

Significance
Hepatitis B virus (HBV) establishes viral persistence in part through the establishment of a minichromosome, covalently closed circular DNA (cccDNA), in the nucleus of hepatocyte upon infection, which currently available antivirals are unable to target. Here, we identify nucleolin as a host factor associated with cccDNA and its role in cccDNA transcriptional regulation. Targeting nucleolin therapeutically could potentially silence cccDNA transcription ultimately leading to a functional cure for chronic HBV.
Keywords: proteomics, HBV minichromosome, viral replication, antiviral, epigenetics
Abstract
Hepatitis B virus (HBV) remains a major public health threat with nearly 300 million people chronically infected worldwide who are at a high risk of developing hepatocellular carcinoma. Current therapies are effective in suppressing HBV replication but rarely lead to cure. Current therapies do not affect the HBV covalently closed circular DNA (cccDNA), which serves as the template for viral transcription and replication and is highly stable in infected cells to ensure viral persistence. In this study, we aim to identify and elucidate the functional role of cccDNA-associated host factors using affinity purification and protein mass spectrometry in HBV-infected cells. Nucleolin was identified as a key cccDNA-binding protein and shown to play an important role in HBV cccDNA transcription, likely via epigenetic regulation. Targeting nucleolin to silence cccDNA transcription in infected hepatocytes may be a promising therapeutic strategy for a functional cure of HBV.
Hepatitis B virus (HBV) remains a major public health threat worldwide (1). The virus infects hepatocytes and establishes itself within the nucleus as a minichromosome referred to as covalently closed circular DNA (cccDNA), which serves as the transcriptional template for all viral products and is capable of replenishing itself (2). Its stability accounts for the long-term persistence of HBV in the liver of infected patients treated with nucleos(t)ide inhibitors, which are effective in blocking viral replication but have little or no impact on eliminating HBV cccDNA (3). Therefore, identifying cccDNA-associated host factors and their functions is not only important in understanding the basic biology of HBV but also relevant to designing novel strategies to target cccDNA for potential curative therapy (3, 4).
Upon HBV infection, the viral genome undergoes chromatinization concurrently with DNA repair in the nucleus (5). Loaded with histone and nonhistone proteins, HBV cccDNA is maintained stably as a minichromosome in infected hepatocytes (6). Its transcription is under the control of two enhancers and four promoters which contain binding sites for ubiquitous and liver-enriched transcription factors and nuclear receptors. In addition, epigenetic modifications of HBV cccDNA, such as DNA methylation and histone modifications, have been implicated in regulating the transcriptional activity of HBV cccDNA (4, 7).
Current knowledge of cccDNA formation, maintenance, and degradation is still rudimentary, yet the emerging cell culture infection systems and other model systems promise opportunities for major advances in the field. We and others previously demonstrated that the HBV cccDNA minichromosome can be established in different HBV infection cell culture models and is associated with the HBV core protein (8, 9). Here, we isolate and purify cccDNA-associated host proteins from HBV-infected cells and identify nucleolin (NCL) as a key cccDNA-binding protein. NCL is a multifunctional protein that participates in a wide range of cellular processes such as ribosome biogenesis, DNA and RNA metabolism, and cell proliferation (10, 11). Here, we demonstrate that NCL plays an important role in HBV cccDNA transcription via epigenetic regulation.
Results
Identification of cccDNA-associated Host Factors.
As HBV core protein (HBc) is associated with cccDNA, we reasoned that pulling-down HBc with an anti-HBc antibody in nuclear extract of HBV-infected HepG2-NTCP cells would result in cccDNA enrichment. As expected, anti-HBc antibody, but not the IgG control, was able to enrich HBV cccDNA from infected cells (SI Appendix, Fig. S1A). To ensure the specificity of anti-HBc to cccDNA, a host gene (PRNP) was also evaluated in parallel. As shown in SI Appendix, Fig. S1B, anti-HBc antibody showed no enrichment for PRNP relative to the IgG control in both infected and uninfected cells. Based on these results, we reason that anti-HBc antibody binds to and can be used to enrich HBV cccDNA minichromosome from infected cells. Previous studies have demonstrated that HBcAg can also bind to rcDNA and a large number of host gene promoters that can potentially disrupt transcriptional expression (5, 12). Thus, our protocol may enrich additional factors that may not bind cccDNA, like rcDNA-associated host factors. In addition, anti-HBc may cross-react with intracellular HBeAg and its derivatives including p22 and p25, and pull down their-associated factors.
Our general workflow to identify cccDNA-associated proteins is shown in Fig. 1A. Nuclei were isolated from HepG2-NTCP cells infected with 1,000 genome equivalent (GE)/cell of HBV genotype D3 cell culture-derived virus (SI Appendix, Fig. S1C) followed by treatment with or without 1% paraformaldehyde to cross-link weak protein–protein interactions. Following lysis of the infected nuclei, cccDNA was immunoprecipitated with anti-HBc and the resulting pull-down samples were digested into peptides for mass spectrometry identification. A parallel experiment with uninfected HepG2-NTCP cells served as a control. The immunoprecipitated samples were electrophoresed on an SDS-PAGE gel and subjected to silver staining to detect enriched proteins (SI Appendix, Fig. S2A). Although protein bands were observed in the uninfected cell controls, HBV-infected samples appeared to harbor specific and enriched protein bands (SI Appendix, Fig. S2A).
Fig. 1.

Identification of HBV cccDNA-associated proteins. (A) The workflow of HBV cccDNA-associated proteins isolation and identification. HepG2-NTCP cells were infected with HBV for 7 d and nuclei were isolated, either cross-linked and lysed or just lysed. Anti-HBc or unrelated antibody was used to specifically immunoprecipitate the cccDNA minichromosome. The resulting pulldown samples were digested with DNAse and proteins identified through mass spectrometry. Uninfected HepG2-NTCP cells were processed similarly as controls. (B) Result of mass spectrometry analysis. Fold enrichments over IgG control sample with or without cross-linking were plotted. In uninfected HepG2-NTCP cells, no specifically enriched cellular proteins were noted. (C) siRNA studies of 17 selected candidate genes in HBV-infected cells. siRNAs targeting the 17 genes and NTCP (positive control), nontargeting (siNT) control were transfected into HepG2-NTCP cells first and then infected with HBV 2 d later. Five days after infection, HBeAg levels were determined by ELISA. All data are normalized to siNT control sample (set as 100) and shown as means + SEM. The results are representative of three independent experiments. ***P < 0.001.
Mass spectrometry identified 586 proteins enriched greater than threefold in the HBV-infected samples after cross-linking and had a ratio of cross-linking over no cross-linking greater than 4 (Fig. 1B and Dataset S1). Of these, 43 nuclear proteins involved in DNA repair, transcriptional regulation, chromatin formation, or chromatin stability were identified including several known cccDNA-binding proteins such as histones, HNF4A, and SETDB1 (highlighted in Fig. 1B), further validating our approach. As expected, a large number of identified proteins were cytoplasmic, indicating that our pull-down strategy also enriched for proteins not directly associated with cccDNA.
Of the identified proteins, 17 candidates were selected and further tested by siRNA knock-down for validation in HBV-infected cells based on their known functions, biological relevance, and potential novelty in the context of HBV infection. siRNA knock-down for each of the 17 identified genes was performed in HepG2-NTCP cells followed by HBV infection. siRNA targeting NTCP was used as a positive control. HBeAg levels in the supernatant and total HBV RNA levels in the cell lysate were determined 5 days postinfection. Knock-down of several genes resulted in either enhancement or reduction of HBeAg levels in the supernatant (Fig. 1C) and cellular total HBV RNA (SI Appendix, Fig. S2B), suggestive of their functions in either restricting or promoting HBV infection, respectively. Among these candidates, we focused on NCL for further mechanistic investigation due to its potential novel role in HBV infection and known diverse functions in both DNA and RNA metabolisms (10, 11, 13). The identified mass spectrum of the enriched NCL peptides in the mass spectrometry analysis is shown in SI Appendix, Fig. S3.
Nucleolin Binds to and Regulates cccDNA Transcription.
Knock-down of NCL resulted in reduction of HBeAg and HBV DNA (~50% and 60%, respectively) in the culture supernatant, and intracellular HBV RNA (~50%) (Fig. 2C). In addition, knock-down of NCL did not show any effect on HBV cccDNA levels (Fig. 2 C and D), suggesting a role of NCL in viral transcription or posttranscriptional regulation. Greater than 90% knock-down of both NCL mRNA and protein levels in the absence of cellular cytotoxicity were achieved following siRNA treatment in HBV-infected HepG2-NTCP cells (Fig. 2 A and B and SI Appendix, Fig. S4A). HBV infection neither affected NCL mRNA levels in both HepG2-NTCP cells and primary human hepatocytes (PHH) (SI Appendix, Fig. S4B), nor changed NCL subcellular distribution (SI Appendix, Fig. S4C). The effect of siNCL knock-down on HBV replication could be rescued by subsequent transfection of an NCL expression plasmid (Fig. 2E). Attempt to generate NCL knock-out cell lines by CRISPR/Cas9 failed, presumably as NCL is essential for cell growth and host DNA replication (11). These data collectively indicate that NCL plays a role in regulating the transcriptional activity of cccDNA.
Fig. 2.

Characterization of nucleolin in HBV replication. siRNA against NCL were transfected into HepG2-NTCP cells; (A) mRNA and (B) protein levels of NCL were evaluated by qPCR (normalized to the siNT control) and western blot, respectively. (C) Five days after HBV infection, various HBV infection markers including cccDNA, HBV RNA (total), HBeAg, and HBV DNA in cell culture supernatant were determined and levels normalized to the siNT control. (D) The amount of HBV cccDNA was further determined by Southern blot. (E) HepG2-NTCP cells were transfected with siRNA against NCL or control and then transfected with or without plasmid overexpressing NCL. After HBV infection, HBeAg in cell culture supernatant were determined by ELISA. *P < 0.05 and ***P < 0.001.
To further investigate the interaction of NCL with cccDNA, we performed chromatin-immunoprecipitation (ChIP) in HBV-infected HepG2-NTCP cells followed by qPCR with primers specific for cccDNA and H3K4me3, a well-known cccDNA binding protein used as a positive control (7). Our results demonstrate a clear enrichment of NCL and H3K4me3 over input and the IgG control (SI Appendix, Fig. S5A), indicating a specific binding of NCL to the HBV minichromosome compared to the host genome as H3K4me3 binding is widely distributed across the human genome (7, 14). Similar results were obtained with HBV-infected PHH and livers of the human liver chimeric HBV-infected Alb-uPA/Scid mice (SI Appendix, Fig. S5 B and C).
Nucleolin Is Enriched on CpG Islands of cccDNA and Involved in Histone Modification.
We next sought to map out where on the cccDNA genome NCL specifically binds to using ChIP-sequencing. As shown in Fig. 3A, the relative abundance of NCL associated with cccDNA was significantly higher and displayed a different pattern than the input in HBV-infected HepG2-NTCP cells, confirming our finding of NCL enrichment on cccDNA by ChIP-qPCR. The main regions of NCL enrichment on cccDNA occur at HBV CpG island I and II regions where DNA methylation can occur and is associated with downregulation of cccDNA transcription (15, 16). We therefore speculated that NCL may be involved in HBV cccDNA methylation; DNA methylation analysis, however, did not reveal any difference in DNA methylation of cccDNA between sample with or without NCL knock-down.
Fig. 3.
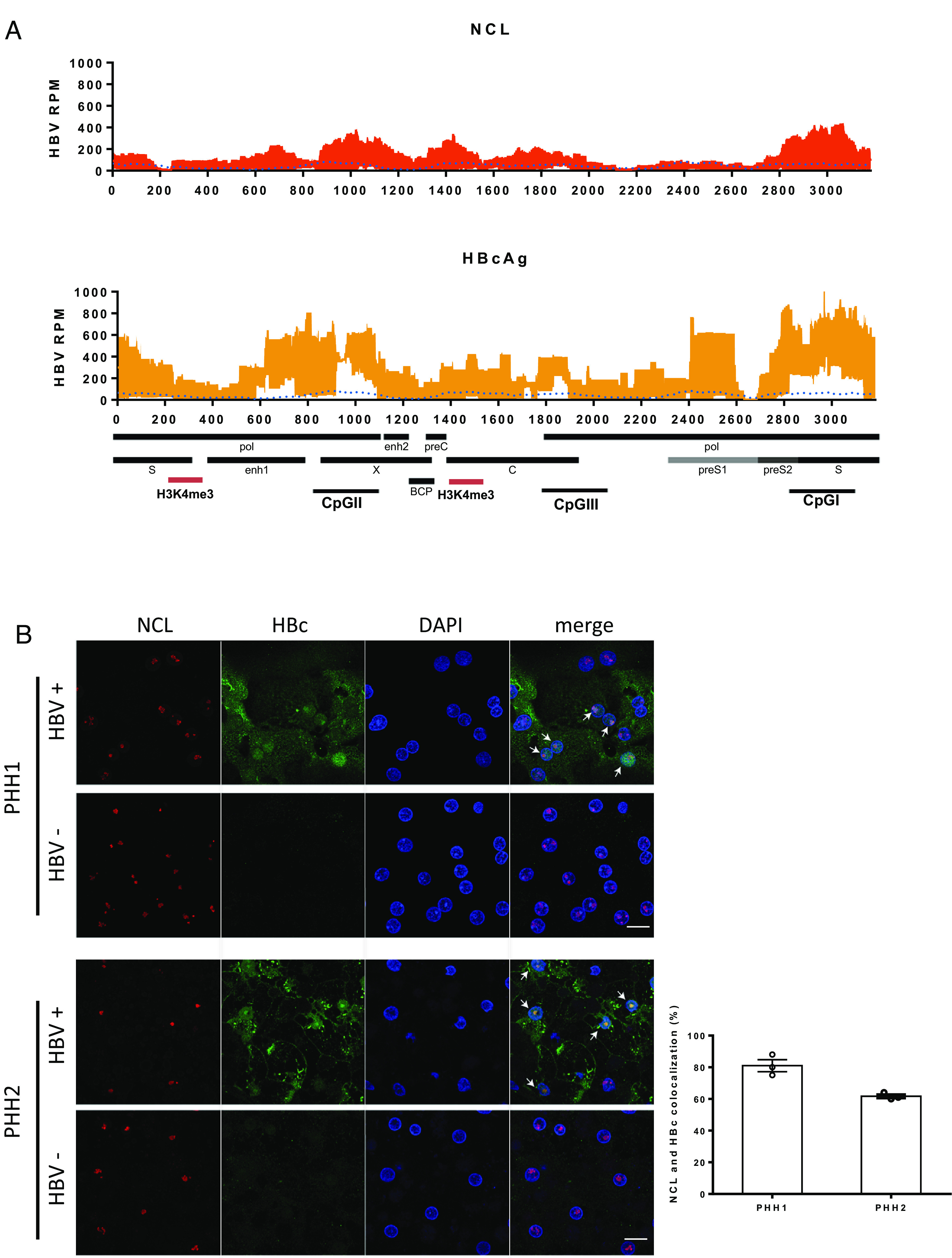
Distribution of nucleolin on cccDNA and in HBV-infected cells. (A) Distribution of nucleosomes and protein binding sites were determined by cccDNA ChIP sequencing across the HBV genome. HBV cccDNA ChIP sequencing experiment was performed by using NCL or HBc antibody. Read density for each track is represented on the y-axis as HBV-derived reads per million total reads (HBV RPM). Averages and SDs from three independent experiments are shown. The signal of input is blotted as the blue dotted line. HBV transcripts, enhancer elements, CpG islands, basal core promoter (BCP), and highly enriched H3K4me3 binding sites (red) on HBV genome are schematically displayed below the x-axis. The results are representative of three independent experiments. (B) Confocal images of immunofluorescence staining for NCL and HBc. PHHs infected with or without HBV from two different donors were fixed and stained with antibodies against NCL (red) and HBc (green). The percentage of cells with NCL and HBc colocalization in HBc-positive cells was determined by counting three random images from each group. The results are representative of three independent experiments (Scale bar, 20 μm).
The distribution of NCL also overlapped with several regions of HBc binding (Fig. 3A; r2 = 0.637). We therefore investigated whether these two proteins indeed colocalize within infected cells by confocal imaging. In HBV-infected PHHs, we found that HBc and NCL colocalized in the nuclei of hepatocytes (Fig. 3B). While they both bound to cccDNA and shared similar distribution on the genome by ChIP-Seq, knock-down of NCL resulted in reduced abundance of NCL on cccDNA but not HBc (SI Appendix, Fig. S6). This result suggested that the binding of HBc to HBV cccDNA is not dependent on NCL.
The NCL binding sites on cccDNA (Fig. 3A) overlap with several previously mapped histone posttranslational modification sites (7). Histone acetyltransferases, such as H3K4me3 and H3K27ac, are highly enriched on transcriptionally active HBV minichromosome and likely enhance HBV transcription via epigenetic modifications (7). We therefore investigated whether NCL regulates cccDNA transcription through interaction with various histones and epigenetic regulation. Proximity ligation assay showed a close association of NCL with the epigenetic markers of transcriptionally active cccDNA (H3K4me3 and H3K27ac) but not the transcriptionally repressive histone marker H3K27me3 (Fig. 4A). Interestingly, HBV cccDNA ChIP-Seq analysis showed that siRNA knock-down of NCL altered the distribution of H3K4me3 binding with increased binding to enh1 and CpG II/III regions but decreased binding to other regions (upstream of enh1 and preS2/S transcription start site) (Fig. 4B). Total H3K4me3 binding to HBV cccDNA as determined by ChIP-qPCR also did not show any difference between control and siNCL samples (Fig. 4C). Binding of inhibitory H3K9me3 to cccDNA was undetectable in both samples (Fig. 4B), suggesting that NCL may affect the binding and distribution of certain histone modifying enzymes on cccDNA for transcriptional regulation.
Fig. 4.
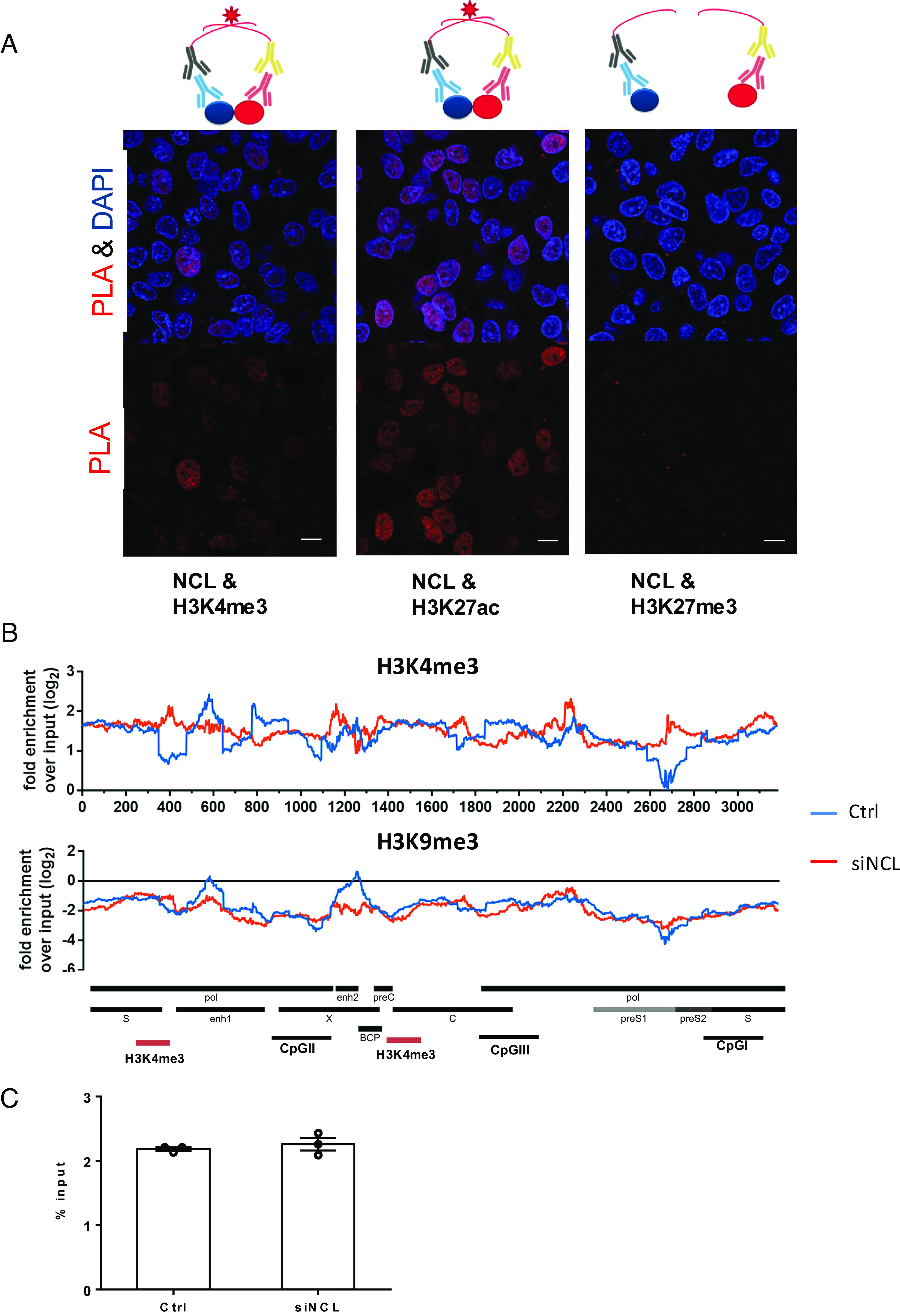
Interaction of nucleolin with HBV cccDNA. (A) Proximity ligation assay (PLA) to analyze the interaction of NCL and histones. The assay is schematically shown on the top of the figure. Antibodies against NCL or different histones were used in proximity ligation assay to determine their interaction in HBV-infected cells. HBV-infected HepG2-NTCP cells were fixed and incubated with NCL or histone antibodies. Secondary antibodies coupled with PLA probes were added and ligated. PLA signals are detected by fluorescent microscopy as discrete spots (in red) and provide the intracellular localization of the protein interaction (Scale bar, 10 μm). (B) Distribution of nucleosomes and H3K4me3 binding sites along the HBV genome in HBV-infected cells with or without NCL knockdown by ChIP-Seq. Read density for each track is represented by height on the y axis scaled to a minimum of 100 reads. Data are shown as fold-enrichment over input for each genome position, and various HBV genomic landmarks are displayed as in Fig. 3A. (C) The overall H3K4me3 signals on the HBV cccDNA were accessed by ChIP-qPCR and shown as % input.
The Effect of Nucleolin on HBV Infection is Not Dependent on HBcAg.
Next, we investigated whether HBc plays a role in the transcriptional regulatory effect of NCL. To investigate this, we utilized the HBVcircle, a close mimic of HBV cccDNA which supports cccDNA-dependent transcription (17, 18). Following siRNA knockdown of NCL, wide-type (WT) or HBc- (HBc-defective) HBVcircle were transfected into Huh7 and HBV viral markers were evaluated. Knock-down was confirmed by qPCR (Fig. 5A) and western blot (Fig. 5B). While the expression of NCL showed no difference between WT and HBc- samples, NCL knockdown resulted in lower HBc protein levels (Fig. 5B) as well as reduced HBsAg and HBeAg levels in the supernatant of both WT and HBc-HBVcircle transfected cells (Fig. 5C). Intracellularly, both WT and HBc- HBVcircle generated similar amounts of HBV RNA and cccDNA (Fig. 5D). NCL knockdown significantly reduced HBV RNA in both wild-type and HBc- HBVcircle transfected cells (Fig. 5D). In addition, NCL showed similar binding by ChIP-qPCR to wild-type and HBc- HBVcircle (Fig. 5E). These data indicated that HBc is dispensable for the transcriptional regulatory effect of NCL.
Fig. 5.
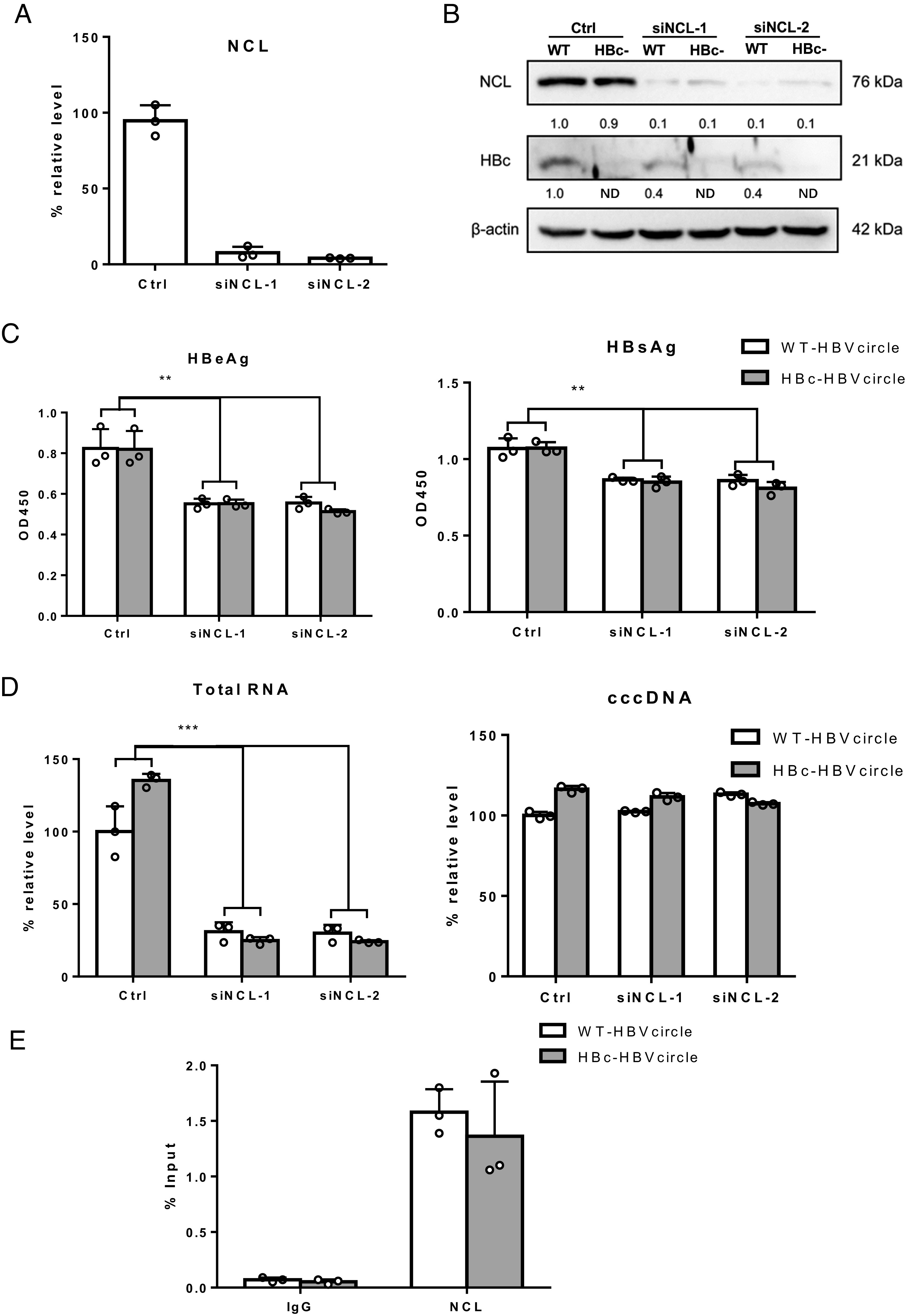
The role of HBc in NCL-mediated transcriptional regulatory effect. (A) Huh7 cells were transfected with siRNA and 2 d later were transfected with WT-HBVcircle or HBc-HBVcircle. Three days later, NCL mRNA were evaluated by qPCR. (B) The expression of HBc and NCL protein was detected by western blot. (C) The levels of HBeAg and HBsAg in supernatants were measured by ELISA. (D) The relative levels of HBV RNA and HBVcircle DNA were determined by qPCR. (E) Chromatin immunoprecipitation was performed using lysates of Huh7 cells transfected with WT-HBVcircle or HBc-HBVcircle. Immunoprecipitation of HBVcircle using antibody against NCL or IgG control was analyzed by qPCR and shown as % input. **P < 0.01 and ***P < 0.001.
To assess whether NCL and HBcAg interact directly in the absence of HBV replication or cccDNA, we transfected NCL and HBcAg expression constructs and performed confocal image analysis for colocalization and coimmunoprecipitation experiments (SI Appendix, Fig. S7), which did not show any direct interaction between the two proteins.
HBx Is Functionally Involved in Nucleolin and HBV Infection.
HBx is essential to initiate and maintain the transcriptional activity of cccDNA via interaction with the SMC5/6 complex after infection (19, 20). To study the role of NCL in HBx-defective HBV mutant (HBVx-) virus infection, cccDNA ChIP-qPCR with either anti-HBc or anti-NCL was performed in both HepG2-NTCP cells and PHHs infected with either HBVwt or HBVx- virus (Fig. 6A). While cccDNA can be established by both HBVwt and HBVx-, HBVx- cccDNA is transcriptionally silent and does not express the viral antigens including HBcAg (19). Interestingly, HBx- cccDNA did not show much binding to NCL nor HBcAg by ChIP-qPCR (Fig. 6A). To further investigate the role of HBcAg in the binding of NCL to cccDNA, we complemented HBVwt and HBVx- infected HepG2-NTCP cells with delivery of HBcAg or control GFP via adenoviral vector and then performed ChIP-qPCR with anti-NCL. HBV RNA and HBeAg levels confirmed the phenotype of lower transcription activity in the absence of HBx that was not restored by HBcAg complementation (Fig. 6B). Consistent with Fig. 6A, NCL binding to cccDNA was also significantly reduced in the HBVx- samples (comparing HBVwt + AdGFP vs. HBVx- + AdGFP) (Fig. 6C). As expected, HBcAg overexpression was not able to restore NCL binding to the HBVx- cccDNA (comparing HBVx- + AdGFP vs. HBVx- + AdHBc)(Fig. 6C). Similar results were obtained in PHHs (Fig. 6D).
Fig. 6.

The role of HBx in NCL-mediated transcriptional regulatory effect. (A) Chromatin immunoprecipitation was performed using lysates of HepG2-NTCP and PHHs infected with wide-type HBV (HBVwt) or X- HBV (HBVx-). Immunoprecipitation of cccDNA minichromosome using antibody against HBc or NCL was analyzed by qPCR. The results are representative of three independent experiments. (B and C) HepG2-NTCP and (D) PHHs were infected with wide-type HBV (HBVwt) or X- HBV (HBVx-) and transduced with adenoviral vectors expressing HBc or GFP control. HBV RNA and HBeAg from infected HepG2-NTCP were determined. Immunoprecipitation of cccDNA minichromosome using antibody against NCL was analyzed by qPCR. (E and F) HepG2-NTCP cells were infection with X- HBV (HBVx-) and transduced with adenoviral vectors expressing HBx or GFP control the next day. Culture supernatant and cells were harvested on day 8 post-HBV infection. (E) HBsAg and HBeAg were determined by ELISA. (F) Immunoprecipitation of cccDNA using antibody against NCL was analyzed by qPCR and shown as % input (subtracting % input of IgG control). *P < 0.05, **P < 0.01, and ***P < 0.001.
Next, we tested whether HBx expression can restore NCL binding to HBVx- cccDNA. We infected HepG2-NTCP cells with HBVx- and reconstituted HBx expression with adenoviral vector. As expected, HBx restored viral protein expression (Fig. 6E). Interestingly, the amounts of NCL binding to cccDNA, as determined by ChIP-qPCR, were also restored by reconstitution of HBx expression in the HBVx- samples (Fig. 6F). Similar results were also observed with the HBVcircle model. WT or HBx-HBVcircle were cotransfected with an HBx expressing or a control plasmid. HBx expression elevated HBeAg levels (SI Appendix, Fig. S8A) and NCL binding to HBVcircle (SI Appendix, Fig. S8B, HBx-HBVcircle + HA vs. HBx-HBVcircle + HA-HBx) in HBVx-HBVcircle transfected cells. Together, these results suggest that NCL binding to HBV cccDNA is dependent on HBx but not HBcAg.
HBx has been shown to exert a promiscuous transcriptional effect on episomal but not chromosomal promoters (21). To assess the role of NCL on general transcription, we tested the effect of NCL knock-down on the HBV enhancer/promoter as well as a heterologous viral promoter (CMV), which has previously been shown to be up-regulated by HBx (22). Knock-down of NCL showed no transcriptional effect on either episomal HBV enhancer/promoter or CMV promoter activity (SI Appendix, Fig. S9A). We also tested the effect of NCL knock-down in HepG2-DE19 cells which contain a chromosomally integrated replication-competent HBV genome under the condition that little or no cccDNA is present (23, 24) and no effect was observed on the chromosomally integrated HBV genome (SI Appendix, Fig. S9B). In addition, ChIP assay did not show any specific enrichment of NCL on chromosomally integrated HBV genome (SI Appendix, Fig. S9C), indicating that NCL likely exerts a specific regulatory effect in the context HBV cccDNA minichromosome.
Finally, to assess whether NCL and HBx interact with each other, we transfected NCL and HBx expression constructs and performed confocal image analysis for colocalization and coimmunoprecipitation experiments (SI Appendix, Fig. S10). We could not observe any interaction between the two proteins.
Discussion
HBV cccDNA plays a central role in the establishment of viral infection and persistence. Its long-term stability is the basis for viral rebound after the cessation of antiviral therapy. There are limited therapeutic treatments for chronic HBV infection, and to date, no available therapies are able to significantly increase the cure rate for chronic HBV infection. The major limitation of current treatment is the failure to eliminate the preexisting cccDNA pool and/or prevent cccDNA formation from low-level viral replication in the presence of antivirals. Thus, there is an urgent need for the development of novel therapeutic agents that directly target cccDNA formation and maintenance (25).
In this study, we utilized HBcAg-specific antibody to enrich cccDNA from HBV-infected hepatocytes as HBcAg has previously been shown to bind to and be an important component of cccDNA minichromosome (6). However, in addition to the nucleus, HBcAg has also been shown to be widely distributed in the cytoplasm of infected cells and known to interact with several host proteins. Thus, proteins which bind to HBcAg, but not cccDNA, could also be enriched (26). In this study, we also identified several HBc-binding proteins reported previously (27, 28). In addition, due to the C-terminal domain of HBcAg’s nucleic acid binding ability, HBcAg also binds to rcDNA (19). The mass spectrometry analysis revealed over 500 potential hits, including many cytoplasmic proteins that are likely not associated with cccDNA. However, of the hits identified, nuclear proteins known to interact with cccDNA, such as histones, HNF4A, and SETDB1 (4), were identified, lending credence to the enrichment of some authentic cccDNA-associated proteins by this protocol. Functional screening by siRNA knock-down confirmed 3 of 17 of the putative cccDNA-interacting hits tested as having a functional role in HBV infection. Among the validated host factors, we selected nucleolin for further study.
NCL is a well-studied multifunctional protein that is mainly localized in the nucleolus but is also found in the cytoplasm and on the cell membrane (13). NCL is involved in several cellular processes, including ribosome biogenesis, DNA and RNA metabolism, regulation of cell proliferation and apoptosis, and cellular stress responses (10, 11). In addition, NCL has been shown to modulate the transcription of host mRNAs in several ways: It represses RNA polymerase II function through its interaction with chromatin components and chromatin remodeling enzymes (29) and by directly interacting with DNA (29).
NCL has also been shown to play a role in the pathogenicity of many viruses. Cell surface NCL has been demonstrated to participate in both the entry and internalization of several viruses into host cells including the human respiratory syncytial virus (30), enterovirus 71 (31), influenza A viruses (32–34), rabbit hemorrhagic disease virus (35), and the HIV type 1 (HIV-1) (36). HB-19 pseudopeptides, which have been developed to target NCL on the cell surface as an anti-NCL agent, can inhibit attachment of HIV-1 to the cell surface as well as subsequent viral entry into the host cell (37). In addition to viral entry, NCL has also been involved in viral replication. Human cytomegalovirus requires NCL to maintain the proper architecture of its replication compartment (38). NCL also seems to be required for episome maintenance, viral transcription, and replication of Epstein–Barr virus (EBV) (39), which is highly analogous to the functional relationship of NCL and HBx in HBV replication as described above. In this study, while greater than 90% knock-down of NCL could be achieved, only a 50% reduction in HBV gene expression was observed (Fig. 2 A–C). In addition, over-expression of NCL did not further increase HBV gene expression (Fig. 2E). It is conceivable that only a low level of NCL is required for the function of NCL in HBV replication and a much higher knock-down level of NCL is necessary to achieve a more potent suppression of HBV expression.
Our ChIP-Seq data demonstrated binding of NCL to various regions of the HBV cccDNA including the enhancer/promoter elements. In cells with NCL knock-down, HBV can still infect and form cccDNA, but its transcriptional activity is much reduced. We showed that histone acetyltransferases, such as H3K4me3 and H3K27ac that are enriched on transcriptionally active HBV minichromosome (7), appear to be functionally involved with NCL. It is interesting to note that NCL knock-down affects the distribution of H3K4me3 binding without affecting the overall binding to HBV cccDNA (Fig. 4B and SI Appendix, Fig. S7). Thus, NCL may exert its positive effect on HBV transcription via selective epigenetic modifications. HBV infection does not alter the subcellular localization of NCL, suggesting that activation of cccDNA may be associated with its relocation in the nucleus as reported recently (40).
Although HBcAg had been thought to play a role in HBV cccDNA activities, accumulating evidence suggests that HBc is not required for cccDNA transcriptional regulation. HepG2-NTCP cells infected with HBVwt and HBc-deficient HBV produce similar amounts of cccDNA, viral RNA and HBsAg during a 3-wk observation period (41, 42). In addition, in cccDNA transfection model and AAV-mediated HBV cccDNA mouse model, HBc is not required for cccDNA transcriptional regulation (43). Consistent with these observations, we showed that similar amounts of HBV RNA and viral antigens were obtained from wide-type and HBc- HBVcircle transfected cells. Furthermore, HBcAg, while showing colocalization with NCL in the infected cells, is not needed for NCL-mediated cccDNA transcription.
Our study demonstrated that HBx expression in HBVx- virus-infected cells can restore NCL composition on the cccDNA. We also showed that knock-down of NCL reduces cccDNA transcription without affecting the binding of HBc to cccDNA. Thus, recruitment of NCL to the cccDNA minichromosomes is likely dependent on HBx. HBx is known to promote viral transcription through the interaction and downregulation of the HBV restriction factor SMC5/6 complex (20). It is not clear whether SMC5/6 is involved here or how HBx interacts with NCL either directly or indirectly to regulate HBV transcription. Future studies are necessary to address this finding.
Here, we show that nucleolin binds to the HBV cccDNA minichromosome and plays an important role in the regulation of HBV transcription. The likely mechanism of epigenetic regulation whereby nucleolin interacts with and functionally modulates the epigenetic machinery of cccDNA remains to be further elucidated. This proposed mechanism is consistent with our finding that HBx mediates the recruitment of nucleolin to a transcriptionally active cccDNA minichromosome. With the goal of silencing cccDNA in infected hepatocytes, targeting nucleolin for epigenetic modulation may be a promising therapeutic strategy for a functional cure of HBV.
Materials and Methods
HBV Infection.
HBV stock was concentrated from the supernatant of HepG2-DE19 cells using centrifugal filter devices (Centricon Plus-70, Millipore Corp.) and quantified by HBV-DNA qPCR. HBx-minus HBV (HBVX-) was generated as described previously (19). For infection, inoculation of cells was performed with multiplicity of infection (MOI) 300 or 1,000, as indicated in the figure legend, in Williams E medium (WEM, 12551032, Gibco|Thermo Fisher Scientific) containing 5% PEG 8000 (Sigma-Aldrich) for 16 h. Then, cells were washed three times with PBS and cultured in WEM medium. Infected cells were cultured with WEM media exchanges as described (8, 44). Cryopreserved PHH (Lot# Hu1894) were purchased from Gibco|Thermo Fisher Scientific.
HBVcircle Synthesis.
For HBVcircle production in vitro, Minicircle DNA Technology (Roche) was performed according to the manufacturer’s protocols (17). Original constructs were gifted by Roche R&D Center (China) Ltd. with material transfer agreement.
Chromatin Isolation for ChIP-qPCR or Sequencing.
Infected HepG2-hNTCP cells were fixed in freshly prepared 1% formaldehyde in PBS (both Thermo Fisher Scientific) for 5 min prior to quenching with 125 mM glycine in PBS. Pelleted cells were either used directly for analysis or snap-frozen and stored at −80 °C until use. Next, cells were washed with cold lysis buffer [PBS with 0.1% Triton X-100, 0.1% NP-40, 1 mM dithiothreitol (DTT), 50 ng/mL trichostatin A (TSA) to inhibit histone deacetylases, and 1x EDTA-free protease inhibitor (Roche)] and lysed in the same buffer for 10 min on ice. Nuclei were pelleted, resuspended in digestion buffer [H2O with 50 mM Tris-HCl pH 7.5, 4 mM MgCl2, 1 mM CaCl2, 10% glycerol, 50 ng/mL TSA, and 1x EDTA-free protease inhibitor (Roche)], and digested with 600 IU/mL micrococcal nuclease (MNase; ThermoFisher Scientific) for 12 min at 37 °C. Digestion was stopped by addition of 10 mM EDTA. Nuclei were pelleted at 6,500×g, and supernatants were collected. The pellet was resuspended in digestion buffer with 10 mM EDTA and 300 mM NaCl and mildly sonicated using a W 375 sonicator (Qsonica) at 50% duty cycle and power setting 3. Nuclei were again pelleted, supernatants combined and mixed with an equal amount of sucrose buffer [H2O with 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM EDTA, 0.01% NP-40, 50 ng/mL TSA, and 1x EDTA-free protease inhibitor (Roche)]. Samples were concentrated using Amicon Ultra-4 100 kDa centrifugal filter units (Millipore-Sigma; Merck KGaA) and spun on a 5–30% continuous sucrose gradient in sucrose buffer for 4 h at 40,000×g and 4 °C using a SW41Ti rotor (Beckman Coulter Inc.). Chemicals were obtained from Millipore-Sigma unless noted otherwise. Mononucleosome-containing fractions were identified by agarose gel-electrophoresis, pooled, and concentrated to ~500 µL prior to addition of 100 ng/µL bovine serum albumin (Thermo Fisher Scientific). Mononucleosomes were either stored at −20 °C or used immediately for ChIP. ChIP-seq raw data were deposited to GEO (GSE123715).
Detection of HBV Antigens by ELISA.
Secreted HBeAg and HBsAg were determined using Human HBeAg Elisa Kit (CD BioScience) and Human HBsAg Elisa Kit (CD BioScience), respectively. The signal-to-noise ratio (S/N) is presented in the figures.
Quantitative Real-time PCR.
The protocol has been described previously (8, 44). For intracellular HBV total DNA and cccDNA quantification, cellular DNA was isolated from infected cells using the NucleoSpin Tissue Tissue kit (Macherey-Nagel). HBV total DNA qPCR was performed using rcDNA1745 fw/rcDNA1844 rev primer pair. DNA samples for cccDNA qPCR were treated with 500 U/mL T5 exonuclease (NEB) at 37 °C for 30 min. cccDNA specific primer pair cccDNA 92 fw and cccDNA 2251 rev were used for qPCR as described. Extracellular HBV DNA quantification was performed with DNA extraction using the DNeasy Blood & Tissue Kit (Qiagen). rcDNA1745 fw / rcDNA1844 rev primer pair were used for qPCR. For qRT-PCR, RNA was extracted using the RNeasy kit (Qiagen) and transcribed into cDNA with the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific) according to the manufacturer’s instructions. qPCR was performed using the LightCycler™ 480 system with SYBR Green Master (Roche) or Probe Master (Roche). A relative unit, defined as the expression ratio of target gene against the reference housekeeping gene (ACTB for RNA; PRNP for DNA), is presented.
siRNA Knock-down.
siRNAs were obtained from Thermo Fisher Scientific Dharmacon®. Cells from one well of a 24-well plate were transfected with 25 nM siRNA using 2 μL RNAiMAX (Invitrogen) according to the manufacturer’s instructions.
Western Blot.
The protocol has been described previously (8, 44). Lysates from PHH cells were obtained by adding 100 μL “M-PER Mammalian Protein Extraction Reagent” (Thermo Scientific) onto cells per well and incubate at 37 °C for 5 min. Fifty microliter 3X SDS sample buffer (Aviva Systems Biology Corporation) was added, and samples were incubated at 99 °C for 5 min. Protein lysates were separated on a NuPAGE 4–12% Bis-Tris Gel (Life Technologies Corporation) and then blotted onto a nitrocellulose membrane (iBlot 2 NC mini Stacks, Thermo Scientific) by using iBlot (Thermo Scientific). The membrane was blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for 1 h at room temperature followed by overnight incubation with primary antibody at 4 °C. Anti-rabbit or anti-mouse secondary antibody conjugated to horseradish peroxidase (Sigma) was used to visualize the stained bands with an enhanced chemiluminescence visualization kit (Thermo Scientific). Antibodies: Nucleolin (Cell Signaling Technology, #14574S), HA (Cell Signaling Technology, #3724S).
Southern Blot.
DNA from cells was extracted by the Hirt procedure as described (45). Briefly, DNA sample was subjected to 1.2% agarose gel electrophoresis and transferred onto Amersham Hybond-N + membrane (GE Healthcare). Then, the membrane was cross-linked by UV, followed by hybridization with HBV probes prepared by Random Primer DNA Labeling Kit Ver. 2 (Takara Bio). Finally, the hybridization signal was captured by a Typhoon FLA 9500 imager (GE Healthcare Lifesciences).
Statistical Analysis.
Student’s unpaired two-tailed t tests were performed with GraphPad Prism 7 (GraphPad Software). Data are mean ± SD. Two-sided P values < 0.05 were considered significant. *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (PDF)
Dataset S02 (PDF)
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH, USA (to T.J.L.), and the National Natural Science Foundation of China (Project No. 81971936, to Y.X.). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD042733 and 10.6019/PXD042733.
Author contributions
Y.X., X.C., and T.J.L. designed research; Y.X., X.C., T.N., M.Z., G.Z., T.I., Y.T., Y.L., D.E.A., and M.H. performed research; T.N., M.Z., T.I., Y.T., Y.L., D.E.A., M.H., and T.J.L. contributed new reagents/analytic tools; Y.X., X.C., T.N., M.Z., G.Z., D.E.A., M.H., and T.J.L. analyzed data; T.J.L. provided funding; and Y.X., X.C., and T.J.L. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Yuchen Xia, Email: yuchenxia@whu.edu.cn.
T. Jake Liang, Email: Jake.Liang@nih.gov.
Data, Materials, and Software Availability
Supporting Information
References
- 1.Hutin Y., et al. , Access to treatment for hepatitis B virus infection-worldwide, 2016. Am. J. Transplant. 18, 2595–2598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liang T. J., et al. , Present and future therapies of hepatitis B: From discovery to cure. Hepatology 62, 1893–1908 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xia Y., Liang T. J., Development of direct-acting antiviral and host-targeting agents for treatment of hepatitis B virus infection. Gastroenterology 156, 311–324 (2018), 10.1053/j.gastro.2018.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xia Y., Guo H., Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antiviral Res. 180, 104824 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiner S., Nassal M., A role for the host DNA damage response in hepatitis B virus cccDNA formation-and beyond? Viruses 9, 125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bock C. T., et al. , Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 307, 183–196 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Tropberger P., et al. , Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. U.S.A. 112, E5715–E5724 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y., et al. , Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus–host interactions. J. Hepatol. 66, 494–503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucifora J., et al. , Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343, 1221–1228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tajrishi M. M., Tuteja R., Tuteja N., Nucleolin: The most abundant multifunctional phosphoprotein of nucleolus. Commun. Integr. Biol. 4, 267–275 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia W., Yao Z., Zhao J., Guan Q., Gao L., New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 186, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Guo Y., et al. , Hepatitis B viral core protein disrupts human host gene expression by binding to promoter regions. BMC Genomics 13, 563 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuteja R., Tuteja N., Nucleolin: A multifunctional major nucleolar phosphoprotein. Crit. Rev. Biochem. Mol. Biol. 33, 407–436 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Bannister A. J., Kouzarides T., Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Y. H., Li Y. N., Zhao J. R., Zhang J., Yan Z., HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics 6, 720–726 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y., et al. , Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS ONE 9, e110442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Z., et al. , HBVcircle: A novel tool to investigate hepatitis B virus covalently closed circular DNA. J. Hepatol. 66, 1149–1157 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Murphy C. M., et al. , Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 16, 2846–2854 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lucifora J., et al. , Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 55, 996–1003 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Decorsiere A., et al. , Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389 (2016). [DOI] [PubMed] [Google Scholar]
- 21.van Breugel P. C., et al. , Hepatitis B virus X protein stimulates gene expression selectively from extrachromosomal DNA templates. Hepatology 56, 2116–2124 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Assogba B. D., Choi B. H., Rho H. M., Transcriptional activation of the promoter of human cytomegalovirus immediate early gene (CMV-IE) by the hepatitis B viral X protein (HBx) through the NF-kappaB site. Virus Res. 84, 171–179 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Guo H., et al. , Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 81, 12472–12484 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai D., et al. , Establishment of an inducible HBV stable cell line that expresses cccDNA-dependent epitope-tagged HBeAg for screening of cccDNA modulators. Antiviral Res. 132, 26–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia Y., Liang T. J., Development of direct-acting antiviral and host-targeting agents for treatment of hepatitis B virus infection. Gastroenterology 156, 311–324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viswanathan U., et al. , Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B. Antiviral Res. 182, 104917 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y., et al. , Hepatitis B virus hijacks TSG101 to facilitate egress via multiple vesicle bodies. PLoS Pathog. 19, e1011382 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim E. S., et al. , Hepatitis B virus X protein counteracts high mobility group box 1 protein-mediated epigenetic silencing of covalently closed circular DNA. PLoS Pathog. 18, e1010576 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdelmohsen K., Gorospe M., RNA-binding protein nucleolin in disease. RNA Biol. 9, 799–808 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tayyari F., et al. , Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 17, 1132–1135 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Nakarai C., et al. , Expression of AKR1C3 and CNN3 as markers for detection of lymph node metastases in colorectal cancer. Clin. Exp. Med. 15, 333–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan Y., Du Y., Wang G., Li K., Non-structural protein 1 of H3N2 influenza A virus induces nucleolar stress via interaction with nucleolin. Sci. Rep. 7, 17761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar D., Broor S., Rajala M. S., Interaction of host nucleolin with influenza A virus nucleoprotein in the early phase of infection limits the late viral gene expression. PLoS ONE 11, e0164146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murayama R., et al. , Influenza A virus non-structural protein 1 (NS1) interacts with cellular multifunctional protein nucleolin during infection. Biochem. Biophys. Res. Commun. 362, 880–885 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Zhu J., et al. , Nucleolin mediates the internalization of rabbit hemorrhagic disease virus through clathrin-dependent endocytosis. PLoS Pathog. 14, e1007383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Said E. A., et al. , Pleiotrophin inhibits HIV infection by binding the cell surface-expressed nucleolin. FEBS J. 272, 4646–4659 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Nisole S., et al. , The anti-HIV pentameric pseudopeptide HB-19 binds the C-terminal end of nucleolin and prevents anchorage of virus particles in the plasma membrane of target cells. J. Biol. Chem. 277, 20877–20886 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Strang B. L., Boulant S., Kirchhausen T., Coen D. M., Host cell nucleolin is required to maintain the architecture of human cytomegalovirus replication compartments. mBio 3, e00301-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y. L., et al. , Nucleolin is important for Epstein–Barr virus nuclear antigen 1-mediated episome binding, maintenance, and transcription. Proc. Natl. Acad. Sci. U.S.A. 111, 243–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang D., et al. , Transcriptionally inactive hepatitis B virus episome DNA preferentially resides in the vicinity of chromosome 19 in 3D host genome upon infection. Cell Rep. 35, 109288 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Qi Y., et al. , DNA Polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 12, e1005893 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu T., Zehnder B., Qu B., Urban S., De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 3, 100195 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhong Y., et al. , Hepatitis B virus core protein Is not required for covalently closed circular DNA transcriptional regulation. J. Virol. 96, e0136222 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Y., et al. , Hepatitis B virus deregulates the cell cycle to promote viral replication and a premalignant phenotype. J. Virol. 92, e00722-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao K., et al. , Limited disassembly of cytoplasmic hepatitis B virus nucleocapsids restricts viral infection in murine hepatic cells. Hepatology 77, 1366–1381 (2022), 10.1002/hep.32622. [DOI] [PubMed] [Google Scholar]
- 46.Xia Y., et al. , Binding of nucleolin to HBV cccDNA. Gene Expression Omnibus (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE123715. Deposited 12 December 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (PDF)
Dataset S02 (PDF)