

Abstract
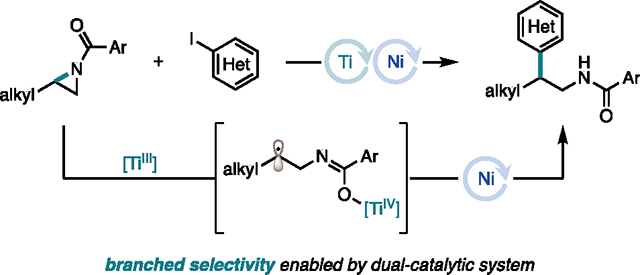
The arylation of 2-alkyl aziridines by nucleophilic ring-opening or transition metal-catalyzed cross-coupling enables facile access to biologically relevant β-phenethylamine derivatives. However, both approaches largely favor C–C bond formation at the less substituted carbon of the aziridine, thus enabling access to only linear products. Consequently, despite the attractive bond disconnection it poses, the synthesis of branched arylated products from 2-alkyl aziridines has remained inaccessible. Herein, we address this long-standing challenge and report the first branched-selective cross-coupling of 2-alkyl aziridines with aryl iodides. This unique selectivity is enabled by a Ti/Ni dual-catalytic system. We demonstrate the robustness of the method by a two-fold approach: an additive screening campaign to probe functional group tolerance and a feature-driven substrate scope to study the effect of the local steric and electronic profile of each coupling partner on reactivity. Furthermore, the diversity of this feature-driven substrate scope enabled the generation of predictive reactivity models that guided mechanistic understanding. Mechanistic studies demonstrated that the branched selectivity arises from a TiIII-induced radical ring-opening of the aziridine.
Graphical Abstract
Introduction
The β-phenethylamine scaffold is an important motif in medicinal chemistry.1,2 A common structural modification of these scaffolds is α- or β-alkyl branching of the phenethylamine backbone, with both regioisomers exhibiting biological activity.3 Owing to the prevalence of both isomers in druglike molecules (Figure 1A), the selective installation of these motifs is of great interest. Traditional methods for the synthesis of this motif include: reduction of β-aryl nitro alkanes or alkenes,4,5 nitriles,6 and enamides;7 hydride ring opening of styrenyl aziridines;8 and hydroaminoalkylation.9 Overall, these methodologies involve early introduction of the β-phenethylamine carbon backbone. Alternatively, arylation of 2-alkyl aziridines presents an attractive retrosynthetic disconnection, as it affords greater modularity in the introduction of both alkyl and aryl substitution to the ethylamine backbone in a single C–C bond-forming step (Figure 1B). Moreover, recent advances in the aziridination of alkenes, as well as classical methods, have rendered 2-alkyl aziridines readily available from abundant organic feedstocks.10,11 Thus, in combination with readily available aryl precursors, aziridines constitute ideal precursors for accessing these high-value β-phenethylamine targets. In addition, depending on the regioselectivity of C–N bond cleavage, aziridines could provide access to both linear and branched regioisomers of β-phenethylamines in a unified approach from a common precursor. However, while methods that facilitate C–N bond cleavage at the less substituted C–N bond to afford linear products are well-established, strategies that enable cleavage at the more substituted C–N bond to form branched products have remained underdeveloped.
Figure 1.

Strategies for the arylation of 2-alkyl aziridines.
Regioselective cleavage of the less sterically hindered (or less substituted) C–N bond can be accessed via both traditional nucleophilic ring-opening strategies as well as transition metal catalysis (Figure 1C). When a Grignard or organolithium reagent is employed in the presence of a copper additive, C–C bond formation occurs via nucleophilic ring-opening, favoring cleavage of the less sterically hindered C–N bond to give the linear isomer (C1).12 While highly enabling, the use of harsh organometallic reagents limits functional group tolerance and restricts the choice of nitrogen protecting group on the aziridine. For example, N-acyl aziridines undergo preferential nucleophilic attack at the acyl carbon over nucleophilic ring-opening.10
Transition metal catalysis has emerged as a mild and selective alternative to traditional substitution reactions for the functionalization of aziridines.13 These strategies take advantage of abundant and readily available aryl coupling partners. The regioselectivity of these processes is determined by the oxidative addition of 2-alkyl aziridines to the metal center. This oxidative addition typically proceeds through an SN2 mechanism,14,15 that favors cleavage of the less sterically hindered C–N bond to form linear products (C2), thereby providing the same structures accessible by direct nucleophilic ring-opening. Our lab has also demonstrated that 2-alkyl aziridines can undergo an in situ halide ring-opening at the less substituted position (C3).16 The resulting alkyl halide then interfaces with Ni catalysis, again resulting in the same regioselectivity as direct oxidative addition.
While both nucleophilic ring-opening and transition metal-catalyzed cross-coupling are highly enabling methods in accessing β-phenethylamines, both strategies favor C–C bond formation at the less substituted carbon of the aziridine, thus providing access to only linear products. While branched products are accessible via the regioselective alkylation of styrenyl aziridines,17–19 formation of these products via the regioselective arylation of 2-alkyl aziridines would benefit from the greater availability of (hetero)aryl coupling partners as compared to styrenyl aziridine precursors. This, in turn, would offer a highly modular and facile approach to accessing branched-selective products with greater structural diversity than what would be attained using more classical alkylation strategies. Such an approach, however, would require overcoming the inherent reactivity profile of 2-alkyl aziridines to substitution reactions in both classic nucleophilic ring-opening and transition metal catalysis. This inherent reactivity has rendered the formation of branched arylated products an unsolved problem in 2-alkyl aziridine functionalization.
To overcome this challenge, we envisioned that we could leverage the reactivity of a Ti co-catalyst to activate the more substituted C–N bond of the 2-alkyl aziridine via either a single-electron20,21 or Lewis acid22 pathway. The Ti catalytic cycle could then be interfaced with Ni catalysis to access branched cross-coupled products (Figure 1D).23,24 Herein, we describe the realization of this goal, which represents the first branched-selective cross-coupling of 2-alkyl aziridines.
Results and Discussion
Reaction Optimization.
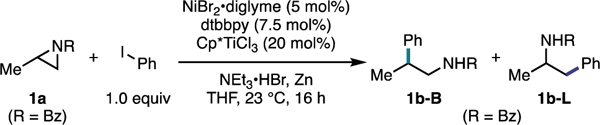
To evaluate the feasibility of this dual-catalytic system, we investigated the coupling of N-protected 2-methyl aziridines with phenyl iodide. We found that the coupling of N-benzoyl-2-methyl aziridine (1a) with phenyl iodide (1.0 equiv) in the presence of NiBr2•diglyme (5 mol%), 4,4’-di-tert-butylbipyridine (dtbbpy) (7.5 mol%), Cp*TiCl3 (Cp* = pentamethylcyclopentadienyl) (20 mol%), NEt3•HBr (2.0 equiv), and Zn (3.0 equiv) in THF (0.15 M with respect to 1a) afforded a 12:1 mixture of 1b-B:1b-L in 81% yield (Table 1, entry 1), thus demonstrating preferential formation of the branched (B) isomer over the undesired linear (L) isomer. Notably, an equimolar amount of aziridine and aryl iodide were employed in this case. This feature is especially attractive for a convergent cross-coupling of late-stage intermediates. Alternative carbonyl-based protecting groups, such as acetyl (Ac) (Table 1, entry 2), tert-butoxycarbonyl (Boc) (Table 1, entry 3), and benzyl carbamate (Cbz) (Table 1, entry 4), afforded the linear product or a mixture of isomers in low yields. Of the Ti catalysts we screened, we found that Cp*TiCl3 (81% yield, 12:1 B:L) (Table 1, entry 1) and CpTiCl3 (Cp = cyclopentadienyl) (40% yield, 3:1 B:L) (Table 1, entry 5) were uniquely able to afford cross-coupled product, with both catalysts preferentially generating the branched product. Lewis acids, such as TMSCl (Table 1, entry 6), or other Ti catalysts (Table 1, entries 7–9) were ineffective in the reaction and provided either trace product or a mixture of isomers in low yields. While we ultimately moved forward with Zn as our optimal reductant, we found that Mn (Table 1, entry 10) and TDAE (Table 1, entry 11) were also effective reductants in the transformation, with both providing the desired cross-coupled product in high yields and with high selectivity, demonstrating broad generality in regard to the reductant and providing evidence against the intermediacy of an organozinc intermediate.
Table 1.
Reaction Optimization
| ||||
|---|---|---|---|---|
| entry | deviation from standard conditionsa | aziridine conversion [%]b | yield [%]b | B:Lb |
| 1 | none | 100 | 81 | 12:1 |
|
| ||||
| 2 | R = Ac | 100 | 31 | 1:6 |
| 3 | R = Boc | 96 | 33 | 1:3 |
| 4 | R = Cbz | 100 | 13 | 1:1 |
|
| ||||
| 5 | CpTiCl3 | 88 | 40 | 3:1 |
| 6 | TMSCl instead of Ti | 36 | < 5 | – |
| 7 | TiCl4•THF2 | 80 | 24 | 1:1 |
| 8 | Cp2TiCl2 | 47 | <5 | – |
| 9 | (Cp*)2TiCl2 | 34 | <5 | – |
|
| ||||
| 10 | Mn | 93 | 71 | 11:1 |
| 11 | TDAE | 100 | 81 | 11:1 |
|
| ||||
| 12 | without Ni/ligand | 100 | 0 | – |
| 13 | without ligand | 100 | 9 | 11:1 |
| 14 | without Cp*TiCl3 | 32 | <5 | – |
| 15 | wtihout NEt3•HBr | 100 | 34 | 9:1 |
| 16 | without Zn | 86 | 0 | – |
|
| ||||
| ||||
NEt3•HBr (2.0 equiv), Zn (3.0 equiv), THF (0.15 M).
Reactions performed on 0.1 mmol scale. Yields and selectivity were determined by GC-FID with dodecane as an internal standard.
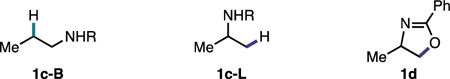
Control experiments (Table 1, entries 12–16) indicated the importance of each reaction component. Specifically, in the absence of Ni and ligand (Table 1, entry 12), 1a was fully consumed; however, it did not undergo the desired C–C bond formation. Instead, the reductive ring-opened products, 1c-B and 1c-L, were formed. Without Ti only 32% conversion of 1a was observed (Table 1, entry 14), and there was only trace cross-coupled product formation. Consistent with our initial mechanistic hypothesis, these results suggest that Ni is likely responsible for C–C bond formation whereas Ti is likely responsible for aziridine activation (vide infra). Throughout optimization of the reaction, we also observed trace amounts of isomerized product 1d which could arise from halide ring-opening at the less-substituted C–N bond followed by displacement of the iodide by oxygen to generate the oxazoline core.25
Scope Design.
Having identified optimal conditions, we sought to explore the scope of the transformation. In the design of our scope, we set out to capture both functional group tolerance and to study how modifications of the local steric and electronic profile of a coupling partner would impact reactivity (Figure 2A).26,27 To this end, we opted to employ a combination of two complementary approaches: additive screening to probe functional group tolerance28 and a steric and electronic feature-driven substrate scope selection to explore the impact of the local environment on reactivity.29,30
Figure 2.

Strategies for substrate scope design. DoE = design of experiments.
Additive Screen.
Glorius and coworkers have developed an additive screening approach to probe the robustness of a reaction to pendant functionalities (Figure 2B).28 In this approach, a model reaction is performed in the presence of an additive containing the functional group of interest. Depending on the yield of the model reaction and additive recovery, a functional group can be classified as either tolerated (i.e., the functional group has no impact on the reaction and the additive is recovered) or incompatible (i.e., the functional group acts either as a catalyst poison or undergoes side reactivity).
To test the functional group tolerance of our method, we employed an additive screen31 in our model reaction of 1a and phenyl iodide to generate benzamide 1b (Figure 3). Functional groups not included in this initial additive screen can be assessed in a prospective additive screen prior to testing an unknown substrate or retrospectively to explain the poor performance of a substrate unaccounted for by a prediction model.30 We found that our method is tolerant (defined as >60% yield and >60% additive recovery) of unactivated and activated alkenes; aliphatic ketones; nitriles; alkyl chlorides; primary alkyl bromides; aryl chlorides, triflates, and boronic esters; anilines; acetals; and protected amines. We were surprised to find that anilines, despite bearing coordinating functionality, were tolerated under the reaction conditions. Potentially problematic functionalities, defined as those with 15–60% yield or additive recovery, include aryl ketones, which are susceptible to reduction by Ti; aryl bromides, which are susceptible to oxidative addition with Ni; and silyl ethers. Functional groups that are incompatible with the method (<15% yield or additive recovery) include alkynes, aldehydes, carboxylic acids, nitro groups, secondary alkyl bromides, alkyl iodides, aliphatic amines, and alcohols. Since the tolerance or intolerance of these functional groups is depicted in the additive screening campaign, we proceeded to substrate scope selection, focusing on the diversity of the local steric and electronic profiles of our selected substrates over the number of functional groups depicted.
Figure 3.

Additive screening to probe functional group tolerance. Groupings were determined by the lower value of the yield or additive recovery. Reactions run on 0.075 mmol scale. Yield and additive recovery were determined by GC-FID with dodecane as an internal standard.
Substrate Scope.
While additive screening provides a wealth of information, it does not account for how the local electronic and steric profile of a substrate will impact reactivity.32 Thus, we moved forward with a feature-driven substrate scope selection with respect to both the aryl iodide and 2-alkyl aziridine coupling partners to capture these intricacies (Figure 2C).
Aryl Iodide Scope.
We began by examining the substrate scope of the reaction with respect to the aryl iodide coupling partner. With aryl iodides, there are several steric and electronic features that may affect reactivity. To navigate this large feature space, we employed a workflow previously developed in our lab that uses uniform manifold approximation and projection (UMAP)33,34 and hierarchal clustering to construct a diverse, but succinct, substrate scope that spans a range of local and global steric and electronic features.30 Accordingly, we defined our chemical space to comprise 4,284 commercially available aryl iodides (see SI for details). In order to describe the steric and electronic profiles of these aryl iodides, we computed density functional theory (DFT) and structural features using Auto-QChem, a program developed by our lab that automates the calculation of these features based on SMILES strings.35 We then performed dimensionality reduction using UMAP to present these computed features in 2D chemical space and performed hierarchal clustering (done with 10 UMAP reduced features), to group compounds with similar steric and electronic features together while placing dissimilar compounds in different clusters (Figure 4A). With our chemical space defined, we filtered out any aryl iodide containing a functionality that is not tolerated as defined by our prior additive screening campaign (see Figure 3 and SI for additional functional group filters). Even upon omission of these functionalities, we have an excellent coverage of feature space, with each cluster being well represented (Figure 4B). From each of these clusters, we selected one aryl iodide (A–P, labeled after the cluster it was selected from) to test in the cross-coupling reaction with 1a.
Figure 4.

(Hetero)aryl iodide scope (0.4 mmol scale). Unless otherwise noted, isolated yields of the mixture of isomers are reported and are the average of two runs. Aryl iodides are labeled based on clusters A–P in chemical space. Cross-coupled products are labeled based on their cluster or class in chemical space (A-1–P-1, het1–het4). a19F NMR yield with an external standard.
The diversity of this substrate scope is highlighted in Figure 4C. Several of our examples represent classic “Hammett” type substrates where the electronics of a single substituent at the meta or para position is modified (A, C–F, H). These substrates revealed that the method is tolerant to both electron-rich and electron-deficient aryl iodides, with the latter giving higher yields but slightly diminished B:L selectivity. Aryl iodide A, which contains a pyrimidine substituent underwent successful cross-coupling in moderate yield, demonstrating promising tolerance of this method to heterocyclic compounds. In addition to classic “Hammett”-type substrates, a number of substrates bearing multiple substituents at the meta and para positions, such as I and J, also underwent successful cross-coupling. Notably, J-1 contains additional aryl chloride functionalities that can be employed for further diversification. In contrast to many literature substrate scopes, this data-science generated aryl iodide scope contained several ortho substituted aryl iodides. Aryl iodides containing a single ortho substituent underwent cross-coupling with excellent B:L selectivity; included among these ortho substituents are: methoxy (B), boronic acid pinacol ester (BPin) (G), sulfonate (K), carbonyl (O), and aniline (P). Of note, aryl iodide G—despite the size of the BPin substituent—underwent successful cross-coupling and maintained the boronate ester functionality for further diversification. As prior additive screening suggested, anilines are compatible with this method; and indeed, we found this to be the case with the successful cross-coupling of P. A greater dependence on the steric profile of the aryl iodide was observed as exemplified by ortho-substituted aryl iodides undergoing cross-coupling in overall lower yields. This data science-driven scope also contained three substrates with di-ortho substitution (L, M, N) that, unsurprisingly, gave 0% yield. Overall, all aryl iodides that yielded cross-coupled product did so with moderate to high levels of B:L selectivity. Lower-yielding substrates tended to provide higher levels of selectivity up to >20:1 selectivity for the branched cross-coupled product.
While not included in our defined aryl iodide chemical space, we also found that the reaction is tolerant to heteroaryl iodides. Specifically, 2-, 3-, and 4-iodopyridines generated cross-coupled products het-1–het-3 in high to moderate yields. In addition, 6-iodoquinoline underwent successful cross-coupling to generate het-4. These examples further highlight the potential utility of this method to be an effective approach toward synthesizing bioactive compounds.
Due to the diversity of aryl iodides selected we sought to quantify the observed dependence on sterics and electronics. We found the yields correlate (R2 = 0.93) with the percent buried volume of the iodide at 3.5 Å (%VBur_I) and the energy of the highest occupied molecular orbital (EHOMO) (Figure 4D). The features %VBur_I and EHOMO capture steric and electronic features respectively. Model robustness was assessed using leave-one-out cross-validation (LOOCV), a k-fold cross-validation technique where a single data point is left out of the data set and the model is trained on the remaining points. Model performance can then be evaluated on the test point as a measure of how the model would perform on an unknown data point. This process is iterated over the size of the data set. The mean absolute error (MAE) values for the LOOCV training and test sets were 7.7% yield and 10.4% yield, respectively. The similar MAEtrain and MAEtest values attest to the robustness of the model in predicting across the entire training set without overfitting certain data points. Outliers A-1 and O-1 were not included in the model or in the cross-validation. Compound A-1 has a lower yield than expected, likely due to the presence of a heterocycle that, while it is sufficiently tolerated to observe reactivity, can poison the Ni catalyst. On the other hand, O-1 had a significantly higher yield than expected. This discrepancy may be attributed to the ortho-carbonyl substituent which could facilitate binding of the aryl iodide to Ni, potentially facilitating oxidative addition.
We aimed to assess the utility of this model in predicting unseen substrates with multiple substituents bearing competing effects on reactivity, which may pose challenges in intuiting reaction performance (Figure 5). For example, product I–2 contains both an electron-withdrawing para substituent and an electron-donating meta substituent. Product L-2 contains an ortho methyl group but an electron-withdrawing para substituent. In both cases, the model predicts the yields within the LOOCV MAEtest value. Thus, although the sixteen selected substrates in the scope do not fully capture the diversity of all aryl iodides within this chemical space, systematic scope design facilitated the generation of a model capable of generalizing to unseen substrates.
Figure 5.

Predicted cross-coupled yields of validation aryl iodides with 1a.
Aziridine Scope.
With the aryl iodide scope, the chemical space was defined based on several steric and electronic features. However, in the case of 2-alkyl aziridines, we hypothesized that that the size of the 2-alkyl substituent would play the most significant role regarding the reactivity and regioselectivity of the transformation. We opted to describe the size of this substituent with the percent buried volume of the substituted carbon calculated at 3.5 Å (%VBur_C). To explore this effect, we selected 2-alkyl substituted aziridines that covered a wide range of %VBur_C values (Figure 6). The selected aziridines contained 2-Me (1a), 2-Et (2a), 2-n-Bu (3a), 2-Bn (4a), 2-i-Bu (5a), 2-i-Pr (6a), 2-Cy (7a), and 2-t-Bu (8a) substitution.
Figure 6.

Alkyl aziridine substrate scope (0.4 mmol scale). Unless otherwise noted, isolated yields of the mixture of isomers are reported and are the average of two runs. a1H NMR yield with an external standard. bBranched isomer isolated in 67% yield (>20:1). cBranched isomer isolated in 74% yield (>20:1). dpyridine•HBr (1.0 equiv) used instead of NEt3•HBr (2 equiv), 3 hours.
These aziridines were then screened under standard reaction conditions to generate cross-coupled products 1b–8b. With the exception of 8a, all reactions proceeded with high yields (i.e., 80–90%) and provided selectivity for the branched cross-coupled product. We noted that as the size of the alkyl substituent increased, the B:L selectivity decreased, ultimately leading to exclusive generation of the linear product in low yield in the case of (8b). This trend can be quantified using univariate linear regression where the B:L ratio is dependent on %VBur_C (R2 = 0.90, MAE = 0.9). We were surprised to see that 5b deviated significantly from this trend proceeding in >20:1 B:L selectivity. Products bearing longer alkyl chains, such as 2-n-Bu (3b), do not display the same effect. Thus, we hypothesize that this phenomenon likely results from a secondary interaction arising from the branching of the 2-i-Bu group, rather than an interaction originating from the polarizability of a longer alkyl chain. We also found the method to be amendable to functional groups on this 2-alkyl chain to generate products 9b and 10b.
While the focus of this study is the coupling of 2-alkyl aziridines, we sought to explore the tolerance of this method to alternate substitution patterns. Our lab has previously demonstrated the cross-coupling of N-tosyl protected cyclic aziridines with aryl iodides.16 Under our Ti/Ni dual-catalytic conditions, we can expand this cyclic scope to include N-benzoyl protected aziridines with 5- and 6-membered rings (11a and 12a respectively) undergoing cross-coupling to generate 11b and 12b in moderate to low yields with trans selectivity. With cyclic aziridines being the exception, we found that aziridines with di- and tri-alkyl substitution patterns did not undergo cross-coupling and would require further reaction optimization (see SI).
In the initial design of this system, we intended for Bz to be employed as a nitrogen protecting group; however, given the importance of the benzamide motif in biologically active compounds, we turned to exploring the sensitivity of the reaction to simple modifications on the benzoyl group to generate benzamides. Indeed, the reaction tolerates ortho substitution on the benzoyl group, generating 13b in moderate yield and selectivity. Both electron-deficient and electron-rich protecting groups were well-tolerated to generate 14b and 15b, with the former giving lower yield but excellent selectivity and the latter giving comparable yield and selectivity to the parent benzoyl protecting group.
Having confirmed the robustness of the method with respect to modifying the aryl iodide and aziridine, we next explored the applications of this method to the synthesis of compounds with reported biological reactivity. Specifically, we sought to synthesize 16b which has been reported to act as a melanin-concentrating hormone (MCH) antagonist.36 In the original synthesis of 16b, the β-aryl group was introduced early in the synthesis, allowing for late-stage diversification of the benzamide component. Complementary to this strategy, our Ti/Ni dual-catalyzed approach, which relies on early introduction of the benzamide component on the aziridine, would allow for late-stage diversification of the β-aryl group. Indeed, even in the presence of the basic pyrrolidine on the aryl iodide and activated aryl bromide on the aziridine, we successfully accessed 16b in two steps from commercially available starting materials. The key Ti/Ni dual-catalyzed step occurred in 46% yield and 10:1 B:L selectivity under slightly modified conditions.
Mechanistic Investigation.
The unique branched selectivity of this transformation, and its dependence on the size of the 2-alkyl substituent, warranted further investigation. Based on our optimization studies, we hypothesized that Ti is responsible for the activation of the aziridine. We envisioned this activation could occur through either a one- or two-electron pathway (Figure 7). Independent reports from the Gansäuer and Lin labs reported that TiIII induces homolytic cleavage of the more substituted C–N bond by a single electron transfer from TiIII to a coordinated aziridine.20,21 Under our reaction conditions, this radical intermediate could then be trapped by Ni to undergo cross-coupling (Figure 7A). Alternatively, prior studies from our group on the linear cross-coupling of 2-alkyl aziridines and aryl iodides demonstrated the intermediacy of a β-haloamine that forms by halide ring-opening.16 While the prior study favored cleavage of the less sterically hindered C–N bond, we envisioned that in our case Ti could act as a Lewis acid to favor cleavage of the more sterically hindered C–N bond. The resulting β-haloamine could then undergo cross-coupling with Ni (Figure 7B).
Figure 7.

Mechanistic possibilities.
To explore the intermediacy of an alkyl radical, substrate 17a, bearing a radical clock, was synthesized and subjected to the reaction conditions (Figure 8A). Indeed, 17a undergoes sequential cyclizations to generate 17b in 30% yield, providing support for a radical pathway.
Figure 8.

Mechanistic studies into a TiIII catalyzed radical ring-opening. Reactions run on 0.1 mmol scale. aDetermined by GC-FID with an external standard. bDetermined by 1H NMR with an external standard. All free energy calculations are in kcal/mol, and were calculated at the UM06/Def2TZVP//UM06/6– 31G(d,p) [LanL2DZ] level of theory with an SMD solvation model (THF).
We next sought to explore the viability of a Ti-induced radical ring-opening to access this alkyl radical intermediate. In the presence of TiIII and 1,4-cyclohexadiene (1,4-CHD), 1a undergoes a reductive ring-opening to generate 1c-B and 1c-L with a B:L ratio of 4:1. In analogy to a reaction reported by the Gansäuer lab with N-acetyl aziridines,20 this process is initiated by a radical ring-opening followed by hydrogen atom transfer (HAT) with 1,4-CHD. Of note, this reactivity indicates that radical ring-opening is a viable pathway for the generation of both branched and linear products. To explore if the selectivity of this step is contributing to the observed B:L ratio under cross-coupling conditions, we subjected 6a to the reductive ring-opening conditions. Even in the presence of the larger alkyl group, 6c-B and 6c-L were generated in similar B:L ratios indicating that the size of the 2-alkyl substituent does not play a large role in the selectivity of the radical ring-opening (Figure 8B).
While these results indicate the feasibility of radical ring-opening to generate both products, they do not account for the steric trends in the 2-alkyl aziridine scope. Instead, we hypothesized that if radical ring-opening is reversible then radical addition to Ni or reductive elimination from Ni could be regioselectivity determining (Figure 8C). In this Curtin–Hammett scenario, as the size of the 2-alkyl substituent increases, the regioselectivity determining step for branched products, either radical addition to Ni or reductive elimination, would become more challenging due to the steric demand of the larger substituent. In these cases, the linear radical ring-opening becomes more favored.
To probe the possibility of this equilibrium we subjected enantioenriched 6a (>99% ee) and phenyl iodide to the standard reaction conditions. Indeed, recovered 6a at 90% conversion exhibits an erosion of % ee (67% ee) (Figure 8D). These results are consistent with a reversible stereoablative step in the catalytic cycle. Furthermore, DFT calculations confirm that generation of the radical ring-opened intermediate is endergonic for both branched and linear isomers (ΔG = 6.3 kcal/mol and 7.1 kcal/mol respectively). Consistent with the observation of both isomers, both transition states are energetically feasible at room temperature (ΔG‡ = 17.9 kcal/mol and 19.6 kcal/mol respectively) (Figure 8E).
Based on the above data, we propose the following mechanism (Figure 9): the dual-catalytic cycle is initiated by reduction of TiIV to TiIII and reduction of the NiII precatalyst to Ni0 or NiI by Zn. Coordination of the 2-alkyl aziridine to TiIII (IntA) primes the aziridine for a reversible radical ring-opening to IntB. In the Ni cross-coupling cycle, LnNiII(Ar)I (IntC) arises from oxidative addition of Ar2I to either LnNi0 or oxidative addition to LnNiI followed by one-electron reduction.23,24,37–40 In a merger of these two cycles, the radical ring-opened aziridine (IntB) adds to NiII (IntC) to access NiIII (IntD). IntD then undergoes a facile reductive elimination yielding the branched cross-coupled product. Radical addition to Ni or reductive elimination is likely the regiodetermining step in this pathway. As the size of the 2-alkyl substituent increases the interface with the Ni catalytic cycle becomes more challenging and the TiIII radical ring-opening to IntE becomes favored. In an analogous catalytic cycle, IntE interfaces with Ni catalysis to form the linear cross-coupled product.
Figure 9.

Proposed mechanism.
Conclusion
The first branched-selective arylation of 2-alkyl aziridines has been achieved using a dual-catalytic system in which Ti induces a radical ring-opening of the aziridine. Through the complementary approaches of additive screening and feature-driven substrate scope selection, we demonstrated the utility of this method on a diverse aryl iodide and aziridine scope. The diversity of features and reaction outcomes in the scope allowed for the generation of reactivity models that helped guide mechanistic understanding. Mechanistic studies indicate that the TiIII radical ring-opening is reversible and that the interface with Ni catalysis is regiodetermining.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this project was provided by NIGMS R35 GM126986. N.E.G.–V. acknowledges financial support from Princeton University’s Office of Undergraduate Research Summer Internship Program (OURSIP). These studies were supported by shared instrumentation grants from the National Science Foundation under equipment grant no. CHE-1048804 and the NIH Office of Research Infrastructure Programs under grant no. S10OD028644. DFT calculations were performed using resources managed and supported by Princeton Research Computing. We thank Prof. Sun Dongbang, Daniel S. Min, and Dr. Andrzej M. Żurański for helpful discussions.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, spectroscopic data, DFT xyz coordinates and energies, aryl iodide chemical space construction (PDF)
REFERENCES
- (1).Irsfeld M; Spadafore M; Prüß BM β-Phenylethylamine, a Small Molecule with a Large Impact. Webmedcentral 2013, 4 (9), 4409. [PMC free article] [PubMed] [Google Scholar]
- (2).Nieto CT; Manchado A; Belda L; Diez D; Garrido NM 2-Phenethylamines in Medicinal Chemistry: A Review. Molecules 2023, 28 (2), 855. 10.3390/molecules28020855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lewin AH; Navarro HA; Mascarella SW Structure–Activity Correlations for β-Phenethylamines at Human Trace Amine Receptor 1. Bioorgan. Med. Chem. 2008, 16 (15), 7415–7423. 10.1016/j.bmc.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Li S; Huang K; Cao B; Zhang J; Wu W; Zhang X Highly Enantioselective Hydrogenation of β,Β-Disubstituted Nitroalkenes. Angew. Chem. Int. Ed. 2012, 51 (34), 8573–8576. 10.1002/anie.201202715. [DOI] [PubMed] [Google Scholar]
- (5).Liu M; Kong D; Li M; Zi G; Hou G Iridium-Catalyzed Enantioselective Hydrogenation of β,Β-Disubstituted Nitroalkenes. Adv. Synth. Catal. 2015, 357 (18), 3875–3879. 10.1002/adsc.201500723. [DOI] [Google Scholar]
- (6).Kendall JD; Rewcastle GW; Frederick R; Mawson C; Denny WA; Marshall ES; Baguley BC; Chaussade C; Jackson SP; Shepherd PR Synthesis, Biological Evaluation and Molecular Modelling of Sulfonohydrazides as Selective PI3K P110α Inhibitors. Bioorg. Med. Chem. 2007, 15 (24), 7677–7687. 10.1016/j.bmc.2007.08.062. [DOI] [PubMed] [Google Scholar]
- (7).Zhang J; Liu C; Wang X; Chen J; Zhang Z; Zhang W Rhodium-Catalyzed Asymmetric Hydrogenation of β-Branched Enamides for the Synthesis of β-Stereogenic Amines. Chem. Commun. 2018, 54 (47), 6024–6027. 10.1039/c8cc02798f. [DOI] [PubMed] [Google Scholar]
- (8).Cabré A; Verdaguer X; Riera A Enantioselective Synthesis of β-Methyl Amines via Iridium-Catalyzed Asymmetric Hydrogenation of N-Sulfonyl Allyl Amines. Adv. Synth. Catal. 2019, 361 (18), 4196–4200. 10.1002/adsc.201900748. [DOI] [Google Scholar]
- (9).Edwards PM; Schafer LL Early Transition Metal-Catalyzed C–H Alkylation: Hydroaminoalkylation for Csp3–Csp3 Bond Formation in the Synthesis of Selectively Substituted Amines. Chem. Commun. 2018, 54 (89), 12543–12560. 10.1039/c8cc06445h. [DOI] [PubMed] [Google Scholar]
- (10).Sweeney JB Aziridines : Epoxides ‘ Ugly Cousins? Chem. Soc. Rev. 2002, 31 (5), 247–258. 10.1039/b006015l. [DOI] [PubMed] [Google Scholar]
- (11).Dequina HJ; Jones CL; Schomaker JM Recent Updates and Future Perspectives in Aziridine Synthesis and Reactivity. Chem 2023, 9, 1658–1701. 10.1016/j.chempr.2023.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hu XE Nucleophilic Ring Opening of Aziridines. Tetrahedron 2004, 60 (12), 2701–2743. 10.1016/j.tet.2004.01.042. [DOI] [Google Scholar]
- (13).Huang C-YD; Doyle AG The Chemistry of Transition Metals with Three-Membered Ring Heterocycles. Chem. Rev. 2014, 114 (16), 8153–8198. 10.1021/cr500036t. [DOI] [PubMed] [Google Scholar]
- (14).Lin BL; Clough CR; Hillhouse GL Interactions of Aziridines with Nickel Complexes: Oxidative-Addition and Reductive-Elimination Reactions That Break and Make C−N Bonds. J. Am. Chem. Soc. 2002, 124 (12), 2890–2891. 10.1021/ja017652n. [DOI] [PubMed] [Google Scholar]
- (15).Duda ML; Michael FE Palladium-Catalyzed Cross-Coupling of N -Sulfonylaziridines with Boronic Acids. J. Am. Chem. Soc. 2013, 135 (49), 18347–18349. 10.1021/ja410686v. [DOI] [PubMed] [Google Scholar]
- (16).Steiman TJ; Liu J; Mengiste A; Doyle AG. Synthesis of Β-Phenethylamines via Ni/Photoredox Cross-Electrophile Coupling of Aliphatic Aziridines and Aryl Iodides. J. Am. Chem. Soc. 2020, 142 (16), 7598–7605. 10.1021/jacs.0c01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Huang C.-Y. (Dennis); Doyle AG. Nickel-Catalyzed Negishi Alkylations of Styrenyl Aziridines. J. Am. Chem. Soc. 2012, 134 (23), 9541–9544. 10.1021/ja3013825. [DOI] [PubMed] [Google Scholar]
- (18).Dongbang S; Doyle AG Ni/Photoredox-Catalyzed C(sp3)–C(sp3) Coupling between Aziridines and Acetals as Alcohol-Derived Alkyl Radical Precursors. J. Am. Chem. Soc. 2022, 144 (43), 20067–20077. 10.1021/jacs.2c09294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kumar GS; Zhu C; Kancherla R; Shinde PS; Rueping M Metal Cations from Sacrificial Anodes Act as a Lewis Acid Co-Catalyst in Electrochemical Cross-Coupling of Aryl Bromides and Aziridines. ACS Catal. 2023, 8813–8820. 10.1021/acscatal.3c01503. [DOI] [Google Scholar]
- (20).Zhang Y; Vogelsang E; Qu Z; Grimme S; Gansäuer A Titanocene-Catalyzed Radical Opening of N-Acylated Aziridines. Angew. Chem. Int. Ed. 2017, 56 (41), 12654–12657. 10.1002/anie.201707673. [DOI] [PubMed] [Google Scholar]
- (21).Hao W; Wu X; Sun JZ; Siu JC; MacMillan SN; Lin S. Radical Redox-Relay Catalysis: Formal [3+2] Cycloaddition of N-Acylaziridines and Alkenes. J. Am. Chem. Soc. 2017, 139 (35), 12141–12144. 10.1021/jacs.7b06723. [DOI] [PubMed] [Google Scholar]
- (22).Ferraris D; Drury WJ; Cox C; Lectka T “Orthogonal” Lewis Acids: Catalyzed Ring Opening and Rearrangement of Acylaziridines. J. Org. Chem. 1998, 63 (14), 4568–4569. 10.1021/jo980558d. [DOI] [Google Scholar]
- (23).Zhao Y; Weix DJ Nickel-Catalyzed Regiodivergent Opening of Epoxides with Aryl Halides: Co-Catalysis Controls Regioselectivity. J. Am. Chem. Soc. 2014, 136 (1), 48–51. 10.1021/ja410704d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Parasram M; Shields BJ; Ahmad O; Knauber T; Doyle AG Regioselective Cross-Electrophile Coupling of Epoxides and (Hetero)Aryl Iodides via Ni/Ti/Photoredox Catalysis. ACS Catal. 2020, 10 (10), 5821–5827. 10.1021/acscatal.0c01199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Heine HW The Isomerization of Aziridine Derivatives. Angew. Chem. Int. Ed. Engl. 1962, 1 (10), 528–532. 10.1002/anie.196205281. [DOI] [Google Scholar]
- (26).Gensch T; Glorius F The Straight Dope on the Scope of Chemical Reactions. Science 2016, 352 (6283), 294–295. 10.1126/science.aaf3539. [DOI] [PubMed] [Google Scholar]
- (27).Kozlowski MC On the Topic of Substrate Scope. Org. Lett. 2022, 24 (40), 7247–7249. 10.1021/acs.orglett.2c03246. [DOI] [PubMed] [Google Scholar]
- (28).Collins KD; Glorius F A Robustness Screen for the Rapid Assessment of Chemical Reactions. Nat. Chem. 2013, 5 (7), 597–601. 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]
- (29).Bess EN; Bischoff AJ; Sigman MS Designer Substrate Library for Quantitative, Predictive Modeling of Reaction Performance. Proc. National. Acad. Sci. U.S.A. 2014, 111 (41), 14698–14703. 10.1073/pnas.1409522111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kariofillis SK; Jiang S; Żurański AM; Gandhi SS; Martinez Alvarado JI; Doyle AG. Using Data Science To Guide Aryl Bromide Substrate Scope Analysis in a Ni/Photoredox-Catalyzed Cross-Coupling with Acetals as Alcohol-Derived Radical Sources. J. Am. Chem. Soc. 2022, 144 (2), 1045–1055. 10.1021/jacs.1c12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).See SI for additives screened.
- (32).Dreher SD; Krska SW Chemistry Informer Libraries: Conception, Early Experience, and Role in the Future of Cheminformatics. Acc. Chem. Res. 2021, 54 (7), 1586–1596. 10.1021/acs.accounts.0c00760. [DOI] [PubMed] [Google Scholar]
- (33).McInnes L; Healy J; Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. 2018–02-09. arXiv. https://arxiv.org/abs/1802.03426. [Google Scholar]
- (34).McInnes L; Healy J; Saul N; Großberger L UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 29 (3), 861. 10.21105/joss.00861. [DOI] [Google Scholar]
- (35).Żurański AM; Wang JY; Shields BJ; Doyle AG Auto-QChem: An Automated Workflow for the Generation and Storage of DFT Calculations for Organic Molecules. React. Chem. Eng. 2022. 10.1039/d2re00030j. [DOI] [Google Scholar]
- (36).Ishihara Y; Kamata M; Takekawa S Amine Derivative. WIPO Patent WO2004072018. August 26, 2004. [Google Scholar]
- (37).Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1, (9), 830–844. 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]
- (38).Ju L; Lin Q; LiBretto NJ; Wagner CL; Hu CT; Miller JT; Diao T Reactivity of (Bi-Oxazoline)Organonickel Complexes and Revision of a Catalytic Mechanism. J. Am. Chem. Soc. 2021, 143 (36), 14458–14463. 10.1021/jacs.1c07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Greaves ME; Humphrey ELBJ; Nelson DJ Reactions of Nickel(0) with Organochlorides, Organobromides, and Organoiodides: Mechanisms and Structure/Reactivity Relationships. Catal. Sci. Technol. 2021, 11 (9), 2980–2996. 10.1039/d1cy00374g. [DOI] [Google Scholar]
- (40).Tang T; Hazra A; Min DS; Williams WL; Jones E; Doyle AG; Sigman MS Interrogating the Mechanistic Features of Ni(I)-Mediated Aryl Iodide Oxidative Addition Using Electroanalytical and Statistical Modeling Techniques. J. Am. Chem. Soc. 2023, 145 (15), 8689–8699. 10.1021/jacs.3c01726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.