

SUMMARY
Dimeric IgA (dIgA) can move through cells via the IgA/IgM polymeric immunoglobulin receptor (PIGR), which is expressed mainly on mucosal epithelia. Here, we studied the ability of dimeric IgA (dIgA) to target commonly mutated cytoplasmic oncodrivers. Mutation-specific dIgA, but not IgG, neutralized KRASG12D within ovarian carcinoma cells and expelled this oncodriver from tumor cells. dIgA binding changed endosomal trafficking of KRASG12D from accumulation in recycling endosomes to aggregation in the early/late endosomes through which dIgA transcytoses. dIgA targeting of KRASG12Dabrogated tumor cell proliferation in cell culture assays. In vivo, KRASG12D-specific dIgA1 limited growth of KRASG12D- mutated ovarian and lung carcinomas in a manner dependent on CD8+ T cells. dIgA specific for IDH1R132H reduced colon cancer growth, demonstrating effective targeting of a cytoplasmic oncodriver not associated with surface receptors. dIgA targeting of KRASG12D restricted tumor growth more effectively than small molecule KRASG12D inhibitors, supporting the potential of this approach for the treatment of human cancers.
Keywords: Immunotherapy, IgA transcytosis, PIGR, therapeutic antibody, KRAS
Graphical Abstract
eTOC
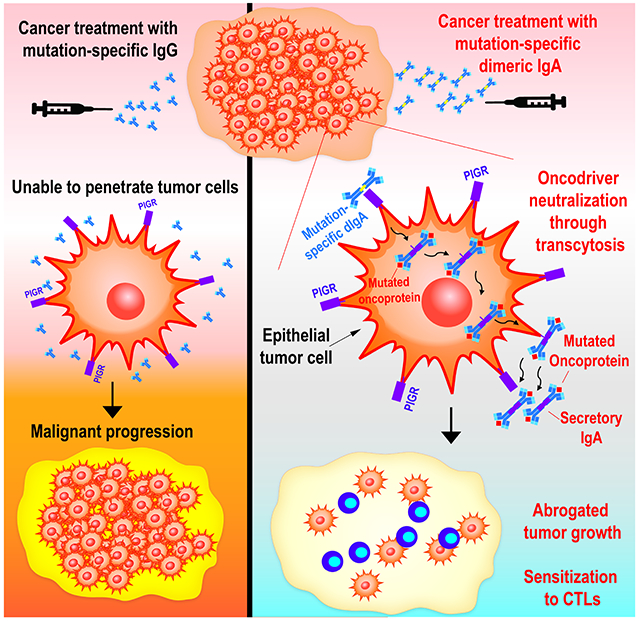
Despite their long half-life, therapeutic antibodies are considered ineffective against intracellular antigens. Biswas et al demonstrate that dimeric IgA undergoes transcytosis through PIGR+ epithelial cancer cells, and can be engineered for specific targeting of intracellular mutated oncodrivers. Binding of mutation-specific IgA expels cells oncodrivers from the cytoplasm and promotes anti-tumor immunity.
INTRODUCTION
Limited by their large size and considered unable to penetrate the cytoplasm, development of antibody-based immunotherapies has focused on transmembrane or extracellular targets. However, most oncodrivers underpinning tumor-promoting pathways are intracellular proteins inaccessible to conventional therapeutic antibodies. Accordingly, considerable efforts have been made to develop small molecules that target these intracellular pathways by inhibiting enzymatic activity or serving as allosteric modulators.
KRAS is considered the most common mutated oncogene driving human cancer1. KRAS mutations are particularly frequent in pancreatic, colorectal, non-small cell lung and endometrial cancer2, which total more than 50,000 cases annually3. KRAS mutations primarily comprise single-base missense mutations in codons 12 (80%), 13 or 611. Following a 40-year quest to target KRAS mutations, Sotorasib4, a specific KRASG12C small molecule inhibitor, was approved by the FDA in 2021. Pharmacological inhibitors against KRASG12D, the more common mutation in pancreatic and colon cancer, are currently under testing5. Small molecule inhibitors that target oncodrivers offer great therapeutic benefits and have made a big impact in oncology. However, the half-life of many small molecules is ~6 hours4, which can limit on target interactions. In contrast, the serum half-life of optimized antibodies is >20 days6, which offers more sustained neutralization and can thus be less frequently dosed. Furthermore, small molecule inhibitors, including Sotorasib7, are inevitably associated with acquired resistance. Some of these mechanisms include locking KRAS in a GTP-bound (activated) form7, which is insensitive to the small molecule inhibitor but could be specifically recognized by an antibody. Furthermore, it is becoming increasingly clear that anti-tumor antibody responses are intrinsically linked to tumor-specific T cell responses, in both preclinical models8 and human cancers9-13.
Although monomeric IgA is found in serum, at mucosal surfaces IgA is predominantly found as a dimer, with 2 subunits bound by a J-chain14. dIgA binds to the IgA/IgM polymeric immunoglobulin receptor (PIGR), a member of the immunoglobulin superfamily that is expressed on the basolateral surface of mucosal epithelia14. Recognition of polymeric IgA (and IgM) by PIGR triggers a process called transcytosis, whereby the PIGR:antibody complex traffics inside the epithelial cell from the basolateral to the apical side, and is then secreted into mucosal fluids15. We previously showed that PIGR is quasi-universally expressed in human ovarian9,16 and endometrial cancer cells12,17, and allows transcytosis of dIgA through these tumor cells. However, it is unclear whether IgA transcytosis also occurs in non-gynecologic tumors. Furthermore, IgA transcytoses through endosomes18, which could prevent direct contact with specific antigens inside tumor cells. Even if transcytosing IgA is not “shielded” by endosomes, targeted antigens could not co-localize inside tumor cells. Experimentation with these processes might determine if it is possible for engineered dIgA to target intracellular molecules, with important therapeutic implications: First, the mechanisms of resistance that invariably follow the administration of small molecule inhibitors19 could be still sensitive to antibody-mediated neutralization. Second, the half-life of unmodified IgAis ~6 days, but can be further optimized to increase stability20. This could lead to a more sustained activity and therefore enhanced effectiveness. For instance, long half-life and occupancy of PD-1 molecules on T cells appear to be important determinant of effectiveness for immune checkpoint inhibitory antibodies6. Third, dIgA is particularly abundant at mucosal surfaces, suggesting that exogenous dIgA could be safely administered to cancer patients21. For instance, oral IgA-IgG treatments and antibody preparations containing >20% IgA have been safely administered to patients22,23. In this study, we investigated the feasibility and therapeutic potential of targeting common mutated oncodrivers using recombinant dIgA. Our results provide a rationale for developing dIgA-based therapeutics to neutralize various intracellular antigens in human cancer and other diseases.
RESULTS
Mutation-specific dIgA neutralizes intracellular KRASG12D and expels it outside tumor cells, without effects on KRASWT cells
To investigate the spectrum of human malignancies that could be susceptible to PIGR-mediated dIgA transcytosis, we first analyzed PIGR expression in tumors in TCGA datasets. We found a clear dichotomy between epithelial malignancies and B cell lymphoma, which express relatively high PIGR mRNA; and non-epithelial tumors such as melanoma or glioblastoma, which express lower amounts of PIGR mRNA (Suppl. Fig.1A). To test whether antigen-specific dIgA, and not non-specific dIgA (Suppl. Fig.1B&C, Data S1), could target specific molecules in the cytoplasm of different carcinomas, we first focused on targeting KRASG12D, a mutational hotspot present in >4% of human cancers, and frequently in pancreatic, colorectal, lung and endometrial carcinomas2,24. Recombinant antibodies generated against GppNHp-bound KRASG12D (equivalent to the oncogenic GTP-bound form25) were produced on a dIgA or monomeric IgG4 backbone, as in anti-PD-1 immune checkpoint inhibitors, using identical variable heavy and variable light sequences (Suppl. Fig.2A-C; Data S1 & Data S2). Although we cannot rule out that GppNHp-KRASG12D-specific Abs recognize GTP-bound conformations driven by other mutations25, both dIgA and IgG Abs specifically recognized ectopic or endogenous mutant KRASG12D, but not other KRAS mutations, in Western-blot analysis (Fig.1A; Suppl. Fig.2D). Notably, only mutation-specific dIgA, but not IgG, penetrated KRASG12D-mutated lung cancer cells (Fig.1B, left), where mutation-specific, but not control dIgA, targeted KRAS (Fig.1B, right). Consistently, KRASG12D-specific dIgA penetrated OVCAR3 (ovarian cancer) cells transduced with KRASWT or KRASG12D fused to photoactivatable (PA)m-Cherry fluorescent protein26, but only disrupts cytoplasmic localization and distribution of intracellular KRASG12D (Fig.1C). Notably, mutant KRAS was not only neutralized inside tumor cells, but also expelled outside the cell through transcytosis, as evidenced by co-occurrence of KRASG12D with secretory IgA in the supernatant of KRASG12D-PAmCherry-transduced OVCAR3 cells upon treatment with KRASG12D-specific dIgA (Fig.1D), with decreased intracellular KRASG12D (Fig.1E & Suppl. Fig2E). Furthermore, PamCherry fragments, fused to KRASG12D, were only found in the supernatant of tumor cells treated with KRASG12D-specific dIgA in 3 independent experiments (Fig.1F, left). In contrast, KRASG12D was not found in supernatants upon treatment with KRASG12D-specific IgG4, irrelevant IgA containing dimers9, or vehicle (Fig.1D&F, left). As expected, PIGR was identified in the supernatants of ovarian cancer cells treated with antigen-specific or control dIgA (Fig.1F, right). Together, these results indicate that tumor cell-penetrating dIgA can indeed target specific mutations in KRAS inside tumor (e.g., ovarian cancer) cells, resulting in intracellular decreased levels and expelling the oncodriver outside the tumor cell, without obvious effects in KRAS unmutated epithelial cells.
Figure 1. KRASG12D mutation-specific dIgA1, but not IgG4, neutralizes KRASG12D inside tumor cells and expels the oncodriver outside tumor cells.

(A) Immunoblots showing that recombinant anti-KRASG12D-IgA1 and anti-KRASG12D-IgG4 antibodies specifically recognize this mutation in KRAS, but not G12C or G13D mutant KRAS, or WT KRAS (n=3). (B) Confocal immunofluorescence microscopy images showing that Alexa Fluor 647-conjugated anti-KRASG12D-IgA1, but not with anti-KRASG12D-IgG4, penetrates KRASG12D-mutant A427 and SK-LU-1 NSCLC cells, while KRASG12D-specific, but not control irrelevant dIgA, co-immunoprecipitated with KRASG12D; Scale bar, 25 μm. (C) OVCAR3 cells were transduced with KRASG12D or WT KRAs fused to PamCherry (Red). Confocal microscopy shows that treatment with Alexa Fluor 488-conjugated anti-KRASG12D-IgA1 (middle panel), but not with anti-KRASG12D-IgG4 (right panel), disarrays KRASG12D intracellular distribution, without effects on KRASWT cells (left panel). (n=3). Scale bar, 50 μm (D) The secretory component of human PIGR was immunoprecipitated from media from KRASG12D-PamCherry-transduced OVCAR3 cells, treated with anti-KRASG12D-IgA1, IgG4, irrelevant IgA or PBS vehicle. Eluted immunocomplexes were ran by SDS-PAGE, stained with Ponceau Red, and immunoblotted for KRASG12D, IgA, and pIgR (n=3). I Immunoblots showing KRASG12D levels in anti-KRASG12D-IgA1/IgG4 or irrelevant IgA-treated KRASG12D-OVCAR3 cell lysates. (F) Supernatants from KRASG12d-PAmCherry-transduced OVCAR3 cells, treated with anti-KRASG12D-IgA1, IgG4, irrelevant IgA or PBS vehicle, were subjected to liquid chromatography with tandem mass spectrometry (LC–MS/MS). Heatmaps showing intensities of mCherry (left panel) and pIgR (right panel) peptides detected (n=3). (G) Left, Transcytosis experiment adding biotinylated anti-KRASG12D-IgA1/IgG4 or irrelevant IgA to the upper chamber of a transwell system, where physical access of antibodies to the basal chamber is prevented by WT or KRASG12D-OVCAR3 cells grown to confluence in transwell inserts. Right, dot blots showing presence of KRASG12D (top panel) or beads (bottom panel) in the streptavidin immunoprecipitates of the basal and upper chamber contents.
KRASG12D and mutation-specific dIgA are found in the same endosomal compartments upon transcytosis
To confirm bona fide unidirectional transcytosis, we used a classical transwell system where ovarian or lung tumor cells prevent the access of dIgA delivered to the upper chamber into the basal chamber, unless it transcytoses through PIGR+ tumor cells27. As shown in Fig.1G & Suppl. Fig.2F, we found an accumulation of KRASG12D antigen in the basal chamber when KRASG12D-mutant ovarian and lung cancer cells were treated with antigen-specific dIgA, but not in wild-type ovarian tumor cells or upon PIGR ablation. However, a frail KRAS signal was detected using a pan-KRAS antibody in the basal chamber of tumor cells with other mutations, suggesting weak recognition of the GTP-bound oncogene25 (not shown). Importantly, only traces of antigen were found in the upper chamber, where dIgA was added and found in similar amounts, supporting that KRASG12D-specific dIgA undergoes “true” transcytosis through PIGR+ ovarian and lung cancer cells. Accordingly, high-resolution confocal microscopy demonstrated that aggregates of KRASG12D-specific dIgA co-localize with KRASG12D-PamCherry inside ovarian tumor cells, while the same antibody on an IgG4 backbone cannot penetrate ovarian cancer cells or alter the preferential location of mutant KRAS on the cell surface (Fig.2A).
Figure 2. KRASG12D-specific dIgA1, but not the same antibody on an IgG4 backbone, captures mutant KRAS during trafficking.

(A) High resolution confocal microscopy showing that aggregates of KRASG12D-specific dIgA, but not KRASG12D-specific IgG, co-localize with KRASG12D-PamCherry after 1 hr of treatment inside KRASG12D-OVCAR3 tumor cells (n=3). No-wash images confirm equivalent positive green fluorescence of added KRASG12D-specific IgA and IgG antibodies outside of the cells (45 min after treatment), Scale bar, 50 μm or 10 μm, as indicated. (B) Top panel, Non-denatured lysates (6 mg) of KRASG12D-OVCAR3 cells treated with anti-KRASG12D-IgA1, anti-KRASG12D-IgG4, or vehicle (PBS) were immunoprecipitated using anti-Rab5A, Rab7A, Rab8A, or Rab11A antibodies. Immunoprecipitates were then blotted for KRASG12D-specific IgA or IgG. Bottom panel, 60 μg lysates (1% of co-IP lysate) of anti-KRASG12D-IgA1, anti-KRASG12D-IgG4, or vehicle (PBS)-treated KRASG12D-OVCAR3 cells were immunoblotted for Rab5A, Rab7A, Rab8A, and Rab11A as input control. (n=3).
To understand the dynamics of interactions between antigen-specific dIgA and mutant KRAS inside KRASG12D-transduced ovarian tumor cells, we immunoprecipitated early (RAB5A+), late (RAB7A+) and recycling (RAB8A+/RAB11A+) endosomes28,29 in KRASG12D-transduced ovarian cancer cells treated with KRASG12D-specific dIgA or IgG. As expected, given the lack of penetration into tumor cells, KRASG12D-specific IgG was undetectable in any endosomes, while dIgA transcytosed through early and late endosomes (Fig.2B). Consistent with known recycling pathways that prevent proteasomal degradation30, mutant KRAS was primarily found in RAB8A+/RAB11A+ recycling endosomes when ovarian tumor cells were treated with vehicle or antigen-specific IgG (Fig.2B). In contrast, treatment with KRASG12D-specific dIgA concentrated mutant KRAS on the same endosomes through which dIgA traffics inside tumor cells (Fig.2B), suggest that transcytosing dIgA encounters the antigen on the cell membrane and hauls it in endosomes, eventually expelling the target through secretory IgA. Although dIgA traffics through endosomes, dIgA retains its capacity to specifically target intracellular antigens, at least those that are located near the cell membrane.
Consistent with capture and extracellular disposal of KRASG12D, treatment of KRASG12D-transduced ovarian cancer cells with mutation-specific dIgA inhibited tumor cell proliferation, without effects on the same cells transduced with KRASWT or untransduced ovarian cancer cells (Fig.3A-C). Treatment of endogenously mutated KRASG12D lung cancer cells with mutation-specific dIgA also inhibited tumor cell proliferation (Fig.3D), while neither antigen specific or control dIgA impacted the proliferation of non-malignant HEK293T cells (Fig.3E). Of note, antigen-specific dIgA dampens proliferation of KRASG12D-transduced ovarian cancer cells without causing apoptosis (Fig.3F). Therefore, antigen-specific dIgA expels oncodrivers outside tumor (e.g., ovarian cancer) cells by preventing endosomal recycling, resulting in KRASG12D accumulation in the same endosomal compartments where dIgA traffics.
Figure 3. KRASG12D-specific dIgA treatment reduces proliferation, without causing apoptosis, of KRASG12D-mutated, but not KRASWT cancer cells.

(A) MTT assay-based absorbance at 570 nm of KRASG12D-transduced OVCAR3 cells treated with irrelevant IgA or anti-KRASG12D IgA1, relative to mean absorbance of vehicle (PBS) treated cells, at different temporal points. (n=8) (B) MTT assay-based absorbance at 570 nm of KRASWT-transduced OVCAR3 cells treated with irrelevant IgA or anti-KRASG12D IgA1, relative to mean absorbance of vehicle (PBS) treated cells, at different temporal points. (n=8). (C) MTT assay-based absorbance at 570 nm of untransduced OVCAR3 cells treated with irrelevant IgA or anti-KRASG12D IgA1, relative to mean absorbance of vehicle (PBS) treated cells, at different temporal points. (n=16). (D) MTT assay-based absorbance at 570 nm of KRASG12D SK-LU-1 cells treated with irrelevant IgA or anti-KRASG12D IgA1, relative to mean absorbance of vehicle (PBS) treated cells, at different temporal points. (n=16). (E) MTT assay-based absorbance at 570 nm of PIGR− HEK293T cells treated with irrelevant IgA or anti-KRASG12D IgA1, relative to mean absorbance of vehicle (PBS) treated cells, at different temporal points. (n=16). (F) Total apoptotic cells, determined by flow cytometry analysis of KRASG12D-transduced OVCAR3 cells, treated with vehicle, or irrelevant IgA, or anti-KRASG12D IgA1, or anti-KRASG12D IgA4 antibodies, and gated on Annexin V+ Propidium iodide+/− cells (n=3). Data are mean ± SEM. ***P ≤ 0.001, NS, not significant; unpaired two-tailed t-test. Data S3 provides details of statistics.
KRASG12D-specific dIgA specifically abrogates the progression of mutant tumors in vivo
To demonstrate the effectiveness of KRASG12D-specific dIgA in vivo, we first performed intra-tumoral infusions of different treatments in KRASG12D-transduced OVCAR3 (ovarian cancer)-bearing Rag1−/− mice. Irrelevant dIgA dampens the RAS pathway by increasing expression of DUSP phosphatases that dephosphorylate ERK1/2, while activating inflammatory pathways associated with IFN and TNF signaling, as well as pro-apoptotic pathways linked to ER stress9,12. Accordingly, non-specific dIgA delayed the growth of established ovarian tumors. However, the anti-tumor effects of KRASG12D-specific dIgA were greater (Fig.4A&B), with no obvious signs of toxicity in any treated mice. Consistently with its inability to penetrate tumor cells, the same variable heavy and light chains on an IgG4 backbone did not elicit significant ovarian cancer reduction, potentially attributable to neutralization of extracellular vesicles (Fig.4B & Suppl. Fig.3A&B). Supporting the specificity of this intervention, no effects on tumor growth were elicited in tumor cells transduced with KRASWT, (Fig.4C & Suppl. Fig.3A&C), beyond the non-antigen-specific effects of IgA transcytosis. As reported5, the KRASG12D-selective inhibitor MRTX1133, currently under preclinical development5, also elicited significant delays in ovarian cancer growth, although daily intraperitoneal administration (20 injections) were required (Suppl. Fig.3A-C).
Figure 4. KRASG12D-specific dIgA1, but not the same antibody on an IgG4 backbone, abrogates KRASG12D tumor growth in vivo.

(A) Schematic of design of experiment shown in B and C. Antibody (100 μg per 20 g body weight) or equal volume of vehicle (PBS) was intratumorally (IT) injected. (B) Tumor growth curves (left), tumor weight (right) in KRASG12D-OVCAR3 tumor-bearing Rag1-deficient mice receiving control IgA, anti-KRASG12D-IgA1, anti-KRASG12D-IgG4 antibodies, or vehicle. Growth curves and tumor weights were pooled from 2 independent experiments (n=10 mice per group, total). (C) Tumor growth curves (left), tumor weight (right) from KRASWT-OVCAR3 tumor-bearing Rag1-deficient mice receiving the same treatments as in B. Growth curves and tumor weights were pooled from 2 independent experiments (n=10 mice per group, total). (D) Schematic of design of experiment. Antibody (100 μg per 20 g body weight), equal volume of vehicle (PBS), or MRTX1133 (200 μg per 20 g body weight) were intraperitoneally (IP) injected every 4 days, or daily, as indicated, into NSG mice growing KRASG12D-OVCAR3 tumors. Tumor growth curves (left) and tumor weight (right) were pooled from 3 independent experiments (n=13 mice per group, total). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, NS, not significant; paired two-tailed t-test for growth curves or unpaired two-tailed t-test for tumor weight (I). (E) Representative immunohistochemical staining of human IgA of KRASG12D-OVCAR3 mice tumors treated intraperitoneally with non-antigen specific or KRASG12D-specific dIgA. Scale bar, 400 or 60 μm, as indicated. Data S3 provides details of statistics.
Selective effectiveness of antigen-specific dIgA was recapitulated when antibodies were intraperitoneally administered in KRASG12D tumor-bearing NSG mice (Fig.4D & Suppl. Fig.4A&B), ruling out the requirement of functional myeloid cells or NK lymphocytes in this model, while supporting systemic distribution of dIgA. Again, 20 (daily) doses of intraperitoneal MRTX1133 or 5 injections of KRASG12D-specific dIgA both delayed tumor progression beyond the effects of irrelevant IgA (Fig.4D & Suppl. Fig.4B). Correspondingly, human IgA was identified in resected tumors (Fig.4E), confirming trafficking of antigen-specific dIgA to tumor beds. These experiments support the potential of tumor cell-penetrating dIgA to specifically target mutated oncodrivers inside carcinoma cells in vivo.
Lung tumors quasi-universally express PIGR and are highly sensitive to mutant oncodriver-neutralizing dIgA
To demonstrate the capacity of dIgA to target human tumors that are spontaneously driven by KRASG12D, as opposed to ectopic KRAS expression, we first focused on non-small cell lung cancer (NSCLC), which is frequently driven by mutant KRAS, including KRASG12D. Analyses of 30 human lung cancer tissues including squamous cell carcinomas and adenocarcinomas, showed PIGR expression in all tumors, along with spontaneous production of IgA and IgG at tumor beds bound to tumor epithelial tissues as well as to stroma (Fig.5A), without differences for histological subtypes (Suppl. Fig.5A). Notably, PIGR and tumor-bound IgA density, IgG coating of tumor cells, or PIGR:IgA interactions were all associated with higher T cell accumulation (Fig.5B). We therefore aimed to demonstrate the effectiveness of mutation-specific dIgA against lung carcinomas endogenously driven by KRASG12D. As shown in Fig.5C&D, 5 intraperitoneal injections of KRASG12D-specific dIgA, or 20 daily treatments of MRTX1133 at high dose (200 μg)5, effectively abrogated the progression of established A427 NSCLC, while the same antibody on an IgG4 backbone had negligible effects (Fig.5C&D; Suppl. Fig.5B&C). Accordingly, treatment with both KRASG12D-specific dIgA and MRTX1133 decreased ERK1/2 phosphorylation in A427 cells in a time-dependent manner (Fig.5E). Anti-tumor effects were not limited to this lung cancer model, because established KRASG12D-mutant SK-LU-1 lung cancers also responded to KRASG12D-specific dIgA with comparable effectiveness, although they were more resistant to MRTX1133 (Fig.5C&F; Suppl. Fig.5B&D). Although established lung tumors were not fully eliminated, different models underwent a significant reduction in tumor growth. These experiments support the potential of dIgA-mediated intracellular targeting of KRASG12D to abrogate the progression of spontaneously mutated NSCLCs, in a mutation-specific manner and without off-target effects in unmutated epithelial cells.
Figure 5. KRASG12D-specific dIgA1 is more effective than small molecule KRASG12D inhibitors at abrogating the progression of KRASG12D NSCLC.

(A) Combined staining of pIgR, IgA, IgG, CD3, Pan cytokeratin (panCK) and DAPI, representative of 30 clinical cases of squamous cell carcinoma or adenocarcinoma of the lung. Scale bar, 200 μm (upper panel), 100 μm (magnified lower panels). (B) Accumulation of CD3+ T cells in human panCK+ NSCLC islets are associated with increased coating of tumor cells with IgA and IgG, expression of pIgR and colocalization of IgA with pIgR. Data are mean ± SEM. **P≤0.01; ***P≤0.001, unpaired two-tailed t-test. (C) Schematic of design of experiment shown in D&E. Antibody (100 μg per 20 g body weight), or equal volume of vehicle (PBS) were administered every 4 days, while MRTX1133 (200 μg per 20 g body weight) was administered daily or every 4 days, as indicated. All treatments were IP injected. (D) Tumor growth curves from KRASG12D A427 tumor-bearing NSG mice receiving (IP) anti-KRASG12D-IgA1, anti-KRASG12D-IgG4 or vehicle every 4 days. MRTX1133 was administered in different mice every 4 days (left) or daily (middle), as indicated. Tumor weight is shown on the right. Growth curves and tumor weights were pooled from 3 independent experiments (n=14 mice per group, toI). (E) Western blot showing phosphorylated and total ERK1/2 in lysates of KRASG12D-mutated A427 cells, treated with anti-KRASG12D-IgA1 or MRTX1133. (F) Identical treatment as in ‘D’ of advanced KRASG12D-mutated SK-LU-1 lung cancers. Growth curves and tumor weights were pooled from 3 independent experiments (n=14 mice per group, total). (G) Left, schematic design of treatments in KPMSH2KIN lung tumor-bearing immunocompetent mice. Antibody (100 μg per 20 g body weight) or equal volume of vehicle (PBS) was IP injected. When indicated, CD8 T cells were depleted with intraperitoneal anti-CD8 antibodies. Right, Tumor growth curves from mice receiving intraperitoneal vehicle or anti-KRASG12D-dIgA1, or anti-IDH1R132H-dIgA1, with or without CD8 T cell depletion. Growth curves were pooled from 2 independent experiments (n=8-10 mice per group). Data are mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; paired two-tailed t-test (D&F) and non-parametric Wilcoxon test (G) for growth curves or unpaired two-tailed t-test for tumor weights. Data S3 provides details of statistics.
dIgA transcytosis sensitizes tumor cells to T cell-mediated killing9. To define the contribution of this mechanism upon KRASG12D-specific dIgA treatment, we utilized a syngeneic, immunogenic KRASG12D-mutant KPMSH2KIN lung tumors, generated by cloning lung cancer cells from C57/BL6 mice with inducible mutations, activated using intra-nasal adenoviral Cre; plus restoring MSH2 through lentiviral transduction. dIgA targeting KRASG12D substantially delayed tumor growth (Fig.5G). Anti-tumor effects in this immunogenic system were dependent on CD8+ T cells (Fig.5G), highlighting the complexity of a coordinated adaptive immune response. Further supporting the specificity of dIgA for cognate antigen, similar anti-tumor effects were not observed upon administration of dIgA targeting a different mutation (IDH1R132H) (Fig.5G). Corresponding effects were observed in another syngeneic KRASG12D breast cancer model (Brpkp11031; Suppl. Fig.5E).
dIgA is effective against mutations located deeper into the cytoplasm of tumor cells, in a PIGR-dependent manner
To demonstrate the feasibility of targeting additional intracellular antigens, located deeper into the cytoplasm of tumor cells; and also the potential of using dIgA to target other epithelial cancers different from ovarian and lung carcinomas, we generated dIgA1 or IgG4 specifically targeting the cytoplasmic32,33 R132H mutation in isocitrate dehydrogenase 1 (IDH1), a common hotspot in gliomas34 and sporadically found in gastroesophageal35, colorectal36 and lung37 carcinomas (Fig.6A; Suppl. Fig.2A-C; Data S2). To further evaluate the role of PIGR abundance for dIgA effectiveness, we procured isogenic IDH1WT and IDH1+/R132H HCT116 colon cancer cells, which express low PIGR levels, and transduced these with PIGR, resulting in expression comparable to those found in multiple carcinoma cell lines (Fig.6A). Low PIGR expression abrogated the non-specific anti-tumor effects of irrelevant IgA intraperitoneally infused into flank colon cancer-bearing mice (Fig.6B-D). In contrast, similar to KRASG12D targeting, IDH1R132H-specific dIgA delayed tumor growth in mutant flank colon tumors, without effect in the absence of IDH1R132H mutations (Fig.6C&D). Notably, anti-tumor effects were dependent on PIGR expression in colon cancer cells, because the administration of IDH1R132H-specific dIgA resulted in a 3.7-fold reduction in flank tumor growth, compared to vehicle, when PIGR-transduced IDH1R132H HCT116 colon cancer cells were treated, compared to a 1.8-fold reduction in low-PIGR-expressing parental IDH1R132H HCT116 colon tumors (Fig.6B,C&E). Importantly, anti-tumor effects were again mutation-specific, because a comparable reduction in colon cancer growth was observed when mice with IDH1WT tumors were treated with irrelevant IgA or IDH1R132H-specifc dIgA (Fig.6F). Dimerization of mutation-specific IgA is also required because the monomeric version of the same IDH1R132H-specific IgA showed no significant anti-tumor effects (Fig.6G). Together with data from targeting KRAS-mutated lung tumors, these results support the potential of targeting carcinomas of multiple histological origins and different mutated oncodrivers, using antigen-specific dIgA. They also support the feasibility of targeting tumors despite low PIGR expression, albeit with possibly decreased effectiveness.
Figure 6. IDH1R132H-specific dIgA1 abrogates IDH1-mutated colon cancer growth in vivo in a PIGR expression-dependent manner.

(A) Immunoblots showing that recombinant anti-IDH1R132H-IgA1 antibodies specifically recognize the mutation in the lysates of IDH1R132H/+ HCT116 cells, but not in IDH1+/+ HCT116 cells, with or without PIGR-transduction. Overexpression of PIGR protein in PIGR-transduced cells is also shown (n=3). (B) Schematic of design of experiment shown in C-F. Antibody (100 μg per 20 g body weight) or equal volume of vehicle (PBS) was injected IP. (C) Untransduced (PIGRlow) mutant-IDH1 (IDH1+/R132H) tumor-bearing NSG mice received IP control IgA, anti-IDH1G12D-IgA1, or vehicle. Tumor growth curves (left), tumor weight (middle) and tumor volume of one representative experiment (right) are shown. Growth curves and tumor weights were pooled from 3 independent experiments (n=14 mice per group, total). (D) Tumor growth curves (left), tumor weight (middle) and tumor volume of one representative experiment (right) from identically treated untransduced (PIGRlow) WT-IDH1 (IDH1+/+) tumor-bearing NSG mice. Growth curves and tumor weights were pooled from 3 independent experiments (n=15 mice per group,Ital). (E) Tumor growth curves (left), tumor weight (middle), tumor volume of one representative experiment (right) from PIGR-transduced mutant-IDH1 (IDH1+/R132H) tumor-bearing NSG mice treated as in C&D. Growth curves and tumor weights were pooled from 3 independent experiments (n=16 mice per group, total). (F) Tumor growth curves (left), tumor weight (middle), tumor volume of one representative experiment (right) from PIGR-transduced WT-IDH1 (IDH1+/+) tumor-bearing NSG mice treated as in C-E. Growth curves and tumor weights were pooled from 3 independent experiments (n=16 mice per group, total). (G) Schematic of design of experiment. Monomeric or dimeric anti-IDH1G12D-IgA1, anti-IDH1G12D-IgG4 antibody or equal volume of vehicle (PBS) were injected IP into mutant-IDH1 (IDH1+/R132H) tumor-bearing NSG mice. All antibodies were administered at 100 μg per 20 g body weight. Growth curves pooled from 2 independent experiments (n=8-9 mice per group, total). Data are mean ± SEM. *P ≤ 0.05, ***P ≤ 0.001, NS, not significant; paired two-tailed t-test for growth curves or unpaired two-tailed t-test for tumor weights. Data S3 provides details of statistics.
DISCUSSION
We found that PIGR-mediated transcytosis of dIgA occurs in non-gynecological carcinomas9, including lung and colorectal cancers. Given that PIGR is expressed in most epithelial malignancies, but only at lower levels in non-epithelial cancers38, this mechanism could be relevant for most human epithelial cancers. This should include recalcitrant tumors such as pancreatic or small cell lung cancer, which our data show quasi-universally express PIGR. PIGR-mediated dIgA transcytosis progresses through endosomal trafficking but their route of trafficking may slightly differ in malignant cells than in non-malignant cells18. Despite trafficking through endosomes, dIgA retains its capacity to specifically target intracellular antigens. Our results show that dIgA transcytosis elicits loss of recycling endosomes carrying oncogenic KRAS and colocalization of antigen and antibody in early and late endosomes. This is consistent with heterotypic fusion of the early endosomes that internalize dIgA with the recycling endosomes bringing back KRAS to the cell surface. However, this activity is not limited to mutated oncodrivers that are located near the cell membrane, such as KRAS, but also mutated oncogenes such as IDH1, which are located deeply inside the cytoplasm. Future studies should examine whether other commonly mutated oncogenes such as PI3K or AKT, or immunosuppressive intracellular pathways such as IDO, could be more effectively targeted with dIgA than with small molecule inhibitors.
dIgA-mediated targeting, along or in combination with small molecules, could have substantial advantages, compared to small molecules: First, our results underscore the specificity of this approach. Second, although the half-life of IgA (~6 days in primates and >15 hrs in mice)39,40 is lower than that of IgG39, and there could be important differences in availability of PIGR-binding dimeric IgA vs. the monomeric form, which does not bind to PIGR but could reach the tumor microenvironment, persistence is nevertheless longer than for small molecule inhibitors. Therefore, the anti-tumor effect could be more sustained as the antibody will remain available to target tumor cells for much longer periods. Moreover, modifications in glycosylation or attaching an albumin-binding domain to the heavy or light chain could increase their half-life20,41. Third, dIgA expels the mutated oncogene outside tumor cells, which is obviously advantageous, compared to temporary neutralization. Nevertheless, our data also show that the effectiveness of intracellular targeting of oncodrivers using dIgA is dependent on the expression levels of PIGR in tumor cells. Therefore, one could envision that resistance to dIgA could arise from PIGR loss by tumor cells. However, PIGR is quasi-universally expressed in most epithelial cancers, suggesting that it provides a survival advantage to tumor cells. The use of dIgA to target KRAs mutations should complement existing and emerging small molecule inhibitors, which inevitably elicit mechanisms of resistance, Antibodies could therefore provide a valid therapy when this occurs, while small molecule inhibitors could be used if PIGR was lost after dIgA targeting. Finally, the prospect of oral secretory IgA treatment42 could make this intervention ideal against tumors of the digestive tract. We therefore propose a rationale for using dIgA as a form of immunotherapy to target intracellular oncodrivers, which could open multiple new avenues to treat otherwise undruggable carcinomas.
Although our dIgA treatment did not fully eliminate xenograft tumors, we administered 5 injections and we only treated established (i.e., palpable) tumors. It is possible that earlier interventions, more sustained antibody administration, or higher doses of dIgA could have resulted in complete tumor rejection. We also found a patchy staining for human IgA, which could reflect suboptimal doses of dIgA, but also different transcytosis timing for distinct tumor areas, or mutational heterogeneity. Furthermore, treatment of an immunogenic syngeneic lung tumor in immunocompetent mice was found to be dependent on T cells, because depletion of CD8+ T cells abrogated therapeutic benefits. This is consistent with our previous report that dIgA transcytosis sensitizes ovarian cancer cells to antigen-specific T cell-mediated killing9. It is therefore possible that combination with immune checkpoint inhibitors synergizes with targeting of mutated intracellular oncodrivers using dIgA, or that our observed effects are maximized in immunocompetent patients. Our reports in ovarian cancer, for instance, support the importance of spontaneous coordinated humoral and cellular adaptive immune responses as predictors of better outcome9.
Our data showing therapeutic effectiveness in immunocompetent mice bearing syngeneic immunogenic carcinomas further supports the concept that the interaction between human dIgA and mouse pIgR mirrors the bioavailability of antigen-specific dIgA in vivo, in the presence of PIGR+ healthy epithelial cells that could compete for the antibody. Furthermore, unlike humans, rats and hamsters, mice do not express CD89, which also binds IgA and could be partially responsible for enlarged persistence of IgA in primates, compared to mice40, and therefore retain dIgA in circulation for much longer.
Finally, an important issue regarding the future translatability of dIgA is potential toxicity. In support of the clinical potential of our approach, oral IgA-IgG treatment has already been safely used to treat children with chronic non-specific diarrhea22, and polyclonal antibody preparations containing >20% IgA can be safely administered in patients with severe pneumonia23. In addition, mutation-specific dIgA had no effect on wild-type cells, while dimeric IgA is the predominant Ig at mucosal secretions21. It is therefore unlikely that IgA administration will cause on-target, off-tumor toxicity. Furthermore, FDA-approved KRASG12C small molecule inhibitors already elicit grade 3-4 toxicity in >11% of patients. New studies should evaluate these issues, but our results provide a rationale for developing dIgA-based therapeutics that specifically target intracellular antigens in human cancer patients.
LIMITATIONS OF THE STUDY
Our study demonstrates that KRASG12D-specific dIgA transcytosis alters the endosomal traffic of mutant KRAS. However, the intricacies of this mechanism remain unclear. One plausible scenario is heterotypic fusion of the early endosomes that internalize dIgA with the recycling endosomes that bring mutant KRAS back to the cell membrane. However, this does not fully explain how dIgA targets deep cytoplasmic oncogenes such as IDH1.
Future studies should also investigate whether mutant tumors that have become resistant to existing small molecule inhibitors (e.g., sotorasib) could be still sensitive to PIGR-mediated interventions. Antibodies designed to recognize the constitutively active (GTP-bound) KRAS structure elicited by different mutations could be particularly effective in this context. Conversely, loss of PIGR could make tumors resistant to dIgA, while remaining sensitive to drug inhibitors.
Similarly, future studies should dissect the mechanisms whereby CD8+ T cells participate in the effects of oncogene-specific dIgA, which may depend on the immunogenicity of each disease (e.g., as a result of different tumor burden).
Finally, generating recombinant dimeric IgA is more challenging than generating IgG, which also has longer half-life. Modifications of the Fc domain of IgG to trigger PIGR-mediated transcytosis, while preserving binding to FcRn to ensure persistence need to be investigated, to pave the way for novel immunotherapies against the most frequent and aggressive human cancers.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jose R. Conejo-Garcia (Jose.Conejo-Garcia@Duke.edu).
Materials availability
Antibody producing plasmids generated in this study will be shared with other research groups only upon receipt of reasonable request on a case-by-case basis with a completed Materials Transfer Agreement (MTA).
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD042914 and PXD044426. This paper does not report original code. The datasets generated during the current study are available from the lead contact upon reasonable request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Human samples
A lung cancer tissue microarray of total 30 cases including squamous cell carcinoma, adenocarcinoma, adenosquamous carcinoma and bronchioloalveolar carcinoma with matched metastatic carcinoma and cancer adjacent tissue, with available pathology grade and survival data was procured from US Biomax.
Cell lines
OVCAR3 (RRID:CVCL_0465), A427 (RRID:CVCL_1055), SK-LU-1 (RRID:CVCL_0629), HEK293T (RRID:CVCL_0063), NCI-H23 (RRID:CVCL_1547), NCI-H647 (RRID:CVCL_1574) cell lines were purchased from ATCC (Manassas, VA). IDH1+/+ and IDH1R132H/+ HCT116 (RRID:CVCL_0291) cell lines were purchased from Horizon discovery. PIGR-ablated OVCAR3 cells was generated in the lab43. Brpkp110 was established in the lab18 and KPMSH2KIN is a new model of lung cancer developed also in our laboratories that carries the same epitope recognized in human G12D-mutated KRAS. Cell lines were routinely cultured in R10 (RPMI-1640, 10% FBS, penicillin (100 IU ml−1), streptomycin (100 μg ml−1), L-glutamine (2 mM), sodium pyruvate (0.5 mM)) media (Thermo). HEK293T cell line was routinely cultured in D10 (DMEM, 10% FBS, penicillin (100 IU ml−1), streptomycin (100 μg ml−1), L-glutamine (2 mM), sodium pyruvate (0.5 mM)) media (Thermo).Cell lines were routinely tested for negative mycoplasma contamination.
Animal models
For mouse models, three different strains were used. Female, 4-6 weeks old Rag1-deficient (Rag1−/−) mice (RRID:IMSR_JAX:003729) and female, 7 weeks old C57BL/6J mice (RRID:IMSR_JAX:000664) were purchased from The Jackson Laboratory. NOD.Cg-Prkdcscid Il2rgtmlWjl/SzJ (NSG) mice (RRID:IMSR_JAX:005557), originally procured from The Jackson Laboratory, were maintained by the Moffitt Cancer Center animal facility. All animals were maintained by the animal facility of the Moffitt Cancer Center and Duke School of Medicine, housed in cages of up to 5 mice per cage, in a temperature controlled (18-23°C), 40-60% humidity, 12 h light/dark cycle facility. Animal studies were performed in accordance with Institutional Animal Care and Use Committee at the University of South Florida Research Integrity and Compliance department (IACUC protocols#IS00006655 and #IS00009457) and at the Duke University and Division of Laboratory Animal Resources (IACUC protocol# A011-23-01)
Method details
Constructs, and transduction
Human PIGR-coding sequence was cloned into pLVX-IRES-ZsGreen1 lentiviral vector (Genscript)44. Constructs encoding Wild-type or G12D mutated KRAS fused with PAmCherry1 (pLenti-TO/CMV-PAmCherry1-KRAS, G12D or WT)26 were procured from Dr. Xiaolin Nan Lab, Oregon Health & Science University. The sequence integrity and accuracy of all constructs was assessed and confirmed by sequencing services from GeneWiz.
Virus production and transduction
Lentiviral particles were generated by co-transfecting HEK 293T/17 (ATCC) with pLVX-IRES-PIGR-ZsGreen, or pLenti-TO/CMV-PAmCherry1-KRAS (G12D mutated KRAS or WT) and packaging/envelope vector pMD2.G (RRID:Addgene_12259) and psPAX2 (RRID:Addgene_12260) using Lipofectamine-3000 (Invitrogen) and the viral supernatant was harvested 48h after. 5×105 HCT116 or OVCAR3 cells were seeded in a 6-well plate with 3mL of the corresponding 0.45 μ filtered viral supernatant with added 10 μg/mL polybrene (Millipore). The plates were spun for 90min at 32°C and 1200g. 12h after transduction the medium was changed to R10 and 48h later the expression of the target gene was verified under microscope for KRAS transductions and selected by FACS-sorted for ZsGreen expression (PIGR).
Tumor Models
Flank tumors were initiated by injecting 1×107 KRASWT or G12D transduced OVCAR3 or A427; or 5×106 IDH1WT or Mut HCT116 cells or 5×105 Brpkp110 cells; or 1 × 106 KPMSH2KIN cells in PBS. SK-LU-1 tumors were initiated by injecting 1×107 cells in PBS admixed with 1:1 Matrigel into flanks.
Tumor volume was calculated as: 0.5 × (L × W2), where L is length and W is width. Intratumoral or peritumoral injections, as well as intraperitoneal injections of antibodies were done on multiple times at 3-4 days interval starting from day 7 for OVACR3, A427, HCT116, Brpkp110 and KPMSH2KIN tumors or from day 20 for SK-LU-1 tumors after the tumor challenge at a dose of 100μg/20g. MRTX1133 (MedChemExpress, #HY-134813) was injected daily or multiple times at 3-4 days interval starting from day 7 for OVACR3 and A427 or from day 20 for SK-LU-1 tumors at a dose of 200μg/20g. CD8 T cell depletion has been performed using αCD8 antibodies (BioXCell), injected at same frequency of injected antibodies, at a dose of 200μg/20g.
Recombinant antibody production
Anti-KRASG12D, and anti-IDH1R132H antibody variable heavy (VH) and variable light (VL) sequences were obtained from the patent US10844136B2 and CN106957367A, respectively. VH sequences followed by human IgA1 or IgG4 heavy chain constant region sequences in frame were cloned into PBMN-I-GFP vector (RRID:Addgene_1736). VL sequences along with human kappa light chain constant region sequences in frame were cloned into pVITRO1 (Genscript). Vectors, coding for all the VH and VL, were produced by Genscript. J-chain encoding pcDNA3.0 vector was procured from Addgene (RRID:Addgene_145146).
For recombinant dimeric and monomeric KRASG12D-IgA1 and KRASG12D-IgG4 antibody production, HEK293T cells were retrovirally transduced with antibody heavy chain encoding PBMN-I-GFP vector and sorted for viable GFP+ cells, followed by transfection with antibody light chain encoding pVitro1 vector and selected using Hygromycin, and finally those cells were transfected with J-chain encoding pcDNA3.0 vector and selected using G418. Heavy chain plus light chain plus J-chain expressing HEK293T cells were grown in suspension for 5-7 days in Free-Style 293 expression media (Thermo) for dimeric KRASG12D-IgA antibody production. Similarly, for monomeric KRASG12D-IgA1 or KRASG12D-IgG4, HEK293T cells expressing only light chain and respective heavy chain were used. Supernatants were concentrated through a 100 kDa membrane, to eliminate any possible contaminants of unassembled J-chains, heavy chains or light chains, and antibodies were purified using IgA or IgG purification columns (Ligatrap). Dimerization of IgA antibodies were confirmed by detecting J-chain in the elutes through mass spectrometry. All transfections were performed using Lipofectamine 3000 (Thermo). Cell lines were routinely tested for negative mycoplasma contamination. Dimeric and monomeric IDH1R132H-IgA1, and monomeric IDH1R132H-IgG4 antibodies and some additional quantity KRASG12D-IgG4 antibodies were procured from GenScript.
Fluorescence-activated cell sorting (FACS)
Sorting of antibody heavy chain-transduced HEK293T cells (GFP expressing) were performed by staining with DAPI (Thermo Scientific) viability dye and gated for DAPI−GFP+ cells. Similarly, PIGR-transduced HCT116 cells were sorted by gating DAPI−GFP+ cells. Samples were subsequently fluorescence-activated cell sorting (FACS) sorted using BD FACS ARIA.
One hundred thousand KRASG12D transduced OVCAR3 cells were placed in 6-well plates. After 12 h wells were washed, and fresh medium was added, and cells were treated with anti-human KRASG12D-IgA1 or KRASG12D-IgG4 or non-specific human IgA (Abcam, ab91025) at 0.5 μg/ml final concentration or vehicle (PBS) and incubated for 16 h in a 37-°C incubator. Total apoptotic cells (annexin V+ propidium iodide+/−) were analyzed by flow cytometry.
MTT assay
Five thousand OVCAR3 (wild-type or G12D mutated KRAS-transduced or untraduced) or SK-LU-1 cells, or five hundred HEK293T cells were seeded in each well of 96-well plates. After 12 h cells were washed with PBS, and fresh medium was added, and were treated with anti-human KRASG12D-IgA1 or non-specific human IgA (Abcam, ab91025) at 0.5 μg/ml final concentration or vehicle (PBS) and the medium was changed after 16 h incubation in a 37-°C incubator to remove the treatment. Proliferation was measure at 3, 5 or 7 days. MTT reagent solution (10ul of 5mg/ml) was added to the media of cultured cells and incubated for 3 hr. The precipitates were dissolved in DMSO, after remove the medium, and absorbance were measured at 570nm. Fold changes were calculated by dividing with the mean absorbance of the replicates of respective vehicle-treated cell lines.
Immunofluorescence and confocal microscopy
Anti-human KRASG12D-IgA1 or KRASG12D-IgG4 antibodies were conjugated with Alexa Fluor 488-conjugation kit (Thermo). Fifty thousand OVCAR3 (transduced with wild-type or G12D mutated KRAS, fused with PAmCherry) cells were placed onto a coverslip within 6-well plates and after 12 h treated with the conjugated antibodies for different temporal points. PAmCherry was photoactivated using the Leica TCS SP8 DAPI bandpass filter cube (Excitation BP 450-490 nm) using the widest aperture. Exposure time was dependent on the day of the experiment and varied between 20 to 30 seconds and was optimized using positive and negative controls. The same exposure time was used for all treatments. The gain for the Hoechst channel was adjusted to compensate for photobleaching due the exposure time required for photoactivating PAmCherry. Images were acquired with a 40X/1.3 NA objective in a confocal microscope (Leica SP8) using LAS X (v.3.5.5.19976) software. Hoechst, Alexa fluor 488, PAmCherry were excited with laser lines 405 nm, 488 nm, 552 nm respectively. Emission was captured through band pass settings 415–480 nm, 500–525 nm, 592–651 nm, respectively. For the highest resolution images, we used the same parameters but we increased the pixel resolution to 2048 x 2048 and took the images with speed=400 and z=0.5um.
Exocytosis of KRASG12D and LC-MS/MS
Five hundred thousand OVCAR3 (transduced with wild-type or G12D mutated KRAS, fused with PAmCherry) cells were placed in 100mm × 20mm cell culture plates and treated with or without anti-human KRASG12D-IgA1 or KRASG12D-IgG4 or non-specific human IgA (Abcam, ab91025) at 0.5 μg/ml final concentration or vehicle (PBS) in low serum media. After 12 h, conditioned medium was collected and filtered for contaminant debris removal. Proteins were extracted from the conditioned medium, reduced by DTT, digested by trypsin, and subjected to mass spectrometry analysis by the Moffitt Cancer Center Proteomics Facility. MaxQuant (version 1.5.2.8) was used to analyze the data, identify, and quantify the proteins45.
Western blot, Native gel electrophoresis and Co-immunoprecipitation (Co-IP)
Cells were lysed in RIPA buffer (Thermo) with protease-phosphatase inhibitor cocktail (CST, #5872S) and cleared by centrifugation. Proteins were quantified by BCA assay (Thermo). Membranes were blotted with recombinantly produced anti-KRASG12D-IgA1 or anti-KRASG12D-IgG4 or anti-IDH1R132H-IgA1 or anti-IDH1R132H-IgG4 antibodies; or commercial anti-mCherry Rockland, #600-401-P16, RRID:AB_2614470), anti-human immunoglobulin heavy alpha chain (Thermo Fisher Scientific, #A18781, RRID:AB_2535558), or anti-human immunoglobulin heavy gamma (plus light) chain (Thermo Fisher Scientific, #A18805, RRID:AB_2535582), or anti-human immunoglobulin kappa chain (R&D Systems, #MAB10050), or anti-human J-chain (Thermo Fisher Scientific, Mc19-9, #MA1-80527, RRID:AB_934333), or anti-human Rab5A (Proteintech, #11947-1-AP, RRID:AB_2269388), or anti-human Rab7A (Proteintech, #55469-1-AP, RRID:AB_11182831), or anti-human Rab8A (Proteintech, #55296-1-AP, RRID:AB_10858398), or anti-human Rab11A (Proteintech, #20229-1-AP, RRID:AB_10666202), or anti-human PIGR (Abcam, #ab96196, RRID:AB_10677612), or anti-human KRASG12D (CST, #14429, D8H7, RRID:AB_2728748) or anti-human β-actin (CST, #5125, 13E5, RRID:AB_1903890), antibodies. Lysates of A427 cells treated with KRASG12D-specific IgA or MRTX1133 for different time points were probed with antibodies against total ERK1/2 (CST, #9102, RRID:AB_330744) and phosphorylated ERK1/2 (CST, D13.14.4E, #9102, RRID:AB_330744). Blots were then probed with appropriate horse radish peroxidase (HRP)-conjugated secondary antibodies (room temperature ,1-2 hour), unless the primary antibodies are HRP-conjugated, and immunoreactive bands were developed using Enhanced Chemiluminescence (ECL) substrate (GE HealthCare).
For the native gel electrophoresis, 5μg of dimeric KRASG12D-IgA1, dimeric IDHR132H-IgA1, monomeric IDHR132H-IgA1 and irrIgA were diluted using Tris-Glycine Native Sample Buffer (ThermoFisher, #LC2673) and ran in a native gel, 12% Tris-Glycine Gel (Invitrogen, # XP00120BOX) for 1 hour at 220V. The gel was stained by overnight shaking with Invitrogen™ Colloidal Blue Staining Kit (Invitrogen, #LC6025). Then, the staining solution was replaced with deionized water, followed by overnight shaking until the gel had a clear background. The gel was scanned in a Canon TR4700 series HTTP.
For co-immunoprecipitation, cells were lysed using non-reducing non-denaturing lysis reagent provided in the co-immunoprecipitation (co-IP) kit (Pierce, Cat#26149) used. Proteins were immunoprecipitated using anti-human PIGR-secretory component (Abcam, SC-05, #ab3924, RRID:AB_2261963) or anti-human Rab5A, or anti-human Rab7A, or anti-human Rab8A, or anti-human Rab11A, or anti-human IgA (Abcam, EPR5367-76, #ab124716, RRID:AB_10976507). Proteins from concentrated conditioned media from anti-human KRASG12D-IgA1 or KRASG12D-IgG4 or non-specific human IgA or vehicle treated OVCAR3 cells were immunoprecipitated using anti-human PIGR-secretory component. The elutes were ran for western blotting. For analyzing changes in intracellular KRASG12D protein level upon antibodies-treatment, lysates of KRASG12D-transduced OVCAR3 or A427 cells, treated every 4 hours for a total of 12 hours, were used for western blotting.
Transwell and transcytosis assay for expelling of KRASG12D
KRASG12D OVCAR3 cells (2×105) were seeded in the upper chamber of a transwell system (Corning® 3413 Transwell® 6.5mm Polycarbonate, membrane Inserts Pre-Loaded in 24-Well Culture Plates, Pore Size: 0.4μm) in 200uL. In addition, 1 mL of fresh medium was added in the basal chamber. The cells were cultured in the incubator at 37°C and 5% of CO2 during 3 days to create a monolayer. KRASG12D IgA1 or KRASG12D IgG4 or non-specific human IgA antibodies were biotinylated using a Biotinylation Kit (Fast, Type B) by following the manufacture’s protocol. Then, 1μg of respective biotinylated antibody was added to the upper chamber. After overnight incubation, the supernatant from the upper and basal chambers were collected into separate microcentrifuge tubes and subjected to streptavidin pull-down using Dynabeads MyOne streptavidin T1. Briefly, 10μl of beads per sample were washed according to the manufacture’s recommendation, added to each sample and then incubated overnight at 4°C with head-over rotation. The beads were collected my magnetic separation for 5 min and washed 3 times in PBS containing 0.1% BSA. Finally, the beads were resuspended in 100uL of PBS and used for performing dot blots.
For dot blots, 10μL of beads were blotted onto a nitrocellulose membrane and dried at room temperature 1-2 hours. Then, membranes were blocked using 5% BSA in TBS-T with a gentle agitation for 1 hour at room temperature. Appropriate primary antibodies (αhuman-IgA-HRP, αhuman-IgG-HRP and αKRASG12D) were added immediately and incubated 1 hour at room temperature with gentle rotation. After 3 washes, blots were directly chemiluminescent-developed when probed with HRP conjugated antibodies αhuman-IgA/IgG antibodies, or first incubated with a secondary antibody and developed.
Multiplex immunohistochemistry
FFPE tissue microarray was immunostained using the PerkinElmer OPAL TM Automation IHC kit (Waltham, MA) on the BOND RX autostainer (Leica Biosystems, Vista, CA) and the following anti-human antibodies: IgA (Abcam, EPR5367-76, ab124716, RRID:AB_10976507; 1:1000), IgG (Abcam, EPR4421, ab109489, RRID:AB_10863040; 1:500), PIGR (Abcam, ab96196, RRID:AB_10677612; 1:100), CD3 (Thermo Fisher Scientific; Cat# MA5-14524, Clone SP7; RRID: AB_10982026, 1:100), and pan-cytokeratin (PCK, Dako, AE1/AE3, M3515, 1:200). Nuclei were stained with DAPI. Precisely, tissues were baked at 65°C for 2 hours then transferred to the BOND RX (Leica Biosystems) followed by automated deparaffinization, antigen retrieval using OPAL IHC procedure (PerkinElmer). Autofluorescence slides (negative control) were included, which use primary and secondary antibodies omitting the OPAL fluors. Slides were scanned and imaged with the PerkinElmer Vectra®3 Automated Quantitative Pathology Imaging System. Multi-layer TIFF images were exported from InForm (PerkinElmer) and loaded into HALO (Indica Labs, New Mexico) for quantitative image analysis. Each fluorescent fluorophore is assigned to a dye color and positivity thresholds were determined per marker based on published nuclear or cytoplasmic staining patterns. Tumor islets and stroma were distinguished by PCK staining.
The Cancer Genome Atlas (TCGA) data analysis
Molecular data for PIGR mRNA expression (RNA Seq V2 RSEM) from TCGA, Firehose Legacy for several human cancer types were analyzed and graphically exported from the cBio Cancer Genomics Portal (http://www.cbioportal.org/).
Quantification and statistical analysis
Unless mentioned otherwise all data are presented as mean ± SEM. Two-tailed t-tests (unpaired and paired, as appropriate) were performed between two groups, and one-way ANOVA were performed for comparisons between more than two groups, unless indicated otherwise. A significance threshold 0.05 for P values was used. Analyses were carried out in Graph Pad Prism (v.9.0).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| non-specific human IgA | Abcam | Abcam Cat#ab91025 |
| anti-β-actin antibody | Cell Signaling Technology | Cell Signaling Technology Cat# 5125, RRID:AB_1903890 |
| anti-Phospho-p44/42 MAPK(ERK1/2) antibody | Cell Signaling Technology | Cell Signaling Technology Cat# 4370, RRID:AB_2315112 |
| anti-p44/42 MAPK(ERK1/2) antibody | Cell Signaling Technology | Cell Signaling Technology Cat# 9102, RRID:AB_330744 |
| anti-RAS (G12D Mutant Specific) (D8H7) antibody | Cell Signaling Technology | Cell Signaling Technology Cat# 14429, RRID:AB_2728748 |
| anti-mCherry antibody | Rockland | Rockland Cat# 600-401-P16, RRID:AB_2614470 |
| Goat anti-Human IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientifi | Thermo Fisher Scientific Cat# A18805, RRID:AB_2535582 |
| Goat anti-Human IgA Secondary Antibody, HRP | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# A18781, RRID:AB_2535558 |
| anti-Human Ig Kappa Light Chain Antibody | R&D Systems | R&D Systems Cat#MAB10050 |
| anti-J Chain Antibody | Thermo Fisher Scientific | Thermo Fisher Scientific Cat# MA1-80527, RRID:AB_934333 |
| anti-PIGR antibody | Abcam | Abcam, ab96196, RRID:AB_10677612 |
| InVivoMAb anti-mouse CD8β (Lyt 3.2) | Bio X Cell | Bio X Cell Cat# BE0223, RRID:AB_2687706 |
| InVivoPlus rat IgG1 isotype control (anti-HRP) | Bio X Cell | Bio X Cell Cat# BE0088, RRID:AB_1107775 |
| anti-Secretory Component antibody | Abcam | Abcam Cat# ab3924, RRID:AB_2261963 |
| anti-IgA antibody | Abcam | Abcam Cat# ab124716, RRID:AB_10976507 |
| anti-IgG antibody | Abcam | Abcam Cat# ab109489, RRID:AB_10863040 |
| Mouse anti-human CD3 antibody | ThemoFisher Scientific | Thermo Fisher Scientific;Cat# MA5-14524, Clone SP7; RRID: AB_10982026 |
| Mouse anti-human PCK antibody | DAKO | Agilent Cat# M3515, Clone AE1/AE3; RRID: AB_2132885 |
| anti-RAB8A antibody | Proteintech | Proteintech Cat# 55296-1-AP, RRID:AB_10858398 |
| anti-RAB5A antibody | Proteintech | Proteintech Cat# 11947-1-AP, RRID:AB_2269388 |
| anti-RAB11A antibody | Proteintech | Proteintech Cat# 20229-1-AP, RRID:AB_10666202 |
| anti-RAB7A antibody | Proteintech | Proteintech Cat# 55469-1-AP, RRID:AB_11182831 |
| Bacterial and Virus Strains | ||
| One Shot™ TOP10 Chemically Competent E.coli | ThermoFisher Scientific | Cat# C404010 |
| Biological Samples | ||
| Lung tumor tissue microarray (TMA) | US BioMax, Inc. | LC953 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 Medium | Gibco™ | Cat# 11875093 |
| Penicillin/Streptomycin | Lonza | Cat# 17602E |
| L-glutamine | Genesee Scientific | Cat# 25509 |
| Sodium pyruvate | ThermoFisher Scientific | Cat# 11360070 |
| 0.25% Trypsin-EDTA | Gibco™ | Cat# 25200056 |
| Dulbecco's Phosphate Buffered Saline 1X | VWR Life Science | Cat# 0201190500 |
| DMEM Medium | Thermo Fisher Scientific | Cat# 11965092 |
| POLYBRENE, 10MG/ML AQUEOUS SOLUTION | Thermo Fisher Scientific | Cat# AB01643-00001 |
| Lipofectamine™ 3000 Transfection reagent | Invitrogen | Cat# L3000015 |
| DAPI | Sigma | Cat# 8417 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat#89900 |
| Protease/Phosphatase Inhibitor Cocktail (100X) | Cell Signaling | Cat#5872 |
| Thiazolyl Blue Tetrazolium Bromide, MTT | Millipore Sigma | Cat#M2128 |
| Hoechst 33342 Solution | Thermo Fisher Scientific | Cat#62249 |
| MRTX1133 | MedChemExpress | Cat#HY-134813 |
| Tris-Glycine Native Sample Buffer | Invitrogen | Cat#LC2673 |
| Novex™ WedgeWell™ 12% Tris-Glycine Gel | Invitrogen | Cat# XP00120BOX |
| Invitrogen™ Colloidal Blue Staining Kit | Invitrogen | Cat#LC6025 |
| ECL™ Prime Western Blotting System | Millipore Sigma | GERPN2232 |
| Dynabeads™ MyOne™ Streptavidin T1 | Invitrogen | Cat#65601 |
| InVivoPure pH 7.0 Dilution Buffer | BioXCell | Cat#IP0070 |
| Critical Commercial Assays | ||
| Biotinylation Kit / Biotin Conjugation Kit (Fast, Type B) - Lightning-Link® | abcam | Cat#ab201796 |
| Alexa Fluor® 488 Conjugation Kit (Fast) - Lightning-Link® | abcam | Cat#ab236553 |
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| Pierce™ Co-Immunoprecipitation Kit | Thermo Fisher Scientific | Cat#26149 |
| APC Annexin V, 100 test | Biolegend | Cat#640920 |
| LigaTrap® Human IgG Purification Kit Product Instructions | LigaTrap Technologies | Cat#LT-095KIT |
| LigaTrap® Human IgA Purification Kit, 10 x 0.10 mL | LigaTrap Technologies | Cat#LT-146KIT |
| Deposited Data | ||
| Mass spectrometry proteomics data | This paper | PRIDE:XXX |
| Experimental Models: Cell Lines | ||
| OVCAR3 | ATCC | (RRID:CVCL_0465) |
| A427 | ATCC | (RRID:CVCL_1055) |
| SK-LU-1 | ATCC | (RRID:CVCL_0629) |
| HEK293T | ATCC | (RRID:CVCL_0063) |
| NCI-H23 | ATCC | (RRID:CVCL_1547) |
| NCI-H647 | ATCC | (RRID:CVCL_1574) |
| IDH1+/+ HCT116 | Horizon Discovery | (RRID:CVCL_0291) |
| IDH1R132H/+ HCT116 | Horizon Discovery | (RRID:CVCL_0291) |
| Brpkp110 | PMID: 27803104 | N/A |
| KPMSH2KIN | This manuscript | N/A |
| PIGR-ablated OVCAR3 | PMID: 33536615 | N/A |
| KRASG12D-PamCherryOVCAR3 | This manuscript | N/A |
| KRASwt-PamCherryOVCAR3 | This manuscript | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD.129S7(B6)-Rag1tm1Mom/J | The Jackson Laboratory | RRID:IMSR_JAX:003729 |
| C57BL/6J mice | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | RRID:IMSR_JAX:005557 |
| Recombinant DNA | ||
| pLVX-IRES-Zs | GreenTakara Bio | Cat# 632187 |
| pLenti-TO/CMV-PAmCherry1-KRASWT | PMID: 26080442 | N/A |
| pLenti-TO/CMV-PAmCherry1-KRASG12D | PMID: 26080442 | N/A |
| pMD2.G | Addgene | Cat# 12259; RRID: Addgene_12259 |
| psPAX2 | Addgene | Cat# 12260; RRID: Addgene_12260 |
| J-chain encoding pcDNA3.0 vector | Addgene | RRID:Addgene_145146 |
| anti-IDH1R132HVH-IgA1 encoding PBMN-I-GFP | Genscript | N/A |
| anti-IDH1R132HVH-IgG4 encoding PBMN-I-GFP | Genscript | N/A |
| anti-IDH1R132HVL-Kappa encoding pVitro1 | Genscript | N/A |
| anti-KRASG12DVH-IgA1 encoding PBMN-I-GFP | Genscript | N/A |
| anti-KRASG12DVH-IgG4 encoding PBMN-I-GFP | Genscript | N/A |
| anti-KRASG12DVL-Kappa encoding pVitro1 | Genscript | N/A |
| Software and Algorithms | ||
| FlowJo v10.7.2 | FlowJoLLC | N/A |
| GraphPad Prism v9 | GraphPad Software Inc. | N/A |
| Adobe Photoshop 2022 | Adobe | N/A |
| Adobe Illustrator 2022 | Adobe | N/A |
HIGHLIGHTS.
Dimeric IgA undergoes bona fide transcytosis through PIGR+ carcinoma cells.
Dimeric IgA specifically neutralizes mutated oncogenes inside tumor cells.
Oncodrivers are expelled outside tumor cells through altered endosomal trafficking.
Mutation-specific dimeric IgA specifically abrogates tumor growth in vivo.
ACKNOWLEDGEMENTS
Support for Shared Resources was provided by Cancer Center Support Grants (CCSG) CA076292 to H. Lee Moffitt Cancer Center and CA014236 to Duke Cancer Institute. We are grateful to Chemical Biology, Analytic Microscopy, Advanced CLIA Tissue Imaging, Proteomics, and Flow Cytometry Shared Resources at Moffitt Cancer Center. This study was supported by the National Institutes of Health R01CA157664, R01CA278907, R01CA124515, R01CA178687, R01CA278907 and U01CA232758 to JRCG; K99CA266947 to SB; R01CA184185 to PCR; K08CA231454 to BAP; and CLIP Award from the Cancer Research Institute to JRCG. SB was supported with Junior Scientist Research Partnership Grant by Moffitt Cancer Center and DBT-Ramalingaswami Re-entry Fellowship (BT/RLF/Re-entry/48/2021) by the Department of Biotechnology, Govt. of India. PI was supported by the Mentored Investigator Grant of the Ovarian Cancer Research Alliance.
Footnotes
DECLARATION OF INTERESTS
JRCG has stock options in Compass Therapeutics, Anixa Biosciences and Alloy Therapeutics; has sponsored research with Anixa Biosciences; receives honorarium from Alloy Therapeutics; and has intellectual property with Compass Therapeutics and Anixa Biosciences; all outside the submitted work. JRCG and SB have filed a patent application for intracellular targeting of oncodrivers using dimeric IgA. B.A.P. has completed Advisory Board with AstraZeneca and has Research Support from BMS. RL: Clinical trial protocol committee – CG Oncology; Scientific advisor/consultant – BMS, Ferring, Fergene, Arquer Diagnostics.
DIVERSITY STATEMENT
CMA, RAC, LULB, BAP, TJC, PCR and JRCG identify themselves as “Hispanic”
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Prior IA, Lewis PD, and Mattos C (2012). A comprehensive survey of Ras mutations in cancer. Cancer Res 72, 2457–2467. 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang L, Guo Z, Wang F, and Fu L (2021). KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther 6, 386. 10.1038/s41392-021-00780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Wagle NS, and Jemal A (2023). Cancer statistics, 2023. CA Cancer J Clin 73, 17–48. 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 4.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. (2020). KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 383, 1207–1217. 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, Dahlke JR, Fell JB, Fischer JP, Gunn RJ, et al. (2022). Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J Med Chem 65, 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- 6.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. (2010). Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 28, 3167–3175. 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al. (2021). Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 384, 2382–2393. 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaurio RA, Anadon CM, Lee Costich T, Payne KK, Biswas S, Harro CM, Moran C, Ortiz AC, Cortina C, Rigolizzo KE, et al. (2022). TGF-beta-mediated silencing of genomic organizer SATB1 promotes Tfh cell differentiation and formation of intra-tumoral tertiary lymphoid structures. Immunity 55, 115–128 e119. 10.1016/j.immuni.2021.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA, Costich TL, Moran C, Harro CM, Rigolizzo KE, et al. (2021). IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature 591, 464–470. 10.1038/s41586-020-03144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conejo-Garcia JR, Biswas S, and Chaurio R (2020). Humoral immune responses: Unsung heroes of the war on cancer. Semin Immunol 49, 101419. 10.1016/j.smim.2020.101419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelhard V, Conejo-Garcia JR, Ahmed R, Nelson BH, Willard-Gallo K, Bruno TC, and Fridman WH (2021). B cells and cancer. Cancer Cell. 10.1016/j.ccell.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Mandal G, Biswas S, Anadon CM, Yu X, Gatenbee CD, Prabhakaran S, Payne KK, Chaurio RA, Martin A, Innamarato P, et al. (2022). IgA-Dominated Humoral Immune Responses Govern Patients' Outcome in Endometrial Cancer. Cancer Res 82, 859–871. 10.1158/0008-5472.CAN-21-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conejo-Garcia JR, Biswas S, Chaurio R, and Rodriguez PC (2023). Neglected no more: B cell-mediated anti-tumor immunity. Semin Immunol 65, 101707. 10.1016/j.smim.2022.101707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strugnell RA (2022). When secretion turns into excretion - the different roles of IgA. Front Immunol 13, 1076312. 10.3389/fimmu.2022.1076312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei H, and Wang JY (2021). Role of Polymeric Immunoglobulin Receptor in IgA and IgM Transcytosis. Int J Mol Sci 22. 10.3390/ijms22052284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harjes U (2021). IgA strikes twice in ovarian cancer. Nat Rev Cancer 21, 215. 10.1038/s41568-021-00342-4. [DOI] [PubMed] [Google Scholar]
- 17.Osorio JC, and Zamarin D (2022). Beyond T Cells: IgA Incites Immune Recognition in Endometrial Cancer. Cancer Res 82, 766–768. 10.1158/0008-5472.CAN-21-4385. [DOI] [PubMed] [Google Scholar]
- 18.Fan X, Zhou D, Zhao B, Sha H, Li M, Li X, Yang J, and Yan H (2021). Rab11-FIP1 and Rab11-FIP5 Regulate pIgR/pIgA Transcytosis through TRIM21-Mediated Polyubiquitination. Int J Mol Sci 22. 10.3390/ijms221910466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, Kim D, Li C, de Stanchina E, Mazutis L, et al. (2020). Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 577, 421–425. 10.1038/s41586-019-1884-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sterlin D, and Gorochov G (2021). When Therapeutic IgA Antibodies Might Come of Age. Pharmacology 106, 9–19. 10.1159/000510251. [DOI] [PubMed] [Google Scholar]
- 21.Woof JM, and Russell MW (2011). Structure and function relationships in IgA. Mucosal Immunol 4, 590–597. 10.1038/mi.2011.39. [DOI] [PubMed] [Google Scholar]
- 22.Casswall TH, Hammarstrom L, Veress B, Nord CE, Bogstedt A, Brockstedt U, and Dahlstrom KA (1996). Oral IgA-IgG treatment of chronic non-specific diarrhoea in infants and children. Acta Paediatr 85, 1126–1128. 10.1111/j.1651-2227.1996.tb14231.x. [DOI] [PubMed] [Google Scholar]
- 23.Welte T, Dellinger RP, Ebelt H, Ferrer M, Opal SM, Singer M, Vincent JL, Werdan K, Martin-Loeches I, Almirall J, et al. (2018). Efficacy and safety of trimodulin, a novel polyclonal antibody preparation, in patients with severe community-acquired pneumonia: a randomized, placebo-controlled, double-blind, multicenter, phase II trial (CIGMA study). Intensive Care Med 44, 438–448. 10.1007/s00134-018-5143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ackermann LW, and Denning GM (2004). Nuclear factor-kappaB contributes to interleukin-4- and interferon-dependent polymeric immunoglobulin receptor expression in human intestinal epithelial cells. Immunology 111, 75–85. 10.1111/j.1365-2567.2004.01773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin SM, Choi DK, Jung K, Bae J, Kim JS, Park SW, Song KH, and Kim YS (2017). Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat Commun 8, 15090. 10.1038/ncomms15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nan X, Tamguney TM, Collisson EA, Lin LJ, Pitt C, Galeas J, Lewis S, Gray JW, McCormick F, and Chu S (2015). Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc Natl Acad Sci U S A 112, 7996–8001. 10.1073/pnas.1509123112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oztan A, Rondanino C, and Apodaca G (2008). Transcytosis of polymeric immunoglobulin a in polarized Madin-Darby canine kidney cells. Methods Mol Biol 440, 157–170. 10.1007/978-1-59745-178-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldenring JR (2015). Recycling endosomes. Curr Opin Cell Biol 35, 117–122. 10.1016/j.ceb.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelabert-Baldrich M, Soriano-Castell D, Calvo M, Lu A, Viña-Vilaseca A, Rentero C, Pol A, Grinstein S, Enrich C, and Tebar F (2014). Dynamics of KRas on endosomes: involvement of acidic phospholipids in its association. Faseb j 28, 3023–3037. 10.1096/fj.13-241158. [DOI] [PubMed] [Google Scholar]
- 30.Schmick M, Kraemer A, and Bastiaens PI (2015). Ras moves to stay in place. Trends in cell biology 25, 190–197. 10.1016/j.tcb.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Allegrezza MJ, Rutkowski MR, Stephen TL, Svoronos N, Perales-Puchalt A, Nguyen JM, Payne KK, Singhal S, Eruslanov EB, Tchou J, and Conejo-Garcia JR (2016). Trametinib Drives T-cell-Dependent Control of KRAS-Mutated Tumors by Inhibiting Pathological Myelopoiesis. Cancer Res 76, 6253–6265. 10.1158/0008-5472.CAN-16-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capper D, Weissert S, Balss J, Habel A, Meyer J, Jäger D, Ackermann U, Tessmer C, Korshunov A, Zentgraf H, et al. (2010). Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol 20, 245–254. 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calvert AE, Chalastanis A, Wu Y, Hurley LA, Kouri FM, Bi Y, Kachman M, May JL, Bartom E, Hua Y, et al. (2017). Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep 19, 1858–1873. 10.1016/j.celrep.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de la Fuente MI (2022). Targeting IDH1/IDH2 mutations in gliomas. Curr Opin Neurol 35, 787–793. 10.1097/WCO.0000000000001111. [DOI] [PubMed] [Google Scholar]
- 35.Li-Chang HH, Kasaian K, Ng Y, Lum A, Kong E, Lim H, Jones SJ, Huntsman DG, Schaeffer DF, and Yip S (2015). Retrospective review using targeted deep sequencing reveals mutational differences between gastroesophageal junction and gastric carcinomas. BMC Cancer 15, 32. 10.1186/s12885-015-1021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Tseng LH, Parini V, Lokhandwala PM, Pallavajjala A, Rodriguez E, Xian R, Chen L, Gocke CD, Eshleman JR, and Lin MT (2021). IDH1 and IDH2 Mutations in Colorectal Cancers. Am J Clin Pathol 156, 777–786. 10.1093/ajcp/aqab023. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez EF, De Marchi F, Lokhandwala PM, Belchis D, Xian R, Gocke CD, Eshleman JR, Illei P, and Li MT (2020). IDH1 and IDH2 mutations in lung adenocarcinomas: Evidences of subclonal evolution. Cancer Med 9, 4386–4394. 10.1002/cam4.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pascal V, Laffleur B, Debin A, Cuvillier A, van Egmond M, Drocourt D, Imbertie L, Pangault C, Tarte K, Tiraby G, and Cogne M (2012). Anti-CD20 IgA can protect mice against lymphoma development: evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica 97, 1686–1694. 10.3324/haematol.2011.061408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leusen JH (2015). IgA as therapeutic antibody. Mol Immunol 68, 35–39. 10.1016/j.molimm.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 40.van Tetering G, Evers M, Chan C, Stip M, and Leusen J (2020). Fc Engineering Strategies to Advance IgA Antibodies as Therapeutic Agents. Antibodies (Basel) 9. 10.3390/antib9040070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer S, Nederend M, Jansen JH, Reiding KR, Jacobino SR, Meeldijk J, Bovenschen N, Wuhrer M, Valerius T, Ubink R, et al. (2016). Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. MAbs 8, 87–98. 10.1080/19420862.2015.1106658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards A, Baranova D, and Mantis NJ (2022). The prospect of orally administered monoclonal secretory IgA (SIgA) antibodies to prevent enteric bacterial infections. Human vaccines & immunotherapeutics 18, 1964317. 10.1080/21645515.2021.1964317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA, Costich TL, Moran C, Harro CM, Rigolizzo KE, et al. (2021). IgA transcytosis and antigen recognition govern ovarian cancer immunity. Nature. 10.1038/s41586-020-03144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandal G, Biswas S, Anadon CM, Yu X, Gatenbee CD, Prabhakaran S, Payne KK, Chaurio RA, Martin A, Innamarato P, et al. (2021). IgA-dominated humoral immune responses govern patients' outcome in endometrial cancer. Cancer Res. 10.1158/0008-5472.CAN-21-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD042914 and PXD044426. This paper does not report original code. The datasets generated during the current study are available from the lead contact upon reasonable request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.