

Summary
ARID1A is among the most commonly mutated tumor suppressor genes in hepatocellular carcinoma (HCC). In this study, we conduct a CRISPR-Cas9 synthetic lethality screen using ARID1A-deficient HCC cells to identify approaches to treat HCC patients harboring ARID1A deficiency. This strategy reveals that the survival of these ARID1A-deficient HCC cells is highly dependent on genes related to the tricarboxylic acid (TCA) cycle. Mechanistically, ARID1A loss represses expression of key glycolysis-related gene PKM, shifting cellular glucose metabolism from aerobic glycolysis to dependence on the TCA cycle and oxidative phosphorylation. Cuproptosis is a recently defined form of copper-induced cell death reported to directly target the TCA cycle. Here, we find that ARID1A-deficient HCC cells and xenograft tumors are highly sensitive to copper treatment. Together, these results offer evidence of the synthetic lethality between ARID1A deficiency and mitochondrial respiration impairment, suggesting that copper treatment constitutes a promising therapeutic strategy for selectively targeting ARID1A-deficient HCC.
Graphical abstract
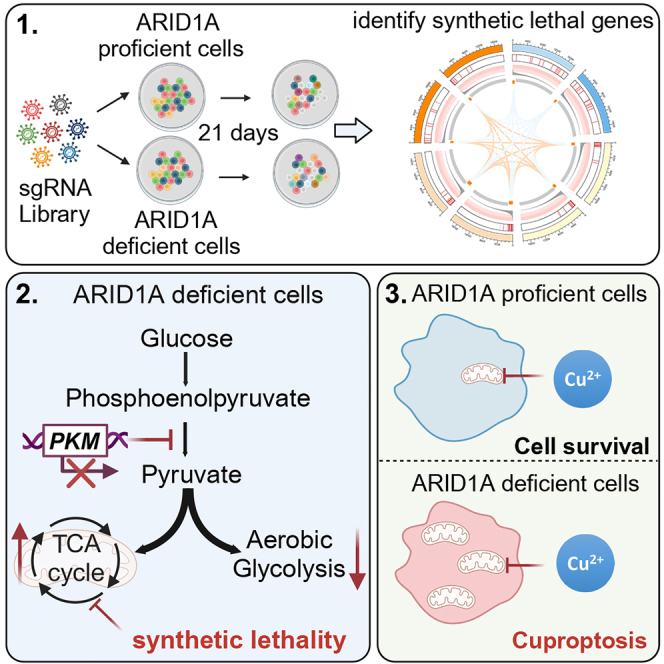
Highlights
-
•
A CRISPR screen identifies dependencies in ARID1A-deficient hepatocellular carcinoma
-
•
ARID1A loss results in impaired glycolysis and increases dependence on the TCA cycle
-
•
ARID1A deficiency suppresses glycolytic activity through repressing PKM transcription
-
•
Copper treatment emerges as a promising strategy against ARID1A-deficient HCC
To uncover therapeutic targets for hepatocellular carcinoma with ARID1A deficiency, Xing et al. undertake synthetic lethality screening and show ARID1A loss shifting cellular glucose metabolism from aerobic glycolysis to dependence on the TCA cycle and oxidative phosphorylation. Furthermore, targeting the TCA cycle through cuproptosis represents a promising therapeutic intervention strategy.
Introduction
Liver cancer is the third leading cause of cancer-associated death and the sixth most common cancer type worldwide, with hepatocellular carcinoma (HCC) accounting for over 90% of liver cancer cases.1,2 Despite extensive research efforts, advanced HCC cases often remain refractory to common multikinase inhibitor or immune checkpoint inhibitor treatment strategies owing to their high levels of genetic heterogeneity and the lack of specific therapeutically targetable mutations.3 Accordingly, there is a clear need to develop novel and effective approaches to treat HCC.
Many newly developed antitumor drugs target oncogenic driver genes that exhibit hyperactivity as a consequence of mutations. However, HCC tumors rarely harbor targetable kinase mutations.4 Tumor suppressor gene loss is another central mechanism by which normal cells undergo malignant transformation. Although gain-of-function mutations in specific oncogenes can generally be targeted relatively easily, efforts to target loss-of-function mutations in tumor suppressor genes are more complex. Synthetic lethality, however, has emerged as a promising means of identifying targets that can be leveraged to effectively target and treat specific cancers.5 This approach is reliant on the ability of certain cancer-specific mutations to lead to the development of acquired vulnerabilities amenable to pharmacologic intervention. These synthetic lethality-based strategies can theoretically be extended to treatment approaches beyond those targeting tumor-associated loss-of-function mutations.
AT-rich interactive domain-containing protein 1A (ARID1A) is a switch in mating type/sucrose non-fermentation (SWI/SNF) complex subunit that is among the most commonly mutated genes in HCC, with 10%–15% of HCC cases harboring ARID1A mutations.6,7 Mechanistically, ARID1A functions as a regulator of chromatin accessibility and associated transcriptional activity; thus, loss of ARID1A expression or function can have a profound impact on the chromatin landscape, altering transcriptional, epigenetic, metabolic, and DNA damage response activity in tumor cells.8,9 As ARID1A loss-of-function mutations are frequently observed in many types of cancer, they represent attractive targets for synthetic lethality-based intervention strategies. Several studies have shown that ARID1A-deficient tumors are susceptible to treatment with inhibitors of DNA damage repair10,11 and epigenetic drugs such as EZH2 inhibitors.12 Evidence also suggests that efforts to target the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway may provide some benefit in the context of ARID1A deficiency.13 However, the efficacy of specific synthetic lethality-based strategies may be dependent on the associated genetic background in a given tumor type, and no such studies focusing on HCC have been published. CRISPR-based high-throughput screening strategies provide a robust means of identifying promising synthetic lethality-related targets in tumor cells.14 In the present study, a genome-wide CRISPR-Cas9 knockout-based screening approach was thus used to identify genetic vulnerabilities in ARID1A-deficient HCC cells and ultimately led to the identification of synthetic lethal interaction between ARID1A and the tricarboxylic acid (TCA) cycle.
Metabolic reprogramming-based approaches to cancer treatment have recently emerged as a focus of intense research interest. Tumor cells have long been known to undergo metabolic reprogramming such that their survival is largely reliant on glycolytic activity.15 However, certain cancer cell types exhibit a higher degree of dependence on other metabolic pathways to support their survival and associated biosynthetic processes. For example, inactivation of the SWI/SNF complex has been reported to lead to dependency on glutamine metabolism in tumors such that they are sensitive to clinically relevant glutamine inhibitor treatment.16 The function of ARID1A as a regulator of tumor metabolic activity, however, remains incompletely documented. In this study, ARID1A deficiency was found to drive reprogramming of glucose metabolism to favor the TCA cycle and oxidative phosphorylation (OXPHOS) over glycolytic activity. In addition, Tsvetkov and colleagues recently defined a form of copper-related cell death termed cuproptosis, demonstrating a close link between copper toxicity and mitochondrial activity.17 Notably, tumor cells with higher levels of mitochondrial respiration activity exhibit greater sensitivity to copper treatment than tumor cells reliant on anaerobic glycolysis. This type of cell death may thus represent an effective means of treating HCC patients with ARID1A deficiency.
Results
Genome-wide CRISPR screening reveals that ARID1A-deficient HCC cells are dependent on the TCA cycle
To define genes required for the survival of ARID1A-deficient HCC cells, the Hep3B and HepG2 cell lines were used to establish ARID1A-deficient models via CRISPR-Cas9-mediated knockout. ARID1A knockout (ARID1A-KO) was confirmed via immunoblot analysis (Figure 1A). In addition, SNU 449 cells harboring a loss-of-function mutation in the ARID1A gene (p.E2033fs) were also used for our study (Figure 1B). The short-term and long-term growth of parental and ARID1A-KO cells was compared, revealing that ARID1A-KO Hep3B and HepG2 cells exhibited increased growth consistent with the increase in tumorigenicity (Figures S1A and S1B).18,19
Figure 1.

Genome-wide CRISPR screening identified a synthetic lethal interaction between ARID1A and the TCA cycle
(A) ARID1A-KO in Hep3B cells and HepG2 cell, as evaluated by immunoblotting.
(B) Immunoblot analysis of ARID1A in three HCC cell lines demonstrating the loss of ARID1A protein expression in SNU449 cells.
(C) CRISPR-Cas9 screening flowchart used in this study.
(D and E) KEGG enrichment analysis of key cellular activity genes (D) and synthetic lethal genes (E).
(F) Circos plots showing the CRISPR screen results. The outermost rim shows the rank of each gene in the screening results; significant screened hits are ranked on the innermost rim (p < 0.05). Heatmap indicating the location of synthetic lethal TCA-related genes in the gene ranking list. The line plots show the −log10 p values for the corresponding genes.
(G) A Sankey plot was used to demonstrate the connection of synthetic lethal TCA-related genes with the screening groups.
(H) Schematic of TCA cycle modules and the locations of synthetic lethal genes.
A genome-wide CRISPR-based screen for synthetic lethal genes was next performed using ARID1A-KO (sg1) Hep3B cells and the Toronto human knockout pooled library (TKOv3) consisting of 70,948 guide RNAs (gRNAs) targeting 18,053 protein-coding genes (four single-guide RNAs [sgRNAs]/gene) (Figure 1C). Parental wild-type (WT) Hep3B cells were also screened as a control. More than 35 million cells infected with library-encoding lentiviral particles were cultured for 3 weeks. Changes in the relative abundance of individual sgRNAs between baseline (T0) and day 21 (T21) were then compared via next-generation sequencing (NGS), and a ranked list of fitness-related genes required for cell survival was generated with the Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) algorithm. Genes with p < 0.01 in both groups were identified as essential genes required for cell survival irrespective of ARID1A deficiency, and 265 genes meeting this criterion were identified. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis indicated that these genes were enriched in essential cellular activity-related pathways, including cell cycle, proteasome, ribosome, spliceosome, RNA transport, RNA polymerase, and aminoacyl-tRNA biosynthesis (Figure 1D). In contrast, those genes that did not affect WT cell survival (p > 0.01) but were lethal in ARID1A-KO cells (p < 0.01) were defined as synthetic lethal genes in the ARID1A-deficient background. With these criteria, 685 genes exhibiting greater lethality in ARID1A-KO cells than in WT cells were identified. KEGG enrichment analysis indicated that these genes were enriched in the nucleotide excision repair, OXPHOS, TCA cycle, mismatch repair, and glutathione metabolism pathways (Figure 1E). Synthetic lethal effects between ARID1A and both glutathione metabolism and mismatch repair-related genes have previously been documented and studied in depth,10,11,16,20 an observation confirming the accuracy of our screening results. To ensure that these effects were not cell-line-specific, synthetic lethality screen was similarly conducted using HepG2 and SNU449 cells. This approach confirmed that TCA cycle-related genes were highly enriched in ARID1A-deficient but not ARID1A-proficient cells. The screening results and connections among pathways and genes were demonstrated using a Circos plot (Figure 1F). As most TCA cycle-related genes were highly ranked in these ARID1A-deficient HCC cells (Figures 1G and 1H), we elected to study the synthetic lethal interaction between ARID1A and TCA cycle-related genes in greater detail.
Validation of the synthetic lethal interactions between ARID1A and TCA cycle-related genes
With respect to synthetic lethal gene pairs, cancers reportedly upregulate one gene when the other gene experiences a loss of function through a compensatory process. To confirm the synthetic lethal interactions between ARID1A and genes associated with the TCA cycle, the public database was next used to profile gene expression, revealing that reductions in ARID1A mRNA (Figure S2A) and protein levels21 (Figure 2A) were significantly correlated with increases in the expression of the majority of TCA cycle-associated genes across cancer types, including HCC. The results of western blotting additionally revealed that key TCA cycle proteins, including aconitase 2 (ACO2), dehydrogenase complex flavoprotein subunit A (SDHA), and fumarate hydratase (FH), were upregulated in ARID1A-KO HCC cells (Figure 2B). Notably, the upregulated levels of TCA cycle proteins were rescued by restoring ARID1A expression in ARID1A-deficient cells, suggesting that these increases were ARID1A expression dependent rather than a potential off-target effect (Figure 2C). Immunohistochemistry (IHC) experiments in 102 human HCC specimens also indicated that the ARID1A protein level was significantly negatively correlated with the protein level of SDHA, which was the top candidate TCA cycle-related gene identified in this screen (Figure 2D). Synthetic lethal genes are presumed to be mutually exclusive given that the loss of function of both of these genes ultimately results in cell death. Of note, patients included in The Cancer Genome Atlas (TCGA) database exhibiting low expression levels of both ARID1A and SDHA, ACO2, or FH exhibited better prognostic outcomes (Figures 2E and S2B), although the relative rarity of mutations in TCA cycle-associated genes in HCC precluded the establishment of any causal or significant link (Figure S2C).
Figure 2.

ARID1A deficiency generated dependence on the TCA cycle-related genes in HCC
(A) A heatmap showing the protein levels of ARID1A, ACO2, SDHA, and FH in samples from HCC patients in CHCC-HBV database (n = 318).
(B and C) The protein expression levels of ARID1A and ACO2, SDHA, and FH were measured by immunoblotting in cells characterized by ARID1A deficiency and proficiency.
(D) HCC specimens from 102 patients were subjected to IHC staining for ARID1A and SDHA; expression was scored by two pathologists. The results suggested a strong correlation between ARID1A and SDHA expression. Scale bar, 100 μm.
(E) Kaplan-Meier curves demonstrating that low ARID1A and SDHA mRNA expression levels were correlated with better prognosis in HCC patients in the TCGA cohort.
(F and I) Silencing of ACO2 (F) or SDHA (I) expression in Hep3B cells by siRNA. α-Tubulin was used as the loading control. The assessment of relative cell viability rates was conducted following the transfection of siRNA in both parental and ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 6 independent experiments).
(G and J) Cell death rates were determined after treatment with ACO2 (G) or SDHA (J) siRNA in parental and ARID1A-KO Hep3B cells (mean ± SD; n = 3 independent experiments).
(H and K) Western blot results demonstrating efficient downregulation of ACO2 (H) or SDHA (K) after transfection of various concentrations of siRNA. The relative cell viability rates were assessed subsequent to transfection with varying concentrations of siRNA in parental and ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 6 independent experiments).
Next, these findings were further validated through in vitro cell-based experiments using small interfering RNA (siRNA) constructs to knock down the expression of ACO2 and SDHA, as they were TCA cycle-associated genes among the top candidates identified in this CRISPR screen. Cell growth and cell death rate were then monitored to assess the synthetic lethality phenotypes associated with these genes. Consistent with the above findings, knockdown of either ACO2 (Figures 2F and 2H) or SDHA (Figures 2I and 2K) resulted in pronounced inhibition of ARID1A-KO HCC cell proliferation relative to that of control cells. When flow cytometry was used to examine the survival of these cells, ARID1A-KO cells transfected with these siRNA constructs were confirmed to exhibit markedly increased cell death rates as compared to WT parental (Figures 2G and 2J). Together, these data confirmed the existence of synthetic lethal interactions between ARID1A and TCA cycle-associated genes.
ARID1A-deficient cells undergo reprogramming of glucose metabolism to rely on the TCA cycle rather than glycolysis
Given that these screening analyses highlighted significant synthetic lethal effects between ARID1A and TCA cycle-associated genes, the role of ARID1A as a regulator of glucose metabolism in HCC cells was next explored in further detail. Seahorse analysis was first performed using ARID1A-KO or SNU449 and parental control cells and showed that glycolytic activity was reduced in ARID1A-deficient cells, whereas the basal mitochondrial respiration and maximal respiration rates were increased (Figures 3A, 3B, S3A, and S3B). To extend the findings from Seahorse analysis, we systematically evaluated the mitochondrial status of ARID1A-WT and ARID1A-KO cells through various approaches. Initially, we conducted an assessment into the expression of OXPHOS proteins using the Total OXPHOS antibody cocktail. In comparison to ARID1A-WT cells, there was a significant upregulation in OXPHOS protein levels within the ARID1A-KO cell (Figure 3C). Employing high-resolution microscopy, we further examined the morphological characteristics of cellular mitochondria. This examination revealed that ARID1A-KO cells exhibited elongated tubular mitochondria alongside a reduction in the abundance of globular mitochondria (Figures 3D and S3C). Upon assessment of the mitochondrial membrane potential within these cells, we found that a significant proportion of ARID1A WT cells had a low mitochondrial membrane potential (Figure S3D). In a parallel vein, we assessed the anti-tumor efficacy of IACS-010759 across the specified cells. These combined findings indicate an increased mitochondrial state within ARID1A-deficient HCC cells. Finally, in line with a prior investigation,22 our findings revealed synthetic lethal interaction between ARID1A and the inhibition of OXPHOS IACS-010759 (Figures 3E and S3E). Collectively, these data establish a robust reliance of ARID1A-deficient HCC cells on mitochondrial function and OXPHOS.
Figure 3.

ARID1A deficiency reprogrammed glucose metabolism from glycolysis to the TCA cycle
(A) Representative traces of extracellular acidification rate (ECAR) values obtained from a glycolytic stress test showing reduced glycolysis and glycolytic capacity in ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 6 independent experiments).
(B) Representative traces of oxygen consumption rate (OCR) values obtained from a mitochondrial stress test, indicating increased basal respiration and maximal respiration rates in ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 6 independent experiments).
(C) The expression of OXPHOS protein levels was evaluated in both parental and ARID1A-KO Hep3B cells, utilizing the Total OXPHOS antibody cocktail.
(D) Representative mitochondrial images of ARID1A WT and ARID1A-KO (sg1) Hep3B cells stained with mitochondrial marker. Scale bar, 2 μm.
(E) Cell viability analyses were conducted on the specified cells subsequent to a 3-day treatment with IACS-010759 (mean ± SD; n = 6 independent experiments).
(F) MSEA demonstrating the differential metabolic pathways in ARID1A-KO (sg1) Hep3B cells.
(G) Different levels of glycolysis intermediates and TCA cycle intermediates in parental and ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 4 independent experiments).
(H) Different levels of pyruvate and lactate were measured by quantitative kit in cells characterized by ARID1A deficiency and proficiency (mean ± SD; n = 3 independent experiments).
(I) Schematic representation of 13C-labeled glucose metabolism.
(J) Fractional enrichment (normalized to glucose m + 6) of glycolytic intermediates and TCA intermediates in parental and ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 3 independent experiments).
(K and L) Decreased pyruvate m + 3/PEP m + 3 ratio (K) and increased citrate m + 2/pyruvate m + 3 ratio (L) in ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 3 independent experiments).
Next, a liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based untargeted metabolomics profiling study was performed to more fully assess the effects of ARID1A-KO on the metabolite profiles in Hep3B cells. Metabolite set enrichment analysis (MSEA) of the differentially abundant metabolites demonstrated a significant degree of enrichment in the galactose metabolism, pyruvate metabolism, glycolysis, and TCA cycle pathways (Figure 3F). Of note, glycolysis intermediates including fructose 6-phosphate, glyceraldehyde 3-phosphate, and phosphoenolpyruvic acid (PEP) accumulated at high levels in ARID1A-KO cells together with increased TCA cycle intermediates and ATP production (Figure 3G). Validating the above results, glycolysis intermediates PEP and lactate exhibited elevated levels in ARID1A-proficient cells, in contrast to ARID1A-deficient cells (Figure 3H). This finding was consistent with the observed reduction in glycolysis and increase in mitochondrial respiration in these ARID1A-deficient HCC cells observed in the Seahorse analysis described above. Stable isotope tracing can be used to effectively monitor the energy sources used by tumor cells, with the metabolite labeling ratio providing insight regarding glycolytic and TCA cycle flux. Thus, WT and ARID1A-KO Hep3B cells were incubated with 13C6-glucose, and metabolites were extracted from these cells 24 h later. After normalization to glucose levels, the levels of labeled pyruvate and lactate were markedly reduced in the ARID1A-KO group, whereas no significant changes were evident for other glycolytic metabolites (Figures 3I and 3J). Pyruvate is generated through PEP dephosphorylation during glycolysis. Relative to WT cells, ARID1A-KO cells exhibited a significantly decreased pyruvate m + 3/PEP m + 3 ratio (Figure 3K), indicating that ARID1A deficiency may influence pyruvate production to regulate glycolytic activity. In a parallel vein, the levels of TCA cycle metabolites were increased (Figures 3J and S3F), along with a notable increase in the citrate m + 2/pyruvate m + 3 ratio in ARID1A-KO cells (Figure 3L), suggesting the influx of pyruvate into the TCA cycle and an increase in mitochondrial respiration. Together, these findings indicated evident reductions in glycolytic activity and increases in mitochondrial respiration in ARID1A-deficient HCC cells, suggesting that loss of functional ARID1A expression results in reprogramming of glucose metabolism to favor the TCA cycle over glycolysis. Loss of expression of TCA cycle-associated genes thus conferred pronounced synthetic lethality on these ARID1A-deficient HCC cells.
ARID1A deficiency suppresses glycolytic activity through inhibition of PKM transcription
The processes through which ARID1A deficiency promotes reprogramming of glucose metabolism in HCC cells were next explored in greater depth. In a prior study, we conducted transcriptomic analyses of HepG2 cells in which we knocked down the expression of ARID1A.23 A further review of these data confirmed that decreased ARID1A expression resulted in pronounced rewiring of the transcriptomic landscape of metabolism-related genes. KEGG enrichment analysis demonstrated that these differentially expressed genes were enriched in the glycolysis, pyruvate metabolism, OXPHOS, and glutathione metabolism pathways (Figure 4A). Gene set enrichment analysis (GSEA) further indicated that OXPHOS-pathway-related genes were significantly upregulated in HepG2 cells with ARID1A knockdown (Figure S4A). Notably, significantly reduced expression of PKM, which catalyzes PEP dephosphorylation to generate pyruvate, was evident in these ARID1A-KO cells (Figures 4B and S4B). Reinstating ARID1A expression in ARID1A-deficient cells likewise yielded a downregulation of PKM expression (Figure 4B). Likewise, a significant reduction in PK activity was validated within the ARID1A-KO cells (Figure 4C). Further analysis of the public database revealed a positive correlation between the mRNA and protein levels of ARID1A and PKM in HCC (Figure 4D). The decreases in PKM expression observed in ARID1A-deficient HCC cells were consistent with the dramatic reductions in pyruvate levels detected through stable-isotope-based tracing. The results of IHC experiments in 102 human HCC specimens also indicated that the ARID1A and PKM protein levels were significantly positively correlated (Figure 4E). The TCGA database suggests a similar correlation also exists for lung adenocarcinoma (Figure S4C). However, we did not observe a notable downregulation of PKM upon ARID1A knockdown in NCI-H1975 lung adenocarcinoma cell lines (Figure S4D). We postulated that the effectiveness of ARID1A regulation might depend on the underlying genetic context within a particular tissue type. Taken together, these findings collectively imply that ARID1A deficiency may lead to the downregulation of glycolytic activity through suppression of PKM expression and pyruvate production in HCC cells.
Figure 4.

ARID1A deficiency suppressed glycolysis by decreasing PKM transcription
(A) KEGG enrichment analysis of genes with significantly altered expression in ARID1A-knockdown HepG2 cells.
(B) Indicated cells were examined for expression of ARID1A and PKM by immunoblot.
(C) Measurement of PK activity in ARID1A-KO and control Hep3B cells (mean ± SD; n = 3 independent experiments).
(D) The mRNA and protein level of PKM was positively correlated with that of ARID1A in the public HCC dataset.
(E) Representative images and quantitation of immunostaining for ARID1A and PKM in HCC specimen sections (n = 102). Scale bar, 100 μm.
(F and G) Representative browser track of CUT&RUN or ChIP-seq (F) and ATAC-seq (G) on the PKM locus in indicated cells.
(H) Representative Integrative Genomics Viewer (IGV) tracks of the peak signals in HIF-1α CUT&RUN peaks at the PKM genomic locus.
(I and J) Quantitative PCR (qPCR) and immunoblot analysis revealed more pronounced PKM upregulation in WT cells than in ARID1A-KO (sg1) cells under CoCl2 stimulation (I) or hypoxia condition (J) (mean ± SD; n = 3 independent experiments).
(K) Alterations in spheroid volume were monitored over a span of 10 days for spheroids derived from ARID1A-KO (sg1) and control Hep3B cells (mean ± SD; n = 3 independent experiments). Scale bar, 50 μm.
(L) qPCR and immunoblot analysis show a more conspicuous upregulation of PKM in WT Hep3B cells compared to ARID1A-KO (sg1) cells within the context of 3D culture system (mean ± SD; n = 3 independent experiments).
Next, the mechanisms through which ARID1A regulates PKM expression were explored in greater detail. SMARCA4 is another SWI/SNF complex subunit that has been reported to target metabolic genes directly, thereby governing the regulation of glucose metabolism.22,24 To determine whether ARID1A can directly regulate PKM, CUT&RUN was used to assess the genomic localization of the ARID1A protein in WT Hep3B cells. Our investigation revealed the direct occupancy of the PKM promoter region by ARID1A (Figures 4F, S4E, and S4F). Consistent with these results, mining of published chromatin immunoprecipitation sequencing (ChIP-seq) data confirmed the ability of ARID1A to directly target PKM in HepG2 cells (Figure 4F). These findings suggest that ARID1A may function as a transcriptional activator of PKM such that loss of ARID1A expression suppresses PKM transcriptional activity. ARID1A encodes an important component of the SWI/SNF chromatin remodeling complex that broadly influences the accessibility of chromatin and its associated transcriptional activity. Thus, loss of functional ARID1A expression may also repress PKM transcription by resulting in altered chromatin accessibility. Assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) is an effective approach for surveying chromatin accessibility in cells. ATAC-seq analysis of WT and ARID1A-KO Hep3B cells was next performed and revealed significant decreases in PKM promoter chromatin accessibility in ARID1A-KO cells (Figures 4G and S4G–S4I).
The transcription factor hypoxia-inducible factor 1 (HIF-1) can bind to the PKM promoter as the primary regulatory protein activating the transcription of this gene. To induce the expression of HIF-1α, cobalt chloride (CoCl2) was applied as a hypoxia mimetic agent. Employing the CUT&RUN, We observed a substantial decrease in the binding affinity of HIF-1α to the PKM promoter region, irrespective of the expression level of HIF (Figures 4H and S4–S4L). The results obtained through both qPCR and western blotting demonstrated that ARID1A-KO cells did not display substantial alterations in HIF-1α expression. Nevertheless, the upregulation of PKM was diminished in ARID1A-KO cells under CoCl2 stimulation (Figure 4I). Similar results were validated within hypoxic environments (Figure 4J). Moreover, in vitro explorations utilizing three-dimensional (3D) culture systems exhibited a heightened fidelity in mimicking the intricate tumor microenvironment in vivo compared to conventional two-dimensional (2D) cultures. Under such conditions, a microenvironment marked by hypoxia is established at the central core of the tumor spheroids. It is noteworthy that the 3D environments introduce a compromise in the growth advantage conferred by ARID1A deficiency (Figure 4K). In line with the above observations, the upregulation of PKM in ARID1A WT cells was more pronounced under a 3D environment (Figure 4L). Together, these findings indicate that ARID1A deficiency drives the reprogramming of glucose metabolism from glycolysis to dependence on the TCA cycle and mitochondrial respiration at least partially via suppression of PKM transcription and the associated decrease in glycolytic activity.
ARID1A deficiency increases HCC cell sensitivity to cuproptosis
Cuproptosis is a recently described type of copper-induced cell death in which cells exhibit characteristic destabilization of Fe-S cluster proteins and aggregation of lipoylation of TCA cycle proteins. Thus, cuproptosis primarily affects cells that rely on the TCA cycle and OXPHOS as their primary modes of energy production. Given that most of the hits identified in our CRISPR screen were Fe-S cluster proteins and that the ARID1A-deficient HCC cells were found to be highly dependent on mitochondrial respiration, we speculated that these ARID1A cells may be more sensitive to cuproptotic cell death than WT cells. To test this possibility, cells were exposed to a range of concentrations of copper and the copper ionophore elesclomol, and the half-maximal inhibitory concentration (IC50) values were found to be significantly reduced in ARID1A-deficient cells (Figures 5A and S5A). In the colony formation assay, ARID1A-KO cells exhibited enhanced copper sensitivity over the 10-day treatment period (Figures 5B and S5B). Elesclomol also significantly inhibited the growth of tumor spheroids formed by ARID1A-KO Hep3B cells but not WT cells cultured in ultra-low-attachment round-bottom 96-well plates (Figure 5C). Then, we tried to determine whether treatment with copper can reduce the expression of TCA cycle proteins and found that copper treatment downregulated the expression of ACO2, SDHA, and succinate dehydrogenase complex iron sulfur subunit B (SDHB). The degree of downregulation was positively correlated with the concentration of copper (Figure 5D). To better explore the forms of cell death induced in response to copper in these ARID1A-deficient cells, ferrostatin-1 (a ferroptosis inhibitor), necrostatin-1 (a necrosis inhibitor), Z-VAD-FMK (an apoptosis inhibitor) and tetrathiomolybdate (a copper chelate) were applied to mitigate the induction of cell death. Treatment with Z-VAD-FMK partially inhibited copper-induced cell death in both WT and ARID1A-KO cells, whereas neither ferrostatin-1 nor necrostatin-1 had an impact on either cell type. However, tetrathiomolybdate significantly inhibited copper-induced cell death in cells (Figure S5C). This pattern revealed that ARID1A deficiency increases the sensitivity of HCC cells to cuproptosis but not to other types of cell death in response to copper treatment. Mitochondrial respiration rates were also assessed in copper-treated cells using a Seahorse analyzer, and copper failed to significantly decrease basal respiration but did moderately decrease the spare respiratory capacity in WT cells. In contrast, significant decreases in both the basal and spare respiratory capacities were evident in ARID1A-KO Hep3B cells (Figure 5E).
Figure 5.

ARID1A deficiency sensitized cells to cuproptosis in vitro
(A) Cell viability of indicated cells after treatment with increasing concentrations of elesclomol (with 1 μM CuCl2 in medium) or CuCl2 (with 10 nM elesclomol in medium) (mean ± SD; n = 6 independent experiments).
(B) Colony formation assay of HepG2 cells treated with different concentrations of elesclomol and CuCl2 for 10–15 days (mean ± SD; n = 3 independent experiments).
(C) Longitudinal changes in the volume of spheroids formed from ARID1A-KO (sg1) and control Hep3B cells under elesclomol or vehicle (with 1 μM CuCl2 in medium) treatment (mean ± SD; n = 3 independent experiments). Scale bar, 50 μm.
(D) ACO2, SDHA, and SDHB protein levels after treatment with different concentrations of elesclomol (with 1 μM CuCl2 in medium) or CuCl2 (with 25 nM elesclomol in medium).
(E) Representative traces of OCR values after treatment with 10 nM elesclomol (with 1 mM CuCl2 in medium) indicating decreased basal respiration and maximal respiration rates in ARID1A-KO (sg1) Hep3B cells (mean ± SD; n = 6 independent experiments).
(F and G) Immunoblotting revealed significant FDX1 (F) and lipoylated proteins (G) upregulation in ARID1A-KO HCC cells.
(H) Images of FDX1 and ARID1A IHC staining in HCC specimen sections (n = 102). Scale bar, 100 μm.
Regarding the specific metabolic pathways involved in mediating copper-related toxicity, Tsvetkov et al. posited that ferredoxin 1 (FDX1) may be a key mediator of copper-induced cell death. By removing the sulfur group from cysteine, the iron-sulfur cluster core complex results in the production of Fe-S clusters in an FDX1-dependent manner. The elesclomol-Cu complex can bind directly to FDX1 and inhibit its function in this Fe-S cluster biosynthetic pathway.25 FDX1 also functions upstream, where it regulates protein lipoylation. Copper can bind to lipoylated proteins, ultimately leading to induction of cuproptosis. Loss of FDX1 function can thus render cells resistant to copper treatment. The increased copper sensitivity of ARID1A-deficient HCC cells suggested that FDX1 and lipoylated proteins may be upregulated in these cells. Consistent with this hypothesis, analysis of the TCGA database revealed that the expression of ARID1A and FDX1 was negatively correlated in HCC and other cancer types (Figure S5D). The results of western blotting further confirmed that FDX1 and lipoylated proteins was upregulated in ARID1A-KO cells (Figures 5F and 5G), and IHC staining for FDX1 and ARID1A in human HCC samples confirmed the strong negative correlation between two proteins (Figure 5H). Notably, further review of our screening data confirmed that FDX1, its ligand FDXR, and the isoenzyme FDX1L all exhibited clear synthetic lethal effects with ARID1A (Figure S5E), indicating that ARID1A-deficient HCC cells exhibit increased reliance on the function of FDX1 for their survival. These results additionally confirmed that ARID1A-deficient HCC cells are more susceptible to copper treatment, owing to the induction of cuproptosis.
Elesclomol can effectively treat ARID1A-deficient HCC in vivo
Given the promising sensitizing effects observed in vitro in this study, the potential therapeutic efficacy of the copper ionophore elesclomol as a treatment for ARID1A-deficient HCC was further explored in vivo using two different mouse model systems. First, the potential difference in the antitumor efficacy of elesclomol in vivo was explored in mice subcutaneously implanted with WT or ARID1A-KO Hep3B cells (cell-derived xenograft [CDX] models) (Figure 6A). In this model, elesclomol treatment was sufficient to significantly inhibit the growth of ARID1A-KO tumors without obviously affecting WT tumors (Figures 6B, 6C, and S6B). These results confirmed the marked selectivity of this therapeutic strategy for treating ARID1A-deficient HCC, with a corresponding improvement in the survival of tumor-bearing mice in the ARID1A-KO group (Figure 6D). Notably, ARID1A-KO significantly increased the expression of the cell proliferation marker Ki67 in tumors formed from ARID1A-KO Hep3B cells, while reduced expression of PKM was also observed in the xenograft tumors (Figure S6A). Given that ARID1A loss-of-function mutations are commonly observed in human HCC patients, patient-derived xenograft (PDX) mouse models were further established to test whether these naturally occurring mutations are similarly associated with dependency on the TCA cycle (Figure 6E). IHC staining and whole-exome sequencing were used to confirm the ARID1A mutation status of these HCC patient specimens (Figure 6F). IHC staining and immunoblots were used to assessed the ARID1A expression of the dissociated tumor derived from the PDX model (Figures 6F and S6C).
Figure 6.

Clinically applicable elesclomol was effective in ARID1A-deficient HCC
(A) Schematic representation of the experimental design and reference time of the CDX model experiment.
(B and C) Longitudinal variations in tumor volume (B) and the ultimate tumor weight measurements (C) were examined in mice treated with elesclomol or vehicle controls (mean ± SD; n = 6 independent experiments).
(D) After treatment cessation, mice from the elesclomol treatment groups were followed for survival analysis by the Kaplan-Meier method (n = 6 independent experiments).
(E) Schematic representation of the experimental design of the PDX model experiment.
(F) The expression of ARID1A in ARID1A WT and ARID1A mutant HCC specimens and PDX models. Scale bar, 100 μm. ARID1A enlarged, 25 μm.
(G) Tumor volumes and weights of mice bearing tumors of PDX models treated with elesclomol or vehicle controls.
(H) Representative images of Ki67, SDHA, and FDX1 staining in PDX tumor samples harvested from mice in the vehicle and elesclomol treatment groups. The H-score was then quantified (mean ± SD; n = 6 independent experiments). Scale bar, 100 μm.
Consistent with the cell-line-based results, elesclomol treatment significantly reduced the tumor burden in mice bearing PDX tumors harboring an ARID1A nonsense mutation (p.E193∗) compared with tumors expressing WT ARID1A (Figure 6G). IHC staining of these tumors additionally revealed that elesclomol treatment significantly decreased the SDHA, FDX1, and Ki-67 levels in these tumors (Figure 6H). Importantly, these mice tolerated elesclomol well, without significant weight loss or apparent morphological changes in major organs as assessed via H&E staining (Figures S6D and S6E). Elesclomol or other copper ionophores may thus be promising tools for treating patients with ARID1A-deficient HCC.
Discussion
Advanced genomic profiling technologies have increasingly enabled the identification and characterization of driver mutations and genes associated with cancer onset and progression,26 allowing clinical efforts to design small-molecule inhibitors or blocking antibodies that target activated oncogenes to achieve clinical benefit. ARID1A has long been known to be a tumor-suppressor gene that is frequently mutated in HCC and other cancer types. Tumor suppressor genes are generally much harder to target than oncogenes; thus, the clinical utility of this knowledge has been limited to date. Synthetic lethality strategies, however, offer an opportunity to define therapeutic targets for cancers driven by mutations in tumor suppressor genes. In this study, we conducted an unbiased CRISPR-Cas9 screen to identify synthetic lethal interaction between ARID1A and the TCA cycle. Specifically, ARID1A deficiency resulted in reprogramming of glucose metabolism from glycolysis to mitochondrial respiration. Based on these results, we proposed that efforts to target the TCA cycle or OXPHOS may lead to the development of a viable means of treating ARID1A-deficient HCC. These results provide a promising approach for the personalized and efficacious treatment of these cancer patients.
Tumor cells undergo metabolic reprogramming to adapt to the increasing demand for energy necessary to support their high level of biosynthetic activity. Metabolic activity has been a major focus of cancer treatment research since the Warburg effect was first documented. While aerobic glycolysis is the primary glucose metabolism pathway active in most cancer cells, accumulating evidence indicates that certain cancer cells generate energy primarily through reliance on other metabolic pathways, particularly in the setting of dysregulated oncogene and tumor suppressor expression. For example, cancers harboring inactivating mutations in the TCA cycle enzyme FH contain defective mitochondria, and inhibiting phosphogluconate dehydrogenase (PGD) in these FH-mutant tumor cells can effectively suppress their proliferative activity.27 In this study, the survival of ARID1A-deficient HCC cells was found to rely heavily on mitochondrial respiration; thus, targeting the TCA cycle may be an attractive treatment strategy. ARID1A deficiency has previously been reported to activate Ang2-dependent angiogenesis and to be correlated with an increase in vascular density in HCC tumors, with these vessels being required to supply adequate levels of oxygen, consistent with the present results.28 Recent studies further found that loss of the SWI/SNF complex component SMARCA4 similarly led to increased oxygen consumption and mitochondrial respiration in lung tumor cells,22,24 suggesting that other processes that interfere with the function of the SWI/SNF complex may similarly drive reprogramming of glucose metabolism in cancer. Approximately 20% of solid tumors are thought to harbor a mutation in at least one component of the SWI/SNF complex,29 although further research is necessary to determine whether these tumors harbor targetable vulnerabilities associated with mitochondrial respiration.
Multi-omics profiling of ARID1A-deficient HCC cells was further performed to clarify the mechanistic basis for glucose metabolism reprogramming following loss of ARID1A expression and to explore the link between ARID1A and transcriptional repression of PKM. Pyruvate kinase is a critical enzyme involved in the regulation of tumor metabolism, and the ability of ARID1A to suppress PKM transcription activates a metabolic switch that results in suppression of glycolytic activity and increased dependence on mitochondrial respiration, as confirmed by the altered metabolome profiles in these HCC cells. Consistent with the present findings, prior research revealed that ARID1A can function as a transcriptional repressor for GLS1, such that loss of ARID1A expression increased the expression of GLS1 and rendered cells dependent on glutamine metabolism.16 The expression of various metabolic genes can also be regulated by other SWI/SNF complex subunits, including BRG1 and SMARCA4, thereby facilitating metabolic reprogramming.22,30 These results suggest that the SWI/SNF complex may serve as a fundamental, evolutionarily conserved mediator of the efficient expression of key metabolism-related genes. HIF-1α has previously been shown to directly target PKM, leading to its transcriptional upregulation and the induction of glycolytic activity. Through a CUT&RUN analysis, ARDI1A deficiency was herein found to impair HIF1α binding to the PKM promoter region, thus suppressing its upregulation and the associated HIF1α-mediated induction of glycolysis. HCC is often treated through transcatheter hepatic arterial embolization (TAE) and transcatheter hepatic arterial chemoembolization (TACE) strategies, in which tumor vessels are embolized,31 thereby exposing tumor cells to ischemic and hypoxic stress that can promote HIF-1α activity and the induction of glycolysis. These events enable these cells to survive and proliferate under these hypoxic conditions and represent a major mechanism underlying tumor therapeutic resistance. Here, ARID1A deficiency was found to interfere with the relationship between HIF-1α upregulation and glycolysis, rendering HCC cells reliant on oxygen consumption to survive. This finding raises the possibility that patients with ARID1A-deficient HCC may exhibit greater sensitivity to TAE or TACE, given that these strategies further limit intratumor oxygen availability and OXPHOS.
Copper is an important enzymatic cofactor necessary for the maintenance of normal cellular activity in mammals. However, intracellular copper levels are extremely low because even small concentrations of free intracellular copper can have harmful effects.32 Efforts to target copper homeostasis have thus been proposed as a means of treating various cancers, although no clinical trial employing copper ionophores or chelators has demonstrated improved patient survival outcomes.33 Thus, further identification of copper-sensitive cancer subtypes may provide a more informed basis for cancer treatment. Tsvetkov and co-workers recently clarified the mechanisms underlying copper-mediated cytotoxicity by determining that copper can directly bind to lipoylated TCA cycle components, thereby resulting in lipoylated protein aggregation, iron-sulfur cluster protein loss, and consequent cell death through a process termed cuproptosis. Given that copper treatment can directly target the TCA cycle, cells that rely on mitochondrial respiration exhibit a 1,000-fold increase in copper sensitivity compared to cells with active glycolytic processes. Given their pronounced dependence on mitochondrial respiration, ARID1A-deficient cells were, unsurprisingly, highly sensitive to copper treatment. Thus, these results provide a promising foundation for the personalized treatment of patients with ARID1A-deficient HCC through the induction of cuproptosis and also emphasize the value of ARID1A as a biomarker when selecting patients likely to benefit from copper therapy.
In conclusion, the present results highlighted a relationship between ARID1A and TCA cycle-related genes that confers synthetic lethality on HCC cells. Subsequent multi-omics profiling demonstrated that loss of ARID1A results in metabolic reprogramming such that these tumor cells are reliant on the TCA cycle rather than glycolysis. Thus, the use of copper to target the TCA cycle is a promising effective means of treating patients with ARID1A-deficient HCC.
Limitations of the study
While we have extensively verified the pivotal role of PKM in driving metabolic reprogramming within ARID1A-deficient HCC cells. It is noteworthy that other key regulators of metabolism, including HK2, HIF2A, GLUT1, and GLS1, have been demonstrated to participate in the metabolic switching induced by altered SWI/SNF subunits.16,22,24 Hence, it is essential to explore further mechanisms underlying this metabolic reprogramming in HCC cells induced by ARID1A deficiency. Approximately 20% of solid malignancies are estimated to harbor mutations in at least one component of the SWI/SNF complex. It is valuable to explore the presence of similar metabolic shifts in other types of tumors. Significantly, our findings indicate that the effectiveness of ARID1A regulation might depend on the underlying genetic context within a particular tissue type. This inconsistency might necessitate a more thorough investigation. Finally, although we have validated the tolerability and efficacy of elesclomol in preclinical models, it remains essential to emphasize the need for further investigation into the safety profile of copper treatment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ARID1A | Cell Signaling Technology | Cat# 12354; RRID:AB_2637010 |
| ACO2 | Abcam | Cat# ab129069; RRID:AB_11144142 |
| SDHA | Abcam | Cat# ab14715; RRID:AB_301433 |
| SDHB | Abcam | Cat# ab175225; RRID:AB_2904585 |
| FH | Abcam | Cat# ab233394; RRID:AB_2892589 |
| PKM | Abcam | Cat# ab150377; RRID:AB_2941747 |
| FDX1 | Proteintech Group Inc | Cat#12592-1-AP; RRID:AB_11182486 |
| HIF-1 alpha | Abcam | Cat# ab179483; RRID:AB_2732807 |
| α-tubulin | Abcam | Cat# ab7291; RRID:AB_2241126 |
| Lipoic Acid | Abcam | Cat# ab58724; RRID:AB_880635 |
| Total OXPHOS Human WB Antibody Cocktail | Abcam | Cat# ab110411; RRID:AB_2756818 |
| ARID1A | Abcam | Cat# ab182560 |
| Bacterial and virus strains | ||
| Toronto human knockout pooled library (TKOv3) | Addgene | Cat# 125517; RRID:Addgene_125517 |
| pLenti-puro-ARID1A | Addgene | Cat# 39478; RRID:Addgene_39478 |
| lentiCas9-Blast | Addgene | Cat# 52962; RRID:Addgene_52962 |
| psPAX2 | Addgene | Cat# 12260; RRID:Addgene_12260 |
| pMD.2G | Addgene | Cat# 12259; RRID:Addgene_12259 |
| Biological samples | ||
| Hepatocellular Carcinoma samples | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| IACS-010759 | MedChemExpress | Cat# HY-112037 |
| ferrostatin-1 | MedChemExpress | Cat# HY-100579 |
| Necrostatin-1 | MedChemExpress | Cat# HY-15760 |
| Z-VAD-FMK | MedChemExpress | Cat# HY-16658B |
| Ammonium tetrathiomolybdate(VI) | MedChemExpress | Cat# HY-W076067 |
| Elesclomol | MedChemExpress | Cat# HY-12040 |
| Puromycin | MedChemExpress | Cat# HY-B1743A |
| Hygromycin B | MedChemExpress | Cat# HY-B0490 |
| Polybrene Infection | Sigma-Aldrich | Cat# TR-1003 |
| Copper(II) chloride | Sigma-Aldrich | Cat# 7447-39-4 |
| MitoBright LT Deep Red | Dojindo | Cat# MT12 |
| TB Green® Premix Ex Taq™ II | Takara | Cat# RR820Q |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat# 11668019 |
| 13C-Glucose | Cambridge Isotopes Laboratories | Cat# CLM-1396 |
| NEBNext® Ultra™ II Q5® Master Mix | New England Biolabs | Cat# M0544S |
| Critical commercial assays | ||
| Pyruvate Assay Kit | Abcam | Cat# ab65342 |
| L-Lactate Assay Kit (Colorimetric) | Abcam | Cat# ab65331 |
| Pyruvate Kinase Assay Kit | Abcam | Cat# ab83432 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent | Cat# 103015-100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103020-100 |
| CUT&RUN Assay Kit | Cell Signaling Technology | Cat# 86652 |
| TIANamp Genomic DNA Kit | TIANGEN | Cat# DP304 |
| Presto Blue™ Cell Viability Reagent | Invitrogen™ | Cat# A13262 |
| DNA Library Prep Kit for Illumina Systems | Cell Signaling Technology | Cat# 56795 |
| JC-1 MitoMP Detection Kit | Dojindo | Cat# MT09 |
| Deposited data | ||
| ChIP-seq sequencing data | Raab et al.34 | GSE69566 |
| CHCC-HBV cohort proteomic data | Gao et al.21 | https://doi.org/10.1016/j.cell.2019.08.052 |
| ATAC sequencing data | This study | GSE228179 |
| CUT&RUN sequencing data | This study | GSE228179 |
| CRISPR/Cas9 screen sequencing data | This study | GSE228179 |
| Experimental models: Cell lines | ||
| Hep3B | Cell Resource Center, IBMS, CAMS/PUMC | Cat# 1101HUM-PUMC000376 |
| HepG2 | Cell Resource Center, IBMS, CAMS/PUMC | Cat# 1101HUM-PUMC000035 |
| 293T | Cell Resource Center, IBMS, CAMS/PUMC | Cat# 1101HUM-PUMC000091 |
| NCI-H1975 | Cell Resource Center, IBMS, CAMS/PUMC | Cat# 1101HUM-PUMC000252 |
| SNU449 | ATCC | Cat# CRL-2234 |
| Experimental models: Organisms/strains | ||
| NOD SCID | Charles River | N/A |
| NOG | Charles River | N/A |
| Oligonucleotides | ||
| ARID1A Knockout sgRNA sequences ARID1A-sg1: 5′- CAGCAGAACTC TCACGACCACGG -3’ (Exon 1) |
This paper | N/A |
| ARID1A Knockout sgRNA sequences ARID1A-sg2: 5′- CCTGTTGACCATACCC GCTGGGG -3’ (Exon 3) |
This paper | N/A |
| siRNA sequences | This paper | Table S1 |
| Primer for qPCR or Cut&Run-qPCR | This paper | Table S1 |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad | https://www.graphpad.com/ |
| ImageJ (version 2.9.0) | NIH | https://ImageJ.nih.gov/ij/download.html |
| FlowJo (version 10.7.2) | BD Biosciences | https://www.flowjo.com/ |
| GATK4 | Broad institute | https://software.broadinstitute.org/gatk/ |
| Fiji software | Schindelin et al.35 | https://ImageJ.net/software/fiji/downloads |
| R (v4.1.1) | R | https://cran.r-project.org/ |
| MAGeCK algorithm (v0.5.9.4) | Li et al.36 | https://sourceforge.net/p/mageck/wiki/ |
| GSEA (v4.1.0) | UC San Diego | https://www.gsea-msigdb.org/gsea/index.jsp |
| LAS X Office | Leica | https://www.leica-microsystems.com.cn/cn/products/microscope-software/p/leica-las-x-ls/downloads/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jun Liang (liangjun1959@aliyun.com).
Materials availability
This study did not generate new reagents.
Data and code availability
-
•
All sequence data have been deposited in the GEO database under accession number GSE228179.
-
•
This study did not generate any novel code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines
Hep3B, HepG2, 293T, and NHI-H1975 cell line were obtained from the Cell Resource Center, Peking Union Medical College (which is part of the National Science and Technology Infrastructure, the National Biomedical Cell-Line Resource, NSTI-BMCR. http://cellresource.cn). SNU449 cell lines was acquired from the American Type Culture Collection (ATCC). Hep3B, HepG2, SNU449 and 293T were cultured in Dulbecco’s modified Eagle’s (DMEM) high glucose medium (DMEM; Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin in a 5% CO2 37°C incubator. NHI-H1975 cell line was cultured in RPMI 1640 (RPMI-1640; Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Murine xenograft models
All mouse studies were conducted in accordance with the Institute of Biophysics, Chinese Academy of Science’s Policy on the Care and Use of Laboratory Animals. Nod-SCID and NOG mice were purchased from Charles River.
CDX models were established by Hep3B ARID1A WT or Hep3B ARID1A-KO (sg1) cells (1 × 107 cells in 200 μL PBS) were implanted subcutaneously into 6–8 weeks old female Nod-SCID mice. After a 1-week period during which tumors were allowed to develop, the mice were randomized into groups (n = 6 mice/group) and treated three times per week for three weeks with either vehicle control (DMSO) or elesclomol (10 mg/kg b.w.). Tumor growth was monitored by slide caliper 3 times a week.Tumor volume was calculated as follows: tumor volume = (length × width2)/2. The survival endpoint for this assay was a maximum tumor diameter of >20 mm.
PDX models were established using HCC tissue samples harvested from patients who had provided written informed consent and undergone surgical liver tumor resection. These tumor tissues were minced into small (∼5 mm3) pieces that were directly subcutaneously implanted into 6–8 weeks old female NOG mice. The resultant xenograft tumors were passaged three times and used for subsequent experiments. As described above, mice were treated three times per week for three weeks using elesclomol (10 mg/kg b.w.). Tumors were then surgically resected, and the tumor burden was assessed based on the measurement of tumor weight.
Method details
CRISPR/Cas9 mediated gene knockout
CRISPR knockout was achieved by using a lentiviral vector encoding the Cas9 plasmid (AddGene; #52962) to establish cells stably expressing Cas9. The lentiviral plasmid for knockout of ARID1A was obtained from GenePharma. Lentiviral production was performed by using polyethylenimine (Proteintech; #PR40001) to cotransfect 293T cells in 10 cm plates with 10 μg of the targeting plasmid, 7.5 μg of psPAX2 (Addgene; #12260), and 2.5 μg of pMD2.G (Addgene; #12259). Initially, HCC cells were infected with the appropriate lentiviral particles in the presence of polybrene (10 μg/mL) (Sigma‒Aldrich; #TR-1003) for 24 h, after which 100–200 μg/mL hygromycin B was used to select for successfully transduced cells. The ARID1A knockout efficiency was confirmed via western blotting. The structures of the lentiviral vectors are presented in Figure S1A. The ARID1A knockout sgRNA sequences were as follows:
ARID1A-sgRNA1: 5′- CAGCAGAACTCTCACGACCACGG -3’ (Exon 1).
ARID1A-sgRNA2: 5′- CCTGTTGACCATACCCGCTGGGG -3’ (Exon 3).
CRISPR/Cas9 screening
The TKOv3 library comprising 70,948 sgRNAs targeting 18,053 genes was a gift from Jason Mofat (Addgene #125517). Protocols provided by the Mofat laboratory were used for library amplification and viral preparation. In brief, cells stably expressing Cas9 were infected at a low multiplicity of infection (0.2–0.3) with this lentiviral library to achieve approximately 500-fold coverage in each group, and the cells were incubated with 2–5 μg/mL puromycin for 3 days to select for successfully transduced cells. A portion of the transfected cells was harvested at baseline (T0), while the remaining cells were expanded for 21 days prior to collection (T21). A TIANamp Genomic DNA Kit (#DP304; TIANGEN) was used to isolate DNA from these cells based on the provided instructions, with sgRNA barcode sequences amplified via PCR(New England Biolabs; #M0544S), followed by deep sequencing of the resultant products. The resultant sgRNA reads were mapped to appropriate sgRNA sequences using Bowtie2, after which a ranked list of fitness-related genes required for the survival of these cells was generated using the MAGeCK algorithm.
siRNA-mediated gene knockdown
All siRNA duplexes and negative control constructs were obtained from GenePharma (Beijing, China). Cells were transfected with either ACO2, SDHA, or ARID1A siRNA constructs using Lipofectamine 2000 (Invitrogen; #11668019) based on the provided instructions. Western blotting was used to assess the knockdown efficiency 48h post-transfection. The siRNA sequences used in this study are listed in Table S1.
ARID1A overexpression
The pLenti-puro-ARID1A plasmid was a kind gift from Ie-Ming Shih (Addgene; #39478) and lentiviral production was performed as described above. The resultant lentiviral particles were used to transduce SNU449 and Hep3B KO (sg1) cells, 2–4 μg/mL puromycin was used to select for cells overexpressing ARID1A. ARID1A overexpression was confirmed by western blotting.
Cell viability assays
For short-term viability assays, cells were plated in 96-well flat-bottom plates (2500–10000 cells/well) and allowed to adhere for 12h prior to treatment. Presto Blue Cell Viability Reagent (Invitrogen; #A13262) was used to assess cell viability based on the provided instructions, with the absorbance at 570 nm in each well measured using a microplate reader (EnSpire; PerkinElmer, CA, USA). Cell viability was calculated by normalizing the absorbance to that of DMSO-treated controls.
Clonogenic assays were performed by plating wells in 6-well or 12-well plates (800/well and 300/well, respectively) followed by drug treatment for 10 days beginning 24 h following seeding or until visible colonies composed of more than 50 cells were identified. Cells were then fixed using 10% formaldehyde and stained using 0.4% crystal violet. At least three replicate samples were analyzed for all assays.
Flow cytometry
Flow cytometric analysis was performed to determine the cell death rate or mitochondrial membrane potential. Cells were collected and washed twice with ice-cold PBS and then incubated with JC-1 Dye (Dojindo; #MT09) or PE Annexin V and 7-AAD(Biolegend; #640934) for 15 min. Finally, cell death rate and mitochondrial membrane potential was detected using FCM (BD Biosciences) and analyzed using FlowJo software.
IncuCyte imaging
Cells were seeded in 96-well plates and images were taken every 12 h using the IncuCyte Live-Cell Analysis Imaging System. Cell proliferation was determined based on cell confluence.
3D culture system
To generate spheroids, ARID1A-KO or vehicle cells suspended in complete DMEM medium were seeded in 96-well round-bottomed ultra-low attachment microplate (Corning) or 60 mm Ultra-Low Attachment Culture Dish (Corning). The plates were incubated for a duration of 10 days, with images captured daily. Spheroid volumes were assessed using a modified ellipsoidal formula. , spheroid volume = (length × width2)/2.
Western blotting
Lysis buffer (KeyGen BioTECH; #KGP250) was used to extract total protein from cells on ice, after which these lysates were separated via 12.5% SDS‒PAGE (70 min, 120 V) and transferred on ice onto PVDF membranes (Merck Millipore; #IPVH00010) using transfer buffer containing 10–20% methanol (1–2.5 h, 300 mA). The membranes were blocked using 5% nonfat milk prior to incubation with appropriate primary and secondary antibodies and imaging with a chemiluminescent substrate (Merck Millipore; #WBKLS0100) and Bio-Rad Gel Doc 2000 analytical software. The membranes were then stripped with Western Blot Fast Stripping Buffer (EpiZyme; # PS107) and incubated with the next antibodies as appropriate.
Antibodies specific for the following proteins were used for western blotting: ARID1A (Cell Signaling Technology; #12354; 1:1,000 dilution), ACO2 (Abcam; #ab129069; 1:1,000 dilution), SDHA (Abcam; #ab14715; 1:1,000 dilution), SDHB (Abcam; #ab175225; 1:1,000 dilution), FH (Abcam; #ab233394; 1:1,000 dilution), PKM(Abcam; #ab150377; 1:1,000 dilution), FDX1 (Proteintech Group Inc.; #12592-1- AP; 1:1,000 dilution), HIF-1 alpha (Abcam; #ab179483; 1:1,000 dilution), Total OXPHOS Human WB Antibody Cocktail (Abcam; #ab110411; 1:1,000 dilution), Lipoic Acid (Abcam; # ab58724; 1:1,000 dilution)and α-tubulin (Abcam; #ab7291; 1:5,000 dilution).
IHC staining
HCC tissue samples were obtained from 102 patients undergoing surgical treatment at Peking University International Hospital. These patients had not undergone any cancer-related treatment prior to surgery. An Immuno-Histo Stainer was used for IHC staining. IHC scoring of tissue samples from these patients was independently performed by two pathologists, with the total immunostaining score computed by multiplying the percentage and intensity scores. ARID1A expression was classified as low, medium or high based on the IHC score.
Primary antibodies against the following proteins were used: ARID1A (Abcam; #182560; 1:250 dilution), SDHA (Abcam; #ab14715; 1:250 dilution), PKM (Abcam; #ab150377; 1:250 dilution), and FDX1 (Proteintech Group Inc; #12592-1- AP; 1:200 dilution).
Mitochondrial staining
Cells were subjected to staining with the mitochondria-specific dye MitoBright LT Deep Red(100 nM) (Dojindo; MT11) for 30 min. High-resolution mitochondrial images were acquired utilizing the Leica TCS SP8 STED 3× confocal microscope (Objective: HC PL APO CS2 100x/1.40 OIL; STED Laser: 775nm). The captured images were deconvoluted using LAS X software (4.5.0) (Leica). The area of mitochondria was quantified using Fiji software (https://imagej.net/software/fiji/downloads).
Seahorse analysis
An XFe96 Extracellular Flux (XF) Analyzer (Seahorse Bioscience) was used to measure the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). The day before this experiment, cells were seeded in XF96 Cell Culture Microplates (10,000 cells/well). The following day, the medium was replaced with XF Glycolysis Stress Test Assay Medium or XF Cell Mito Stress Test Assay Medium, and the cells were incubated for 1 h in a 37°C CO2-free incubator to allow the equilibration of pH and temperature values. Glycolytic stress testing was performed by sequentially injecting glucose, oligomycin, and 2-deoxy-D-glucose at final concentrations of 10 mM, 2 μM, and 50 mM, respectively. Mitochondrial stress testing was performed by sequentially injecting oligomycin, FCCP, and a mixture of antimycin and rotenone at final concentrations of 2 μM, 1 μM, and 4 μM, respectively. OCR and ECAR values were normalized based on cell numbers after completion of these experiments.
Metabolic analyses
For untargeted metabolomics, cells were collected in an EP tube and resuspended with ice-cold 80% methanol, followed by sonication for 5 min at centrifugation at 14,000 xg at 4°C. Supernatants were subsequently freeze-dried, dissolved using 10% methanol, and injected into an LC‒MS/MS system. Raw data were processed with Compound Discoverer 3.1 (Thermo Fisher) to facilitate peak alignment, peak selection, and quantitation for individual metabolites. Differentially abundant metabolites were identified based on a threshold value of <0.05 and a fold change of ≥1.5. Metabolite set enrichment analysis (MSEA) was conducted with MetaboAnalyst 5.0 (https://www.metaboanalyst.ca) to assess the enrichment of these differentially abundant metabolites.
Metabolic flux analyses were performed by seeding cells in standard growth media for 24 h, after which these cells were rinsed two times with PBS and media was exchanged for glucose-free media for 12 h. This media was subsequently exchanged for media containing 20 mM of U-13C glucose (99% pure, Cambridge Isotope Laboratories; #CLM-1396). After incubation for 36 h, cells were rinsed two times using chilled saline and metabolite extraction and analysis were performed as above.
The MS measurement of isotopologue distribution is analyzed via a Thermo Q-Exactive plus hybrid quadrupole–orbitrap mass spectrometer coupled to a Thermo Vanquish UPLC system.Data processing and ion annotation based on accurate mass were performed in TraceFinder 5.0 (Thermo Fisher) and Xcalibur 4.0 (Thermo Fisher).Correction for natural abundance is required prior to meaningful interpretation. The correction used both general applicable correction matrix based on Eqn and isotopic correction matrix that was generated by treating a control group of unlabeled sample.
L-lactate and pyruvate assay
L-lactate and pyruvate levels were determined using The L- Lactate Assay Kit (Abcam, # ab65331) and the Pyruvate Assay Kit (Abcam, # ab65342), respectively, according to the provided instructions. The samples were measured using a microplate reader and the amount of lactate was calculated according to a standard curve with normalization to the number of cells.
PK activity assay
A Pyruvate Kinase Assay (#ab83432; Abcam) was used based on the provided instructions. In brief, 2x106 cells were extracted with 4 volumes of assay buffer and centrifuged to yield a clarified extract. Then, 50 μL of the reaction mixture was added per well, and the absorbance at 570 nm was measured at baseline (T1) and following incubation for 20 min in the dark at 25°C (T2). An increase in the signal was considered to result from PK-mediated pyruvate generation. PK activity was then calculated based on the provided instructions.
ATAC-seq
ATAC-seq was performed as described in prior reports. In brief, 1 × 104 cells cultured under normal conditions were harvested and rinsed two times with chilled PBS, and unfixed nuclei from these cells were tagged for 30 min with tn5 transposase (TruePrep DNA Library Prep Kit V2 for Illumina) at 37°C. Library fragments were then generated through 10–12 PCR cycles, and sequencing was then conducted with an Illumina NovaSeq 6000 instrument. Bowtie2 was used to align the clean paired-end reads to the reference genome (GRCh38/hg38), and peak distributions on genes of interest were visualized with IGV. Two biological replicates were performed for each sample group.
CUT&RUN
CUT&RUN was conducted using a CUT&RUN Assay Kit (Cell Signaling Technology; #86652). In brief, 1 × 105 freshly cultured cells were harvested, rinsed two times with cold PBS, and washed once with 1 mL of wash buffer. Concanavalin A Bead Activation Buffer was also used to wash Concanavalin A Magnetic Beads two times. For each reaction, 10 μL of these activated beads was added to washed cells prior to the addition of 1 μg of antibody and incubation for 2 h at 4°C. The tubes were then transferred to a magnetic rack to separate the beads from the liquid fraction. The beads were rinsed using 1 mL of digitonin buffer, and 50 μL of pAG-MNase premix was added to each reaction prior to incubation for 1 h at 4°C. After the addition of 3 μL of cold calcium chloride to activate the pAG-MNase in each tube, the samples were incubated for an additional 30 min at 4°C. Then, 150 μL of 1× Stop Buffer was added to each sample prior to incubation for 10 min at 37°C. The tubes were then centrifuged for 2 min at 16,000 x g and 4°C and placed onto magnetic racks until the solution had clarified. Supernatants containing enriched chromatin samples were then transferred into new tubes, and DNA was purified from these samples with DNA Purification Buffer and Spin Columns. A DNA Library Prep Kit for Illumina (#56795; Cell Signaling Technology) was utilized to prepare an NGS sequencing library, which was then subjected to 150 bp paired-end sequencing with an Illumina NovaSeq 6000 instrument. Bowtie2 was used to map the reads to the GRCh38/hg38 reference genome, and SAMtools was used to remove duplicate PCR sequences. Enriched peaks were identified using the MACS2 peak calling algorithm, and peak distributions along genomic regions of interest were assessed using Integrative Genomics Viewer (IGV). Two biological replicates were analyzed for each sample group.
qPCR and CUT&RUN–qPCR analyses
TRIzol (Invitrogen) was used to extract total cellular RNA based on the provided instructions, after which PrimeScript RT Master Mix (TaKaRa; # RR036A) was used to prepare cDNA. All qPCRs were performed in a 20 μL volume containing TB Green reagent (#RR820A; TaKaRa). β-Actin was used for normalization of mRNA expression. CUT&RUN–qPCR was used to quantify ARID1A and HIF-1 enrichment at the PKM promoter. Enrichment results were analyzed by calculating the amount of immunoprecipitated DNA as a percentage of the input DNA for individual samples. The primers used in these analyses are listed in Table S1. The ΔΔCt method was used to quantify gene expression.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 9. Differences between groups were evaluated using Student’s t test, with p < 0.05 as the significance threshold. Statistical significance was denoted as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Methods for statistical tests, number of independent experiments, and definition of error bars were indicated in figure legends. All experiments were repeated at least three times unless otherwise noted in the legend. Correlational relationships were examined using Spearman correlation analysis, while the Kaplan‒Meier method with the log rank test was used to analyze survival-related data.
Acknowledgments
We thank the Center of Biomedical Analysis of Tsinghua University for experimental and technical support. This study was supported by grants from the Natural Science Foundation of Beijing Municipality (no. 7232227), the National Natural Science Foundation of China (no. 82202882, no. 82303727), Foundation of Peking University International Hospital (no. YN2022ZD01), and Beijing Science and Technology Association, Youth Talent Promotion Project (no. BYESS2022226).
Author contributions
J.L., D.X., L.L., and T.X. designed the study. T.X. performed experiments with assistance from L.L., Y.C., G.J., J.Z., Z.C., and Y.L. Y.R., X.Z., and G.L. performed the IHC staining and scoring. T.X. and L.L. wrote the manuscript and all authors critically revised the manuscript.
Declaration of interests
All the authors declare no competing interests.
Published: November 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101264.
Contributor Information
Da Xu, Email: daxu@bjmu.edu.cn.
Jun Liang, Email: liangjun1959@aliyun.com.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Llovet J.M., Zucman-Rossi J., Pikarsky E., Sangro B., Schwartz M., Sherman M., Gores G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers. 2016;2 doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 3.Hilmi M., Neuzillet C., Calderaro J., Lafdil F., Pawlotsky J.M., Rousseau B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: current knowledge and future research directions. J. Immunother. Cancer. 2019;7:333. doi: 10.1186/s40425-019-0824-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guichard C., Amaddeo G., Imbeaud S., Ladeiro Y., Pelletier L., Maad I.B., Calderaro J., Bioulac-Sage P., Letexier M., Degos F., et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaelin W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 6.Fujimoto A., Totoki Y., Abe T., Boroevich K.A., Hosoda F., Nguyen H.H., Aoki M., Hosono N., Kubo M., Miya F., et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 7.Nault J.C., Martin Y., Caruso S., Hirsch T.Z., Bayard Q., Calderaro J., Charpy C., Copie-Bergman C., Ziol M., Bioulac-Sage P., et al. Clinical Impact of Genomic Diversity From Early to Advanced Hepatocellular Carcinoma. Hepatology. 2020;71:164–182. doi: 10.1002/hep.30811. [DOI] [PubMed] [Google Scholar]
- 8.Mittal P., Roberts C.W.M. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020;17:435–448. doi: 10.1038/s41571-020-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morel D., Almouzni G., Soria J.C., Postel-Vinay S. Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann. Oncol. 2017;28:254–269. doi: 10.1093/annonc/mdw552. [DOI] [PubMed] [Google Scholar]
- 10.Shen J., Peng Y., Wei L., Zhang W., Yang L., Lan L., Kapoor P., Ju Z., Mo Q., Shih I.M., et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015;5:752–767. doi: 10.1158/2159-8290.CD-14-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williamson C.T., Miller R., Pemberton H.N., Jones S.E., Campbell J., Konde A., Badham N., Rafiq R., Brough R., Gulati A., et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016;7 doi: 10.1038/ncomms13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bitler B.G., Aird K.M., Garipov A., Li H., Amatangelo M., Kossenkov A.V., Schultz D.C., Liu Q., Shih I.M., Conejo-Garcia J.R., et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehman H., Chandrashekar D.S., Balabhadrapatruni C., Nepal S., Balasubramanya S.A.H., Shelton A.K., Skinner K.R., Ma A.H., Rao T., Agarwal S., et al. ARID1A-deficient bladder cancer is dependent on PI3K signaling and sensitive to EZH2 and PI3K inhibitors. JCI Insight. 2022;7 doi: 10.1172/jci.insight.155899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang A., Garraway L.A., Ashworth A., Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020;19:23–38. doi: 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Wu S., Fukumoto T., Lin J., Nacarelli T., Wang Y., Ong D., Liu H., Fatkhutdinov N., Zundell J.A., Karakashev S., et al. Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat. Cancer. 2021;2:189–200. doi: 10.1038/s43018-020-00160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsvetkov P., Coy S., Petrova B., Dreishpoon M., Verma A., Abdusamad M., Rossen J., Joesch-Cohen L., Humeidi R., Spangler R.D., et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–1261. doi: 10.1126/science.abf0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J.N., Roberts C.W.M. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun X., Wang S.C., Wei Y., Luo X., Jia Y., Li L., Gopal P., Zhu M., Nassour I., Chuang J.C., et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell. 2017;32:574–589.e6. doi: 10.1016/j.ccell.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogiwara H., Takahashi K., Sasaki M., Kuroda T., Yoshida H., Watanabe R., Maruyama A., Makinoshima H., Chiwaki F., Sasaki H., et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell. 2019;35:177–190.e8. doi: 10.1016/j.ccell.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Gao Q., Zhu H., Dong L., Shi W., Chen R., Song Z., Huang C., Li J., Dong X., Zhou Y., et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell. 2019;179:561–577.e22. doi: 10.1016/j.cell.2019.08.052. [DOI] [PubMed] [Google Scholar]
- 22.Lissanu Deribe Y., Sun Y., Terranova C., Khan F., Martinez-Ledesma J., Gay J., Gao G., Mullinax R.A., Khor T., Feng N., et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018;24:1047–1057. doi: 10.1038/s41591-018-0019-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao J., Xu W., Zhang Y., Lv X., Chen Y., Ju G., Yang F., Lin L., Rao X., Guo Z., et al. Decreased expression of ARID1A invasively downregulates the expression of ribosomal proteins in hepatocellular carcinoma. Biomark. Med. 2021;15:497–508. doi: 10.2217/bmm-2020-0464. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X., Fu Z., Chen S.Y., Ong D., Aceto G., Ho R., Steinberger J., Monast A., Pilon V., Li E., et al. Alanine supplementation exploits glutamine dependency induced by SMARCA4/2-loss. Nat. Commun. 2023;14:2894. doi: 10.1038/s41467-023-38594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsvetkov P., Detappe A., Cai K., Keys H.R., Brune Z., Ying W., Thiru P., Reidy M., Kugener G., Rossen J., et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019;15:681–689. doi: 10.1038/s41589-019-0291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stine Z.E., Schug Z.T., Salvino J.M., Dang C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022;21:141–162. doi: 10.1038/s41573-021-00339-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun Y., Bandi M., Lofton T., Smith M., Bristow C.A., Carugo A., Rogers N., Leonard P., Chang Q., Mullinax R., et al. Functional Genomics Reveals Synthetic Lethality between Phosphogluconate Dehydrogenase and Oxidative Phosphorylation. Cell Rep. 2019;26:469–482.e5. doi: 10.1016/j.celrep.2018.12.043. [DOI] [PubMed] [Google Scholar]
- 28.Hu C., Li W., Tian F., Jiang K., Liu X., Cen J., He Q., Qiu Z., Kienast Y., Wang Z., et al. Arid1a regulates response to anti-angiogenic therapy in advanced hepatocellular carcinoma. J. Hepatol. 2018;68:465–475. doi: 10.1016/j.jhep.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 29.Shain A.H., Pollack J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8 doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romero O.A., Setien F., John S., Gimenez-Xavier P., Gómez-López G., Pisano D., Condom E., Villanueva A., Hager G.L., Sanchez-Cespedes M. The tumour suppressor and chromatin-remodelling factor BRG1 antagonizes Myc activity and promotes cell differentiation in human cancer. EMBO Mol. Med. 2012;4:603–616. doi: 10.1002/emmm.201200236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golfieri R., Bargellini I., Spreafico C., Trevisani F. Patients with Barcelona Clinic Liver Cancer Stages B and C Hepatocellular Carcinoma: Time for a Subclassification. Liver Cancer. 2019;8:78–91. doi: 10.1159/000489791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ge E.J., Bush A.I., Casini A., Cobine P.A., Cross J.R., DeNicola G.M., Dou Q.P., Franz K.J., Gohil V.M., Gupta S., et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat. Rev. Cancer. 2022;22:102–113. doi: 10.1038/s41568-021-00417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Day S.J., Eggermont A.M.M., Chiarion-Sileni V., Kefford R., Grob J.J., Mortier L., Robert C., Schachter J., Testori A., Mackiewicz J., et al. Final results of phase III SYMMETRY study: randomized, double-blind trial of elesclomol plus paclitaxel versus paclitaxel alone as treatment for chemotherapy-naive patients with advanced melanoma. J. Clin. Oncol. 2013;31:1211–1218. doi: 10.1200/JCO.2012.44.5585. [DOI] [PubMed] [Google Scholar]
- 34.Raab J.R., Resnick S., Magnuson T. Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet. 2015;11 doi: 10.1371/journal.pgen.1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All sequence data have been deposited in the GEO database under accession number GSE228179.
-
•
This study did not generate any novel code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.