

Summary
A deep understanding of immunotherapy response/resistance mechanisms and a highly reliable therapy response prediction are vital for cancer treatment. Here, we developed scCURE (single-cell RNA sequencing [scRNA-seq] data-based Changed and Unchanged cell Recognition during immunotherapy). Based on Gaussian mixture modeling, Kullback-Leibler (KL) divergence, and mutual nearest-neighbors criteria, scCURE can faithfully discriminate between cells affected or unaffected by immunotherapy intervention. By conducting scCURE analyses in melanoma and breast cancer immunotherapy scRNA-seq data, we found that the baseline profiles of specific CD8+ T and macrophage cells (identified by scCURE) can determine the way in which tumor microenvironment immune cells respond to immunotherapy, e.g., antitumor immunity activation or de-activation; therefore, these cells could be predictive factors for treatment response. In this work, we demonstrated that the immunotherapy-associated cell-cell heterogeneities revealed by scCURE can be utilized to integrate the therapy response mechanism study and prediction model construction.
Keywords: immunotherapy, single-cell RNA-seq, therapy response prediction models, cancer
Graphical abstract
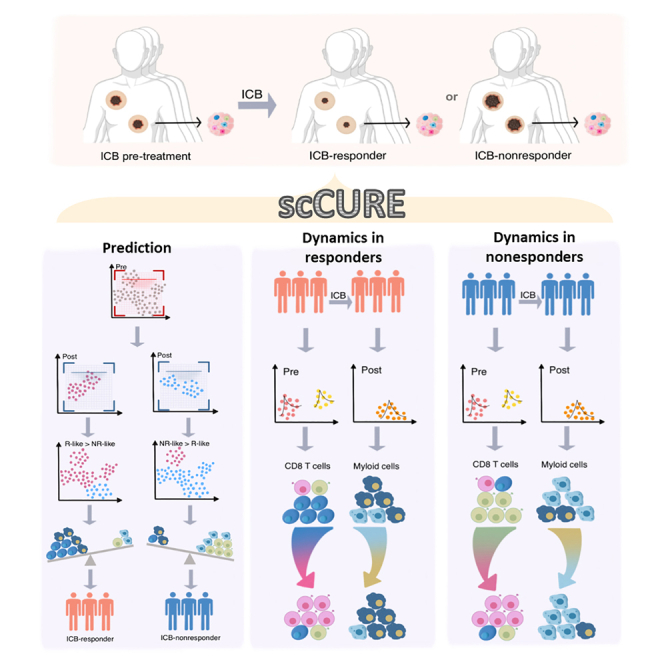
Highlights
-
•
scCURE identifies cell types that are changed or unchanged during immunotherapy
-
•
Unchanged cells enable immunotherapy outcome prediction
-
•
Changed cells demonstrate immune dynamics driven by immunotherapy
-
•
scCURE can be applied to analyze unmatched pre- and post-treatment samples
Motivation
Single-cell RNA sequencing (scRNA-seq) has significantly advanced our understanding of how different types of cells respond to immunotherapy. However, inconsistencies persist in our understanding of the mechanistic underpinnings and in predictive models of immunotherapy, with studies reporting different cell subtypes contributing to mechanisms of response and prediction models without clear explanation. Such inconsistencies might originate from uncharacterized cellular heterogeneity. To tackle this issue, we present scCURE, which leverages pattern recognition modeling to differentiate between changed and unchanged cells during the course of immunotherapy.
Zou et al. develop scCURE, a method to identify cell types that are changed or unchanged by immunotherapy. Being robust to batch effects, scCURE can effectively leverage pre- and post-treatment datasets to enable predictive models for immunotherapy response and to suggest mechanism of response.
Introduction
The successful development of cancer immunotherapies targeting T cells and their immune checkpoint proteins in recent years is expected to provide promising clinical responses to cancer.1,2 However, clinical benefit is only limited to a small subset of patients, and not all patients can be treated with immune checkpoint blockade (ICB) to reduce recurrence rates and prolong survival.3,4 Given the high cost of treatment, understanding the precise cellular and molecular mechanisms of therapy response and resistance and assessing immune function prior to clinical treatment to predict patients most likely to respond positively to ICB therapy remain critical tasks to improve immunotherapies and develop new treatment pathways.
Recently, much effort has been made to interpret the cell dynamics associated with immunotherapy using single-cell techniques, including melanoma,4,5,6 breast cancer,7 renal cell cancer (RCC),8 non-small cell lung cancer (NSCLC),9 hepatocellular carcinoma (HCC),10 acute myeloid leukemia (AML),11 colorectal cancer,12 and head and neck squamous cell carcinoma (HNSCC).13 Generally, the purposes of previous single-cell RNA sequencing (scRNA-seq)-based immunotherapy studies can be categorized into two types: mechanism investigation and prediction model construction. In terms of prediction, prediction models have been constructed for melanoma6 and RCC8 by combining scRNA-seq and bulk RNA-seq datasets. In HCC, Shi et al. identified CXCR3+CD8+ effector memory T cells and type 1 conventional CD11c+ dendritic cells (DCs) from peripheral blood mononuclear cells (PBMCs) as immunotherapy response factors.10 In breast cancer, Zhang et al. identified that high levels of CXCL13+ T cells at baseline could predict a positive response to combination therapy.7 At the level of pan-cancer, Zhang et al. develop a stemness-based signature to predict immunotherapy response.14 Other studies focused on mechanism investigation. In AML, Abbas et al. identified the expansion of T cell receptor (TCR) repertoires and the emergence of GZMK+CD8+ T cell clonotypes in responders during ICB treatment.11 Liu et al. reported a phenomenon termed clonal revival during lung cancer ICB treatment.9 Li et al. revealed that ICB changed the inflammatory features of colorectal tumors.12 There are also some studies that have simultaneously investigated mechanisms and prediction models. In HNSCC, Obradovic et al. investigated both immunotherapy response/resistance mechanisms and therapy response prediction, but the two aspects were linked with different cancer-associated fibroblast (CAF) cell subtypes, and the reason is still unclear.13 Until now, it has been a challenge to comprehensively integrate the above two aspects. It is reasonable to speculate that such inconsistency may be due to uncharacterized cell-cell heterogeneities. For example, in our previous studies, we successfully extracted prediction markers from post-treatment RCC samples.8 Such a preliminary result implies that there must exist cells that remain unchanged during the course of therapy, and those cells may contain predictive features for therapy outcomes. In addition, the ways in which immunotherapy reshapes immune systems in various patients could be interpreted based on the cells responding to treatment interventions. However, the profiles of changed and unchanged cells have yet to be fully depicted.
Deep mining of single-cell data using machine learning can lead to further understanding of cellular heterogeneity and the underlying immune response mechanisms. To achieve this, a variety of unsupervised clustering methods have been developed, including the k-means-based methods pcaReduce,15 SAIC,16 SC3,17 SCUBA,18 scVDMC,19 etc.; the hierarchical-clustering-based methods BackSPIN,20 cellTree,21 CIDR,22 DendroSplit,23 ICGS,24 RCA,25 etc.; the graph-based clustering methods TCC,26 SIMLR,27 SNN-Cliq,28 SCANPY,29 etc.; and mixture-model-based clustering methods including BISCUIT,30 DTWScore,31 TSCAN,32 etc. However, unsupervised methods usually fail to specifically reveal the cell heterogeneities related to the problems being studied (e.g., pre- and post-treatment or response and non-response). In addition, supervised methods, e.g., differential expression gene identification,33 also cannot be applied to investigate unknown cell heterogeneities.
To specifically characterize immunotherapy-related cell heterogeneities, we here present scCURE (scRNA-seq-based Changed and Unchanged cell Recognition during immunotherapy). scCURE combines the Gaussian mixture model (GMM) and Kullback-Leibler (KL) divergence, which endows the algorithm with high noise robustness. On simulated data, scCURE outperformed multiple existing methods in discriminating immunotherapy-induced gene expression variations from irrelevant interferences. We demonstrate the utility of scCURE in constructing more reliable therapy outcome prediction models and better interpreting the associated response/resistance mechanisms on melanoma and breast cancer scRNA-seq datasets. We envisage that our work may promote clinical applications of single-cell techniques and shed light on the development of more precise, efficient, and broadly suitable tumor treatment strategies.
Results
Principles of scCURE
We hypothesize that the putative cell heterogeneities can be categorized into two major types: (1) changed cells. The dynamics of these cells reflect the way in which patients respond to immunotherapy. (2) Unchanged cells. Those cells from the pre- and post-treatment conditions have similar cellular and molecular functions, and the differences are mainly caused by irrelevant factors, e.g., batch effects and random noise. The baseline profile of unchanged cells may provide prediction feature for therapy outcomes.
To appropriately capture such heterogeneity, it is key to discriminate immunotherapy-induced cell-state transitions from irrelevant variations. To achieve this, scCURE consists of the following major steps: (1) model construction. The GMM is trained using the expectation maximization (EM) method on the cells from pre- and post-treatment conditions, respectively, with pre-defined K numbers of Gaussian models (see STAR Methods for parameter optimization). Each cell is assigned to a specific Gaussian model based on the maximum likelihood criterion. Assuming that the dataset consists of cells with K categories of subtypes/functions, within each subtype/functional category, cells are homogeneous. Therefore, each subtype/functional category can be modeled as a Gaussian distribution. The combination of multiple Gaussian distributions with various parameters can be used to approximate the real-world single-cell data. Assuming that the number of Gaussian models is sufficiently high to characterize intra-condition cell heterogeneities, a Gaussian model represents either changed or unchanged cells. (2) Changed and unchanged cell discrimination. To discriminate the Gaussian models representing changed and unchanged cells, we exhaust all possible model pairs between the two conditions and calculate KL divergences between them. If a pair of Gaussian models from two conditions are mutually closest to each other in terms of KL divergence, the cells assigned to the two Gaussian models should not contain systematic variations, and these cells are annotated as unchanged; otherwise, they are annotated as changed (see STAR Methods, proof of concept).
Based on the characteristics of unchanged cells and a self-defined prediction score, a new therapy outcome prediction model can be constructed. In addition, the immunotherapy response/resistance mechanisms interpreted from changed cells can be well integrated with the prediction models. The schematic diagram of scCURE is illustrated in Figure 1.
Figure 1.

Flowchart of scCURE-based immunotherapy scRNA-seq data analysis
“scRNA-seq” demonstrates the data acquiring procedure. “scCURE” illustrates the model construction and discrimination of changed and unchanged cells. “Unchanged cells for prediction” illustrates the cell proportion characteristics are related to immunotherapy outcomes. In clinical application scenarios, an scCURE model can be constructed using training data and applied on testing patient to identify unchanged cells. “Changed cells for mechanisms interpretation” illustrates how to interpret immunotherapy mechanisms on changed cells.
Validation of scCURE on simulated datasets
Simulated datasets mimicking pre- and post-treatment conditions were generated (see STAR Methods for details), on which the ability of scCURE in identifying unchanged cells was evaluated in terms of sensitivity, specificity, F1 score, area under the curve (AUC) of the receiver operating characteristic curve (ROC), and positive prediction value (PPV) (see STAR Methods for definition). The proposed algorithm was benchmarked against multiple leading methods that are able to identify unchanged cells, i.e., mutually nearest neighbor (MNN)34 and canonical correlation analysis (CCA)35 (STAR Methods). As the number of neighbors set up in MNN and the CCA may affect the final results, various parameters were tested, i.e., 5, 10, 20, and 50. At the same time, various numbers of Gaussian models K in scCURE were also evaluated.
The simulated datasets with signal-to-noise ratios (SNRs) from low to high were generated, i.e., FCs (fold changes) from 1.1 to 2 (Figure 2; see STAR Methods simulated datasets). For low-SNR datasets, the MNN and CCA methods had trouble in simultaneously improving sensitivity and specificity. A larger number of neighbors resulted in higher sensitivity but lower specificity and as such resulted in compromised PPV, F1 score, and AUC. Satisfactory results were only obtained by MNN when the SNR was high enough, i.e., FC = 2, but the CCA had poor performance in all circumstances. If K is no less than the actual number of cell subclusters, the highest sensitivity, specificity, PPV, F1 score, and AUC could be achieved by scCURE, even in low-SNR circumstances. We noticed that if K was not large enough to cope with data heterogeneity, specificity, PPV, F1 score, and AUC were obviously affected. Moreover, we evaluated the impact of different numbers of principal components (5, 10, 20) on the model performance using these metrics (Figure S1). We found that scCURE was not obviously affected by the choice of different numbers of principal components, but using top 5 principal components (PCs) could obtain the best AUC in the case of 4 subclusters. Therefore, 5 PCs were used on the subsequent analyses.
Figure 2.

scCURE evaluation on simulated data
The values of K for pre- and post-treatment cells were set to equal.
In summary, simulated data demonstrated that the proposed scCURE algorithm is able to reliably capture treatment-associated cell heterogeneity in the presence of strong interferences. The results were also insensitive to varied parameters as long as the model was sufficiently comprehensive to cope with data heterogeneity. In the subsequent sections, we will introduce a strategy to optimize the choice of K in experimental applications.
Baseline CD8+ T cell composition profiles predict ICB outcome in patients with melanoma
We then demonstrate how scCURE can be used to reveal immunotherapy response/non-response characteristics using a melanoma scRNA-seq dataset.5 The dataset contained cells collected from pre- and post-ICB-treated patients with melanoma who showed response or non-response phenotypes. The cells were reannotated, and CD8+ T cells were selected for analysis. There were 2,538 CD8+ T cells collected from pre-treatment patients (including responders and non-responders), 1,074 cells from post-treatment responders, and 3,228 cells from post-treatment non-responders. Unchanged CD8+ T cells were identified for ICB outcome prediction purposes. Specifically, all pre-treatment cells were compared to the cells from post-treatment responders and post-treatment non-responders using scCURE, respectively. The scCURE parameter K was optimized by maximizing the prediction capability of the model (see STAR Methods for details). We annotated the pre-treatment cells as R-like if they were identified as unchanged cells when comparing all pre-treatment cells and post-treatment responder cells. Similarly, non-responder (NR)-like cells were the pre-treatment unchanged cells when compared to post-treatment NRs. We defined an ICB outcome prediction score for each individual patient as the ratio number of cells between responder (R)-like/NR-like cells. Using this score, the prediction capability of scCURE with various parameter K values was evaluated using leave-one-out cross validation (see STAR Methods for details). According to the results, K = 5 was set for pre-treatment and K = 3 for post-treatment (both Rs and NRs) in unchanged/changed discrimination. According to Seurat36 standard analysis process, all the identified pre-treatment unchanged cells were reclustered into 3 main clusters using t-distributed stochastic neighbor embedding (t-SNE) (without cross validation; Figure 3B). Based on the results of FindAllMarker and the expression of canonical markers, we annotated the three clusters as CD8-C2-SELL (naive state), CD8-C1-HAVCR2 (exhausted state), and CD8-C3-GNLY (intermediate state) (Figure 3C). Moreover, we observed that CD8-C2-SELL was dominated by R-like cells and the other two clusters were dominated by NR-like cells (Figure 3D). Figure 3E illustrates that the R-like/NR-like ratios could be used to well predict the response patterns (the ratios were calculated using leave-one-out cross validation as described in the STAR Methods section). Moreover, Rs had a higher mean expression of the CD8-C2-SELL signatures than NRs in the PREJEB23709 dataset (Rs = 49, NRs = 42) (Figure 3F; STAR Methods). Gene set enrichment analysis (GSEA) revealed that significant enrichment of the immune-related pathways in CD8-C2-SELL clusters, such as the interferon α/γ (IFN-α/γ) response, complement signaling pathways, whereas the p53, apoptosis, and transforming growth factor β (TGF-β) signaling pathways were activated in the CD8-C1-HAVCR2 cluster and the tumor necrosis factor α (TNF-α) signaling via the nuclear factor κB (NF-κB) and MTORC1 signaling pathways were activated in the CD8-C3-GNLY cluster (Figure 3G). Interestingly, the changed cells in pre-treatment could also predict the ICB outcome. The 1,709 changed cells were identified as CD8-C1-HAVCR2, CD8-C2-SELL, and CD8-C3-GNLY (Figures S2A and S2B). Like the result of the unchanged cells, the ratios of R change and NR change could also discriminate Rs and NRs (Figure S2C).
Figure 3.

scCURE identified R-like and NR-like CD8+ T cells from pre-treatment patients with melanoma
(A) The AUC and p values of prediction models obtained using different choices of scCURE parameters.
(B) t-SNE plot of the scCURE-identified unchanged CD8+ T cells colored by cell type, cell group, and ICB response pattern.
(C) Heatmap of canonical CD8+ T cell functional markers.
(D) Chord diagram showing the association between cell clusters and R-like/NR-like categories by hypergeometric distribution test.
(E) Bar plot showing the well distribution of R-like/NR-like ratios between Rs and NRs.
(F) Signature scores of CD8-C2-SELL cluster computed on the PREJEB23709 dataset (Rs = 49, NRs = 42) and compared between cell groups with a two-sided Wilcoxon test. Signature scores were defined by the average expression of the top 30 CD8-C2-SELL markers. Statistical significance indicated by asterisks is as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(G) Hallmark pathways enriched in different CD8+ T cell clusters (Benjamini-Hochberg [BH]-adjusted p < 0.05).
For comparison purposes, we also tried to use MNN and CCA to identify the R-like cells and NR-like cells (Figure S3). For most patients, MNN failed to assign R-like or NR-like cells; therefore, the ratio cannot be calculated. The R/NR ratios for the rest of the patients were irrelevant to response pattern, which failed to predict the ICB outcome. CCA was unable to predict the response status of the cells, as it could not identify any specific R-like or NR-like cells (data not shown).
ICB treatment induces dual CD8+ T cell state transition paths in both Rs and NRs
The dynamic patterns of changed CD8+ T cells induced by ICB treatment were comprehensively interrogated to investigate the mechanisms of the ICB response and non-response.
First, 1,478 CD8+ T cells from pre-treatment responders and 582 CD8+ T cells from post-treatment Rs were identified as changed. All 2,060 cells were categorized into 7 main clusters. Among these clusters, CD8-C1-PDCD1, CD8-C2-CTLA4, CD8-C4-LAYN, and CD8-C6-GNLY mainly expressed co-inhibitory receptors (PDCD1, CTLA4, HAVCR2, LAYN, TIGIT), while CD8-C3-CCR7, CD8-C5-IL7R, and CD8-C7-FGFBP2 enriched in naive/co-stimulation molecules (SELL, IL7R, CCR7, TNF, FGFBP2, GNLY) (Figures 4A and 4B). Additionally, the proportion of CD8-C2-CTLA4 and CD8-C5-IL7R clusters had increased and the proportion of CD8-C1-PDCD1, CD8-C4-LAYN, CD8-C3-CCR7, CD8-C6-GNLY, and CD8-C7-FGFBP2 clusters had decreased after treatment, which indicated that ICB might reverse the exhausted state of CD8+ T cells. Pseudotime trajectory analysis37 (STAR Methods) illustrated that there may exist two transitional pathways from pre- to post-treatment samples, i.e., from two pre-treatment start nodes, the terminal exhausted cluster (CD8-C1-PDCD1) and naive/effector clusters (CD8-C7-FGFBP2 and CD8-CD3-CCR7), to one post-treatment end node, CD8-C5-IL7R (Figure 4C). It is reasonable to deduce that naive/effector and terminal exhausted CD8+ T cells might be activated into the progenitor exhausted state, which is responsible for tumor killing, as illustrated by survival analysis using the top 30 markers of CD8-C5-IL7R (Figure 4H). Previous studies showed that exhausted CD8+ T cells can be further categorized into progenitor and terminal cells.38,39 Here, we evaluated the expression distribution of the terminal and progenitor exhaustion markers reported in Bi et al.38 and found that the expression of progenitor exhaustion markers tended to improve after treatment, and terminal exhaustion features were the opposite (Figure 4D). This transition was also demonstrated by showing the expression of progenitor and terminal exhausted markers within different cell clusters along the trajectory (Figure 4E). We then investigated the molecular functions of these clusters by hallmark pathway enrichment analysis. Compared with CD8-C1-PDCD1 and CD8-C7-FGFBP2, immune-related pathways were enriched in CD8-C5-IL7R, such as IFN-γ, complement pathways. We discovered that CD8-C1-PDCD1 was dominated by p53 and epithelial-mesenchymal transition (EMT) pathways and that CD8-C7-FGFBP2 was dominated by TGF-β or estrogen response late pathways (Figure 4F).
Figure 4.

The ICB treatment response mechanism explained in the scCURE identified changed CD8+ T cells between pre-treatment samples and post-treatment Rs
(A) Heterogeneity of the dynamical CD8+ T cells shown in the t-SNE scatterplot. Cell cluster frequency shown as a fraction of total cells in pre-treatment and post-treatment.
(B) Heatmap shows the expression of canonical T cell functional markers across cell clusters.
(C) Pseudotime trajectory reconstruction and its association with cell clusters and sample labels.
(D) Heatmap in t-SNE space showing the signature scores for terminally exhausted and progenitor exhausted CD8+ T cells.
(E) Terminally exhausted and progenitor exhausted CD8+ T cell signatures across CD8+ T cell subtypes. Asterisks indicate statistical significance by Kruskal-Wallis test between groups for each signature as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(F) Enriched GSEA hallmarks of two representative CD8+ T clusters of pre- and post-treatment samples (Benjamini-Hochberg [BH]-adjusted p < 0.05).
(G) Signature scores for the top 30 markers of CD8-C5-IL7R in bulk RNA-seq samples from the GEO: GSE91061 melanoma cohort.
(H) Survival analysis using the top 30 markers of CD8-C5-IL7R on TCGA melanoma data.
To validate the above observation, we examined an independent cohort of patients (GEO: GSE91061) undergoing therapy with the anti-PD-1 antibody nivolumab for whom pre- and post-therapy bulk transcriptomes were obtained (STAR Methods). In these biopsies, we observed significantly increased levels of the top 30 marker genes in the post-treatment samples (Seurat FindAllMarkers function) of the single-cell dataset CD8-C5-IL7R (Figure 4G).
Similar analyses were conducted for the changed cells between pre-treatment and post-treatment NRs (Figure S4). Surprisingly, we observed that the antitumoral characteristics of naive/effector/terminal exhausted CD8+ T cells were also activated. The major difference between Rs and NRs lies in that the upregulation trend of progenitor exhausted markers was relatively less obvious in NRs.
In summary, similar CD8+ T cell activation procedures were revealed in both Rs and NRs, but the activation seemed less effective in NRs. Taking the prediction model into consideration, it is reasonable to deduce that the activation path from naive/effector to progenitor exhausted CD8+ T cells may be more effective than the other path. As a result, patients with more naive and effector CD8+ T cells at baseline were more prone to respond. This speculation is also consistent with a previous study,40 which suggests that responsiveness to immune checkpoint inhibitors is less dependent on the reinvigoration of exhausted T cells using scTCR-seq.
Baseline macrophage subtype compositions affect melanoma ICB outcomes
In addition to adaptive immune cells, the influence of innate immune cells, e.g., macrophages, on ICB outcomes was also evaluated. There were 405 macrophage cells collected from pre-treatment patients, 98 cells from post-treatment Rs, and 905 cells from post-treatment NRs.
Using K = 2 for pre-treatment and K = 2 for post-treatment (both Rs and NRs) (Figure 5A), a total of 351 unchanged pre-treatment macrophage cells were obtained, including 85 R-like and 266 NR-like cells (Figure 5B). For cell-subtype annotation purposes, the cells were categorized into 5 main clusters, among which Macro-C2-IL27RA was dominated by R-like cells and Macro-C1-CCL17, Macro-C3-CXCL10, Macro-C4-CCL23, and Macro-C5-IL2RA were associated with NR-like cells (Figures 5B and 5D). The expression pattern of canonical markers of M1 and M2 macrophages38 indicated that the R-like cluster Macro-C2-IL27RA might be unpolarized macrophage cells and NR-like clusters Macro-C4-CCL23 and Macro-C5-IL2RA might be M2 macrophages (Figure 5C).
Figure 5.

scCURE identified predictive R-like and NR-like macrophage cells from pre-treatment patients with melanoma
(A) The AUC and p values of prediction models obtained based on different choices of scCURE parameters.
(B) t-SNE plot of the scCURE-processed macrophage cells colored by cell type, cell group, and ICB response.
(C) Heatmap of M1 and M2 macrophage cell markers.
(D) The association between clusters and response patterns by hypergeometric test.
(E) The ratio of R-like and NR-like can well discriminate Rs and NRs.
(F) Circos plot depicting the performance of signatures of each cluster in three bulk melanoma cohorts. The vertical axis indicates AUC values.
(G) Enriched GSEA hallmarks of three predictive macrophage clusters (Benjamini-Hochberg [BH]-adjusted p < 0.05).
With the same results as for CD8+ T cells, the ratio of R-like/NR-like of each patient could be a potential predictor for ICB outcomes (Figure 5E). The therapeutic outcome prediction capabilities of the different unchanged macrophage clusters (top 10 markers by Seurat FindAllMarkers function) were evaluated on three independent bulk datasets (GEO: GSE78220 and GSE91061 and PREJEB23709) in terms of prediction AUC (Figure 5F). scCURE_C1 had high prediction capability in three datasets, and scCURE_C3 and scCURE_C4 possessed potential predictive in some datasets. We also evaluated prediction capabilities of each cluster by cancerclass41 and lasso regression (Figure S5; STAR Methods). In summary, all five clusters showed prediction potentials (AUC > 0.6) on various independent bulk datasets using various machine learning methods, which explains why the ratio between R-like/NR-like cells can be used as a predictor. GSEAs were then performed on the three clusters with higher predictive capability, C1-CCL17, C3-CXCL10, and C4-CCL23. It can be seen that C1-CCL17 was dominated by allograft rejection, IFN-α, and fatty acid metabolism pathways, while inflammatory response, EMT, and interleukin-2 (IL-2) STAT5 signaling pathways were activated in C3-CXCL10 and coagulation and MTORC1 signaling pathways were activated in C4-CCL23 (Figure 5G).
Macrophage polarization/depolarization is associated with ICB response/non-response
Similar to CD8+ T cells, the changed macrophage cells were characterized to interpret the mechanisms of Rs/NRs.
To probe the cell kinetics in Rs, 279 cells from pre-treatment and 42 cells from post-treatment Rs were identified as changed. All 321 cells were categorized into 3 clusters, among which Macro-C4-CXCR4 was dominated by post-samples and the other two clusters by pre-samples (Figures 6A and 6C). Canonical M1 and M2 macrophage markers indicated that Macro-C4-CXCR4 may be unpolarized macrophage cells (Figure 6B). Trajectory analysis implied that ICB therapy may reprogram M2 macrophage cells to M0 phenotype (from Macro-C2-CCL18 to Macro-C4-CXCR4) (Figure 6D). GSEA of the C4-CXCR4 markers illustrated that proinflammatory functions, e.g., the IFN-α/γ response, were activated in Rs after treatment (Figure 6E). Survival analysis of TCGA data revealed that ICB may improve the overall survival of patients with melanoma (Figure 6F).
Figure 6.

Analyses of the changed macrophage cells in Rs show that antitumoral functions are activated by depolarization of M2 macrophages, which coordinates with the activation of CD8+ T cells
(A) Heterogeneity of the dynamical macrophage cells shown in the t-SNE scatterplot. Cell cluster frequency shown as a fraction of total cells in pre-treatment and post-treatment.
(B) Heatmap showing the signature scores for M1 and M2 macrophages.
(C) Enrichment of different cell clusters in pre- and post-samples.
(D) Pseudotime trajectory reconstruction and its association with cell clusters and sample labels.
(E) Enriched GSEA hallmarks of two representative macrophage clusters of pre- and post-treatment samples (Benjamini-Hochberg [BH]-adjusted p < 0.05).
(F) Survival analysis using the top 30 markers of Macro-C2-CCL18 and Macro-C3-CXCR4.
(G) Nichnet cell-cell communication between macrophages and CD8+ T cells in NRs reveals that the decrease in TNF and IFN-γ on CD8+ T cells is correlated with macrophage depolarization and proinflammatory function activation.
To further investigate how immune cells coordinate in response to ICB treatment, the NicheNet R package42 was adopted. It was noticed that the ligands TNF and IFN-γ from CD8+ T cells were outstandingly highly correlated with a few target markers on macrophage cells, including the M2 markers CXCR4 and CCL2 (Figure 6G). Considering that TNF and IFN-γ were relatively high in pre-treatment CD8+ T cells and tended to decrease after ICB treatment (Figure 4B), we hypothesized that in Rs, the activation of CD8+ T cells may coordinate with the depolarization of M2 macrophage cells; as a result, antitumoral inflammatory functions are activated.
In contrast, we found a reverse macrophage transition pattern in NRs. In NRs, the antitumoral unpolarized macrophage cells were polarized toward the M2 macrophage subtype, and the antitumoral functionality was lost. We did not observe coordination between macrophages and CD8+ T cells in NRs (Figure S6).
The ICB response characteristics shared between melanoma and breast cancer
To further validate scCURE, the data from a triple-negative breast cancer (TNBC) chemoimmunotherapy study were adopted.7 In this study, the CD8+ T cells collected from chemoimmunotherapy-treated tissue samples were reanalyzed, i.e., scCURE was used to compare 11,599 pre-treatment CD8+ T cells with 3,906 cells from post-treatment Rs and 3,468 cells from post-treatment NRs. According to the maximization of AUC criteria, K = 3 was set for pre-treatment and K = 4 for post-treatment (Figure 7A). As a result, 2,014 and 6,418 pre-treatment cells were identified as R-like and NR-like, respectively. The identified unchanged cells were categorized into 6 main clusters, among which CD8-C1-SELL, CD8-C2-PDCD1, CD8-C5-GNLY, and CD8-C6-HAVCR2 were dominated by R-like cells and CD8-C3-CX3CR1 and CD8-C4-IFN-γ were dominated by NR-like cells (Figures 7B and 7C). Among all clusters, CD8-C1-SELL had obvious naive (high SELL and CCR7) and progenitor exhaustion (high GZMK and moderate high TIGIT and PDCD1) characteristics. The R-associated CD8-C1-SELL, CD8-C2-PDCD1, and CD8-C6-HAVCR2 clusters specifically highly expressed CXCL13, which was consistent with the findings reported in the original study (Figure 7D). Rs and NRs can be well separated based on the ratio of R-like to NR-like cells (Figure 7E). We then evaluated predictive performance of each cluster in the I-SPY2 trial cohort. By using top 10 markers, the AUC of each cluster revealed that scCURE_C1, scCURE_C2, and scCURE_C3 had higher predictive potential (Figure 7F). The result demonstrated again that CD8-C2-PDCD1 had stronger correlation with ICB response. We also performed GSEA to investigate their molecular functions (Figure 7G). In R-like cells, CD8-C1-SELL was dominated by protein folding and response to oxidative stress and CD8-C2-PDCD1 was dominated by cell adhesion, and regulation of cell differentiation and cell activation were enriched in CD8-C3-CX3CR1 of NR-like cells.
Figure 7.

scCURE identified predictive R-like and NR-like CD8+ T cells from pre-treatment patients with TNBC
(A) The AUC and p values of prediction models obtained using different choices of scCURE parameters.
(B) t-SNE plot of the scCURE-processed CD8+ T cells colored by cell type, cell group, and ICB response.
(C) Chord diagram showing the enrichment result between cell type and cell group by hypergeometric distribution.
(D) Heatmap of canonical CD8+ T cell functional markers.
(E) Histogram showing the well distribution of R-like/NR-like ratios between Rs and NRs.
(F) The multiple ROC plot depicting the predictive performance of the average expression of top 10 markers of each cluster in I-SPY2 cohort.
(G) Hallmark pathways enriched in the predictive macrophage clusters. Hypergeometric test. BH-adjusted p < 0.05.
Discussion
scRNA-seq has made it possible to systematically profile cell types and cell molecular functions in patients with tumor receiving immunotherapy. Although scRNA-seq may provide a detailed modality of data, high noise levels and limited a priori knowledge hamper the discovery of immunotherapy-associated mechanisms from scRNA-seq data. Here, we presented a novel algorithm, scCURE, which leveraged pattern recognition modeling to discriminate changed and unchanged cells during the course of immunotherapy. By elucidating the usage of changed and unchanged cells in immunotherapy investigation, we shed light on the application of single-cell techniques in precision immunotherapy.
The whole work was conducted based on the assumption that undiscovered immunotherapy-associated cell-cell heterogeneities exist and that such heterogeneities are the main cause of inconsistencies between previous prediction model construction and mechanism exploring studies. In conventional studies, some investigators focused on linking specific cell characteristics, e.g., stemness,14 with immunotherapy outcomes. This kind of study can be categorized as supervised. However, in most studies, cell heterogeneities are usually revealed by unsupervised methods, e.g., clustering,35 pseudotime trajectory analysis,37,43 and cell-cell interaction.42,44 Then, the heterogeneities associated with immunotherapy outcome were identified, which can be referred to as unsupervised. The supervised methods are limited by the availability of a priori knowledge and therefore cannot exhaust all informative characteristics contained in data. In contrast, unsupervised methods do not rely on a priori knowledge, but the analyses are easily distracted by irrelevant factors. Different from the aforementioned conventional methods, scCURE can specifically extract the cell heterogeneities related to immunotherapy intervention; at the same time, no additional a priori information is required. Given the robustness of scCURE against batch effects, the patients contained in the pre- and post-treatment datasets are not necessarily matched. Therefore, by leveraging existing post-treatment data, scCURE can calculate R-like/NR-like ratios of treatment-naive patients.
In this article, we have demonstrated two strategies in which scCURE can help to construct immunotherapy prediction models, i.e., signature identification for bulk RNA-seq data-based prediction and a new prediction score for scRNA-seq data. Although the former can achieve promising prediction results in many practical circumstances, the characteristics of specific cell subtypes cannot be extracted, which may compromise the prediction performance. The latter can make full use of cell-subtype information, but the high costs of single-cell techniques may hamper their clinical applications. Since unchanged cells can profile cell-subtype characteristics of Rs and NRs, deconvolution methods, e.g., CIBERSORT,45 Batman,46 and EPIC,47 might be applied to combine the merits of the above two strategies. Specifically, scCURE can be applied to extract cell-subtype characteristics from scRNA-seq data, and such characteristics can then be profiled from bulk RNA-seq data by deconvolution methods and used to guide cancer treatment. In the future, we will systematically explore the potential of scCURE in more clinical applications.
We noticed that the choice of cell clusters K was usually relatively small, and such small numbers may not be enough to account for the complexity of scRNA-seq data. However, as we are only interested in the cell heterogeneities related to treatment interventions, some cell subtypes/functional categories may be merged without affecting the final results.
Although scCURE has been demonstrated to have potential in immunotherapy investigation, it mathematically does not contain any specifications for immunotherapy. Therefore, we envisage that it can be applied in other circumstances, in which immune profile changes may be associated, e.g., chemotherapy.48,49
Different from other methods that investigate cell abundance changes between experimental conditions, e.g., Milo,50 scCURE is designed to detect cell functional alterations between conditions, i.e., whether and change between pre- and post-treatment conditions in Equation 5. Although scCURE is not designed to directly investigate cell abundance changes, we found that the abundance of cells with specific functional characteristics is associated with phenotypes, i.e., response and non-response. We envisage that the combination of scCURE and existing methods can provide a profile of the problem of interest with more biologically and clinically meaningful details.
Limitations of the study
Upon application of scCURE on melanoma and breast cancer immunotherapy scRNA-seq data, preliminary results reveal that the baseline profiles of scCURE-identified CD8+ T and macrophage cells can determine the way in which tumor microenvironment immune cells respond to immunotherapy, e.g., antitumor immunity activation or de-activation. Despite three bulk RNA-seq datasets being leveraged to validate the dynamic molecular change after treatment, there remains a pressing need for larger immunotherapy cohorts and an expanded array of immunotherapy datasets to further elucidate the potential treatment mechanism. Furthermore, the application of scCURE across different tumor types is essential for the construction of a comprehensive predictive model for immunotherapy at the pan-cancer level.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Processed single-cell RNA-seq data in human Melanoma patients | Sade-Feldman et al.5 | GEO: GSE120575 |
| Processed single-cell RNA-seq data in human TNBC patients | Zhang et al.7 | GEO: GSE169246 |
| Anti-PD1-treated breast cancer-I-SPY2 | Wolf et al.51 | GEO: GSE196096 |
| Anti-PD1-treated melanoma-Hugo | Hugo et al.52 | GEO: GSE78220 |
| Anti-PD1-treated advanced melanoma-Riaz | Riaz et al.53 | GEO: GSE91061 |
| Anti-PD1, anti-PD1+anti-CTLA-4-treated melanoma- Gide | Gide et al.54 | ENA: PREJEB23709 |
| Software and algorithms | ||
| scCURE | GitHub | https://doi.org/10.5281/zenodo.8418094 |
| Seurat-4.9.9 | CRAN | https://satijalab.org/seurat/ |
| R-4.1.2 | The R Foundation | https://www.r-project.org/ |
| Batchelor-1.6.3 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/batchelor.html |
| Monocle-2.28.0 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/monocle.html |
| MATLAB R2021a | MathWorks | https://www.mathworks.com |
| NicheNet-v2.0.0 | Browaeys et al.42 | https://github.com/saeyslab/nichenetr |
| GSEA Analysis | Mootha et al.55 Subramanian et al.56 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Survival Analysis | Tang et al.57 | http://gepia2.cancer-pku.cn/#survival |
| pheatmap-v1.0.12 | CRAN | https://cran.r-project.org/web/packages/pheatmap/ |
| VISION-v3.0.1 | Github | https://github.com/YosefLab/VISION |
| ggplot2-3.4.3 | CRAN | https://ggplot2.tidyverse.org |
| circlize-0.4.15 | Gu et al.58 | https://cran.r-project.org/web/packages/circlize/ |
| glmnet-4.1-8 | CRAN | https://cran.r-project.org/web/packages/glmnet/ |
| cancerclass-1.44.0 | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/cancerclass.html |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xin Zou (xzou@fudan.edu.cn)
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All of the sequencing data used in this paper were obtained from publicly available sources, and are listed in the key resources table.
-
•
The scCURE has been deposited at Github and Zenodo (see key resources table). The scCURE original code and full tutorials are available at https://github.com/Hao-Zou-lab/scCURE.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Proof of concept
Let’s denote X and Y were scRNA-seq expression data matrices with genes as rows and cell as columns, from pretreatment group and post-treatment group, respectively. It is reasonable to assume that both X and Y are composed of common basic cell subtypes or biological functions, each of which can be characterized as variable expression patterns of specific genes. Therefore, define a matrix , each column vector represents a gene expression pattern of cell subtype of cell function. Each single cell data can be represented as a combination of those fundamental functions with various weights. Furthermore, we introduce an additional variable to describe the mismatch between two datasets caused by non-biological factors, e.g., random technique noise or batch effect. Although may occasionally be correlated with B, on a large scale, the noise and signal components can be considered independent. In summary, the data X and Y can be described as:
| (Equation 1) |
| (Equation 2) |
where and are weights representing the cell subtype and/or cell function shifting between the pre- and post-treatment conditions, i.e., therapy-induced biological changes. Our aim is to find a group of cells Y, modeled by Gaussian distribution , which is most similar to the given X, modeled by , given fixed B. KL divergence can be used to measure the distances between the two Gaussian distributions,
| (Equation 3) |
Unfortunately, the evaluation of requires integrating over all possible values of X, which is generally intractable. To simplify the calculation, we assume that the function inside the integral (i.e., ) has a maximum, then the integral may be approximated by evaluating the function at its maximum. Therefore, our goal becomes:
| (Equation 4) |
Because is random and supposed to have the same distribution across cells, the term can be considered a constant. As and are linearly related, the probability density functions of them have for any fixed (https://stats.libretexts.org/Bookshelves/Probability_Theory/Probability_Mathematical_Statistics_and_Stochastic_Processes_(Siegrist)/03%3A_Distributions/3.07%3A_Transformations_of_Random_Variables). Therefore, the minimization of Equation 4 can be transformed as:
| (Equation 5) |
where is the determinant of the matrix. Equation 5 indicates that the KL divergence between the two groups of cells, X and Y, can be minimized to zero (as the KL divergence is nonnegative) if and only if there are no biological differences between X and Y.
The above derivation clearly indicates that if cells are characterized by multiple Gaussian distributions, unchanged cells from pre- and post-treatment groups can be identified by finding the mutually nearest Gaussian models in terms of KL divergence.
GMM construction
The Gaussian mixture modeling (GMM) method is applied to profile intra-heterogeneities of cells from each group due to its strong capability in dealing with complicated data structures.59 The GMM contains a series of Gaussian models, each of which represents a cluster of cells. The data from pre- and post-treatment groups are first pooled together and PCA was conducted for dimension reduction purposes. The top 5 PCs with the highest eigenvalues were used. After that GMM models were constructed from the cells from pre- and post-treatment samples separately. First, the K-means clustering method was applied to categorize all cells into K clusters as an initial clustering result (https://smorbieu.gitlab.io/gaussian-mixture-models-k-means-on-steroids/). The mean and variance of each cluster were calculated as the initial parameters of the GMM. The initial weight of each Gaussian model was the relative size of each cluster to the whole dataset, denoted by .
The expectation maximization method iteratively updates the GMM parameters , and until maximum likelihood is obtained.
| (Equation 6) |
| (Equation 7) |
| (Equation 8) |
| (Equation 9) |
where n indicates cells, k specifies index of Gaussian model, and denotes the Gaussian distribution model. After the GMM is constructed, each cell is assigned to a Gaussian model based on the maximum likelihood criterion,
| (Equation 10) |
The KL divergence between two Gaussian models and can be calculated as:
| (Equation 11) |
where tr() is the matrix trace, −1 is the matrix inverse and T is the matrix transpose. We define as paired if and . After paired Gaussian models are identified and cells have been assigned to specific Gaussian models, unchanged and changed cells from pretreatment samples can be discriminated by
| (Equation 12) |
Therapy outcome prediction score definition
The pretreatment cells that are identified as unchanged when compared with post-treatment responders are denoted as R-like cells. NR-like cells were identified in a similar way. We assume that the unchanged cells from pretreatment samples contain predictive information for immunotherapy outcomes. Based on such an assumption, a therapy outcome prediction model can be constructed on a self-defined prediction score, i.e., the ratio of R-like and NR-like cells. Based on a hard threshold of the ratio, patients can be discriminated into responders and non-responders.
Using the score as prediction factor, the number of Gaussian models for pre- and post-treatment samples can be optimized by maximizing the area under the curve (AUC) of the receiver operating characteristic curve (ROC). To avoid overfitting, leave-one-out cross validation is applied. Specifically, the cross validation is performed in the following steps: 1) in each iteration, assign a patient from pre-treatment dataset as test one, and remove all cells belonging to the test patient from post-treatment data if there are any. As scCURE is theoretically insensitive to batch effects, the patients contained in the pre- and post-treatment datasets are not necessarily matched. 2) we use scCURE to compare pre- (including the test patient) and post-treatment (excluding the test patient) datasets, and assign each cell from the pre-treatment dataset as either R-like or NR-like. By doing so, we can count the numbers of R-like and NR-like cells from the test patient. As the whole procedure calculates the ratio of R-like/NR-like without requiring post-treatment data of the patient, it mimics a real clinical application scenario. (3) repeat the two steps for each patient contained in the pre-treatment dataset, and we can calculate the R-like/NR-like ratio for each patient. Each obtained AUC is evaluated on a null hypothesis model obtained by randomly shuffling the response patterns of all individuals 100 times, and a p value is obtained.
In application scenarios, the R-like/NR-like ratio cutoff values may vary across tumor and immune cell types. The authors suggest that for a specific tumor type and immune cell type, leave-one-out cross validation (as described in the last paragraph) should be performed using training data to determine the most appropriate cutoff value.
Simulated datasets
The protocol presented in Haghverdi et al.34 is adopted to generate simulated datasets to benchmark the proposed algorithm against existing methods. The simulated datasets contain two groups that mimic case-control studies, and the datasets contain batch effects and random and biological factors. In the simulated dataset, each group contains 1000 cells characterized by 100 genes. Random noise is equally distributed across all 2000 cells, and batch effect factors are evenly distributed within groups. Both random noise and batch effect factors are simulated using the default parameters of the original protocol. The heterogeneity with the case groups was generated by manipulating the fold changes (FCs = 1.1, 1.2, 1.3, 1.4, 1.5, 2) of randomly selected case group cells and differentially expressed (DE) genes. In the case group, 2 to 4 subclusters were generated to represent cell heterogeneity, and the proportions of subclusters were equal. Among the subclusters in the case group, there was always one subcluster without manipulated gene fold changes, which mimics the unchanged cells in the case group compared to the control group. Each subcluster contained 25 DE genes, and each simulated dataset was generated 50 times.
Evaluation metrics
The ability of the scCURE to reliably differentiate cells with unchanged/dynamic characteristics is compared with that of the MNN method in terms of three metrics, i.e., sensitivity, specificity, positive prediction value (PPV), F1-score and AUC of ROC. These are calculated using the equations shown below:
| (Equation 13) |
| (Equation 14) |
| (Equation 15) |
| (Equation 16) |
| (Equation 17) |
| (Equation 18) |
| (Equation 19) |
True positives (TPs) were defined as the cells correctly identified as changed between groups and false positives (FPs) indicate spectral variables incorrectly identified as changed cells. True negatives (TNs) indicate cells were correctly identified as unchanged between groups and false negatives (FNs) were defined as the spectral variables incorrectly identified as unchanged cells. P is the total number of changed cells, and N is the total number of cells within the dataset. R is the cells correctly identified as changed between groups in the true changed cells.
Quantification and statistical analysis
Reprocessing published datasets
The key resources table elucidates the diverse datasets employed throughout this manuscript. In this study, two scRNA-seq datasets (GSE120575 and GSE169246) were reprocessed for developing scCURE. We applied Seurat36 (4.9.9) to generate SeuratObject containing UMI counts, normalized gene expression, reduction information, cluster identities and cell type annotations. Subsequently, CD8+ T and macrophage cells were divided into changed and unchanged cells by scCURE. Then, changed and unchanged cells of each cell type were reanalyzed by Seurat, respectively.
Comparison of scCURE with alternative uncovering cellular heterogeneity approaches
To better estimate the performance of scCURE, we compare it with CCA and MNN through real data. As the number of neighbors set up in the MNN and CCA may affect the final results, various parameters were tested, i.e., 5, 10, 20, and 50. At the same time, various numbers of Gaussian models K in scCURE were also evaluated. Then, the ratios of R-like/NR-like cells were calculated and observed their distribution in barplot.
Single cell pseudotime analysis
To analyze the trajectory of changed CD8+ T and macrophage cells based on scRNA-seq expression data, we utilized Moncle237 to determine the potential lineage differentiation. The 2000 highly variable genes (HVGs) were selected to reconstruct “trajectory skeleton graph”, indicating the differentiation trajectories. In CD8+ T cells, based on previous knowledge, we selected two pre-treatment start nodes terminal exhausted cluster (CD8-C1-PDCD1) and naive/effector clusters (CD8-C7-FGFBP2 and CD8−CD3-CCR7) to one post-treatment end node CD8-C5-IL7R. Simailarly, M2 macrophage cells to M0 phenotype (from C2-CCL18 to C4-CXCR4) were selected. All functions were performed with default parameters to characterize the data.
Calculation of signature score
Single cell signature scoring was performed using the VISION v3.0.1 R package.60 We used progenitor exhausted CD8 and terminally exhausted CD8 signature5 to calculate progenitor exhausted signature score and terminally exhausted signature score, which improve the accuracy of progenitor/terminal exhausted cell identification.
Functional enrichment analysis
The FindAllMarker function in the Seurat package was used to obtain the differentially expressed genes in clusters utilizing the Wilcoxon rank-sum test. A Bonferroni false discovery rate (FDR) correction less than 0.05 was used as a cutoff for identifying statistically significant DEGs. Then, we used GSEA to perform GO biological process and hallmark enrichment analysis on the differentially expressed genes within each subset that was associated with before or after treatment. Gene sets with a significance level of FDR of <0.05 were considered significant.
Survival analysis
A standard Kaplan-Meier survival analysis was used to analyze the top 30 markers of CD8-C5-IL7R, Macro-C2-CCL18 and Macro-C4-CXCR4 clusters with overall survival in TCGA datasets. The website is here: http://gepia2.cancer-pku.cn/#survival.
CD8+ T cell – Macrophage cell interaction analysis
NicheNet analysis was performed on cells from changed CD8+ T and macrophage cells according to the code deposited in GitHub (https://github.com/saeyslab/nichenetr). We used post-treatment group as reference datasets. The list of prioritized ligands for each “sender” cell type was identified on the basis of the top DEGs that were found in the “receiver” cell type.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (82170045 to J.H.); the Translational Medicine Cross Research Fund of Shanghai Jiao Tong University (ZH2018QNB29 to J.H.); and the Shanghai Pujiang program (16PJ1405100 to J.H.). We thank all members of the Hao-Zou lab for their support and help.
Author contributions
X.Z., J.H., and L.G. conceived the study and the experimental setup, performed and analyzed the experiments, wrote the manuscript with input from all authors, and are responsible for the overall content of this manuscript; M.W. and Y.L. performed the experiments and the data analysis and wrote the manuscript; X.Z., Y.L., J.Z., Y.S., and X.S. performed the data analysis of scRNA-seq; and J.X., H.H.Y.T., and Y.J. assisted with the review and editing of the draft.
Declaration of interests
The authors declare that they have no competing interests.
Published: November 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100643.
Contributor Information
Xin Zou, Email: xzou@fudan.edu.cn.
Lv Gui, Email: 18930815666@189.cn.
Jie Hao, Email: jhao@fudan.edu.cn.
Supplemental information
References
- 1.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Littman D.R. Releasing the Brakes on Cancer Immunotherapy. Cell. 2015;162:1186–1190. doi: 10.1016/j.cell.2015.08.038. [DOI] [PubMed] [Google Scholar]
- 3.Reck M., Rodríguez-Abreu D., Robinson A.G., Hui R., Csőszi T., Fülöp A., Gottfried M., Peled N., Tafreshi A., Cuffe S., et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019;37:537–546. doi: 10.1200/JCO.18.00149. [DOI] [PubMed] [Google Scholar]
- 4.Vokes E.E., Ready N., Felip E., Horn L., Burgio M.A., Antonia S.J., Arén Frontera O., Gettinger S., Holgado E., Spigel D., et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann. Oncol. 2018;29:959–965. doi: 10.1093/annonc/mdy041. [DOI] [PubMed] [Google Scholar]
- 5.Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M., et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiong D., Wang Y., You M. A gene expression signature of TREM2(hi) macrophages and gammadelta T cells predicts immunotherapy response. Nat. Commun. 2020;11:5084. doi: 10.1038/s41467-020-18546-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y., Chen H., Mo H., Hu X., Gao R., Zhao Y., Liu B., Niu L., Sun X., Yu X., et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell. 2021;39:1578–1593.e8. doi: 10.1016/j.ccell.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Zheng K., Gao L., Hao J., Zou X., Hu X. An immunotherapy response prediction model derived from proliferative CD4(+) T cells and antigen-presenting monocytes in ccRCC. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.972227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B., Hu X., Feng K., Gao R., Xue Z., Zhang S., Zhang Y., Corse E., Hu Y., Han W., Zhang Z. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer. 2022;3:108–121. doi: 10.1038/s43018-021-00292-8. [DOI] [PubMed] [Google Scholar]
- 10.Chuah S., Lee J., Song Y., Kim H.D., Wasser M., Kaya N.A., Bang K., Lee Y.J., Jeon S.H., Suthen S., et al. Uncoupling immune trajectories of response and adverse events from anti-PD-1 immunotherapy in hepatocellular carcinoma. J. Hepatol. 2022;77:683–694. doi: 10.1016/j.jhep.2022.03.039. [DOI] [PubMed] [Google Scholar]
- 11.Abbas H.A., Hao D., Tomczak K., Barrodia P., Im J.S., Reville P.K., Alaniz Z., Wang W., Wang R., Wang F., et al. Single cell T cell landscape and T cell receptor repertoire profiling of AML in context of PD-1 blockade therapy. Nat. Commun. 2021;12:6071. doi: 10.1038/s41467-021-26282-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J., Wu C., Hu H., Qin G., Wu X., Bai F., Zhang J., Cai Y., Huang Y., Wang C., et al. Remodeling of the immune and stromal cell compartment by PD-1 blockade in mismatch repair-deficient colorectal cancer. Cancer Cell. 2023;41:1152–1169.e7. doi: 10.1016/j.ccell.2023.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Obradovic A., Graves D., Korrer M., Wang Y., Roy S., Naveed A., Xu Y., Luginbuhl A., Curry J., Gibson M., et al. Immunostimulatory Cancer-Associated Fibroblast Subpopulations Can Predict Immunotherapy Response in Head and Neck Cancer. Clin. Cancer Res. 2022;28:2094–2109. doi: 10.1158/1078-0432.CCR-21-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z., Wang Z.X., Chen Y.X., Wu H.X., Yin L., Zhao Q., Luo H.Y., Zeng Z.L., Qiu M.Z., Xu R.H. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 2022;14:45. doi: 10.1186/s13073-022-01050-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Žurauskienė J., Yau C. pcaReduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinf. 2016;17:140. doi: 10.1186/s12859-016-0984-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L., Liu J., Lu Q., Riggs A.D., Wu X. SAIC: an iterative clustering approach for analysis of single cell RNA-seq data. BMC Genom. 2017;18:689. doi: 10.1186/s12864-017-4019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiselev V.Y., Kirschner K., Schaub M.T., Andrews T., Yiu A., Chandra T., Natarajan K.N., Reik W., Barahona M., Green A.R., Hemberg M. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods. 2017;14:483–486. doi: 10.1038/nmeth.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marco E., Karp R.L., Guo G., Robson P., Hart A.H., Trippa L., Yuan G.C. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc. Natl. Acad. Sci. USA. 2014;111:E5643–E5650. doi: 10.1073/pnas.1408993111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H., Lee C.A.A., Li Z., Garbe J.R., Eide C.R., Petegrosso R., Kuang R., Tolar J. A multitask clustering approach for single-cell RNA-seq analysis in Recessive Dystrophic Epidermolysis Bullosa. PLoS Comput. Biol. 2018;14 doi: 10.1371/journal.pcbi.1006053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeisel A., Muñoz-Manchado A.B., Codeluppi S., Lönnerberg P., La Manno G., Juréus A., Marques S., Munguba H., He L., Betsholtz C., et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 21.duVerle D.A., Yotsukura S., Nomura S., Aburatani H., Tsuda K. CellTree: an R/bioconductor package to infer the hierarchical structure of cell populations from single-cell RNA-seq data. BMC Bioinf. 2016;17:363. doi: 10.1186/s12859-016-1175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin P., Troup M., Ho J.W.K. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 2017;18:59. doi: 10.1186/s13059-017-1188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J.M., Fan J., Fan H.C., Rosenfeld D., Tse D.N. An interpretable framework for clustering single-cell RNA-Seq datasets. BMC Bioinf. 2018;19:93. doi: 10.1186/s12859-018-2092-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsson A., Venkatasubramanian M., Chaudhri V.K., Aronow B.J., Salomonis N., Singh H., Grimes H.L. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016;537:698–702. doi: 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Courtois E.T., Sengupta D., Tan Y., Chen K.H., Goh J.J.L., Kong S.L., Chua C., Hon L.K., Tan W.S., et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017;49:708–718. doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 26.Ntranos V., Kamath G.M., Zhang J.M., Pachter L., Tse D.N. Fast and accurate single-cell RNA-seq analysis by clustering of transcript-compatibility counts. Genome Biol. 2016;17:112. doi: 10.1186/s13059-016-0970-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang B., Zhu J., Pierson E., Ramazzotti D., Batzoglou S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods. 2017;14:414–416. doi: 10.1038/nmeth.4207. [DOI] [PubMed] [Google Scholar]
- 28.Xu C., Su Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics. 2015;31:1974–1980. doi: 10.1093/bioinformatics/btv088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prabhakaran S., Azizi E., Carr A., Pe'er D. Dirichlet Process Mixture Model for Correcting Technical Variation in Single-Cell Gene Expression Data. JMLR Workshop Conf. Proc. 2016;48:1070–1079. [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z., Jin S., Liu G., Zhang X., Wang N., Wu D., Hu Y., Zhang C., Jiang Q., Xu L., Wang Y. DTWscore: differential expression and cell clustering analysis for time-series single-cell RNA-seq data. BMC Bioinf. 2017;18:270. doi: 10.1186/s12859-017-1647-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji Z., Ji H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016;44:e117. doi: 10.1093/nar/gkw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou J., Deng F., Wang M., Zhang Z., Liu Z., Zhang X., Hua R., Chen K., Zou X., Hao J. scCODE: an R package for data-specific differentially expressed gene detection on single-cell RNA-sequencing data. Brief. Bioinform. 2022;23 doi: 10.1093/bib/bbac180. [DOI] [PubMed] [Google Scholar]
- 34.Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bi K., He M.X., Bakouny Z., Kanodia A., Napolitano S., Wu J., Grimaldi G., Braun D.A., Cuoco M.S., Mayorga A., et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell. 2021;39:649–661.e5. doi: 10.1016/j.ccell.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller B.C., Sen D.R., Al Abosy R., Bi K., Virkud Y.V., LaFleur M.W., Yates K.B., Lako A., Felt K., Naik G.S., et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019;20:326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Durante M.A., Rodriguez D.A., Kurtenbach S., Kuznetsov J.N., Sanchez M.I., Decatur C.L., Snyder H., Feun L.G., Livingstone A.S., Harbour J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020;11:496. doi: 10.1038/s41467-019-14256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jan B., Kosztyla D., von Törne C., Stenzinger A., Darb-Esfahani S., Dietel M., Denkert C. cancerclass: An R Package for Development and Validation of Diagnostic Tests from High-Dimensional Molecular Data. J. Stat. Software. 2014;59:1–19. doi: 10.18637/jss.v059.i01. [DOI] [Google Scholar]
- 42.Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 43.La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 45.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao J., Astle W., De Iorio M., Ebbels T.M.D. BATMAN--an R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a Bayesian model. Bioinformatics. 2012;28:2088–2090. doi: 10.1093/bioinformatics/bts308. [DOI] [PubMed] [Google Scholar]
- 47.Racle J., de Jonge K., Baumgaertner P., Speiser D.E., Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6 doi: 10.7554/eLife.26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu X., Tian X., Ji L., Zhang X., Cao Y., Shen C., Hu Y., Wong J.W.H., Fang J.Y., Hong J., Chen H. A tumor microenvironment-specific gene expression signature predicts chemotherapy resistance in colorectal cancer patients. npj Precis. Oncol. 2021;5:7. doi: 10.1038/s41698-021-00142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng H., Liu H., Ge Y., Wang X. Integrated single-cell and bulk RNA sequencing analysis identifies a cancer associated fibroblast-related signature for predicting prognosis and therapeutic responses in colorectal cancer. Cancer Cell Int. 2021;21:552. doi: 10.1186/s12935-021-02252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dann E., Henderson N.C., Teichmann S.A., Morgan M.D., Marioni J.C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 2022;40:245–253. doi: 10.1038/s41587-021-01033-z. [DOI] [PubMed] [Google Scholar]
- 51.Wolf D.M., Yau C., Wulfkuhle J., Brown-Swigart L., Gallagher R.I., Lee P.R.E., Zhu Z., Magbanua M.J., Sayaman R., O'Grady N., et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: Predictive biomarkers across 10 cancer therapies. Cancer Cell. 2022;40:609–623.e6. doi: 10.1016/j.ccell.2022.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., Hodi F.S., Martín-Algarra S., Mandal R., Sharfman W.H., et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gide T.N., Quek C., Menzies A.M., Tasker A.T., Shang P., Holst J., Madore J., Lim S.Y., Velickovic R., Wongchenko M., et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell. 2019;35:238–255.e6. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 56.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang Z., Kang B., Li C., Chen T., Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 59.Jančovič P., Zou X., Köküer M. Speech enhancement based on Sparse Code Shrinkage employing multiple speech models. Speech Commun. 2012;54:108–118. doi: 10.1016/j.specom.2011.07.005. [DOI] [Google Scholar]
- 60.DeTomaso D., Jones M.G., Subramaniam M., Ashuach T., Ye C.J., Yosef N. Functional interpretation of single cell similarity maps. Nat. Commun. 2019;10:4376. doi: 10.1038/s41467-019-12235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All of the sequencing data used in this paper were obtained from publicly available sources, and are listed in the key resources table.
-
•
The scCURE has been deposited at Github and Zenodo (see key resources table). The scCURE original code and full tutorials are available at https://github.com/Hao-Zou-lab/scCURE.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.