6.15–5.93 (m, 1H), 5.63–5.47 (m, 1H), 5.34–5.21 (m, 1H), 3.78–3.55 (m, 4.4 Hz, 1H), 2.43–2.27 (m, 1H), 2.23–2.02 (m, 1H), 1.83 (d, J = 5.8 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 165.3, 157.6, 149.3, 142.9, 141.3, 140.6, 138.5, 136.1, 129.7, 128.6, 125.1, 123.3, 122.6, 121.9, 120.1, 115.5, 115.3, 84.3, 82.8, 60.5, 30.0, 16.7. HRMS (ESI) calcd. for C28H25NO6S [M + Na]+ 526.1294, found 526.1296.
Phenyl-6-(4-hydroxyphenyl)-5-(4-(3-methylbut-2-enamido)phenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (28c, mixture of 5:1 isomers). 67% yield as yellow solid. 1H NMR (400 MHz, CD3OD) δ 7.51–7.43 (m, 2H), 7.33–7.05 (m, 10H), 6.73–6.63 (m, 2H), 5.81 (d, J = 5.2 Hz, 1H), 5.60–5.50 (m, 1H), 5.36–5.22 (m, 1H), 3.71–3.56 (m, 1H), 2.43–2.29 (m, 1H), 2.20–2.06 (m, 4H), 1.85 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 166.3, 157.8, 153.1, 149.4, 142.7, 140.7, 138.8, 138.3, 136.2, 129.7, 128.6, 123.3, 122.6, 121.9, 119.9, 118.4, 115.5, 115.3, 84.3, 82.8, 60.1, 30.0, 29.4, 26.2, 18.8. HRMS (ESI) calcd. for C29H27NO6S [M + Na]+ 540.1451, found 540.1452.
Phenyl-6-(4-(2-chloroacetamido)phenyl)-5-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (28d, mixture of 3:1 isomers). 43% yield as yellow solid. 1H NMR (400 MHz, acetone-d6) δ 9.57–9.50 (m, 1H), 8.74–8.66 (m, 1H), 7.67 (t, J = 8.9 Hz, 2H), 7.47–7.23 (m, 10H), 6.89–6.80 (m, 2H), 5.73–5.69 (m, 1H), 5.49 (td, J = 4.1, 1.2 Hz, 1H), 4.28–4.26 (m, 2H), 3.89–3.76 (m, 1H), 2.49–2.41 (m, 1H), 2.39–2.26 (m, 1H). 13C NMR (100 MHz, acetone-d6) δ 164.6, 157.8, 149.6, 143.1, 140.9, 138.9, 138.2, 136.6, 129.9, 127.6, 123.7, 122.9, 122.3, 119.6, 115.8, 115.6, 84.4, 82.7, 60.5, 30.6. HRMS (ESI) calcd. for C26H22ClNO6S [M + Na]+ 534.0749, found 524.0752.
Phenyl-5-(4-(2-bromoacetamido)phenyl)-6-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (28e, mixture of 3:1 isomers). 55% yield as yellow solid. 1H NMR (400 MHz, CD3OD) δ 7.57–7.47 (m, 2H), 7.38–7.10 (m, 10H), 6.79–6.69 (m, 2H), 5.56–5.66 (m, 1H), 5.42–5.29 (m, 1H), 3.96 (d, J = 5.3 Hz, 2H), 3.78–3.62 (m, 1H), 2.46–2.36 (m, 1H), 2.25–2.17 (m, 1H). 13C NMR (100 MHz, CD3OD) δ 166.2, 157.7, 155.1, 149.4, 143.2, 140.5, 138.8, 137.9, 136.0, 129.7, 128.6, 123.2, 122.4, 121.9, 120.0, 115.5, 115.3, 84.3, 82.7, 60.4, 28.4. HRMS (ESI) calcd. for C26H22BrNO6S [M + Na]+ 578.0243, found 578.0246.
4-Hydroxyphenyl-5-(4-hydroxyphenyl)-6-(4-(3-methylbut-2-enamido)phenyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonate (28f, mixture of 3:1 isomer). 40% yield as yellow solid, mp 95–104 °C. 1H NMR (400 MHz, CD3OD) δ 7.54 (d, J = 8.5 Hz, 2H), 7.23 (dd, J = 26.4, 8.5 Hz, 4H), 6.99 (d, J = 8.9 Hz, 2H), 6.75 (dd, J = 36.6, 8.7 Hz, 4H), 5.88 (s, 1H), 5.64 (s, 1H), 5.42 (d, J = 3.4 Hz, 1H), 3.64 (dd, J = 8.3, 4.4 Hz, 1H), 2.48–2.43 (m, 1H), 2.20 (s, 4H), 1.93 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 166.3, 157.8, 156.3, 153.1, 141.8, 140.7, 138.6, 138.3, 129.2, 128.0, 127.2, 122.9, 122.6, 119.6, 118.3, 115.6, 115.5, 84.4, 82.6, 59.4, 30.2, 29.4, 26.2, 18.8. HRMS (ESI) calcd. for C29H27NO7S [M + Na]+ 556.1400, found 556.1401.
N-(4-(3-(4-Hydroxyphenyl)-5-(N-(4-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)acrylamide (29a). 55% yield as yellow solid, mp 110–111 °C. 1H NMR (400 MHz, CD3OD) δ 7.65 (d, J = 8.6 Hz, 2H), 7.35–7.24 (m, 2H), 7.21–7.06 (m, 4H), 6.76–6.62 (m, 4H), 6.51–6.37 (m, 2H), 5.84–5.77 (m, 1H), 5.51 (d, J = 1.2 Hz, 1H), 5.36 (dd, J = 4.4, 1.2 Hz, 1H), 4.40 (q, J = 8.6 Hz, 2H), 3.56–3.50 (m, 1H), 2.28–2.20 (m, 1H), 2.07–2.00 (m, 1H). 13C NMR (125 MHz, CD3OD) δ 164.8, 157.6, 157.6, 142.9, 138.3, 136.3, 130.9, 130.2, 130.2, 128.3, 128.3, 128.1, 126.7, 124.2 (d, J = 279.7 Hz), 123.4, 120.2, 115.5, 115.2, 84.3, 82.7, 61.2, 52.1 (d, J = 34.0 Hz), 30.0. HRMS (ESI) calcd. for C31H29F3N2O6S [M + Na]+ 609.1277, found 609.1279.
(E)-N-(4-(3-(4-Hydroxyphenyl)-5-(N-(4-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)but-2-enamide (29b). 81% yield as yellow solid, mp 131–133 °C. 1H NMR (400 MHz, CD3OD) δ 7.59 (d, J = 8.6 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 7.12 (d, J = 8.7 Hz, 4H), 6.98–6.89 (m, 1H), 6.68 (t, J = 9.0 Hz, 4H), 6.13 (dd, J = 15.2, 1.7 Hz, 1H), 5.49–5.46 (m, 1H), 5.31 (d, J = 3.9 Hz, 1H), 4.37 (q, J = 8.5 Hz, 2H), 3.48–3.55 (m, 1H), 2.25–2.16 (m, 1H), 2.05–1.96 (m, 1H), 1.91 (dd, J = 6.9, 1.5 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 165.4, 157.6, 157.5, 142.7, 141.4, 138.4, 136.2, 130.3, 130.2, 128.4, 128.1, 128.0, 125.6, 125.0, 124.2 (d, J = 281.8 Hz), 123.4, 122.8, 120.2, 115.6, 115.3, 84.3, 82.7, 61.3, 52.4 (d, J = 33.4 Hz), 30.1, 16.7. HRMS (ESI) calcd. for C30H27F3N2O6S [M + Na]+ 623.1431, found 623.1434.
N-(4-(3-(4-Hydroxyphenyl)-5-(N-(4-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29c). 82% yield as yellow solid, mp 113–115 °C. 1H NMR (400 MHz, CD3OD) δ 7.56 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 7.12 (d, J = 8.8 Hz, 4H), 6.68 (t, J = 8.7 Hz, 4H), 5.88 (s, 1H), 5.31 (d, J = 4.1 Hz, 1H), 4.37 (q, J = 8.5 Hz, 2H), 3.49–3.54 (m, 1H), 2.39–2.15 (m, 4H), 2.04–1.95 (m, 1H), 1.92 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 166.3, 157.6, 157.5, 153.2, 142.6, 138.7, 136.3, 130.3, 130.2, 128.3, 128.1, 127.6, 124.2 (d, J = 280.8 Hz), 123.5, 119.9, 118.3, 115.6, 115.2, 84.3, 82.7, 61.3, 52.2 (d, J = 31.5 Hz), 30.1, 26.2, 18.8. HRMS (ESI) calcd. for C31H29F3N2O6S [M + Na]+ 637.1590, found 637.1592.
N-(4-((3-(4-Hydroxyphenyl)-6-(N-(4-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29c′). 12% yield as yellow solid, mp 109–111 °C. 1H NMR (400 MHz, CD3OD) δ 7.50 (d, J = 8.5 Hz, 2H), 7.26–7.14 (m, 4H), 7.10 (d, J = 8.9 Hz, 2H), 6.80 (d, J = 8.7 Hz, 2H), 6.74–6.62 (m, 4H), 5.88–5.84 (m, 1H), 5.53–5.47 (m, 1H), 5.30 (dd, J = 4.4, 1.2 Hz, 1H), 4.37 (q, J = 8.5 Hz, 2H), 3.53–3.44 (m, 1H), 2.28–2.17 (m, 4H), 2.00–2.09 (m, 1H), 1.90 (s, 3H). 13C NMR (100 MHz, CD3OD) δ 166.3, 157.7, 157.6, 153.2, 149.8, 140.6, 138.5, 130.4, 130.1, 129.4, 128.2, 127.1, 124.2 (d, J = 278.8) 122.7, 119.7, 118.3, 115.7, 115.5, 115.5, 84.4, 82.6, 61.0, 52.2 (d, J = 34.3 Hz), 30.3, 26.2, 18.9. HRMS (ESI) calcd. for C31H29F3N2O6S [M + Na]+ 637.1590, found 637.1592.
N-(4-(3-(4-Hydroxyphenyl)-6-(N-(p-tolyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29d). 35% yield as yellow solid, mp 85–87 °C. 1H NMR (400 MHz, acetone-d6) δ 9.20 (s, 1H), 8.83 (d, J = 2.3 Hz, 1H), 7.69–7.64 (m, 2H), 7.37–7.11 (m, 8H), 6.91–6.81 (m, 2H), 5.92–5.84 (m, 1H), 5.56 (d, J = 1.2 Hz, 1H), 5.38 (dd, J = 4.3, 1.3 Hz, 1H), 4.56 (q, J = 8.7 Hz, 2H), 3.57 (dd, J = 8.3, 4.5 Hz, 1H), 2.32 (s, 3H), 2.24–2.18 (m, 4H), 2.15–2.09 (m, 1H), 1.88 (d, J = 1.3 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 166.2, 157.8, 153.0, 140.6, 138.6, 138.5, 136.4, 129.7, 129.3, 128.5, 128.1, 127.1, 124.1 (d, J = 279.2 Hz), 122.7, 119.5, 118.3, 115.4, 84.4, 82.6, 61.1, 60.2 (d, J = 31.3 Hz), 30.2, 26.1, 19.7, 18.7. HRMS (ESI) calcd. for C32H31F3N2O5S [M + Na]+ 635.1797, found 635.1798.
N-(4-(3-(4-Hydroxyphenyl)-6-(N-phenyl-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29e). 35% yield as yellow solid, mp 88–90 °C. 1H NMR (400 MHz, CD3OD) δ 7.54–7.48 (m, 2H), 7.36–7.27 (m, 5H), 7.24–7.09 (m, 4H), 6.81–6.75 (m, 2H), 5.87 (d, J = 1.3 Hz, 1H), 5.51–5.48 (m, 1H), 5.35–5.29 (m, 1H), 4.53–4.40 (m, 2H), 3.50–3.45 (m, 1H), 2.25–2.18 (m, 4H), 2.07–2.00 (m, 1H), 1.94–1.90 (m, 3H). 13C NMR (101 MHz, CD3OD) δ 166.3, 157.7, 153.1, 140.6, 139.1, 138.6, 138.4, 129.3, 129.2, 128.7, 128.3, 128.1, 127.1, 124.1 (d, J = 280.8 Hz), 122.7, 119.6, 118.3, 115.5, 84.4, 82.6, 61.3, 51.9 (d, J = 34.3 Hz), 30.2, 26.2, 18.8. HRMS (ESI) calcd. for C31H29F3N2O5S [M + Na]+ 621.1641, found 621.1643.
N-(4-(3-(4-Hydroxyphenyl)-5-(N-(4-methoxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29f). 77% yield as yellow solid, mp 114–117 °C. 1H NMR (400 MHz, CD3OD) δ 7.52 (d, J = 8.7 Hz, 2H), 7.25–7.16 (m, 6H), 6.80 (d, J = 8.8 Hz, 4H), 5.89–5.86 (m, 1H), 5.50–5.48 (m, 1H), 5.36–5.33 (m, 1H), 4.47–4.38 (m, 2H), 3.78 (s, 3H), 3.47–3.42 (m, 1H), 2.29–2.23 (m, 1H), 2.20 (d, J = 1.3 Hz, 3H), 2.10–2.03 (m, 1H), 1.93 (d, J = 1.4 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 166.2, 159.6, 157.8, 153.0, 140.6, 138.5, 138.5, 131.3, 130.2, 129.4, 127.0, 126.8 (d, J = 283.0 Hz), 122.75, 119.58, 118.3, 115.5, 114.2, 84.4, 82.6, 66.9, 60.9, 54.6, 51.4 (d, J = 35.2 Hz), 30.2, 26.2, 18.7. HRMS (ESI) calcd. for C31H29F3N2O5S [M + Na]+ 621.1641, found 621.1645.
N-(4-(5-(N-(4-Fluorophenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-3-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29g). 44% yield as yellow solid, mp 90–92 °C. 1H NMR (400 MHz, CD3OD) δ 7.52 (d, J = 8.7 Hz, 2H), 7.39–7.34 (m, 2H), 7.24 (d, J = 8.6 Hz, 2H), 7.20–7.16 (m, 2H), 7.08–7.02 (m, 2H), 6.80 (d, J = 8.7 Hz, 2H), 5.89–5.86 (m, 1H), 5.52–5.50 (m, 1H), 5.37 (d, J = 4.3 Hz, 1H), 4.48 (q, J = 8.5 Hz, 2H), 3.50–3.46 (m, 1H), 2.26–2.19 (m, 4H), 2.11–2.04 (m, 1H), 1.94 (d, J = 1.4 Hz, 3H). 13C NMR (101 MHz, CD3OD) δ 166.2, 162.2 (d, J = 247.0 Hz), 157.8, 153.1, 140.6, 138.6, 138.4, 135.2, 135.1, 131.0, 130.9, 129.3, 128.1, 127.1, 126.9 (d, J = 289 Hz), 122.7, 119.6, 118.3, 116.0, 115.7, 115.5, 84.4, 82.6, 61.5, 52.0 (d, J = 35.0 Hz), 30.2, 26.2, 18.7. HRMS (ESI) calcd. for C31H28F4N2O5S [M + Na]+ 639.1547, found 639.1549.
N-(4-(3-(4-Hydroxyphenyl)-5-(N-(3-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29h). 77% yield as yellow solid, mp 114–117 °C. 1H NMR (400 MHz, CD3OD) δ 7.56 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.6 Hz, 2H), 7.18–7.07 (m, 3H), 6.96–6.91 (m, 1H), 6.83–6.75 (m, 2H), 6.72 (d, J = 8.6 Hz, 2H), 5.90 (s, 1H), 5.51 (s, 1H), 5.35 (d, J = 4.2 Hz, 1H), 4.47 (q, J = 9.0, 8.5 Hz, 2H), 3.60–3.53 (m, 1H), 2.27–2.17 (m, 4H), 2.04–1.92 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 166.3, 158.0, 157.5, 153.1, 142.6, 140.1, 138.7, 136.3, 129.8, 128.4, 127.8, 127.5, 124.1 (d, J = 280.8), 123.4, 119.9, 118.7, 118.3, 115.9, 115.3, 115.2, 84.3, 82.8, 61.7, 51.8 (d, J = 34.3 Hz), 30.1, 26.2, 18.8. HRMS (ESI) calcd. for C31H29F3N2O6S [M + Na]+ 637.1590, found 623.1594.
N-(4-(5-(N-Ethyl-N-(4-hydroxyphenyl)sulfamoyl)-3-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29i). 82% yield as yellow solid, mp 121–123 °C. 1H NMR (400 MHz, CD3OD) δ 7.57 (d, J = 8.6 Hz, 2H), 7.26 (d, J = 8.7 Hz, 2H), 7.19–7.13 (m, 2H), 7.11–7.06 (m, 2H), 6.76–6.69 (m, 4H), 5.92–5.87 (m, 1H), 5.51–5.46 (m, 1H), 5.34 (d, J = 3.7 Hz, 1H), 3.80–3.69 (m, 2H), 3.52–3.47 (m, 1H), 2.28–2.19 (m, 4H), 2.06–2.00 (m, 1H), 1.93 (s, 3H), 1.06 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 166.3, 157.4, 157.1, 153.2, 142.3, 138.7, 136.5, 130.5, 129.8, 128.4, 127.9, 127.7, 123.6, 119.9, 118.3, 115.4, 115.2, 84.4, 82.8, 61.0, 46.6, 30.0, 26.2, 18.8, 13.6. HRMS (ESI) calcd. for C31H32N2O6S [M + Na]+ 583.1873, found 583.1876.
N-(4-(5-(N-Ethyl-N-(4-methoxyphenyl)sulfamoyl)-3-(4-hydroxyphenyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbut-2-enamide (29j). 69% yield as yellow solid, mp 92–94 °C. 1H NMR (400 MHz, CD3OD) δ 7.57 (d, J = 8.7 Hz, 2H), 7.31–7.21 (m, 2H), 7.22–7.12 (m, 4H), 6.88–6.81 (m, 2H), 6.75–6.69 (m, 2H), 5.90 (p, J = 1.4 Hz, 1H), 5.47 (d, J = 1.2 Hz, 1H), 5.34 (dd, J = 4.4, 1.2 Hz, 1H), 3.79–3.72 (m, 7H), 3.48 (dd, J = 8.4, 4.4 Hz, 1H), 2.29–2.19 (m, 4H), 2.04 (dd, J = 12.1, 8.4 Hz, 1H), 1.94 (d, J = 1.4 Hz, 3H), 1.07 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 166.3, 159.3, 157.5, 153.1, 142.3, 138.7, 136.5, 131.0, 130.4, 128.4, 127.9, 127.7, 123.6, 119.8, 118.3, 115.2, 114.0, 84.4, 82.8, 67.5, 61.0, 54.6, 29.9, 26.2, 25.1, 18.8, 13.6. HRMS (ESI) calcd. for C32H34N2O6S [M + Na]+ 597.2029, found 597.2033.
N-(4-((1R,4R)-3-(4-Hydroxyphenyl)-5-(N-(4-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)sulfamoyl)-7-oxabicyclo[2.2.1]hept-2-en-2-yl)phenyl)-3-methylbutanamide (29k). 60% yield as yellow solid, mp 105–107 °C. 1H NMR (400 MHz, CD3OD) δ 7.56 (d, J = 8.7 Hz, 2H), 7.25 (d, J = 8.7 Hz, 2H), 7.21–7.06 (m, 4H), 6.78–6.62 (m, 4H), 5.54–5.42 (m, 2H), 5.33 (d, J = 3.7 Hz, 1H), 4.39 (q, J = 8.5 Hz, 2H), 3.59–3.50 (m, 1H), 2.29–2.13 (m, 4H), 2.06–1.96 (m, 1H), 1.03 (d, J = 6.5 Hz, 6H). 13C NMR (100 MHz, CD3OD) δ 172.8, 157.6, 157.5, 142.7, 138.3, 136.3, 130.3, 130.2, 128.3, 128.1, 127.9, 124.2 (d, J = 280.8 Hz), 123.4, 120.2, 115.6, 115.2, 84.3, 82.7, 61.3, 52.1 (d, J = 34.3 Hz), 45.8, 30.1, 26.2, 21.4. HRMS (ESI) calcd. for C31H31F3N2O6S [M + Na]+ 639.1747, found 639.1749.
5-(4-Aminophenyl)-N,6-bis(4-hydroxyphenyl)-N-(2,2,2-trifluoroethyl)-7-oxabicyclo[2.2.1]hept-5-ene-2-sulfonamide (29l, mixture of 2:1 isomer). 82% yield as yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 7.25–7.09 (m, 4H), 7.02 (t, J = 9.3 Hz, 2H), 6.82–6.66 (m, 4H), 6.54 (dd, J = 22.7, 8.2 Hz, 2H), 5.46 (d, J = 17.1 Hz, 1H), 5.32 (dd, J = 14.8, 4.1 Hz, 1H), 4.46 (d, J = 9.2 Hz, 2H), 3.54–3.38 (m, 1H), 2.12–1.84 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 157.6, 141.6, 137.6, 134.9, 130.7, 130.4, 129.4, 129.1, 124.7 (d, J = 281.8 Hz), 124.3, 123.7, 120.0, 116.1, 115.8, 84.0, 82.3, 61.3, 52.4 (d, J = 29.2), 25.6. HRMS (ESI) calcd. for C26H23F3N2O5S [M + Na]+ 555.1171, found 555.1174.
4-((5,6-bis(4-Hydroxyphenyl)-N-(2,2,2-trifluoroethyl)-7-oxabicyclo[2.2.1]hept-5-ene)-2-sulfonamido)phenyl 3-methylbut-2-enoate (29m). 55% yield as yellow solid, mp 108–111 °C. 1H NMR (400 MHz, CD3OD) δ 7.41–7.31 (m, 2H), 7.22–7.09 (m, 4H), 7.08–6.98 (m, 2H), 6.87–6.67 (m, 4H), 5.93 (dd, J = 2.8, 1.4 Hz, 1H), 5.47 (d, J = 1.2 Hz, 1H), 5.32–5.27 (m, 1H), 4.47 (q, J = 8.5 Hz, 2H), 3.58–3.45 (m, 1H), 2.28–2.15 (m, 4H), 2.08–1.95 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 164.5, 161.3, 157.4, 157.2, 150.5, 141.0, 136.6, 136.3, 129.9, 129.4, 129.4, 129.2, 129.0, 128.2, 123.8, 123.1, 122.7, 122.6, 125.0 (d, J = 280.1 Hz), 115.4, 115.1, 114.8, 114.5, 114.2, 84.4, 82.7, 61.7, 52.0 (d, J = 34.3 Hz), 30.2, 26.3, 19.3. HRMS (ESI) calcd. for C31H28F3NO7S [M + Na]+ 638.1430, found 623.1436.
4.3. Estrogen receptor binding affinity
RBA values were detected via a competitive fluorometric binding experiment. Fluorescence tracer (40 nmol/L), human ERα or ERβ LBD (0.8 μmol/L) and bovine gamma globulin (100 μg/mL) were dissolved in 100 mmol/L K3PO4 buffer (pH 7.4). All targeted compounds were diluted in different concentration with 100 mmol/L K3PO4 buffer and was mixed with the fluorescence tracer/ER complex solution. The mixture was excluded from light incubating for 2 h. Fluorescence polarization values were measured by BioTek reader. The values were mean ± SD of at least three independent determinations.
4.4. Cell culture and cell viability assay
Human breast cancer cells MCF-7 and MCF-10A cells was purchased from the National Collection of Authenticated Cell Cultures (NCACC) and ATCC, respectively. Human tamoxifen-resistant breast cancer cell line (LCC-2), and T-47D/T-47DD538G/Y537S breast cancer cell lines were donated from the Wuhan University Medical Research Institute and Kaiwei Liang group at Wuhan University School of Basic Medical Sciences, respectively. MCF-7 or MCF-10A cell lines were cultured in DMEM medium (1% penicillin-streptomycin and 10% Fetal Bovine Serum). LCC-2, T-47D, T-47DD538G and T-47DY537S cell lines were cultured in RPMI-1640 medium (1% penicillin‒streptomycin and 10% Fetal Bovine Serum). Cells were planted in 96-well plates overnight and then incubated with different concentration of target compounds for 72 h. The cell lines viability was examined by Kit-8 (Biosharp, China). The IC50 was analyzed by GraphPad Prism 6.0 software. The procedure of apoptosis experiment was as follows: after a 48-h exposure to drug solutions, the cells were collected and subjected to staining with the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences), as directed by the manufacturer. Subsequently, flow cytometry analysis was performed using a Beckman Coulter instrument.
4.5. Mass spectrometry to assess covalency of 29c
ERα Y537S and C381S–C417S–Y537S proteins were incubated in 25 mmol/L Tris (pH = 8.0), 150 mmol/L NaCl, and 1 mmol/L TCEP with a 20-fold excess of compound (20 μmol/L compound 29c DMSO solution:1 μmol/L ERα protein solution) at 4 °C overnight respectively. Intact protein was analyzed using an Agilent 1290 Infinity II LC coupled with a 6545-XT QTOF mass spectrometer. Mobile Phase A was water with 0.1% formic acid and Mobile Phase B was acetonitrile with 0.1% formic acid. Intact protein samples were separated with an PLRP-S biomolecules columns (1.0 mm × 50 mm, 5 μm) employing 15 min gradient at a flow rate of 0.300 mL/min (5% B for 5 min, 5%–95% B for 5 min, 95% for 2 min, 95%–5% B for 0.1 min, 5% B for 2.9 min). The 6545-XT QTOF mass instrument parameters were set as the following: the dry gas flow rate was set 10.0 L/min at 325 °C, the nebulizer was set at 40 psig, the capillary voltage was set at 3.5 kV and the scan range was from 350 to 3000 m/z at 1 Hz. Data were collected with the MassHunter B.06.01 software.
4.6. Transcriptional activity assay
Human HEK-293T (ATCC) cells were seeded into 48-well plates with about 1 × 106 cells per well before transfection for 24 h. A solution of CaCl2 (2.5 mol), pGL3-3ERE-tk (150 ng), pHAGE-puro-ERα (5 ng) or ERβ (50 ng) or pET46EK-ERαC530S, and pRL-cmv (1 ng) was adjusted to 12.5 μL and then mixed with an equal volume of 2 × HBS buffer. Refreshed medium after 24 h of transfection which contained the desired concentrations of detected compounds. For detection of antagonistic activity, medium of dilute compound containing 10 nmol/L 17β-estradiol was used to make the above concentrations for 24 h, and transcriptional activity was measured by a Stop & Glo Reagent.
4.7. Western blot assay
Western blot was conducted to analyze ERα protein levels on MCF-7, LCC-2, T-47D, T-47DY537S, T-47DD538G cells. Cells were planted in six-well plate and incubated with target compound (1 or 5 μmol/L) for indicated time. Cellular proteins were extracted using RIPA lysates and were separated by electrophoresis on SDS-PAGE gels. After the gel was transferred to the polyvinylidene fluoride (PVDF) membrane, it was blocked with 5% skim milk. Then PVDF membranes were incubated with mouse Anti-β-Actin antibody and rabbit anti-ERα antibody overnight. Membranes were washed and incubated with goat anti rabbit secondary antibody (Wuhan Feiyi Group, China) for 1 h. After washing, membranes were developed by ECL. For IP of tagged protein, Flag-ERα and HA-Ub plasmids (donated from the Wuhan University Medical Research Institute and Kaiwei Liang group at Wuhan University School of Basic Medical Sciences) were transfected into HEK-293T cells using Lipo2000 transfection reagent for 8–12 h. The basic medium (Opti-MEM) was replaced with complete medium (10% FBS) and incubated for another 12 h and the cells were collected and lysed with 1 mL of lysis buffer, followed by centrifugation at 21,000 rcf for 10 min at 4 °C. The supernatant was collected and the beads were mixed and washed twice with 1 mL of lysis buffer. The simples were then transferred to each EP tube and followed by electrophoresis on SDS-PAGE gels as previously described.
4.8. Protein crystallization and data acquisition
Purifications of ERα and methods of crystallization were summarized in supporting information. All graphics for the protein structures were prepared by PyMOL. Atomic coordinates for the crystal structures of ERα with compounds 29c (7YMK) can be accessed from the RCSB Protein Data Bank (www.rcsb.org).
4.9. PK studies in mice
PK Studies were conducted in BALB/c female mice (n = 3 mice/group). A solution of 29c was prepared in Corn Oil and DMSO (95%:5%) for po administration (20 mg/kg), or in DMSO, PEG400, β-cyclodextrin, and saline (5%:40%:10%:45%) for iv administration (2 mg/kg), or in DMSO, PEG400 and saline (5%:40%:55%) for ip. administration (4 mg/kg). After administration, blood samples were collected at indicated time points of 0.25, 0.5, 1, 2, 4, 8, 12, 24 and 48 h respectively. The plasma samples were deproteined with MeCN and concentration of each compound in supernatant was analyzed by LC/MS/MS.
4.10. Pharmacodynamic studies in nude mice
Female BALB/c nude mice (15–18 g, 5 weeks old) were purchased from Beijing HFK Bioscience Co. Ltd. All animal experiments are strictly followed the Guide for the Care and Use Committee at Wuhan University (permit no. S01320070A, Wuhan, China). 100 μL PBS containing 1 × 107 MCF-7 cells were injected in the right axillary mammary fat pad area of mice. Once the tumor volume reached ∼100 mm3, mice were randomly divided into four groups (n = 5/group) and the conducted drug administration. The mice were intraperitoneally administered with vehicle control (PEG-400:DMSO:PBS = 40:5:55), Tam (4 mg/kg), Ful (2 mg/kg) or compound 29c (2 or 4 mg/kg) every other day. The weight of the mice and the tumor volume were measured every 2 days. The formula for tumor size calculation: Volume = (Width)2 × (Length) × 0.5. On Day 33, the mice were sacrificed. Tumor tissue sections Ki-67 were analyzed through immunohistochemistry and Western blot analysis. The heart, liver, spleen and kidney of mice were stained with H&E.
Acknowledgments
The work was supported by National Key R&D Program of China (2020YFA0908800, 2021YFC2100300), National Natural Science Foundation of China (82273774, 82073690, 81773557, 82173676, 82103994), the Fundamental Research Funds for the Central Universities of China (2042022kf0056), and the China Postdoctoral Science Foundation (2020M672435).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.05.005.
Contributor Information
Rey-Ting Guo, Email: guoreyting@hubu.edu.cn.
Chune Dong, Email: cdong@whu.edu.cn.
Hai-Bing Zhou, Email: zhouhb@whu.edu.cn.
Author contributions
Chune Dong and Hai-Bing Zhou conceived and supervised the project. Yubo Wang, Xiangping Deng, Tian Feng, Hebing Hu, Xinyi Guo, Yan Chen, Baohua Xie and Yu Yang performed experiments. Yubo Wang, Jian Min, Xiangping Deng and Tian Feng are involved in acquisition of data, analysis and interpretation of data. Yubo Wang, Jian Min, Chun-Chi Chen, Rey-Ting Guo, Chune Dong and Hai-Bing Zhou analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics. Ca - Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. 2020. [DOI] [PubMed] [Google Scholar]
- 2.Tong C.W.S., Wu M., Cho W.C.S., To K.K.W. Recent advances in the treatment of breast cancer. Front Oncol. 2018;8:227. doi: 10.3389/fonc.2018.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson W.F., Katki H.A., Rosenberg P.S. Incidence of breast cancer in the United States: current and future trends. J Natl Cancer Inst. 2011;103:1397–1402. doi: 10.1093/jnci/djr257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckhardt B.L., Francis P.A., Parker B.S., Anderson R.L. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat Rev Drug Discov. 2012;11:479–497. doi: 10.1038/nrd2372. [DOI] [PubMed] [Google Scholar]
- 5.Ariazi E.A., Ariazi J.L., Cordera F., Jordan V.C. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6:181–202. [PubMed] [Google Scholar]
- 6.Gandhi N., Das G.M. Metabolic reprogramming in breast cancer and its therapeutic implications. Cells. 2019;8:89. doi: 10.3390/cells8020089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciruelos E., Pascual T., Arroyo Vozmediano M.L., Blanco M., Manso L., Parrilla L., et al. The therapeutic role of fulvestrant in the management of patients with hormone receptor-positive breast cancer. Breast. 2014;23:201–208. doi: 10.1016/j.breast.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Lin X., Xiang H., Luo G. Targeting estrogen receptor α for degradation with PROTACs: a promising approach to overcome endocrine resistance. Eur J Med Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112689. [DOI] [PubMed] [Google Scholar]
- 9.Lu Y., Liu W. Selective estrogen receptor degraders (SERDs): a promising strategy for estrogen receptor positive endocrine-resistant breast cancer. J Med Chem. 2020;63:15094–15114. doi: 10.1021/acs.jmedchem.0c00913. [DOI] [PubMed] [Google Scholar]
- 10.Lai A., Kahraman M., Govek S., Nagasawa J., Bonnefous C., Julien J., et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58:4888–4904. doi: 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- 11.Hu J., Hu B., Wang M., Xu F., Miao B., Yang C.Y., et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER) J Med Chem. 2019;62:1420–1442. doi: 10.1021/acs.jmedchem.8b01572. [DOI] [PubMed] [Google Scholar]
- 12.Lu Z., Cao Y., Zhang D., Meng X., Guo B., Kong D., et al. Discovery of thieno[2,3-e]indazole derivatives as novel oral selective estrogen receptor degraders with highly improved antitumor effect and favorable druggability. J Med Chem. 2022;65:5724–5750. doi: 10.1021/acs.jmedchem.2c00008. [DOI] [PubMed] [Google Scholar]
- 13.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robertson J.F.R., Bondarenko I.M., Trishkina E., Dvorkin M., Panasci L., Manikhas A., et al. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet. 2016;388:2997–3005. doi: 10.1016/S0140-6736(16)32389-3. [DOI] [PubMed] [Google Scholar]
- 15.Howell A., Sapunar F. Fulvestrant revisited: efficacy and safety of the 500-mg dose. Clin Breast Cancer. 2011;11:204–210. doi: 10.1016/j.clbc.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 16.McDonnell D.P., Wardell S.E., Norris J.D. Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J Med Chem. 2015;58:4883–4887. doi: 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernando C., Ortega-Morillo B., Tapia M., Moragón S., Martínez M.T., Eroles P., et al. Oral selective estrogen receptor degraders (SERDs) as a novel breast cancer therapy: present and future from a clinical perspective. Int J Mol Sci. 2021;22:7812. doi: 10.3390/ijms22157812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fanning S.W., Mayne C.G., Dharmarajan V., Carlson K.E., Martin T.A., Novick S.J., et al. Estrogen receptor α somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. 2016;5 doi: 10.7554/eLife.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L., Sharma A. The quest for orally available selective estrogen receptor degraders (SERDs) ChemMedChem. 2020;15:2072–2097. doi: 10.1002/cmdc.202000473. [DOI] [PubMed] [Google Scholar]
- 20.Kiely-Collins H., Winter G.E., Bernardes G.J.L. The role of reversible and irreversible covalent chemistry in targeted protein degradation. Cell Chem Biol. 2021;28:952–968. doi: 10.1016/j.chembiol.2021.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Abdeldayem A., Raouf Y.S., Constantinescu S.N., Moriggl R., Gunning P.T. Advances in covalent kinase inhibitors. Chem Soc Rev. 2020;49:2617–2687. doi: 10.1039/c9cs00720b. [DOI] [PubMed] [Google Scholar]
- 22.Singh J., Petter R.C., Baillie T.A., Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 23.De Vita E. 10 years into the resurgence of covalent drugs. Future Med Chem. 2021;13:193–210. doi: 10.4155/fmc-2020-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boike L., Henning N.J., Nomura D.K. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022:1–18. doi: 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furman C., Hao M.H., Prajapati S., Reynolds D., Rimkunas V., Zheng G.Z., et al. Estrogen receptor covalent antagonists: the best is yet to come. Cancer Res. 2019;79:1740–1745. doi: 10.1158/0008-5472.CAN-18-3634. [DOI] [PubMed] [Google Scholar]
- 26.Furman C., Puyang X., Zhang Z., Wu Z.J., Banka D., Aithal K.B., et al. Covalent ERα antagonist H3B-6545 demonstrates encouraging preclinical activity in therapy-resistant breast cancer. Mol Cancer Therapeut. 2022;21:890–902. doi: 10.1158/1535-7163.MCT-21-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puyang X., Furman C., Zheng G.Z., Wu Z.J., Banka D., Aithal K., et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERαWT and ERαMUT breast cancer. Cancer Discov. 2018;8:1176–1193. doi: 10.1158/2159-8290.CD-17-1229. [DOI] [PubMed] [Google Scholar]
- 28.Hamilton E.P., Wang J.S., Pluard T.J., Johnston S.R.D., Morikawa A., Dees E.C., et al. Phase I/II study of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2‒) advanced breast cancer. J Clin Oncol. 2021;39:1018. [Google Scholar]
- 29.Zhou H.B., Comninos J.S., Stossi F., Katzenellenbogen B.S., Katzenellenbogen J.A. Synthesis and evaluation of estrogen receptor ligands with bridged oxabicyclic cores containing a diarylethene motif: estrogen antagonists of unusual structure. J Med Chem. 2005;48:7261–7274. doi: 10.1021/jm0506773. [DOI] [PubMed] [Google Scholar]
- 30.Ning W., Hu Z., Tang C., Yang L., Zhang S., Dong C., et al. Novel hybrid conjugates with dual suppression of estrogenic and inflammatory activities display significantly improved potency against breast cancer. J Med Chem. 2018;61:8155–8173. doi: 10.1021/acs.jmedchem.8b00224. [DOI] [PubMed] [Google Scholar]
- 31.Tang C., Li C., Zhang S., Hu Z., Wu J., Dong C., et al. Novel bioactive hybrid compound dual targeting estrogen receptor and histone deacetylase for the treatment of breast cancer. J Med Chem. 2015;58:4550–4572. doi: 10.1021/acs.jmedchem.5b00099. [DOI] [PubMed] [Google Scholar]
- 32.Deng X., Xie B., Li Q., Xiao Y., Hu Z., Deng X., et al. Discovery of novel bicyclic bhenylselenyl-containing hybrids: an orally bioavailable, potential, and multiacting cass of estrogen receptor modulators against endocrine-resistant breast cancer. J Med Chem. 2022;65:7993–8010. doi: 10.1021/acs.jmedchem.2c00525. [DOI] [PubMed] [Google Scholar]
- 33.Aliau S., Mattras H., Richard E., Borgna J.L. Cysteine 530 of the human estrogen receptor alpha is the main covalent attachment site of 11β-(aziridinylalkoxyphenyl)estradiols. Biochemistry. 1999;38:14752–14762. doi: 10.1021/bi991176k. [DOI] [PubMed] [Google Scholar]
- 34.Katzenellenbogen J.A. The 2010 Philip S. Portoghese medicinal chemistry lectureship: addressing the "core issue" in the design of estrogen receptor ligands. J Med Chem. 2011;54:5271–5282. doi: 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weerapana E., Wang C., Simon G.M., Richter F., Khare S., Dillon M.B., et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nacht M., Qiao L., Sheets M.P., St Martin T., Labenski M., Mazdiyasni H., et al. Discovery of a potent and isoform-selective targeted covalent inhibitor of the lipid kinase PI3K. α. J Med Chem. 2013;56:712–721. doi: 10.1021/jm3008745. [DOI] [PubMed] [Google Scholar]
- 37.Hu Z., Li Y., Xie B., Ning W., Xiao Y., Huang Y., et al. Novel class of 7-oxabicyclo[2.2.1]heptene sulfonamides with long alkyl chains displaying improved estrogen receptor α degradation activity. Eur J Med Chem. 2019;182 doi: 10.1016/j.ejmech.2019.111605. [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Zhang S., Zhang J., Hu Z., Xiao Y., Huang J., et al. Exploring the PROTAC degron candidates: OBHSA with different side chains as novel selective estrogen receptor degraders (SERDs) Eur J Med Chem. 2019;172:48–61. doi: 10.1016/j.ejmech.2019.03.058. [DOI] [PubMed] [Google Scholar]
- 39.Zheng Y., Zhu M., Srinivasan S., Nwachukwu J.C., Cavett V., Min J., et al. Development of selective estrogen receptor modulator (SERM)-like activity through an indirect mechanism of estrogen receptor antagonism: defining the binding mode of 7-oxabicyclo[2.2.1]hept-5-ene scaffold core ligands. ChemMedChem. 2012;7:1094–1100. doi: 10.1002/cmdc.201200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srinivasan S., Nwachukwu J.C., Bruno N.E., Dharmarajan V., Goswami D., Kastrati I., et al. Full antagonism of the estrogen receptor without a prototypical ligand side chain. Nat Chem Biol. 2017;13:111–118. doi: 10.1038/nchembio.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fanning S.W., Jeselsohn R., Dharmarajan V., Mayne C.G., Karimi M., Buchwalter G., et al. The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. Elife. 2018;7 doi: 10.7554/eLife.37161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C., Li C., Zhou H., Huang J. High-throughput screening assays for estrogen receptor by using coumestrol, a natural fluorescence compound. J Biomol Screen. 2014;19:253–258. doi: 10.1177/1087057113502673. [DOI] [PubMed] [Google Scholar]
- 43.Min J., Nwachukwu J.C., Min C.K., Njeri J.W., Srinivasan S., Rangarajan E.S., et al. Dual-mechanism estrogen receptor inhibitors. Proc Natl Acad Sci U S A. 2021;118 doi: 10.1073/pnas.2101657118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.