

Abstract
Cells devoid of cytosolic superoxide dismutase (SOD) suffer enzyme inactivation, growth deficiencies, and DNA damage. It has been proposed that the scant superoxide (O2−) generated by aerobic metabolism harms even cells that contain abundant SOD. However, this idea has been difficult to test. To determine the amount of O2− that is needed to cause these defects, we modulated the O2− concentration inside Escherichia coli by controlling the expression of SOD. An increase in O2− of more than twofold above wild-type levels substantially diminished the activity of labile dehydratases, an increase in O2− of any more than fourfold measurably impaired growth, and a fivefold increase in O2− sensitized cells to DNA damage. These results indicate that E. coli constitutively synthesizes just enough SOD to defend biomolecules against endogenous O2− so that modest increases in O2− concentration diminish cell fitness. This conclusion is in excellent agreement with quantitative predictions based upon previously determined rates of intracellular O2− production, O2− dismutation, dehydratase inactivation, and enzyme repair. The vulnerability of bacteria to increased intracellular O2− explains the widespread use of superoxide-producing drugs as bactericidal weapons in nature. E. coli responds to such drugs by inducing the SoxRS regulon, which positively regulates synthesis of SOD and other defensive proteins. However, even toxic amounts of endogenous O2− did not activate SoxR, and SoxR activation by paraquat was not at all inhibited by excess SOD. Therefore, in responding to redox-cycling drugs, SoxR senses some signal other than O2−.
The discovery of superoxide dismutase (SOD) was accidental (51), and it has been a long road to an understanding of its role in cell physiology. Its wide distribution among aerobic organisms (50) suggested both that superoxide (O2−) is formed inside all cells that grow in air and that O2− is toxic. This hypothesis has been extended to predict that enough O2− might evade SOD to generate chronic oxidative damage, the gradual accumulation of which may contribute to age-associated pathologies (2, 7, 11, 15). However, the physiological role of SOD was actively debated for many years because it was difficult to discern intracellular sources and targets of O2−.
The construction of Escherichia coli mutants lacking the cytosolic SODs gave the first insight into the significance of intracellular O2− (6). These mutants exhibit several defects when grown aerobically: they are auxotrophic for branched-chain, aromatic, and sulfur-containing amino acids, and they catabolize only fermentable carbon sources (6, 35). The SOD− mutants also show high rates of spontaneous mutagenesis (12). Severe phenotypes of SOD− mutants, ranging from growth defects to decreased fertility and life spans, were subsequently observed in higher organisms as well (56, 65).
Analysis of the E. coli mutants illuminated the molecular targets of O2−. The branched-chain auxotrophy and the requirement for a fermentable carbon source arise because O2− inactivates dihydroxyacid dehydratase (13, 41), aconitase (20), and fumarases A and B (14, 46). These dehydratases, as well as 6-phosphogluconate dehydratase of E. coli (18) and similar enzymes of other organisms (14, 16, 31, 32), each contain a distinctive [4Fe-4S] cluster that provides a local positive charge to help bind and dehydrate the substrate. In the absence of substrate, O2− can oxidize and thereby destabilize the exposed cluster. Iron then dissociates, resulting in the loss of enzyme activity. Reactivation of the clusters occurs in vivo, and it is likely that labile enzymes are repeatedly damaged and repaired during oxidative stress, so that the steady-state activity is a balance of the two processes. When iron from the damaged cluster spills into the cytosol, it is available to participate in Fenton chemistry (38, 47) and catalyzes oxidative damage to DNA (40). This causes the high rate of mutagenesis that characterizes SOD mutants.
An important question is whether the O2− production during aerobic metabolism is sufficient to cause damage in a cell that contains SOD. Unfortunately, the intracellular O2− concentration of either SOD-proficient or SOD-deficient cells is below detection by current techniques capable of measuring O2− (34, 43). Flavoproteins of the electron transport chain generate an estimated 3 μM O2−/s during exponential growth (34). Yet E. coli contains ca. 3,000 U of SOD per ml, enough to restrict the calculated steady-state O2− concentration to 10−10 M, or about 0.1 molecule per cell. In fact, the concentration of SOD exceeds that of O2− in vivo by about 100,000 to 1, which is an unprecedented relation between enzyme and substrate (1). Thus, it may seem unlikely that such large amounts of SOD are necessary to prevent damage from endogenous O2− sources.
An alternative is that E. coli synthesizes so much SOD solely as a preemptive defense against the O2− that is produced during exposures to redox-cycling drugs, many of which are made by other microorganisms as a means to defend their habitat. Redox-cycling drugs are able to enter bacterial cells and generate O2− through interactions with flavoproteins. The rate of O2− production by redox-cycling drugs can approach the rate of respiration, exceeding the normal rate of endogenous O2− formation by orders of magnitude (28, 33). E. coli mutants that are deficient in SOD activity are hypersensitive to these drugs (6, 33).
SOD synthesis is positively regulated by the SoxRS regulon (25, 64), which responds to redox-cycling drugs. This raised the question of whether the signal to which SoxRS responds is O2− itself or some other effect of the drug (18, 22, 44, 48, 54). If the signal were O2− itself, then it is possible that the SoxRS regulon also responds to metabolically generated O2− levels, adjusting SOD synthesis in order to keep the intracellular O2− concentration within a narrow range. Such an adaptive mechanism has been proposed for the OxyR-dependent induction of catalase in response to endogenous hydrogen peroxide (23).
To determine whether abundant SOD is needed to defend cells against damage by endogenous O2−, we constructed a strain in which the cytosolic SOD activity could be modulated. The extent of O2− damage was then assessed over a range of SOD concentrations. The analysis of our results will lead us to conclusions regarding the ability of cells to tolerate increases in O2−.
MATERIALS AND METHODS
Reagents.
Manganese (II) chloride tetrahydrate was obtained from Aldrich Chemical Company, Inc., Milwaukee, Wis. Beef liver catalase was purchased from Boehringer Mannheim, Indianapolis, Ind. Coomassie protein assay reagent was obtained from Pierce, Rockford, Ill. All other chemicals were purchased from Sigma Chemical Co., St. Louis, Mo. Water used in all reagents was from the house deionized system and was further purified by a Labconco Water Pro PS system to minimize metal contamination.
Strain construction.
The strains that were used in this study are shown in Table 1. During strain construction, the introduction of chromosomal null mutations was achieved by P1 transduction with selection for linked antibiotic resistance markers.
TABLE 1.
Strains, plasmids, and phage
| Strain, plasmid, or phage | Relevant genotype | Source or reference |
|---|---|---|
| Strains | ||
| AB1157 | F−thr-1 leuB6 proA2 his-4 thi-1 argE2 lacY1 galK2 rpsL surE44 ara-14 xyl-15 mtl-1 tsx-33 | 37 |
| AS240 | Same as that for AB1157 plus (sodA::Mu d PR13)25 (sodB-kan)1-Δ2 zdg-299::Tn10 λAS2 | This study |
| AS241 | Same as that for AS240 plus pCKR101 | This study |
| AS290 | Same as that for AB1157 plus pCKR101 | This study |
| AS291 | Same as that for JI132 plus pCKR101 | This study |
| AS356 | Same as that for JI131 plus pCKR101 | This study |
| AS357 | Same as that for JI130 plus pCKR101 | This study |
| AS358 | Same as that for TN530 plus (sodA::Mu d PR13)25 (sodB-kan)1-Δ2 | This study |
| AS360 | Same as that for AS358 plus pDT1.16 | This study |
| AS364 | Same as that for TN530 plus pDT1.16 | This study |
| AS370 | Same as that for AS240 plus pMS421 | This study |
| AS372 | Same as that for JI131 plus pMS421 | This study |
| AS374 | Same as that for AB1157 plus pMS421 | This study |
| AS376 | Same as that for JI130 plus pMS21 | This study |
| AS395 | Same as that for TN530 plus (sodA::Mu d PR13)25 | This study |
| AS396 | Same as that for AS395 plus pKK1 | This study |
| JI130 | Same as that for AB1157 plus (sodA::Mu d PR13)25 | 37 |
| JI131 | Same as that for AB1157 plus (sodB-kan)1-Δ2 | 37 |
| JI132 | Same as that for AB1157 plus (sodA::Mu d PR13)25 (sodB-kan)1-Δ2 | 37 |
| TN530 | λ soxS::lacZ ΔsoxR ΔlacU169 rpsL | 55 |
| Plasmids | ||
| pCKR101 | pBR328 derivative containing lacIq | Jeff Gardner |
| pMS421 | pSC101 derivative containing lacIq | 24 |
| pDT1.16 | pBR322 derivative containing tac promoter fused to sodA | 63 |
| pKK1 | pBR328 derivative containing sodB | 38 |
| Phage λAS2 | lacA lacY′ Ptac-sodA kan | This study |
To construct a strain in which the expression of SOD could be modulated, the EcoRI/MscI fragment from pDT1.16 (63) containing sodA under control of the tac promoter was cloned into the vector pRS551 (61), which had been digested with EcoRI and SnaBI. The resultant plasmid was designated pAS1. The regions of pAS1 which flank the sodA insert have homology to λRS45 and permitted marker exchange onto the λ phage (61). The insert on this phage is preceded by four terminator sequences, preventing transcriptional readthrough from upstream genes. Single-copy lysogens were recovered in E. coli by selection for phage-encoded kanamycin resistance. Null mutations of sodA and sodB were introduced into the lysogen by transduction. Thus, synthesis of cytosolic SOD was under the control of the tac promoter and responded to the addition of isopropyl-β-d-thiogalactopyranoside (IPTG). The presence of the lacY1 mutation in all these strains circumvented the difficulties that occur when a titrating molecule induces synthesis of its own transporter (53, 60).
Media and cell growth.
Defined media contained minimal A salts and either 0.2% glucose, 0.2% gluconate, or 40 mM fumarate as a carbon source. Histidine, leucine, threonine, arginine, and proline were present in all media to satisfy the genetic auxotrophies of AB1157 derivatives; supplemented media additionally contained either a 0.5 mM concentration of the other 15 amino acids or 0.25% Casamino Acids. Luria broth (LB) was used for some experiments. pCKR101 was maintained with 50 μg of ampicillin per ml, and pMS421 was maintained with 100 μg of spectinomycin per ml and 50 μg of streptomycin per ml.
The same media were used for preliminary anaerobic cultures as were used in the aerobic portions of the experiments, except when cultures were grown in fumarate medium. For the experiments with fumarate medium, cells were first cultured anaerobically and then were cultured aerobically in minimal glucose medium before being washed and diluted into fumarate medium. Overnight cultures of SOD-deficient strains were always incubated in a Coy anaerobic chamber (85% N2, 10% H2, 5% CO2) to prevent the accumulation of phenotypically suppressed mutants. The stationary-phase cultures were typically diluted to an optical density at 600 nm (OD600) of 0.010 in anaerobic medium and cultured for at least four generations before being diluted again to an OD600 of 0.010 in a series of flasks containing 300 to 500 ml of aerobic media. The flasks were supplemented with a range of concentrations of IPTG (typically 5 to 50 μM) and MnCl2 (0.1 to 1 μM) to vary manganese SOD (MnSOD) activity. Cell density was monitored by absorbance during growth to an OD600 of 0.1 to 0.2. At an OD600 of 0.10, portions of the cultures were harvested for SOD and enzyme assays. The growth rates that are reported were determined by using density measurements made before and after some cells had been harvested for SOD assays. Some imprecision in the correlation between growth rate and SOD activity may have occurred due to a slight drift in SOD content during the period of growth measurements.
Assays of labile enzymes.
The lysis buffer used for aconitase assays contained 50 mM Tris (pH 7.4), 0.6 mM MnCl2, and 20 μM fluorocitrate; that for 6-phosphogluconate dehydratase assays contained 50 mM Tris buffer (pH 7.6); and that for fumarase assays contained 50 mM potassium phosphate buffer (pH 7.8). Exponentially growing cultures were centrifuged at 15,000 × g for 3 min, resuspended in 1 ml of buffer, and lysed by either sonication or passage through a French pressure cell without any difference in the results. The lysates were clarified by centrifugation in a Fisher Scientific microcentrifuge for 1 min at 12,000 rpm and immediately frozen in a dry ice-ethanol bath to avoid loss of enzyme activity before assay. After the lysates were thawed, aconitase assays and 6-phosphogluconate assays were performed (19, 20). Fumarase was assayed by monitoring the conversion of 50 mM l-malate to fumarate at an OD of 250 nm (ɛ[fumarate] = 1.62 mM−1 cm−1) in sodium phosphate buffer (pH 7.3). Superoxide-resistant fumarase C activity was measured after fumarase A activity had been inactivated by O2−. To this end, 1 ml of diluted extract was incubated with 2.8 mU of bovine xanthine oxidase and 55 μM xanthine for 15 min at room temperature. l-Malate was then added, and the sample was assayed.
Reactivation of aconitase was achieved in cell suspensions prior to lysis. Tetracycline (100 μg/ml) was added to aerobic cultures to block protein synthesis, the cells were centrifuged, and the cell pellets were transferred into the anaerobic chamber. There, the pellets were resuspended in anaerobic lysis buffer containing 100 μg of tetracycline per ml. At time points, aliquots of the cell suspension were removed from the chamber, lysed, clarified, and frozen as detailed above. The assays were performed as described previously (20). Control experiments confirmed that inactive enzymes were not detectably reactivated during their few moments in anaerobic cell pellets nor were active enzymes inactivated during storage prior to assay (data not shown).
Other methods.
Induction of SoxRS was achieved by the addition of the indicated concentration of paraquat followed by a 45-min incubation at 37°C. Assays for β-galactosidase were done as described previously (52). SOD activity was assayed after overnight dialysis against 50 mM potassium phosphate buffer (pH 7.8) containing 1 mM EDTA (51). Protein assays were performed with chicken egg albumin as a protein standard and Coomassie protein assay reagent.
Sensitivity to killing by H2O2 was determined by using cultures growing exponentially in glucose-Casamino Acids medium (OD600 = 0.10). Hydrogen peroxide was added to a final concentration of 2.5 mM, and the cultures were shaken for 10 min at 37°C. Dilutions were made into LB containing catalase (130 U/ml) and plated. Colonies were counted after a 16-h incubation, and the rate of killing (k) was calculated by the equation k = 1/t × ln(N0/N1), where N0 and N1 are the initial and final numbers of viable cells, respectively, and t is time.
Measurements of free iron were made by electron paramagnetic resonance (EPR) analysis (40), using exponentially growing cultures (OD600 = 0.10) in 2 liters of aerobic glucose-Casamino Acids medium. Iron levels were quantitated by normalizing the amplitude of the iron signal to iron standards, and internal concentrations were calculated by using the intracellular volume (34).
RESULTS
Construction of the experimental strain.
To determine the amount of cytosolic SOD that is sufficient to prevent oxidative damage, we constructed an E. coli strain in which the amount of SOD could be modulated. The major challenge which we anticipated was the need to minimize SOD expression sufficiently so that phenotypes of oxidative injury could emerge, and therefore it was important to reduce the construct to a single copy. We subcloned a Ptac-sodA insert from pDT1.16 onto plasmid pRS551 and allowed it to recombine onto a lambda phage (see Material and Methods). The lambda phage was then used to infect a SOD-proficient strain, and a lysogen in which the lambda had stably integrated into the chromosome was isolated. Chromosomal null mutations in sodA and sodB were introduced by P1 transduction. The resulting lysogen was then transformed with either pCKR101 or pMS421, medium-copy-number plasmids that overexpressed the lac repressor and further reduced the basal expression of the sodA gene to approximately 3% of that of the wild type.
In this background, MnSOD, the sole cytosolic SOD, could be induced by the administration of IPTG in the presence of Mn2+. The concentration of manganese added was kept below 1 μM to prevent SOD-independent dismutation of O2− (8), which became significant at higher Mn2+ concentrations (data not shown). The level of maximum induction with IPTG and this manganese supplementation was roughly 30% the activity of a wild-type strain; however, since phenotypes are apparent only at lower SOD titers (see below), this level of expression was sufficient for this work. Strains that retained either the chromosomal sodA or sodB genes were also constructed. In most media, these single mutants contained >30% of wild-type SOD activity and exhibited wild-type behavior. The lone exception was the sodB mutant grown in fumarate, because the sodA gene is poorly expressed in fumarate medium (see below).
Inhibition of growth by endogenous superoxide.
Strains of E. coli that lack both cytosolic SODs cannot grow in minimal medium without amino acid supplements (6, 35). Cells containing 25% of the wild-type SOD level grew at 75% of the rate measured for wild-type cells in unsupplemented medium (Fig. 1A). Cells containing less than 10% grew at rates that approached zero.
FIG. 1.

Effect of SOD activity on growth rate in minimal medium. Cultures of strains AS290 (SOD+) (○), AS356 (sodB) (◊), AS291 (sodA sodB) (⊞), and AS241 (sodA sodB λ Ptac-sodA) (□) were grown in unsupplemented (A) or amino acid-supplemented (B) minimal A medium containing glucose. SOD activity in AS241 was modulated with a range of both IPTG and MnCl2 concentrations as described in Materials and Methods. The data shown for growth in unsupplemented minimal medium is a combination of the data from two separate experiments, with the relative SOD activity normalized to 9.2 U/mg, the average specific activity found in wild-type cells. SOD activities for the cultures grown in supplemented minimal medium were normalized to the specific activity of the wild-type culture, 5.8 U/mg.
SOD mutants require branched-chain, aromatic, and sulfurous amino acids. At limiting SOD concentrations, the separate addition of these amino acid groups was unable to restore rapid growth (data not shown). However, when media were supplemented with all 20 amino acids, cultures grew as rapidly as wild-type cells unless SOD levels were reduced to less than 10% of the wild-type level, and even then growth continued at approximately 60% of the rate of wild-type cells (Fig. 1B). Therefore, the decreased growth rate of the unsupplemented cells occurred primarily because of damage to multiple amino acid biosynthetic pathways, whereas a less-sensitive, unknown target limited the growth rate of supplemented cells.
Inactivation of [4Fe-4S] dehydratases.
The effects of O2− can be more precisely observed by measuring the activities of the enzymes that it directly damages. We assayed aconitase and 6-phosphogluconate dehydratase over a range of SOD levels. Cultures were grown with amino acid supplements and fermentable carbon sources so that low SOD activities had only minor effects on growth rates and presumably on the rates of metabolic O2− production.
The data are shown in Fig. 2. Both aconitase and 6-phosphogluconate dehydratase activities were progressively lower in cells containing less than 40% of wild-type SOD activity. At very low SOD activities, the 6-phosphogluconate dehydratase activity approached zero, whereas about 10% of the wild-type aconitase activity remained active. Gruer et al. have reported that E. coli contains two or three aconitase isozymes (26, 27); we are investigating the possibility that a minor isozyme is resistant to O2−.
FIG. 2.

Effect of SOD activity on aconitase and 6-phosphogluconate dehydratase activities. Cells for aconitase (A) and 6-phosphogluconate dehydratase (B) assays were grown in minimal medium containing Casamino Acids and either glucose or gluconate, respectively. SOD activities were modulated and extracts were prepared as detailed in Materials and Methods. The relative SOD activity is normalized to wild-type specific activity. The wild-type SOD activity in glucose medium was 6.1 U/mg; in gluconate medium, it was 9.8 U/mg. For identification of symbols, see the legend to Fig. 1.
It seemed possible, albeit unlikely, that the low dehydratase activities in SOD-deficient cells reflected a lower rate of synthesis rather than enzyme damage. To determine whether the SOD-deficient cells contained inactivated dehydratases, we added inhibitors of protein synthesis to a fraction of the SOD-deficient cells and incubated them under anaerobic conditions prior to harvesting. The cluster repair processes that are active during such an incubation restored the aconitase activity to that of a wild-type strain within 10 min (data not shown). Thus, the low dehydratase activities of SOD-limited cells accurately reflect enzyme damage.
These results indicate that E. coli requires nearly its normal complement of SOD to prevent growth deficiencies from endogenous O2−. Furthermore, it is possible that the negative effect of O2− may have been underestimated due to a reduction in the metabolic O2− production as the growth rate declined.
Effect of superoxide on DNA damage and free iron.
The oxidation of [4Fe-4S] dehydratase clusters by O2− causes the release of iron into the cell cytoplasm. This free iron can catalyze the Fenton reaction and thereby rapidly generate DNA damage in SOD− mutants. The vulnerability to DNA damage is reflected by a high rate of killing when SOD-deficient cells are exposed to exogenous hydrogen peroxide (37). Sensitivity to hydrogen peroxide increased greatly when cells contained less than 15% the wild-type level of SOD (Fig. 3A). This dosimetry agreed with the sensitivity of the dehydratases to decreases in SOD (Fig. 2).
FIG. 3.

Sensitivity to DNA damage and detection of internal free iron. (A) Rates of killing were determined during exposure to 2.5 mM hydrogen peroxide. Cell death is due to DNA damage (36). Cultures were grown and challenged in minimal medium containing Casamino Acids and glucose. A value of 8.0 U of specific SOD activity per mg for the SOD+ strain was used to normalize the SOD activities. (B) The EPR spectra are shown for a SOD+ strain, a sodA sodB (SOD−) strain, and a sodA sodB strain containing λ Ptac-sodA (SOD+/−) without further induction by IPTG. The peak at g equal to 4.3 is from Fe3+ complexed to deferoxamine mesylate, a chelator of free iron. For identification of symbols, see the legend to Fig. 1.
To verify that this heightened sensitivity reflected a change in iron homeostasis, we used a whole-cell EPR technique to measure the internal concentration of free iron (Fig. 3B). An SOD-proficient strain contained only a modest amount of free iron (6 μM), while a strain devoid of SOD contained almost 10-fold more (52 μM), consistent with a previous report (40). Cells that expressed 6% of the wild-type level of SOD activity had substantially more free iron than did SOD-proficient cells (25 μM), which readily accounts for the DNA damage data (Fig. 3A).
It is striking that the rate of DNA damage was not reduced to zero as SOD levels increased. Apparently, even unstressed E. coli contains a pool of free iron, and so it is only when the flux of iron from damaged dehydratases is large that O2− has an appreciable effect on the pool size. Because of that basal pool of iron, E. coli could not fully avoid oxidative DNA damage by producing higher amounts of SOD. In fact, SOD overproduction has been shown to have no effect upon oxidative DNA damage in a wild-type cell (38).
The SoxRS regulon is induced by drugs but not by superoxide.
Since the cell makes just enough SOD to protect itself from metabolic O2−, it seemed plausible that the SOD concentration might be continually adjusted in response to O2− levels. The SoxRS regulon, which controls MnSOD synthesis, has been proposed to respond directly to O2− and is efficiently induced by redox-cycling drugs that generate O2−. The activated SoxR protein induces synthesis of SoxS (55, 67), which in turn positively regulates a set of genes whose products may be protective, including sodA. If SoxRS were to modulate SOD in response to O2−, then the regulon should be highly induced in SOD-deficient strains. Using strains containing soxS::lacZ fusions on a lambda prophage, we assayed β-galactosidase to monitor the degree of SoxRS induction in wild-type strains and in SOD mutants (Fig. 4). The striking result was that soxS was not induced in SOD mutants, despite the fact that they contained enough O2− to completely inactivate metabolic pathways. Glucose-6-phosphate dehydrogenase, a member of the SoxRS regulon, was also minimally induced in SOD mutants (data not shown). In contrast, soxS was highly expressed when cells were exposed to paraquat (Fig. 4).
FIG. 4.

Endogenous O2− is a poor inducer of SoxRS. SOD-proficient and SOD-deficient lysogens of a λ containing a soxS::lacZ fusion were assayed for β-galactosidase activity in minimal medium containing Casamino Acids and glucose (shaded bars) and in LB medium (open bars). Cultures of each strain were grown to an OD near 0.050. The cultures were each split into two flasks, paraquat was added to one to a final concentration of 10 μM, and all cultures were incubated for an additional 45 min. Cells grown in LB were washed and resuspended in minimal medium containing Casamino Acids and glucose prior to assay. The data shown have been normalized to the β-galactosidase activity present in the uninduced cultures. The error bars represent the ranges of activity measured for four independent cultures.
We considered the possibility that O2− had failed to induce the regulon because the SOD mutants had been necessarily cultured in medium that did not require the function of superoxide-sensitive enzymes. That is, SoxRS might only be activated when the enzymes which it defends are essential for growth. Therefore, we monitored the expression of fumarase C, a member of the regulon (45), in cells that contained limiting amounts of SOD during growth in fumarate medium. Fumarase function is essential for the catabolism of fumarate. Fumarase A, the major fumarase activity of aerobic cells (66), utilizes a [4Fe-4S] cluster that O2− rapidly inactivates (14, 46). The gene encoding fumarase B is expressed only under anaerobic conditions; we observed that a fumA fumC mutant had less than 3% of the normal fumarase activity when grown aerobically (data not shown). Fumarase C is a minor isozyme that has no iron-sulfur cluster. It is resistant to O2− and it is induced by SoxRS.
The growth rate in fumarate medium declined when cells contained less than 20% of the wild-type SOD activity (Fig. 5A). Fumarase A activity fell in parallel (Fig. 5B). Importantly, as fumarase A activity declined, fumarase C activity increased only slightly. Whether SoxRS activation is responsible for the modest induction of fumarase C was not tested. When SOD activity was limited to 10% of the wild-type level, fumarase A was 80% inactive and fumarase C was induced only twofold. This was not enough to restore total fumarase activity or to permit normal growth in the medium. In contrast, paraquat was a much more effective inducer: at a dose that generated enough O2− (in a SOD-proficient strain) to inactivate fumarase A by 80%, it induced soxS expression 11-fold (Table 2). This induction, which is mediated by SoxRS (45), was sufficient to maintain normal total fumarase activity. The implication was that SoxR does not sense toxic levels of O2− but does sense some other signal that is provided by the paraquat.
FIG. 5.

Growth rates (A) and fumarase activities (B) of cultures with modulated SOD activity. Cultures of strains AS370 (sodA sodB λ Ptac-sodA) (□, ▪), AS372 (SOD+) (○, •), AS374 (sodB) (◊, ⧫), and AS376 (sodA) (▵, ▴) were grown aerobically in unsupplemented minimal medium containing fumarate. The relative SOD activity was determined by normalizing to a wild-type activity of 5.2 U/mg.
TABLE 2.
Paraquat induction of fumarase C compensates for the inactivation of fumarase A
| Cell culture | Activity (U/mg)
|
||
|---|---|---|---|
| Fumarase A | Fumarase C | Total fumarase | |
| SOD+ | 1.27 | 0.09 | 1.36 |
| SOD+ + 10 μM paraquat | 0.24 | 0.94 | 1.18 |
| 10% of Wild-type SOD | 0.20 | 0.19 | 0.39 |
To test this idea more directly, we examined whether excess SOD could inhibit soxS induction by paraquat. The soxS::lacZ fusion was placed into a strain expressing only sodB (to avoid the complications of sodA induction) and then that strain was transformed with a plasmid bearing sodB. There was no difference in the induction profiles of the two strains despite a 20-fold difference in SOD activities (Fig. 6). If O2− were the inducer, one would have expected the half-inducing dose to be 20 times higher in the overproducer. Similar paraquat treatment was unable to induce soxS in a soxR deletion mutant (data not shown), confirming that in paraquat-treated cells soxS induction responds exclusively to SoxR. These experiments collectively demonstrated that O2− is neither sufficient nor necessary for SoxRS induction.
FIG. 6.

Paraquat induction of SoxRS does not require O2−. Strains AS395 (sodA λsoxS::lacZ) (◊) and AS396 (sodA λsoxS::lacZ psodB) (▪) were grown aerobically in LB medium to an OD of 0.050. The culture was aliquoted into tubes containing paraquat and further incubated at 37°C for 45 min. The data were normalized to the activity present in the uninduced cultures (Miller units). SOD activity was assayed, with AS395 having 9 U/mg and AS396 having 216 U/mg.
DISCUSSION
Our results show that E. coli can tolerate only small decreases in SOD content. A decrease in SOD of more than twofold led to significant dehydratase inactivation, and further decreases in SOD both lowered the growth rate in unsupplemented medium and resulted in sensitivity to DNA damage. Clearly, enough O2− is made during normal metabolism to require the synthesis of abundant SOD.
For wild-type cells, it is useful to determine the extent by which O2− production must increase before toxicity results. The answer can be inferred from this study if the steady-state O2− concentration varies inversely with SOD activity. That has been generally assumed to be true (17, 34) because SOD functions as a first-order, unsaturable enzyme at physiological concentrations of O2− (4, 5, 57) and because no other significant scavenger of O2− has been found in the cytosol of E. coli (35). Although O2− can spontaneously dismute, it will do so at an appreciable rate only in cells containing <0.1% of wild-type SOD activity (calculated from reference 34). Labile iron sulfur clusters are too scarce (100 μM) and too slowly reactivated (half-life [t1/2] = 7 min) (39) to consume more than 5% of the 200 μM O2− flux/min. The absence of SOD-independent scavenging mechanisms is supported by the fact that O2− toxicity continues to worsen when even very low SOD activities are further diminished (Fig. 2).
Therefore, during steady-state conditions, the formation of O2− is balanced by its dismutation by SOD. The following equations describe the relation between O2− concentration and SOD concentration, where kf and kSOD denote the rate of O2− formation and the rate constant for O2− dismutation by SOD, respectively:
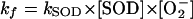 |
1 |
 |
2 |
Thus, the intracellular O2− concentration varies inversely with the amount of SOD. A threefold decrease in SOD results in a threefold increase in the steady-state O2− concentration. Notably, this increase could also result from a threefold increase in the rate of O2− formation. Even if the assumption that most O2− were scavenged by SOD were wrong, the consequence would be that our estimate of O2− sensitivity would be too conservative. In that circumstance, an additional term would be included in the denominator of equation 2, so that the toxic effects of O2− would ensue from less than a threefold increase in its formation.
By using the relation between SOD and O2− concentrations, the data can be reevaluated. Substantial enzyme damage will result when O2− levels increase by more than twofold, and growth deficits will become pronounced upon an increase of more than fourfold. In fact, because the measurements of growth rates were less exacting than assays of dehydratase activity, we suspect that more-precise measurements might indicate that growth declines at O2− concentrations even closer to those of wild-type cells. Clearly, E. coli is poised near the brink of toxicity from endogenous oxidants. This fact is remarkable given the extremely low concentration of O2− in living cells and attests to both the high rate and specificity of its reactions with dehydratase iron-sulfur clusters. It is also clear that additional O2− formation by redox-cycling drugs would be detrimental if SOD synthesis were not augmented.
Concordance between calculated and observed enzyme damage.
The fractional activity of a dehydratase population within the cell is determined by the balance between the rate of intracellular O2− formation and the rate constant for O2− inactivation of the dehydratase, on the one hand, and the concentration of SOD and the speed of enzyme reactivation, on the other hand. These four parameters have all been measured independently, making it possible to test whether they fit our results.
Superoxide is formed primarily in E. coli by the reaction between oxygen and reduced components of the respiratory chain. In air-saturated minimal medium, the rate was estimated to be 3 μM O2−/s (34). Based on the SOD content of these cells, the steady-state O2− concentration was calculated to be 2 × 10−10 M. It has since been shown that only half of the electron flux passes through the auto-oxidizable NADH dehydrogenase, which lowers the best estimate of O2− concentration to 10−10 M (unpublished data).
The rate constants for the inactivation by O2− of several dehydratases, namely, fumarase A, fumarase B, dihydroxyacid dehydratase, and beef heart aconitase, were each determined in vitro to lie between 1 × 106 and 6 × 106 M−1 s−1 (14). These rates were measured by two indirect techniques that produced similar but not equivalent results. A higher value has been reported for purified E. coli aconitase, 3 × 107 M−1 s−1 (29). In all studies, the inactivation rates were lowered somewhat by the presence of substrate (14, 20), which partially shields the active site; this might particularly affect aconitase, since substrate more effectively protects aconitase and since citrate levels may be saturating in vivo (20, 42, 49). Based on the values determined by Flint, we use 3 × 106 M−1 s−1 as a representative value for the inactivation rate constants (kinactivation) of the dehydratases in the following equation:
 |
3 |
where E is the amount of active enzyme. By integration where E1 and E2 represent amounts of active enzyme at times separated by an interval (+), we calculate
 |
giving t1/2 = 39 min. These calculations predict that the labile enzymes of E. coli are likely to be damaged at least once per generation (55 min) in glucose-saturated medium. The dehydratases are likely to cycle between active and inactive forms. Damage to the enzymes may occur even more often per generation when E. coli is in its natural habitat, which supports a much lower growth rate.
Under steady-state conditions, this inactivation rate will be equal to the reactivation rate. The reactivation half-time was measured to be 7 min (kreactivation = 0.00165 s−1) (39):
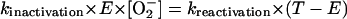 |
4 |
where T is the total (active plus inactive) enzyme. In a wild-type strain,
 |
giving E/T = 0.85. Therefore, SOD-proficient cells growing in air-saturated medium are expected to contain enough O2− that the labile dehydratases are only 85% active. This agrees with the observation that aconitase activity increased by about 13% when aerobic cells were shifted to anaerobic conditions in the presence of protein synthesis inhibitors (20). Comparisons of the amounts of SOD and labile clusters inside cells (12 and 100 μM, respectively) and of their rate constants for reaction with O2− (2 × 109 and 3 × 106 M−1 s−1) suggest that SOD scavenges about 99% of the O2− before it damages dehydratases.
More generally, one can predict the fraction of active enzyme as a function of SOD content. Using equation 4, we calculate
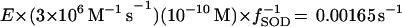 |
5 |
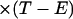 |
where fSOD is the SOD activity as a fraction of that found in wild-type cells, and
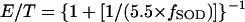 |
6 |
Consequently, dehydratases would be half inactive when SODs were present at 20% of the wild-type activity. This result is in good agreement with Fig. 2 and 5. Because the values used in this calculation were approximations, the concordance seen between the calculated and observed behaviors should not be considered precise. Nevertheless, the in vivo results support the published in vitro measurements of O2− formation and of dehydratase sensitivity. Apparently, the field has achieved a detailed, coherent view of the physiology of oxidative stress. It seems that E. coli has evolved so that the production of O2− and its reactivity with cellular components is just balanced by the O2− scavenging and repair activities. This balance ensures that the labile dehydratases are almost fully active in air.
Induction of the SoxRS regulon.
Upon its discovery, the SoxRS regulon was thought to respond to O2−, since the regulon is fully activated by superoxide-forming drugs and is able to induce MnSOD. The inefficiency of induction under anaerobic conditions appeared to support the idea that O2− was the direct inducer. However, subsequent work has shown that paraquat can partially activate the SoxRS regulon even anaerobically, where there is no chance of O2− formation (58, 59), and oxygen may have enhanced the induction merely by chemically oxidizing the reduced paraquat and ensuring that a sufficient amount was in the oxidized form. Gaudu et al. and Hidalgo et al. elegantly demonstrated that the activation of SoxR occurs when its [2Fe-2S] cluster is oxidized (22, 30). It is clear that a number of low-molecular-weight oxidants, including O2−, oxygen, and even redox-cycling drugs themselves (which were used in determining the cluster midpoint potential) (10, 21), are capable of oxidizing the cluster directly when present in high concentrations. The problem is to identify the predominant oxidant in vivo. Our data show that physiological concentrations of O2− do not efficiently induce the SoxRS regulon. The modest induction that occurred at high O2− concentrations was too slight to be physiologically effective. In contrast, paraquat activated the regulon in a superoxide-independent manner, and the resulting fumarase C activity fully compensated for the inactivation of fumarase A.
The simplest interpretation of our results is that SoxRS exists as a general defense against exogenous redox-cycling drugs rather than against O2− per se. Superoxide is a component of the stress imposed by these drugs, and the induction of SOD is an appropriate response, since basal SOD synthesis is inadequate to preserve enzyme activity if O2− formation is accelerated. However, these drugs also have superoxide-independent mechanisms of toxicity (3, 33, 62). At least two elements of the SoxRS response—the reduction of outer membrane pores (9) and the activation of the NADPH-producing pentose phosphate pathway—can be more easily rationalized as defenses against exogenous redox-cycling drugs than against O2− itself. If so, then it is reasonable that SoxR might sense an effect of the antibiotic other than O2− stress. Were SoxR focused on O2− levels, the response might fail to be activated in microaerobic conditions or might be short-circuited upon SOD induction. Potential signals include the direct oxidation of SoxR by the drugs and the diminution of SoxR rereduction as the NAD(P)H pool is depleted (18, 44, 48).
ACKNOWLEDGMENTS
We thank Bruce Demple and Daniele Touati for providing strains for this work. We are very grateful to Alex I. Smirnov, Tatyana I. Smirnov, and R. Linn Belford (University of Illinois) for their assistance with the EPR experiments conducted at the Illinois EPR Research Center, a National Institutes of Health Biomedical Research and Technology Resource (P41-RR01811).
This work was supported by a National Institutes of Health grant (GM49640) and an American Cancer Society grant (CN-146). A.S.G. was partially supported by a Cell and Molecular Biology training grant from the National Institutes of Health (T32 GM07283-20).
REFERENCES
- 1.Albe K R, Butler M H, Wright B E. Cellular concentrations of enzymes and their substrates. J Theor Biol. 1990;143:163–195. doi: 10.1016/s0022-5193(05)80266-8. [DOI] [PubMed] [Google Scholar]
- 2.Ames B N, Shigenaga M K, Hagen T K. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7914–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunmark A, Cadenas E. Redox and addition chemistry of quinoid compounds and its biological implications. Free Radic Biol Med. 1989;7:435–477. doi: 10.1016/0891-5849(89)90126-3. [DOI] [PubMed] [Google Scholar]
- 4.Bull C, Fee J. Steady-state kinetic studies of superoxide dismutases: properties of the iron containing protein from Escherichia coli. J Am Chem Soc. 1985;107:3295–3304. [Google Scholar]
- 5.Bull C, Niederhogger E, Yoshida T, Fee J. Kinetic studies of superoxide dismutases: properties of the manganese-containing protein from Thermus thermophilus. J Am Chem Soc. 1991;113:4069–4076. [Google Scholar]
- 6.Carlioz A, Touati D. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 1986;5:623–630. doi: 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerutti P A. Prooxidant states and tumor promotion. Science. 1985;227:375–381. doi: 10.1126/science.2981433. [DOI] [PubMed] [Google Scholar]
- 8.Chang E C, Kosman D J. Intracellular Mn(II)-associated superoxide scavenging activity protects Cu, Zn superoxide dismutase-deficient Saccharomyces cerevisiae against dioxygen stress. J Biol Chem. 1989;264:12172–12178. [PubMed] [Google Scholar]
- 9.Chou J H, Greenberg J T, Demple B. Posttranscriptional repression of Escherichia coli OmpF protein in response to redox stress: positive control of the micF antisense RNA by the soxRS locus. J Bacteriol. 1993;175:1026–1031. doi: 10.1128/jb.175.4.1026-1031.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding H, Hildalgo E, Demple B. The redox state of the [2Fe-2S] clusters in SoxR protein regulates its activity as a transcription factor. J Biol Chem. 1996;271:33173–33175. doi: 10.1074/jbc.271.52.33173. [DOI] [PubMed] [Google Scholar]
- 11.Farmer K J, Sohal R S. Relationship between superoxide anion radical generation and aging in the housefly, Musca domestica. Free Radic Biol Med. 1989;7:23–29. doi: 10.1016/0891-5849(89)90096-8. [DOI] [PubMed] [Google Scholar]
- 12.Farr S B, D’Ari R, Touati D. Oxygen-dependent mutagenesis in Escherichia coli lacking superoxide dismutase. Proc Natl Acad Sci USA. 1986;83:8268–8272. doi: 10.1073/pnas.83.21.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flint D H, Emptage M H. Dihydroxyacid dehydratase: isolation, characterization as Fe-S proteins, and sensitivity to inactivation by oxygen radicals. In: Chipman D, Barak Z, Schloss J V, editors. Biosynthesis of branched chain amino acids. New York, New York: VCH Publishers; 1990. pp. 285–314. [Google Scholar]
- 14.Flint D H, Tuminello J F, Emptage M H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J Biol Chem. 1993;268:22369–22376. [PubMed] [Google Scholar]
- 15.Fridovich I. The biology of oxygen radicals. Science. 1978;201:875. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 16.Gardner P R, Raineri I, Epstein L B, White W C. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13406. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 17.Gardner P R, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- 18.Gardner P R, Fridovich I. NADPH inhibits transcription of the Escherichia coli manganese superoxide dismutase gene (sodA) in vitro. J Biol Chem. 1993;268:12958–12963. [PubMed] [Google Scholar]
- 19.Gardner P R, Fridovich I. Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J Biol Chem. 1991;266:1478–1483. [PubMed] [Google Scholar]
- 20.Gardner P R, Fridovich I. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 21.Gaudu P, Weiss B. SoxR, a [2Fe-2S] transcription factor, is active only in its oxidized form. Proc Natl Acad Sci USA. 1996;93:10094–10098. doi: 10.1073/pnas.93.19.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaudu P, Moon N, Weiss B. Regulation of the soxRS oxidative stress regulon. J Biol Chem. 1997;272:5082–5086. doi: 10.1074/jbc.272.8.5082. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Flecha B, Demple B. Homeostatic regulation of intracellular hydrogen peroxide concentration in aerobically growing Escherichia coli. J Bacteriol. 1997;179:382–388. doi: 10.1128/jb.179.2.382-388.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grana D, Gardella T, Susskind M. The effects of mutations in the ant promoter of prophage P22 depend on context. Genetics. 1988;120:319–327. doi: 10.1093/genetics/120.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenberg J T, Monach P, Chou J H, Josephy P D, Demple B. Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in E. coli. Proc Natl Acad Sci USA. 1990;87:6181–6185. doi: 10.1073/pnas.87.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruer M J, Bradbury A J, Guest J R. Construction and properties of aconitase mutants of Escherichia coli. Microbiology. 1997;143:1837–1846. doi: 10.1099/00221287-143-6-1837. [DOI] [PubMed] [Google Scholar]
- 27.Gruer M J, Guest J R. Two genetically-distinct and differentially-regulated aconitases (AcnA and AcnB) in Escherichia coli. Microbiology. 1994;140:2531–2541. doi: 10.1099/00221287-140-10-2531. [DOI] [PubMed] [Google Scholar]
- 28.Hassan H M, Fridovich I. Intracellular production of superoxide radical and of hydrogen peroxide by redox active compounds. Arch Biochem Biophys. 1979;196:385–395. doi: 10.1016/0003-9861(79)90289-3. [DOI] [PubMed] [Google Scholar]
- 29.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 30.Hidalgo D, Ding H, Demple B. Redox signal transduction: mutations shifting [2Fe-2S] clusters of the SoxR sensor-regulator to the oxidized form. Cell. 1997;88:121–129. doi: 10.1016/s0092-8674(00)81864-4. [DOI] [PubMed] [Google Scholar]
- 31.Hofmeister A E, Albracht S P, Buckel W. Iron-sulfur cluster-containing l-serine dehydratase from Peptostreptococcus asaccharolyticus: correlation of the cluster type with enzymatic activity. FEBS Lett. 1994;351:416–418. doi: 10.1016/0014-5793(94)00901-5. [DOI] [PubMed] [Google Scholar]
- 32.Hofmeister A E, Grabowoski R, Linder D, Buckel W. l-Serine and l-threonine dehydratase from Clostridium propionicum. Two enzymes with different prosthetic groups. Eur J Biochem. 1993;215:341–349. doi: 10.1111/j.1432-1033.1993.tb18040.x. [DOI] [PubMed] [Google Scholar]
- 33.Imlay J A, Fridovich I. Exogenous quinones directly inhibit the respiratory NADH dehydrogenase in Escherichia coli. Arch Biochem Biophys. 1992;296:337–346. doi: 10.1016/0003-9861(92)90581-g. [DOI] [PubMed] [Google Scholar]
- 34.Imlay J A, Fridovich I. Assay of metabolic superoxide production in Escherichia coli. J Biol Chem. 1991;266:6957–6965. [PubMed] [Google Scholar]
- 35.Imlay J A, Fridovich I. Suppression of oxidative envelope damage by pseudoreversion of a superoxide dismutase-deficient mutant of Escherichia coli. J Bacteriol. 1992;174:953–961. doi: 10.1128/jb.174.3.953-961.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imlay J A, Linn S. Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J Bacteriol. 1986;166:519–527. doi: 10.1128/jb.166.2.519-527.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imlay J A, Linn S. Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J Bacteriol. 1987;169:2967–2976. doi: 10.1128/jb.169.7.2967-2976.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keyer K, Gort A S, Imlay J A. Superoxide and the production of oxidative DNA damage. J Bacteriol. 1995;177:6782–6790. doi: 10.1128/jb.177.23.6782-6790.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keyer K, Imlay J A. Inactivation of dehydratase [4Fe-4S] clusters and disruption of iron homeostasis upon cell exposure to peroxynitrite. J Biol Chem. 1997;272:27652–27659. doi: 10.1074/jbc.272.44.27652. [DOI] [PubMed] [Google Scholar]
- 40.Keyer K, Imlay J A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci USA. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuo C F, Mashino T, Fridovich I. Alpha, beta-dihydroxyisovalerate dehydratase: a superoxide-sensitive enzyme. J Biol Chem. 1987;262:4724–4727. [PubMed] [Google Scholar]
- 42.Lakshmi T M, Helling R B. Selection for citrate synthase deficiency in icd mutants of Escherichia coli. J Bacteriol. 1976;127:76–83. doi: 10.1128/jb.127.1.76-83.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liochev S I, Fridovich I. Lucigenin luminescence as a measure of intracellular superoxide dismutase activity in Escherichia coli. Proc Natl Acad Sci USA. 1997;94:2891–2896. doi: 10.1073/pnas.94.7.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liochev S I, Fridovich I. Effects of overproduction of superoxide dismutases in Escherichia coli on inhibition of growth and on induction of glucose-6-phosphate dehydrogenase by paraquat. Arch Biochem Biophys. 1992;294:138–143. doi: 10.1016/0003-9861(92)90147-o. [DOI] [PubMed] [Google Scholar]
- 45.Liochev S I, Fridovich I. Fumarase C, the stable fumarase of Escherichia coli, is controlled by the soxRS regulon. Proc Natl Acad Sci USA. 1992;89:5892–5896. doi: 10.1073/pnas.89.13.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liochev S I, Fridovich I. Modulation of the fumarases of Escherichia coli in response to oxidative stress. Arch Biochem Biophys. 1993;301:379–384. doi: 10.1006/abbi.1993.1159. [DOI] [PubMed] [Google Scholar]
- 47.Liochev S I, Fridovich I. The role of superoxide in the production of hydroxyl radical: in vitro and in vivo. Free Radic Biol Med. 1994;16:29–33. doi: 10.1016/0891-5849(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 48.Liochev S I, Hausladen A, Beyer W F, Fridovich I. NADPH:ferridoxin oxidoreductase acts as a paraquat diaphorase and is a member of the soxRS regulon. Proc Natl Acad Sci USA. 1994;91:1328–1331. doi: 10.1073/pnas.91.4.1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowry O H, Carter J, Ward J B, Glaser L. The effect of carbon and nitrogen sources on the level of metabolic intermediates in Escherichia coli. J Biol Chem. 1971;246:6511–6521. [PubMed] [Google Scholar]
- 50.McCord J M, Keele B B, Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proc Natl Acad Sci USA. 1971;28:1024–1027. doi: 10.1073/pnas.68.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCord J M, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 52.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 53.Novick A, Weiner M. Enzyme induction as an all-or-none phenomenon. Proc Natl Acad Sci USA. 1957;43:553–566. doi: 10.1073/pnas.43.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nunoshiba T, Derojaswalker T, Wishnok J S, Tannenbaum S R, Demple B. Activation by nitric oxide of an oxidative stress response that defends Escherichia coli against activated macrophages. Proc Natl Acad Sci USA. 1993;90:9993–9997. doi: 10.1073/pnas.90.21.9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nunoshiba T, Hidalgo E, Amabilecuevas C F, Demple B. Two-stage control of an oxidative stress regulon: the Escherichia coli SoxR protein triggers redox-inducible expression of the soxS regulatory gene. J Bacteriol. 1992;174:6054–6060. doi: 10.1128/jb.174.19.6054-6060.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phillips J P, Campbell S D, Michaud D, Charbonneau M, Hilliker A J. Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proc Natl Acad Sci USA. 1989;86:2761–2765. doi: 10.1073/pnas.86.8.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pick M, Rabani J, Yost F, Fridovich I. The catalytic mechanism of the manganese-containing superoxide dismutase of Escherichia coli studied by pulse radiolysis. J Am Chem Soc. 1974;96:7329–7333. doi: 10.1021/ja00830a026. [DOI] [PubMed] [Google Scholar]
- 58.Privalle C T, Fridovich I. Inductions of superoxide dismutases in Escherichia coli under anaerobic conditions. Accumulation of an inactive form of the manganese enzyme. J Biol Chem. 1988;263:4274–4279. [PubMed] [Google Scholar]
- 59.Schiavone J R, Hassan H M. The role of redox in the regulation of manganese-containing superoxide dismutase biosynthesis in Escherichia coli. J Biol Chem. 1988;263:4269–4273. [PubMed] [Google Scholar]
- 60.Siegele D A, Hu J C. Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci USA. 1997;94:8168–8172. doi: 10.1073/pnas.94.15.8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Simons R W, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- 62.Siwecki, G., and O. R. Brown. Overproduction of superoxide dismutase does not protect Escherichia coli from stringency-induced growth inhibition by 1 mM paraquat. Biochem. Int. 20:191–199. [PubMed]
- 63.Touati D. Transcriptional and posttranscriptional regulation of manganese superoxide dismutase biosynthesis in Escherichia coli, studied with operon and protein fusions. J Bacteriol. 1988;170:2511–2520. doi: 10.1128/jb.170.6.2511-2520.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsaneva I R, Weiss B. soxR, a locus governing a superoxide response regulon in Escherichia coli K-12. J Bacteriol. 1990;172:4197–4205. doi: 10.1128/jb.172.8.4197-4205.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Loon A P G M, Pesold-Hurt B, Schatz G. A yeast mutant lacking mitochondrial manganese-superoxide dismutase is hypersensitive to oxygen. Proc Natl Acad Sci USA. 1986;83:3820–3824. doi: 10.1073/pnas.83.11.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Woods S A, Schwartzbach S D, Guest J R. Two biochemically distinct classes of fumarase in Escherichia coli. Biochim Biophys Acta. 1988;954:14–26. doi: 10.1016/0167-4838(88)90050-7. [DOI] [PubMed] [Google Scholar]
- 67.Wu J, Weiss B. Two-stage induction of the soxRS (superoxide response) regulon of Escherichia coli. J Bacteriol. 1992;174:3915–3920. doi: 10.1128/jb.174.12.3915-3920.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]