

Abstract
Parkinson’s disease (PD) is a neurological disorder characterized by the progressive accumulation of neuronal α-synuclein (αSyn) inclusions called Lewy bodies. It is believed that Lewy bodies spread throughout the nervous system due to the cell-to-cell propagation of αSyn via cycles of secretion and uptake. Here, we investigated the internalization and intracellular accumulation of exogenous αSyn, two key steps of Lewy body pathogenesis, amplification and spreading. We found that stable αSyn fibrils substantially accumulate in different cell lines upon internalization, whereas αSyn monomers, oligomers, and dissociable fibrils do not. Our data indicate that the uptake-mediated accumulation of αSyn in a human-derived neuroblastoma cell line triggered an adaptive response that involved proteins linked to ubiquitin ligases of the S-phase kinase-associated protein 1 (SKP1), cullin-1 (Cul1), and F-box domain–containing protein (SCF) family. We found that SKP1, Cul1, and the F-box/LRR repeat protein 5 (FBXL5) colocalized and physically interacted with internalized αSyn in cultured cells. Moreover, the SCF containing the F-box protein FBXL5 (SCFFBXL5) catalyzed αSyn ubiquitination in reconstitution experiments in vitro using recombinant proteins and in cultured cells. In the human brain, SKP1 and Cul1 were recruited into Lewy bodies from brainstem and neocortex of patients with PD and related neurological disorders. In both transgenic and nontransgenic mice, intracerebral administration of exogenous αSyn fibrils triggered a Lewy body–like pathology, which was amplified by SKP1 or FBXL5 loss of function. Our data thus indicate that SCFFXBL5 regulates αSyn in vivo and that SCF ligases may constitute targets for the treatment of PD and other α-synucleinopathies.
INTRODUCTION
Progressive accumulation of α-synuclein (αSyn) aggregates into neuronal inclusions termed Lewy bodies (LBs) and Lewy neurites (LNs) characterizes Parkinson’s disease (PD) and related disorders, collectively called α-synucleinopathies (1). The role of αSyn in PD pathology is supported by the facts that LBs/LNs are present in the brains of virtually all sporadic and familial forms of the disease (2), that autosomal dominant mutations and multiplications in the gene encoding human αSyn (SNCA) lead to early PD onset (3–5), and that SNCA polymorphisms are associated with higher risk of PD (6, 7).
αSyn is a relatively small (14 kDa) protein found as soluble monomers or small oligomers that play important roles in vesicle trafficking and neurotransmitter release (8–12). αSyn physiological roles in vesicular homeostasis include interfering with the endosomal sorting complex required for transport (ESCRT) (13) and promoting the assembly of the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex (8, 10–12), among others. Under pathological conditions, αSyn undergoes marked conformational changes, leading to its aggregation into insoluble β-sheet–enriched amyloid fibrils, which are highly neurotoxic (14), and are thought to play important roles in the pathogenesis of LBs/LNs (15, 16).
αSyn accumulation into LBs/LNs is invariably observed in PD and other α-synucleinopathies (5, 17), suggesting that higher quantities of this protein are linked to disease development. αSyn accumulation can result from its increased biosynthesis or diminished degradation. Increased αSyn biosynthesis has been reported in familial PD with multiplication of SNCA, a process recapitulated in multiple experimental animal models based on ectopic αSyn expression [reviewed in (18, 19)]. Inhibition of αSyn degradation, which occurs via the ubiquitin-proteasomal, autophagy-lysosomal, and endosomal-lysosomal pathways (20–28), can also lead to a αSyn accumulation with the concomitant PD-like phenotype, as shown in cell cultures and in rodents (29). In addition, a third mechanism for the intracellular accumulation of αSyn in synucleinopathies has been recently proposed; using in vitro and in vivo models, it has been shown that extracellular αSyn proteopathic seeds are taken up by brain cells, accumulate intracellularly, and template the aggregation of the endogenous counterpart of recipient neuronal and glial cells (30, 31). For example, it has been proposed that in multiple system atrophy, extracellular αSyn is internalized and accumulates within glial cells that do not normally express this protein (32, 33). Uptake-mediated accumulation of αSyn in glia triggers neuroinflammation and the formation of intracytoplasmic inclusions that resemble neuronal LBs/LNs (34, 35).
The cell-to-cell propagation of αSyn in PD is also supported by early neuroanatomical studies, showing that αSyn inclusions appear first in the lower brainstem or olfactory bulb and ascend to susceptible areas of the midbrain, setting the foundations for the hypothesis that LB pathology self-propagates and spreads through the central nervous system in a temporally and topologically sequential manner (36). Spreading of LBs results from the cell-to-cell transmission of pathological forms of αSyn, which is first released from cells harboring LBs and then internalized by anatomically and functionally interconnected neurons (37). Internalized αSyn promotes the structural corruption of its endogenous counterpart in healthy neurons, a process that amplifies the aggregation cascade that leads to LB formation (38). Because of the intercellular transmission of αSyn, embryonic mesencephalic neurons grafted into PD patient’s brains acquire LBs (39, 40).
The efficient clearance of αSyn reduces its accumulation and associated cytotoxicity (25, 27, 41), suggesting that promoting αSyn degradation is crucial to counteract disease pathology (42). What determines αSyn degradation susceptibility and how cells respond to αSyn accumulation and deposition are, however, still open questions. Here, we carried out a systematic study on the uptake of different αSyn species and found that αSyn fibrils, but not monomers or oligomers, accumulated intracellularly upon internalization. Internalization of insoluble αSyn fibrils triggered an orchestrated cellular response involving pathways that had previously been associated to αSyn such as those related to vesicle trafficking, as well as modules whose link with αSyn remains unexplored such as SKP1 (S-phase kinase-associated protein 1)–Cul1 (cullin-1)–based ubiquitin ligases. We found that the SCF (SKP1/Cul1/F-box protein) ligase containing FBXL5 (F-box/leucine-rich repeat protein 5) (SCFFBXL5) ubiquitinated internalized αSyn fibrils and promoted their degradation in a proteasome- and lysosome-dependent manner. Thereby, SCFFBXL5 protected cells from αSyn cell-to-cell propagation. SKP1 and Cul1 colocalized with αSyn inclusions in cell cultures and in LBs from patients with PD and dementia with LBs (DLB). Depletion of SKP1 or FBXL5 in the mouse brain induced the formation of αSyn inclusions, suggesting that SCFFBXL5 plays protective roles in the initiation and spreading of LB-like pathology in mice.
RESULTS
Insoluble αSyn fibrils are internalized and accumulate within neuronal cells
On the basis of the high structural diversity of the αSyn species found in vitro and in vivo, we hypothesized that the structure of different species of αSyn could be a critical factor in determining internalization and accumulation properties. To test this hypothesis, we produced a set of structurally different αSyn variants by incubating monomeric αSyn (M-αSyn) for different periods (Fig. 1A and fig. S1A) (15). We obtained small oligomers (<100 kDa) after 6 hours of incubation (sO-αSyn), oligomers greater than 100 kDa after 3 days (O-αSyn), and two types of high–molecular weight (HMW) fibrils (>200 kDa) of different stabilities after 10 and 20 days of incubation. In contrast to fibrils obtained at 10 days of incubation, which were disassembled by detergents such as Triton X-100 and sarkosyl and therefore referred as F-αSyn, fibrils obtained after a 20-day incubation were detergent-resistant, and we called them iF-αSyn (fig. S1C). Because of their increased resistance to detergents, iF-αSyn was retained in the upper part of denaturing SDS–polyacrylamide gel electrophoresis (PAGE) even after boiling and expressed epitopes of fibrils as shown by Western blot (WB) analysis using the conformational antibody OC that recognizes amyloid fibrils (Fig. 1A) (43).
Fig. 1. Internalization of insoluble fibrils leads to αSyn accumulation and proteome alterations.
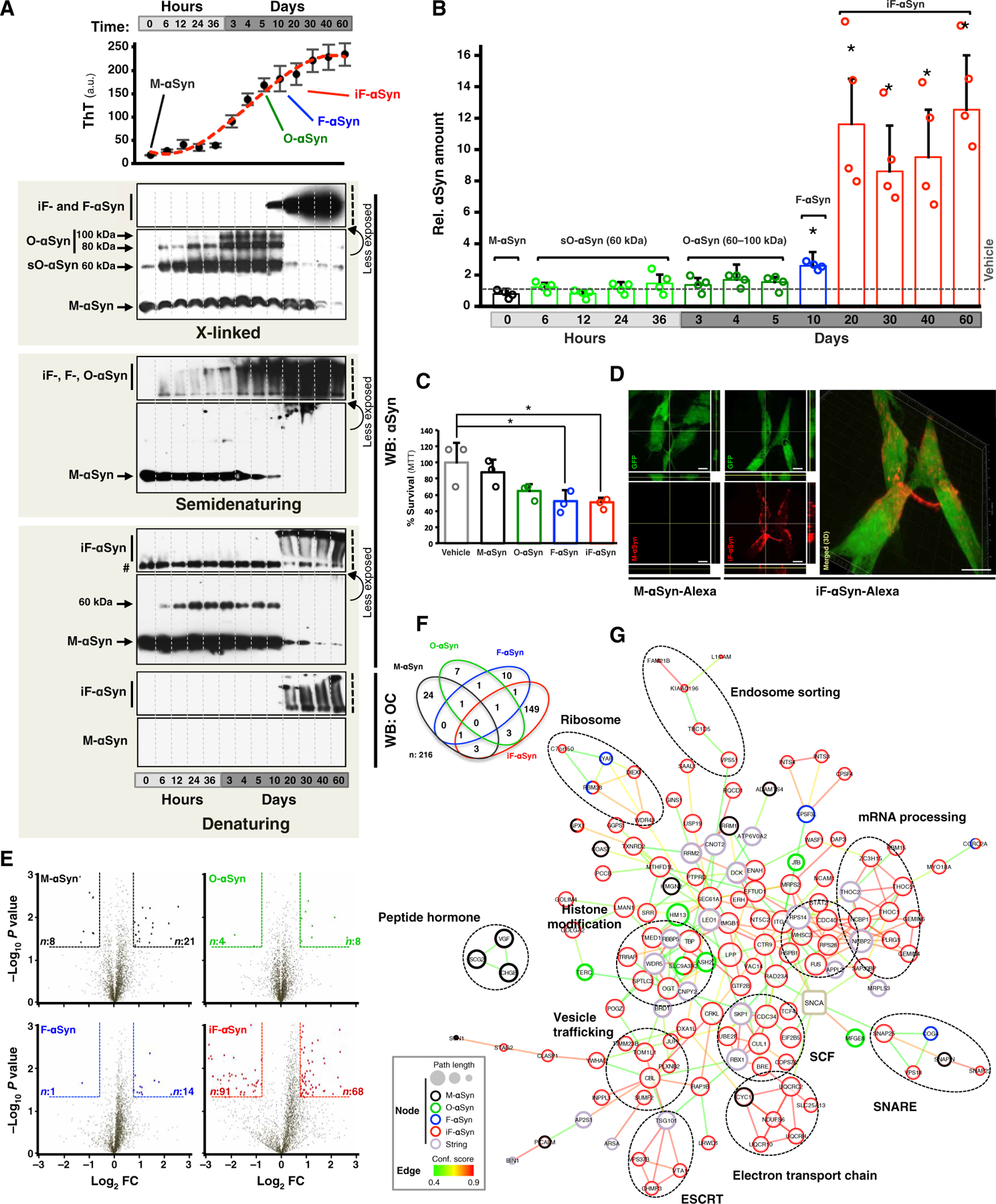
(A) Thioflavin T (ThT) fluorescence and WB analyses of αSyn species obtained by incubating recombinant M-αSyn for the indicated times. Representative M-αSyn, O-αSyn, F-αSyn, and iF-αSyn species are indicated. Number sign (#) indicates uncharacterized αSyn immunoreactive band. To show stacking gels (represented by black vertical dashed lines) at nonsaturating levels, a less exposed blot of the same membrane is shown (indicated by a curved arrow). a.u., arbitrary units. (B) Bar graph showing the amount of αSyn contained in cells treated with the species obtained in (A). (C) Cell viability of SH-SY5Y cells treated with the indicated αSyn using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. (D) Confocal microscopy and three-dimensional (3D) image reconstruction analyses of green fluorescent protein (GFP)–expressing SH-SY5Y cells treated with Alexa Fluor–labeled M-αSyn or iF-αSyn (both in red). GFP was used to delimitate cell morphology. Scale bars, 10 μm. (E) Volcano plots showing global protein quantities of SH-SY5Y cells treated with M-αSyn (black), O-αSyn (green), F-αSyn (blue), or iF-αSyn (red). Within dashed lines are shown differentially expressed proteins (DEPs), defined as those with fold change (FC) > 1.75 and P < 0.05 (unpaired, two-tailed distribution Student’s t test) compared to vehicle. Numbers of down- and up-regulated DEPs are indicated in left and right corners, respectively. For visualization purposes, only proteins with log10 P < 3 and log2 fold change < 3 and > −3, which represent >99% of the quantified proteins, are displayed. (F) Venn diagram of the overlap of 216 DEPs obtained from SH-SY5Y cells treated with the four species of αSyn. (G) Protein-protein interaction network analysis of DEPs from αSyn-treated cells using the STRING database. Functionally related DEPs are indicated by dashed ovals. In (A) and (C), the results are expressed as means + SD. *P < 0.05 compared to vehicle [one-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test].
Subsequently, human neuroblastoma SH-SY5Y cells were treated with the obtained αSyn species for 16 hours, then extensively washed to remove noninternalized αSyn (fig. S1E), and finally harvested to quantify the amount of internalized αSyn by selected reaction monitoring (SRM) mass spectrometry (MS) (Fig. 1B and fig. S1D). Similar low amounts of αSyn were found in cells treated with vehicle or M-αSyn, sO-αSyn, and O-αSyn (Fig. 1B), indicating that these αSyn species do not accumulate in vitro in this cell line. Compared to vehicle-treated cells, αSyn quantities were 2- and 10-fold higher (P < 0.05) in cells exposed to F-αSyn and iF-αSyn, respectively (Fig. 1B). Moreover, both F-αSyn and iF-αSyn led to a significant (P < 0.05) reduction in cell viability at both 24 and 72 hours after treatment, whereas O-αSyn was cytotoxic only at later time points (Fig. 1C and fig. S1B) compared to vehicle-treated cells. Intracellular accumulation of exogenous αSyn was also observed in GFP-expressing SH-SY5Y cells incubated with fluorescently labeled iF-αSyn but not M-αSyn (Fig. 1D). Uptake-mediated intracellular accumulation was significantly (P < 0.001) reduced when iF-αSyn was physically dissociated into monomers, suggesting that iF-αSyn stability was critical for accumulation in neuroblastoma cells (fig. S1, F and G). Thus, exogenous insoluble and detergent-resistant αSyn fibrils efficiently accumulated within neuronal SH-SY5Y cells.
Cell responses to extracellular αSyn revealed by proteome-wide analyses
We next investigated the cellular responses triggered by exogenous αSyn and evaluated global proteome changes that occur in neuronal SH-SY5Y cells exposed to M-αSyn, O-αSyn, F-αSyn, and iF-αSyn. A shotgun proteomics approach coupled to label-free quantification was used to quantify the proteomes of SH-SY5Y cells in two independent experiments (table S1 to S3). Based on shotgun proteomics data, iF-αSyn–treated cells displayed 10-fold αSyn increase compared to controls (fig. S1H), supporting SRM data and the unbiased approach used for proteome quantification. Significant differences (P < 0.05) were observed in cells treated with each αSyn species relative to vehicle-treated controls (Fig. 1E and fig. S1I). iF-αSyn led to the most pronounced cellular response with more than 130 DEPs identified. Most DEPs were specific to one αSyn variant (Fig. 1F and fig. S1J). Protein-protein interaction analyses using the STRING database revealed functional associations among DEPs with the majority among those of iF-αSyn (Fig. 1G and fig. S1K). Similar interactions were found in the two independent experiments (Fig. 1G and fig. S1K) where pathways such as vesicle trafficking, ribosome metabolism, and SCF ubiquitin ligases were consistently identified. Proteins belonging to vesicle trafficking systems included those of the ESCRT, SNARE, and the endosome sorting cellular machineries, and the fact that all these pathways have been previously linked to αSyn (8, 28) supported the validity of our approach. Additional interactions that were identified in the two experiments and whose link with αSyn has not been studied in detail included ribosome metabolism and SCF ubiquitin ligases. Modules that were identified in only one experiment, on the other hand, included protein aggregation, histone modification, peptide hormone and transcription, and mRNA processing. Gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses revealed significant enrichments (P < 0.05) among iF-αSyn–related DEPs (fig. S1, L and M, and tables S4 and S5). These included ubiquitin-mediated proteolysis and several pathways related to neurodegenerative diseases in which protein aggregation plays central roles.
An SCF ubiquitin ligase promotes degradation and ubiquitination of internalized αSyn
SCF belongs to the family of cullin-RING ligases (CRLs), the largest class of E3 ubiquitin ligases in mammals (44). CRLs are multisubunit complexes where cullins constitute the scaffold that brings together the E2 conjugase with the E3 subunits involved in substrate recognition. To date, eight cullin proteins (Cul) were identified in humans: Cul1, Cul2, Cul3, Cul4A/Cul4B, Cul5, Cul7, and Cul9. In the case of SCF, Cul1 is the CRL scaffold and SKP1 and a variable F-box domain–containing protein constitute the heterodimeric substrate-recognition module (45, 46). Because SCF ubiquitin ligases control a multitude of processes at the cellular and organismal levels, their dysregulation has been associated to many human pathologies (47). Because very little is known on how SCF ubiquitin ligases might contribute to PD, and because SKP1 was recently proposed a risk gene for PD (48) and its absence leads to a PD-like phenotype in mice (49), we aimed at evaluating a possible effect of SCF ubiquitin ligases on internalized αSyn. First, SKP1 and Cul1 were silenced in neuronal SH-SY5Y cells, glial BV-2, and kidney-derived Cos7 cells by short interfering RNAs (siRNAs). BV-2 and Cos7 do not express detectable endogenous αSyn (Fig. 2A and fig. S2A) (33), allowing us to quantify the exogenously acquired protein. We found that SKP1- and Cul1-depleted cells displayed higher quantities of internalized αSyn, which accumulated as HMW species in all cell types assayed [Fig. 2A (lanes 5 and 6 versus lane 4 and lanes 11 and 12 versus lane 10) and fig. S2, A and B (lanes and bars 5 and 6 versus lane and bar 4)]. Glial cells are the main scavengers of αSyn aggregates in the brain (41), and internalized αSyn did not accumulate in control BV-2 glial cells (Fig. 2A, lane 4). In agreement with MS data, higher quantities of Cul1 were observed in cells treated with iF-αSyn compared to vehicle-treated cells (Fig. 2A and fig. S2, A and C).
Fig. 2. An SCF E3 ubiquitin ligase targets internalized αSyn for ubiquitination and degradation.
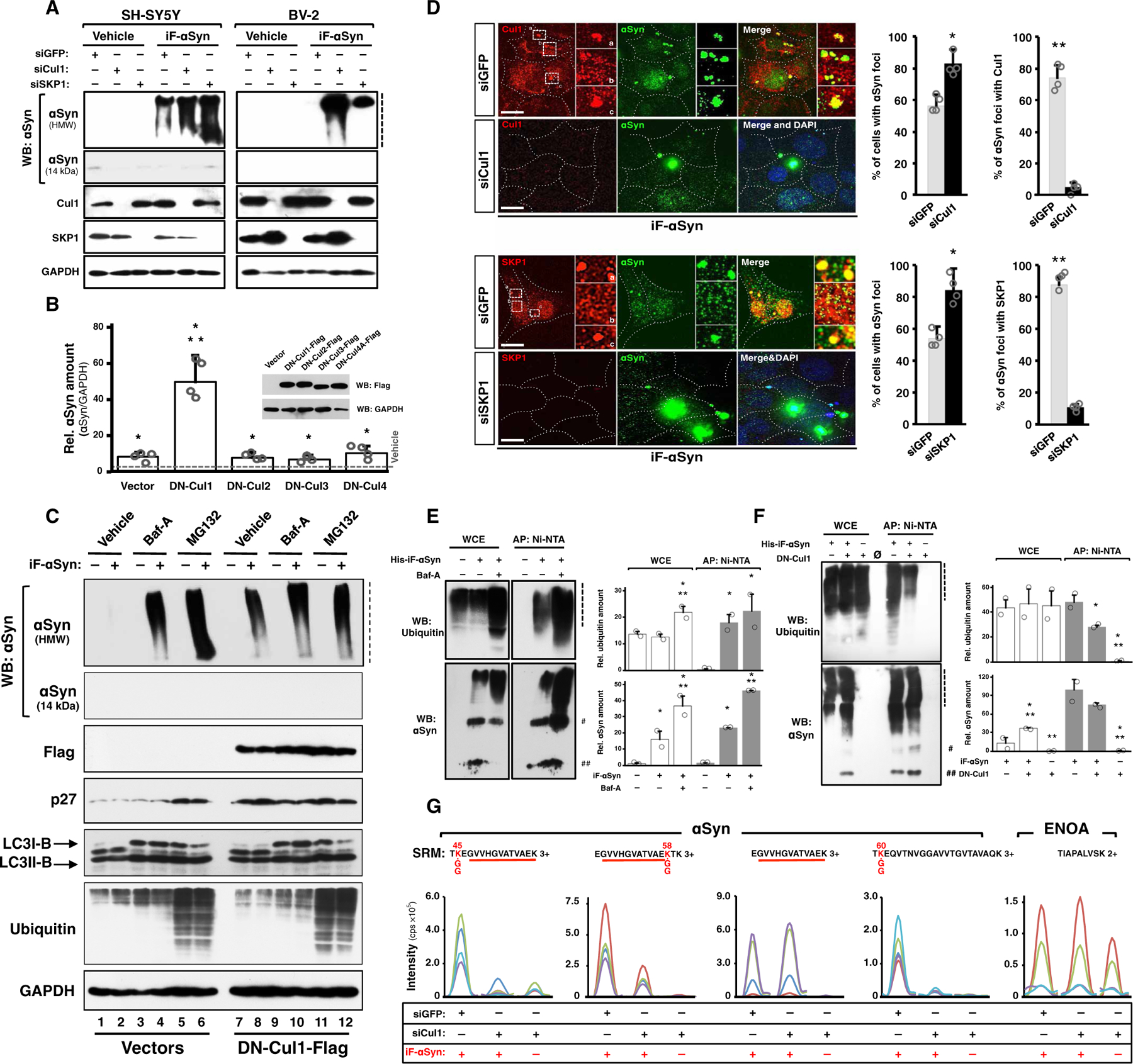
(A) WB of neuronal SH-SY5Y (left) and glial BV-2 (right) cells transfected with siGFP (control), siCul1, or siSKP1 and treated with iF-αSyn or vehicle. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) Bar graph showing the amount of αSyn contained in Cos7 cells expressing DN-Cul1, DN-Cul2, DN-Cul3, and DN-Cul4A (see WB inset) and treated with iF-αSyn. Results show means + SD. *P < 0.01 compared to vehicle, **P < 0.05 compared to the rest of the treatments (one-way ANOVA followed by Tukey’s post hoc test). (C) WB showing the effects of DN-Cul1, Baf-A (bafilomycin-A), or MG132 (N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal) on the degradation (but not uptake) of internalized αSyn. DN-Cul1 expression, as well as the Baf-A and MG132 treatments, was initiated in cells pretreated with iF-αSyn (+) (see also fig. S2H). (D) Immunofluorescence and confocal microscopy analyses of Cos7 cells transfected with siGFP, siCul1, or siSKP1 and treated with iF-αSyn. Quantification shows the percentage of cells with αSyn-positive foci (right) and the percentage of αSyn-positive foci that also contained Cul1 or SKP1 (left). Results show means + SEM. *P < 0.05 and **P < 0.01 compared to siGFP-transfected cells (unpaired, two-tailed distribution Student’s t test). Scale bars, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. (E and F) WB of Cos7 cells pretreated with (E) Baf-A or (F) stably expressing DN-Cul1 and treated with His-iF-αSyn (+). Cells were harvested under denaturing conditions, and whole-cell extracts (WCE) were subjected to metal-affinity purification (AP: Ni-NTA) to isolate His-iF-αSyn. ## and # indicate two αSyn immunoreactive bands of 15 and 60 kDa observed in all experiments with His-iF-αSyn. Quantification of the blots shows data expressed as means + SEM. *P < 0.05 and **P < 0.05 compared to vehicle and Baf-A (E) or DN-Cul1 (F), respectively (one-way ANOVA followed by Fisher’s post hoc test). (G) Quantification of ubiquitinated His-iF-αSyn purified from control cells (siGFP) or from cells depleted of Cul1 (siCul1). In this experiment, the cells were treated with Baf-A to avoid αSyn degradation. SRM traces of the αSyn peptides embedding K45, K58, and K60 bearing a GG-tag mass shift (indicated in red together with the modified lysine) resulting from trypsinized ubiquitinated proteins are shown. The unmodified αSyn peptide EGVVHGATVAEK and ENOA were analyzed as controls. Dashed lines indicate stacking gels.
To confirm that αSyn accumulates upon Cul1 loss of function, we used a dominant-negative Cul1 mutant (DN-Cul1) that, when expressed in mammalian cells, efficiently inhibits the endogenous protein. Dominant-negative mutants of different cullins such as Cul2, Cul3, and Cul4A were expressed similarly to evaluate whether other CRLs could also affect αSyn. Compared to vehicle-treated cells, αSyn accumulated in cells treated with iF-αSyn and expressing any of the dominant-negative cullins assayed (Fig. 2B). An accumulation of around 10-fold was observed in iF-αSyn–treated cells transfected with an empty vector and with expression vectors for DN-Cul2, DN-Cul3, or DN-Cul4, indicating that these cullins did not affect αSyn. In cells expressing DN-Cul1, on the contrary, αSyn accumulation reached 50-fold (Fig. 2B), therefore indicating that the effect on αSyn is specific for Cul1-based ligases. Increased αSyn levels by SCF inhibition were confirmed by a pretreatment of Cos7 cells with MLN4924, an inhibitor of CRLs, including Cul1 (50), before addition of iF-αSyn (fig. S2D). SCF loss of function by either DN-Cul1 expression (fig. S2E) or siRNA-mediated silencing (fig. S2F) failed to promote intracellular accumulation of exogenously applied M-αSyn or recombinant amyloid-β42 fibrils, suggesting that SCF specifically targets αSyn fibrils. HeLa cells treated with 50 or 250 nM iF-αSyn for different time points showed the kinetics of αSyn turnover and also revealed that the amount of αSyn increased in a time- and dose-dependent manner and, after reaching a maximum (4 and 12 hours for 50 and 250 nM, respectively), decreased over time (fig. S2G).
The decrease in αSyn accumulation suggested the presence of cellular degradation mechanisms able to degrade αSyn fibrils. To test this hypothesis, we uncoupled αSyn uptake from its degradation using stably transfected HeLa cells with doxycycline-inducible expression of DN-Cul1. These cells were pretreated with iF-αSyn for 12 hours and then incubated for 16 hours in medium devoid of αSyn but containing doxycycline, which induced a robust expression of DN-Cul1 (fig. S2H). As shown by WB, very low quantities of αSyn were detected in control cells, indicating that internalized αSyn was efficiently degraded when a recovery phase followed αSyn administration (Fig. 2C and fig. S2I, lane 2). In contrast, accumulation of αSyn was observed in cells with delayed expression of DN-Cul1 (Fig. 2C and fig. S2I, lane 8 versus lane 2), confirming that Cul1 was involved in αSyn degradation. Exogenous αSyn was previously shown to be degraded by the proteasome and lysosome (25, 51), and treatment with the proteasome inhibitor MG132 or the lysosome inhibitor Baf-A led to a substantial increase of αSyn amount (Fig. 2C and fig. S2I, lanes 4 and 6 versus lane 2). The effects of DN-Cul1 alone and its combination with Baf-A or MG132 were similar in magnitude, suggesting that Cul1 promoted both proteasomal and lysosomal degradation of αSyn. Neither iF-αSyn (Fig. 2C, lane 2 versus lane 1) nor DN-Cul1 (Fig. 2C, lane 7 versus lane 1) affected overall proteasome or lysosome activity, and therefore, no effect was observed on the amount of ubiquitinated proteins or LC3B as observed by WB (Fig. 2C and fig. S2I). The expression of DN-Cul1 stabilized p27, a known SCF substrate (Fig. 2C and fig. S2I) (52). In control experiments, neither Cul1 silencing nor the iF-αSyn treatment affected uptake or degradation of fluorescently labeled dextran (fig. S2J) or epidermal growth factor receptor (fig. S2K), indicating that neither Cul1 depletion nor the treatment with iF-αSyn causes massive disturbances in endocytic or degradative pathways.
Internalized αSyn localized within intracytoplasmic foci that were present in about 60% of the cells (Fig. 2D and fig. S2L). These foci also contained Cul1 and SKP1 and were more abundant in Cul1- and SKP1-depleted cells than in control cells. To confirm the association between αSyn and SCF in cells, we carried out immunoprecipitation assays in cells treated with iF-αSyn and expressing SKP1 fused to a short V5 tag. Using anti-V5 antibodies, we found SKP1 and HMW species of αSyn in these immunoprecipitates, which confirmed that SKP1 and internalized αSyn physically interact in cells (fig. S2M). Given the role of SCF in protein ubiquitination, we next tested whether this enzymatic complex could promote ubiquitin attachment to internalized αSyn. We treated SH-SY5Y cells with polyhistidine-tagged iF-αSyn (His-iF-αSyn) because it can be purified under denaturing conditions, making identification of the ubiquitinated forms by WB amenable. His-iF-αSyn, which was retained in stacking gels and showed kinetics of aggregation similar to iF-αSyn (fig. S2, N and O), was used in internalization experiments and then purified under denaturing conditions from whole-cell extracts of cells pretreated with Baf-A or MG132. Ubiquitin immunoreactive bands were detected in His-iF-αSyn precipitates, indicating that this protein was ubiquitinated (Fig. 2E). The amount of ubiquitinated αSyn increased upon treatment of cells with either Baf-A or MG132, confirming that ubiquitinated αSyn is degraded by the lysosome and proteasome. Ubiquitination of His-iF-αSyn was reduced in DN-Cul1 cells (Fig. 2F) and, likewise, in cells depleted of Cul1 (Fig. 2G), indicating that SCF is critical for αSyn ubiquitination. To identify the lysine residues on αSyn targeted by SCF, we analyzed purified His-iF-αSyn by shotgun and targeted MS. With the exception of K6, αSyn was ubiquitinated at all N-terminal lysines (K10 to K45), such as K45, K58, and K60 (Fig. 2G and fig. S2Q). The purified αSyn also contained peptide signatures of K48-K63 branched ubiquitin chains (fig. S2R), confirming that ubiquitinated αSyn is targeted to proteolytic pathways. Thus, our results suggest that SCF promotes ubiquitination and degradation of internalized αSyn fibrils.
An SCF inhibits the prion-like properties of extracellular αSyn
Like prions, αSyn fibrils can template the aggregation of homotypic molecules through a nucleation-dependent mechanism known as seeding (30). Thus, we studied whether SCF could inhibit iF-αSyn seeding on cellular GFP-tagged αSyn. We found that the treatment with iF-αSyn led to a relocalization of GFP-αSyn into cytoplasmic foci (Fig. 3A), a phenomenon attributed to aggregate formation (38, 51). Supporting aggregation of cellular αSyn, GFP-αSyn foci were immunostained with the OC antibody (fig. S3A). Foci increased in number in cells expressing DN-Cul1, which colocalized with cellular GFP-αSyn in cytosolic inclusions. No relocalization of GFP alone was observed in iF-αSyn–treated DN-Cul1–expressing cells (Fig. 3A). Seeded aggregation of GFP-αSyn was confirmed by WB in neuronal SH-SY5Y cells, where Cul1 or SKP1 depletion led to higher amounts of HMW GFP-αSyn with a concomitant reduction of both monomeric GFP-αSyn and endogenous αSyn (Fig. 3B and fig. S3B, lanes 11 and 12 versus lane 10). Similar results were obtained in non-neuronal HeLa cells expressing DN-Cul1 because they displayed higher amount of GFP-αSyn aggregates compared to controls (fig. S3C). We next examined whether SCF inhibited cell-to-cell propagation of αSyn species and generated a cellular model to evaluate αSyn transcellular spreading. We first generated SH-SY5Y cell clones that stably expressed oligomers and aggregates of GFP-αSyn (fig. S3, D to G), which are released to the culture media and are taken up by acceptor cells (fig. S3F). Control, Cul1-deficient, or DN-Cul1–expressing cells were used as αSyn acceptors and treated with these culture media for 16 hours. We observed higher quantities of αSyn in acceptor cells lacking Cul1 or expressing DN-Cul1 (Fig. 3C), suggesting that Cul1 is involved in GFP-αSyn degradation. In addition, these results also indicated that SCF specificity is not restricted to recombinant iF-αSyn.
Fig. 3. An SCF inhibits the prion-like properties of extracellular αSyn.
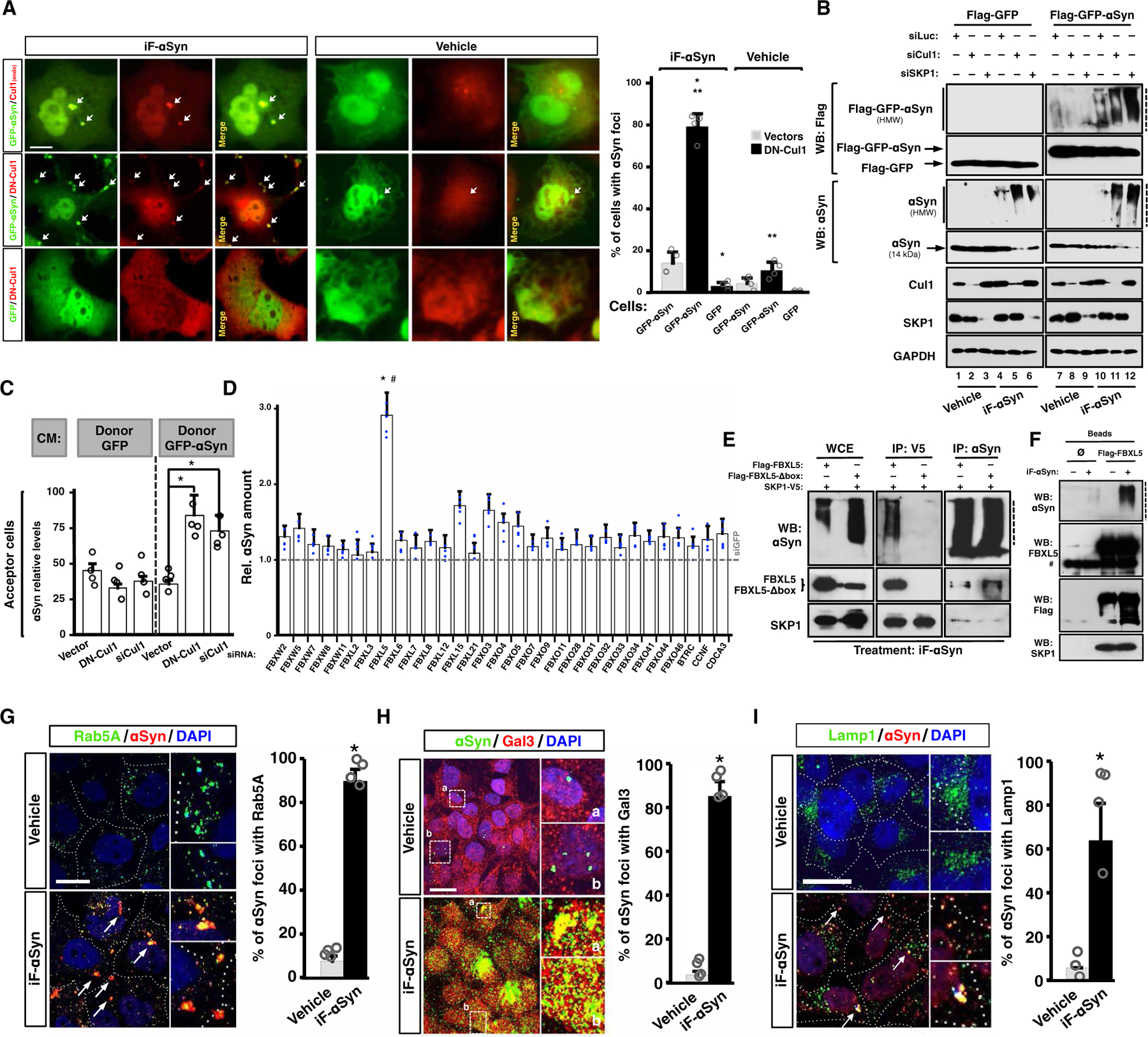
(A) Confocal microscopy images of Cos7 cells expressing GFP, GFP-αSyn, and/or DN-Cul1–Flag and treated with iF-αSyn. Arrows indicate GFP-αSyn–containing foci. Quantification shows means + SD. *P < 0.05 and **P < 0.05 compared to DN-Cul1 or GFP-αSyn, respectively (one-way ANOVA and the Tukey’s test). Scale bar, 10 μm. (B) WB analyses of GFP or GFP-αSyn–expressing SH-SY5Y cells transfected with siLuc, siCul1, or siSKP1 and treated with iF-αSyn. (C) Bar graph showing the amount of αSyn determined by SRM in Cos7 acceptor cells transfected with an empty vector, DN-Cul1 vector, or siCul1. In this experiment, exogenous αSyn was obtained from conditioned media (CM) from GFP- or GFP-αSyn–expressing SH-SY5Y cells (referred as donors). Data show means + SD. *P < 0.05 compared to vector (one-way ANOVA followed by Dunnett’s post hoc test). (D) Bar graph showing the amount of αSyn determined by SRM in HeLa cells transfected with siRNAs targeting 31 different F-box domain–containing proteins or GFP (siGFP; dashed horizontal line) and treated with iF-αSyn. Data show means + SD. *P < 0.05 by one-way ANOVA followed by Tukey’s post hoc test; #P < 0.05 by unpaired, two-tailed distribution Student’s t test compared to siGFP. (E) WB of whole-cell extracts (WCE) immunoprecipitates (IP: V5 and IP: αSyn) obtained from Cos7 cells expressing SKP1-V5 and Flag-FBXL5 or Flag-FBXL5-Δbox. In this experiment, the cells were pretreated with Baf-A and then exposed to iF-αSyn. (F) Flag-FBXL5 was expressed and immunoprecipitated from cells treated with iF-αSyn (+). A control immunoprecipitation (Ø) using an unrelated antibody is shown. #, heavy immunoglobulin chain. (G) Colocalization analyses between internalized iF-αSyn and Rab5A by immunofluorescence and confocal microscopy. Scale bar, 10 μm. Arrows show foci containing both αSyn and Rab5A. Quantification is shown on the right. (H and I) Colocalization analyses between internalized iF-αSyn and (H) galectin-3 (Gal3) and (I) lysosome-associated membrane protein-1 (Lamp1) by immunofluorescence and confocal microscopy. Scale bars, 10 μm. Arrows indicate foci positive for αSyn. Quantification is shown on the right. (G to I) Data show means + SEM. *P < 0.05 compared to vehicle (unpaired, two-tailed distribution Student’s t test). Dashed lines indicate stacking gels.
FBXL5 physically links αSyn with SKP1/Cul1
SCF E3 ubiquitin ligases are composed of a constitutive Cul1/SKP1 scaffold heterodimer and a variable F-box domain–containing protein (F-box) with substrate-recognition function (46, 53). To identify the F-box protein that recognizes internalized αSyn, we silenced 31 F-box proteins that physically interact with SKP1 in eukaryotic cells (http://thebiogrid.org/). Increased amount (threefold) of internalized αSyn was observed upon knockdown of FBXL5 in HeLa cells (P < 0.05; Fig. 3D). This was confirmed by WB in Cos7 cells (fig. S3H). Like SKP1 and Cul1, FBXL5 colocalized with internalized αSyn within foci (fig. S3I). Moreover, FBXL5 and SKP1 coimmunoprecipitated with αSyn in iF-αSyn–treated cells (Fig. 3, E and F, and fig. S3J). Confirming that FBXL5 physically interacts with internalized αSyn, the SCF/αSyn physical interaction was lost when a FBXL5 mutant unable to interact with SKP1 (FBXL5-Δbox) was expressed (Fig. 3E and fig. S3J) (54). Further, we expressed and purified SKP1-V5 and Flag-FBXL5 from transiently transfected cells. These proteins were then incubated with recombinant Cul1, ubiquitin, and all the E1/E2 enzymes required for in vitro ubiquitination reactions. Thus, this approach allowed us to study whether the reconstituted SKP1-Cul1-FBXL5 complex (SCFFBXL5) could catalyze ubiquitin attachment to αSyn in vitro. As indicated by WB, we observed the formation of HMW species of αSyn only when all these proteins were included in the reaction mix (fig. S3K), indicating that the in vitro reconstituted SCFFBXL5 catalyzed αSyn ubiquitination. Next, we asked whether internalized αSyn was stabilized upon FBXL5 degradation, which naturally occurs upon iron depletion (55). We treated Cos7 cells with either the iron source ferric ammonium citrate (FAC) or the iron chelator deferoxamine (DFO) and found reduced amounts of αSyn in cells treated with FAC, which stabilized FXBL5 (fig. S3L). On the contrary, the treatment with the iron chelator DFO led to decreased FBXL5 quantities with the concomitant accumulation of αSyn. The effect of FAC was abolished upon FBXL5 silencing (fig. S3L, lane 6 versus lane 3), indicating that the observed αSyn up-regulation in FAC-treated cells was attributable to FBXL5. We next tested whether endogenous FXBL5 could physically interact with internalized αSyn and conducted immunoprecipitation assays using anti-αSyn antibodies. In these experiments, the cells were pretreated with Baf-A to avoid αSyn degradation. We found that FBXL5 coprecipitated with αSyn in cells treated with FAC and iF-αSyn (fig. S3M). No interaction was observed in either vehicle- or DFO-treated cells, indicating that αSyn physically interacted with endogenous FBXL5. Last, FBXL5 silencing phenocopied SKP1 and Cul1 depletion on αSyn accumulation and ubiquitination (fig. S3N). Thus, the iron-regulated SCF substrate receptor FBXL5 physically connects αSyn with SKP1/Cul1.
Endocytosed αSyn is released into the cytoplasm by vesicle rupture
We next studied the mechanisms of iF-αSyn uptake and intracellular processing. αSyn did not accumulate in iF-αSyn–treated SH-SY5Y cells incubated at 4°C, suggesting active mechanisms of uptake (25). Internalized iF-αSyn associated with the early endosome marker Rab5A (Fig. 3G), in line with previous works, showing that extracellular αSyn seeds are endocytosed (24, 25). We then treated SH-SY5Y cells with iF-αSyn for 1, 2, and 6 hours and analyzed αSyn amount in endosomal, lysosomal, and cytosolic fractions. We found that after 1 and 2 hours, αSyn was exclusively present in endosomes (fig. S3O), confirming that iF-αSyn is taken up by endocytosis. After 6 hours of treatment, αSyn and SCFFBXL5 were detected in lysosomal and cytosolic fractions (fig. S3O, lane 12). This suggested that (i) at this time point endocytosed αSyn is released to the cytoplasm presumably by inducing vesicle rupture, as recently shown for neurotoxic aggregates besides αSyn (56–59), and (ii) SCFFBXL5 is recruited to areas of ruptured lysosomes to encounter recently released αSyn. Supporting lysosome rupture as the main mechanism of release of endocytosed αSyn into the cytoplasm, internalized αSyn colocalized with Gal3, a marker of ruptured vesicles (Fig. 3H) (59), caused signal intensity attenuations of LysoTracker, a dye that specifically stains acidic vesicles such as late endosomes and lysosomes (fig. S3P), and negatively affected the activity of lysosomal enzymes such as cathepsin-B and cathepsin-D (fig. S3Q). Moreover, the effect of iF-αSyn on acidic vesicles was reduced in cells expressing FBXL5 and increased by DN-Cul1 (fig. S3P). Additional evidence of a disruptive effect of iF-αSyn on endosomal-lysosomal vesicles were the presence of Lamp1 into αSyn-containing foci (Fig. 3I) and its mislocalization in iF-αSyn–treated cells (fig. S3R). Last, we knocked down SCFFBXL5 in cells overexpressing αSyn and confirmed that this enzymatic complex targets the αSyn that localizes in the cytoplasm (fig. S3S). Because this cellular model is devoid of recombinant fibrils, these data also indicated that SCF might be important for the cell-autonomous degradation of αSyn.
SCFFBXL5 counteracts LB-like pathology in vivo
To understand the pathological relevance of the link between SCF and αSyn in PD and related neurological disorders, we analyzed brain tissue from patients with PD and DLB. Our results showed that both Cul1 and SKP1 significantly (P < 0.05) colocalized with the αSyn contained in LBs of brainstem and neocortex (Fig. 4A). In sharp contrast, no colocalization of αSyn with Cul1 or SKP1 was observed in healthy individuals. Because this suggested an important role of SCF in synucleinopathies, we asked whether SCF could inhibit LB formation in vivo. We first used αSyn transgenic mice that develop a spontaneous LB-like pathology (60), which can be accelerated by intracerebral administration of αSyn preformed fibrils (61, 62). A lentivirus encoding short hairpin RNAs (shRNAs) targeting GFP (LV-shGFP) or SKP1 (LV-shSKP1) (fig. S4A) was injected unilaterally into the hippocampus of these mice, and 5 weeks later, a single unilateral dose of iF-αSyn was administered at the same site (fig. S4B). Animals were sacrificed 10 weeks after iF-αSyn administration, and brain slices were immunostained with antibodies specific for αSyn and pSer129-αSyn, two histopathological markers of LB (63). We observed prominent inclusions near the injection site and distant from it in striatum, cortex, and the CA2 region of the hippocampus (Fig. 4, B and C). αSyn and pSer129-αSyn immunoreactivity was detected in cell bodies and neurite-like projections (Fig. 4, B and C). In striatum and CA2, both αSyn and pSer129-αSyn–positive inclusions were more abundant and widespread in LV-shSKP1–injected animals than in animals that received LV-shGFP (Fig. 4C). In cortex, no differences were observed for total αSyn, whereas increased amounts of the phosphorylated protein were observed. These results indicated that SCF counteracts LB pathology induced by exogenous αSyn fibrils in transgenic animals.
Fig. 4. SCFFBXL5inhibits LB-like pathology induced by extracellular αSyn fibrils.
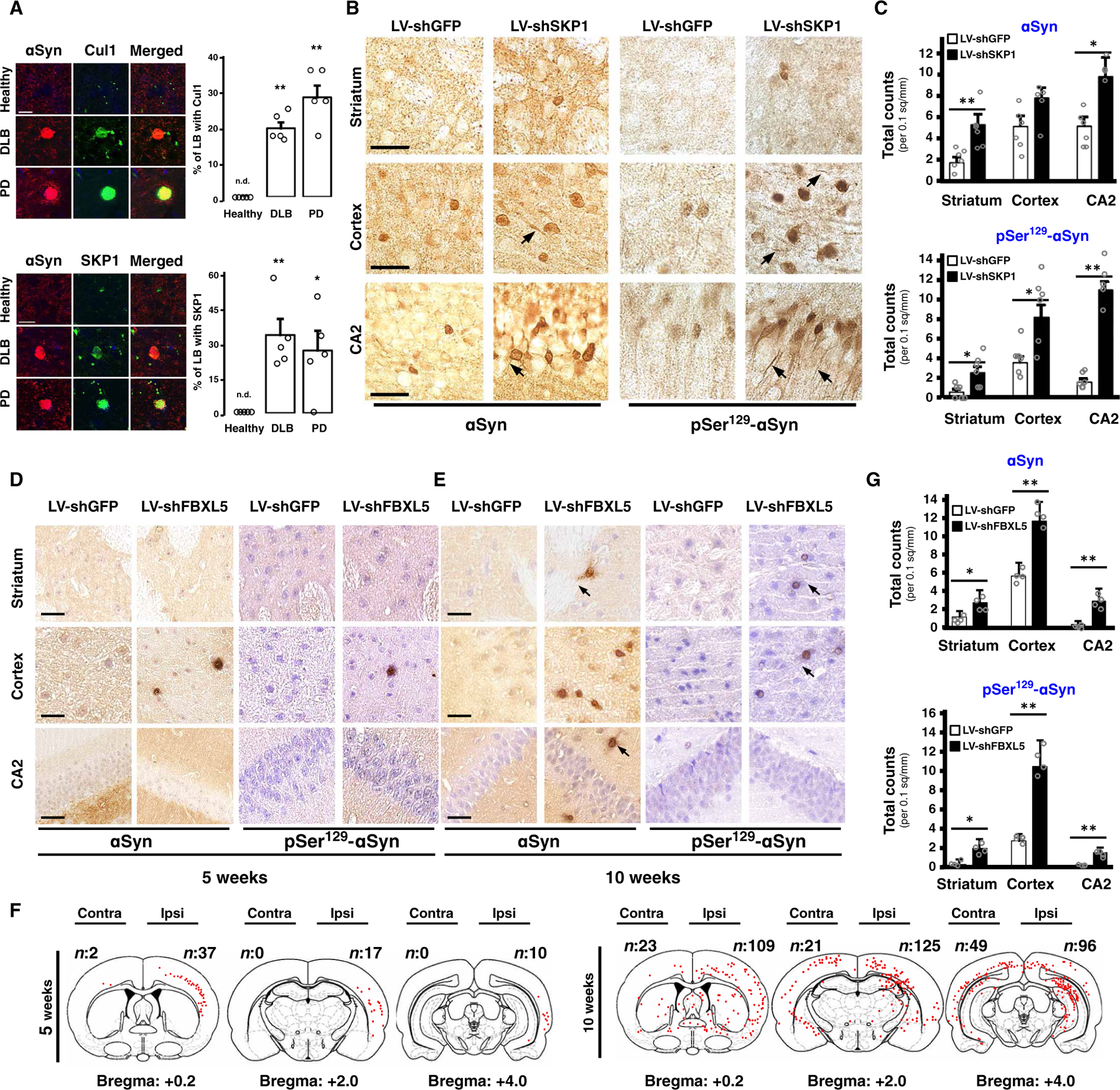
(A) Representative confocal microscopy images of brain slices from healthy subjects (n = 5) and from DLB (n = 5) and PD (n = 5) patients. Immunofluorescence shows αSyn in red and Cul1 and SKP1 in green. Data show means + SEM. *P < 0.05 and **P < 0.01 (unpaired, two-tailed distribution Student’s t test). n.d., nondetected. Scale bars, 10 μm. (B) Representative images of paraffin-embedded sections of striatum, cortex, and hippocampus (CA2) of αSyn transgenic mice injected with LV-shGFP (n = 6) or LV-shSKP1 (n = 6) and then with iF-αSyn. Immunohistochemistry was carried out with αSyn (clone LB509) or pSer129-αSyn antibodies. Scale bars, 20 μm. (C) Quantification of αSyn- and pSer129-αSyn–positive inclusions of the αSyn mice of (B). (D and E) Representative images of paraffin-embedded sections of striatum, cortex, and CA2 from wild-type nontransgenic mice injected with LV-shGFP (n = 6) and LV-shFBXL5 (n = 6) (left and right hemispheres, respectively) and subsequently with iF-αSyn. Animals were sacrificed at (D) 5 or (E) 10 weeks after iF-αSyn injection. Immunohistochemistry was carried out with αSyn (clone 509) and pSer129-αSyn antibodies. Arrows indicate neuronal projections. Scale bars, 25 μm. (F) Schematic representation of three brain sections of wild-type mice of (D) and (E). αSyn-positive inclusions are indicated by red dots, and quantification is indicated in the upper corner. (G) Quantification of αSyn- and pSer129-αSyn–positive inclusions of the mice of (E). (C and G) Data show means + SEM. *P < 0.05 and **P < 0.01 compared to LV-shGFP–injected mice (C) or hemispheres (G) (paired, two-tailed distribution Student’s t test).
Next, we asked whether SCFFBXL5 could inhibit LB pathogenesis and spreading in wild-type nontransgenic animals. To quantify the effect of SCF on LB spreading, we injected LV-shSKP1 or LV-shFBXL5 into the right cortex and LV-shGFP contralaterally (fig. S4C). Five weeks later, iF-αSyn was injected bilaterally at the same sites, and 5 or 10 weeks after iF-αSyn administration, the animals were sacrificed. At both 5 and 10 weeks after iF-αSyn administration, αSyn-positive inclusions were observed in LV-shSKP1– and LV-shFBXL5–injected hemispheres and rarely found in contralateral areas (Fig. 4, D, E, and G, and fig. S4D). Inclusions contained both human and mouse αSyn (fig. S4E), suggesting recruitment of host αSyn and providing evidence of aggregate amplification in vivo. Inclusions were positive for ubiquitin (fig. S4F) and for the anti-OC antibody (fig. S4G), and they were stained with ThT (fig. S4H). Moreover, human αSyn was detected as HMW species in brain extracts from LV-shSKP1–injected mice (fig. S4I). αSyn inclusions were found in neurons and glia (fig. S4J). Human αSyn was not detected in control experiments in which the primary antibody was omitted, confirming the specificity of the staining (fig. S4K). When amplification and spreading were analyzed, at 5 weeks after iF-αSyn administration, the inclusions were found mainly in LV-shSKP1– and LV-shFBXL5–injected cortices. They were abundant near the injection site and decreased gradually with distance from the site of injection (Fig. 4F), suggesting spreading along the cerebral anteroposterior axis. This was confirmed in animals injected at 10 weeks after iF-αSyn administration, where amplification and spreading of the inclusions were observed, mainly in the brains where SKP1 or FBXL5 was silenced (Fig. 4E). Likely because of the lateral spreading of the ipsilateral pathology, LB-like pathology was also observed in contralateral hemispheres of mice sacrificed at 10 weeks after iF-αSyn administration (Fig. 4, F and G). The results suggest that SCFFBXL5 inhibits initiation and spreading of the LB-like pathology induced by extracellular αSyn seeds in mice.
DISCUSSION
In PD and other α-synucleinopathies, neuronal degeneration is accompanied by intracellular αSyn accumulation and deposition, which may arise from non–cell-autonomous αSyn uptake from the extracellular milieu (64, 65). αSyn is a major genetic risk factor of PD (7), and its accumulation and aggregation have long been proposed to play causative roles in the disease due to the neurotoxic properties of high-order assemblies such as the amyloid fibrils used in this work. Our study revealed that extracellular αSyn fibrils showed increased uptake-mediated accumulation compared to monomers and oligomers. High stability is a key biochemical feature of the αSyn aggregates found in LBs (66, 67) and a determinant for αSyn proteolysis and accumulation (16, 68). However, previous studies have shown that upon inhibition of major degradation systems, internalized αSyn monomers and oligomers can also accumulate within cells (25, 27), suggesting that accumulation of internalized αSyn would depend not only on the biochemical properties of exogenous αSyn species (33, 69) but also on the pathways involved in αSyn clearance.
Using a human-derived neuroblastoma cell line, we showed that internalization of αSyn fibrils triggers an orchestrated cellular response that includes up-regulation of SCF ubiquitin ligases. Although future works are needed to elucidate whether SCF directly catalyzes ubiquitin attachment to αSyn in vivo, data suggest that SCFFBXL5 is critical for αSyn ubiquitination and degradation in cultured cells. This, together with the fact that depletion of SKP1 and FBXL5 in the mouse brain led to a substantial amplification of the LB-like pathology triggered by exogenous αSyn fibrils, suggested that SCF might be part of an adaptive cellular response of neurons and glia to counteract αSyn deposition in the mammalian brain.
Because the multiplication of SCNA and αSyn up-regulation are linked to PD, reducing αSyn quantities has been proposed as a logical therapeutic approach for synucleinopathies. Inhibiting the overall synthesis of αSyn has been successfully achieved in cell cultures, rodents, and nonhuman primates (70–74), but the neuroprotective effects of this approach are still controversial presumably because of the loss of the physiological function of this protein (75–77). Therefore, targeting the αSyn species that are responsible for toxicity is preferable, and induction of both the autophagy and proteasomal systems is appealing. Induction of autophagy causes a significant reduction αSyn load, aggregation, and toxicity in various cellular and animal models based on αSyn overexpression (78). However, the potential use of such modulators for PD is limited because the autophagy and proteasomal systems lack specificity and multiple essential pathways are also affected upon their perturbation. In light of these caveats, the selectivity of E3 ubiquitin ligases confers unique advantages. The facts that ubiquitination is critical for the clearance of pathological forms of αSyn and that ubiquitinated αSyn is massively found in LBs of PD patients but not in healthy subjects (27, 79–85) support this notion. SCFFBXL5 targets αSyn fibrils acquired from the extracellular milieu, whereas Parkin, an E3 ubiquitin ligase linked to autosomal recessive juvenile PD, ubiquitinates a glycosylated form of αSyn in cultured cells (86). The ubiquitin ligases C terminus of Hsc70-interacting protein (CHIP) and seven-in-absentia homolog (SIAH) ubiquitinate endogenous αSyn monomers in vitro and in cultured cells (87, 88), whereas the neuronal-precursor cell-expressed developmentally down-regulated gene 4 (Nedd4) ubiquitinates endogenous αSyn and internalized αSyn monomers that freely diffuse through the plasma membrane (27, 83). Thus, distinct ubiquitin ligases play nonoverlapping and complementary roles by targeting structurally and potentially pathologically different forms of αSyn, and data showing that residue-specific ubiquitin attachment to αSyn depends on biological context support this idea (84, 89). In this sense, no physiological function has been assigned to extracellular αSyn aggregates to date, and thereby, the selectivity of SCFFBXL5 toward neurotoxic exogenous αSyn fibrils would be a crucial advantage over other ubiquitin ligases such as Parkin, CHIP, SIAH, and Nedd4. As an example of the clinical importance that targeting extracellular αSyn aggregates might have in synucleinopathies, neutralization of this protein by immunotherapy has been shown to prevent αSyn uptake and downstream pathological events in mice (28, 90, 91).
Our data suggest that iF-αSyn is internalized by endocytosis, trafficked through the endolysosomal pathway, and released into the cytosol by inducing rupture of endolysosomal vesicles. A similar mechanism of release has been recently shown for neurotoxic extracellular seeds of αSyn (59), tau (56), Huntingtin (58), and SOD (superoxide dismutase) (57), suggesting that this may be a general mechanism by which exogenous entities with prion-like properties reach the cytoplasm. This mechanism allows exogenous aggregates not only to seed host molecules residing in the cytoplasm, an essential step in the aggregate amplification and spreading loop (92), but also to encounter SCF and proteolytic enzymes in the cytoplasm (93). Thus, vesicle rupture might be an obligate step for infectivity of prion-like proteins, therefore constituting an attractive target for therapeutic intervention (58).
A growing body of clinical and experimental evidence supports an important role of SCF ligases in PD and related disorders. SKP1 quantities are lower in patients with sporadic PD than in healthy controls (48, 94), genetic variations of the SKP1 gene increase PD risk (95), and increased susceptibility to neuronal death and behavioral deficits is observed in mice when SKP1 expression is silenced in the brain (49). Moreover, Cul1 and SKP1 are down-regulated in animal models of Huntington’s and Machado-Joseph diseases (96), two neurodegenerative disorders characterized by accumulation of polyglutamine-containing protein aggregates. In these models, Cul1 or SKP1 silencing results in aggregate accumulation and enhanced polyglutamine-induced toxicity in vivo.
Although additional F-box domain–containing proteins might also contribute to the clearance of certain αSyn species including fibrils, we demonstrated that FBXL5 loss of function promoted accumulation of internalized αSyn fibrils. FBXL5 is regulated by iron and highly expressed in the human brain (54, 55), and its dysregulation leads to cellular and systemic iron accumulation and oxidative stress (97), two features of the degenerating midbrain of patients with PD and related disorders (98). Whether the increased amount of iron observed in PD represents an adaptive strategy aiming at the stabilization of SCFFBXL5 or whether they result from SCFFBXL5 loss of function remains to be elucidated.
By promoting degradation of pathologic forms of αSyn, SCFFBXL5 counteracted LB-like pathology initiation and spreading in both transgenic and wild-type mice, therefore supporting the existence of a “biological barrier” that regulates the in vivo induction of LB-like pathology induced by extracellular αSyn seeds (99). However, several limitations related to our study need to be further investigated in light of potential therapeutic applications. For instance, the sole recruitment of SKP1 and Cul1 into LBs from PD and DLB patients does not warrant an involvement of SCF in the clearance of pathological αSyn in the human brain, and therefore, additional clinical-oriented studies will be needed to support this hypothesis. In this sense, it has not been systematically addressed how representative are the recombinant αSyn fibrils used in this and other works to the aggregates found in synucleinopathies. SKP1 and Cul1 constitute the core SCF module that is shared among multiple SCF complexes, limiting the rationale for therapeutic developments to modulate exclusively FBXL5, a protein that is constitutively degraded by the proteasome (55). Stabilization of FXBL5 can be achieved by iron (54, 55), which is itself not compatible with therapies in the brain due to potential cellular damage through hydroxyl radical production and the resulting oxidative stress. Thus, the development of alternative stabilizers would be needed, and in this sense, several drug discovery programs aimed to modulate SCF for therapeutic applications have been launched (47). Strategies that enhance SCFFBXL5 activity in vivo will be powerful tools to investigate the therapeutic potential of this enzymatic complex in synucleinopathies.
MATERIALS AND METHODS
Study design
The aim of this study was to identify the pathways involved in the clearance of exogenous αSyn. We first studied the structural determinants for the efficient uptake and intracellular accumulation of αSyn and generated several αSyn species of distinct structural properties. We speculated that soluble low–molecular weight species such as monomers and small oligomers might be taken up by the cells and rapidly degraded by intracellular machineries. On the contrary, certain HMW species such as fibrils might resist degradation and thereby accumulate intracellularly upon uptake. The different αSyn species were formed by incubating recombinant M-αSyn at different time points, as it has been shown that αSyn oligomers and fibrils are formed in these experimental conditions (15). We then used a well-established cellular model in which cultured human-derived neuroblastoma SH-SY5Y cells are treated with the generated αSyn species and intracellular αSyn accumulation is evaluated. For the sensitive and accurate quantification of αSyn amounts, we used targeted proteomics, which was then complemented by multiple techniques such as WB, shot gun proteomics, and microscopy. To investigate the cellular responses to exogenous αSyn monomers, oligomers, and fibrils, we quantified the proteomes of SH-SY5Y cells treated with these four different αSyn species by an approach that combines shotgun proteomics and label-free quantification of proteins using the software Progenesis. These analyses were replicated in two independent experiments. The protein quantities from αSyn-treated cells were compared to those obtained from cells treated with vehicle. By using this approach, proteins that were differentially expressed (either down- or up-regulated) in cells treated with αSyn were identified. Functional associations among these DEPs were investigated using protein-protein interaction network analyses, gene ontology, and pathway enrichment analyses. The databases used for such analyses were STRING, Panther, and DAVID.
On the basis of these functional associations, we validated SCFFBXL5 as ubiquitin ligase for exogenous αSyn fibrils in multiple experiments that involved WB, immunofluorescence, and (shotgun and targeted) MS. All the data shown were replicated in at least two independent experiments.
On the basis of the in vitro data, we studied the subcellular localization of SCF core subunits in the neocortex and brainstem from patients with PD (n = 5) and DLBs (n = 5) and healthy age-matched subjects (n = 5). Because the colocalization of these two proteins with αSyn in LBs suggested that SCF might play important roles in synucleinopathies, we investigated whether SCFFBXL5 could be an important modulator of the LB-like pathology triggered by exogenous αSyn fibrils in the mammalian central nervous system. To test this hypothesis, we selected two different mice lines: the αSyn transgenic mice “line 61,” in which the gene encoding human αSyn is under the control of Thy-1 promoter (60), and nontransgenic wild-type mice. The foundation of our choice is that the transgenic mice develop a robust and progressive LB-like pathology spontaneously, which allowed us to investigate whether SCFFBXL5 loss of function could enhance an incipient αSyn pathology that these mice develop at 14 weeks of age. A lentivirus carrying an shRNA specific for SKP1 was injected into the hippocampus of these transgenic animals (n = 6). Control animals (n = 6) were injected similarly with a lentivirus carrying a control shRNA. Animals were kept for 5 weeks (a time lapse where the shRNA is expected to exert its action), and then iF-αSyn was administrated at the same site. The animals were sacrificed 10 weeks after iF-αSyn administration based on a previous work (37). In transgenic mice, lentivirus as well as αSyn fibril administration was restricted to hippocampus based on a previous work (37). Nontransgenic wild-type mice were used to answer the question of whether SCFFBXL5 silencing could promote initiation of the LB-like pathology triggered by exogenous αSyn fibrils. Because nontransgenic animals do not develop αSyn pathology spontaneously, this model allowed us to study not only SCFFBXL5 effect on pathology initiation but also amplification and spreading. In this case, the control shRNA and the shRNA specific for SKP1 or FBXL5 were injected in different hemispheres of the same animal (n = 6) because this experimental design minimizes the animal-to-animal variability. Five weeks after lentivirus injection, a single dose of iF-αSyn was administrated in the same site, and the animals were sacrificed at two time points (5 or 10 weeks after iF-αSyn administration) to evaluate the spreading and amplification of the LB-like pathology. We did not use statistics to predetermine sample size a priori. Instead, sample size in experiments involving mice was estimated on the basis of our recent studies (28, 100). Randomization was used in assignment of mice to different treatment groups, and investigators were not blinded during experimental treatment and data collection.
Expression, purification, and preparation of human αSyn species
Escherichia coli cells (strain BL21, DE3) were transformed with the pT7–7–αSyn plasmid [provided by Stöckl et al. (101)] and grown in LB medium at 37°C to an A600 (absorbance at 600 nm) of 0.6 followed by induction of protein expression by addition of 1 M isopropyl-β-d-thiogalactopyranoside for 4 hours. Purification of the recombinant protein was conducted as described previously (102). The purity of the sample was assessed by high-performance liquid chromatography (RESOURCE Q, GE Healthcare), and the identity of the eluted material was confirmed by SDS-PAGE with Coomassie blue staining, by WB, and by MS. The purified αSyn was lyophilized and stored at −20°C. N-terminal 6× histidine-tagged human αSyn was purchased from Sigma-Aldrich (S7820). Monomeric αSyn was obtained by dissolving the lyophilized protein in phosphate-buffered saline (PBS) buffer supplemented with 0.05% of sodium azide. Preexisting HMW species were removed by ultracentrifugation and filtration with a Millex-GV, low protein binding Durapore (polyvinylidene difluoride) membrane (Millipore). αSyn oligomers and fibrils were obtained by incubating the monomeric protein in a final concentration of 250 μM at 37°C for the indicated times in a thermomixer set at 700 rpm. Aliquots of the sample were subjected to the ThT binding assay, circular dichroism measurements, and transmission electron microscopy (TEM) imaging as described below.
αSyn internalization assays
Before use in internalization experiments, F-αSyn and iF-αSyn species prepared from the recombinant M-αSyn were subjected to a mild sonication step in a bath sonicator (30) (Elmasonic P, Elma; ultrasound efficiency 100%; temperature, 20°C; time, 10 min). Only for the experiment of fig. S1 (F and G), αSyn fibrils obtained after 60 days of incubation were subjected to repeated cycles of sonication using an ultrasonic cell disruptor Misonix sonicator 3000 (settings, no temperature control; output level, 10; pulse on, 5 s to 5 min; pulse off, 5 s to 5 min) using a microtip. Sonications were performed on aliquots of 150 μl. Unless indicated, cells were treated with 250 nM of extracellular αSyn for 16 hours in Dulbecco’s modified Eagle’s medium supplemented with 2% fetal calf serum, 4 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 mg/ml). To remove membrane-bound extracellular αSyn, the cells were washed four times with ice-cold PBS supplemented with 0.001% NP-40. For MS analyses, traces of detergent were removed by four additional washes with ice-cold PBS. For αSyn cell-to-cell propagation experiments, αSyn expression was induced in SH-SY5Y cell clones for 7 days, and conditioned medium was collected and centrifuged at 1000g to remove detached cells. The conditioned medium was then used to treat Cos7 cells for 16 hours. After washing, Cos7 cells were lysed for MS analyses. The data shown are representative of at least three independent experiments.
Protein identification and label-free quantification by shotgun MS
The peptide samples were analyzed on a quadrupole-orbitrap mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific) equipped with a nanoelectrospray ion source and coupled to EASY-nLC 1000 liquid chromatography system (Thermo Fisher Scientific). Peptides were loaded and chromatographically separated using a linear gradient from 2 to 35% acetonitrile over 160 min at a flow rate of 300 nl/min. Twenty MS/MS spectra were acquired per each MS scan, at 70,000 full width at half maximum (FWHM) resolution settings. One microscan was acquired per each MS/MS scan at 17,500 FWHM resolution. Charge state screening was used including all multiple charged ions for triggering MS/MS attempts, excluding all singly charged precursor ions, and ions for which no charge state could be determined. Only peptide ions exceeding a threshold of 1300 ion counts were allowed to trigger MS/MS scans, followed by dynamic exclusion for 30 s. Repeat count was set to 1. For protein identification and label-free quantification, the obtained spectra were analyzed using Progenesis LC-MS software (version 3.0; Nonlinear Dynamics). This software was used to generate extracted peptide intensity (MS1) features using .Raw data files as input. Selection of the best reference run was conducted manually. Automatic processing was used for spectral alignment (based on retention times of each spectra) and peak picking. Last, only spectra with rank < 3 and charge < 5 were used to generate the .mgf output file for protein identification using Mascot (Matrix Science). The UniProt/SwissProt human database (accessed November 2012) was used, and trypsin was set as the digesting protease, allowing up to one missed cleavage and no cleavages of KP (lysine followed by a proline) and RP (arginine followed by a proline) bonds. The monoisotopic peptide and fragment mass tolerances were set to 1.2 and 0.6 Da, respectively. Carboxyamidomethylation of cysteines (+57.0214 Da) was defined as fixed modification and oxidation of methionines (+15.99492) as a variable modification. Protein identifications were statistically analyzed using the automatically generated decoys plus manual filtering to a false discovery rate (FDR) of <1%. For detection of ubiquitinated αSyn, the collected spectra were searched against the human protein database (UniProt/SwissProt) with Sorcerer-SEQUEST (Thermo Electron) including ubiquitination of lysines (+114.0429 Da) as a variable modification. Peptide identifications were statistically analyzed with PeptideProphet (version 3.0) and filtered to a cutoff of 0.9 PeptideProphet probability, which, in this particular case, corresponds to an FDR of <1%, calculated on the basis of a target-decoy approach (103). For spectra visualization, Proteome Discoverer 1.4 (Thermo Fisher Scientific) was used. Protein quantities (normalized abundance) obtained from Progenesis were averaged, and the fold change was calculated using the average of the protein quantities for a particular αSyn treatment divided by the corresponding averaged quantities of vehicle-treated cells and/or empty vector–transfected cells.
Animal experiments
Mouse experiments were performed under the license number 41/2012 and according to the regulations of the Veterinary Office of the Canton Zurich. Mice used in this study include the αSyn transgenic mouse line 61, in which the gene encoding human αSyn is under the control of Thy-1 promoter (60), and nontransgenic wild-type C57BL6/C3H mice. Six animals were used per treatment. Fourteen-month-old αSyn transgenic mice and 9-week-old female C57BL6/C3H mice were anesthetized with isoflurane and placed in a motorized stereotaxic frame controlled by software with a three-dimensional brain map (NeuroStar), allowing for real-time monitoring of intracerebral injection. After the skull was exposed by cutting along the midline, a small hole was drilled using a surgical drill and the needle was placed at the following coordinates from bregma: anterior-posterior (AP), +2.0 mm; medial-lateral (ML), −1.5 mm; dorsal-ventral (DV), −1.3 mm for αSyn transgenic mouse (line 61), and AP, +0.2 mm; ML, ±2.0 mm; DV, −1.6 mm for wild-type nontransgenic mice. Injections with the lentiviruses were performed using a 10-μl syringe (Hamilton) at a rate of 0.5 μl/min (6 μl total per site) with the needle in place for >5 min at each target. αSyn transgenic mice (six animal per experimental group) were subjected to a single injection with LV-shGFP or LV-shSKP1 and, 5 weeks later, to a single inoculation with iF-αSyn. Nontransgenic animals (four animals per group) were injected bilaterally: on the left side with LV-shGFP and on the right with LV-shSKP1 or LV-shFBXL5. Five weeks later, iF-αSyn was injected in both hemispheres. The skin was sutured with Vicryl 6–0. iF-αSyn (250 nM) was injected 5 weeks after lentivirus injections using a 10-μl syringe (Hamilton) at a rate of 0.5 μl/min (10 μl total per site) with the needle in place for >5 min at each target. During the intervention, mice were treated with subcutaneous injections of buprenorphin (0.1 mg/kg) and flunixin (5 mg/kg) to alleviate postoperative complications. All mice were maintained under highly hygienic conditions, monitored regularly after recovery from surgery, and sacrificed at predetermined time points. For histological studies, the brain was removed after transcardial perfusion with PBS. Brains were fixed in ethanol-NaCl for 48 hours. Fixed tissues were treated with concentrated formic acid for 60 min to inactivate potentially infectious particles and were embedded in paraffin. Paraffin sections (5 mm) of brains were stained with rabbit anti-αSyn (#2642 for total αSyn and D37A6 for mouse αSyn, Cell Signaling Technology) or rabbit anti-pSer129–αSyn (#59264, Abcam). αSyn-positive inclusions were quantified from six control and six treated animals and/or three consecutive brain sections.
Statistics
For the identification and label-free quantification of proteins by shotgun MS, we used Student’s t test (unpaired, two-tailed distribution) to calculate P values. Data shown are means ± SDs. DEPs were arbitrarily defined as those with a fold change of >1.75 and P < 0.05. For in vitro studies, we used unpaired two-tailed Student’s t test when only two groups were compared, ANOVA with a Dunnett’s test for multigroup comparisons where every group/treatment is compared with a single control, one-way ANOVA followed by a Fisher test for multicomparisons within experiments of small sample sizes, and one-way ANOVA followed by a Tukey’s test for all possible pairwise comparisons. P value of 0.05 or lower was considered statistically significant. Additional information is provided in figure legends. For studies in mice, we used paired two-tailed Student’s t test for experiments where only two groups of the same animal (not independent) are compared. Differences among means were assessed by one-way ANOVA with Dunnett’s post hoc test when compared to LV-shGFP–injected animals/hemispheres. The null hypothesis was rejected at the 0.05 level. All results are expressed as means + SEM. Statistical analyses were performed with STATISTICA v7 (StatSoft) and GraphPad Prism. Original data are provided in data file S1.
Supplementary Material
Data file S1. Source data (Excel file).
Table S2. Label-free quantification of peptides from SH-SY5Y cells treated with the αSyn species.
Table S1. Label-free quantification of proteins from SH-SY5Y cells treated with the αSyn species.
Table S3. DEPs from SH-SY5Y cells treated with the αSyn species.
Materials and Methods
Fig. S1. Internalization of insoluble fibrils leads to αSyn accumulation and proteome alterations.
Fig. S2. An SCF E3 ubiquitin ligase targets internalized αSyn for ubiquitination and degradation.
Fig. S3. An SCF inhibits the prion-like properties of extracellular αSyn.
Fig. S4. SCFFBXL5 inhibits LB-like pathology induced by extracellular αSyn fibrils.
Table S4. Gene ontology enrichment analysis according to the Panther database.
Table S5. KEEG pathway enrichment analysis according to the STRING database.
Acknowledgments:
We are grateful to P. Nanni and R. Schlapbach from the Functional Genomics Centre Zurich for access to MS instrumentation, to the Light Microscopy Center (LMC) at ETH Zurich, and to D. Roderer (ETH Zurich) for technical assistance with CD measurements.
Funding:
P.P. was supported by an EU FP7-ERC starting grant (FP7-ERC-StG-337965), an FP7 Reintegration grant (FP7-PEOPLE-2010-RG-277147), a “Foerderungsprofessur”and Sinergia grants from the Swiss National Science Foundation (SNSF) (grants PP00P3-133670 and CRSII5_177195), a Personalized Health and Related Technologies (PHRT) grant (PHRT-506), and a Promedica Stiftung (grant 2-70669-11). J.A.G. was supported by an EMBO postdoctoral fellowship (ALTF-254-2012), a “Wilhelm Hurka” Stiftung, and a grant from the SNSF (Sinergia 154461). N.C.P. is supported by a Wilhelm Hurka Stiftung. M.P., R.I.E., and T.C. were supported by an ERC consolidator grant, the SNSF, and the ETH Zurich. R.I.E. was supported by a Marie Curie postdoctoral fellowship.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data used for the preparation of the manuscript are present in the main text or in the Supplementary Materials. Materials generated in this work such as plasmids or recombinant proteins are available through a material transfer agreement.
REFERENCES AND NOTES
- 1.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M, α-synuclein in Lewy bodies. Nature 388, 839–840 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Poulopoulos M, Levy OA, Alcalay RN, The neuropathology of genetic Parkinson’s disease. Mov. Disord 27, 831–842 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL, Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O, AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet 18, 106–108 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K, α-synuclein locus triplication causes Parkinson’s disease. Science 302, 841 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T, Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet 41, 1303–1307 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T, Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet 41, 1308–1312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC, α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH, Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartels T, Choi JG, Selkoe DJ, α-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D, In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem 288, 6371–6385 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Sudhof TC, Properties of native brain α-synuclein. Nature 498, E4–E6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spencer B, Kim C, Gonzalez T, Bisquertt A, Patrick C, Rockenstein E, Adame A, Lee S-J, Desplats P, Masliah E, α-synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum. Mol. Genet 25, 1100–1115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, Jiang L, Arbing MA, Nannenga BL, Hattne J, Whitelegge J, Brewster AS, Messerschmidt M, Boutet S, Sauter NK, Gonen T, Eisenberg DS, Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conway KA, Harper JD, Lansbury PT, Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med 4, 1318–1320 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Conway KA, Harper JD, Lansbury PT Jr., Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39, 2552–2563 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A, α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Bezard E, Yue Z, Kirik D, Spillantini MG, Animal models of Parkinson’s disease: Limits and relevance to neuroprotection studies. Move. Disord 28, 61–70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chesselet M-F, In vivo alphα-synuclein overexpression in rodents: A useful model of Parkinson’s disease? Exp. Neurol 209, 22–27 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM, Degradation of α-synuclein by proteasome. J. Biol. Chem 274, 33855–33858 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Tofaris GK, Layfield R, Spillantini MG, α-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509, 22–26 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC, α-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem 278, 25009–25013 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D, Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Sung JY, Kim J, Paik SR, Park JH, Ahn YS, Chung KC, Induction of neuronal cell death by Rab5A-dependent endocytosis of α-synuclein. J. Biol. Chem 276, 27441–27448 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, Lee S-J, Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol 40, 1835–1849 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Zhang J-P, Shi M, Quinn T, Bradner J, Beyer R, Chen S, Zhang J, Rab11a and HSP90 regulate recycling of extracellular α-synuclein. J. Neurosci 29, 1480–1485 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugeno N, Hasegawa T, Tanaka N, Fukuda M, Wakabayashi K, Oshima R, Konno M, Miura E, Kikuchi A, Baba T, Anan T, Nakao M, Geisler S, Aoki M, Takeda A, Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4–1 facilitates endosomal sequestration of internalized α-synuclein. J. Biol. Chem 289, 18137–18151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K, Shen J, Rockenstein E, Patrick C, Adame A, Gonzalez T, Sierks M, Masliah E, ESCRT-mediated uptake and degradation of brain-targeted α-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol. Ther 22, 1753–1767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK, Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci 31, 14508–14520 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volpicelli-Daley LA, Luk KC, Lee VM-Y, Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc 9, 2135–2146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM-Y, Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tu P.-h., Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM-Y, Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann. Neurol 44, 415–422 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E, Alphα-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Lee H-J, Suk J-E, Patrick C, Bae E-J, Cho J-H, Rho S, Hwang D, Masliah E, Lee S-J, Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem 285, 9262–9272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong J-S, Zhang J, Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J 19, 533–542 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E, Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y, Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM-Y, Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U.S.A 106, 20051–20056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW, Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med 14, 504–506 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Li J-Y, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, Widner H, Revesz T, Lindvall O, Brundin P, Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med 14, 501–503 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Lee H-J, Suk J-E, Bae E-J, Lee S-J, Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun 372, 423–428 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Vekrellis K, Stefanis L, Targeting intracellular and extracellular alphα-synuclein as a therapeutic strategy in Parkinson’s disease and other synucleinopathies. Expert Opin. Ther. Targets 16, 421–432 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG, Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener 2, 18 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metzger MB, Pruneda JN, Klevit RE, Weissman AM, RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 1843, 47–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA, Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell 134, 995–1006 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saha A, Deshaies RJ, Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol. Cell 32, 21–31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skaar JR, Pagan JK, Pagano M, SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov 13, 889–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molochnikov L, Rabey JM, Dobronevsky E, Bonucelli U, Ceravolo R, Frosini D, Grünblatt E, Riederer P, Jacob C, Aharon-Peretz J, Bashenko Y, Youdim MB, Mandel SA, A molecular signature in blood identifies early Parkinson’s disease. Mol. Neurodegener 7, 26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mandel SA, Fishman-Jacob T, Youdim MB, Modeling sporadic Parkinson’s disease by silencing the ubiquitin E3 ligase component, SKP1A. Parkinson. Relat. Disord 15 (suppl. 3), S148–S151 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP, An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Desplats P, Lee H-J, Bae E-J, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee S-J, Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U.S.A 106, 13010–13015 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carrano AC, Eytan E, Hershko A, Pagano M, SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol 1, 193–199 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Skaar JR, Pagan JK, Pagano M, Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol 14, 369–381 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salahudeen AA, Thompson JW, Ruiz JC, Ma H-W, Kinch LN, Li Q, Grishin NV, Bruick RK, An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA, Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calafate S, Flavin W, Verstreken P, Moechars D, Loss of Bin1 promotes the propagation of tau pathology. Cell Rep 17, 931–940 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Zeineddine R, Pundavela JF, Corcoran L, Stewart EM, Do-Ha D, Bax M, Guillemin G, Vine KL, Hatters DM, Ecroyd H, Dobson CM, Turner BJ, Ooi L, Wilson MR, Cashman NR, Yerbury JJ, SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol. Neurodegener 10, 57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, Kordower JH, Melki R, Campbell EM, Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol 134, 629–653 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM, Alphα-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLOS ONE 8, e62143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E, Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res 68, 568–578 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y, Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med 209, 975–986 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sacino AN, Brooks M, McGarvey NH, McKinney AB, Thomas MA, Levites Y, Ran Y, Golde TE, Giasson BI, Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol. Commun 1, 38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T, α-synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol 4, 160–164 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Steiner JA, Angot E, Brundin P, A deadly spread: Cellular mechanisms of α-synuclein transfer. Cell Death Differ 18, 1425–1433 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luk KC, Lee VM-Y, Modeling Lewy pathology propagation in Parkinson’s disease. Parkinson. Relat. Disord 20, S85–S87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T, Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol 152, 879–884 (1998). [PMC free article] [PubMed] [Google Scholar]
- 67.Kahle PJ, Neumann M, Ozmen L, Muller V, Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ, Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar HA, Haass C, Selective insolubility of α-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am. J. Pathol 159, 2215–2225 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sacino AN, Brooks MM, Chakrabarty P, Saha K, Khoshbouei H, Golde TE, Giasson BI, Proteolysis of α-synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J. Neurochem 140, 662–678 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rey NL, Petit GH, Bousset L, Melki R, Brundin P, Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126, 555–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, Braithwaite A, He Z, Ogholikhan S, Hinkle K, Kent C, Toudjarska I, Charisse K, Braich R, Pandey RK, Heckman M, Maraganore DM, Crook J, Farrer MJ, In vivo silencing of α-synuclein using naked siRNA. Mol. Neurodegener 3, 19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sapru MK, Yates JW, Hogan S, Jiang L, Halter J, Bohn MC, Silencing of human α-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol 198, 382–390 (2006). [DOI] [PubMed] [Google Scholar]
- 72.McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA, α-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLOS ONE 5, e12122 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, El Ayadi A, Hastings TG, Greenamyre JT, Burton EA, shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest 125, 2721–2735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ, Abo KM, Long E, Jin M, Xu B, Xiang YK, Rochet J-C, Engeland A, Rizzu P, Heutink P, Bartels T, Selkoe DJ, Caldarone BJ, Glicksman MA, Khurana V, Schule B, Park DS, Riise T, Scherzer CR, β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 357, 891–898 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, Meyers C, Manfredsson FP, Muzyczka N, In vivo RNAi-mediated α-synuclein silencing induces nigrostriatal degeneration. Mol. Ther 18, 1450–1457 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanaan NM, Manfredsson FP, Loss of functional α-synuclein: A toxic event in Parkinson’s disease? J. Parkinsons Dis 2, 249–267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collier TJ, Redmond DE Jr., Steece-Collier K, Lipton JW, Manfredsson FP, Is alphα-synuclein loss-of-function a contributor to parkinsonian pathology? Evidence from non-human primates. Front. Neurosci 10, 12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC, Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem 282, 5641–5652 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ, Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem 281, 29739–29752 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM-Y, Trojanowski JQ, Mann D, Iwatsubo T, Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem 277, 49071–49076 (2002). [DOI] [PubMed] [Google Scholar]
- 81.Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT, McLean PJ, CHIP targets toxic α-synuclein oligomers for degradation. J. Biol. Chem 283, 17962–17968 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rott R, Szargel R, Haskin J, Bandopadhyay R, Lees AJ, Shani V, Engelender S, α-synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. U.S.A 108, 18666–18671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tofaris GK, Kim HT, Hourez R, Jung J-W, Kim KP, Goldberg AL, Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. U.S.A 108, 17004–17009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abeywardana T, Lin YH, Rott R, Engelender S, Pratt MR, Site-specific differences in proteasome-dependent degradation of monoubiquitinated α-synuclein. Chem. Biol 20, 1207–1213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haj-Yahya M, Fauvet B, Herman-Bachinsky Y, Hejjaoui M, Bavikar SN, Karthikeyan SV, Ciechanover A, Lashuel HA, Brik A, Synthetic polyubiquitinated α-synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. U.S.A 110, 17726–17731 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, Schneider R, Mizuno Y, Kosik KS, Selkoe DJ, Ubiquitination of a new form of α-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 293, 263–269 (2001). [DOI] [PubMed] [Google Scholar]
- 87.Kalia LV, Kalia SK, Chau H, Lozano AM, Hyman BT, McLean PJ, Ubiquitinylation of α-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLOS ONE 6, e14695 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, Bornemann A, Riess O, Ross CA, Rott R, Engelender S, Ubiquitylation of synphilin-1 and α-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A 101, 5500–5505 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meier F, Abeywardana T, Dhall A, Marotta NP, Varkey J, Langen R, Chatterjee C, Pratt MR, Semisynthetic, site-specific ubiquitin modification of α-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc 134, 5468–5471 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tran HT, Chung CH-Y, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM-Y, α-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep 7, 2054–2065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D, Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46, 857–868 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Prymaczok NC, Riek R, Gerez J, More than a rumor spreads in Parkinson’s disease. Front. Hum. Neurosci 10, 608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM-Y, Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem 288, 15194–15210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grunblatt E, Ruder J, Monoranu CM, Riederer P, Youdim MBH, Mandel SA, Differential alterations in metabolism and proteolysis-related proteins in human Parkinson’s disease substantia nigra. Neurotox. Res 33, 560–568 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MBH, Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J. Neural Transm 111, 1543–1573 (2004). [DOI] [PubMed] [Google Scholar]
- 96.Bhutani S, Das A, Maheshwari M, Lakhotia SC, Jana NR, Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis 3, e428 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moroishi T, Nishiyama M, Takeda Y, Iwai K, Nakayama KI, The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab 14, 339–351 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Ayton S, Lei P, Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. BioMed Res. Int 2014, 581256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sacino AN, Brooks M, Thomas MA, McKinney AB, McGarvey NH, Rutherford NJ, Ceballos-Diaz C, Robertson J, Golde TE, Giasson BI, Amyloidogenic α-synuclein seeds do not invariably induce rapid, widespread pathology in mice. Acta Neuropathol 127, 645–665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bae E-J, Lee H-J, Rockenstein E, Ho D-H, Park E-B, Yang N-Y, Desplats P, Masliah E, Lee S-J, Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci 32, 13454–13469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stöckl M, Fischer P, Wanker E, Herrmann A, α-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J. Mol. Biol 375, 1394–1404 (2008). [DOI] [PubMed] [Google Scholar]
- 102.De Franceschi G, Frare E, Bubacco L, Mammi S, Fontana A, de Laureto PP, Molecular insights into the interaction between α-synuclein and docosahexaenoic acid. J. Mol. Biol 394, 94–107 (2009). [DOI] [PubMed] [Google Scholar]
- 103.Elias JE, Gygi SP, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Source data (Excel file).
Table S2. Label-free quantification of peptides from SH-SY5Y cells treated with the αSyn species.
Table S1. Label-free quantification of proteins from SH-SY5Y cells treated with the αSyn species.
Table S3. DEPs from SH-SY5Y cells treated with the αSyn species.
Materials and Methods
Fig. S1. Internalization of insoluble fibrils leads to αSyn accumulation and proteome alterations.
Fig. S2. An SCF E3 ubiquitin ligase targets internalized αSyn for ubiquitination and degradation.
Fig. S3. An SCF inhibits the prion-like properties of extracellular αSyn.
Fig. S4. SCFFBXL5 inhibits LB-like pathology induced by extracellular αSyn fibrils.
Table S4. Gene ontology enrichment analysis according to the Panther database.
Table S5. KEEG pathway enrichment analysis according to the STRING database.