

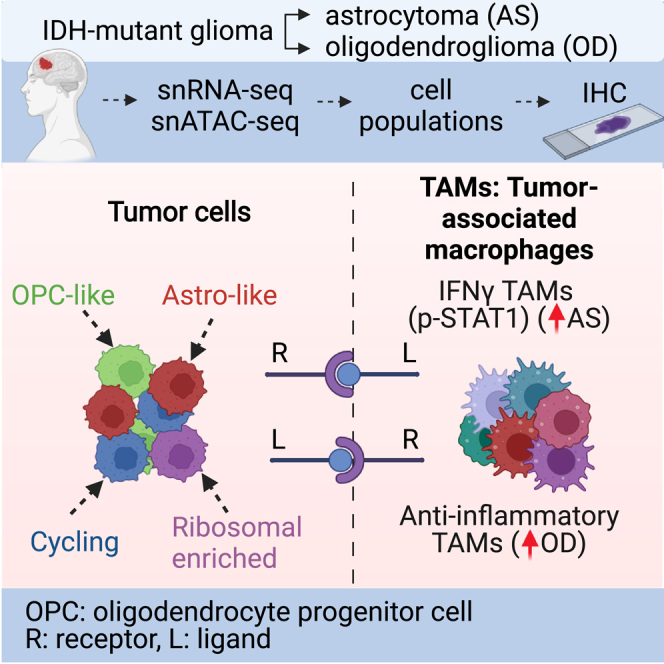
Summary
The isocitrate dehydrogenase (IDH) gene is recurrently mutated in adult diffuse gliomas. IDH-mutant gliomas are categorized into oligodendrogliomas and astrocytomas, each with unique pathological features. Here, we use single-nucleus RNA and ATAC sequencing to compare the molecular heterogeneity of these glioma subtypes. In addition to astrocyte-like, oligodendrocyte progenitor-like, and cycling tumor subpopulations, a tumor population enriched for ribosomal genes and translation elongation factors is primarily present in oligodendrogliomas. Longitudinal analysis of astrocytomas indicates that the proportion of tumor subpopulations remains stable in recurrent tumors. Analysis of tumor-associated microglia/macrophages (TAMs) reveals significant differences between oligodendrogliomas, with astrocytomas harboring inflammatory TAMs expressing phosphorylated STAT1, as confirmed by immunohistochemistry. Furthermore, inferred receptor-ligand interactions between tumor subpopulations and TAMs may contribute to TAM state diversity. Overall, our study sheds light on distinct tumor populations, TAM heterogeneity, TAM-tumor interactions in IDH-mutant glioma subtypes, and the relative stability of tumor subpopulations in recurrent astrocytomas.
Keywords: IDH mutation, glioma, astrocytoma, oligodendroglioma, recurrent glioma, snRNA-seq, snATAC-seq, microglia, bone-marrow-derived macrophages, tumor microenvironment
Graphical abstract
Highlights
-
•
A tumor population shows increased expression of ribosomal genes and elongation factors
-
•
Longitudinal analysis of astrocytomas reveals stable tumor populations during recurrence
-
•
Receptor-ligand analysis infers interactions between tumor and TAM subpopulations
-
•
Compositions of TAM transcriptional states differ in oligodendrogliomas and astrocytomas
Blanco-Carmona et al. apply single-nucleus RNA-seq and ATAC-seq to profile the cellular heterogeneity in adult IDH-mutant gliomas. They identify a range of transcriptional states within tumor cells and tumor-associated macrophage subpopulations in these gliomas. Comparing the relative abundance of these populations sheds light on the distinctions between IDH-mutant glioma subtypes.
Introduction
Diffuse gliomas exhibit recurrent mutations in the isocitrate dehydrogenase (IDH) gene.1 IDH-mutant gliomas are classified into two subtypes: oligodendrogliomas featuring chromosome arm 1p/19q co-deletion and astrocytomas characterized by euploid 1p/19q.2 Intratumoral heterogeneity is a feature of IDH-mutant gliomas,3,4 showcasing a hierarchy of cellular phenotypes wherein a neural stem cell-like population gives rise to tumor subpopulations resembling expression profiles of astrocytes and oligodendrocytes. Nevertheless, a comprehensive comparative analysis of transcriptional and epigenomic heterogeneity in oligodendrogliomas versus astrocytomas remains elusive. Furthermore, IDH-mutant gliomas manifest distinct activation states of tumor-associated microglia/macrophages (TAMs),5,6 although it remains uncertain whether TAM composition differs between oligodendrogliomas and astrocytomas, how tumor subpopulations interact with TAMs, and to what extent tumor grade and recurrence influence TAM diversity. Therefore, analyses of tumor heterogeneity and tumor-host interactions are important for our understanding of IDH-mutant gliomas and their evolution.
Here, we performed high-throughput single-nucleus RNA and ATAC sequencing (snRNA- and snATAC-seq) on primary IDH-mutant gliomas and snRNA-seq on a cohort of primary and recurrent astrocytoma pairs. This effort generated a comprehensive resource for resolving tumor diversity and TAM states. Our findings reaffirm previously described differentiation hierarchies and unveil a distinct subgroup of non-cycling, ribosomal-enriched stem-like tumor cells characterized by unique epigenetic and transcriptional signatures. We identify significant transcriptional differences in TAM states between oligodendrogliomas and astrocytomas. By mapping receptor-ligand interactions between tumor and TAM subpopulations, we highlight a notable interaction between inflammatory TAMs and astrocytic tumor subpopulations in astrocytomas, subsequently validated through immunohistochemistry in independent cohorts. Results from the clinical trial with vorasidenib, the brain-penetrant mutant IDH inhibitor, has shown promising results in patients with grade 2 IDH-mutant gliomas, significantly improving progression-free survival and delaying time to next intervention.7 Moving forward, molecular studies focused on higher grade and recurrent IDH-mutant gliomas may uncover molecular alterations suitable for targeted therapeutic strategies.
Results
Tumor metasignatures in IDH-mutant gliomas reveal the presence of a ribosomal-enriched population
To investigate intratumor heterogeneity in adult IDH-mutant gliomas, we generated snRNA-seq from 14 snap-frozen primary tissues, comprising 8 oligodendrogliomas and 6 astrocytomas (Figure 1A; Tables S1A–S1D), encompassing 76,680 and 72,385 nuclei, respectively. We used a multistep approach to delineate cell types based on their genetic and transcriptional states. To distinguish nuclei as tumor or tumor microenvironment (TME)-derived, we merged, normalized, and clustered the datasets, assigning initial cell-type classifications to each cluster using literature-derived marker signatures.4,8,9,10 This approach identified major cell types, including microglia, oligodendrocytes, neurons, astrocytes, endothelial cells, pericytes, and T cells (Figures S1A and S1B). Using microglia and oligodendrocytes as non-malignant references, we inferred genome-wide copy-number alterations (CNAs) from snRNA-seq data and compared CNA profiles to distinguish malignant and non-malignant cells. In oligodendrogliomas, we detected the broad-arm 1p/19q co-deletion, whereas astrocytomas exhibited CNA heterogeneity, with glioma-specific amplifications (e.g., chromosome 7) and deletions (e.g., chromosome 10) observed in several patients but not all (Figures S1C and S1D). Following data integration with Harmony,11 we labeled tumor cells based on enrichment of previously described6 astrocyte (astro)-like, oligodendrocyte progenitor cell (OPC)-like (oligo), and cycling programs (Figures 1B and 1C).
Figure 1.

snRNA-seq identifies gene expression programs driving cell states in IDH-mutant gliomas
(A) Clinical and molecular characteristics of the IDH-mutant glioma cohort for single-nuclei sequencing.
(B and C) UMAP representation and initial cluster assignments for integrated snRNA-seq samples of oligodendrogliomas (n = 8) (B) and astrocytomas (n = 6) (C). Nuclei labeled as “excluded” in (B) are omitted from downstream analyses due to technical reasons (tumor cells clustering with microglia and originating from a single patient). UMAP1, x axis; UMAP2, y axis. Colors indicate cell-type assignments.
(D and E) Pearson’s correlation scores for individual NMF programs in oligodendrogliomas (n = 8) (D) and astrocytomas (n = 6) (E). Metaprograms were identified by hierarchical clustering of individual NMF programs.
(F) Dot plot displaying five marker genes for each tumor population.
(G and H) UMAP embedding of oligodendrogliomas (G) and astrocytomas (H), colored by NMF metaprograms, gradient cells, and TME (dark gray).
(I) Bar plot showing tumor population proportions across individual samples, grouped by subtype and grade. Bars are arranged by descending RE proportion.
(J) Representative IHC staining for H&E and two RE population markers, EEF2 and EEF1A1, for oligodendroglioma and astrocytoma samples, separated by grade. Oligodendroglioma (OD) n = 22 (11 grade 2, 11 grade 3); astrocytoma (AS) n = 15 (10 grade 2, 5 grade 3). Scale bars represent 50 μm.
(K) Spatial distribution of EEF2 and EEF1A1 (40×). Scale bars represent 50 μm.
(L) Plots showing semi-quantitative histological scores (0 = 0%, 1 = 0%–5%, 2 = 6%–29%, 3 = 30%–69%, and 4 >70% positive staining) for EEF2 (top) and EEF1A1 (bottom). Top: a qualitative increase in EEF2 staining in grade 3 compared with grade 2 AS tumors. Bottom: a qualitative increase in EEF1A1 staining in grade 2 OD tumors compared with grade 2 AS tumors. Wilcoxon rank-sum test was performed to test for significance, ∗p < 0.05.
We then characterized tumor cells using unbiased non-negative matrix factorization (NMF) independently for each patient’s tumor cells, revealing shared tumor-associated transcriptional programs (NMF programs). Hierarchical clustering of these programs revealed four major clusters (NMF metaprograms) in oligodendrogliomas (Figure 1D) and three in astrocytomas (Figure 1E), with strong pairwise correlations. Two metaprograms resembled previously described4 astro-like (NRG3, ADGRV1, SPARCL1) and oligo-like (OPCML, DSCAM) programs. We also identified a cycling metaprogram (MKI67, CENPK) and a fourth metaprogram enriched in elongation factors (EEF2, EEF1A1), stemness genes (OLIG1), oncogenes (ETV1), and ribosomal genes (RPL13), which we termed the ribosomal-enriched (RE) program (Figures 1F–1H; Tables S2A and S2B). Enrichment analysis of cells for these metaprograms revealed a gradient nature rather than a clear association with specific clusters. To statistically categorize cells enriched for each metaprogram, we implemented a permutation testing approach to label tumor cells. Specifically, cells enriched (adjusted p ≤ 0.0125) for either OPC-like or astro-like metaprograms received a primary assignment. Subsequently, cells were assigned to the RE metaprogram and then to the cycling metaprogram, replacing previous labels. Finally, tumor cells that were not statistically enriched for any metaprogram were labeled as “gradient” (Figures 1F and 1G). The gradient population shared some characteristics with the four metaprograms but to a lesser extent (Figures S1E and S1F). In the integrated uniform manifold approximation and projection (UMAP), gradient cells showed proximity to the OPC-like population in oligodendrogliomas (Figure 1G) and the astro-like population in astrocytomas (Figure 1H). When isolated and clustered, the gradient population was enriched for both astro-like and OPC-like populations in both tumor types (Figures S1G and S1H), suggesting partial resemblance to both populations without definitive metaprogram assignment.
Cell-type proportion analysis showed a higher OPC-like population in astrocytomas compared with oligodendrogliomas, whereas the RE population was variable and particularly high in one patient (Figure 1I). To ensure the robustness of the RE metaprogram, we performed additional NMF analyses in oligodendrogliomas without ribosomal genes and excluding the patient with high RE (Figures S2A–S2D). This analysis yielded a metaprogram with the same key marker genes and statistically enriched cells, confirming its reliability (Table S3). Furthermore, nuclei assigned to this metaprogram had high unique molecular identifiers (UMIs) and gene counts (Figures S2E and S2F), confirming its high-quality composition.
To validate the RE population, we used SPOTlight12 to deconvolute IDH-mutant glioma cohorts from The Cancer Genome Atlas (TCGA) and the Chinese Glioma Genome Atlas (CGGA) (Figures S3). We detected varying proportions of the RE population in oligodendrogliomas in both cohorts, except for one tumor in the CGGA cohort (Figures S3E and S3G). However, not all astrocytomas harbored the RE population (Figures S3F and S3H). Additionally, we queried IDH-mutant glioma single-cell datasets3,4 for enrichment of the RE population (Figures S4A and S4B). Cells were considered RE if the likelihood of finding an enrichment score higher than the empirical value was less than 5%. We then computed cell activities using decoupleR13 with a custom network consisting of our NMF metaprograms and literature-derived6 astro and oligo programs. This analysis revealed significant RE metaprogram activity in the selected RE population in both oligodendrogliomas (Figure S4A) and astrocytomas (Figure S4B), indicating a clear association. To exclude random enrichment effects, we computed cell activities based on a network containing the RE metaprogram and 50 gene sets with randomly selected genes with similar expression levels to those in the RE metaprogram. The results demonstrated high activity of the RE metaprogram compared with random gene sets (Figure S4A, right, and S4B, right).
To support our findings, we performed immunohistochemistry (IHC) with RE markers (EEF2 and EEF1A1) in a validation cohort of 22 oligodendrogliomas and 15 astrocytomas. The results revealed more positively stained tumor cells in primary oligodendrogliomas than in astrocytomas (Figures 1J–1L). Notably, EEF2 levels increased significantly with tumor grade in astrocytomas, and EEF1A1 levels were higher in grade 2 oligodendrogliomas than in grade-matched astrocytomas (Figures 1J and 1L). Both proteins exhibited spatial heterogeneity, more so in astrocytomas than in oligodendrogliomas (Figures 1K and S5).
To gain deeper insights into tumor subpopulations, we focused on stemness-associated genes4 in IDH-mutant gliomas and computed a diffusion map using these genes and the astro-like and oligo-like programs (Figures S4C and S4D). Ordering cells along diffusion component 2 (DC2) revealed a correlation between stemness-associated gene enrichment and the positive end of DC2, predominantly driven by RE cells, indicating their stem-like properties (Figure 2A). Functional enrichment analysis14 revealed depletion of the p53 signature and enrichment of the proliferation-associated FOXM115 signature in cycling cells but not in the RE population (Figures 2B and 2C), suggesting the presence of non-cycling progenitor-like cells in IDH-mutant gliomas. To contextualize the tumor populations with respect to normal development and glioblastoma, we performed reference mapping with Azimuth16 against two reference datasets: GBmap,17 the single-cell atlas of glioblastoma (Figures S6A and S6B), and a human fetal development atlas (Figures S6C and S6D). In both datasets, the RE population was highly enriched for oligodendrocytes/OPC-like cells (Figure S6), suggesting similarities to an oligo-like program.
Figure 2.

Integrated snRNA-seq and snATAC-seq indicates a stem-like signature in the RE tumor population
(A) Diffusion map visualization of tumor cells in OD and AS tumors, based on publicly available4 astro-like, oligo-like, and stemness program markers. Right: scaled and centered enrichment scores for NMF metaprograms. Cells are ordered along diffusion components 1 (DC_1) and 2 (DC_2); gradient cells were excluded for clarity.
(B) Scaled and centered pathway activity scores (normalized weighted mean from snRNA-seq) for OD and AS tumors.
(C) Scaled and centered regulon activity scores (normalized weighted mean from snRNA-seq) for OD and AS tumors.
(D and E) UMAP embedding of OD (D) and AS (E) snATAC-seq data (OD n = 4, AS n = 5) colored by labels transferred from snRNA-seq datasets. UMAP1, x axis; UMAP2, y axis.
(F) Bar plot showing proportion of cell types in snATAC-seq data.
(G) Pearson correlation of highly variable peaks from snATAC-seq data in OD and AS tumors.
(H and I) Heatmap showing the top significantly enriched transcription factor motifs in OD (H) and AS tumors (I). Scores are scaled and centered for clarity.
To determine whether oligodendrogliomas and astrocytomas exhibited unique chromatin accessibility profiles, we generated snATAC-seq on 4 oligodendrogliomas (total nuclei = 7,817) and 5 astrocytomas (total nuclei = 14,214) (Figures 1A and S7; Tables S1E–S1H, S2C, and S2D). The nuclei used for snATAC-seq were paired with snRNA-seq samples.18 For annotation, we performed label transfer from snRNA-seq to snATAC-seq data, excluding the gradient population that did not fit any NMF metaprogram. We identified NMF-derived tumor and TME populations in oligodendrogliomas (Figure 2D) and astrocytomas (Figure 2E). Cell-type proportions revealed a higher prevalence of OPC-like cells in oligodendrogliomas and an astro-like predominance in astrocytomas (Figure 2F). The RE population was consistently present in the snATAC-seq data across all tumors, suggesting an epigenetically distinct RE population in both tumor types. However, the RE population was transcriptionally more prominent in oligodendrogliomas. The astro-like and OPC-like populations formed separate clusters with negatively correlated chromatin accessibility profiles (Figure 2G). Enrichment analysis using Genomic Regions Enrichment of Annotations Tool (GREAT)19 showed that OPC-like populations in both oligodendrogliomas and astrocytomas were enriched in oligodendrocyte differentiation and glial cell-fate commitment, whereas the astro-like population of oligodendrogliomas was enriched in Notch signaling pathway regulation and blood vessel development (Table S4). Chromatin-accessibility-based correlation analysis indicated an association between RE and OPC-like populations, with a weaker association with the cycling population (Figure 2G). Transcription factor (TF) motif enrichment analysis using chromVAR20 showed significantly enriched motifs in all clusters of both oligodendrogliomas (Figure 2H) and astrocytomas (Figure 2I). Motifs enriched in the RE cluster displayed similarities to OPC-like populations in both tumor types (Figures 2H and 2I). Although E2F1 was not transcriptionally overrepresented in the RE population, its regulon showed slight enrichment in oligodendrogliomas and, to a lesser degree, in astrocytomas (Figure 2C). Coupled with the E2F1 motif accessibility, this regulatory program may be poised for activation in gliomas.
The immune microenvironment of IDH-mutant gliomas
In gliomas, TAMs21 consisting of tissue-resident microglia, bone-marrow-derived (BMD) macrophages, and border-associated macrophages (BAMs) are the most abundant non-tumor cells. To determine differences in TAM composition between oligodendrogliomas and astrocytomas, we integrated, clustered, and annotated TAM subpopulations from both tumor types, using literature-derived marker genes.22,23 Each TAM subpopulation was further annotated as microglia (Mg), BMD macrophages, or BAMs based on marker gene enrichment. We identified ten TAM subpopulations: Mg homeostatic (PTPRC, ITGAM, P2RY12); Mg activated (CX3CR1, GPR34, FOXP2); Mg resident-like (PLCL1, CEBPD, VIM); Mg stressed (HSPA1A, HSPA1B, SORCS2); Mg phagocytic (GPNMB, RGCC, PPARG); Mg inflammatory (CCL4, IL1B, CCL3); Mg inflammatory ICAM1+ (ICAM1, RELB, TNFAIP3); Mg interferon γ (IFNγ; IFIT2, IFIT3, STAT1); BAMs (F13A1, LYVE1, CD36); and BMD anti-inflammatory (CD163, SELENOP, MRC1) (Figures 3A and S8A; Table S2E). Oligodendrogliomas and astrocytomas showed significantly different proportions of TAM subpopulations (Figure 3B). Mg homeostatic TAMs (p = 0.01, Wilcoxon test) were more abundant in astrocytomas, whereas BAM (p = 0.048, Wilcoxon test), BMD anti-inflammatory (p = 0.048, Wilcoxon test), and Mg phagocytic (p = 0.012, Wilcoxon test) TAMs were more abundant in oligodendrogliomas (Figure 3C).
Figure 3.

Integrated TAM states highlight differences between AS and OD tumors
(A) UMAP embedding of integrated microglia from snRNA-seq data of primary OD and AS tumors, colored by assigned TAM subpopulations. UMAP1, x axis; UMAP2, y axis.
(B) Bar plots indicating TAM subpopulation proportions, separated by tumor subtype and grade.
(C) Boxplots showing TAM subpopulation proportions by tumor type. OD, oligodendroglioma; AS, astrocytoma. Wilcoxon rank-sum test was performed to test for significance.
(D) Representative IHC staining for TAM markers IBA1, CD74, and CD163 and double staining for p-STAT1 and IBA1 (60×) in OD and AS tumors. Images are captured at either 40× (scale bar, 50 μm) or 60× original magnification (scale bar, 30 μm)
(E) Plots showing semi-quantitative histological scores (0 = 0%, 1 = 0%–5%, 2 = 6%–29%, 3 = 30%–69%, 4 >70% positive staining) for p-STAT1+IBA1+ TAMs in OD and AS tumors, separated by grade (n = 3 per group).
(F and G) Select statistically significant receptor-ligand interactions between OPC-like and astro-like tumor populations (source) and the TAM subpopulations (target) (F) and TAM subpopulations (source) and OPC-like and astro-like tumor cells (target) (G). Dot size represents significance (adjusted p values); dot color reflects expression magnitude (means of average expression level of the interacting pair of genes).
Using marker genes, we scored each TAM subpopulation as pro- or anti-inflammatory, finding no distinct differences between oligodendrogliomas and astrocytomas (Figures S8B–S8D). We performed IHC with several TAM markers including IBA1 (TAM marker), CD74 (marker of pro-inflammatory M1-like phenotype24), and CD163 (marker of anti-inflammatory M2-like phenotype25) on grade 2 and 3 astrocytomas and oligodendrogliomas (n = 5 per group). We observed diffuse and intense positive immunostaining for IBA1 in astrocytomas, mainly corresponding to activated ramified Mg-like cells, along with a small number of macrophages, independent of tumor grade (Figure 3D). In oligodendroglioma, fewer IBA1+ cells were observed compared with astrocytomas, primarily showing a rounded ameboid-like TAM morphology. Astrocytoma TAMs mainly expressed CD74, with a small number of anti-inflammatory CD163+ TAMs. In oligodendroglioma, CD74+ cells were barely detected, with few immunoreactive CD163+ cells, all exerting a macrophage phenotype. Double IHC staining showed the presence of phospho-STAT1 (p-STAT1) on a subset of IBA1+ TAMs in astrocytomas (n = 6) but not in oligodendrogliomas (n = 6) (Figures 3D and 3E). Double IHC staining with IDH1 R132H and p-STAT1 antibodies indicated that a proportion of p-STAT1+/IBA1− cells were tumor cells (Figure S9A).
We then explored whether TAM diversity might result from interactions of TAMs with tumor cells. Using LIANA,26 we predicted enriched receptor-ligand interactions between tumor populations (astro-like and OPC-like) and TAM subpopulations (Figures 3F and 3G). These interactions included BMP pathway proteins in the OPC-like population and PGF-NRP1/2 interactions between the astro-like population and TAMs in oligodendrogliomas, although these interactions were significant in both tumor types. Other interactions were more ubiquitous, such as SIRPA-CD47, suggesting that tumor cells may block the phagocytic capacity of innate immune cells.27 WNT5A-FZD3 and WNT5A-PTPRK interactions were common across TAM subpopulations, and the astro-like population of astrocytomas exhibited stronger CSF1-CSF1R interactions with TAMs (Figure 3F), although this was less pronounced in oligodendrogliomas. Receptor-ligand interactions from TAMs to tumor populations included DLL1 in astrocytomas and tumor necrosis factor (TNF) in oligodendrogliomas (Figure 3G).
We validated two receptor-ligand pairs, CSF1-CSF1R and PGF-NRP1, via IHC on two formalin-fixed, paraffin-embedded grade 2 astrocytomas with >60% tumor content. We qualitatively confirmed CSF1 and CSF1R (in serial sections) (Figure S9A) and PGF and NRP1 (in separate sections) (Figure S9B) in primary tissues. CSF1 and CSF1R showed a wider staining pattern, whereas PGF and NRP1 staining patterns were scattered. CSF1/CSF1R regulates macrophage development and survival,28,29 while the PGF/NRP1 signaling axis is a potential therapeutic target in pediatric medulloblastoma30 and intrahepatic cholangiocarcinoma.31 Taken together, these results highlight the heterogeneity of receptor-ligand pairs between TAMs and tumor cells, suggesting potential pathways favored by oligodendrogliomas or astrocytomas. Further studies are needed to elucidate their functional consequences.
IDH-mutant gliomas have low baseline infiltration by T cells.32,33 We also observed a small number of T cells in our cohort (86 in oligodendrogliomas and 171 in astrocytomas). TOX, a critical TF for T cell exhaustion,34,35 was expressed in both tumor types in our cohort (Figure S8E), although this remains an observation that needs to be confirmed in larger IDH-mutant glioma datasets.
Grade- and recurrence-related changes in IDH-mutant gliomas
To assess grade- and recurrence-related differences in tumor and TAM subpopulations, we conducted snRNA-seq on six patients with paired primary and recurrent astrocytomas. This cohort included two untreated patients and four treated with radiotherapy (RT), temozolomide (TMZ), or both (12 tumors in total) (Figure 4A; Tables S1K and S1L). Integrating and clustering of these paired datasets, totaling 48,723 nuclei, revealed all major cell types, including the gradient and RE populations (Figures 4B, S1I, S2D, and S2G; Table S2F). Proportions of astro-like and OPC-like tumor populations showed no significant changes at recurrence (Figures 4C and S10B). However, we observed a grade-dependent increase in cycling and gradient populations along with a decrease in OPC-like and astro-like populations (Figure 4C). Using EEF2 and EEF1A1 as RE markers, we performed IHC in cohorts of paired primary and recurrent oligodendrogliomas (6 patients, 12 tumors) and paired primary and recurrent astrocytomas (6 patients, 12 tumors; Figure 4D). Recurrent astrocytomas exhibited higher EEF2 immunostaining and an increase in EEF1A1+ cells compared with primary astrocytomas (Figure 4E). In oligodendrogliomas, EEF2+ and EEF1A1+ cells were highly expressed in both primary and recurrent tumors (Figure 4E).
Figure 4.

snRNA-seq of paired primary and recurrent AS cohorts indicates shifts in tumor populations associated with grade
(A) Clinical and molecular characteristics of 6 paired primary and recurrent IDH-mutant AS cohorts for snRNA-seq.
(B) UMAP embedding of integrated snRNA-seq data from paired AS tumors, colored by assigned cell types. UMAP1, x axis; UMAP2, y axis.
(C) Bar plots showing tumor population proportions, separated by pairs, relapse status, and grade.
(D) Representative IHC staining for RE markers EEF2 and EEF1A1 in paired primary (grade 2) and relapse (grade 3) AS tumors. Images are captured at 40x original magnification (scale bar, 50 μm)
(E) Heatmap showing semi-quantitative EEF1A1 histological scores (0 = 0%, 1 = 0%–5%, 2 = 6%–29%, 3 = 30%–69%, 4 >70% positive staining) for 6 pairs of primary and recurrent OD and AS tumors. Wilcoxon signed-rank test for paired samples was performed to test for significance, ∗p < 0.05.
(F) UMAP embedding of integrated microglia population of paired primary and recurrent AS cohorts, colored by assigned TAM subpopulations. UMAP1, x axis; UMAP2, y axis.
(G) Bar plots showing TAM subpopulation proportions separated by pairs, relapse status, and grade.
(H) Boxplots showing TAM subpopulation proportions separated by relapse status. P, primary; R, relapse.
Pooling Mg cells from paired astrocytomas revealed nine distinct TAM subpopulations: Mg homeostatic, Mg activated, Mg resident-like, Mg inflammatory ICAM1+, Mg inflammatory, Mg hypoxic, Mg phagocytic, Mg IFNγ, and BMD anti-inflammatory macrophages (Figure 4F; Table S2G). Similar to primary astrocytoma snRNA-seq data, BAM subpopulations were not identified in paired astrocytomas. We excluded the primary tumor of pair 2 and the recurrent tumor of pair 5 from Figure 4H due to their low Mg numbers. Cell-type proportion analysis showed a trend toward increased proportions of Mg hypoxic TAMs, BMD anti-inflammatory macrophages, and Mg IFNγ TAMs at recurrence and decreased Mg homeostatic TAMs at recurrence (Figures 4G and 4H). Furthermore, treated relapsed astrocytoma exhibited an increase in the proportion of BMD anti-inflammatory macrophages (pairs 3, 4, and 6), while untreated pairs (pairs 1 and 2) showed a reduction or a slight increase (Figures 4G and 4H). TAM proportion changes may also be associated with tumor grade more than relapse status (Figure S10A). These findings highlight complex interactions between tumor subtype, grade, and TAM states in IDH-mutant gliomas.
Discussion
Recent studies have expanded our understanding of diffuse glioma heterogeneity. However, a comprehensive comparison between oligodendrogliomas and astrocytomas remains incomplete. In this study, we aimed to address these gaps by generating snRNA-seq/ATAC-seq datasets from primary IDH-mutant gliomas (oligodendroglioma and astrocytoma) and snRNA-seq data from paired primary and recurrent astrocytomas. Additionally, we integrated our genomics data with IHC analysis in separate cohorts of primary and recurrent IDH-mutant gliomas for comprehensive tumor and TAM composition comparisons.
We identified a distinct tumor cell population in IDH-mutant gliomas, referred to as RE. This population exhibited stem cell characteristics, lacked proliferation-associated genes, and showed high expression of ribosomal genes and elongation factors. We confirmed the presence of this population by verifying the expression of marker proteins EEF1A1 and EEF2 in separate tissue cohorts of IDH-mutant gliomas. While the RE population constituted a relatively low fraction of cell types in the snRNA-seq data, IHC indicated a broader distribution of EEF1A1 and EEF2. This discrepancy in distribution may arise from the limited number of cells expressing the complete RE metaprogram or from associations with spatial features within tumors. Ribosome composition has been linked to spatial heterogeneity in glioblastoma,36 and the role of ribosome biogenesis in IDH-mutant gliomas warrants further exploration.
Our results reveal an enriched MYC/MYCN regulon within the RE population, suggesting increased biosynthetic properties due to the known targeting of EEF1A1 by MYC.37 The combined analysis of single-nuclei and IHC data consistently demonstrated a higher RE prevalence in oligodendrogliomas compared with astrocytomas. Deconvolution of bulk RNA-seq datasets corroborated this finding, suggesting a widespread expression of the RE population in nearly all oligodendrogliomas. Targeting this population with differentiation-promoting therapies may hold promise,38,39 as biosynthetic capacity diminishes during tumor cell differentiation.40
Understanding longitudinal changes in IDH-mutant glioma progression is necessary to identify drivers of progression. Spearheaded by the GLASS (Glioma Longitudinal Analysis) consortium, major efforts are underway to capture and analyze glioma dynamics.41,42 In our study, we investigated longitudinal changes in paired primary and recurrent astrocytomas to gain insights into glioma progression. Our findings indicated that transcriptional subpopulations identified in primary tumors persist in both treated (RT and/or TMZ) and untreated recurrent tumors. Importantly, tumor grade, rather than recurrence or treatment, may influence tumor and TAM subpopulations. This aligns with recent studies in gliomas where cell compositions remained relatively stable during recurrence.43 These persistent cell states may represent potential therapeutic targets. For instance, combining master regulator analysis44 with functional studies on transcriptional states could yield insights into key mechanisms maintaining these cellular identities. Additionally, uncovering the bidirectional interactions between tumor populations and the TME is critical for understanding glioma biology and developing effective therapies. While gliomas harbor tumor cells with expression signatures resembling astros and OPCs, their functions remain poorly understood. Here, we characterized receptor-ligand interactions between tumor and TAM subpopulations, and our findings suggest that the astro-like tumor subpopulation may orchestrate a pro-inflammatory microenvironment in astrocytomas driven by IFNγ signaling, which induces STAT1 phosphorylation. These distinctions highlight the role of IFNγ signaling in astrocytomas and raise the possibility that targeting tumor transcriptional states may reshape the TME landscape.
While TAM composition has been studied in IDH-mutant gliomas,5,45 a direct comparison between TAMs in oligodendrogliomas and astrocytomas is lacking. Our results, in line with previous studies,45 show a higher proportion of Mg-derived TAMs in IDH-mutant gliomas compared with BMD-derived TAMs. However, we also observed significant differences in TAM transcriptional states between oligodendrogliomas and astrocytomas. Our combined snRNA-seq and IHC data indicate that astrocytomas contain inflammatory TAMs expressing p-STAT1, a key regulator of TAM inflammatory response and neuronal damage.46,47 Considering the distinct molecular alterations in oligodendrogliomas and astrocytomas, it remains to be determined whether any tumor-specific alterations contribute to myeloid diversity. Receptor-ligand analysis inferred potential interactions between tumor and TAM subpopulations, including a notable interaction between PGF (a VEGF family member) and NRP1. PGF is involved in immune modulation and cancer pathogenesis48 and supports medulloblastoma growth through increased PGF signaling via Nrp1.30 While we confirmed the presence of both proteins in primary tissues through IHC, further studies are needed to characterize and prioritize these candidate interaction pairs.
In summary, our study provides insights into heterogeneity of IDH-mutant gliomas by conducting a comprehensive comparison of oligodendrogliomas and astrocytomas. Our results shed light on distinct tumor populations, heterogeneity of TAM states, TAM-tumor interactions, and the relative stability of tumor and TAM transcriptional states in recurrent astrocytomas. We also emphasize the importance of categorizing IDH-mutant gliomas by subtype in both tumor and TME studies. These findings pave the way for further research to enhance our understanding of the complexity of diffuse gliomas.
Limitations of the study
We acknowledge several limitations of our study. First, it will be important to validate the roles of different tumor subpopulations in IDH-mutant glioma pathology. Second, although our study identified potential receptor-ligand pairs that may influence the diversity of myeloid states within these tumors, it remains to be determined whether and to what extent these inferred pairs interact in situ and contribute to the TAM landscape in gliomas. We validated several key findings using IHC on separate cohorts of primary and paired glioma tissues. However, addressing both of these aspects from a translational perspective will necessitate functional studies. Unfortunately, the scarcity of available bona fide preclinical models for IDH-mutant gliomas, particularly tumorspheres derived from untreated primary IDH-mutant gliomas, presents a challenge. Developing such models will facilitate the translational application of genomic studies and enhance our understanding of IDH-mutant gliomas.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-CD74 (clone LN2) | BioLegend | Cat# 326802; RRID: AB_893401 |
| Mouse monoclonal anti-CD163 (clone 10D6) | ThermoFisher Scientific | Cat# MA5-11458; RRID: AB_10982556 |
| Mouse monoclonal anti-STAT1 (pY701) (clone 14/P-STAT1) | BD Biosciences | Cat# 612132; RRID: AB_399503 |
| Rabbit monoclonal anti-EEF2 (EP880Y) | Abcam | Cat# ab75748; RRID: AB_1310165 |
| Rabbit monoclonal anti-EEF1A1 (EPR9470) | Abcam | Cat# ab140632; RRID: AB_2687995 |
| Mouse monoclonal anti-CSF1 (clone 2D10) | Sigma-Aldrich | Cat# MABF191 |
| Rabbit polyclonal anti-CSF1R | Proteintech | Cat# 25949-1-AP; RRID: AB_2880306 |
| Rat monoclonal anti- PIGF (clone #358905) | R&D | Cat# MAB2642; RRID:AB_10718412 |
| Mouse mouse monoclonal NRP1 (clone 2H3F6) | Proteintech | Cat# 60067-1-Ig; RRID:AB_2150840 |
| Biological samples | ||
| Adult brain tumor tissue (fresh frozen) | Division of Experimental Neurosurgery, Department of Neurosurgery, University Hospital Heidelberg; Acıbadem Mehmet Ali Aydınlar University, School of Medicine, Department of Neurosurgery | N/A |
| Adult brain tumor tissue (FFPE) | Department of Pathology, Spedali Civili of Brescia; Department of Neuropathology, Heidelberg University Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Strepatidine HRP | Vector | Cat# SA-5005 |
| ImmPress HRP Universal Antibody (Horse Anti-Mouse/Rabbit IgG) Polymer detection Kit, Peroxidase | Vector | Cat# MP7500 |
| Dako REAL™ Detection System, Alkaline Phosphatase/RED, rabbit/mouse, | Biocompare | Cat# K5005 |
| MACH 4 Universal AP Polymer Kit | Biocare Medical | Cat# M4U536 |
| Dako EnVision+ System-HRP Labeled Polymer anti-mouse or anti rabbit | Dako | Cat# K4001 |
| Novolink™ Polymer Detection System (Novocastra™) | Leica Biosystems | Cat# RE7280-K |
| Critical commercial assays | ||
| Chromium system using the Single Cell 3′ Reagent Kit v3 or v3.1 | 10x Genomics | Cat# 1000121 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10x Genomics | Cat# 1000175 |
| Deposited data | ||
| Processed snRNA-seq and snATAC-seq data | This paper | GEO: GSE205771 |
| TCGA LGG dataset | Genomics Data Common | https://portal.gdc.cancer.gov/projects/TCGA-LGG |
| Chinese glioma genome atlas (CGGA) | CGGA | http://www.cgga.org.cn/download.jsp#mRNAseq_693 |
| Software and algorithms | ||
| Cellranger version 3.1 | Cellranger v3.1 10x Genomics https://support.10xgenomics.com/ | Cellranger v3.1 10x Genomics https://support.10xgenomics.com/ |
| R (version 3.6.1–4.2.0) | https://www.r-project.org/ | https://www.r-project.org/ |
| Seurat (version 3.1.0–4.3.0) | Hao et al.16; Stuart et al.49 | https://satijalab.org/seurat/ |
| scrublet version 0.2.1 | Wolock et al.50 | https://github.com/swolock/scrublet |
| inferCNV version 1.5.0 | Tickle et al.51 | https://github.com/broadinstitute/inferCNV |
| Signac version 1.0 | Stuart et al.52 | https://stuartlab.org/signac/ |
| LIANA version 0.1.10 | Dimitrov et al.26 | https://saezlab.github.io/liana/index.html |
| Other | ||
| Code used in this study | This paper | https://github.com/gcapture/glioma-het |
| HTML book containing code used in this study | This paper | https://gcapture.github.io/glioma-het/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Şevin Turcan. (sevin.turcan@med.uni-heidelberg.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participants details
Human patient samples
A total of 14 fresh frozen archival IDH-mutant glioma samples (8 oligodendroglioma and 6 astrocytoma) were used for snRNA-seq and snATAC-seq. Additionally, 6 fresh frozen paired primary and recurrent samples (IDH-mutant astrocytoma) were used for snRNA-seq. Patient information and tumor characteristics are provided in Figures 1A and 4A. Fresh frozen samples were collected by the Division of Experimental Neurosurgery, Department of Neurosurgery, University Hospital Heidelberg, and the Department of Neurosurgery, Acıbadem Mehmet Ali Aydınlar University, School of Medicine, Istanbul. Samples used as validation cohort for immunohistochemistry were retrieved from the institutional databases of the Department of Pathology, Spedali Civili of Brescia, and the Department of Neuropathology, Heidelberg University Hospital. All patients provided written informed consent, in accordance with the Declaration of Helsinki. All samples were reviewed and received approval from the respective Institutional Review Boards and local authorities at the institutions where samples were originally collected. Specifically, the samples were approved by the Ethics Committee of Spedali Civili of Brescia, Institutional Review Board at the Medical Faculty of Acıbadem Mehmet Ali Aydınlar University, and the Ethics Committee of Heidelberg University.
Method details
Nucleus isolation from fresh frozen samples for snRNA-seq and snATAC-seq
Isolation of nuclei for snRNA-seq and snATAC-seq was performed as previously described.18 Briefly, fresh frozen tissue samples were cut into small pieces and homogenized using a Dounce homogenizer in EZ lysis buffer (Sigma Aldrich). Tissue was homogenized 20 times with pestle A and 20 times with pestle B. This was followed by centrifugation, filtration, and buffer-mediated gradient centrifugation to obtain pure single nuclei, which were then used for snRNA-seq and snATAC-seq.
Construction of libraries and generation of cDNA on the 10x genomics platform
Nuclei were counted using a hemocytometer, and their concentration adjusted as needed to meet the optimal range for loading on the 10x Chromium chip. The nuclei were then loaded into the 10x Chromium system using the Single Cell 3′ Reagent Kit v3 or v3.1 (for snRNA-seq) and Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 (for snATAC-seq) according to the manufacturer’s protocol. We aimed to load ∼20,000 nuclei for each snRNA-seq run and ∼10,000 nuclei for each snATAC-seq run. Following library construction, libraries were sequenced on the Illumina NovaSeq 6000 system.
Immunohistochemistry
2 μm sections were cut from formalin-fixed paraffin-embedded (FFPE) tissue samples provided by the Pathological Department of Spedali Civili of Brescia. Sections were de-waxed and rehydrated. Endogenous peroxidase activity was blocked with 0.3% H2O2 in methanol for 20 min. Antigen retrieval was performed using a microwave oven or a thermostatic bath in 1.0 mM EDTA buffer (pH 8.0) or in 1.0 mM Citrate buffer (pH 6.0). Sections were then washed in tris-buffered saline (TBS, pH 7.4) and incubated for 1 h with the specific primary antibody diluted in TBS 1% bovine serum albumin. The reaction was revealed by using Dako EnVision System-HRP Labeled Polymer anti-mouse or anti rabbit (Dako) or Novolink Polymer Detection System (Novocastra) followed by diaminobenzydine (DAB) as chromogen and hematoxylin as counterstain. For double immunohistochemistry, after completing the first immune reaction, the second one was revealed by using MACH4 Universal AP Polymer kit (Biocare Medical) followed by Ferangi Blue Chromogen kit (Biocare Medical) and nuclei were counterstained with hematoxylin. Images were acquired with a Nikon DS-Ri2 camera (4908 x 3264 full-pixel) mounted on a Nikon Eclipse 50i microscope equipped with Nikon Plan lenses (x10/0.25; x20/0.40; x40/0.65; x100/1.25) using NIS-Elements 4.3 imaging software (Nikon Corporation). The following primary antibodies were used: anti-Iba1 rabbit polyclonal (1:300, Wako), anti-CD74 mouse monoclonal (clone LN2) (1:100, BioLegend), anti-CD163 mouse monoclonal (clone 10D6) (1:50, ThermoFisher Scientific), anti-STAT1 (pY701) mouse monoclonal (clone 14/P-STAT1) (1:500, BD Biosciences), anti-EEF2 rabbit monoclonal (EP880Y) (1:250, Abcam), and anti-EEF1A1 rabbit monoclonal (EPR9470) (1:50, Abcam).
For IHC staining of human astrocytomas, 5 μm FFPE sections were obtained from the Department of Neuropathology, Heidelberg University Hospital, and stained with the following antibodies: mouse monoclonal CSF1 antibody (1:25, clone 2D10, Sigma-Aldrich #MABF 191), rabbit polyclonal CSF1R antibody (1:50, Proteintech #25949-1-AP), rat monoclonal human PIGF antibody (1:25, clone #358905, R&D #MAB 2642), mouse monoclonal NRP1 antibody (1:50, clone 2H3F6, Proteintech #60067-1-Ig). For CSF1, a pre-treatment steamer with Tris buffer pH 9.0 was performed followed by overnight incubation at room temperature. Detection was performed with ImmPress HRP, universal antibody (horse, anti-mouse) polymer detection kit, peroxidase (Vector # MP 7500). For CSF1R, we performed the same incubation and antigen retrieval as for CSF1 and detected with Dako REAL Detection System, Alkaline Phosphatase/RED, Rabbit/Mouse (Biocompare #K5005). For PIGF, pre-treatment with citrate buffer pH 6.0 was performed, followed by overnight incubation at room temperature. Detection was performed using the secondary antibody - biotinylated anti-rat IgG (Vector # BA-4001, 1:200) for 30′ at 37°C, followed by HRP Streptavidin (1:200, Vector #SA-5004), for 30' at 37°C, and DAB (brown color) was used as a chromogen (ImmPress HRP Universal Antibody (Horse Anti-Mouse/Rabbit IgG) Polymer detection Kit, Peroxidase, (Vector # MP 7500). For NRP1 IHC, samples were pretreated with citrate buffer pH 6.0 and incubated for 2 h at 37°C, and detected using the Dako REAL Detection System, Alkaline Phosphatase/RED, rabbit/mouse, (Biocompare #K5005). Samples were imaged at 40x magnification with an Olympus VS.200 slide scanner (Olympus Corporation).
Single-nucleus RNA-seq data analysis
Raw sequencing data from all samples were processed using the Cell Ranger workflow (version 3.1.0) using a combined intron-exon reference generated following the manufacturer-provided “Generating a Cell Ranger compatible ‘pre-mRNA’ Reference Package” guidelines (https://support10xgenomics.com/single-cell-gene-expression/software/pipelines/3.1/advanced/references). In brief, the “pre-mRNA” reference was derived using “cellranger mkfastq” with a modified GTF annotation for which all introns were counted as exons, and therefore accounted for the count matrix generation. Subsequent data analysis was conducted using R (versions 3.6.1–4.2.0) package Seurat16,49 (version 3.1.0–4.3.0).
To remove doublets, a two-step quality control cutoff was applied, which involved the number of unique mRNAs per nucleus (Unique Molecular Identifier, UMI), the number of genes per nucleus, and the percentage of mitochondrial RNA in the nuclei. Nuclei with less than 1000 UMIs/nucleus or 500 genes/nucleus were removed from the analysis, according to pre-established best practices.53 Nuclei with >5% of mitochondrial RNA were also excluded. To remove doublets, we used scrublet v0.2.1,50 which assigns a doublet probability score to each nucleus, with a higher score indicating a higher likelihood of being a doublet. For each sample, individual inspection of the doublet scoring distribution was conducted to determine sample-specific cutoffs.
Data normalization was performed using Regularized Negative Binomial Regression (RNBR) as described in the Seurat package using Seurat::SCTransform() function. 3000 features with high variable expression across the nuclei were identified, which were used in a subsequent Principal Component Analysis (PCA) using the Seurat::RunPCA() function. To remove biases arising from different samples, integration based on the PCA embedding was computed using Harmony,11 which generated a new dimensional reduction embedding. For this, the most relevant principal components (PCs) were used, selected based on the elbow plot showcasing the standard deviations comprised accounted in each PC. Further dimensional reduction using Uniform Manifold Approximation and Projection (UMAP) is performed to visualize the cells. The UMAPs and clusters were generated using the Seurat::FindNeighbors(), Seurat::FindClusters(), and Seurat::RunUMAP() functions. All plots shown in figures have been generated using SCpubr54 and ggplot2. In brief, dimensional reduction plots were generated using SCpubr::do_DimPlot(), feature plots using SCpubr::do_FeaturePlot(), dot plots using SCpubr::do_DotPlot(), bar plots using SCpubr::do_BarPlot(), violin plots using SCpubr::do_ViolinPlot() and heatmaps using SCpubr::do_ExpressionHeatmap(), SCpubr::do_EnrichmentHeatmap(), SCpubr::do_CorrelationPlot(), SCpubr::do_PathwayActivityPlot(), SCpubr::do_TFActivityPlot(), SCpubr::do_MetadataPlot(), SCpubr::do_DiffusionMapPlot().
Determination of malignant cells
To distinguish between tumor cells and tumor microenvironment we inferred copy number variation (CNV) signatures, using the R package inferCNV v.1.5.0.51 Since 10x snRNA-seq data is very sparse, we opted to use meta-cells instead of single cells to increase the overall sensitivity. Meta-cells are generated computationally by aggregating the expression values of five cells from the same cluster. As the microglia/macrophage and the oligodendrocyte populations were easily distinguished by marker gene inspection (PTPRC, CD163 and CD14 for microglia/macrophages, and MBP and MOG for oligodendrocytes), these were used as references. Cells were classified as either tumor cells or tumor microenvironment (TME) cells based on their CNV profiles. For oligodendrogliomas, we used a combination of 1p/19q co-deletion status and enrichment for previously published lists of marker genes,4 whereas for astrocytomas, CNVs were not clearly recovered by inferCNV and samples did not share the same CNV event. For astrocytoma samples, tumor cells were characterized based on the enrichment of marker gene lists.
Deconvolution
To deconvolute the TCGA and CGGA bulk RNA sequencing data, we used the single-cell dataset generated in this study as a reference. Deconvolution was performed using SPOTlight12 version 1.0.3. To train the model, we used all cell populations from our snRNA-seq data. Marker genes for each cell type were identified using the Seurat function Seurat::FindAllMarkers() with the following parameters: test.use = "MAST", min.pct = 0.5, logfc.threshold = 0.5, only.pos = TRUE. We then used the avg_log2FC as the weights to initialize the W NMF matrix for each topic prior to initialization.
Single-nucleus ATAC-seq data analysis
snATAC-seq data analysis was carried out using the R (version 3.6.1) package Signac (version 1.0).52 Peak count matrices were generated using Cell Ranger ATAC version 1.2, with each row representing a chromatin region and each column representing a different cell. To merge multiple datasets together, the standard protocol from Signac was followed. For peak annotation and further downstream analysis, we used the GRCh38 annotation retrieved from EnsDb.Hsapiens.v86 R package.55 To assess the quality of the nuclei, we defined the following variables: total number of fragments in the nuclei, the percentage of reads falling on peaks, the percentage of reads falling in blacklist regions,56 the chromosome binding pattern referred to as “nucleosome signal” computed with Signac::NucleosomeSignal() and the enrichment of the peaks in transcription start sites (TSS) computed with Signal::TSSEnrichment(). We then removed the nuclei for which a) total number of fragments were lower than 3000 and/or higher than 20,000, b) the percentage of reads in peaks were lower than 15% or reads in blacklist regions were higher than 5%, c) nucleosome signal was higher than 10%, and d) TSS enrichment was lower than 2%.
To normalize and perform linear dimensionality reduction, we used term frequency-inverse document frequency (TF-IDF) normalization with Signac:RunTFIDF(), followed by feature selection with Signac::FindTopFeatures() paired with “q0” as argument to select all peaks. Then, we ran singular value decomposition (SVD) on the TD-IDF matrix, with reduction name “LSI”. The first LSI component captured technical variation corresponding to sequencing depth. Therefore, we excluded it from downstream analyses.
We then computed non-linear dimensionality reduction of the data with Seurat::RunUMAP() and clustered the cells using Seurat::FindNeighbors() and Seurat::FindClusters() under default parameters, yielding 30 components for oligodendroglioma and astrocytoma samples.
To estimate the gene activity for each gene based on their associated accessibility, we used Signac::GeneActivity() and normalized the resulting assay data with Seurat::SCTransform(), as gene activity intends to resemble an snRNA-seq experiment. To further integrate the results with our snRNA-seq datasets, we identified shared correlation patterns between the normalized gene activity assay from snATAC-seq data and the normalized count data assay from snRNA-seq data. For this, shared anchors between both datasets are identified, which correspond to matching patterns between the two modalities. As the snRNA-seq dataset is already annotated, nuclei with shared anchors in the snATAC-seq data will receive the same annotation.49 This is computed with Signac::FindTransferAnchors() and Signac::TransferData(), following Signac’s standard protocol for multimodal integration. To further remove patient-specific effects, we integrated the snATAC-seq datasets using Harmony with harmony::RunHarmony(), and recomputed the UMAP embedding using the same number of LSI components as previously.
Next, to infer gain or loss of accessibility of peaks sharing the same transcription factor (TF) motifs, we used Signac::AddMotifs(), a wrapper from chromVAR version 1.14.057 following Signac protocol for motif analysis. This returns a motif activity score per nucleus. To compare accessibility and expression we computed differentially accessible peaks between the identities retrieved after label transfer using Seurat::FindAllMarkers() with default parameters, filtered by p value ≤0.05 and annotated to their closest gene with Signac::ClosestFeature(). Differentially enriched motifs across tumor populations were correlated using Pearson’s correlation and the score is displayed as a heatmap for both tumor subtypes. Top differentially enriched motifs across tumor populations are displayed as a heatmap.
Single nucleus RNA-seq data analysis of the tumor microenvironment
snRNA-seq data analysis of the tumor microenvironment (TME) was carried out using the R (version 4.1.0) package Seurat16 (version 4.0.3). To obtain a finer-grained resolution of cell states, we extracted TAM cells from the integrated snRNA-seq dataset, and recomputed the integration workflow, downstream analyses and clustering: TAMs were log-normalized using a scale factor of 1e4. Then, we selected the top 3000 variable genes and integrated using Harmony (version 0.1.0) and computed a new UMAP embedding. New clusters were called based on the top 30 principal components with a resolution of 0.7 using the Louvain algorithm. Marker genes were retrieved by differential expression analysis of each microglia subpopulation against the rest using Wilcoxon Rank-Sum test, selecting the genes that presented an average of 0.25-fold difference (log2 scale) between the two groups (adjusted p value ≤0.05). For cluster annotation, we used matchSCore258 R package and SCpubr::do_FeaturePlot().
Non-negative matrix factorization (NMF) of primary samples
To unveil the major cell states within our samples, we applied Non-Negative Matrix Factorization (NMF) as described.59 NMF was performed individually to the tumor cells in each patient. Nuclei from the TME were excluded from the analysis. The normalized count matrix was scaled and centered. For each patient, NMF was computed with factorization ranks ranging from two to ten, resulting in computed NMF programs for each iteration and individual patient. Each NMF program consists of a vector of NMF scores for each gene in the count matrix. Higher NMF scores indicate a stronger contribution of the gene to the NMF program. The top 30 scored genes in each signature were selected, with prior exclusion of the mitochondrial genes. Next, to assess the overall enrichment of each nucleus to the NMF program, a scoring system was applied. For each gene in the NMF program, a control set of genes was defined as the top 100 genes closest in expression. This was achieved by first computing a vector of averaged expression values for each gene across all nuclei. Then, genes with similar expression patterns were selected by computing the absolute value of the difference between the average expression value of the gene across all the nuclei and the vector of averaged scores. After sorting these values, the top 100 genes closest to zero, excluding the first one (which is zero, corresponding to the given gene itself), were selected. Once the reference set is generated, for each nucleus, the difference in averaged expression between the given gene and the averaged expression values for the reference set of genes is calculated. This returns a vector of the difference in averaged expression values between the gene and the control set per nucleus, multiplied by each of the genes in the NMF program. This matrix, containing each individual nucleus as columns and each gene in the NMF program as rows, is transformed into a vector by averaging the values in each of the columns (i.e., genes). This gave the average score across all genes for each nucleus, indicating how much each nucleus is enriched in the given set of genes in the NMF program.
To detect similarities in the scoring of the NMF signatures across nuclei, Pearson’s correlation was computed on a matrix formed by the different scoring of the NMF program. This resulted in distinct groups of NMF programs highly similar to each other in terms of how enriched each nucleus in the tumor bulk is in the gene sets forming these programs, referred to as NMF metaprograms. This process was repeated for each value of rank computed, and the one that returned the clearest and highest number of NMF metaprograms was selected. To define the top 30 genes driving each of the NMF metaprograms, an almost identical scoring approach as the previously described was applied, using all the unique genes in the NMF programs contained in the NMF metaprogram, but averaging the final scores by rows (i.e., nuclei) instead. This returns a vector with a score for each gene, indicating how much weight each gene carries in the NMF metaprogram across all nuclei. The top 30 genes with the highest scores were selected. Each group of genes was functionally characterized using the enrichR60 package, which computes the enrichment of the set of genes in different term databases, such as GO, KEGG, etc. Seurat enrichment scores were computed with the function Seurat::AddModuleScore() for all the nuclei to assess how the enrichment on the NMF metaprograms is distributed across the tumor cells of the integrated dataset.
Assigning meta-programs to cells
To avoid defining tumor bulk populations based on manually selected cutoffs of enrichment scores, especially when the enrichment patterns occur in a gradient fashion rather than specific clusters, we developed a permutation testing method to statistically assign cells to a given NMF metaprogram. The empirical distribution is defined as the vector of enrichment scores for a list of genes (i.e., NMF metaprogram), and the null distribution is generated by permuting the expression values of each of the genes queried for enrichment across the nuclei, thereby disrupting any enrichment pattern arising from the combined expression of the entire set of genes. The resulting vector of enrichment scores represents individual permuted scores that represent what the enrichment scores for the set of genes would be if the list of genes were not correlated, defined as the null distribution. This method assumes differential gene expression across the nuclei; otherwise, permuting the expression values would not have the desired effect. To determine whether a nucleus is statistically enriched for the given list of genes, we compute the fraction of values in the null distribution that are higher than the enrichment score for the selected nucleus plus one, both in the numerator and denominator, thus avoiding infinite values (representing the p value). Therefore, the number of computed permutations in the null distribution will determine the lowest possible p value. For this, a million permutations are performed to achieve a minimum possible p value of 1e−6, and p values are corrected for multiple testing using Benjamini-Hochberg method61 (Benjamini and Hochberg, 1995), generating adjusted p values. Nucleus for which the adjusted p value is lower than 0.05 (FDR = 5%) are selected as statistically enriched in the list of genes provided. However, if the same set of nuclei is queried for different list of genes, the choice of FDR should be further corrected by the number of comparisons to avoid inflation of the alpha error. This is achieved by dividing the desired FDR by the total number of comparisons performed to the same group of nuclei. This step is particularly important if nuclei are being labeled based on a combination of results (i.e., being enriched for a given list of genes and not for the others).
Pathway enrichment analysis and transcription factor-target interactions
To infer pathway activity from normalized gene expression data for 14 pre-defined pathways (Androgen, Estrogen, EGFR, Hypoxia, JAK-STAT, MAPK, NFkB, PI3K, p53, TGFβ, TNFα, Trail, VEGF, and WNT) we used PROGENy62 in combination with decoupleR.13 To ensure comparability across both subtypes, we merged the raw counts for both subtypes and re-normalized the data using RNBR. The normalized counts were then used as input for PROGENy, which returns a Seurat object with an independent score for each cell. Similar to PROGENy, we used the same approach to compute TF-target interactions with DoRothEA63 in combination with decoupleR,13 We applied DoRothEA in combination with the statistical method VIPER on matrices of single nuclei RNA-seq samples (genes in rows and either bulk samples or single cells in columns) containing normalized gene expression scores scaled gene-wise to a mean value of 0 and standard deviation of 1 or on contrast matrices (genes in rows and summarized perturbation experiments into contrasts in columns) containing log fold changes (FC). In the case of single-nuclei RNA-seq sample analysis, the contrasts were built based on TF activity matrices yielding the change in TF activity (perturbed samples - control sample) summarized as logFC. TFs with less than four targets listed in the corresponding gene expression matrix were discarded from the analysis. VIPER provides a normalized enrichment score (NES) for each TF which we consider as a metric for the activity. We used the R package viper (version 1.17.0) to run VIPER in combination with DoRothEA.
Inferring receptor-ligand interactions
To assess receptor-ligand interactions between the two major tumor populations (OPC-like and astro-like cells) and the newly defined TAM subpopulations, we used LIANA version 0.1.1022 with five different methods: ‘natmi’, ‘connectome’, ‘logfc’, ‘sca’ and ‘cellphonedb’, and filtered out the interactions to those with p value (aggregated consensus rank) ≤ 0.05. We then selected the most relevant interactions and plotted them as a dot plot.
Quantification and statistical analysis
Statistics
GraphPad Prism 9 software was used for statistical analysis of the IHC data. Unpaired two-sided t tests were used to determine statistical significance (p values < 0.05). For IHC, expression levels of markers were semi-quantitatively scored on representative tumor regions based on percentage [score ranges: 0 (no expression), 1 (0–5%, moderate intensity), 2 (6–29%, moderate intensity), 3 (30–69%, high intensity), 4 (≥70%, high intensity)] of immunoreactive (IR) cells.
Acknowledgments
We would like to thank Volker Hovestadt for his input and the base code for NMF analysis and Martin Sill for his input and statistical advice on the permutation testing method design. This work was supported by the German Cancer Aid, Max Eder Program grant number 70111964 (S.T.), DFG Project-ID 404521405, SFB 1389 – UNITE Glioblastoma, work package A04 (S.T.), and the Baden-Württemberg Foundation project number ID27/DEFOG (S.T). For the publication fee, we acknowledge financial support by Deutsche Forschungsgemeinschaft (DFG) within the funding program "Open Access Publikationskosten" as well as by Heidelberg University.
Author contributions
E.B.-C., A.N., I.H., J.C.N., H.H., and S.T. designed the study. A.N. performed snRNA-seq and snATAC-seq sample preparation with assistance from X.S. E.B.-C., A.N., I.H., J.C.N., and M.E.-B. conducted snRNA-seq and snATAC-seq computational analysis. N.P., K.O., and C.H.-M. provided fresh-frozen tissue samples. C.S., F.P., and M.C. performed the IHC experiments. A.v.D. and P.L.P. provided formalin-fixed, paraffin-embedded (FFPE) samples for IHC. J.T., M.C., and P.L.P. analyzed and scored the IHC data. J.T., W.W., M.R., and M.S. provided intellectual input. E.B.-C., A.N., I.H., J.C.N., H.H., and S.T. wrote the manuscript with comments from all authors. E.B.-C. generated all figures, as well as the associated GitHub code repository and GitHub pages. H.H. and S.T. supervised the project.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 25, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101249.
Contributor Information
Holger Heyn, Email: holger.heyn@cnag.eu.
Şevin Turcan, Email: sevin.turcan@med.uni-heidelberg.de.
Supplemental information
Data and code availability
-
•
Processed single-nucleus RNA-seq and single-nucleus ATAC-seq data have been deposited at GEO (GSE205771) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code has been deposited at GitHub (https://github.com/gcapture/glioma-het) and GitHub pages (https://gcapture.github.io/glioma-het/), and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Yan H., Parsons D.W., Jin G., McLendon R., Rasheed B.A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G.J., et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis D.N., Perry A., Wesseling P., Brat D.J., Cree I.A., Figarella-Branger D., Hawkins C., Ng H.K., Pfister S.M., Reifenberger G., et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23:1231–1251. doi: 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tirosh I., Venteicher A.S., Hebert C., Escalante L.E., Patel A.P., Yizhak K., Fisher J.M., Rodman C., Mount C., Filbin M.G., et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venteicher A.S., Tirosh I., Hebert C., Yizhak K., Neftel C., Filbin M.G., Hovestadt V., Escalante L.E., Shaw M.L., Rodman C., et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klemm F., Maas R.R., Bowman R.L., Kornete M., Soukup K., Nassiri S., Brouland J.P., Iacobuzio-Donahue C.A., Brennan C., Tabar V., et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell. 2020;181:1643–1660.e17. doi: 10.1016/j.cell.2020.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedrich M., Sankowski R., Bunse L., Kilian M., Green E., Ramallo Guevara C., Pusch S., Poschet G., Sanghvi K., Hahn M., et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer. 2021;2:723–740. doi: 10.1038/s43018-021-00201-z. [DOI] [PubMed] [Google Scholar]
- 7.Mellinghoff I.K., van den Bent M.J., Blumenthal D.T., Touat M., Peters K.B., Clarke J., Mendez J., Yust-Katz S., Welsh L., Mason W.P., et al. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. N. Engl. J. Med. 2023;389:589–601. doi: 10.1056/NEJMoa2304194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenzie A.T., Wang M., Hauberg M.E., Fullard J.F., Kozlenkov A., Keenan A., Hurd Y.L., Dracheva S., Casaccia P., Roussos P., Zhang B. Brain Cell Type Specific Gene Expression and Co-expression Network Architectures. Sci. Rep. 2018;8:8868. doi: 10.1038/s41598-018-27293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M., et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weng Q., Wang J., Wang J., He D., Cheng Z., Zhang F., Verma R., Xu L., Dong X., Liao Y., et al. Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell. 2019;24:707–723.e8. doi: 10.1016/j.stem.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elosua-Bayes M., Nieto P., Mereu E., Gut I., Heyn H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 2021;49:e50. doi: 10.1093/nar/gkab043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Badia-I-Mompel P., Vélez Santiago J., Braunger J., Geiss C., Dimitrov D., Müller-Dott S., Taus P., Dugourd A., Holland C.H., Ramirez Flores R.O., Saez-Rodriguez J. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinform. Adv. 2022;2:vbac016. doi: 10.1093/bioadv/vbac016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland C.H., Tanevski J., Perales-Patón J., Gleixner J., Kumar M.P., Mereu E., Joughin B.A., Stegle O., Lauffenburger D.A., Heyn H., et al. Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 2020;21:36. doi: 10.1186/s13059-020-1949-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wierstra I., Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 2007;388:1257–1274. doi: 10.1515/BC.2007.159. [DOI] [PubMed] [Google Scholar]
- 16.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz-Moreno C., Salas S.M., Samuelsson E., Brandner S., Kranendonk M.E.G., Nilsson M., Stunnenberg H.G. Harmonized single-cell landscape, intercellular crosstalk and tumor architecture of glioblastoma. bioRxiv. 2022 doi: 10.1101/2022.08.27.505439. Preprint at. 2022.2008.2027.505439. [DOI] [Google Scholar]
- 18.Narayanan A., Blanco-Carmona E., Demirdizen E., Sun X., Herold-Mende C., Schlesner M., Turcan S. Nuclei Isolation from Fresh Frozen Brain Tumors for Single-Nucleus RNA-seq and ATAC-seq. J. Vis. Exp. 2020 doi: 10.3791/61542. [DOI] [PubMed] [Google Scholar]
- 19.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hambardzumyan D., Gutmann D.H., Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pombo Antunes A.R., Scheyltjens I., Lodi F., Messiaen J., Antoranz A., Duerinck J., Kancheva D., Martens L., De Vlaminck K., Van Hove H., et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021;24:595–610. doi: 10.1038/s41593-020-00789-y. [DOI] [PubMed] [Google Scholar]
- 23.Paolicelli R.C., Sierra A., Stevens B., Tremblay M.E., Aguzzi A., Ajami B., Amit I., Audinat E., Bechmann I., Bennett M., et al. Microglia states and nomenclature: A field at its crossroads. Neuron. 2022;110:3458–3483. doi: 10.1016/j.neuron.2022.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeiner P.S., Preusse C., Blank A.E., Zachskorn C., Baumgarten P., Caspary L., Braczynski A.K., Weissenberger J., Bratzke H., Reiß S., et al. MIF Receptor CD74 is Restricted to Microglia/Macrophages, Associated with a M1-Polarized Immune Milieu and Prolonged Patient Survival in Gliomas. Brain Pathol. 2015;25:491–504. doi: 10.1111/bpa.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S., Zhang C., Maimela N.R., Yang L., Zhang Z., Ping Y., Huang L., Zhang Y. Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. OncoImmunology. 2019;8 doi: 10.1080/2162402X.2019.1601478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimitrov D., Türei D., Garrido-Rodriguez M., Burmedi P.L., Nagai J.S., Boys C., Ramirez Flores R.O., Kim H., Szalai B., Costa I.G., et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun. 2022;13:3224. doi: 10.1038/s41467-022-30755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrissey M.A., Kern N., Vale R.D. CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity. 2020;53:290–302.e6. doi: 10.1016/j.immuni.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hume D.A., MacDonald K.P.A. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119:1810–1820. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]
- 29.Chitu V., Stanley E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Snuderl M., Batista A., Kirkpatrick N.D., Ruiz de Almodovar C., Riedemann L., Walsh E.C., Anolik R., Huang Y., Martin J.D., Kamoun W., et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell. 2013;152:1065–1076. doi: 10.1016/j.cell.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aoki S., Inoue K., Klein S., Halvorsen S., Chen J., Matsui A., Nikmaneshi M.R., Kitahara S., Hato T., Chen X., et al. Placental growth factor promotes tumour desmoplasia and treatment resistance in intrahepatic cholangiocarcinoma. Gut. 2022;71:185–193. doi: 10.1136/gutjnl-2020-322493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berghoff A.S., Kiesel B., Widhalm G., Wilhelm D., Rajky O., Kurscheid S., Kresl P., Wöhrer A., Marosi C., Hegi M.E., Preusser M. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017;19:1460–1468. doi: 10.1093/neuonc/nox054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohanbash G., Carrera D.A., Shrivastav S., Ahn B.J., Jahan N., Mazor T., Chheda Z.S., Downey K.M., Watchmaker P.B., Beppler C., et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest. 2017;127:1425–1437. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott A.C., Dündar F., Zumbo P., Chandran S.S., Klebanoff C.A., Shakiba M., Trivedi P., Menocal L., Appleby H., Camara S., et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571:270–274. doi: 10.1038/s41586-019-1324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larionova T.D., Bastola S., Aksinina T.E., Anufrieva K.S., Wang J., Shender V.O., Andreev D.E., Kovalenko T.F., Arapidi G.P., Shnaider P.V., et al. Alternative RNA splicing modulates ribosomal composition and determines the spatial phenotype of glioblastoma cells. Nat. Cell Biol. 2022;24:1541–1557. doi: 10.1038/s41556-022-00994-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li M., Yang L., Chan A.K.N., Pokharel S.P., Liu Q., Mattson N., Xu X., Chang W.H., Miyashita K., Singh P., et al. Epigenetic Control of Translation Checkpoint and Tumor Progression via RUVBL1-EEF1A1 Axis. Adv. Sci. 2023;10 doi: 10.1002/advs.202206584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park J.W., Sahm F., Steffl B., Arrillaga-Romany I., Cahill D., Monje M., Herold-Mende C., Wick W., Turcan Ş. TERT and DNMT1 expression predict sensitivity to decitabine in gliomas. Neuro Oncol. 2021;23:76–87. doi: 10.1093/neuonc/noaa207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turcan S., Fabius A.W.M., Borodovsky A., Pedraza A., Brennan C., Huse J., Viale A., Riggins G.J., Chan T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013;4:1729–1736. doi: 10.18632/oncotarget.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morral C., Stanisavljevic J., Hernando-Momblona X., Mereu E., Álvarez-Varela A., Cortina C., Stork D., Slebe F., Turon G., Whissell G., et al. Zonation of Ribosomal DNA Transcription Defines a Stem Cell Hierarchy in Colorectal Cancer. Cell Stem Cell. 2020;26:845–861.e12. doi: 10.1016/j.stem.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GLASS Consortium Glioma through the looking GLASS: molecular evolution of diffuse gliomas and the Glioma Longitudinal Analysis Consortium. Neuro Oncol. 2018;20:873–884. doi: 10.1093/neuonc/noy020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barthel F.P., Johnson K.C., Varn F.S., Moskalik A.D., Tanner G., Kocakavuk E., Anderson K.J., Abiola O., Aldape K., Alfaro K.D., et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576:112–120. doi: 10.1038/s41586-019-1775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varn F.S., Johnson K.C., Martinek J., Huse J.T., Nasrallah M.P., Wesseling P., Cooper L.A.D., Malta T.M., Wade T.E., Sabedot T.S., et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell. 2022;185:2184–2199.e16. doi: 10.1016/j.cell.2022.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paull E.O., Aytes A., Jones S.J., Subramaniam P.S., Giorgi F.M., Douglass E.F., Tagore S., Chu B., Vasciaveo A., Zheng S., et al. A modular master regulator landscape controls cancer transcriptional identity. Cell. 2021;184:334–351.e20. doi: 10.1016/j.cell.2020.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friebel E., Kapolou K., Unger S., Núñez N.G., Utz S., Rushing E.J., Regli L., Weller M., Greter M., Tugues S., et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell. 2020;181:1626–1642.e20. doi: 10.1016/j.cell.2020.04.055. [DOI] [PubMed] [Google Scholar]
- 46.Butturini E., Boriero D., Carcereri de Prati A., Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019;669:22–30. doi: 10.1016/j.abb.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 47.Hu X., Herrero C., Li W.P., Antoniv T.T., Falck-Pedersen E., Koch A.E., Woods J.M., Haines G.K., Ivashkiv L.B. Sensitization of IFN-gamma Jak-STAT signaling during macrophage activation. Nat. Immunol. 2002;3:859–866. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- 48.Smith G.T., Radin D.P., Tsirka S.E. From protein-protein interactions to immune modulation: Therapeutic prospects of targeting Neuropilin-1 in high-grade glioma. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.958620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tickle T.T.,I., Georgescu C., Brown M., Haas B. Klarman Cell Observatory. Broad Institute of MIT and Harvard; 2019. inferCNV of the Trinity CTAT Project.https://github.com/broadinstitute/inferCNV [Google Scholar]
- 52.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Slyper M., Porter C.B.M., Ashenberg O., Waldman J., Drokhlyansky E., Wakiro I., Smillie C., Smith-Rosario G., Wu J., Dionne D., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020;26:792–802. doi: 10.1038/s41591-020-0844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanco-Carmona E. Generating publication ready visualizations for Single Cell transcriptomics using SCpubr. bioRxiv. 2022 doi: 10.1101/2022.02.28.482303. Preprint at. 2022.2002.2028.482303. [DOI] [Google Scholar]
- 55.Rainer J. 2017. EnsDb.Hsapiens.v86: Ensembl Based Annotation Package. R package version 2.99.0. [Google Scholar]
- 56.Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mereu E., Lafzi A., Moutinho C., Ziegenhain C., McCarthy D.J., Álvarez-Varela A., Batlle E., Sagar Grun D., Grün D., Lau J.K., et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020;38:747–755. doi: 10.1038/s41587-020-0469-4. [DOI] [PubMed] [Google Scholar]
- 59.Hovestadt V., Smith K.S., Bihannic L., Filbin M.G., Shaw M.L., Baumgartner A., DeWitt J.C., Groves A., Mayr L., Weisman H.R., et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–79. doi: 10.1038/s41586-019-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 62.Schubert M., Klinger B., Klünemann M., Sieber A., Uhlitz F., Sauer S., Garnett M.J., Blüthgen N., Saez-Rodriguez J. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 2018;9:20. doi: 10.1038/s41467-017-02391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia-Alonso L., Holland C.H., Ibrahim M.M., Turei D., Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29:1363–1375. doi: 10.1101/gr.240663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Processed single-nucleus RNA-seq and single-nucleus ATAC-seq data have been deposited at GEO (GSE205771) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code has been deposited at GitHub (https://github.com/gcapture/glioma-het) and GitHub pages (https://gcapture.github.io/glioma-het/), and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.