

The ALPK3 gene encodes the alpha (α)-protein kinase 3, a muscle-enriched protein. Various ALPK3 variants have been implicated in cardiomyopathies1. Additionally, Alpk3 knockout mice rapidly develop dilated cardiomyopathy, leading to premature mortality before adulthood 2, suggesting that ALPK3 is essential for cardiac function.
ALPK3 belongs to the α-kinase family, characterized by unique structural features and regulatory mechanisms that set them apart from canonical eukaryotic protein kinases. Recent studies present conflicting findings on ALPK3’s kinase activity2,3. Agarwal et al. performed phosphoproteomic studies using human pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), comparing wildtype ALPK3, ALPK3 null mutant, and ALPK3 mutants of key conserved residues predicted to be essential for its putative kinase activity. Notably, no significant changes in phosphorylation were observed across the proteome for any of these variants. Additionally, even upon massive overexpression of ALPK3 in HEK293T cells, no significant alterations in phosphorylation were detected across the proteome. Thus, they concluded that ALPK3 does not have kinase activity2. However, McNamara et al. reported a significant increase in phosphorylated proteins when the recombinant ALPK3 kinase domain is added to lysates from ALPK3-deficient hiPSC-CMs, and ALPK3 directly phosphorylates SQSTM1 at the T269/S272 site3. It is important to note that both studies assessed the kinase activity of ALPK3 by measuring phosphorylation events in cultured cells or in vitro. Whether ALPK3 functions as a true kinase in heart remains unknown.
To determine if ALPK3 has potential protein kinase activity, we conducted a 3D alignment of murine ALPK3 catalytic domain with that of murine TRPM7, a closely related member of the α-kinase family with experimentally confirmed protein kinase activity. The alignment was performed using the previously published crystal structure of murine TRPM7 bound to ADP and the AlphaFold model of murine ALPK3 (Figure 1A). Eighteen nucleotide-interacting residues superimpose very well between ALPK3 and TRPM7 (RMSDCα < 1Å), allowing us to identify K1420 as the invariant catalytic lysine required for the kinase activity of vast majority of protein kinases including TRPM74 (Figure 1A). Despite sharing an invariant lysine, ALPK3 and TRPM7 nucleotide binding pockets consist of largely non-identical residues, as have been noticed in a previous report 2 and seen in our 3D superimposition of ALPK3 and TRPM7 catalytic domains (Figure 1A). Although the presence of an invariant lysine residue is essential, it is not the sole determinant of the kinase activity of protein kinases. Numerous catalytically inactive pseudokinases maintain this residue, highlighting the responsibility of other nucleotide-interacting residues for the catalytic inactivity of pseudokinases5. Importantly, human and murine ALPK3 share a significant 93% sequence identity. Furthermore, a detailed analysis of ATP binding residues in both catalytic domains reveals a close alignment, with an RMSDCα value of 1.86 Å from the 3D AlphaFold models.
Figure 1. ALPK3 functions as a pseudokinase.
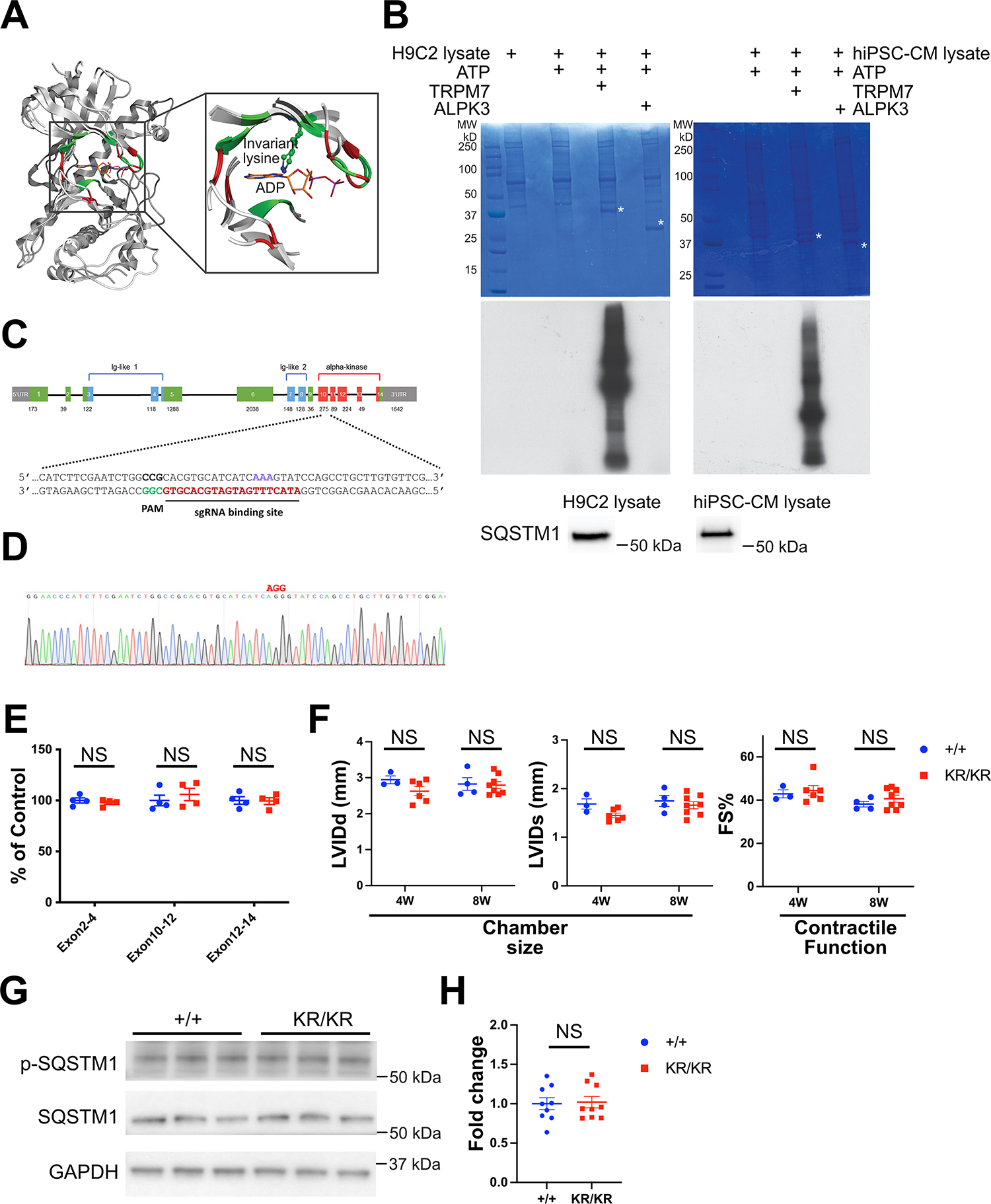
(A) 3D alignment of catalytic domains of ALPK3 (AlphaFold) and TRPM7 (PDB ID: 1IAH). Positions of identical ADP-interacting residues are in green, differing ones in red. Position of the invariant catalytic lysine residue is marked in the enlarged nucleotide binding pocket. (B) In vitro phosphorylation assay of murine TRPM7 and murine ALPK3 catalytic domains on heat inactivated H9c2 (left panel) and hiPSC-CM (right panel) cell lysates. Coomassie blue stain (top) and autoradiography of γ-32P (middle) showing phosphorylated cell lysate substrate are shown. White asterisks indicate the position of the added recombinant protein kinase bands on the gel. Presence of SQSTM1 in H9c2 and hiPSC-CM cell lysates is confirmed by western blotting (bottom). (C) Schematics of CRISPR-Cas9 strategy to generate ALPK3 K1420R knock-in mice. (D) Sanger sequencing confirming mutation of AAA (encoding lysine) to AGG (encoding arginine) in ALPK3 KR knock-in mice. (E) qRT-PCR assessment of ALPK3 mRNA levels in the hearts of +/+ (N = 4) and KR/KR (N = 4) mice using primers targeting exons 2–4, 10–12, and 12–14 was performed. Each dot represents the value of ALPK3 mRNA levels obtained with qRT-PCR, with the average value of ALPK3 levels in +/+ mouse hearts arbitrarily set as 100%. Mean ± standard error of the mean (SEM) is shown. NS, not significant by Student’s t-test. (F) Echocardiography assessment of cardiac chamber size and contractile function of +/+ (N = 3–5) and KR/KR (N = 4–8) mice. Left ventricle internal dimension at diastole (LVIDd) and left ventricle internal dimension at systole (LVIDs) are used for measurement of chamber size. Fraction shortening (FS) is used for measurement of contractile function. Mean ± standard error of the mean (SEM) was shown. NS, not significant by Student’s t-test. (G) Representative western blotting of phosphorylated (T269/S272) and total SQSTM1 in +/+ and KR/KR mice. GAPDH is shown for loading control. (H) Quantification of p-SQSTM1 (T269/S272) to total SQSTM1 ratio from western blotting of phosphorylated (T269/S272) and total SQSTM1 in +/+ and KR/KR mice (N = 9 for each group). Mean ± standard error of the mean (SEM) was shown. NS, not significant by Student’s t-test.
As the alignment data suggests that ALPK3 may or may not possess kinase activity, we first evaluated the kinase activity of murine ALPK3 catalytic domain (1339–1608 UniprotID: Q924C5) using an in vitro phosphorylation assay, along with the catalytic domain of murine TRPM7 (residues 1548–1863 of UniprotID: Q923J1) as a positive control on heat-inactivated H9c2(2–1) and hiPSC-CM lysates containing SQSTM1, a direct ALPK3 substrate identified by McNamara et al. 3 (Figure 1B). However, our experimental results revealed the absence of detectable kinase activity in ALPK3 under the tested conditions (Figure 1B). In stark contrast, TRPM7 demonstrated effective phosphorylation of the cellular lysate when [γ-32P] ATP was presented, consistent with prior reports4.
To exclude the possibility that factors absent in our in vitro assay might be required for ALPK3 catalytic activity and to assess the functional significance of the putative kinase activity of ALPK3 in vivo, we utilized CRISPR/Cas9 technology to generate knock-in mice in which the invariant K1420 of ALPK3 is mutated to arginine(R) (Figure 1C–D). All mouse protocols were approved by the Institutional Animal Care and Use Committee. Mutation of this invariant K to R has been experimentally shown to inactivate numerous eukaryotic protein kinases including TRPM7 4. qRT-PCR analysis showed comparable levels of ALPK3 mRNA between the control and the homozygous knock-in (Alpk3 KR/KR) mice (Figure 1E). In contrast to the published Alpk3 null mutant mice2, the homozygous Alpk3 KR/KR mice did not exhibit premature mortality and maintained normal cardiac function into adulthood (Figure 1F). Additionally, we investigated the phosphorylation status of the T269/S272 site of SQSTM1 by western blotting, but we did not observe a significant difference in T269/S272 phosphorylation to total SQSTM1 ratios between the hearts of the control mice and those carrying homozygous ALPK3KR/KR mutation (Figure 1G–H).
In conclusion, our study presents compelling new evidence from both in vitro and in vivo experiments, supporting that ALPK3 functions as a pseudokinase. The ALPK3 variants associated with cardiomyopathy may affect its protein levels, interaction with binding partners and/or subcellular localization, rather than its putative kinase activity. Detailed data supporting these findings can be obtained from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
The authors thank the Transgenic Core at UCSD for generating the ALPK3 knock-in mice.
Sources of Funding
J. Chen is funded by National Heart, Lung, and Blood Institute (NHLBI) Grants and holds an American Heart Association Endowed Chair in Cardiovascular Research.
Non-standard abbreviation:
- hiPSC-CM
human induced pluripotent stem cell-derived cardiomyocytes
- RMSDCα
root-mean-square deviation Of C alpha atoms
Footnotes
Disclosures
J. Chen consults for LEXEO Therapeutics and Morphic Therapeutic.
J.B. consults for Rocket Pharmaceuticals on image analysis.
References Cited
- 1.Walsh R, Bezzina CR. ALPK3: a full spectrum cardiomyopathy gene? Eur Heart J. 2021;42:3074–3077. doi: 10.1093/eurheartj/ehab415 [DOI] [PubMed] [Google Scholar]
- 2.Agarwal R, Wakimoto H, Paulo JA, Zhang Q, Reichart D, Toepfer C, Sharma A, Tai AC, Lun M, Gorham J, et al. Pathogenesis of Cardiomyopathy Caused by Variants in ALPK3, an Essential Pseudokinase in the Cardiomyocyte Nucleus and Sarcomere. Circulation. 2022;146:1674–1693. doi: 10.1161/CIRCULATIONAHA.122.059688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNamara JW, Parker BL, Voges HK, Mehdiabadi NR, Bolk F, Ahmad F, Chung JD, Charitakis N, Molendijk J, Zech ATL, et al. Alpha kinase 3 signaling at the M-band maintains sarcomere integrity and proteostasis in striated muscle. Nature Cardiovascular Research. 2023;2:159–173. doi: 10.1038/s44161-023-00219-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaitsuka T, Katagiri C, Beesetty P, Nakamura K, Hourani S, Tomizawa K, Kozak JA, Matsushita M. Inactivation of TRPM7 kinase activity does not impair its channel function in mice. Sci Rep. 2014;4:5718. doi: 10.1038/srep05718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwon A, Scott S, Taujale R, Yeung W, Kochut KJ, Eyers PA, Kannan N. Tracing the origin and evolution of pseudokinases across the tree of life. Sci Signal. 2019;12. doi: 10.1126/scisignal.aav3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.