

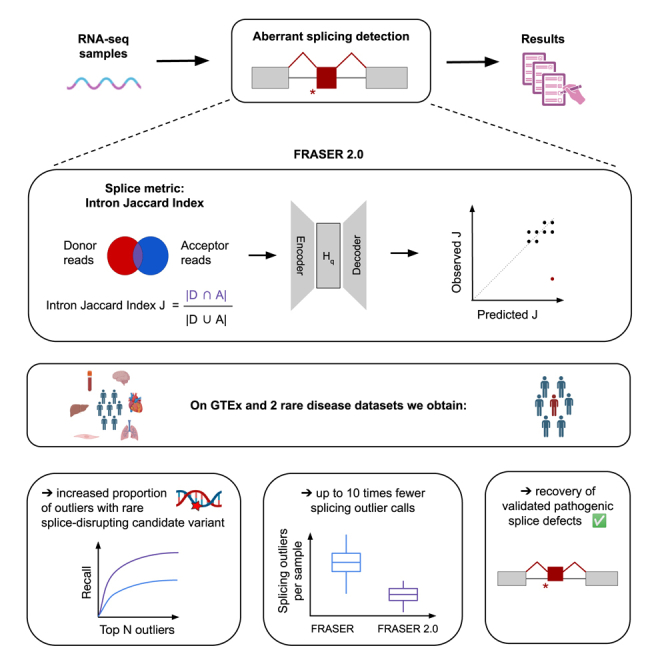
Summary
Detection of aberrantly spliced genes is an important step in RNA-seq-based rare-disease diagnostics. We recently developed FRASER, a denoising autoencoder-based method that outperformed alternative methods of detecting aberrant splicing. However, because FRASER’s three splice metrics are partially redundant and tend to be sensitive to sequencing depth, we introduce here a more robust intron-excision metric, the intron Jaccard index, that combines the alternative donor, alternative acceptor, and intron-retention signal into a single value. Moreover, we optimized model parameters and filter cutoffs by using candidate rare-splice-disrupting variants as independent evidence. On 16,213 GTEx samples, our improved algorithm, FRASER 2.0, called typically 10 times fewer splicing outliers while increasing the proportion of candidate rare-splice-disrupting variants by 10-fold and substantially decreasing the effect of sequencing depth on the number of reported outliers. To lower the multiple-testing correction burden, we introduce an option to select the genes to be tested for each sample instead of a transcriptome-wide approach. This option can be particularly useful when prior information, such as candidate variants or genes, is available. Application on 303 rare-disease samples confirmed the relative reduction in the number of outlier calls for a slight loss of sensitivity; FRASER 2.0 recovered 22 out of 26 previously identified pathogenic splicing cases with default cutoffs and 24 when multiple-testing correction was limited to OMIM genes containing rare variants. Altogether, these methodological improvements contribute to more effective RNA-seq-based rare diagnostics by drastically reducing the amount of splicing outlier calls per sample at minimal loss of sensitivity.
Keywords: Aberrant splicing, outlier detection, rare disease diagnostics, RNA-seq, rare variant, rare disease
Graphical abstract
Introduction
The regulation of splicing is important to control isoform expression and cellular function.1,2,3 Defects at the level of the pre-mRNA splicing process represent a major cause of human disease. It is estimated that 15%–50% of all variants leading to disease in humans alter splicing.4,5 Different methods that predict the impact of a variant in splicing from sequence alone have been developed.6,7,8,9,10,11,12 However, even with increasing precision, their accuracy remains imperfect, especially for diagnostics and for variants located far from the splice sites.9,12 Even the ACMG guidelines for variant pathogenicity require additional functional evidence, such as RNA-seq for variants predicted to disrupt splicing.13 Importantly, current prediction models do not provide information about the consequence of a potential splicing defect on the resulting transcript isoform (e.g., frameshift or exon truncation).
Identifying splicing aberrations on RNA sequencing (RNA-seq) data provides more direct evidence of the presence of splicing defects and reveals the resulting transcript isoforms. This approach has been successfully used in the diagnosis of patients with rare genetic disorders in large cohorts.14,15,16,17,18,19,20 After this initial success, computational tools such as LeafCutterMD,21 SPOT,22 and FRASER23 that are specialized in detecting aberrant splicing from RNA-seq data have been developed.
Both LeafCutterMD and SPOT model the counts of clusters of both annotated and not previously annotated introns with shared splice sites by using the Dirichlet-Multinomial distribution. SPOT identifies abnormal splicing by computing the Mahalanobis distance of each sample to the fitted distribution and calculates an empirical p value by comparing it to the Mahalanobis distance of samples simulated from the fitted distribution.22 LeafCutterMD implements a one-versus-all splicing outlier test by using the estimated Dirichlet-Multinomial parameters of each cluster to compute p values with the beta-binomial distribution for each intron in the cluster.21 One limitation of these approaches is that they do not account for sources of covariation in the split-read counts. However, Frésard et al.16 showed that such covariations can be strong in RNA-seq splicing measurements and may be related both to biological confounders such as age and to technical biases such as batch effects or RNA integrity levels.
FRASER models three metrics that are computed from split reads and unsplit reads detected at all splice sites identified in the RNA-seq data, allowing it to consider expressed splice sites regardless of whether they have been previously annotated.23 These three metrics are (1) the percent spliced in for testing the splice acceptor site (ψ5) and (2) donor site (ψ3) and (3) splicing efficiency (θ). These metrics capture different types of aberrant splicing, namely aberrant acceptor-site usage, aberrant donor-site usage, and aberrant splicing efficiency. FRASER uses a denoising-autoencoder approach to control for potentially unknown sources of covariation between samples and calculates beta-binomial p values to identify splicing outliers. Benchmarks on rare-variant enrichment among reported splicing outliers showed that FRASER outperformed LeafCutterMD and SPOT.
Despite FRASER’s improvements, the number of outlier calls per sample often remains very large. Notably, these splice-site-centric metrics can lead to reporting outliers that reflect local aberrations that might only have minor effects on the abundance of the canonical splicing isoform. Figure 1A illustrates such a case, where an exon elongation appearing in one sample is supported by 1,017 reads, whereas there are 32,018 supporting the annotated intron. Taking as reference the newly created donor site, this exon elongation, which is present in only six out of 582 samples, appears as an outlier event. Consistently, this event results in a high differential value of 0.99 in the ψ5 metric of FRASER. However, this event does not strongly affect the canonical isoform distribution because the vast majority of the reads still support the annotated intron.
Figure 1.

Intron Jaccard index improves splicing outlier calling
(A) Sashimi plot of three GTEx skin not-sun-exposed RNA-seq samples showing exons 4 and 5 of KRT1. A splicing outlier was detected in the top sample with the ψ5 metric of FRASER (red). The position of the donor site of the outlier intron is indicated with a blue dashed line. Although this intron is not expressed in most other samples (dark blue) and is therefore detected with a high Δψ5 value as shown in the table on the right, its functional impact is probably minor because the canonical intron remains largely dominant.
(B) Schematic definition of the intron Jaccard index for an intron of interest (purple) defined by a donor site d and acceptor site a. The set of donor-associated reads D (red) and acceptor-associated reads A (blue) are highlighted.
(C) Representation of different types of aberrant splicing events that can be captured with the intron Jaccard index. The right column contains the formulae to compute the intron Jaccard index of the canonical intron (black dotted line) from the split (s) and non-split (u) reads of the involved introns in each scenario.
(D) Recall of rare splice-disrupting candidate variants as defined by VEP (canonical splice-site variants, n = 1,544), MMSplice (n = 3,395), SpliceAI (n = 2,971), and AbSplice (n = 2,265) versus the rank of nominal p values from FRASER (light blue) and from an adaptation of FRASER using the intron Jaccard index (dark blue) on the GTEx skin not-sun-exposed dataset (n = 582). Different nominal p value cutoffs are indicated with shapes.
To address this issue, we here introduce an intron-based metric that we named the intron Jaccard index. It is defined as the proportion of reads supporting the splicing of an intron of interest among all reads associated with either splice site of the intron (Figure 1B). The intron Jaccard index is computed from both split and non-split reads, thus allowing it to capture several types of aberrant splicing, including exon skipping, exon truncation, exon elongation, exon creation, and full intron retention (Figure 1C). The intron Jaccard index is conceptually similar to the intron-excision-ratio concept underpinning LeafCutter.24 However, the statistical model of LeafCutter models multiple introns of a locus jointly, which leads to modeling complications and does not allow for modeling sample co-variation. Here we model the intron Jaccard index of individual introns separately by using the same beta-binomial autoencoder approach as in FRASER. We furthermore perform a systematic evaluation of model parameters, including pseudocounts and filtering criteria. We refer to this new method as FRASER 2.0 (Figure S1). We next benchmark FRASER 2.0 against FRASER, LeafCutterMD, and SPOT by using the multi-tissue GTEx dataset. Finally, we apply FRASER 2.0 to independent rare-disease cohorts and validate it on previously reported pathogenic splice defects.
Material and methods
Datasets
The GTEx dataset consists of 17,350 RNA-seq samples from 54 tissues of 948 assumed-to-be-healthy individuals of the Genotype-Tissue Expression Project V8.25 The GTEx data used for the analyses described in this manuscript were obtained from dbGaP: phs000424.v8.p1. Samples with an RNA integrity number (RIN) <5.7 and tissues with less than 100 samples were discarded. This resulted in a total of 16,146 samples and 48 tissues.
The Yépez et al. dataset consists of 303 individuals affected with a rare mitochondrial disorder described in Yépez et al.20 The intron counts were downloaded from Zenodo.26,27
The Undiagnosed Diseases Network (UDN) dataset consists of data from individuals suffering from a rare genetic disorder, as well as data collected from unaffected control individuals by different centers in the United States.28 We downloaded it from dbGaP (dbGaP: phs001232; v.4.p2). It contains 821 RNA-seq samples extracted from blood (n = 370), fibroblasts (n = 398), and other tissues (n = 53) that were not further considered. All RNA-seq samples are stranded except for one that was removed. Poly(A)-sequenced samples with a high-quality exonic rate lower than 0.7 were removed. Samples with size factors, which are a sequencing-depth metric,29 larger than 3 or less than 0.25 were also discarded. The resulting dataset consists of 252 poly(A) blood, 104 total RNA blood, and 391 fibroblast samples.
Splicing-outlier detection with FRASER
We used the integrated workflow DROP v.1.1.330 to count split and non-split reads from the BAM files (from the GTEx and UDN datasets) and to detect splicing outliers with FRASER v.1.2.223 on all datasets. Default cutoffs (FDR < 0.1, |Δψ| ≥ 0.3, minimal intron coverage ≥ 5 reads) and default intron filtering settings (95% of samples with n ≥ 1 and at least one sample with an intron count ≥20) were used.
Splicing quantification with the intron Jaccard index
We defined the intron Jaccard index (J) as the Jaccard index of the sets of donor-associated reads D and acceptor-associated reads A. For a given sample i and intron j, the set of donor-associated reads Dij was defined as the set of all split reads with the same donor site as intron j as well as all non-split reads spanning the exon-intron boundary at that donor site of sample i. The set of acceptor-associated reads Aij was analogously defined for the split and non-split reads at the acceptor site of intron j (Figure 1B).
The intron Jaccard index Jij for sample i and intron j was calculated as follows:
where sij denotes the count of split reads mapping to intron j in sample i, dj is the donor site of intron j, aj is the acceptor site of intron j, Lj is the set of introns using dj, Rj is the set of introns using aj, and uit denotes the count of non-split reads spanning the exon-intron boundary at a splice site t.
Denoising autoencoder
As for the original FRASER, we modeled and controlled for sample covariation by using an autoencoder that takes as input a matrix X consisting of the logit-transformed splice metrics of each intron i and sample j by using the following formula:
where kij and nij are the numerator and the denominator of the intron Jaccard index of intron j in sample i, respectively, and α corresponds to a pseudocount needed to avoid taking the logarithm of or dividing by zero. Originally, in FRASER a pseudocount of 1 was used as a default. Here we assessed various possible pseudocount values.
Annotation of genes to introns
To report results on the gene level, FRASER uses a user-provided gene annotation file to assign introns to genes. Each intron is assigned to the gene(s) overlapping either the donor or the acceptor site of the intron in a strand-specific manner. As such, an intron might contribute to the gene-level p value of several genes. Genes fully contained in an intron but not overlapping either splice site are not assigned to the intron.
Multiple-testing correction and effect-size cutoff
FRASER 2.0 obtains p values for each intron by using the beta-binomial distribution to evaluate the significance of the observed split-read count given the intron Jaccard index value predicted from the autoencoder latent space.23 We used the same strategy as in FRASER to obtain gene-level p values. Specifically, intron-level p values are corrected via Holm’s method per gene and sample, and the minimal corrected p value is selected. Then, false-discovery rate (FDR) correction of those selected p values is applied across genes per sample by the Benjamini-Yekutieli method. This FDR correction step is by default done transcriptome-wide across all expressed genes. Genes are considered to be expressed if they have at least one intron with enough supporting reads to pass the filtering step assigned to them in the dataset. FRASER 2.0 introduces the additional option to restrict the FDR correction to user-provided lists of genes. The provided genes can differ between samples. To define outlier status, we set the FRASER 2.0 default cutoffs at FDR ≤0.1 and effect size at ΔJ ≥ 0.1, where ΔJ = Jij − and is the predicted intron Jaccard index value as modeled by the autoencoder. FRASER 2.0 additionally introduces an option to flag results located in blacklist regions of the genome31 because those results might be less trustworthy.
Rare-variant-recall benchmarks
To perform analyses of rare-variant recalls, we considered five different sets of splice-affecting variants, each defined by a different variant-effect prediction tool. The first two sets are canonical splice-site variants (the first two bases into the intron) and splice-region variants (corresponding to the first three bases into an exon and from the third to the eighth bases into an intron) as defined by VEP.32 For the third set, we used SpliceAI9 and considered variants with a SpliceAI score ≥0.5. We used the masked SpliceAI scores for this analysis because those are the recommended scores for variant interpretation. For the fourth set, we used MMSplice7 and considered variants with an |Δ logit Ψ| score ≥2. The fifth set contained variants predicted to cause aberrant splicing by AbSplice;12 specifically, it contained variants with a maximum score across tissues except testis ≥0.05, which corresponds to the suggested "medium" cutoff of AbSplice. Rare variants were defined by having a minor-allele frequency (MAF) of <0.1%, obtained from gnomAD,33 and by being present in at most two individuals of GTEx.
In each GTEx tissue, genes were ranked based on the nominal gene-level p value, and the recall of rare splice-affecting variants in expressed genes was calculated at each rank. To facilitate comparison across tissues and ensure that the reported outliers are relevant, we used two different measures of performance for each tissue: (1) the recall and precision of ranking the nominal p values of FDR significant results, and (2) the recall at the rank corresponding to a mean of 20 outliers per sample for each tissue.
Correlation with mapped reads
The number of mapped reads was computed using the function idxstats from SAMtools v.1.11.34 Then, the Spearman correlation coefficient and p value between the mapped reads and its splicing outliers per sample were calculated with the cor.test function in R.
Tissue reproducibility analysis
To assess the reproducibility of our splicing-outlier calls, we called aberrant splicing events across all 48 GTEx tissues with both FRASER and FRASER 2.0, as well as SPOT v.1.0.2 and LeafCutterMD v.0.2.9, under default settings for all methods. We performed this analysis on the set of individuals with at least 20 out of the 48 tissues and genes with available p values in at least 25 out of the 48 tissues. This led to 14,707 genes for FRASER 2.0, 16,104 genes for FRASER, 12,154 genes for SPOT, and 14,001 genes for LeafCutterMD. For all methods, we applied three nominal p value cutoffs, of 10−5, 10−7, and 10−9, and for each gene-sample combination that passed this cutoff in at least one tissue, we computed in how many other tissues it could be reproduced with a nominal p value <10−3.
Benchmark on previously reported pathogenic splice events
For the Yépez et al. dataset (n = 303), 26 cases were found to have a splice defect in the disease-causal gene with FRASER. Further information about the pathogenic variants of those cases is available in Tables S2 and S4 of Yépez et al.20 The list of OMIM genes was downloaded from the website www.omim.org.
Ethics declaration
The study was approved by the ethical committee of the Technical University of Munich (#200/15s, #5360/13, and #2341/09). We adhered to the German Genetic Diagnostics Act (GenDG) and the international guidelines for good clinical practice (GCP). The research conformed to the principles of the Declaration of Helsinki.
Results
Introduction of the intron Jaccard index as a robust splice metric
To be less sensitive to local splice-site aberrations that do not have a strong effect on the canonical splice isoform, we introduce the intron Jaccard index, an intron-centric metric that integrates the alternative donor, alternative acceptor, and intron retention signal (Figure 1B). We define the intron Jaccard index Jij as the Jaccard index of the set of donor-associated reads Dij and the set of acceptor-associated reads Aij for a given sample i and intron j:
where Dij contains, for sample i, both the split reads with the same donor site as intron j and the non-split reads spanning the exon-intron boundary at that donor site and analogously for Aij (Figure 1B, materials and methods). The intersection of those two sets represents the split reads mapping exactly to intron j, whereas the union captures all reads mapping to any intron with the same donor or acceptor site as intron j in addition to non-split reads at those splice sites.
The inclusion of both split and non-split reads in the metric allows retaining FRASER’s capability to capture several types of aberrant splicing, including partial or full intron retention, within a single metric (Figure 1C). As in FRASER, we model the intron Jaccard index with a beta-binomial denoising autoencoder and calculate p values with the beta-binomial distribution to assess the statistical significance of each intron Jaccard index value. Outliers are then called on the basis of the multiple-testing-corrected p values and effect size (ΔJ).
We ran FRASER with the intron Jaccard index as the splice metric on several GTEx tissues. Following the rationale that rare variants in the vicinity of splice sites are likely to disrupt splicing,35 we evaluated our new metric on the recall of rare (MAF < 0.1%) splice-disrupting candidate variants in expressed genes by using a combination of variant annotation tools (materials and methods). On this benchmark, adopting the intron Jaccard index increased the recall at each rank in comparison to the three metrics from FRASER together and individually (Figures 1D and S2).
Optimization of FRASER 2.0 parameters
After having established the intron Jaccard index, we evaluated several FRASER parameters to identify their optimal values with respect to the new metric. Because FRASER’s autoencoder works with values in the logit space, which is defined for values greater than 0 and less than 1, we needed to add a pseudocount to both the numerator and denominator when calculating each metric on raw read counts. So far we had set the pseudocount to 1. Here we investigated a range of possible values on a subset of 15 selected representative GTEx tissues because this analysis is computing intensive. Reducing the pseudocount improved the recall of rare splice-disrupting candidate variants until an optimal performance was reached at a pseudocount of 0.1, after which further reduction of the pseudocount decreased the performance again (Figures S3A and S3B).
We further investigated the effect-size cutoff. The highest recall of splice-disrupting candidate variants was achieved for a ΔJ of 0.2 consistently across different filtering settings, where ΔJ denotes the difference between the observed value and the expected value (Figure S4). However, the more permissive cutoff ΔJ = 0.1 was similarly optimal. We therefore decided to adopt the Δ = 0.1 cutoff as the default (Figures S3C and S3D).
Furthermore, we investigated the intron filtering criteria. FRASER uses three parameters, denoted k, n, and q, to filter out lowly expressed introns. The parameter k is the minimal value of the splice-metric numerator required in at least one sample. The parameter n is the minimal value of the splice-metric denominator at percentile q across samples. Among parameter values with optimal performance, we opted for the most permissive setting (k = 20, q = 25%, and n = 10, Figure S3E). This parameter setting means that to be considered by the algorithm in the first place, an intron must be supported by at least 20 split reads in at least one sample (intron Jaccard index numerator) and shall have a total of more than 10 donor-site- and acceptor-site-related reads (intron Jaccard index denominator, Figure 1B) in at least 25% of the samples. Adopting these cutoffs resulted in testing 287,943 introns and 16,548 coding and non-coding genes on average for each GTEx tissue (Figure S3F).
Finally, we investigated whether discarding outlier calls in introns with a poor goodness-of-fit would improve the performance further. Generally, a poor fit can indicate a violation of the modeling assumption, making the p value estimates not trustworthy. FRASER models the data of each individual intron by using a beta-binomial regression on the latent space. We observed some instances for which the modeling assumption was violated—in particular, cases of splicing quantitative trait loci (sQTL) in cis that were not captured by the latent space (Figures S5C and S5D). To systematically capture those cases, we considered the beta-binomial overdispersion parameter (ρ) as a measure of the goodness-of-fit. However, filtering out introns with a high overdispersion parameter had minimal effects on the overall recall of rare variants. Therefore, we decided not to implement this filtering (Figures S5A and S5B).
Overall, we call this new approach FRASER 2.0. The user can change all the parameters for which we explored the optimal default values here.
Improved performance and robustness of FRASER 2.0 on GTEx
We ran FRASER 2.0 on 48 GTEx (V8) tissues and benchmarked it against FRASER,23 LeafCutterMD,21 and SPOT.22 All the analyses are performed independently for each tissue. Notable sample-sample correlations were present in raw intron Jaccard index values, analogous to what has been previously described in FRASER, and the autoencoder of FRASER 2.0 was able to correct for them (Figure S6). Introns that were detected as outliers in FRASER but not with FRASER 2.0 tended to have a high Ψ5 or Ψ3 value and a low intron Jaccard index value, whereas the intron splice-metric values that were found as outliers by both methods were similar (Figure S7). FRASER 2.0 p values were better calibrated than FRASER’s p values for each metric (Figures 2A and S8), confirming that the intron Jaccard index is more robust. We then evaluated each method on its ability to identify splicing outliers in genes predicted to be affected by a rare splice-disrupting candidate variant. FRASER 2.0 consistently outperformed FRASER, SPOT, and LeafCutterMD and increased the recall of such genes throughout all ranks and across tissues (10-fold increase in precision at FDR = 0.1, Figures 2B, S9, and S10). Importantly, FRASER 2.0 reported significantly fewer outliers per sample than FRASER on all tissues (10.0 ± 2.6 times less, Figures 2C and 2D). Although the overlap of outliers from FRASER and FRASER 2.0 is only 8% across all tissues (Figure 2C), outliers identified only by FRASER 2.0 were more enriched for candidate rare splice-disrupting variants than those identified by FRASER only (Figure S11), highlighting that FRASER 2.0 reports fewer outlier calls that are not supported by such variants than FRASER does. In addition, splicing outliers from FRASER, LeafCutterMD, and SPOT were more likely to arise with a higher sequencing depth than ones detected with FRASER 2.0 (Figure 3), thus confirming the robustness of the latter. FRASER 2.0 outliers were also more often reproducible across tissues than competitor methods (Figure S12). Using skeletal muscle, the GTEx tissue with the largest available sample size (n = 782), we explored the effect of the sample size on the amount of detected outliers by subsampling to smaller dataset sizes. The median number of outliers detected per sample initially increases with increasing sample size but plateaus at sample sizes of 300 or more (Figure S13).
Figure 2.

FRASER 2.0 increases recall of rare splice-disrupting candidate variants on GTEx
(A) Quantile-quantile plots of the p values for the different splice metrics from FRASER (ψ3, ψ5, θ, shown in shades of blue) and the intron Jaccard index from FRASER 2.0 (purple) on the GTEx skin not-sun-exposed dataset. The red line depicts the diagonal, and the gray ribbon around it depicts the 95% confidence interval.
(B) Recall of rare splice-disrupting candidate variants as defined by the variant annotation tools VEP, MMSplice, SpliceAI, and AbSplice (facets) versus the rank of nominal p values combined across GTEx tissues for FRASER (blue), FRASER 2.0 (purple), LeafCutterMD (yellow), and SPOT (green). Nominal p value cutoffs are indicated with shapes.
(C) Venn diagram of the overlap of gene-level splicing outliers found with FRASER (blue) and FRASER 2.0 (purple).
(D) Box plots of the number of splicing outliers (gene level) per sample (y axis) called by FRASER (blue) and FRASER 2.0 (purple) for each GTEx tissue (x axis). All brain tissues have been combined for readability.
Figure 3.

FRASER 2.0 is less sensitive to sequencing depth than previous methods
(A) Scatterplot of the number of splicing outliers at the gene level against the total mapped reads per sample on the GTEx skin not-sun-exposed dataset for LeafCutterMD, SPOT, FRASER, and FRASER 2.0 (facets). Spearman correlation coefficients (rho) are shown. All are significant (Spearman test, p < 3 × 10−3).
(B) Box plots of the Spearman correlation coefficients (y axis) between the mapped reads and the number of gene-level splicing outliers called by LeafCutterMD, SPOT, FRASER, and FRASER 2.0 (x axis) for each GTEx tissue (n = 48). p values of Wilcoxon tests are shown above brackets.
To evaluate the sensitivity of FRASER 2.0 to splicing defects giving rise to isoforms rapidly degraded by NMD, we also compared the overlap with gene underexpression outliers for all methods. The proportion of underexpression outliers among its splicing outlier calls is strikingly higher for FRASER 2.0 than for all other methods (5-fold on median across GTEx tissues, Figure S14A). In absolute numbers, LeafCutterMD called the most splicing outliers in underexpression outlier genes, however, proportionally those constitute only a very minor fraction of LeafCutterMD’s total results (Figure S14B). FRASER 2.0 also runs about a third faster than FRASER, with a typical FRASER 2.0 run (after generating the split and non-split count matrices) on a cohort of about 200 samples taking less than 2.5 h on a Rocky Linux 8.8-based server with 128 physical cores and 512 GB memory utilizing 15 cores in parallel for each run (Figure S15). Overall, FRASER 2.0 reports less, but more relevant splicing outliers supported by a rare variant, reproduced across tissues and less influenced by sequencing depth than FRASER, LeafCutterMD, and SPOT.
Application of FRASER 2.0 to rare-disease cohorts
To evaluate FRASER 2.0 in a diagnostic setting, we applied it to 747 samples from the Undiagnosed Diseases Network28 and to the cohort composed of RNA-seq samples from 303 individuals suspected to be affected with a mitochondrial Mendelian disorder; we refer to this cohort as the Yépez et al. dataset.20 As with GTEx, FRASER 2.0 reported fewer splicing outliers than FRASER in all cohorts (a median of 3–9.5 times fewer outliers; Figure 4A).
Figure 4.

Application of FRASER 2.0 to rare-disease cohorts
(A) Distribution of the splicing outliers per sample at the gene level on the UDN (n = 391, n = 252, n = 104 for fibroblasts, blood poly(A), and blood total RNA, respectively) and the Yépez et al. dataset (n = 303) for FRASER (blue) and FRASER 2.0 applied to three gene sets considered for FDR correction: expressed genes (dark purple), expressed OMIM genes (light purple), and expressed OMIM genes with a rare variant (violet red; see material and methods).
(B) Number of events (bars) in all non-empty intersections (linked dots) between four splicing outlier sets from the Yépez et al. dataset: (1) the 26 originally reported pathogenic events, (2) the transcriptome-wide significant FRASER 2.0 calls, (3) the significant FRASER 2.0 calls when only OMIM genes with a rare variant are considered, and (4) the transcriptome-wide significant FRASER calls. Intersections with the set of pathogenic events are highlighted in red.
(C) Fraction of recovered pathogenic splicing outliers from the Yépez et al. dataset (y axis, total n = 26) when subsampling to different sample sizes (x axis) was performed. Each sample size was randomly sampled five times. RV: rare variant.
Frequently in diagnostics, researchers are not interested in testing all genes, but only those that could cause the disease (e.g., from OMIM or curated lists for each disease). In addition, interpretation of variants revealed by panel, whole-exome sequencing (WES), or whole-genome sequencing (WGS) is often performed prior to RNA sequencing, yielding a restricted list of candidate genes per sample. Such prior information has the potential to reduce the multiple-testing burden during analysis of splicing-outlier calls from RNA-seq. To implement this strategy, we extended the FDR correction step of FRASER 2.0 to allow consideration of a subset of genes specific for each sample. Testing OMIM36 genes on both cohorts and OMIM genes harboring at least one rare variant (gnomAD MAF <0.1%, in cohort allele frequency <1%, within the gene body except for the UTRs) called by WES in the Yépez et al. dataset consistently led to fewer outliers to be manually inspected per sample (median of 2 outliers per sample, Figure 4A). The latter approach resulted in a median of 5,427 introns and 149 OMIM genes to be tested per sample, in comparison to 140,230 introns and 16,846 genes in the transcriptome-wide setting (Figure S16).
In the Yépez et al. dataset, 26 cases with aberrant splicing on the disease causal gene were identified by FRASER. FRASER 2.0 reported 22 out of those 26 diagnosed cases with the default cutoffs (Figure 4B; Table S1). Two missing cases were exon truncations caused by synonymous variants and showed a large deviation from canonical splicing (ΔJ = −0.55 and ΔJ = −0.39), which was reflected in low nominal p values (1.08 × 10−4 and 1.87 × 10−5), but due to the multiple-testing burden from testing all expressed introns and genes, the resulting FDR was 1 in both cases. Another case missed by FRASER 2.0 is caused by a heterozygous 6.5 Kbp deletion that results in the skipping of multiple exons. This splicing defect is hard to detect because it triggers nonsense-mediated decay (NMD), resulting in the aberrant expression of this gene (−74% expression reduction computed by OUTRIDER37). In this case, the novel intron does not pass our default intron filter settings in FRASER 2.0; only 7 split reads map to it. The last missed case is a case in which a deletion results in exon creation and truncation. Although FRASER 2.0 detects a large deviation from canonical splicing for this case (ΔJ = 0.69), it also fits a high value for the beta-binomial overdispersion parameter of this intron (ρ = 0.80). This gives a nominal p value of 0.00294, which results in an FDR of 1 after multiple-testing correction.
Testing only OMIM genes with rare variants during multiple-testing correction recovered two of these four cases at 10% FDR (FDR = 0.087 and 0.094). However, six other cases could not be detected with this approach because the gene affected by the splice defect was not in the list of tested genes. In one case, the causal variant had a gnomAD33 MAF of 0.96%, which is above the 0.1% cutoff we adopted here, and in the other five cases, the disease-causing variants were either deletions or intronic variants missed by WES (Figure 4B). Calling variants from WGS could help to overcome this issue and further improve the usefulness of this feature.
Finally, we investigated the sensitivity of FRASER 2.0 to sample size because the number of samples is often limited in rare-disease cohorts. We used the Yépez et al. dataset and the 26 known pathogenic splicing events to estimate the dataset size required for reaching significance for most of the clinically relevant events. As expected, the percentage of recovered pathogenic events dropped with reduced sample size (Figures 4C and S17). With already 50 samples, we recovered 69% of cases (18 out of 26, on average). Additionally, we investigated the effect of combining the small subset of samples with pathogenic events with samples from an external dataset. We used samples from the UDN dataset for this analysis. There was, however, no difference in the sample size required for recovering the pathogenic events, although the total number of outlier calls per sample was on median 1.47 times higher when the samples were combined with external samples (Figure S18).
Altogether, these results show the general applicability of FRASER 2.0 to various RNA-seq cohorts and show that it reports considerably fewer outliers than FRASER. In addition, the new functionality of testing a preselected number of genes per sample can further help to reduce the search space. FRASER 2.0 is publicly available at github.com/gagneurlab/fraser and integrated into the workflow DROP.30
Discussion
Here we introduced FRASER 2.0, a method for detecting aberrant splicing via an intron-centric splice metric, the intron Jaccard index. In a single metric, the intron Jaccard index captures former metrics of splicing efficiency as well as alternative donor and acceptor site choice. The use of a single metric in FRASER 2.0 translates into easier interpretation of results, as well as reduced runtime, computational resources, and storage. Benchmarks on assumed-healthy samples of the multi-tissue dataset GTEx showed that FRASER 2.0 decreases the number of reported splicing outliers by one order of magnitude, recovers splicing outliers associated with candidate splice-disrupting rare variants more accurately than competitor methods and is more robust to variations in sequencing depth. Application to two unrelated rare disease cohorts further confirmed the relative advantage of FRASER 2.0 over its predecessors. These results are relevant because in a diagnostic research setting, the vast reduction of splicing outliers per sample and of noise that FRASER 2.0 provides will substantially minimize the amount of manual inspection required to evaluate candidate splice defects and streamline the analysis of results. Often, obtaining a high number of potential candidates from an analysis providing additional evidence, such as aberrant splicing detection, can be a major barrier to a more widespread implementation of these approaches in practice.
Motivated by applications with various collaborators, we have here introduced the option to filter genes on the basis of prior information during multiple-testing correction. In practice, it is often the case that RNA sequencing is performed after an inconclusive WES or WGS, which might have yielded variants of unknown significance for which RNA-seq constitutes the needed functional assay. The possibility of focusing on candidate genes and variants in such a “DNA-first, RNA-second” mode of operation can be valuable, as we have shown for the mitochondrial rare-disease dataset application.
Initially, we introduced the intron Jaccard index to focus on more functionally relevant outliers than the original three splicing metrics of FRASER could identify. However, our benchmarks are based on candidate splice-disrupting variants without consideration for canonical splice-isoform abundance. Perhaps these benchmarks, along with a stronger robustness to sequencing depth, show that the main advantage of the intron Jaccard index is merely statistical. An explanation for this could be that the intron Jaccard index might be more stable because, compared to the denominators of the three splicing metrics of FRASER, its denominator accounts for a larger set of reads.
One concern with moving from the original splicing metrics of FRASER to the intron Jaccard index is that some genuinely pathogenic isoforms can turn out to represent a minor fraction of the expressed splice isoforms and therefore could now be discarded. This includes rapidly degraded splice isoforms. To capture those, we recommend additionally using an expression outlier caller such as OUTRIDER.37 Our analysis here, however, suggests that FRASER 2.0 does not lose sensitivity to detect such cases in comparison to the previous version. Moreover, it is possible to experimentally inhibit NMD, for instance by using the translation inhibitor cycloheximide, in case of a suspected NMD-introducing splicing defect to allow detection of splice defects otherwise undergoing NMD.38 Generally, however, NMD-introducing splicing defects pose a general problem of detection for all methods based on RNA-seq samples not treated by a translation inhibitor, not just for FRASER 2.0. Other examples that could benefit from the use of FRASER’s splice-site-centric metrics are aberrant isoforms leading to gain of function and aberrant splicing on dosage-sensitive genes. In our results, however, the noise reduction achieved by moving to the intron Jaccard index outweighs the benefit of detecting such minor aberrant isoforms. Nonetheless, the software of FRASER 2.0 still supports calling outliers with FRASER’s splice metrics, if the user wishes to include them. One limitation of FRASER 2.0 and other methods designed to detect outliers is that a sufficiently large cohort is needed. In the rare-disease field, attaining these minimal requirements can be especially challenging. Integrating unaffected samples or samples from individuals suffering from other disorders can help to overcome this. In addition, count matrices are provided for a variety of tissues in DROP, and these can be downloaded and integrated with the local samples. In any case, the aggregated samples should have been probed from the tissue and sequenced with a similar protocol. Another limitation of calling aberrant splicing in RNA-seq data is that the gene might not be sufficiently expressed in the probed tissue, and the choice of tissue is usually limited to clinically accessible ones (e.g., blood or skin). One could consider using FRASER 2.0 output, however, as an input on the recently developed AbSplice-RNA method12 to predict aberrant splicing in multiple tissues.
Another limitation of this study is that we used the GTEx dataset for tuning FRASER 2.0’s parameters as well as comparing it to competitor methods. However, even the version of FRASER 2.0 without optimized parameters clearly outperformed previous methods (Figure 1D). Moreover, as a result of insufficient available data, we have not assessed in other datasets whether the parameters fitted on GTEx remain optimal. As we share our analysis pipeline, users with a large enough cohort could reconduct the parameter optimization on their own dataset.
Long-read sequencing shall eventually lead to more direct measurements of isoform expression and to more accurate quantifications of the expressed isoforms, especially at complex loci.39 Such direct estimations of the abundance of functional splice isoforms on every locus hold the promise of advantageous replacement of local splicing metrics, including the intronic Jaccard index. However, because of the higher cost and limited sequencing depth of long-read sequencing, which is particularly limiting for RNA-seq as a result of the high dynamic range of gene expression, short-read-based methods such as FRASER 2.0 will probably remain relevant for rare-disease diagnostics in the coming years.
In conclusion, FRASER 2.0 can be readily used in the context of rare-disease diagnostics because it has the same sensitivity as FRASER and reports fewer, but more relevant, outliers and thus facilitates manual candidate gene inspection. We anticipate that the option to include prior knowledge and allow the restriction of the FDR correction to sets of candidate genes will be especially useful in diagnostic strategies where DNA sequencing is routinely done as a first step and RNA-seq is used as a follow-up. As splicing-based therapeutics are on the rise,3,40 we hope FRASER 2.0 will be a useful tool for the identification of the exact aberrant splicing event for these new targeted therapies.
Data and code availability
This study did not generate new data. For a description of the used datasets, refer to the materials and methods. FRASER 2.0 is an open-source R/Bioconductor41,42 package available in the GitHub repository https://github.com/gagneurlab/fraser (version 1.99.0 and above). It is also integrated into the workflow DROP30 (version 1.3.0 and above) available at https://github.com/gagneurlab/drop. The code to reproduce the figures in this manuscript can be found at https://github.com/gagneurlab/FRASER-2.0-analysis.
Acknowledgments
We would like to thank Nicholas Smith and Felix Brechtmann for advice throughout the project, as well as the different people who have tested the software. Some icons for the graphical abstract were created with BioRender. This work was supported by the German Bundesministerium für Bildung und Forschung (BMBF) through the ERA PerMed project PerMiM (01KU2016B to I.S., V.A.Y., and J.G.]; and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via the projects “Identification of host genetic variation predisposing to severe COVID-19 by genetics, transcriptomics and functional analyses” (466168909 to V.A.Y. and J.G.), “Identification and Characterization of Long COVID-19 patients using whole-blood transcriptomics” (466168626 to K.L. and J.G.), and Nationale Forschungsdateninfrastruktur (NFDI) 1/1 “GHGA - German Human Genome-Phenome Archive” (441914366 to C.M. and J.G.). The Genotype-Tissue Expression (GTEx) project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the National Cancer Institute, National Human Genome Research Institute, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. The Undiagnosed Diseases Network (UDN) is supported in part by the Intramural Research Program of the National Human Genome Research Institute and by grants U01HG007674, U01HG007703, U01HG007709, U01HG007672, U01HG007690, U01HG007708, U01HG007530, U01HG007942, U01HG007943, U01TR002471, U54NS093793, U54NS108251, U01HG010215, U01HG010233, U01HG010217, U01HG010230, U01HG010219, and U01TR001395 from the National Institutes of Health Common Fund, through the Office of Strategic Coordination and the Office of the NIH Director. The content is solely the responsibility of the authors and does not necessarily represent the official views of UDN investigators or the NIH.
Author contributions
Conceptualization: I.S., C.M., V.A.Y., J.G.; methodology: I.S., C.M., V.A.Y., J.G.; software: I.S.; validation: I.S., K.L., V.A.Y.; formal analysis: I.S., K.L., V.A.Y.; data curation: I.S., C.M., V.A.Y.; writing—original draft: I.S., V.A.Y., J.G.; writing—review and editing: all authors; visualization: I.S., C.M., V.A.Y., J.G.; supervision: C.M., V.A.Y., J.G.; project administration: V.A.Y., J.G.; funding acquisition: J.G.
Declaration of interests
The authors declare no competing interests.
Published: November 24, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.10.014.
Contributor Information
Vicente A. Yépez, Email: yepez@in.tum.de.
Julien Gagneur, Email: gagneur@in.tum.de.
Supplemental information
This table contains information about the 26 pathogenic splice defects identified in the Yépez et al. study; it is annotated with the results of FRASER 2.0 for these samples
References
- 1.Kelemen O., Convertini P., Zhang Z., Wen Y., Shen M., Falaleeva M., Stamm S. Function of alternative splicing. Gene. 2013;514:1–30. doi: 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vuong C.K., Black D.L., Zheng S. The neurogenetics of alternative splicing. Nat. Rev. Neurosci. 2016;17:265–281. doi: 10.1038/nrn.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogalska M.E., Vivori C., Valcárcel J. Regulation of pre-mRNA splicing: roles in physiology and disease, and therapeutic prospects. Nat. Rev. Genet. 2022:1–19. doi: 10.1038/s41576-022-00556-8. [DOI] [PubMed] [Google Scholar]
- 4.López-Bigas N., Audit B., Ouzounis C., Parra G., Guigó R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–1903. doi: 10.1016/j.febslet.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 5.Baralle D., Buratti E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017;131:355–368. doi: 10.1042/CS20160211. [DOI] [PubMed] [Google Scholar]
- 6.Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K.C., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R., et al. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347 doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng J., Duong Nguyen T.Y., Cygan K.J., Çelik M.H., Fairbrother W., Avsec Ž., Gagneur J. Modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 2019:1–15. doi: 10.1186/s13059-019-1653-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng J., Çelik M.H., Kundaje A., Gagneur J. MTSplice predicts effects of genetic variants on tissue-specific splicing. Genome Biol. 2021;22:94. doi: 10.1186/s13059-021-02273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Danis D., Jacobsen J.O.B., Carmody L.C., Gargano M.A., McMurry J.A., Hegde A., Haendel M.A., Valentini G., Smedley D., Robinson P.N. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am. J. Hum. Genet. 2021;108:1564–1577. doi: 10.1016/j.ajhg.2021.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rentzsch P., Schubach M., Shendure J., Kircher M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13:31. doi: 10.1186/s13073-021-00835-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner N., Çelik M.H., Hölzlwimmer F.R., Mertes C., Prokisch H., Yépez V.A., Gagneur J. Aberrant splicing prediction across human tissues. Nat. Genet. 2023:1–10. doi: 10.1038/s41588-023-01373-3. [DOI] [PubMed] [Google Scholar]
- 13.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cummings B.B., Marshall J.L., Tukiainen T., Lek M., Donkervoort S., Foley A.R., Bolduc V., Waddell L.B., Sandaradura S.A., O’Grady G.L., et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kremer L.S., Bader D.M., Mertes C., Kopajtich R., Pichler G., Iuso A., Haack T.B., Graf E., Schwarzmayr T., Terrile C., et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017;8 doi: 10.1038/ncomms15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frésard L., Smail C., Ferraro N.M., Teran N.A., Li X., Smith K.S., Bonner D., Kernohan K.D., Marwaha S., Zappala Z., et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat. Med. 2019;25:911–919. doi: 10.1038/s41591-019-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonorazky H.D., Naumenko S., Ramani A.K., Nelakuditi V., Mashouri P., Wang P., Kao D., Ohri K., Viththiyapaskaran S., Tarnopolsky M.A., et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 2019;104:466–483. doi: 10.1016/j.ajhg.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamanaka K., Miyatake S., Koshimizu E., Tsurusaki Y., Mitsuhashi S., Iwama K., Alkanaq A.N., Fujita A., Imagawa E., Uchiyama Y., et al. RNA sequencing solved the most common but unrecognized NEB pathogenic variant in Japanese nemaline myopathy. Genet. Med. 2019;21:1629–1638. doi: 10.1038/s41436-018-0360-6. [DOI] [PubMed] [Google Scholar]
- 19.Lee M., Kwong A.K.Y., Chui M.M.C., Chau J.F.T., Mak C.C.Y., Au S.L.K., Lo H.M., Chan K.Y.K., Yépez V.A., Gagneur J., et al. Diagnostic potential of the amniotic fluid cells transcriptome in deciphering mendelian disease: a proof-of-concept. NPJ Genom. Med. 2022;7:74. doi: 10.1038/s41525-022-00347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yépez V.A., Gusic M., Kopajtich R., Mertes C., Smith N.H., Alston C.L., Ban R., Beblo S., Berutti R., Blessing H., et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022;14:38. doi: 10.1186/s13073-022-01019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenkinson G., Li Y.I., Basu S., Cousin M.A., Oliver G.R., Klee E.W. LeafCutterMD: an algorithm for outlier splicing detection in rare diseases. Bioinformatics. 2020;36:4609–4615. doi: 10.1093/bioinformatics/btaa259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferraro N.M., Strober B.J., Einson J., Abell N.S., Aguet F., Barbeira A.N., Brandt M., Bucan M., Castel S.E., Davis J.R., et al. Transcriptomic signatures across human tissues identify functional rare genetic variation. Science. 2020;369 doi: 10.1126/science.aaz5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mertes C., Scheller I.F., Yépez V.A., Çelik M.H., Liang Y., Kremer L.S., Gusic M., Prokisch H., Gagneur J. Detection of aberrant splicing events in RNA-seq data using FRASER. Nat. Commun. 2021;12:529. doi: 10.1038/s41467-020-20573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y.I., Knowles D.A., Humphrey J., Barbeira A.N., Dickinson S.P., Im H.K., Pritchard J.K. Annotation-free quantification of RNA splicing using LeafCutter. Nat. Genet. 2018;50:151–158. doi: 10.1038/s41588-017-0004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.GTEx Consortium. Laboratory Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups. NIH Common Fund. Jo B., Mohammadi P., Park Y., Parsana P., et al. Biospecimen Collection Source Site—NDRI Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yépez V.A., Gusic M., Kopajtich R., Meitinger T., Gagneur J., Prokisch H. Gene expression and splicing counts from the Yepez, Gusic et al study - non-strand specific. Zenodo. 2021 [Google Scholar]
- 27.Yépez V.A., Gusic M., Kopajtich R., Meitinger T., Gagneur J., Prokisch H. Gene expression and splicing counts from the Yepez, Gusic et al study - strand specific. Zenodo. 2021 [Google Scholar]
- 28.Gahl W.A., Wise A.L., Ashley E.A. The Undiagnosed Diseases Network of the National Institutes of Health: A National Extension. JAMA. 2015;314:1797–1798. doi: 10.1001/jama.2015.12249. [DOI] [PubMed] [Google Scholar]
- 29.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yépez V.A., Mertes C., Müller M.F., Klaproth-Andrade D., Wachutka L., Frésard L., Gusic M., Scheller I.F., Goldberg P.F., Prokisch H., Gagneur J. Detection of aberrant gene expression events in RNA sequencing data. Nat. Protoc. 2021;16:1276–1296. doi: 10.1038/s41596-020-00462-5. [DOI] [PubMed] [Google Scholar]
- 31.Amemiya H.M., Kundaje A., Boyle A.P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci. Rep. 2019;9:9354. doi: 10.1038/s41598-019-45839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rivas M.A., Pirinen M., Conrad D.F., Lek M., Tsang E.K., Karczewski K.J., Maller J.B., Kukurba K.R., DeLuca D.S., Fromer M., et al. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348:666–669. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amberger J.S., Bocchini C.A., Scott A.F., Hamosh A. OMIM.org: leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res. 2019;47:D1038–D1043. doi: 10.1093/nar/gky1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brechtmann F., Mertes C., Matusevičiūtė A., Yépez V.A., Avsec Ž., Herzog M., Bader D.M., Prokisch H., Gagneur J. OUTRIDER: A Statistical Method for Detecting Aberrantly Expressed Genes in RNA Sequencing Data. Am. J. Hum. Genet. 2018;103:907–917. doi: 10.1016/j.ajhg.2018.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carter M.S., Doskow J., Morris P., Li S., Nhim R.P., Sandstedt S., Wilkinson M.F. A Regulatory Mechanism That Detects Premature Nonsense Codons in T-cell Receptor Transcripts in Vivo Is Reversed by Protein Synthesis Inhibitors in Vitro. J. Biol. Chem. 1995;270:28995–29003. doi: 10.1074/jbc.270.48.28995. [DOI] [PubMed] [Google Scholar]
- 39.Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21:30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E., et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 41.Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.R Core Team . R Foundation for Statistical Computing; 2021. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table contains information about the 26 pathogenic splice defects identified in the Yépez et al. study; it is annotated with the results of FRASER 2.0 for these samples
Data Availability Statement
This study did not generate new data. For a description of the used datasets, refer to the materials and methods. FRASER 2.0 is an open-source R/Bioconductor41,42 package available in the GitHub repository https://github.com/gagneurlab/fraser (version 1.99.0 and above). It is also integrated into the workflow DROP30 (version 1.3.0 and above) available at https://github.com/gagneurlab/drop. The code to reproduce the figures in this manuscript can be found at https://github.com/gagneurlab/FRASER-2.0-analysis.