

Abstract
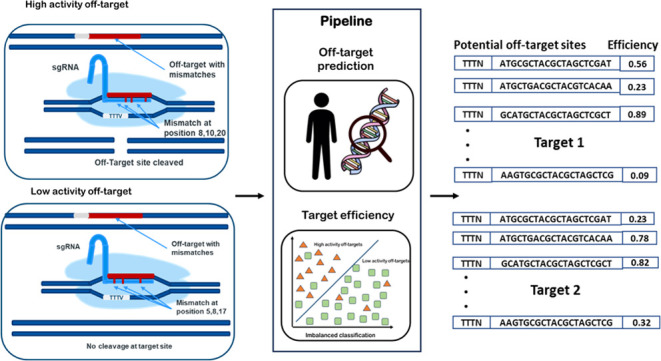
Targeted nucleases are widely used for altering the specific location of the genome with precision. The endonucleases facilitate efficient genome editing via designing a guide RNA (gRNA) consisting of a 20-nucleotide target sequence. gRNA preferably binds to the target location, but the on- and off-target activities of gRNAs vary widely. The off-target activity due to mismatch tolerance in the CRISPR-Cas system is a major factor inhibiting its clinical applications. Ensuring on-target efficiency and minimizing off-targets for a target sequence are the major objectives of this study. A pipeline has been designed to predict potential off-target sites in the human genome for a target sequence, and a multilayer perceptron (MLP) has been used to predict the cleavage efficiency of the potential off-target sites. An MLP-based classifier was trained with sequence- and base-dependent binding energy-associated features for AsCpf1 and LbCpf1 to predict the target efficiencies. Positional preferences of nucleotides, distribution of mismatches, and classification-dependent feature importance between high-activity and low-activity off-targets were also studied. Positional preference of nucleotides revealed that thymine is highly disfavored at positions adjacent to Protospacer Adjacent Motif (PAM), whereas guanine is favored in high-activity off-targets. Mismatch distribution analysis revealed that mismatches were more prominent in the trunk region (16, 17, 18 nucleotides from PAM sequence), and the promiscuous region and transition type mismatch were more preferred at 16, 17, and 18 nucleotides positions. The distribution of mismatches was a distinctive feature between high-activity and low-activity off-targets. Thermodynamics-associated features such as low to moderate melting temperature of the nonseed region and base-dependent PAM binding energy were predicted as best predictors by the multilayer perceptron for high-activity off-targets. GC content, some types of dinucleotide frequencies, number of bulges, and mismatches in the seed and trunk regions were other characteristic features between high-activity and low-activity off-targets for both LbCpf1 and AsCpf1.
1. Introduction
Targeted genome editing is a powerful technology used for gene modification to assess and alter gene function. Various genome editing technologies such as zinc finger nucleases, transcription activator-like effector nucleases, and, more recently, RNA-guided endonucleases are used to introduce double-stranded breaks at the location of choice for precise editing or gene disruption.1,2 Different repair mechanisms of the cell incorporate either indels without template strands or precise editing using template strands at that location. Currently, the adaptive bacterial immune system CRISPR/Cas has taken a lead as an RNA-guided targetable nuclease for widespread application in pharmaceutics, agriculture, and genetics. The CRISPR system is advantageous over ZFNs and TALENs because of its simplicity in designing a guide, controlled delivery, and cost-effectiveness. Many studies have successfully employed the CRISPR system not only in bacteria3 but also in nematodes,4 mice,5 zebrafish,6 and human pluripotent stem cells7 for targeted mutation since its discovery. However, there are various challenges associated with the clinical application of the CRISPR-Cas system, and among them, one of the major challenges is off-target effects. Cas9 nuclease has been engineered to precisely control the off-target activity, gene regulation, and transcriptional repression.8
In 2015, a Cas nuclease named Cpf19 was characterized and reported for lower off-target activity than most Cas-associated nucleases. Another characteristic feature of Cpf1 that distinguishes it from the popular Cas9 is that Cpf1 recognizes a T-rich PAM which ensures stringency in PAM recognition and processes the precursor CRISPR RNAs into mature CRISPR RNAs. Cpf1 also results in staggered ends at cleavage sites which allow easy designing of a template strand with the identity of sticky ends of the double-stranded break.
Single-guide RNA (sgRNA) of CRISPR-Cpf1 consists of CRISPR RNA (crRNA) and guide RNA (gRNA). This gRNA consists of the seed region (1–6 nucleotides including the PAM sequence), the trunk region (7–18 nucleotides), and the promiscuous region (19–23 nucleotides).10 While searching for a target sequence, gRNA mostly tolerates mismatches in the PAM-distal region rather than in the PAM-proximal region. CRISPR-Cpf1 system is found to be highly specific in mammals,11−14 human cells,12 and plant cells.15−18 CRISPR-Cpf1 has also been used to correct genetic mutations in human cells.19,20 The T-rich PAM sequence that Cpf1 mostly favors is TTTV where V is A, C, or G in the IUPAC codes.
The major challenge associated with the CRISPR-Cas system is that the target specificity and efficiency of sgRNAs vary widely resulting in off-target effects which can be controlled by designing and selecting an optimal gRNA.10,21 Currently, there are very limited computational approaches that exist for sgRNA designing and prediction of off-targets,21−23 and INDEL frequencies for Cpf1.10 There are many existing tools for alignment-based guide RNA design24−27 and machine learning-based off-target prediction for Cas9.28−30 However, there is a need for a better performing algorithm for the selection of sgRNAs for Cpf1. Existing studies for Cpf1 have attempted to correlate gRNA features with the target efficiency of the CRISPR-Cpf1 system.10,21 We have previously worked on Cas9 for the prediction of off-targets using sequence-based features and alignment-based approaches.25,28 Further, a machine learning model has been developed to predict target efficiencies with sequence features and binding energies.28,30,31 Therefore, in this study, sequence-associated and base-dependent binding energy-associated features were used to study its biological importance in off-target cleavage activity by Cpf1 extracted from Acidaminococcus species Cpf1 (AsCpf1) and Lachnospiraceae bacterium Cpf1 (LbCpf1) species. Additionally, a potential off-target prediction pipeline and a target efficiency prediction pipeline using sequence- and binding energy-associated features for AsCpf1 and LbCpf1 have been developed.
2. Results
2.1. Performance of Sequence Search and Alignment Tools
Blastn,32 bowtie,33 and Fasta3634 were optimized to predict the maximum number of potential off-target sites in the human reference genome. FASTA36 mapped a large number of highly divergent sequences compared to BLASTn. BOWTIE predicted a maximum number of ungapped off-target sites with up to 9 mismatches. BLASTn overlooked diverse hits predicted by using BOWTIE and FASTA36. The number of such sites predicted using BLASTn, BOWTIE, and FASTA36 for AsCpf1 targets was 124, 483,546, and 58,431. Similarly, the same methods estimated 141, 1,454,619, and 80,346 potential off-targets for LbCpf1. The number of potential off-target sites predicted with the optimized FASTA36, BLASTn, and BOWTIE are summarized in Table 1. Although BOWTIE mapped the maximum number of off-target sites because of the flexibility of the algorithm to map off-target sites, it did not allow for gaps in the calculation. On the other hand, BLASTn was found to be stringent in mapping highly divergent sequences to the human genome and predicted the least number of potential off-target sites. Considering all of these factors, FASTA36 was selected for the pipeline to predict potential off-target sites over BLASTn and BOWTIE since it predicted all of the experimental off-target sites along with other potential off-target locations. The FASTA36 algorithm was optimized to predict off-target sites with 9 mismatches and 3 gaps. Further, the performance of the developed pipeline was benchmarked with existing tools using a randomly chosen target sequence, as shown in Table 2. The pipeline developed in this study performed better compared to the existing tools in predicting potential off-target sites. Also, it could identify most off-target sites that have already been experimentally validated. The list of experimental off-targets predicted by the optimized pipeline for all of the target sequences of AsCpf1 and LbCpf1 are mentioned in the Supporting Information file 2.
Table 1. Number of Potential Off-targets Predicted in the Human Genome Using FASTA36, BLASTn, and BOWTIE.
| predicted
number of similar sites |
|||
|---|---|---|---|
| species | FASTA36 | BLASTn | BOWTIE |
| Lachnospiraceae Bacterium Cas12a (LbCas12a) | 80,346 | 141 | 1,454,619 |
| Acidaminococcus sp. Cas12a (AsCas12a) | 58,431 | 124 | 483,546 |
Table 2. Performance Comparison of Developed Pipeline with the Existing Methods.
| target-TTTCCCTCACTCCTGCTCGGTGAATTT | ||||
|---|---|---|---|---|
| method/pipeline | potential off-target sites predicted | number of experimental off-targets covered | number of experimental off-targets known | time taken to predict off-targets (minutes) |
| developed pipeline | 13,399 | 8 | 12 | 15 |
| Casoff-finder | 9581 | 0 | 12 | 5 |
| CHOPCHOP | 0 | 0 | 12 | 2 |
| CRISPRscan | 0 | 0 | 12 | 2–3 |
| CC-TOP | 468 | 5 | 12 | 2–5 |
2.2. Target Efficiencies of Optimized Models
The positive and negative off-targets were collected for LbCpf1 and AsCpf1, respectively. The gRNA off-target pairs for LbCpf1 and AsCpf1 were used to train LinearSVC, RandomForest, AdaboostClassifier, MLPClassifier, LogisticRegression, and DecisionTreeClassifier algorithms. Hyperparameter tuning of all of the mentioned algorithms was performed. The best set of parameters was selected for prediction. A similar approach was repeated for all of the algorithms and both data sets. The list of hyperparameters for all of the machine learning models are listed in Supporting Information Table 1.
2.2.1. AsCpf1 Target Efficiency Prediction Model
Five different machine learning algorithms were optimized on hybrid feature sets of the AsCpf1 data set, and the best model was selected based on their performance on an unseen test data set. The best-performing prediction models with and without undersampling were also evaluated using bias, variance, and MSE with 25% split, and it was found that all of the optimized models with undersampled data were overfitting the train data. The bias, variance, and MSE of all of the models are given in the Supporting Information Table 2. Therefore, the different machine learning algorithms were trained and optimized without the use of sampling techniques. The performance of the models was evaluated using the precision, recall, F1 score, and the Matthews correlation coefficient. The performances of all of the optimized models on each feature set on the test split (25%) are summarized in Supporting Information Table 3. Further, precision, recall, and f1 score of the best model on each feature set are given in Table 3.
Table 3. Precision–Recall and F1 Score of the Best Models on Test and Train Data of the Hybrid Feature Sets.
| sequence-based features |
base-dependent binding energy-based features |
sequence
and base-dependent binding energy-based
features |
|||||
|---|---|---|---|---|---|---|---|
| species | performance metrics | score on test data | overall score | score on test data | overall score | score on test data | overall score |
| AsCpf1 | precision | 0.92 | 0.93 | 0.95 | 0.94 | 0.92 | 0.98 |
| recall | 0.90 | 0.89 | 0.89 | 0.88 | 0.91 | 0.88 | |
| F1 score | 0.91 | 0.91 | 0.92 | 0.91 | 0.92 | 0.93 | |
| MCC | 0.82 | 0.81 | 0.84 | 0.82 | 0.83 | 0.86 | |
| LbCpf1 | precision | 0.92 | 0.94 | 0.94 | 0.94 | 0.91 | 0.91 |
| recall | 0.90 | 0.92 | 0.90 | 0.89 | 0.92 | 0.93 | |
| F1 score | 0.91 | 0.93 | 0.92 | 0.91 | 0.91 | 0.92 | |
| MCC | 0.82 | 0.86 | 0.83 | 0.83 | 0.82 | 0.84 | |
Optimized AdaBoostClassifier performed best with sequence-based features and base-dependent binding energy-based features, while the optimized MLPClassifer performed best with a data set consisting of the combined feature set. The optimized MLPClassifier model performed the best among all of the algorithms on the unseen test split of the data set as well as on the 5-fold cross-validation run. The test data set contained 14,739 off-targets after a test split, out of which 134 were positive off-targets and 14,605 were negative. The average precision of the optimized MLPClassifier using a test split was 0.90, and the confusion matrix also suggested that 83% of positive off-targets were correctly classified and 100% of negative off-targets were correctly classified. The precision–recall curve and confusion matrix for the comparison of model performances on three different feature sets for AsCpf1 are shown in Figure 1. Both performance metrics indicate that the MLPClassifier model that is trained on a combined feature set of AsCpf1 data has high predictive performance in the classification of positive and negative off-targets. The 5-fold cross-validation was also performed to evaluate the performance of the MLPClassifier using precision–recall curves for all folds, as shown in Figure 2a. The mean area under the curve of all of the folds was 0.82. The optimized MLP-based classifier trained on the combined feature set of the AsCpf1 data set was used to develop a pipeline for the prediction of target efficiency.
Figure 1.

Comparison of performance of the optimized model on the hybrid feature sets. The performance of best models was evaluated with PR-AUC trained on (A) AsCpf1 and (E) LbCpf1 data sets. MLP-based classifier improved the performance with a combined feature set in both LbCpf1 and AsCpf1. Classification performance of the best-performing model using (B) sequence-based feature set, (C) base-dependent binding energy-associated feature set, and (D) sequence and base-dependent binding energy-associated feature set on AsCpf1 data set using a confusion matrix. Classification performance of LbCpf1 data set on (F) sequence-based feature set, (G) base-dependent binding energy-associated feature set, and (H) sequence and mismatch-energy-associated feature set using a confusion matrix.
Figure 2.

Performance validation of the best-performing model on combined feature set. The PR-AUC was evaluated on 5-fold cross-validation of best models on (A) AsCpf1 data set and (B) LbCpf1 data set. The comparison of ROC–AUC of all of the best models trained on the hybrid feature sets with DeepCpf1, CRISPR-DT, and CINDEL on a 25% independent test split of the (C) AsCpf1 data set and (D) LbCpf1 data set. The blue bar represents the best-performing models for both the data sets and the red, green, and yellow bars represent the AUC of DeepCpf1, CRISPR-DT, and CINDEL, respectively.
2.2.2. LbCpf1 Target Efficiency Prediction Model
Similarly, five different machine learning algorithms were optimized on hybrid feature sets of the LbCpf1 data set, and the best model was selected based on the performance of the unseen test data set. The best-performing prediction models with and without undersampling were evaluated using bias, variance, and MSE with 25% split, and it was found that all of the optimized models with undersampled data were overfitting the train data. The bias, variance, and MSE of all of the models are given in Supporting Information Table 2. Therefore, the different machine learning algorithms were trained and optimized without employing sampling techniques. The performances of all of the optimized models on each feature set using a 25% independent test split are summarized in Supporting Information Table 4. The precision, recall, and F1 score of the best model on each feature set are summarized in Table 3. While optimized MLPClassifier performed best with sequence-based features, optimized AdaBoostClassifier performed best with base-dependent binding energy-based features. Similarly, optimized MLPClassifer performed best with a data set consisting of a combined feature set on the LbCpf1 data set. These models were then selected for the development of a pipeline for the prediction of target efficiency for the LbCpf1 data set. The MLPClassifier model on the combined feature set performed best among all of the algorithms on the unseen test split as well as on 5-fold cross-validation. The test data set contained 20,207 off-targets, of which 129 were positive and 20,078 were negative. The average precision of the optimized MLPClassifier using a test split was observed to be 0.90. On the same lines, the confusion matrix also depicted that 83% of positive off-targets were correctly classified and 100% of negative off-targets were correctly classified. The precision–recall curve and confusion matrix for the comparison of model performances on three different feature sets for LbCpf1 are shown in Figure 1. Both performance metrics indicated that the MLPClassifier model trained on a combined feature set of AsCpf1 data has high predictive performance in the classification of positive and negative off-targets. The 5-fold cross-validation was also performed to evaluate the performance of the MLPClassifier using precision–recall curves for all folds, as shown in Figure 2b. The mean area under the curve of all of the folds was 0.81. The target efficiency prediction pipeline for LbCpf1 was developed by using an optimized MLP-based classifier on the combined feature set.
2.2.3. Benchmarking Other Existing Models
The performance of all six optimized algorithms on the AsCpf1 and LbCpf1 data sets was compared with CRISPR-DT,23 DeepCpf1,21 and CINDEL10 using a 25% test split of training data. All of the best-performing models on different feature sets for AsCpf1 and LbCpf1 performed better than the existing models, as shown in Figure 2c,d. MLPClassifier optimized on combined feature sets was selected as the best classifier for AsCpf1 and LbCpf1, respectively, based on different performance measures discussed above. All of the generated models on different feature sets showed better performance than DeepCpf1, CRISPR-DT, and CINDEL in classifying positive from negative off-targets. The developed models on a combined feature set optimized on LbCpf1 and AsCpf1 data had high performance in classifying positive and negative off-targets, which can help control off-targets and ensure target efficiency compared to the existing ones. Different performance metrics taken into consideration for benchmarking the performance of the existing models with the developed model are summarized in Supporting Information Table 5.
2.3. Feature Importance Analysis
Feature importance analysis was performed to study the features that can distinguish between positive and negative off-targets. Positional preference of nucleotides, mismatch distribution analysis, and SHAP1 were employed to understand the biological relevance of position-specific and other thermodynamics-related features in the classification of positive and negative off-targets.
2.3.1. Position-Specific Preference of Nucleotides
Position-specific preference analysis of nucleotides was performed to understand the preference of nucleotides at a particular position using the Welch t test between the positive and negative off-targets of AsCpf1 and LbCpf1. In addition to that, positional preference of favored and disfavored nucleotides was also understood using the enrichment score of a nucleotide at all locations given in eq 1. Enrichment scores greater than 0.75 were considered favored nucleotides, and those less than 0.75 were considered disfavored nucleotides. Position-wise enrichment score and their respective P-values of each nucleotide are given in Supporting Information Table 6.
| 1 |
It was found that thymine in the fourth place of the PAM sequence was highly disfavored in positive off-targets of both AsCpf1 and LbCpf1 compared to negative off-targets (P < 0.005). Furthermore, guanine was favored at a position adjacent to the PAM sequence, whereas thymine was highly disfavored at the location. This is further supported by other studies that arrive at a similar conclusion.12 Both findings in our study were found to be similar for AsCpf1 and LbCpf1.
Based on the p-value from the Welch t test, it is reported that guanine is highly favored at the third position from the PAM sequence (P < 0.005), and thymine is disfavored (P < 0.005). Further, adenine is highly favored (P < 0.005) at the eleventh position from the PAM sequence. Similarly, while three positions adjacent to the eleventh favor the presence of cytosine (P < 0.005), the sixth position in the seed region disfavors thymine (P < 0.005). It was also found that thymine is mostly disfavored in the seed region of both LbCpf1 and AsCpf1 positive off-targets compared to negative off-targets. All of the findings reported in this study were consistent in both AsCpf1 and LbCpf1, which suggests that both the enzymes have a very similar sequence preference. The p-values of both position-specific favored and disfavored nucleotides for both LbCpf1 and AsCpf1 off-targets are compiled in Figure 3.
Figure 3.

Position-specific favored and disfavored nucleotides in positive off-targets. The positive and negative Y-axis denotes the P-values for favored and disfavored nucleotides in (A) AsCpf1 and (B) LbCpf1 positive off-targets.
2.3.2. Mismatch Distribution Analysis of Off-Targets
Position-specific nucleotide mismatches between target and off-target pairs were studied because the nucleotides favored or disfavored at different positions vary between positive and negative off-targets. Mismatch distribution analyses were performed for positive and negative off-targets by using the Welch t test. This revealed that the positive off-targets of both LbCpf1 and AsCpf1 had more prominent mismatches in the trunk region (nucleotides 16, 17, 18) and the promiscuous region, as shown in Figure 4a,b. The transition type of mismatches at the trunk region is more preferred at positions 16, 17, and 18 in positive off-targets for AsCpf1 as shown in Supporting Information Figure 1a–d and for LbCpf1 in Supporting Information Figure 2a–d. Similar results were obtained for both AsCpf1 and LbCpf1. It has been reported that the transition type of mismatches is more compatible in the trunk region and all types of mismatches are preferred in the promiscuous region.10 In this study, it was found that the C-G type of mismatch at position 23 was tolerated more in comparison to other mismatch types in both AsCpf1 and LbCpf1 off-targets. As shown in Figure 4a,b, PAM-Distal regions of both AsCpf1 and LbCpf1 positive off-targets had a high tolerance for mismatches, whereas negative off-targets of both Ascpf1 and LbCpf1 had equal frequencies of mismatches at all of the positions. In previous studies, the PAM-Distal region with a higher tolerance for mismatches has also been reported.10
Figure 4.

Mismatch distributions in the positive and negative off-targets of (A) LbCpf1 and (B) AsCpf1. The white bars and gray bars depict the distribution of mismatches in negative off-targets and positive off-targets respectively.
2.3.3. Classifier-Associated Feature Importance
A SHAP (Shapley Additive exPlanations) is used to estimate Shapley values for individual features to study their importance in model prediction.35 The main goal of the SHAP approach is to explain each prediction by computing the contribution of each feature. The top 20 features were ranked based on the Shapley values for the best-performing models on three different feature sets of AsCpf1 and LbCpf1 data sets. The higher the Shapley Values, the greater the contribution of each feature in model prediction and classification of off-targets.
The stacked bar plot was constructed based on mean SHAP values for each best-performing model trained in different feature sets, and the top 20 features of each of them are shown in Figure 5a–f. Common features predicted from both AsCpf1 and LbCpf1 models trained with a feature set 1 are some bulges, GC content, a mismatch at position 5 (position adjacent to PAM), dinucleotide frequencies such as AG, CC, a mismatch at position 8, and melting temperature of a nonseed region of target and off-target pairs.36 The importance of these features in the classification of AsCpf1 and LbCpf1 off-targets is shown in Figure 5a,d. Mean Shapley values of melting temperature of the nonseed region suggest that the negative off-targets have a high melting temperature as compared to positive off-targets. It can also be concluded from the summary plot shown in Supporting Information Figure 3a,d that the presence of mismatch at a position adjacent to PAM is favored in positive off-targets. Also, it can be inferred that thymine is generally substituted by guanine, which suggests that thymine is disfavored at the position adjacent to PAM in gRNA. Similar findings are also reported in previous studies10 and discussed using statistical analysis. The high frequency of AG in the target location is indicative of a negative off-target site, whereas the high frequency of CC in the target location is common in positive off-target. It has also been predicted from mean SHAP values that low to moderate GC content is the key feature of positive off-targets, similar to cas9 as reported in a previous study.37 Mean SHAP values of the best-performing model of AsCpf1 and LbCpf1 on the feature set 2 suggest that base-dependent PAM binding energy,38 total energy weights of mismatches in seed, trunk, and promiscuous regions of gRNA, and base-dependent binding energy of the fourth nucleotide of the PAM region play important roles in the classification of off-targets, and they are highly correlated with a high target efficiency. The importance of these features in the classification can be visualized in Figure 5b,e. The summary plot shown in the Supporting Information Figure 3b,e suggests that the PAM binding energy is moderate to high in negative off-targets. Total energy weights for mismatches in the seed are predicted to be higher in most of the positive off-targets, whereas energy weights for mismatches in the trunk and promiscuous regions are mostly high in the case of negative off-targets. However, mean SHAP values of the best-performing models trained on the feature set 3 suggest that the relative PAM binding energy, PAM binding energy, GC content, number of bulges, and CC frequency are prominent in the classification of positive and negative off-targets of both AsCpf1 and LbCpf1. The importance of these features based on mean SHAP values is shown in Figure 5c,f. The summary plot shown in the Supporting Information Figure 3c,f suggests that high relative PAM binding energy is the feature of negative off-targets, whereas positive off-targets have medium relative PAM binding energy. Mean SHAP values also suggest that bulges are less likely to be tolerated in positive as well as negative off-targets, but positive off-targets can tolerate a greater number of bulges than negative off-targets. In addition to the above-discussed features, high GC content and low to moderate melting temperature of complete gRNA and nonseed region is also a feature of positive off-targets, whereas negative off-targets have high melting temperature and GC content. Various biochemical and structural studies reveal that gRNA binds to target DNA by intermittent contact before getting associated with the cleavage site, suggesting that thermodynamic properties such as melting temperature are very useful in the classification of experimental cleavage sites.39,40 A high amount of thymine in off-targets may also indicate that its probability of being a positive off-target is more. However, the occurrence of thymine in the seed region of positive off-targets is highly disfavored, as discussed above. It can be concluded from stacked bar plots and summary plots of different feature sets that PAM binding energy, energy weights of mismatches, melting temperature, GC content, and bulges are the important features in the activity prediction of gRNAs and therefore highly correlated in the prediction of target efficiency.
Figure 5.

Stacked bar plot depicting top 20 features of best models on hybrid feature sets. The mean shap values indicate the magnitude of importance of each feature in the classification of off-targets. Top 20 features based on mean SHAP values of models trained on AsCpf1 data set using (A) sequence-based feature set, (B) base-dependent binding energy-associated feature set, and (C) sequence and base-dependent binding energy-associated feature set. Similarly, the stacked bar plot values for the LbCpf1 data set using (D) sequence-based feature set, (E) base-dependent binding energy-associated feature set, and (F) sequence and base-dependent binding energy-associated feature set.
3. Discussion
The CRISPR/Cas technology can precisely manipulate specific locations of the genome. The clinical application of CRISPR technology is still a challenge due to numerous concerns regarding the efficacy and safety of the system. Recently, CRISPR-Cpf1 system was identified9 and is known to have comparatively fewer off-targets. Cpf1 recognizes T-rich PAM, which ensures higher target specificity in human and plant cells.10,12,15,17,18 There are two important aspects of the CRISPR-Cas system: target specificity and target efficiency. To ensure target specificity, we designed a pipeline to predict potential off-target sites in the human genome for a target sequence. The pipeline was developed for both AsCpf1 and LbCpf1 to predict potential off-target sites. It allows the user to select the desired target sequence. Further, the predicted potential off-target sites have been classified into positive and negative off-targets using a machine learning model to predict the target efficiency of all of the potential off-target sites. ML-based classifiers were developed for both AsCpf1 and LbCpf1. Different machine learning models were optimized with and without undersampling by using various hyperparameters. Machine learning models with undersampled data were trained, and their bias, variance, and MSE were compared with the developed models in pipeline for different feature sets. It was concluded that the models trained with undersampled data were overfitting. The conclusion made from the above finding is that there is a disadvantage of undersampling, as it may discard potentially relevant data and not represent the variability of negative off-targets in such a short sample space. Therefore, data set without undersampling was selected for the development of pipeline for both the species. The MLPClassifier performed best on both data sets with a feature set 3. Here, we analyzed the AsCpf1 and LbCpf1 positive and negative off-targets to understand the position-specific nucleotide preferences and mismatch distribution between positive and negative off-targets. In this study, it was found that thymine is highly disfavored at a position adjacent to PAM whereas guanine is favored.10 The distribution of mismatches is a distinctive feature between positive and negative off-targets. Mismatch distribution analysis reveals that the positive off-targets have a high tolerance for mismatches in the PAM-distal region,12,13 whereas negative off-targets have a uniform distribution of mismatches at all locations. High tolerance for mismatches is mostly visualized in the trunk region and promiscuous region. After analyzing the position-specific mismatch type preference between positive and negative off-targets, transition type mismatch was found to be prominent in positive off-targets at 16, 17, and 18 positions of nucleotides including PAM sequence. On the other hand, at position 23, the C-G type of mismatch was found to be preferred. As is known from previous studies, all types of mismatches are favored in the promiscuous region. A machine learning model generated on AsCpf1 and LbCpf1 for different feature sets revealed the importance of features in the classification of respective off-targets using mean SHAP values of individual features. The top 20 features were short-listed based on mean SHAP values. All of the best models on different feature sets had different top 20 features for the classification of off-targets. Interestingly, the melting temperature of a nonseed region of gRNA, base-dependent PAM binding energy, GC content, and the number of bulges were the most common features between the models. It was found that the mismatches at position 9 were preferred in the seed region of LbCpf1 positive off-targets than other positions of the seed region, and similar findings were reported in a previous study. It was also found that in the case of AsCpf1 positive off-targets, T-G type of mismatches were more preferred at a position adjacent to PAM. The above predictions suggested that the seed region does not tolerate thymine in positive off-target sites, especially at positions 1 and 5 from the PAM sequence, whereas it is preferred in the trunk and promiscuous regions of positive off-targets.
4. Conclusions
In this study, a pipeline was generated to predict possible off-target sites in a human genome for a target sequence, and these can be further fed into developed machine learning-based models for LbCpf1 and AsCpf1 to evaluate target efficiency. This is important since all probable off-target sites in the host genome do not undergo cleavage under experimental conditions. Therefore, an MLP-based classifier was developed using sequence- and base-dependent binding energy-associated features for AsCpf1 and LbCpf1 to predict the target efficiency of potential off-target sites. Both of these models were trained on experimental (positive data set) and predicted target and off-target pairs (negative data set). An MLP-based classifier model trained on the LbCpf1 and AsCpf1 data sets using sequence and base-dependent binding energy-associated features was found to have higher predictive performance as compared to other ML-based classifiers and existing tools using a 25% test split and 10-fold cross-validation evaluation criteria. Mismatch distribution analysis reveals that mismatches were more prominent in the trunk region (16, 17, 18 nucleotides from PAM sequence). On the other hand, promiscuous region and transition type mismatch were more preferred at 16, 17, and 18 nucleotides position (including PAM sequence). Other features such as the low to moderate melting temperature of the nonseed region and base-dependent PAM binding energy were also predicted as important features in the classification of off-targets. Some other features that are characteristic of LbCpf1 and AsCpf1 off-targets were the GC content, some types of dinucleotide frequencies, number of bulges, and mismatches in the seed and trunk region. The study also adds to the information available for rule-based guide design and target selection for Cpf1-based experiments. The results from this study can be used to design better Cpf1-based experiments with fewer off-target effects.
5. Methodology
5.1. Pipeline to Improve On-Target Specificity
Among the multiple orthologues of Cpf1, the ones extracted from Acidaminococcus sp. (AsCpf1), Lachnospiraceae bacterium (LbCpf1), and Francisella novicida (FnCpf1) are most commonly exploited to manipulate the human genome. This study focuses on AsCpf1 and LbCpf1 because of their higher efficiencies in human cells than other orthologues of Cpf1. The pipeline was developed by optimizing parameters such as mismatch penalty, gap penalty, and E-score of BLAST,32 BOWTIE2,33 and FASTA3634 to predict the maximum potential off-target sites in the human genome for AsCpf1 and LbCpf1 species. The performances were also benchmarked based on the correlation between the number of predicted off-target sites and experimentally known off-target sites. The optimized FASTA36 predicted a large number of potential off-target sites with 9 mismatches and 3 gaps compared to BOWTIE and BLAST. Therefore, an optimized FASTA36 algorithm was used to design a pipeline for the prediction of potential Cpf1 off-target sites in the human genome. The developed pipeline to predict potential off-target sites in the human genome involved two steps: (1) target sequences were mapped to the Human reference genome (hg37) using FASTA3634 to search for possible off-target locations with a default value of eight mismatches and three gaps that Cpf1 is known to tolerate when searching for a target, and (2) the SeqIO module of the Bio package41 was utilized to filter the alignments. The number of mismatches for the prediction of off-target sites in this pipeline was set to 9 mismatches and 3 gaps. Custom python scripts were used in this pipeline to filter output based on various parameters. A schematic of the workflow to predict the CRISPR target sites and their efficiencies implemented in this study is summarized in Figure 6.
Figure 6.
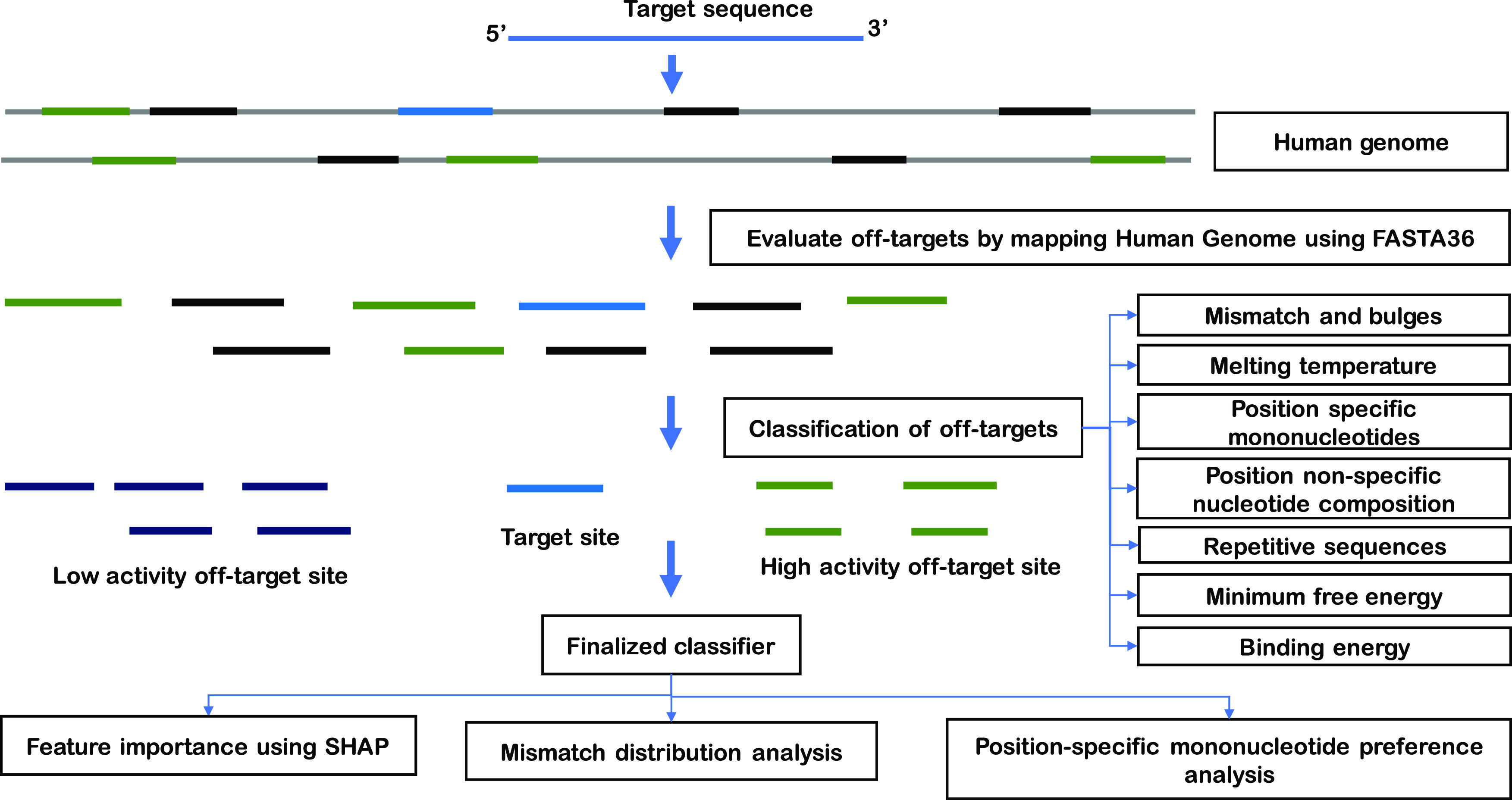
Workflow employed to predict potential off-target sites and classify positive and negative off-targets.
5.2. Off-Target Prediction Model
5.2.1. Data Retrieval and Visualization
Training data for the prediction of target efficiencies consisted of positive and negative off-targets. The target and experimentally detected off-targets pairs collected from Guide-seq,13,42 targeted deep sequencing,13,43 digenome seq,12 and DNA-Seq44 studies were labeled as positive off-targets. Other potential off-target sites present in the human genome but not reported to be cleaved under the experimental conditions were predicted using the pipeline mentioned above and are labeled as negative off-targets. The training data for LbCpf1 consisted of 540 positive and 80,844 negative off-targets associated with 42 target sites. Similarly, training data for AsCpf1 had 491 positive and 58,907 negative off-targets associated with 41 targets. The test data sets include the known and predicted off-targets of 7 genomic targets of each of the data sets. The data imbalance in the case of positive and negative off-targets of the CRISPR-Cas system could be attributed to the fact that off-targets recognized in experimental conditions are much smaller as compared to the possibility of similar sites in the human genome.45 Therefore, negative off-targets were undersampled, and each set of negative off-targets is trained with all positive off-targets to balance the data set. The best-performing models were optimized by tuning the hyperparameters, and bias, variance, and mean-squared error (MSE) were estimated to understand the prediction errors in the models. In addition to that, precision, recall, F1 score, and the Matthews correlation coefficient were used as evaluation metrics to understand the classification accuracy of developed models.
5.2.2. Feature Extraction
Various studies on other Cpf1 nucleases utilized sequence-based features, and the performance of the existing models indicated a need to incorporate more diverse features for the classification of off-targets. Therefore, in this study, a total of 713 features were calculated for positive and negative off-targets of both AsCpf1 and LbCpf1 falling under the following categories: (1) position-specific nucleotide composition, (2) position nonspecific nucleotide composition, (3) mismatches and bulge-associated features, (4) melting temperature, (5) minimum free energy, (6) occurrence of repetitive sequences, and (7) base-dependent binding energy.38 Position-specific mononucleotides were encoded for the target and off-target sequences. The detailed structure of all of the aforementioned categories is given in Supporting Information Table 7. In this study, three feature sets were constructed: (i) sequence-associated feature set (feature set 1), (ii) base-dependent binding energy-associated feature set (feature set 2), and (iii) combined feature set (feature set 3). All three types of feature sets were obtained for both the AsCpf1 and LbCpf1 target and off-target pairs. The minimum free energy of all of the off-targets was calculated using the RNAfold module of the ViennaRNA package,46 and melting temperature was calculated using Tm_NN function of the BioSeqUtils module with thermodynamic values from the DNA_NN2 table.41,47,48
5.2.3. Optimization of Machine Learning Algorithm
Multiple classification algorithms were employed to classify positive and negative off-targets present in LbCpf1 and AsCpf1 data sets on three different feature sets. These algorithms were optimized on both data sets to improve the performances of the models. A 25% test split of the data set and 5-fold cross-validation were employed to optimize the various machine learning algorithms on both data sets. The optimization process involved the tuning of hyperparameters of each of the algorithms using a 25% test split and 10-fold cross-validation. The partitioning of training data into 5-fold was performed by using a stratified sampling method with a 25% split of data in each iteration. Hyperparameter tuning was performed to select the optimal parameters to classify the data sets accurately by calculating the performance at each iteration of different sets of values for each algorithm. The set of values that best classified the data set were considered optimal parameters and were used to generate a model for all of the feature sets. A similar approach was used for both data sets. Precision, recall, F1 score, and the Matthews correlation coefficient were used to evaluate the performance of the models. Of all of the generated models on different feature sets, the optimized MLP-based classifier resulted in maximum classification accuracy on the combined feature set on both data sets.
5.2.4. Feature Importance and Statistical Analysis
The statistical significance of position-specific features such as the presence of a nucleotide at a specific position, position of mismatch, and frequency of mismatch type at a specific location was evaluated using Welch’s t test using Python.49
The contribution of features in the classification of positive and negative off-targets was analyzed by using mean SHAP values. The SHAP approach allows all possible permutations of the features to compute mean values of individual features to indicate their contribution to prediction.35 The top 20 features were short-listed based on mean SHAP values to study the biological importance of these features in the off-target cleavage activity of Cpf1. SHAP values were calculated from the best-performing models trained on the AsCpf1 and LbCpf1 data sets. SHAP value gives detailed insights into the contribution of an individual feature that models used for the classification.
6. Implementation
All of the models were developed using the Scikit-learn package. The model hyperparameters were optimized using extensive grid search, and all of the best models, analysis, and prediction results are available at the GitHub repository (https://github.com/TeamSundar/CRISPR-Cpf1_study).
Acknowledgments
P.K. is supported by GSK’s “Trust in Science” initiative in partnership with RCB.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c05691.
Presence of one-base mismatch type in positive and negative off-targets of AsCpf1 (Figure S1), LbCpf1 (Figure S2) at position 16, 17, 18, 23 from PAM; summary plot depicting top 20 features of best models on hybrid feature sets (Figure S3); hyperparameters tuned recursively to optimize machine learning models for both AsCpf1 and LbCpf1 (Table S1); comparison of prediction errors of the best-performing models of three different feature sets with and without undersampling (Table S2); performances of optimized machine learning models on 25% test split of AsCpf1 (Table S3) and LbCpf1 (Table S4) data sets with hybrid feature set; comparison of performance metrics of the best-performing model with the existing models (Table S5); highly significant position-specific mononucleotides with enrichment score and P-values calculated using a Welch t test (Table S6); complete set of features used for training model for the prediction of on-target efficiencies (Table S7) (PDF)
Data source and number of potential off-targets (XLSX)
Author Contributions
P.K.: Conceptualization, pipeline design, data analysis, writing original draft; D.S.V.: Conceptualization, data curation, writing-reviewing and editing; and D.S.: Conceptualization, supervision, writing-reviewing and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Ran F. A.; Hsu P. D.; Wright J.; Agarwala V.; Scott D. A.; Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8 (11), 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T.; Gersbach C. A.; Barbas C. F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31 (7), 397–405. 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.; Bikard D.; Cox D.; Zhang F.; Marraffini L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31 (3), 233–239. 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland A. E.; Tzur Y. B.; Esvelt K. M.; Colaiácovo M. P.; Church G. M.; Calarco J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 2013, 10 (8), 741–743. 10.1038/nmeth.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Qiu Z.; Shao Y.; Chen Y.; Guan Y.; Liu M.; Li Y.; Gao N.; Wang L.; Lu X.; et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31 (8), 681–683. 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- Hwang W. Y.; Fu Y.; Reyon D.; Maeder M. L.; Tsai S. Q.; Sander J. D.; Peterson R. T.; Yeh J. J.; Joung J. K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31 (3), 227–229. 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q.; Regan S. N.; Xia Y.; Oostrom L. A.; Cowan C. A.; Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013, 12 (4), 393. 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora D. S.; Dhanjal J. K.; Sundar D.. Engineering of Cas9 for improved functionality. In Genome Engineering via CRISPR-Cas9 System; Elsevier, 2020; pp 111–122. [Google Scholar]
- Zetsche B.; Gootenberg J. S.; Abudayyeh O. O.; Slaymaker I. M.; Makarova K. S.; Essletzbichler P.; Volz S. E.; Joung J.; Van Der Oost J.; Regev A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163 (3), 759–771. 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. K.; Song M.; Lee J.; Menon A. V.; Jung S.; Kang Y.-M.; Choi J. W.; Woo E.; Koh H. C.; Nam J.-W.; Kim H. In vivo high-throughput profiling of CRISPR–Cpf1 activity. Nat. Methods 2017, 14 (2), 153–159. 10.1038/nmeth.4104. [DOI] [PubMed] [Google Scholar]
- Hur J. K.; Kim K.; Been K. W.; Baek G.; Ye S.; Hur J. W.; Ryu S.-M.; Lee Y. S.; Kim J.-S. Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat. Biotechnol. 2016, 34 (8), 807–808. 10.1038/nbt.3596. [DOI] [PubMed] [Google Scholar]
- Kim D.; Kim J.; Hur J. K.; Been K. W.; Yoon S.-h.; Kim J.-S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34 (8), 863–868. 10.1038/nbt.3609. [DOI] [PubMed] [Google Scholar]
- Kleinstiver B. P.; Tsai S. Q.; Prew M. S.; Nguyen N. T.; Welch M. M.; Lopez J. M.; McCaw Z. R.; Aryee M. J.; Joung J. K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34 (8), 869–874. 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Cheong S.-A.; Lee J. G.; Lee S.-W.; Lee M. S.; Baek I.-J.; Sung Y. H. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 2016, 34 (8), 808–810. 10.1038/nbt.3614. [DOI] [PubMed] [Google Scholar]
- Hu X.; Wang C.; Liu Q.; Fu Y.; Wang K. Targeted mutagenesis in rice using CRISPR-Cpf1 system. J. Genet. Genomics 2017, 44 (1), 71–73. 10.1016/j.jgg.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Kim H.; Kim S.-T.; Ryu J.; Kang B.-C.; Kim J.-S.; Kim S.-G. CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 2017, 8 (1), 14406 10.1038/ncomms14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Lowder L. G.; Zhang T.; Malzahn A. A.; Zheng X.; Voytas D. F.; Zhong Z.; Chen Y.; Ren Q.; Li Q. A CRISPR–Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat. Plants 2017, 3 (3), 17103 10.1038/nplants.2017.103. [DOI] [PubMed] [Google Scholar]
- Xu R.; Qin R.; Li H.; Li D.; Li L.; Wei P.; Yang J. Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol. J. 2017, 15 (6), 713–717. 10.1111/pbi.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Wei H.; Wang Y.; Deng J.; Tang Y.; Zhou L.; Guo G.; Tong A. Targeted disruption of V600E-mutant BRAF gene by CRISPR-Cpf1. Mol. Ther.--Nucleic Acids 2017, 8, 450–458. 10.1016/j.omtn.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Long C.; Li H.; McAnally J. R.; Baskin K. K.; Shelton J. M.; Bassel-Duby R.; Olson E. N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3 (4), e1602814 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. K.; Min S.; Song M.; Jung S.; Choi J. W.; Kim Y.; Lee S.; Yoon S.; Kim H. H. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 2018, 36 (3), 239–241. 10.1038/nbt.4061. [DOI] [PubMed] [Google Scholar]
- Luo J.; Chen W.; Xue L.; Tang B. Prediction of activity and specificity of CRISPR-Cpf1 using convolutional deep learning neural networks. BMC Bioinf. 2019, 20 (1), 332 10.1186/s12859-019-2939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H.; Liang C. CRISPR-DT: designing gRNAs for the CRISPR-Cpf1 system with improved target efficiency and specificity. Bioinformatics 2019, 35 (16), 2783–2789. 10.1093/bioinformatics/bty1061. [DOI] [PubMed] [Google Scholar]
- Montague T. G.; Cruz J. M.; Gagnon J. A.; Church G. M.; Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42 (W1), W401–W407. 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanjal J. K.; Radhakrishnan N.; Sundar D. CRISPcut: a novel tool for designing optimal sgRNAs for CRISPR/Cas9 based experiments in human cells. Genomics 2019, 111 (4), 560–566. 10.1016/j.ygeno.2018.03.011. [DOI] [PubMed] [Google Scholar]
- Bae S.; Park J.; Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30 (10), 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F.; Kerr G.; Boutros M. E-CRISP: fast CRISPR target site identification. Nat. Methods 2014, 11 (2), 122–123. 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- Dhanjal J. K.; Dammalapati S.; Pal S.; Sundar D. Evaluation of off-targets predicted by sgRNA design tools. Genomics 2020, 112 (5), 3609–3614. 10.1016/j.ygeno.2020.04.024. [DOI] [PubMed] [Google Scholar]
- Haeussler M.; Schönig K.; Eckert H.; Eschstruth A.; Mianné J.; Renaud J.-B.; Schneider-Maunoury S.; Shkumatava A.; Teboul L.; Kent J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17 (1), 148 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora D. S.; Verma Y.; Sundar D. A Machine Learning Approach to Identify the Importance of Novel Features for CRISPR/Cas9 Activity Prediction. Biomolecules 2022, 12 (8), 1123. 10.3390/biom12081123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora D. S.; Yadav S.; Sundar D. Hybrid Multitask Learning Reveals Sequence Features Driving Specificity in the CRISPR/Cas9 System. Biomolecules 2023, 13 (4), 641. 10.3390/biom13040641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215 (3), 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Langmead B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinf. 2010, 32 (1), 11.7 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson W. R.Using the FASTA Program to Search Protein and DNA Sequence Databases. In Computer Analysis of Sequence Data: Part I; Springer, 1994; pp 307–331. [DOI] [PubMed] [Google Scholar]
- Lundberg S. M.; Lee S.-I. In A Unified Approach to Interpreting Model Predictions, Proceedings of the 31st International Conference on Neural Information Processing Systems, 2017; pp 4768–4777.
- Doench J. G.; Fusi N.; Sullender M.; Hegde M.; Vaimberg E. W.; Donovan K. F.; Smith I.; Tothova Z.; Wilen C.; Orchard R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34 (2), 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Wei J. J.; Sabatini D. M.; Lander E. S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343 (6166), 80–84. 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht D. A.; Xu Y.; Lambert G. Massively parallel CRISPRi assays reveal concealed thermodynamic determinants of dCas12a binding. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (21), 11274–11282. 10.1073/pnas.1918685117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M.; Jiang F.; Taylor D. W.; Sternberg S. H.; Kaya E.; Ma E.; Anders C.; Hauer M.; Zhou K.; Lin S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343 (6176), 1247997 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg S. H.; Redding S.; Jinek M.; Greene E. C.; Doudna J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507 (7490), 62–67. 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P. J. A.; Antao T.; Chang J. T.; Chapman B. A.; Cox C. J.; Dalke A.; Friedberg I.; Hamelryck T.; Kauff F.; Wilczynski B.; de Hoon M. J. L. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25 (11), 1422–1423. 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth E.; Varga É.; Kulcsár P. I.; Kocsis-Jutka V.; Krausz S. L.; Nyeste A.; Welker Z.; Huszár K.; Ligeti Z.; Tálas A.; Welker E. Improved LbCas12a variants with altered PAM specificities further broaden the genome targeting range of Cas12a nucleases. Nucleic Acids Res. 2020, 48 (7), 3722–3733. 10.1093/nar/gkaa110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S.-H.; Lee W.-j.; An J.-H.; Lee J.-H.; Kim Y.-H.; Kim H.; Oh Y.; Park Y.-H.; Jin Y. B.; Jun B.-H.; et al. Prediction-based highly sensitive CRISPR off-target validation using target-specific DNA enrichment. Nat. Commun. 2020, 11 (1), 198 10.1038/s41467-020-17418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Zhou J.; Wan Y.; Liu H.; Li Y.; Liu Z.; Wang H.; Lei J.; Zhao K.; Zhang Y.; et al. A Cas12a ortholog with stringent PAM recognition followed by low off-target editing rates for genome editing. Genome Biol. 2020, 21 (1), 78 10.1186/s13059-020-01989-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Chuai G.; Yu W.; Qu S.; Liu Q. Data imbalance in CRISPR off-target prediction. Briefings Bioinf. 2020, 21 (4), 1448–1454. 10.1093/bib/bbz069. [DOI] [PubMed] [Google Scholar]
- Lorenz R.; Bernhart S. H.; Höner zu Siederdissen C.; Tafer H.; Flamm C.; Stadler P. F.; Hofacker I. L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6 (1), 26 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novère N. MELTING, computing the melting temperature of nucleic acid duplex. Bioinformatics 2001, 17 (12), 1226–1227. 10.1093/bioinformatics/17.12.1226. [DOI] [PubMed] [Google Scholar]
- Sugimoto N.; Nakano S.-i.; Yoneyama M.; Honda K.-i. Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 1996, 24 (22), 4501–4505. 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P.; Gommers R.; Oliphant T. E.; Haberland M.; Reddy T.; Cournapeau D.; Burovski E.; Peterson P.; Weckesser W.; Bright J.; et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17 (3), 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.