

Abstract
More than a hundred genes have been identified that, when disrupted, impart large risk for autism spectrum disorder (ASD). Current knowledge about the encoded proteins — although incomplete — points to a very wide range of developmentally dynamic and diverse biological processes. Moreover, the core symptoms of ASD involve distinctly human characteristics, presenting challenges to interpreting evolutionarily distant model systems. Indeed, despite a decade of striking progress in gene discovery, an actionable understanding of pathobiology remains elusive. Increasingly, convergent neuroscience approaches have been recognized as an important complement to traditional uses of genetics to illuminate the biology of human disorders. These methods seek to identify intersection among molecular-level, cellular-level and circuit-level functions across multiple risk genes and have highlighted developing excitatory neurons in the human mid-gestational prefrontal cortex as an important pathobiological nexus in ASD. In addition, neurogenesis, chromatin modification and synaptic function have emerged as key potential mediators of genetic vulnerability. The continued expansion of foundational ‘omics’ data sets, the application of higher-throughput model systems and incorporating developmental trajectories and sex differences into future analyses will refine and extend these results. Ultimately, a systems-level understanding of ASD genetic risk holds promise for clarifying pathobiology and advancing therapeutics.
Autism spectrum disorder (ASD) constitutes a group of aetiologically and symptomatically heterogeneous, early-onset developmental disorders that are defined by core deficits in social communication and the presence of repetitive, stereotyped behaviours1. Although these features are the diagnostic sine quo non, individuals come to clinical attention representing a very wide range of severity and often with co-occurring signs and symptoms that can include (but are not limited to) intellectual disability, anxiety, depression, sleep disturbance, epilepsy, specialized talents (also known as savant skills), delayed motor development and neurological soft signs. The inherent breadth of the presentation has led to an often-stated clinical pearl that “if you have seen one person with ASD, you have seen one person with ASD”. Indeed, the notion of an autism spectrum is a central feature of the diagnostic nosology.
However, despite the wide variety of clinical manifestations and the absence of reliable diagnostic biomarkers2, ASD persistently ranks at the very top of the list of neuropsychiatric disorders with regard to relative genetic contribution3–20. In fact, over the past 15 years, despite an extraordinary degree of aetiological and clinical heterogeneity, the pursuit of specific genetic contributors to ASD has been remarkably successful. Similarly to that of other relatively common disorders, the genetic architecture of ASD is now confirmed to include a wide range of variation with regard to type, frequency and effect size. However, the greatest promise for insight into the pathobiology of ASD presently is the result of striking progress in gene discovery based on the identification of large-effect, rare (often de novo) damaging variants in the coding portion of the genome21–34.
The success in characterizing many rare large-effect mutations and concomitant gene discovery would reasonably be expected to set the stage for the rapid identification of key biological features and potential treatment targets. However, the extensive locus heterogeneity and biological pleiotropy of ASD risk genes, the developmental dynamism of the human brain along with its structural and functional complexity, the wide range of clinical outcomes associated with even large-effect genetic risks, the presence of marked sexual dimorphism and the inherent limitations of relying on evolutionarily distant model system to illuminate distinctly, if not uniquely, human characteristics have posed formidable barriers to translating genetic insights into an actionable understanding of underlying mechanisms.
However, as gene identification has progressed over the past decade, these successes have also ushered in a generation of ‘convergent neuroscience’ analyses aimed at illuminating core aspects of ASD pathology. By convergent neuroscience, we refer here to analyses that address the overlap, or intersection, of diverse ASD genetic risks with respect to molecular-level, cellular-level and circuit-level function as well as across multiple dimensions of analysis including anatomical localization and developmental timing35–40. These conceptual approaches originated with the earliest successful efforts at ‘idiopathic’ ASD gene discovery41 and have often relied on Gene Ontology terms to offer initial insights, although contemporary approaches have begun to integrate other data, such as gene expression patterns from the human brain and maps of interactions between proteins and/or genes.
These studies have proven valuable in generating hypotheses about functional commonalities among ASD risk genes, providing consistent evidence for chromatin modification, synapse structure and function, RNA-binding proteins and several other points of mechanistic overlap29,42–44. They have also generated hypotheses about spatial-level, temporal-level and cellular-level convergence of ASD genetic risk, repeatedly implicating mid-gestational developmental stages, the frontal cortex and developing cortical excitatory neurons. However, in evaluating the current state of the field with regard to the understanding of mechanism, it is important to consider the potential for cryptic biases in these types of analyses related to the selection and the sources of the underlying data. Nonetheless, with the accelerating development of foundational large-scale biological data sets, particularly those that derive from direct experimental evidence and include human data and developmental trajectories, convergence neuroscience analyses hold increasing promise for identifying core biological features and offer avenues to constrain key experimental parameters in investigations of the pathological sequelae of individual genes and mutations — a key challenge for studies of the developing brain that confront an extraordinarily complex and developmentally dynamic biological landscape37. In addition to offering relatively hypothesis-free insights into genetic risks and related pathobiology, newer convergent approaches relying on highly parallelized modelling of rare variants carrying large risk for ASD also have the potential to identify previously unappreciated, yet widely applicable, mechanisms of resilience39,40,45, a potentially promising avenue for the development of treatments impacting broad populations of individuals who do not necessarily carry mutations in the same gene or genes. In this Review, we summarize progress in the genetics and genomics of ASD, assess the state of the field with respect to findings from analyses employing a convergent neuroscience framework and consider the implications of progress in genomics and convergent neuroscience for the future of translational research.
Gene and locus discovery in ASD
Whole exome-based discovery.
The past two decades have been a period of striking progress in the identification of specific genes and loci carrying individually large risks for ‘idiopathic’ ASD (BOX 1). The first successful efforts at isolating bona fide risk genes can be traced to just after the turn of the millennium, with the identification of heterozygous damaging and putative loss-of-function mutations in the genes NLGN3 and NLGN4, through traditional cytogenetic mapping strategies46 and parametric linkage analysis47 followed by DNA sequencing. Soon thereafter, the rapid evolution of genomic technologies allowed for cost-effective, comprehensive scanning of the genome for rare and de novo mutations at increasingly high resolution (BOX 2). The critical observations that de novo structural and sequence variants contribute writ large to common forms of ASD21,30,31,34, together with the insight that even a small number of exceedingly rare de novo variants mapping to the same gene or genomic interval in unrelated individuals offers sufficient statistical power to reliably identify risk loci22,24,33,48–51 and genes23–30,32,42 with limited confounds due to ancestry, constituted a critical turning point for the field, setting the stage for systematic and reliable gene discovery (FIG. 1). At present, microarray and whole-exome sequencing (WES) studies focused on rare variants have yielded convincing statistical support for the association of about a dozen copy number variant (CNV) loci24 and more than 100 genes23 (FIG. 1a), with this list rapidly growing (for example, see REF.52) and many more of each predicted to exist22,23,28,30,33,53.
Box 1 |. Idiopathic versus syndromic ASD.
There is debate about the definition of ‘syndromic’ forms of autism spectrum disorder (ASD) versus ‘idiopathic’ presentations. Syndromic generally refers to rare and severe conditions with a highly characteristic presentation that may have co-occurring ASD — for example, fragile X syndrome, Angelman syndrome, Rett syndrome, tuberous sclerosis, neurofibromatosis and PTEN hamartoma tumour syndrome. In practice, syndromic ASD tends to have a greater contribution of features apart from social communication deficits and repetitive behaviours, shows greater penetrance and tends towards monogenic causality. By contrast, idiopathic refers to cases that are not obviously accompanied by characteristic physical features or a pathognomic natural history. Although there may be considerable overlap in symptoms among these groups, and an increasing appreciation of intersecting biology, the history of gene identification efforts has diverged at the extremes of these distinctions. For example, there was substantial progress in the identification of syndromic loci and genes leveraging classic genetic methods, before the sequencing of the human genome and the genomic era227–247. By contrast, reliable, systematic gene discovery in the larger group of individuals who commonly present clinically with ASD was highly dependent on the development of high-throughput genomic technologies focused on rare and de novo mutations, as discussed in the main text.
As gene discovery has progressed, and larger groups of affected individuals with rare variants in the same gene have been characterized, the boundaries between syndromic and non-syndromic have become less clear. Careful retrospective clinical analyses of patients whose ASD is apparently idiopathic and who share a mutated ASD gene or chromosomal segment have not infrequently led to the recognition of distinctive physical features and co-morbidities suggesting the presence of previously unappreciated syndromes248–254. Therefore, at present, syndromic versus idiopathic ASD may be most productively thought of existing on a continuum both with regard to relative contribution of core features of the ASD diagnosis as well as with regard to the reliability of genotype–phenotype correlations.
Box 2 |. Major methods to identify ASD risk genes and loci.
Rare variant-based approaches (allele frequency <1%)
Whole-exome sequencing (WES).
This approach aims to identify rare, especially de novo, coding variants recurring in the same gene in unrelated patients more often than expected by chance (based on theoretical mutation rates, unaffected siblings or population frequency). These approaches have historically focused on ‘simplex’ families (unaffected parents with one child with autism), which tend to be enriched for de novo variants. However, rare inherited variants and case–control data from simplex, multiplex or unknown family types have increasingly contributed to gene discovery, especially as databases accumulate information on population variant frequency and the ability to stratify the most deleterious variants improves58–60. As these databases grow, the definition of rare is beginning to shift to <0.1% or even <0.01%. Genes identified by this approach tend to carry large effect sizes (FIG. 1).
Whole-genome sequencing (WGS).
Conceptually similar to WES, these analyses expand to include non-coding variants. Identifying non-coding loci requires very large cohort sizes and is challenging due to incomplete characterization of regulatory loci (that is, functional boundaries, target gene), the relative inability to prioritize deleterious variants (especially compared with coding variants) and the tendency for these effects to be restricted to particular (yet currently unknown) developmental epochs, tissues and/or cell types. Accordingly, specific non-coding loci have not yet been identified at genome-wide significant thresholds.
Copy number variant (CNV) detection.
CNVs are deletions or duplications of DNA (also known as structural variants). The approach to detecting CNVs is conceptually similar to WES and WGS: identify rare and/or de novo CNVs from genotyping or sequencing data, and then search for recurrent variants overlapping the same locus in multiple unaffected individuals. Associated loci tend to overlap genes and carry large effects (FIG. 1). When combined with WES and WGS, small coding CNVs, especially deletions, can help identify individual risk genes24.
Common variant-based approach (allele frequency >1%)
Genome-wide association.
This approach relies on common variants (also known as single-nucleotide polymorphisms (SNPs)), occurring at a frequency greater than 1% in the population. Variants are generally identified with genotyping arrays. Cases and controls are compared to identify SNPs with an allele that occurs more frequently in cases. Very large cohorts are required to identify risk loci as they carry small effect sizes (FIG. 1). Only five genome-wide significant loci for autism spectrum disorder (ASD) have been identified to date92 but this number is expected to grow as sample sizes increase.
Fig. 1 |. Relationship between effect size and allele frequency for loci discovered in autism spectrum disorder or schizophrenia.
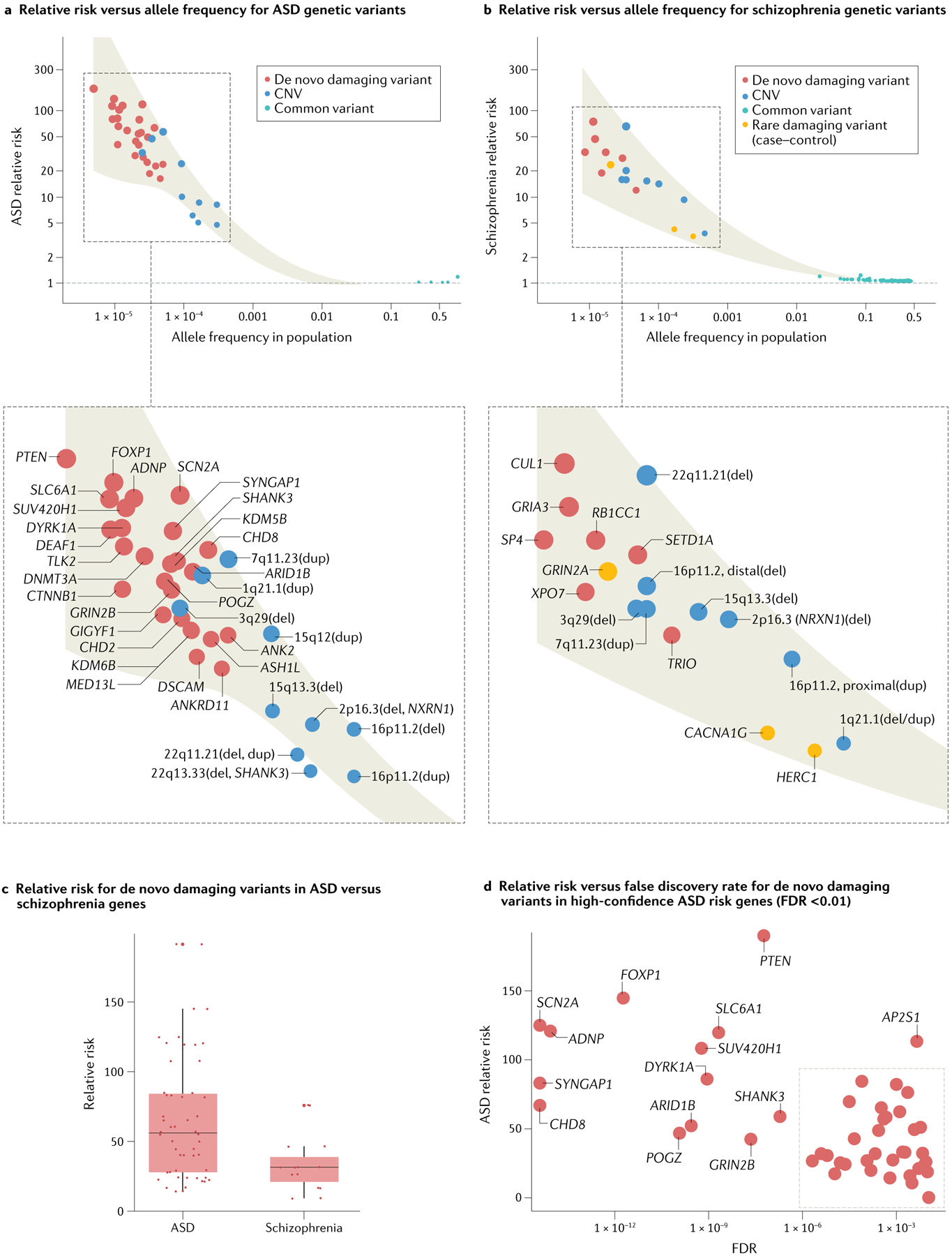
a,b | Systematic studies of rare and common variants have identified risk loci for autism spectrum disorder (ASD) and for schizophrenia, but the trajectory of discovery differs between these conditions. In ASD, the majority of loci discovered to date are genes and have been identified by exome-wide sequencing studies of rare, generally de novo, coding variants (P < 2.5 × 10−6; red dots; for ASD, 26 genes23; for schizophrenia, 10 genes66), whereas in schizophrenia the majority are single-nucleotide polymorphisms (SNPs) and have been discovered based on genome-wide association studies (GWAS) of common variants (P < 5 × 10−8; teal dots; for ASD, 5 SNPs92; for schizophrenia, 270 SNPs130, of which only the 132 SNPs with relative risk >1 are shown). None of these genes (red) or SNPs (teal) overlap between ASD and schizophrenia. Both disorders have a similar number of loci associated based on rare, generally de novo, copy number variants (CNVs; blue dots; for ASD, permutation-based false discovery rate (FDR) ≤ 0.02, 10 loci24; for schizophrenia, Benjamini–Hochberg FDR ≤ 0.02, 8 loci222). In contrast to rare coding variants and common variants, most of the top CNVs overlap between ASD and schizophrenia. In general, rare variants carry substantially higher relative risks than common variants. c | De novo damaging variants in the top 26 ASD risk genes tend to carry higher relative risk than de novo damaging variants within the top 7 schizophrenia risk genes (de novo damaging variants have been identified in only 7 out of the 10 schizophrenia risk genes), underscoring the particularly large contribution of rare variants to ASD. d | Utilizing a conservative FDR threshold instead of an exome-wide significant P value (FDR ≤ 0.01 versus P < 2.5 × 10−6) highlights 47 high-confidence ASD risk genes23, with varying relative risks. In general, the most strongly associated genes carry the largest relative risks. See Supplementary Table 1 for a complete list of gene names, FDRs and effect sizes; see REF.223 for a broader list of ASD risk genes as determined by FDR. Note that we defined relative risk as the ratio of the frequency of a given variant in cases versus unaffected controls. Dot size is proportional to relative risk. Shaded area represents 95% confidence interval of the locally weighted least squares regression. Location on x axis in parts a,b is based on frequency in unaffected controls. As there are no associations in ASD for alleles with frequencies between 0.001 and 0.1, no curve is shown in this interval in part a. Damaging variants consist of protein-truncating variants (frameshift, canonical splice-acceptor, canonical splice-donor and nonsense variants) and missense 3 (Mis3) variants (those predicted to be probably damaging by PolyPhen-2 (REF.62)). To estimate the per gene relative risk for de novo damaging variants, we defined the frequency in cases as the total number of de novo damaging variants observed in a given gene23,66 and the frequency in controls based on estimates of the number of mutations expected per generation23. All 26 genes identified in ASD have multiple de novo damaging variants in probands23. However, only seven of ten genes identified in schizophrenia have de novo variants (the rest were identified by rare damaging variants in case–control data only)66. Therefore, we estimated relative risk for de novo damaging variants when possible (red dots) and for rare damaging variants when necessary (orange dots). To estimate relative risk for rare damaging variants (case–control, schizophrenia only), we defined the frequency of each allele in cases based on reported frequency for schizophrenia66 and the frequency in controls based on the frequency observed in the non-psychiatric subset of the gnomAD v2 data set58. To estimate relative risk for rare CNVs, we defined the frequency in cases based on reported frequencies for ASD24 and for schizophrenia222 and the frequency in controls based on frequency observed in the Database of Genomic Variants (DGV)224 (ASD) or directly from controls (schizophrenia)222. We estimated the 3q29(del) frequency in schizophrenia as 0.00005 because no 3q29(dels) were observed in 20,227 controls. The 15q13.3(del) is referred to as 15q13.2–13.3 in REF.24 and we estimated its frequency as 0.000092 based on structural variants in gnomAD225 because of inconsistent frequencies reported in smaller studies in DGV. We estimated relative risk for common variants based on reported odds ratios for ASD92 or for schizophrenia130. Part b adapted from REF.66, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
The initial progress in ASD gene discovery relied heavily on the identification of rare heterozygous de novo mutations leading to putative loss of function of the encoded protein (also referred to as protein-truncating variants). However, missense de novo mutations30 as well as rare inherited and somatic mutations (also known as mosaic mutations) have now also been demonstrated to carry risk23,24,29,53–57. In addition, the dramatic expansion of databases of control exome and genome sequences has enabled the identification of ultra-rare genetic variation, the characterization of evolutionarily constrained genes (that is, those likely intolerant to variation) and the prioritization of missense variants most likely to be deleterious (for example, as determined by the MPC or ‘missense badness, PolyPhen-2 and constraint’ score)58–62, all of which have demonstrated utility in parsing risk for ASD and, consequently, the potential to improve gene discovery23,29,55,60,63–65. Indeed, characterizing ultra-rare variants among highly conserved genes in case–control data alone has emerged as an efficient, viable alternative to family-based studies designed to identify rare de novo mutations (for example, FIG. 1b; also REF.66).
Likewise, there have been long-standing efforts to identify rare recessive variants and compound heterozygous variants contributing to ASD. Based on the strong track record of success relying on homozygosity mapping of consanguineous pedigrees in a wide range of severe neurological and neurodevelopmental disorders (NDDs), including brain malformation syndromes, severe intellectual disability and epilepsy67–69, it has been somewhat surprising that the yield of novel genes from similar efforts have been relatively limited for the ASD phenotype, although there have been some notable exceptions70–74. Additionally, several more recent studies have demonstrated an overall modest contribution of recessive inheritance, including compound heterozygous damaging mutations in non-consanguineous families74–77.
Studies have also supported the contribution of rare somatic variation to ASD, suggesting that these variants may contribute to risk in approximately 5–22% of simplex cases56,57,78–83. However, gene discovery based on somatic variants alone has been underpowered. Additionally, because these studies generally use data derived from the DNA of clinically accessible tissues such as peripheral blood and saliva57,79–82, brain-specific somatic mutations will, by their nature, be missed. Identifying somatic variants within brain tissue from individuals with ASD is an active area of investigation56, but the obstacles to this as a systematic approach to identifying novel ASD risk genes remain formidable.
Collectively, rare coding variants identified from WES not only have been a particularly valuable resource for biological studies but also have been shown to contribute to a substantial minority of individuals presenting for clinical evaluation. The estimates of clinical contribution range from a low of about 10% to a high of approximately one half of the clinical ASD population, depending in part on the ascertainment criteria. For example, increasing parental age, low IQ relative to mid-parental expectation, dysmorphology, epilepsy, congenital heart disease, female sex and simplex status — along with a larger number of unaffected siblings — are all known to lead to higher positive yields in genetic testing21,22,24,28–31,33,44,64,84–89.
Whole-genome sequencing.
To date, the contribution of whole-genome sequencing (WGS) studies to identifying risk genes and loci for ASD has been limited compared with the approaches noted above. Capturing the full complement of variation across the entire genome, including both sequence and structural mutations, clearly represents the future of genomic analyses for ASD and, indeed, all of medicine. However, several factors limit its current utility: broadly, that the analysis and storage of WGS data require extensive computational infrastructure and confer substantial costs; the ‘search space’ for WGS is 100-fold greater than for WES, yet is less well characterized; the ability to interpret the consequences of non-coding variation is far more challenging than assigning functional significance to canonical coding mutations; and, as a group, rare non-coding variation is estimated to carry relatively more modest risks for ASD as compared to rare damaging coding mutations90. Consequently, the cohort sizes needed to identify associated loci with confidence are estimated to be an order of magnitude or more larger than for contemporary WES studies, and, currently, no published investigations have yet realized sufficient numbers to identify, definitively, individual non-coding risk variants (BOX 2).
Common variant-based discovery.
Although most of the ASD risk loci confirmed to date have come from studies of rare coding variants of large effect, there is strong evidence that common variants of small effect carry the majority of population risk4,5. Despite this, initial ASD genome-wide association studies (GWAS) yielded disappointing results owing to insufficient statistical power91. However, more recent studies, involving more than 18,000 cases and almost 28,000 controls, have successfully identified five common risk alleles of modest effect (BOX 2; FIG. 1a), meeting rigorous statistical criteria for association92. Undoubtedly, this number will continue to grow as case–control cohorts further increase in size91. Moreover, despite the relatively small number of loci implicated to date, polygenic risk scores, which are a means to quantify cumulative common genetic risks, have already demonstrated a modest ability to differentiate cases versus controls92. Additionally, polygenic risk scores appear to differ by sex93,94, with a higher genetic load observed in females, providing evidence for a ‘female protective effect’ (FPE; discussed below) — a phenomenon that has also been widely observed with respect to rare variants22,24,27,28,30,32,33,95. There has also been some success in identifying shared common genetic risks that cross a wide range of apparently disparate psychiatric diagnoses96.
There is also evidence that among individuals carrying large-effect rare risk variants, common alleles and polygenic inheritance likely contribute to idiopathic ASD97,98. Indeed, the question of the extent to which polygenic ‘background’ may shape the impact of rare mutations and help define an individual’s clinical presentation and natural history has been long-standing99. The potential for very large, well-powered cohort studies in ASD makes this an increasingly tractable area of investigation93,100. Similarly, as the yield of SNPs from ASD GWAS grows, important questions regarding the genetic architecture underlying various groups of individuals with autism, including those with so-called high-functioning autism, may well be clarified. Finally, the discovery of a much larger set of genome-wide significant common alleles will undoubtedly empower studies of convergence similar to those currently successfully leveraging rare, large-effect mutations.
The number of ASD-associated genes.
Contemporary association analyses of common, genetically complex and heterogeneous phenotypes such as ASD rely heavily on the application of rigorous statistical methods that effectively protect against false positive findings, which have historically bedevilled psychiatric genetic studies. Over the past decade, two broad approaches to the analysis of rare variants, using either a Bayesian or a frequentist framework, have emerged, providing the field with robust and consistent results. For WES studies specifically, it has become commonplace to require a discovery P value of less than or equal to 2.5 × 10–6 — effectively correcting for the multiple comparisons among the approximately 20,000 genes comprising the human exome. Widely used alternative methods estimate false discovery rates (FDRs) per gene using a Bayesian approach, assigning significant association typically to genes showing FDR ≤ 0.1 and reserving a ‘highest-confidence’ designation to those with FDR ≤ 0.01. By contrast, GWAS arrived relatively quickly at a single consensus standard and threshold, namely a discovery P value of less than 5 × 10−8 followed by the finding of nominal significance in a replication cohort101. Importantly, the fact that there remain alternative accepted statistical approaches to WES studies of rare variants contributes to some degree of variability in gene ‘accounting’. For example, the recent omnibus analysis by the Autism Sequencing Consortium23 is commonly cited as identifying 102 ASD risk genes, based on the Bayesian FDR threshold of 0.1 noted above. Using the alternative accepted exome-wide significant P value threshold (2.5 × 10−6) yields 26 risk genes (FIG. 1a) and restricting to the highest-confidence genes based on FDR ≤ 0.01 identifies 47 risk genes (FIG. 1d).
Additional definitional issues contribute to a range of views on what constitutes the definitive list of bona fide risk genes. For example, some widely used databases (for example, SFARI Gene102,103) include genes discovered in studies of syndromic forms of ASD. Such approaches have the advantage of being more complete than those relying solely on large-scale association studies restricted to ‘idiopathic’ clinical samples (BOX 1). However, consensus approaches also introduce a degree of subjectivity and potential ascertainment bias that may be largely avoided by focusing only on genes that exceed predetermined statistical thresholds23,24. The task of building a singular high-confidence ASD risk gene list is similarly made more challenging by studies that cross diagnostic categories. For instance, although approaches that combine ASD and other NDD cohorts increase the yield of gene discovery, they also complicate our understanding of the relative risk that each identified gene carries for ASD versus other NDDs. Further, results from studies that rely on targeted sequencing can be difficult to integrate with findings from WES or WGS, given the challenges of arriving at a single applicable statistical approach or threshold.
ASD versus other NDDs.
With recent success in gene identification across multiple NDDs and psychiatric disorders, the question of overlap of genetic risks among apparently distinct clinical syndromes has emerged as a key area of inquiry, for both conceptual and pragmatic reasons. Some of the areas currently of greatest interest in this regard include the degree to which ASD risks are distinguishable from the risks for schizophrenia or from intellectual disability. Both questions have historical relevance as well: autism was initially conceptualized clinically as a form of childhood-onset schizophrenia104,105, but conventional wisdom subsequently shifted and the notion was formally rejected after the second edition of the Diagnostic and Statistical Manual of Mental Disorders106. Similarly, it is now clear that early diagnostic criteria for autism tended to enrich for individuals with co-existing intellectual disability, resulting in a long-standing overestimate of the prevalence of intellectual disability in the population with ASD107.
The question of whether and to what degree there is specificity to ASD genetic risk versus other developmental phenotypes remains a controversial topic. Successive generations of systematic CNV studies have shown that identical structural variations can contribute to multiple distinct diagnostic outcomes, including, but not limited to, epilepsy, schizophrenia, bipolar disorder, intellectual disability, attention deficit hyperactivity disorder, Tourette disorder and ASD (see also FIG. 1a,b). Moreover, as noted above, the yield of rare risk mutations from high-throughput sequencing studies of ASD cohorts has consistently been found to increase as IQ declines. Not surprisingly, these kinds of observations have fuelled debates over whether these diagnoses are truly separable at the genetic level6,92,108–112. For CNVs, this variability in phenotypic outcomes has been particularly dramatic. From over a decade of progress in studies of ASD, schizophrenia, epilepsy and intellectual disability, it has become increasingly difficult to identify any structural variant associated with one of these diagnoses that is not also associated with some risk for one or more of the others — and the list of additional clinical outcomes showing overlap continues to grow113–116. Although there is some evidence for differing relative representations of clinical manifestations for individual CNVs, including for duplications versus deletions of the same genomic interval24, overall, the data have consistently pointed to the absence of diagnostic specificity6,108,110–112 (FIG. 1a,b).
These findings, not surprisingly, led to concern that the pursuit of biological mechanisms related to any given CNV would lack the ability to inform development of ASD therapeutics, as well as to speculation that individual genes of large effect would pose similar challenges. A related concern was, and remains, that the wide range of potential clinical outcomes from any given variant would pose serious obstacles to the design and execution of early intervention trials, including those targeting individuals with large-effect genetic risks. For example, it is not obvious how one would assess efficacy in a group of high genetic-risk individuals when the clinical course might evolve over weeks, months or years and include symptoms that vary dramatically from one person to the next, irrespective of treatment.
As both rare and common variant studies have progressed, a more nuanced view of the genetic architecture and loci contributing to ASD versus other neurodevelopmental syndromes is beginning to emerge. For example, in comparison with schizophrenia, much smaller cohorts have been required to identify large-effect risk genes in ASD. Conversely, much larger sample sizes have been required to identify the first genome-wide significant common variants in ASD versus in schizophrenia92,117–119. Consistent with these observations, rare variants show, on average, a larger relative risk in ASD than in schizophrenia (FIG. 1a–c). Moreover, in a recent study of schizophrenia focused on identifying ultra-rare coding variants, none of the 10 genes meeting the most rigorous cut-off for statistical significance (P ≤ 2.5 × 10–6) overlaps with the 26 genes identified at the same threshold in prior studies of ASD23,66 (FIG. 1a,b). Moreover, an examination of a broader set of genes from this recent study that fell just below a threshold of FDR ≤ 0.1 for schizophrenia risk demonstrates that only 3 out of these 34 probable schizophrenia risk genes overlap with the 102 high-confidence ASD risk genes identified based on the same statistical cut-off23,66. In a similar vein, recent detailed investigations of diverse mutations in a high-confidence ASD risk gene have shown a stronger relationship between individual alleles and specific phenotypic outcomes than would be anticipated based on the findings from genotype–phenotype studies of high-confidence CNVs120. Moreover, given strong evidence that rare and common alleles combine to contribute to both disorders93,100 and the current state of locus discovery overall — with a paucity of common alleles identified in ASD and rare alleles in schizophrenia — definitive conclusions about the presence or absence of specificity between ASD and schizophrenia genetic risks are likely premature.
There has also been long-standing interest in the overlap, both clinically and aetiologically, of ASD and intellectual disability. As noted, early diagnostic criteria for autism were biased towards the ascertainment of individuals with concomitant intellectual impairment. However, as the wider spectrum of ASD clinical manifestations has been increasingly appreciated, the percentage of individuals with ASD and without intellectual disability has increased markedly and now reflects the majority of those who carry an ASD diagnosis. Nonetheless, the frequent co-occurrence clinically of intellectual disability and ASD has prompted an ongoing debate over the separability of social versus intellectual functioning and has raised key questions as to whether recent gene discovery efforts in ASD cohorts have the potential to offer insight into the core biology of social disability110–112. There has been a long-standing hypothesis that social ability is continuously distributed in the population and that higher IQs buffer behaviourally against autism symptoms121,122. One contemporary interpretation of this idea is that the recent spate of large-effect mutations identified in WES studies primarily lead to lower IQ, which, in turn, limits an individual’s ability to compensate for social impairment mediated by common alleles. Along these lines, multiple analyses have noted that the rate of rare de novo mutations in ASD cohorts increases as IQ declines33,44,64,88 and some have concluded that rare de novo sequence mutations are over-represented only in individuals with IQ below the mean64.
By contrast, multiple studies have also shown that rare sequence variants and CNVs contributing large amounts of risk are found across the full range of IQ23,24,33. Moreover, one of these studies evaluated ASD genes with regard to whether they showed evidence for being either ‘ASD-predominant’ or ‘ASD/NDD-predominant’ and found evidence for differing distributions of phenotypes corresponding to specific genes23. Although the authors of the study did not conclude that any genes exclusively carried risk for ASD alone, their analysis was consistent with numerous observations suggesting that the rare variant genetics of human social and intellectual functioning are separable to some degree. For example, one study showed that duplications versus deletions of the identical Williams syndrome region on chromosome 7q11.23 leads, on average, to similar levels of intellectual impairment but also, typically, to strikingly divergent social phenotypes33.
As progress in both common and rare variant studies paints a more complete picture of the allelic architecture of ASD, intellectual disability, schizophrenia and other psychiatric disorders, multiple related questions will become increasingly tractable. Studies examining the correlation of specific genes with differing distributions of diagnoses will have increased power to draw definitive conclusions. Moreover, the relationships between diagnosis, variant class (for example, protein-truncating variants versus missense variants123), direction of effect (for example, duplication versus deletion and loss of function versus gain of function120) and patient sex are likely to provide key insights into the mediators and moderators of whatever specificity is found to be present. Finally, although there is understandable consternation regarding the possibility that genetic risks for ASD, intellectual disability, epilepsy and other NDDs are inseparable, the high degree of comorbidity that is also observed, including for individuals with large-effect rare mutations in the highest-confidence ASD genes, may turn out to facilitate early therapeutic trials. For example, phenotypes apart from social functioning, such as seizures, motor development and overall intellectual functioning, are presently more amenable to quantification and to reliable longitudinal assessment, particularly early in development.
Future work in ASD genetics.
For those who recall the early days of gene discovery efforts in psychiatric disorders writ large, and in ASD in particular, the past 15 years of progress has been nothing short of spectacular. However, considerable opportunities remain: the methods that have been most successful in elaborating the growing list of risk genes introduce some biases owing, in part, to differential detection of types of variants. For example, the sex chromosomes have been largely unexplored to date, owing to complexities in accurate variant calling at scale. In addition, their distinctive inheritance patterns and selection pressures preclude the use of current metrics (for example, the probability of being loss-of-function intolerant) that have accelerated gene discovery on the autosomes. It is also likely that the relative sparsity of female samples in existing research cohorts, especially from simplex families, has limited the ability to detect sex-specific or sex-biased risk associated with specific large-effect variants. Similarly, missense mutations, gain-of-function mutations, non-canonical splice site mutations, somatic mutations and the universe of non-coding variants remain far more difficult to interpret at scale than canonical loss-of-function mutations. Recessive variants, inherited variants and variants with low probability of being loss-of-function intolerant also remain difficult to identify, although network-based approaches have begun to fill in these gaps, by identifying putative novel risk genes based on proximity to strongly associated risk genes54,124,125.
Similarly, efforts to identify genotype–phenotype relationships have so far been met with limited success24,126 but this is a highly promising area for future investigation. Hypotheses regarding the utility of endophenotypes to increase the power of gene discovery efforts have not panned out127. Thankfully, however, the well-known limitations of categorical psychiatric diagnoses combined with the extreme clinical heterogeneity of ASD have not thwarted the identification of dozens of large-effect genes and the first set of common risk alleles. The combination of increasingly large cohorts combined with the ability to work from a well-established list of definitive genes and mutations promises to provide insight into questions, such as the relative specificity of genetic risks, that have long been vexing to investigators. As foundational network data characterizing the relationship between diverse risk genes and the proteins they encode are generated, so-called ‘network–phenotype’ analyses may increase power to extract meaningful relationships between risk variants co-localizing in network space and patient phenotypes37,124,128. That being said, despite the abundance of genes identified based on rare and de novo mutations, there is a strong rationale to continue to pursue common variant analyses. Although studying individual small-effect non-coding alleles may not be as direct a path to exploring core biology, they account for a substantial proportion of population risk and the interplay of common and rare variant risk is now a tractable area of inquiry that could have profound importance124,129,130. The ability to evaluate the combined contributions of variation across the frequency and effect spectrum promises to yield critical insights into natural history and treatment response, and potentially to contribute to diagnostic strategies especially aimed at very early detection.
Progress in convergent neuroscience
The progress made to date in gene and locus discovery in ASD has set the stage for understanding the role of these genes and loci in human brain development and function, and for clarifying how disruptions can lead to the emergence of social impairments. However, the extensive biological and locus heterogeneity revealed by contemporary genomic analyses has, simultaneously, led to concern that any individual gene or mutation, and consequently any related therapy, might be relevant only for a fleetingly small number of individuals with autism. At the same time, from the earliest successful studies, there has also been evidence that a larger number of apparently functionally diverse ASD risk genes tend to converge on a smaller number of biological pathways, developmental stages, brain regions and cell types. Indeed, steady progress has been made in specifying and defining these points of biological overlap through reliance on relatively hypothesis-free approaches and on the expanding armamentarium of developmentally contextualized neurobiological data resources, spanning levels of analysis, construct-valid models and human brain tissue.
The problem of pleiotropy.
A major challenge in translating successful gene discovery in ASD into a reliable understanding of pathobiological mechanisms — and the identification of related treatment targets — has been the pleiotropy of genes, that is, their tendency to be involved in different roles throughout brain development and function, potentially dependent on, for example, cell type, brain region and/or developmental stage35,37,38. Consequently, although an identified risk gene may have a well-studied and well-characterized role in a biologically plausible process, it does not necessarily follow that either this is the only function that the gene plays or it is necessarily the role that contributes to the emergence of ASD.
The challenge of pleiotropy is, of course, also highly relevant in the study of animal models. Recapitulating a mutation in a human ASD risk gene in, for example, rodents, has become routine, as has the ability to characterize a very wide range of neurobiological sequelae, including in awake behaving animals. However, the observation of even the most plausibly ASD-relevant phenotype in a construct-valid mutant does not, on its own, provide reassurance that the observation is pointing to causal mechanisms in the human syndrome — a problem that is compounded by the well-established challenges of relying on face-valid behavioural phenotypes in evolutionarily distant species to illuminate psychiatric syndromes35. Similarly, even for a single gene, limitations in the ability to assay molecular and cellular phenotypes in a hypothesis-free manner across the entire brain at high resolution in more complex models introduces inherent analytical biases. In this regard, having strong evidence regarding when and where to look for ASD-related pathobiology is potentially invaluable in sorting through an immensely complex and dynamic biology. These types of considerations have motivated convergent neuroscience approaches over the past several years. These studies assess risk genes in parallel, in a hypothesis-free manner, to identify and confirm shared developmental epochs, brain regions, specific cell types and biological pathways associated with human vulnerability to social impairment as a prelude to resolving the pleiotropy of ASD risk genes37.
Incomplete gene ontologies.
Convergent neuroscience approaches are based on the hypothesis that subsets of risk genes for a given disorder, such as ASD, will point to shared vulnerability somewhere along the path from gene to complex behaviour. This logic is reflected in the earliest sequencing studies in ASD as successful gene discovery was immediately followed by a search for functional overlap. Many of the first genes associated with ASD based on targeted sequencing studies were known to encode proteins localized to the synapse (for example, NRXN1, SHANK3, NLGN3 and NLGN4)46,131,132. Not surprisingly, as a result, the field focused on the structure and function of the synapse as a convergent point of ASD-relevant biology41,133,134. As systematic gene and locus discovery proceeded, prima facie evidence for convergence was also found for genes involved in chromatin modification and transcriptional regulation27,28,30,33,43. As risk gene lists have grown and the vast degree of allelic and locus heterogeneity has been revealed, enrichment analyses (for example, Gene Ontology enrichment analysis and gene set enrichment analysis) have been invoked to identify statistical over-representation of molecular functions, cellular components, biological processes and other gene lists from various pathway databases43,135–140. These analyses have repeatedly highlighted gene targets of FMRP and the β-catenin pathways28,34,42 and confirmed significant enrichment of Gene Ontology terms associated with chromatin modification and synaptic function23,24,29,41,43,133,134. However, it is important to note that the underlying data that support Gene Ontology terms and other annotations may offer an incomplete and biased view of gene function and that these approaches are generally unable to account for pleiotropy and/or missing data37,141. The annotations underlying these terms are often created by aggregating existing data, a process that can skew towards categories of function and, potentially, cell types and developmental time points that are more commonly or readily studied, while minimizing or omitting others142,143. For example, given several decades of effort aimed at characterizing neurons and a particular emphasis on illuminating the structure and function of the synapse, it is not surprising that ontologies tend to be relatively enriched for related annotation terms. These types of inherent, and often cryptic, ascertainment biases can cascade into self-reinforcing, but potentially incomplete, biological interpretations.
Indeed, recent analyses of some of these same genes annotated at the synapse have shown experimental evidence of functional convergence that precedes synapse formation — for example, in neurogenesis40,144. Of course, these genes probably have multiple roles during brain development with their observed function dependent, in part, on spatiotemporal parameters. In addition, it is possible that both processes are relevant to ASD and interrelated, with synapse dysfunction following from earlier developmental derangements. To address the inherent limitations of Gene Ontology analyses, recent work has increasingly focused on the development of highly parallelized, systematic, experimental characterizations of relevant genes and/or proteins across developmental stages, tissues and cell types. Similarly, data sets developed through the manipulation of known ASD genes within intact biological systems also offer a basis for convergence analysis that mitigates some of the limitations of ontology-based approaches. These efforts are described in more detail below.
Examining neurotypical brains.
Several groups have taken advantage of the growing list of reproducibly associated risk genes to identify anatomical regions, cell types and/or developmental stages of the typically developing brain of particular relevance to ASD. Conceptually, these studies are based on two underlying hypotheses: first, genes expressed in a highly coordinated spatiotemporal fashion probably share functions; and, second, identifying points of significant overlap among a group of disparate ASD risk genes in the context of typical development is a means of triangulating key parameters associated with core pathobiology. These hypotheses highlight the importance of the nature of the input data: these analyses are dependent on gene lists derived in a systematic, unbiased fashion. For example, the potential for circular reasoning introduced by investigating convergence among a set of ‘candidate’ genes identified based, in part or in whole, on biological plausibility is obvious. For this reason, the emergence of widely accepted statistical thresholds in genome-wide and exome-wide studies of idiopathic ASD cohorts to identify gene lists has been a key to the development of meaningful convergence analyses.
Some of the earliest efforts in this vein used network-based approaches to analyse data from the BrainSpan Atlas of the Developing Human Brain, which generated transcriptomic data from early gestation to late adult stages across multiple anatomical regions of ‘neurotypical’ human brains145. Several studies leveraged these data to develop spatio-temporally defined co-expression networks and then examined their relationship to a set of ASD genes meeting specified statistical thresholds for association. The observation of a statistically significant over-representation of risk genes mapping with co-expression networks or of greater than expected expression correlation between ASD genes within networks was considered evidence for convergent vulnerability. Three groups published the first examples of these approaches leveraging somewhat different gene lists and approaches to network generation. Strikingly, despite these differences and the sparse lists of associated genes at the time, all three groups identified evidence for convergence in prenatal development25,146,147, with two resolving this further to mid-gestational prefrontal cortex (PFC) and developing excitatory neurons25,146. More recent studies — using similar or divergent methods, rapidly expanding gene lists and more comprehensive foundational transcriptomic resources — have consistently replicated these findings and, additionally, provided evidence for involvement of the cerebellum and striatum126,148–153 (FIG. 2a).
Fig. 2 |. Emerging patterns of ASD convergence identified from gene expression data.
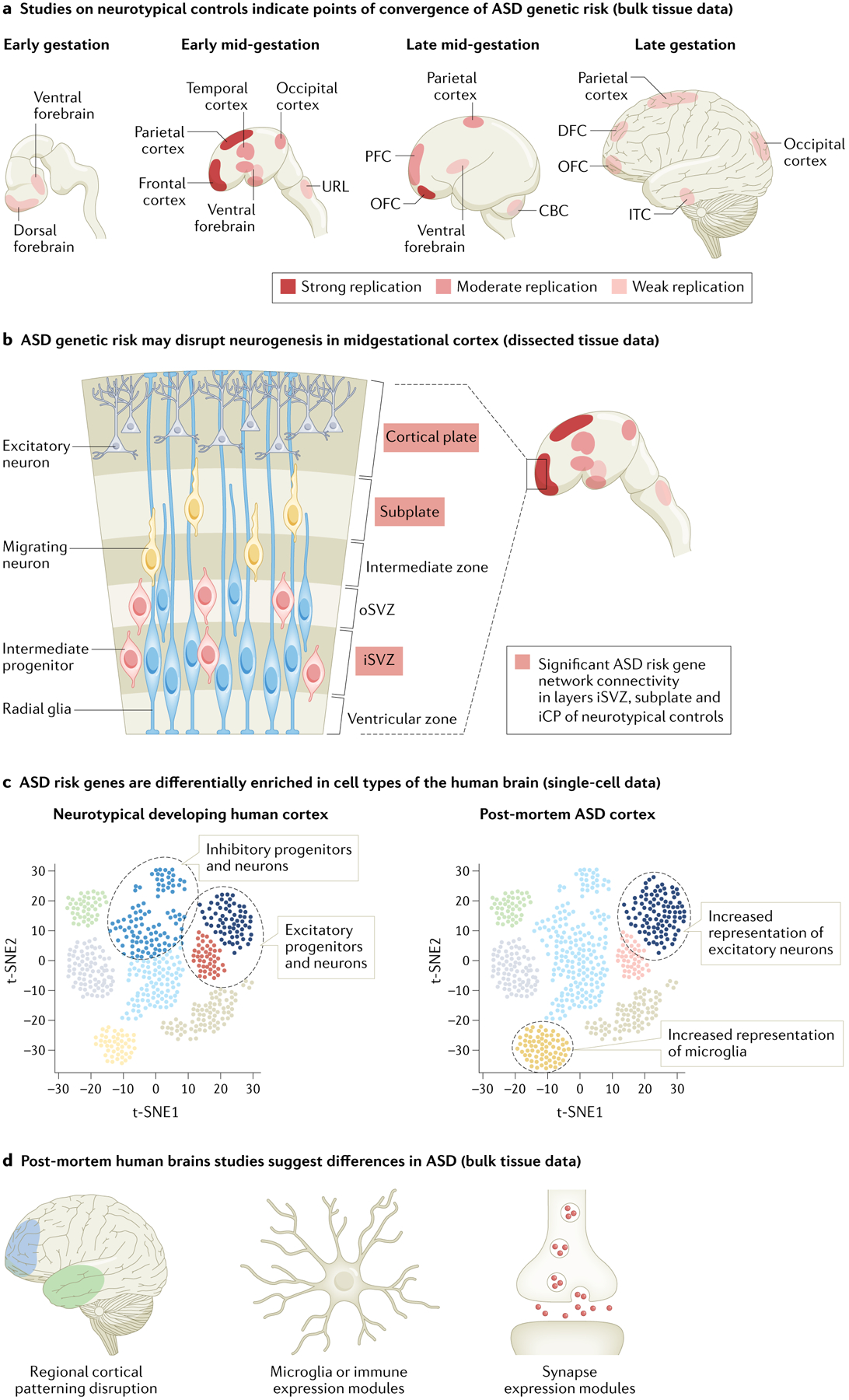
Numerous studies investigating human gene expression data sets for convergence related to autism spectrum disorder (ASD) risk have been conducted, spanning so-called ‘neurotypical’ (control) brain samples (parts a–c) as well as post-mortem brain samples from individuals with ASD (parts c,d). These data sets encompass bulk tissue from major regions of the brain (parts a,d), bulk tissue from more finely dissected regions of the developing cortex (part b) and single cells from the cortex (part c). Neurotypical data tend to include prenatal and postnatal samples across the entire spectrum of human brain development whereas patient-derived data include postnatal samples only (ASD diagnosis requires behavioural assessment). Generally, approaches utilizing neurotypical expression data seek to identify convergence by searching for ‘enrichment’ of ASD risk genes in particular developmental epochs, brain regions and/or cell types. In this case, enrichment is indicated by the ‘strength’ of co-expression of ASD risk genes or by ‘high’ or ‘specific’ expression of ASD risk genes. By contrast, approaches utilizing patient expression data generally aim to identify convergence by characterizing systematic differences between cases and matched controls (that is, genes consistently differentially expressed in brain tissue from individuals with ASD). Although data and approaches differ substantially across studies, the developing frontal cortex and excitatory neurons are recurring points of enrichment. a | Studies investigating spatiotemporal convergence of ASD genetic risk in gene expression data from bulk samples of neurotypical brains have consistently highlighted mid-gestational frontal and parietal cortices23,25,126,129,146,148,149,153. We summarize the extent of replication by color intensity, with the strongest replication indicated by dark red and the weakest by light red. These studies varied widely in granularity of developmental periods and brain regions studied, and therefore we considered studies that examined two or fewer regional (for example, grouping all cortical regions, studying only one brain region) or temporal (for example, prenatal versus postnatal) contexts to provide ‘limited’ evidence. Strong replication denotes a significant finding in at least three ‘non-limited’ studies, moderate replication indicates a significant finding in two non-limited studies and weak replication corresponds to a significant finding in one non-limited study and at least one limited study. We define early gestation as 8–10 post-conception weeks (PCW), early mid-gestation as 10–16 PCW, late mid-gestation as 16–24 PCW and late gestation as 24–38 PCW. b | Based on findings summarized in part a, several studies have examined laminar convergence of ASD genetic risk in neurotypical early mid-gestational frontal cortex. An initial study assessing preservation of spatiotemporal co-expression networks by layer implicated the inner cortical plate25 whereas a contemporaneous study assessing specificity of expression of ASD gene lists by layer indicated a relative lack of layer-specific enrichment146. A later study incorporating orthogonal gene interaction data and assessing ASD gene network connectivity by layer replicated the inner cortical plate finding and newly implicated the inner subventricular zone and subplate40. c | Analyses of single-cell gene expression data from neurotypical controls and individuals with ASD have consistently highlighted excitatory neurons. In neurotypical mid-gestational cortex, ASD risk genes tend to be highly and/or specifically expressed in developing excitatory and inhibitory neurons (progenitors and maturing neurons)23,54,151,157,159. Analyses of single-cell gene expression data from (postnatal) cortex of individuals with ASD have similarly identified consistent differences between cases and controls in excitatory neurons, as well as potential differences in microglia and oligodendrocytes166,167. d | Analyses of bulk samples of brain tissue from individuals with ASD compared with those from matched neurotypical controls support the major findings from parts a–c. These studies implicate frontal cortex, excitatory and inhibitory neurons, and microglia, and additionally highlight differences in genes associated with astrocytes and in expression modules related to synaptic function and inflammation162–165,226. CBC, cerebellar cortex; DFC, dorsal frontal cortex; iCP, inner cortical plate; iSVZ, inner subventricular zone; ITC, inferior temporal cortex; OFC, orbital frontal cortex; oSVZ, outer subventricular zone; PFC, prefrontal cortex; URL, upper rhombic lip. Some of the brain images in parts a,b and d are adapted with permission from REF.199
Based on the aforementioned reproducible association of the developing mid-gestational frontal cortex, analyses of the more fine-grained BrainSpan prenatal laser micro-dissected data set154 have similarly been used to identify points of laminar convergence for ASD risk genes in the developing cortex, through the analysis of the preservation of network connections by layer25, the over-representation of associated gene lists146 or the degree of connectivity of ASD-associated interaction networks40 (FIG. 2b). Early analyses implicated the inner cortical plate (iCP)25,146, and a recent analysis using more than 100 ASD genes and a comprehensive database of molecular interactions confirmed this finding and further identified the inner subventricular zone (iSVZ) and subplate40. Developmentally, a key process occurring in mid-gestational cortex is neurogenesis, with neural progenitors actively undergoing cell division and differentiation in the subventricular zone, leading to the generation of the postmitotic excitatory neurons of the cortex (migrating through the subplate and into the cortical plate) (FIG. 2b). The findings from the biological analyses of these systems are also consistent with findings from orthogonal enrichment analyses23,25,29,146,148, as well as emerging evidence from model system studies, as noted in later sections.
Recent advances in single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA sequencing technology have enabled the generation of high-resolution expression data at the level of single cells or nuclei from the typically developing prenatal cortex151,155–158. Analyses of scRNA-seq data tend to first identify the cell types present based on clustering patterns of individual cells and expression of cell type-specific marker genes, and then assess each cell type for concurrent (that is, co-expression), high or specific expression of ASD risk genes. These studies have again implicated excitatory progenitor cells and maturing excitatory cortical neurons, and have additionally implicated inhibitory progenitor cells and maturing inhibitory neurons of the cortex23,54,151,157,159 (FIG. 2c, left).
Finally, these observations of convergent expression of ASD risk genes in excitatory and inhibitory neurons and the developing cortex have also been reproduced in analyses of expression data from wild type model organisms. In one of the earliest studies, analysis of layer-specific expression data from developing mouse brains supported the idea that ASD-associated genetic risk converges in developing deep layer cortical excitatory neurons25. A contemporaneous study leveraging gene expression data from adult primate cortex160 observed similar enrichment in excitatory neurons, although the signal was stronger for more superficial layers146. Two additional studies in mice generated and analysed a data set with gene expression profiles for 25 distinct CNS cell types and observed enrichment of ASD-associated genes in cortical excitatory neurons, as well as inhibitory neurons of the cortex and striatum126,148,161. A recent study using Xenopus tropicalis observed that ten ASD risk genes chosen solely based on the strength of statistical evidence for association are co-expressed in the developing telencephalon at time points that correspond to human mid-gestational periods in the frontal cortex40. These non-human studies suggest that expression convergence among ASD genes is at least partially conserved through evolution, although this needs to be examined in more detail and across additional models.
Comparative post-mortem studies.
An alternative approach to identifying convergence of ASD risk involves comparing the molecular profiles of post-mortem brain tissue from individuals with ASD and from neurotypical controls. One of the first published studies of this type profiled the transcriptomes of ASD and typically developing brains across the PFC, temporal cortex and cerebellum, and observed that regional expression differences identified in control brains were attenuated in the frontal and temporal lobes of individuals with an ASD diagnosis162 (FIG. 2d). This observation was replicated in a subsequent PsychENCODE study with larger cohort sizes163. These studies and others have also identified downregulation of neuron and synapse-related genes and upregulation of microglia and immune-related genes in the brains of individuals with ASD compared with controls162–166 (FIG. 2d).
scRNA-seq has provided some additional granularity to the observed differences between brains of individuals with ASD and neurotypical brains (FIG. 2c, right). Experiments leveraging data from scRNA-seq to deconvolve bulk RNA-sequencing (RNA-seq) expression data have shown that the majority of expression variation across brain tissue samples is attributable to varying proportions of basic cell types, and that that the cortex of individuals with ASD may have higher fractions of excitatory neurons, microglia and astrocytes, and lower fractions of oligodendrocytes167. This suggests that prior observations, from comparative bulk RNA-seq studies, of alterations in synapse-related and immune-related genes may derive from changes in cell representation rather than from large alterations in gene expression within given cell types162–164.
Overall, studies that compare the transcriptome of ASD and neurotypical brains have repeatedly noted that there may be altered cortical patterning and changes in relative abundance and/or function of excitatory neurons and microglia (FIG. 2d). As noted above, these findings are largely congruent with data from studies searching for convergence of ASD risk gene expression in neurotypical brain tissue, in that both implicate the frontal cortex and excitatory neurons in ASD risk. However, they differ in that neurotypical data implicate inhibitory neurons but not microglia23,25,54,126,129,146,148,151,153,157,159 (FIG. 2c). There are multiple potential explanations for these divergent findings, including that they may reflect some of the inherent practical limitations of studies involving comparisons of affected versus unaffected post-mortem tissue. For example, brain bank resources from individuals with ASD remain quite limited and, therefore, most studies involve very small sample sizes and have attendant challenges with statistical power. Additionally, those samples that are available are often highly heterogeneous in characteristics such as age and sex, as well as with regard to important covariates such as medication history, post-mortem interval, agonal events and/or time of collection165,168–170. Moreover, as the diagnosis of ASD is based on behaviour, these studies involve postnatal samples only, generally from patients 2 years of age or older, raising the critical issue of how to relate postnatal changes in cases versus controls to pathobiology of a developmental disorder with strong evidence for a nexus of prenatal risk, such as ASD. Given the natural history, with symptoms emerging typically in the first 1–2 years of life, as well as the evidence for the relevance of prenatal events in syndrome aetiology, it is plausible and, indeed, likely that observations in postnatal tissue may capture events downstream of primary pathobiology and may be offering insights into ‘effect’ rather than ‘cause’. Although it would be ideal to perform differential expression analyses in prenatal affected versus neurotypical tissue, these types of experiments pose formidable logistical challenges, including as a result of ASD being a clinical and behavioural diagnosis that can only formally be made postnatally. Although one might consider experiments leveraging prenatal post-mortem tissue carrying large-effect ASD-associated mutations without relying on clinical diagnosis, the observation of the wide range of potential diagnostic and phenotypic outcomes from even the largest-effect risk genes would place limits on the ability to interpret findings with respect to ASD pathology specifically.
Patient-derived model systems.
Functional and transcriptional profiling has also been conducted on human induced pluripotent stem cell (iPSC)-derived monolayer models of cell types from the developing brain as well as in 3D models of human brain development (also known as organoids and spheroids)171. Over the past few years, there have been several studies comparing neurons generated from iPSCs derived from donors with ASD versus those from neurotypical individuals. Some recurrent findings in iPSC-derived neurons from patients with ASD and macrocephaly, compared with controls, include increased cellular proliferation, accelerated neuronal differentiation, impaired synapse development and decreased spontaneous and synchronous neuronal activity172–174. Interestingly, a recent study showed that iPSCs derived from individuals with ASD but without macrocephaly exhibited impairments in neurogenesis compared with those from neurotypical individuals175. Thus far, systematic profiling of iPSCs derived from individuals with idiopathic autism have been limited by small sample sizes (on the order of ten individuals carrying an ASD diagnosis) and varied timing and methods of profiling cellular phenotypes172–178. Given the aetiological heterogeneity of the syndrome, the presence of comorbid phenotypes (such as macrocephaly), the diverse genetic backgrounds of probands and controls, and the variability in derivation of iPSC lines and downstream cell types, it is not yet clear whether these findings will replicate in larger, better-powered analyses.
Transcriptomic and epigenetic profiling of iPSC-derived telencephalic organoids has identified ASD-associated co-regulated gene modules related to synaptic function that overlap with those previously identified in studies using post-mortem brain tissue146,163,178. Recently, organoid models of developing forebrain generated from individuals with ASD and from neurotypical controls were profiled for chromatin accessibility and gene expression over time179. Overall, ASD genetic risk mapped to glial progenitor cells, mid-stage excitatory neurons and late-stage excitatory and inhibitory neurons. These results are therefore largely consistent with the aforementioned findings from human brain. It is important to note, however, that numerous distinctions exist between organoids and primary tissue, including limited spatial organization, impaired cell-type maturation, the absence of cell types found in primary tissue and the fact that organoids may be significantly impacted by factors related to oxygen diffusion and stress180–182. As organoid technology continues to develop (for example, see REFS183–185), it will be an increasingly promising avenue for a more granular understanding of the molecular-level and cell type-level changes taking place during early neurodevelopment, including with respect to ASD.
Parallelized modelling of genetic risk.
The efforts to identify where and when ASD pathology arises have already provided important insights into underlying biology and now offer opportunities for parallel investigations of ASD risk genes that constrain or define key experimental parameters including cell types and developmental stages of interest. In addition, in recent years, advances in CRISPR-based technology have facilitated the parallelized study of multiple ASD-associated genes in vitro and in vivo to identify points of functional convergence. Initial in vitro studies have characterized the effect of perturbing roughly a dozen ASD-associated risk genes in neurons derived from human iPSCs or embryonic neuronal precursor cells186,187. These initial studies suggest that perturbation of ASD genes results in impaired neuronal differentiation and reduced spontaneous synaptic activity, and highlight the feasibility of assessing the functional consequence of multiple ASD genes in parallel. Going forward, advances in pooled CRISPR screening will enable hypothesis-naive functional screening of larger numbers of genes with a wide range of read-outs, including proliferation, differentiation and cellular function as well as gene expression and chromatin accessibility at single-cell resolution188–195. Patient-derived iPSC model systems still tend to suffer from small sample sizes in addition to variability among iPSC lines and heterogeneity in the derived cell populations196. In this regard, systems with isogenic or comparatively homogeneous genetic backgrounds are an important addition to the experimental armamentarium, as they moderate some of the challenges of interpreting subtle biological effects across varied genetic backgrounds39,40,197 — although the generalizability of findings in isogenic backgrounds needs further investigation, as does the effect of sex (see below).
Studying ASD risk genes in vivo in intact biological systems is critical to model dynamic neurodevelopmental contexts, circuit-level function and higher-order phenotypes. However, generating animal models of individual ASD genes is labour and time-intensive. Consequently, historically, studying multiple ASD genes in parallel to identify points of functional convergence has been challenging. Increasingly, these challenges can be mitigated through the use of pooled screening approaches or using model organisms that allow for scalable arrayed screening. In a recent study, an in vivo pooled CRISPR–scRNA-seq system (in vivo Perturb-seq) that combines genetic editing in the developing mouse brain with postnatal scRNA-seq was used to identify the cell type-specific functional consequences of perturbing 35 ASD risk genes during brain development39. The authors observed overlapping gene expression changes linked to perturbation of small subsets of ASD genes (up to four) in different cell types, hinting at cell type-specific functional convergence. Importantly, this study highlights the feasibility of pooled in vivo CRISPR screens with single-cell profiling to interrogate many risk genes for functional convergence across multiple cell types. However, there were no clear convergent findings involving large numbers of genes. Larger-scale experiments with greater numbers of cells and multiple time points across brain development may be better powered to identify convergent gene modules within specific cell types.
Our group recently leveraged CRISPR alongside the X. tropicalis model system to study the effects of loss-of-function mutations in ten ASD risk genes in parallel in an arrayed format40. Strikingly, perturbation of all ten ASD risk genes altered telencephalic size and impaired neurogenesis. This convergent neurogenesis phenotype, spanning ASD genes with apparently disparate cellular functions, was conserved in human 2D and 3D in vitro models of brain development. Overall, these observations are in line with prior findings that ASD risk converges in mid-gestational telencephalic excitatory neurons, and provide strong evidence for impaired neurogenesis as a point of vulnerability, as has been previously hypothesized198. These findings underscore the power of Xenopus for productive functional genomics studies, in particular highlighting the ability of medium-throughput parallelized in vivo studies to identify convergent mechanisms that can then be prioritized for validation and extension in human systems199.
Sex differences and genetic risk
Although, in general, the field is enjoying success in elaborating points of functional and spatiotemporal convergence among a rapidly expanding list of autosomal ASD genetic risks, far less is understood about the nature of the contribution of sex to ASD aetiology. A strong male sex bias has consistently been observed for ASD, but this remains largely unexplained200. The male to female ratio has been consistently estimated to be around 4:1 (REF.201), although estimates of male predominance for individuals with autism with normal to high IQ tend to be even higher. Similarly, although the findings have been roughly consistent across studies, it is also clear that these estimates are vulnerable to several potential confounds beyond IQ, including ascertainment methods and potential diagnostic bias200–202. Still, studies that have endeavoured to account for these issues have, nonetheless, found consistent evidence for male bias in ASD risk95,202,203. One putative avenue of explanation for these findings is the FPE, evidence for which derives mostly from genetic studies that show enrichment of risk in females versus males — that is, female probands tend to carry a relatively increased burden of rare damaging de novo and transmitted mutations, including CNVs and sequence variants23,24,29,31,33,44, as well as rare complete knockouts76. Additionally, females — including unaffected mothers of children with autism — also appear to be enriched for common variant risk93,94. Similarly, unaffected female siblings of children diagnosed with ASD appear to carry an increased rate of rare variants22–24,27,28,30,32,33,95 and appear to inherit more polygenic risk93,94, as compared with male siblings. These observations support the hypothesis that females are more resilient to genetic risks, resulting in the observation of higher thresholds for females to become symptomatic and meet diagnostic criteria. Interestingly, to date, there has been no evidence of sex-biased risk for individual autosomal genes24 and rare genetic risks mapping to the sex chromosomes have been relatively unexplored owing to technical and methodological barriers.
In contrast to the types of data noted above, family-based and twin-based studies of recurrence rates have provided less clear evidence for the FPE. Younger siblings of females diagnosed with ASD may be more likely to receive an ASD diagnosis than younger siblings of males diagnosed with ASD94,204; however, there are conflicting data in this regard and these analyses are constrained by limited statistical power owing to the relatively small female cohort sizes95,153,203.
There is also the possibility of male-specific ‘risk’, which is not mutually exclusive with the FPE model. For example, males with ASD have an increased rate of hemizygous rare loss-of-function variants on chromosome X76. However, the sex chromosomes, in general, appear to be a minor factor in determining risk versus resilience31,205,206, although they also tend to be omitted from large-scale genetic studies owing to technical difficulties and complexities in accurate variant calling due to confounding sequence homology between sex chromosomes and differences in ploidy by sex. Therefore, it is possible that there are as yet undiscovered factors on the sex chromosomes.
These findings suggest that sex differences are a critical aspect of the biology of ASD and a potentially promising avenue for the development of broadly applicable therapeutics, especially in light of the possibility that unravelling underlying mechanisms may illuminate resilience as well as risk factors. The lack of mechanistic understanding of sex bias is probably a consequence of the relative paucity of knowledge regarding differential molecular and cellular mechanisms in male and female brain development, inherent limitations in commonly employed model systems for interrogating these differences and the lack of any immediately obvious explanation for sex bias in otherwise highly successful gene discovery efforts. Identifying robust differences in ‘neurotypical’ male and female brain development will probably facilitate understanding of risk for many psychiatric disorders beyond ASD, as many show some degree of sex bias207. Rich developmental human brain omics data sets from both males and females (see TABLE 1 for a list of currently available gene expression data) will be a critical resource in efforts to identify these differences.
Table 1 |.
Available human developmental brain expression data sets for studying sex differences
| Resource | Brain regions | Developmental age | Sample size (female, male) | Technology | Number of cells | Year |
|---|---|---|---|---|---|---|
| Colantuoni et al.218 | PFC | 12–18 PCW, birth–78 years | 269 (92, 177) | Microarray | NA | 2011 |
| BrainSpan microarray145 | 16 (12 cortical) | 5 PCW–82 years | 57 (26, 31) | Microarray | NA | 2011 |
| BrainSpan LMD154 | ~300 cortical regions (LMD of 6 layers) | 15–21 PCW | 4 (3, 1) | Microarray | NA | 2014 |
| BrainSpan RNA-seqa | 16 (12 cortical) | 8 PCW–40years | 42 (19, 23) | BulkRNA-seq | NA | 2013 |
| O’Brien et al.219 | Entire brain hemisphere | 12–19 PCW | 120 (50, 70) | BulkRNA-seq | NA | 2018 |
| Li et al. (PsychENCODE)151 | 16 regions (11 cortical) | 8 PCW–40years | 41 (18, 23) | BulkRNA-seq | NA | 2018 |
| Walker et al.152 | Unspecified cortex | 14–21 PCW | 201 (84, 117) | BulkRNA-seq | NA | 2019 |
| Werling et al. (BrainVar)153 | 1 region (DPFC) | 6 PCW–20 years | 176 (72, 104) | BulkRNA-seq | NA | 2020 |
| Pollen et al.155 | Unspecified cortex (MD of 2 layers) | 14–16 PCW | 3b(3, 0) | scRNA-seq | 393 | 2015 |
| Nowakowski et al.156 | 2 cortical regions (MD of 2 layers), MGE | 5–37 PCW | 48b (21, 27) | scRNA-seq | 4,261 | 2017 |
| Zhong et al.220 | PFC | 6–24 PCW | 12b (9, 3) | scRNA-seq | 2,309 | 2018 |
| Fan et al.159 | 22 regions (20 cortical) | 22–23 PCW | 3 (2, 1) | scRNA-seq | 4,213 | 2018 |
| Li et al. (PsychENCODE)151 | Cortex (fronto-parietal) | 5–20 PCW | 9 (3, 6) | scRNA-seq | 1,195 | 2018 |
| Polioudakis et al.157 | Unspecified cortex (MD of 2 groups of cortical layers) | 15–16 PCW | 4 (3, 1) | scRNA-seq | 33,976 | 2019 |
| Bhaduri et al.182 | 7 regions (all cortical) | 4–20 PCW | 5b(4,1) | scRNA-seq | 189,409 | 2020 |
| Eze et al.158 | 18 regions (3 cortical) | 4–8 PCW | 10b (7, 3) | scRNA-seq | 289,000 | 2021 |
| Trevino et al.221 | Unspecified cortex | 16–24 PCW | 4b(3, 1) | scRNA-seq | 57,868 | 2021 |
DPFC, dorsolateral prefrontal cortex; LMD, laser micro-dissection; MD, micro-dissection; MGE, medial ganglionic eminence; NA, not applicable; PCW, post-conception weeks; PFC, prefrontal cortex; RNA-seq, RNA sequencing; scRNA-seq, single-cell RNA sequencing.
BrainSpan RNA-seq data can be accessed at http://brainspan.org/static/download.html.
Sex inferred by X-inactive specific transcript expression.
In considering points of intersection between ASD risk factors and biological sex differences, there have been some recent data pointing to the potential contribution of the sex hormones, testosterone and oestrogen, to ASD sex bias95,208–210. Indeed, two independent in vivo resilience screens identified oestrogen as a protective factor in the context of modelled ASD genetic risks40,45, implying that oestrogen could be an endogenous protective factor if it is active at higher levels in females than in males. However, testosterone excess has also been hypothesized to be a risk factor for ASD209,211,212. Further research into the precise manner by which sex hormones intersect with ASD risk is needed, especially characterizing developmental differences in sex hormone levels in the brain and whether they result in sex differences in brain development and function. With the recent discovery that oestrogen inhibits sonic hedgehog (SHH) signalling endogenously in the developing brain40, critical future areas of research will include determining whether SHH signalling is sexually dimorphic during prenatal development and whether it is a convergent pathway in ASD risk. SHH has key roles throughout brain development, spanning patterning, proliferation, arealization and differentiation213–215, suggesting that there may be many points of potential contribution to sexually dimorphic outcomes just within this single pathway.
Perspective
We are at a highly opportune moment in the effort to identify core pathobiological processes underlying ASD. There is a long and growing list of reliably associated large-effect genes accompanied by an expanding armamentarium of multidimensional omics data sets, higher-throughput model systems and CRISPR-based genetic tools that are empowering studies focused on convergence. Although Gene Ontology enrichment analyses highlighting chromatin modification and synapse function have been a readily accessible resource for developing evidence of shared biology, newer hypothesis-free system biological approaches are important complements to these types of studies and have already provided strong evidence for a nexus of ASD risk, particularly in developing excitatory neurons in the mid-gestational frontal cortex.
Of course, critical challenges remain. First, it is not clear how to span the gap from molecular and cellular mechanisms of convergence to differences in the clinical presentation of ASD. Animal models provide an opportunity to observe and manipulate behaviours, as well as the ability to identify underlying circuit-level changes. However, the limitations of anthropomorphizing and relying on face-valid behaviours for complex neuropsychiatric phenotypes are well known35. Second, despite the long-observed male predominance in ASD, the field still lacks a detailed understanding of how this phenomenon is generated and therefore, going forward, it will be critical to investigate sexual dimorphism in brain development and to intersect this with genetic risk.
Despite these challenges, there are already ongoing efforts to exploit successful gene discovery directly to develop novel therapies35. These include gene replacement, gene editing and antisense oligonucleotide strategies. Particularly with regard to the most common forms of syndromic ASD, these initiatives are gaining momentum and clinical trials are on the horizon. Similar efforts for the most highly penetrant high-confidence ASD genes are unlikely to be far behind. The pursuit of convergence neuroscience approaches can contribute to these and related efforts, in part, by clarifying when and where vulnerability to ASD risk emerges, and therefore can help answer key remaining questions regarding when interventions addressing ASD would need to be delivered and where in the brain37,38. At the same time, points of convergence, with regard to risk and resilience, have the potential to support more traditional therapeutics development by identifying biological mechanisms and targets that are ‘downstream’ of individual genes and applicable to more than one gene at a time. These points of convergence may also have diagnostic utility. Also, there are critical practical and ethical issues that still confront the design and execution of clinical trials in ASD, particularly early in development. Nonetheless, progress in other areas of medicine, including with regard to early-onset neurodegenerative disorders216,217, combined with the progress in multiple areas noted above points to several viable paths towards more effective, personalized and rational treatments for ASD, particularly for individuals with severe autism.
Supplementary Material
Autism spectrum disorder.
(ASD). A group of developmental disorders characterized by deficits in social communication and social interaction, and restrictive and repetitive patterns of behaviour, interests or activities.
Intellectual disability.
A developmental disorder characterized by deficits in intellectual functioning (including reasoning, problem-solving and academic learning) and adaptive functioning (including communication and independent living).
Genetic architecture.
The characteristics of genetic variation that contribute to a specific phenotype, including variant frequency, effect size and their interaction with each other and the environment.
Locus heterogeneity.
Variants at different gene loci result in a similar phenotype, individually (for monogenic diseases) or in combination (for complex traits).
Pleiotropy.
A phenomenon in which a single gene contributes to multiple processes or phenotypic traits.
Convergent neuroscience.
Studies that address the overlap, or intersection, of genetic risk for a psychiatric disorder with respect to molecular-level, cellular-level and circuit-level function as well as across multiple dimensions of analysis, including anatomical localization and developmental timing.
Gene Ontology terms.
A hierarchical set of terms that aim to define the universe of possible descriptors a gene can have, including properties such as molecular function, cellular component and biological process.
Penetrance.
The probability that an individual with a given genotype will exhibit the associated phenotype.
Resilience.
The capacity of an individual to have a ‘better than expected outcome’ (for example, being unaffected despite having multiple autism spectrum disorder-associated genetic variants).
Protein-truncating variants.
(Also referred to as loss-of-function or likely gene disrupting variants). Sequence variants that are predicted to shorten the protein-coding sequence of a gene — usually to the point of resulting in a non-functional protein or no protein at all due to nonsense-mediated decay — including nonsense, frameshift and essential splice site variants.
Somatic mutations.
(Also known as mosaic mutations). Variants that are present in fewer than 100% of the cells of an individual (for example, present in brain cells but not present in germ-line cells), generally because the mutation occurred after fertilization.
Compound heterozygous variants.
Variants similar to recessive variants, in that both alleles of a gene are mutated; however, in this case, the two alleles are different.
Neurodevelopmental disorders.
(NDDs). A group of conditions (including autism spectrum disorder, attention deficit hyperactivity disorder, tic disorders and intellectual disability) characterized by developmental onset, atypical brain development and resultant impairments in cognition, communication, behaviour and/or motor skills.
Polygenic risk scores.
Estimates of an individual’s genetic predisposition for a disorder or disease based on the collective effects of many genetic variants.
SFARI Gene.
An evolving database compiled by the Simons Foundation Autism research initiative (SFARI) research program that includes a list of genes and copy number variants (CNVs) associated with autism spectrum disorder curated from human studies and animal models.
Missense variants.
Single base-pair coding variants that result in altered amino acids, for which several metrics have been developed to predict the functional consequence, including the PolyPhen-2 (Polymorphism Phenotyping v2) and MPC (missense badness, PolyPhen-2 and constraint) metrics, for which a categorization of PolyPhen-2 missense 3 (Mis3) or an MPC score ≥2 reflects a probably damaging variant.
Gene Ontology enrichment analysis.
A statistical approach that assesses whether specific Gene Ontology terms are statistically over-represented in a (generally large) set of genes or proteins (for example, among autism spectrum disorder risk genes).
Gene set enrichment analysis.
Like Gene Ontology enrichment analysis, this statistical approach assesses whether specific Gene Ontology terms are statistically over-represented in a large set of genes or proteins; however, unlike Gene Ontology enrichment analysis, gene set enrichment analysis also incorporates the rank of genes within the set into the statistical test (for example, differentially expressed genes ranked based on the fold-change between cases and controls).
Pathway databases.
Databases that annotate genes with an ontological approach to capture functional relationships, including molecular interactions, regulation and phenotype associations; common pathway databases include Gene Ontologies (GO) and Kyoto encyclopedia of Genes and Genomes (KeGG).
Neurogenesis.
The process by which new neurons are generated, in humans most active during gestational weeks 10–25.
BrainSpan Atlas of the Developing Human Brain.
A foundational resource that includes bulk transcriptome profiling of up to 16 cortical and subcortical structures across human brain development (prenatal to adult).
Neurotypical.
Description of individuals with apparently typical intellectual and cognitive development (for example, not affected by a neurodevelopmental or psychiatric disorder).
PsychENCODE.
A consortium-based project that aims to produce multidimensional genomic data from human post-mortem brain tissue from neurotypical and patient donors to begin to functionally characterize risk variants in model systems, with an initial focus on autism spectrum disorder, bipolar disorder and schizophrenia.
Induced pluripotent stem cell.
(iPSC). A pluripotent cell obtained by reprogramming somatic cells through ectopic expression of defined pluripotency factors and/or treatment with small molecules.
Organoids.
(Also known as spheroids) 3D structures, derived in vitro from primary tissue, embryonic stem cells or induced pluripotent stem cells (iPSCs), that self-organize and recapitulate aspects of organ development, anatomy, cellular composition, physiology and function.
Gene modules.
Typically, a set of genes with similar expression profiles that are inferred to be functionally related and co-regulated, and that can be identified through various module-detection methods (for example, gene co-expression).
Functional convergence.
A situation in which seemingly disparate entities are associated with an overlapping function, which can occur at various levels of investigation, including molecular, cell taxonomic, morphological, neural circuit or phenotype level.
Sex differences.
Differences between individuals of different sex in the same species, often including secondary sex characteristics, size and behavioural or cognitive traits.
Acknowledgements
The authors thank S. Wang for data analysis and graphical representation for Fig. 1 and S. Pyle for graphic design. This work was supported by a gift from the Overlook International Foundation and grant support from the National Institutes of Mental Health (NIMH) (U01MH116487, R01MH115963, 1U01MH115747, R25MH06048) and the Psychiatric Cell Map Initiative (pcmi.ucsf.edu).
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41583-022-00576-7.
RELATED LINKS
Brainspan Atlas of the Developing Human Brain: https://brainspan.org
References
- 1.Lord C et al. Autism spectrum disorder. Nat. Rev. Dis. Prim 6, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Díaz-Caneja CM et al. A white paper on a neurodevelopmental framework for drug discovery in autism and other neurodevelopmental disorders. Eur. Neuropsychopharmacol 48, 49–88 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Folstein S & Rutter M Infantile autism: a genetic study of 21 twin pairs. J. Child. Psychol. Psychiatry 18, 297–321 (1977). [DOI] [PubMed] [Google Scholar]
- 4.Gaugler T et al. Most genetic risk for autism resides with common variation. Nat. Genet 46, 881–885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klei L et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism 3, 9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross-Disorder Group of the Psychiatric Genomics Consortium. et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet 45, 984–994 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey A et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med 25, 63–77 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Devlin B & Scherer SW Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev 22, 229–237 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Hallmayer J et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68, 1095–1102 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steffenburg S et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child. Psychol. Psychiatry 30, 405–416 (1989). [DOI] [PubMed] [Google Scholar]
- 11.Ozonoff S et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics 128, 488–495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Constantino JN, Zhang Y, Frazier T, Abbacchi AM & Law P Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry 167, 1349–1356 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg RE et al. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch. Pediatr. Adolesc. Med 163, 907–914 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Le Couteur A et al. A broader phenotype of autism: the clinical spectrum in twins. J. Child. Psychol. Psychiatry 37, 785–801 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Tick B, Bolton P, Happé F, Rutter M & Rijsdijk F Heritability of autism spectrum disorders: a meta-analysis of twin studies. J. Child. Psychol. Psychiatry 57, 585–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritvo ER, Freeman BJ, Mason-Brothers A, Mo A & Ritvo AM Concordance for the syndrome of autism in 40 pairs of afflicted twins. Am. J. Psychiatry 142, 74–77 (1985). [DOI] [PubMed] [Google Scholar]
- 17.Taniai H, Nishiyama T, Miyachi T, Imaeda M & Sumi S Genetic influences on the broad spectrum of autism: study of proband-ascertained twins. Am. J. Med. Genet. B Neuropsychiatr. Genet 147B, 844–849 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Lichtenstein P, Carlström E, Råstam M, Gillberg C & Anckarsäter H The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry 167, 1357–1363 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Nordenbæk C, Jørgensen M, Kyvik KO & Bilenberg N A Danish population-based twin study on autism spectrum disorders. Eur. Child. Adolesc. Psychiatry 23, 35–43 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Colvert E et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 72, 415–423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebat J et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study conducts comparative genomic hybridization in simplex and multiplex families with ASD and establishes that de novo CNVs significantly contribute to ASD risk.
- 22.Levy D et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Satterstrom FK et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work presents the largest published ASD WES study to date, which identifies 102 high-confidence ASD risk genes.
- 24.Sanders SJ et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This large study combines WES data from De Rubeis et al. (2014) and Iossifov et al. (2014) as well as de novo CNV data to identify 65 ASD risk genes and 6 ASD risk loci.
- 25.Willsey AJ et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first studies to assess spatiotemporal convergence of ASD risk genes, this study performs a co-expression network analysis that integrates BrainSpan developmental expression data with genetic data from simplex families with ASD to identify deep layer cortical projection neurons in prefrontal and primary motor-somatosensory cortical regions during mid-gestational development as a critical nexus of ASD risk.
- 26.O’Roak BJ et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet 43, 585–589 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first WES studies of people with ASD and their families.
- 27.Neale BM et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with refs. 28, 30 and 34, this paper cemented the contribution of de novo sequence variants to ASD risk.
- 28.Iossifov I et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This large ASD WES study, contemporaneous with ref. 44, confirms that de novo loss-of-function mutations contribute significantly to ASD, identifies 33 ASD risk genes and implicates ASD risk genes in chromatin remodelling, transcription and splicing, and synaptic function.
- 30.Sanders SJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; As well as contributing as noted above, this paper outlined a key paradigm for the statistical assessment of a gene’s association to ASD, based on the number and type of recurrent de novo variants.
- 31.Jacquemont S et al. A higher mutational burden in females supports a ‘female protective model’ in neurodevelopmental disorders. Am. J. Hum. Genet 94, 415–425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong S et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 9, 16–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders SJ et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of several early studies associating de novo CNVs with ASD risk, this paper also generated a statistical framework for quantifying the significance of recurrent de novo CNVs. This framework was later adapted to early studies of de novo sequence variants.
- 34.O’Roak BJ et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; As well as contributing as noted above, this paper was one of the first to demonstrate that genes with damaging mutations in individuals with ASD are highly connected in protein–protein interaction modules, suggesting that they may converge on similar biological functions.
- 35.Sestan N & State MW Lost in translation: traversing the complex path from genomics to therapeutics in autism spectrum disorder. Neuron 100, 406–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.State MW & Sestan N The emerging biology of autism spectrum disorders. Science 337, 1301–1303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review describes the rationale for and approaches to leveraging convergence among ASD genes to identify not only relevant molecules and pathways but also when and where they act in the developing human brain.
- 37.Willsey AJ et al. The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell 174, 505–520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review describes convergent system biological approaches to study genes implicated in neuropsychiatric disorders such as ASD, emphasizing the importance of parallel investigations of multiple genes and mutations, in as unbiased a fashion as possible, to identify points of convergence and reveal high-order (molecular, cellular and circuit-level) phenotypes.
- 38.Willsey AJ & State MW Autism spectrum disorders: from genes to neurobiology. Curr. Opin. Neurobiol 30, 92–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin X et al. In vivo Perturb-seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 370, eaaz6063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper conducted in vivo transcriptional screening of a large number of ASD risk genes using single-cell transcriptomics and CRISPR technology.
- 40.Willsey HR et al. Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 109, 1409 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study conducts parallel in vivo analysis of the ten highest-confidence ASD risk genes in Xenopus, finding that the functional consequences of mutations in these genes converge upon impaired neurogenesis in the developing forebrain — a phenotype that could be rescued by exogenous oestrogen, suggesting oestrogen is a resilience factor that may mitigate various ASD genetic risks.
- 41.Zoghbi HY Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302, 826–830 (2003). [DOI] [PubMed] [Google Scholar]; This early commentary proposes that ASD genetic risk may mechanistically converge at the synapse.
- 42.O’Roak BJ et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilman SR et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study develops a framework (NETBAG) that integrates multiple levels of molecular data to demonstrate that genes affected by CNVs in ASD are functionally interconnected.
- 44.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This large ASD WES study, contemporaneous with ref. 29, confirms that de novo missense and likely gene-disrupting mutations contribute significantly to ASD and identifies 27 ASD risk genes. It also carefully quantifies the contributions of different types of rare variants to ASD risk by comparing normalized rates in probands versus unaffected sibling controls.
- 45.Hoffman EJ et al. Estrogens suppress a behavioral phenotype in zebrafish mutants of the autism risk gene, CNTNAP2. Neuron 89, 725–733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first studies to identify a functional interaction between oestrogen and an ASD risk gene.
- 46.Jamain S et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet 34, 27–29 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work uses linkage analysis and targeted DNA sequencing to identify two ASD-associated genes, NLGN3 and NLGN4, which are among the earliest individual genes with robust evidence of association with idiopathic (non-syndromic) ASD.
- 47.Laumonnier F et al. X-Linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet 74, 552–557 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar RA et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet 17, 628–638 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Marshall CR et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet 82, 477–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss LA et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med 358, 667–675 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Pinto D et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Consortium SPARK. SPARK: a US cohort of 50,000 families to accelerate autism research. Neuron 97, 488–493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He X et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 9, e1003671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work develops a Bayesian model (transmission and de novo association (TADA)) that integrates information from multiple types of genetic variation from large-scale human gene sequencing studies to improve the power to detect novel risk genes. A variation of this model is still used to discover ASD risk genes. This model has also been applied to other psychiatric disorders, such as Tourette syndrome.
- 54.Ruzzo EK et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell 178, 850–866.e26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satterstrom FK et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci 22, 1961–1965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodin RE et al. The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat. Neurosci 24, 176–185 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the largest studies to date assessing the frequency of somatic (mosaic) variants in brain tissue from people with ASD and controls.
- 57.Sherman MA et al. Large mosaic copy number variations confer autism risk. Nat. Neurosci 24, 197–203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study aggregates and analyses high-quality WES data for 60,706 individuals of diverse ancestries generated as part of the Exome Aggregation Consortium (ExAC), providing a key foundation for the field (which has since grown to include more than 100,000 individuals). Together with several other studies, this work develops a paradigm to prioritize genes on the basis of ‘tolerance’ to genetic variation. Intolerant genes tend to carry higher risk for ASD and other human disorders, so tolerance is a key metric for prioritizing functional mutations. The associated resource also facilitates the identification of ultra-rare genetic variants — a similarly critical metric for prioritizing mutations.
- 60.Petrovski S, Wang Q, Heinzen EL, Allen AS & Goldstein DB Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9, e1003709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with several other studies, this work develops a paradigm to prioritize genes based on “tolerance” to genetic variation. Intolerant genes tend to carry higher risk for ASD and other human disorders, and is therefore a key metric for prioritizing functional mutations.
- 61.Huang N, Lee I, Marcotte EM & Hurles ME Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 6, e1001154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work develops a paradigm to prioritize genes based on likelihood of haploinsufficiency (i.e. likelihood that a heterozygous loss of function variant results in a clinical phenotype). Haploinsufficient genes tend to carry higher risk for ASD and other human disorders, and is therefore a key metric for prioritizing functional mutations.
- 62.Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; A longstanding and widely used resource for predicting the severity of missense mutations.
- 63.Kosmicki JA et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet 49, 504–510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents a frequentist framework to evaluate excesses of de novo mutations at the level of individual genes, for which simple case–control comparisons cannot achieve meaningful levels of significance due to the rarity of de novo events; when applied to ASD WES data, this method highlights several risk genes at a genome-wide level of statistical significance.
- 65.Samocha KE et al. Regional missense constraint improves variant deleteriousness prediction. Preprint at bioRxiv 10.1101/148353 (2017). [DOI] [Google Scholar]
- 66.Singh T et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 10.1038/s41586-022-04556-w (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; The largest WES study published on schizophrenia to date, this paper identifies 10 exome-wide significant schizophrenia risk genes of large effect size.
- 67.Bilgüvar K et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilmore EC & Walsh CA Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol 2, 461–478 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dias CM et al. Homozygous missense variants in NTNG2, encoding a presynaptic Netrin-G2 adhesion protein, lead to a distinct neurodevelopmental disorder. Am. J. Hum. Genet 105, 1048–1056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strauss KA et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med 354, 1370–1377 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Novarino G et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 338, 394–397 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morrow EM et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manzini MC et al. CC2D1A regulates human intellectual and social function as well as NF-κB signaling homeostasis. Cell Rep. 8, 647–655 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu TW et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron 77, 259–273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doan RN et al. Recessive gene disruptions in autism spectrum disorder. Nat. Genet 51, 1092–1098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lim ET et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 77, 235–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper characterizes the rate of homozygous knockout mutations in ASD and is one of the first studies to characterize the contribution of rare mutations on chromosome X to ASD risk.
- 77.Schmitz-Abe K et al. Homozygous deletions implicate non-coding epigenetic marks in autism spectrum disorder. Sci. Rep 10, 14045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.D’Gama AM et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88, 910–917 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Freed D & Pevsner J The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. 12, e1006245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krupp DR et al. Exonic mosaic mutations contribute risk for autism spectrum disorder. Am. J. Hum. Genet 101, 369–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lim ET et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci 20, 1217–1224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dou Y et al. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum. Mutat 38, 1002–1013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.D’Gama AM & Walsh CA Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci 21, 1504–1514 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Kong A et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leppa VM et al. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am. J. Hum. Genet 99, 540–554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin SC et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Capkova Z et al. Differences in the importance of microcephaly, dysmorphism, and epilepsy in the detection of pathogenic CNVs in ID and ASD patients. PeerJ 7, e7979 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Robinson EB et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl Acad. Sci. USA 111, 15161–15165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo H et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Genet. Med 21, 1611–1620 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Werling DM et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet 50, 727–736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study develops an analytical framework for WGS termed a category-wide association study, which mirrors the statistical rigour of GWAS with annotation categories in place of SNPs.
- 91.Sullivan PF et al. Psychiatric genomics: an update and an agenda. Am. J. Psychiatry 175, 15–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work is the largest ASD GWAS published to date, including ~18,000 cases and ~28,000 controls, and identifies 5 genome-wide significant loci.
- 93.Antaki D et al. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Preprint at medRxiv 10.1101/2021.03.30.21254657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wigdor EM et al. The female protective effect against autism spectrum disorder. Preprint at medRxiv 10.1101/2021.03.29.21253866 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Werling DM The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex. Differ 7, 58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This review explores the magnitude of male bias in ASD prevalence, describes the FPE and highlights sex-differential pathways that may underlie sex bias in ASD.
- 96.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; A critical study that assesses the common variant risk shared across ASD, attention deficit-hyperactivity disorder, bipolar disorder, major depressive disorder and schizophrenia.
- 97.Weiner DJ et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet 49, 978–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iakoucheva LM, Muotri AR & Sebat J Getting to the cores of autism. Cell 178, 1287–1298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.State MW & Levitt P The conundrums of understanding genetic risks for autism spectrum disorders. Nat. Neurosci 14, 1499–1506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Davies RW et al. Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat. Med 26, 1912–1918 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jannot A-S, Ehret G & Perneger T P < 5 × 10−8 has emerged as a standard of statistical significance for genome-wide association studies. J. Clin. Epidemiol 68, 460–465 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Banerjee-Basu S & Packer A SFARI Gene: an evolving database for the autism research community. Dis. Model. Mech 3, 133–135 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Abrahams BS et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bleuler E in Dementia Praecox or the Group of Schizophrenias (ed. Zinkin J) 548 (International Universities Press, 1950). [Google Scholar]
- 105.Evans B How autism became autism: the radical transformation of a central concept of child development in Britain. Hist. Hum. Sci 26, 3–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spitzer RL, Williams JB & Skodol AE DSM-III: the major achievements and an overview. Am. J. Psychiatry 137, 151–164 (1980). [DOI] [PubMed] [Google Scholar]
- 107.Thurm A, Farmer C, Salzman E, Lord C & Bishop S State of the field: differentiating intellectual disability from autism spectrum disorder. Front. Psychiatry 10, 526 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Consortium Brainstorm. et al. Analysis of shared heritability in common disorders of the brain. Science 360, eaap8757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Malhotra D & Sebat J CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148, 1223–1241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Myers SM et al. Insufficient evidence for ‘autism-specific’ genes. Am. J. Hum. Genet 106, 587–595 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Buxbaum JD et al. Not all autism genes are created equal: a response to Myers et al. Am. J. Hum. Genet 107, 1000–1003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Myers SM, Challman TD, Martin CL & Ledbetter DH Response to Buxbaum et al. Am. J. Hum. Genet 107, 1004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gudmundsson OO et al. Attention-deficit hyperactivity disorder shares copy number variant risk with schizophrenia and autism spectrum disorder. Transl. Psychiatry 9, 258 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Martin J et al. Biological overlap of attention-deficit/hyperactivity disorder and autism spectrum disorder: evidence from copy number variants. J. Am. Acad. Child. Adolesc. Psychiatry 53, 761–70.e26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zarrei M et al. A large data resource of genomic copy number variation across neurodevelopmental disorders. NPJ Genom. Med 4, 26 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Williams NM et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 376, 1401–1408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shi J et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stefansson H et al. Common variants conferring risk of schizophrenia. Nature 460, 744–747 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.International Schizophrenia Consortium. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ben-Shalom R et al. Opposing effects on Na1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82, 224–232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies a link between putative loss-of-function and gain-of-function mutations in ASD and epilepsy, respectively, providing one of the first successful examples of understanding how a single gene (SCN2A) contributes risk to multiple disorders.
- 121.Skuse DH Rethinking the nature of genetic vulnerability to autistic spectrum disorders. Trends Genet. 23, 387–395 (2007). [DOI] [PubMed] [Google Scholar]
- 122.Skuse DH et al. Social communication competence and functional adaptation in a general population of children: preliminary evidence for sex-by-verbal IQ differential risk. J. Am. Acad. Child. Adolesc. Psychiatry 48, 128–137 (2009). [DOI] [PubMed] [Google Scholar]
- 123.Rees E et al. Schizophrenia, autism spectrum disorders and developmental disorders share specific disruptive coding mutations. Nat. Commun 12, 5353 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rosenthal SB et al. A convergent molecular network underlying autism and congenital heart disease. Cell Syst. 12, 1094–1107 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu L et al. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol. Autism 5, 22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chang J, Gilman SR, Chiang AH, Sanders SJ & Vitkup D Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci 18, 191–198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chaste P et al. A genome-wide association study of autism using the Simons Simplex Collection: does reducing phenotypic heterogeneity in autism increase genetic homogeneity? Biol. Psychiatry 77, 775–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Huang JK et al. Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst. 6, 484–495.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ben-David E & Shifman S Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet. 8, e1002556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first studies to assess spatiotemporal convergence of ASD risk genes, highlighting the prenatal period as well as chromatin regulation as putative factors in ASD pathogenesis.
- 130.Trubetskoy V et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 10.1038/s41586-022-04434-5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Durand CM et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet 39, 25–27 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim H-G et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet 82, 199–207 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bourgeron T A synaptic trek to autism. Curr. Opin. Neurobiol 19, 231–234 (2009). [DOI] [PubMed] [Google Scholar]
- 134.Walsh CA, Morrow EM & Rubenstein JLR Autism and brain development. Cell 135, 396–400 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ashburner M et al. Gene Ontology: tool for the unification of biology. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]; A marker paper for the Gene Ontology Consortium, this manuscript outlines their goal to produce a structured, precisely defined, common, controlled vocabulary for describing the roles of genes and gene products in any organism. These ‘GO terms’ have formed the foundation of many systems biological analyses conducting GO enrichment or gene set enrichment analysis.
- 137.Gene Ontology Consortium. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mi H, Muruganujan A, Ebert D, Huang X & Thomas PD PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen J, Bardes EE, Aronow BJ & Jegga AG ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dutkowski J et al. A Gene Ontology inferred from molecular networks. Nat. Biotechnol 31, 38–45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schnoes AM, Ream DC, Thorman AW, Babbitt PC & Friedberg I Biases in the experimental annotations of protein function and their effect on our understanding of protein function space. PLoS Comput. Biol 9, e1003063 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Haynes WA, Tomczak A & Khatri P Gene annotation bias impedes biomedical research. Sci. Rep 8, 1362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper analyses multiple annotation databases and finds significant inequalities across genes that have become more prominent over time, highlighting a self-perpetuating cycle that may be driven by the tendency of researchers to focus their efforts on richly annotated genes rather than those with the strongest molecular data.
- 144.Zhao H et al. Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res. 27, 1293–1297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kang HJ et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work presents a large gene expression database that includes human brain samples from before birth to late adulthood in 16 brain regions, which allows spatially and temporally informed analyses. This key resource has been widely used to generate critical insights into convergence of genetic risk for ASD and other disorders of the human brain.
- 146.Parikshak NN et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first studies to assess spatiotemporal convergence of ASD risk genes, this study conducts a weighted gene co-expression network analysis using BrainSpan developmental expression data and assesses modules for enrichment of a broad range of ASD-associated genes, implicating the early and late foetal periods and upper layer glutamatergic neurons in ASD.
- 147.Ben-David E & Shifman S Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol. Psychiatry 18, 1054–1056 (2013). [DOI] [PubMed] [Google Scholar]
- 148.Xu X, Wells AB, O’Brien DR, Nehorai A & Dougherty JD Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci 34, 1420–1431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Uddin M et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat. Genet 46, 742–747 (2014). [DOI] [PubMed] [Google Scholar]
- 150.Lin GN et al. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13–Cul3–RhoA pathway in psychiatric diseases. Neuron 85, 742–754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li M et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Walker RL et al. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell 179, 750–771.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Werling DM et al. Whole-genome and RNA sequencing reveal variation and transcriptomic coordination in the developing human prefrontal cortex. Cell Rep. 31, 107489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Miller JA et al. Transcriptional landscape of the prenatal human brain. Nature 508, 199–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study presents gene expression data from laser-microdissected human mid-gestational brain tissue. These data have been central to increasing the resolution of systems biological analyses of ASD genetic risk.
- 155.Pollen AA et al. Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nowakowski TJ et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Polioudakis D et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ & Kriegstein AR Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci 24, 584–594 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fan X et al. Spatial transcriptomic survey of human embryonic cerebral cortex by single-cell RNA-seq analysis. Cell Res. 28, 730–745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bernard A et al. Transcriptional architecture of the primate neocortex. Neuron 73, 1083–1099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Doyle JP et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Voineagu I et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first papers comparing gene expression profiles in postmortem brain tissue of neurotypical controls versus ASD patients, this study identifies several molecular signatures that have now been well replicated (e.g. downregulation of neuron- and synapse-related genes and upregulation of microglia-related genes).
- 163.Parikshak NN et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gupta S et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun 5, 5748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gandal MJ et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Velmeshev D et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, eaat8464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes molecular data resources generated by the PsychENCODE Consortium from adult human brain tissue, and conducts a deconvolution analysis of bulk and single-cell expression data that finds that the majority of expression variation across bulk brain tissue samples is attributable to varying proportions of basic cell types.
- 168.Jaffe AE et al. qSVA framework for RNA quality correction in differential expression analysis. Proc. Natl Acad. Sci. USA 114, 7130–7135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhu Y, Wang L, Yin Y & Yang E Systematic analysis of gene expression patterns associated with postmortem interval in human tissues. Sci. Rep 7, 5435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li JZ et al. Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum. Mol. Genet 13, 609–616 (2004). [DOI] [PubMed] [Google Scholar]
- 171.Chiaradia I & Lancaster MA Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat. Neurosci 23, 1496–1508 (2020). [DOI] [PubMed] [Google Scholar]
- 172.Mariani J et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162, 375–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schafer ST et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci 22, 243–255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Marchetto MC et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 22, 820–835 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first studies comparing iPSC-derived neural cells from neurotypical controls to those from people with ASD and macrocephaly.
- 175.Adhya D et al. Atypical neurogenesis in induced pluripotent stem cells from autistic individuals. Biol. Psychiatry 89, 486–496 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.DeRosa BA et al. Convergent pathways in idiopathic autism revealed by time course transcriptomic analysis of patient-derived neurons. Sci. Rep 8, 8423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Griesi-Oliveira K et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol. Psychiatry 26, 1589–1605 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Amiri A et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362, eaat6720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Trevino AE et al. Chromatin accessibility dynamics in a model of human forebrain development. Science 367, eaay1645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pollen AA et al. Establishing cerebral organoids as models of human-specific brain evolution. Cell 176, 743–756.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Andrews MG & Nowakowski TJ Human brain development through the lens of cerebral organoid models. Brain Res. 1725, 146470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bhaduri A et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Andersen J et al. Generation of functional human 3D cortico-motor assembloids. Cell 183, 1913–1929.e26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Miura Y et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol 38, 1421–1430 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Birey F et al. Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Deneault E et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Rep. 11, 1211–1225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lalli MA, Avey D, Dougherty JD, Milbrandt J & Mitra RD High-throughput single-cell functional elucidation of neurodevelopmental disease-associated genes reveals convergent mechanisms altering neuronal differentiation. Genome Res. 30, 1317–1331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dixit A et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Adamson B et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jaitin DA et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896.e15 (2016). [DOI] [PubMed] [Google Scholar]
- 191.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rubin AJ et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tian R et al. CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 104, 239–255.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kampmann M CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol 16, 465–480 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tian R et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci 24, 1020–1034 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Volpato V & Webber C Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model. Mech 13, dmm042317 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Genç Ö et al. Homeostatic plasticity fails at the intersection of autism-gene mutations and a novel class of common genetic modifiers. eLife 9, e55775 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Packer A Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci. Biobehav. Rev 64, 185–195 (2016). [DOI] [PubMed] [Google Scholar]; This comprehensive review details the potential convergence of ASD genetic risk around neurogenesis.
- 199.Exner CRT & Willsey HR Xenopus leads the way: frogs as a pioneering model to understand the human brain. Genesis 59, e23405 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Werling DM & Geschwind DH Sex differences in autism spectrum disorders. Curr. Opin. Neurol 26, 146–153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Loomes R, Hull L & Mandy WPL What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child. Adolesc. Psychiatry 56, 466–474 (2017). [DOI] [PubMed] [Google Scholar]
- 202.Kim YS et al. Prevalence of autism spectrum disorders in a total population sample. Am. J. Psychiatry 168, 904–912 (2011). [DOI] [PubMed] [Google Scholar]
- 203.Bai D et al. Inherited risk for autism through maternal and paternal lineage. Biol. Psychiatry 88, 480–487 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Palmer N et al. Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatr. 171, 1107–1112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gockley J et al. The female protective effect in autism spectrum disorder is not mediated by a single genetic locus. Mol. Autism 6, 25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Martin HC et al. The contribution of X-linked coding variation to severe developmental disorders. Nat. Commun 12, 627 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.McCarthy MM Multifaceted origins of sex differences in the brain. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Baron-Cohen S The extreme male brain theory of autism. Trends Cogn. Sci 6, 248–254 (2002). [DOI] [PubMed] [Google Scholar]
- 209.Baron-Cohen S et al. Elevated fetal steroidogenic activity in autism. Mol. Psychiatry 20, 369–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Manoli DS & Tollkuhn J Gene regulatory mechanisms underlying sex differences in brain development and psychiatric disease. Ann. N. Y. Acad. Sci 1420, 26–45 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ingudomnukul E, Baron-Cohen S, Wheelwright S & Knickmeyer R Elevated rates of testosterone-related disorders in women with autism spectrum conditions. Horm. Behav 51, 597–604 (2007). [DOI] [PubMed] [Google Scholar]
- 212.Auyeung B et al. Fetal testosterone and autistic traits. Br. J. Psychol 100, 1–22 (2009). [DOI] [PubMed] [Google Scholar]
- 213.Komada M et al. Hedgehog signaling is involved in development of the neocortex. Development 135, 2717–2727 (2008). [DOI] [PubMed] [Google Scholar]
- 214.Britto J, Tannahill D & Keynes R A critical role for sonic hedgehog signaling in the early expansion of the developing brain. Nat. Neurosci 5, 103–110 (2002). [DOI] [PubMed] [Google Scholar]
- 215.Echelard Y et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75, 1417–1430 (1993). [DOI] [PubMed] [Google Scholar]
- 216.Finkel RS et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med 377, 1723–1732 (2017). [DOI] [PubMed] [Google Scholar]
- 217.Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med 377, 1713–1722 (2017). [DOI] [PubMed] [Google Scholar]
- 218.Colantuoni C et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.O’Brien HE et al. Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 19, 194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhong S et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018). [DOI] [PubMed] [Google Scholar]
- 221.Trevino AE et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell 184, 5053–5069.e23 (2021). [DOI] [PubMed] [Google Scholar]
- 222.Marshall CR et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Searles Quick VB, Wang B & State MW Leveraging large genomic datasets to illuminate the pathobiology of autism spectrum disorders. Neuropsychopharmacology 46, 55–69 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.MacDonald JR, Ziman R, Yuen RKC, Feuk L & Scherer SW The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Collins RL et al. A structural variation reference for medical and population genetics. Nature 581, 444–451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Purcell AE, Jeon OH, Zimmerman AW, Blue ME & Pevsner J Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 57, 1618–1628 (2001). [DOI] [PubMed] [Google Scholar]
- 227.Sutcliffe JS et al. The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 7, 368–377 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Greenberg F & Ledbetter DH Deletions of proximal 15q without Prader–Willi syndrome. Am. J. Med. Genet 28, 813–820 (1987). [DOI] [PubMed] [Google Scholar]
- 229.Kaplan LC et al. Clinical heterogeneity associated with deletions in the long arm of chromosome 15: report of 3 new cases and their possible genetic significance. Am. J. Med. Genet 28, 45–53 (1987). [DOI] [PubMed] [Google Scholar]
- 230.Magenis RE, Brown MG, Lacy DA, Budden S & LaFranchi S Is Angelman syndrome an alternate result of del(15)(q11q13)? Am. J. Med. Genet 28, 829–838 (1987). [DOI] [PubMed] [Google Scholar]
- 231.Williams CA, Gray BA, Hendrickson JE, Stone JW & Cantú ES Incidence of 15q deletions in the Angelman syndrome: a survey of twelve affected persons. Am. J. Med. Genet 32, 339–345 (1989). [DOI] [PubMed] [Google Scholar]
- 232.Vilain A, Apiou F, Vogt N, Dutrillaux B & Malfoy B Assignment of the gene for methyl-CpG-binding protein 2 (MECP2) to human chromosome band Xq28 by in situ hybridization. Cytogenet. Cell Genet 74, 293–294 (1996). [DOI] [PubMed] [Google Scholar]
- 233.D’Esposito M et al. Isolation, physical mapping, and northern analysis of the X-linked human gene encoding methyl CpG-binding protein, MECP2. Mamm. Genome 7, 533–535 (1996). [DOI] [PubMed] [Google Scholar]
- 234.Kremer EJ et al. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252, 1711–1714 (1991). [DOI] [PubMed] [Google Scholar]
- 235.Verkerk AJ et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). [DOI] [PubMed] [Google Scholar]
- 236.Pieretti M et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822 (1991). [DOI] [PubMed] [Google Scholar]
- 237.Li J et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997). [DOI] [PubMed] [Google Scholar]
- 238.Li DM & Sun H TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res. 57, 2124–2129 (1997). [PubMed] [Google Scholar]
- 239.Steck PA et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet 15, 356–362 (1997). [DOI] [PubMed] [Google Scholar]
- 240.van Slegtenhorst M et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808 (1997). [DOI] [PubMed] [Google Scholar]
- 241.Chromosome European 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993). [DOI] [PubMed] [Google Scholar]
- 242.Marchuk DA et al. cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1 gene product. Genomics 11, 931–940 (1991). [DOI] [PubMed] [Google Scholar]
- 243.Cawthon RM et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 62, 193–201 (1990). [DOI] [PubMed] [Google Scholar]
- 244.Viskochil D et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62, 187–192 (1990). [DOI] [PubMed] [Google Scholar]
- 245.Wallace MR et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 249, 181–186 (1990). [DOI] [PubMed] [Google Scholar]
- 246.Rouleau GA et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363, 515–521 (1993). [DOI] [PubMed] [Google Scholar]
- 247.Trofatter JA et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 72, 791–800 (1993). [DOI] [PubMed] [Google Scholar]
- 248.Guo H et al. NCKAP1 disruptive variants lead to a neurodevelopmental disorder with core features of autism. Am. J. Hum. Genet 107, 963–976 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Mirzaa GM et al. De novo and inherited variants in ZNF292 underlie a neurodevelopmental disorder with features of autism spectrum disorder. Genet. Med 22, 538–546 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Guo H et al. Disruptive mutations in TANC2 define a neurodevelopmental syndrome associated with psychiatric disorders. Nat. Commun 10, 4679 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Van Dijck A et al. Clinical presentation of a complex neurodevelopmental disorder caused by mutations in ADNP. Biol. Psychiatry 85, 287–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Blackburn ATM et al. DYRK1A-related intellectual disability: a syndrome associated with congenital anomalies of the kidney and urinary tract. Genet. Med 21, 2755–2764 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ji J et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet 23, 1473–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bernier R et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158, 263–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first studies to recognize distinctive physical features and co-morbidities shared across patients with rare mutations in the same ‘idiopathic’ ASD risk gene — opening the door to the presence of previously unappreciated syndromes within idiopathic ASD.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.