

Summary
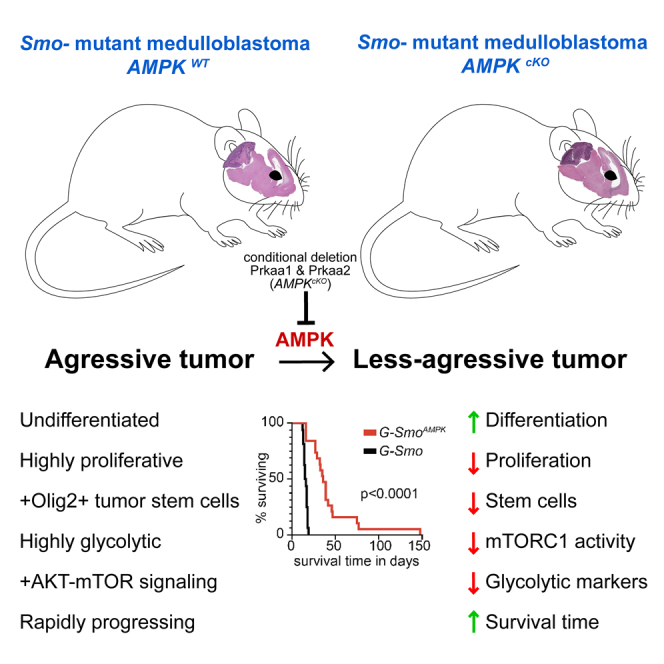
We show that inactivating AMPK in a genetic medulloblastoma model depletes tumor stem cells and slows progression. In medulloblastoma, the most common malignant pediatric brain tumor, drug-resistant stem cells co-exist with transit-amplifying cells and terminally differentiated neuronal progeny. Prior studies show that Hk2-dependent glycolysis promotes medulloblastoma progression by suppressing neural differentiation. To determine how the metabolic regulator AMPK affects medulloblastoma growth and differentiation, we inactivated AMPK genetically in medulloblastomas. We bred conditional Prkaa1 and Prkaa2 deletions into medulloblastoma-prone SmoM2 mice and compared SmoM2-driven medulloblastomas with intact or inactivated AMPK. AMPK-inactivation increased event-free survival (EFS) and altered cellular heterogeneity, increasing differentiation and decreasing tumor stem cell populations. Surprisingly, AMPK-inactivation decreased mTORC1 activity and decreased Hk2 expression. Hk2 deletion similarly depleted medulloblastoma stem cells, implicating reduced glycolysis in the AMPK-inactivated phenotype. Our results show that AMPK inactivation disproportionately impairs medulloblastoma stem cell populations typically refractory to conventional therapies.
Subject areas: Molecular biology, Cell biology, Sequence analysis, Cancer, Model organism
Graphical abstract
Highlights
-
•
Genetic AMPK inactivation slows tumor progression in SHH medulloblastoma in mice
-
•
AMPK-deleted tumors show increased differentiation and reduced proliferation
-
•
Disabling AMPK reduces mTORC1 activity and HK2-dependent glycolysis
-
•
AMPK inactivation disproportionately impairs medulloblastoma stem cell populations
Molecular biology; Cell biology; Sequence analysis; Cancer; Model organism
Introduction
Medulloblastoma is the most common malignant pediatric brain tumor and recurrence after treatment is the major cause of morbidity for patients with medulloblastoma. Medulloblastoma, is a heterogeneous group of cancers with 4 major subgroups, each with different recurrence risk,1,2,3,4 and individual tumors in each subgroup show cellular heterogeneity.5,6 Cellular heterogeneity may contribute to recurrence by increasing overall robustness to overcome selective pressures of therapy, and by generating specific resistant populations that drive recurrence. Understanding how different conditions affect the diversity of cell types within medulloblastomas is critical to designing therapies that can reduce the chance of recurrent disease. We previously showed that disrupting energy metabolism by conditionally deleting Hk2 in transgenic SHH medulloblastoma-prone mice slowed tumor progression and increased differentiation.7 These data show that metabolic patterns regulate clinically relevant aspects of medulloblastoma phenotype. Here, we examine how reducing metabolic adaptability alters medulloblastoma growth by disabling the intracellular energy sensor AMP-activated kinase (AMPK) in a genetic mouse model of SHH medulloblastoma. We then compare the phenotype of tumors with AMPK inactivation or Hk2 deletion.
SHH (Sonic Hedgehog) subgroup tumors, the most frequent subgroup of medulloblastoma, can be recapitulated in mice genetically engineered to hyperactivate SHH signaling in the brain, producing tumors that resemble human SHH medulloblastoma in site, pathology, gene expression, and cellular heterogeneity.5,8,9 Studies of SHH-driven medulloblastomas in mice show that cellular heterogeneity contributes to tumor recurrence as individual cell types within tumors show different sensitivity or resistance to specific therapies and cells that survive treatment drive recurrence.10,11 Tumor stem cells, marked by the expression of OLIG2, are specifically resistant to cytotoxic treatments currently used in medulloblastoma therapy and initiate post-treatment recurrence.10 Currently, 20% of patients with SHH subgroup medulloblastoma experience incurable recurrence despite optimal up-front therapy. Identifying ways to target therapy-resistant tumor stem cells may allow conventional treatment to be effective for more patients.
Tumor cells in medulloblastoma show a range of differentiation states that reflect the developmental progression in cerebellar development: from undifferentiated stem cells to committed cerebellar granule neuron progenitors (CGNPs) to differentiated neurons, including unipolar brush cells (UBCs) and cerebellar granule neurons (CGNs).12 However, medulloblastomas also contain tumor-derived cells with glial differentiation, and this divergence from the neural lineage of CGNPs provides indirect evidence of medulloblastoma stem cells with glio-neuronal potency.11,13
AMPK is a multi-subunit complex that is a primary regulator of cellular energy homeostasis,14 and disrupting AMPK function may produce important anti-tumor effects.15 AMPK-mediated coordination of energy metabolism may be particularly important in SHH medulloblastomas, because SHH induces specialized metabolic features that support malignant growth,16 including HK2-dependent aerobic glycolysis,7,17 lipogenesis,18 HIF1⍺ stabilization19 and mitochondrial fragmentation.20 These metabolic adaptations may increase the need for AMPK to sense energy scarcity in order to maintain energy homeostasis.
While AMPK is activated by the tumor suppressor LKB1, many lines of evidence show that the role of AMPK in cancer is more complex [20]. In contrast to the multiple inactivating LKB1 mutations that have been found in diverse human cancers,21,22,23 mutations in AMPK subunits causing human cancer are unknown. Divergent roles of LKB1 and AMPK are demonstrated by the KrasG12D mouse lung tumor model, in which Lkb1 deletion accelerates tumor growth while the co-deletion of the two AMPK catalytic subunits Prkaa1 and Prkaa2 inhibits tumorigenesis.24 Apart from operating downstream of LKB1, numerous studies show that AMPK functions to increase metabolic adaptability in normal cells within the brain.25,26 AMPK-mediated metabolic adaptability may be advantageous to brain tumors.27,28 Importantly, AMPK can be inactivated in the mouse brain or whole body without producing discernable developmental abnormalities.28,29,30 While chronic, sustained AMPK inactivation through Prkab1/1 co-deletion in astrocytes results in neurodegeneration in aged mice,26 therapies that transiently disrupt AMPK may be safe in pediatric patients with medulloblastoma.28,31
Prior studies show that AMPK acts in medulloblastoma to support tumor growth, as the deletion of Prkaa2, the gene encoding the AMPK⍺2 catalytic domain slows tumor progression in mice engineered to develop SHH-driven medulloblastoma.31 One process contributing to this effect is loss of AMPK-mediated phosphorylation of the zinc finger protein CNBP, which decreases the translation of Ornithine Decarboxylase (ODC) and the production of polyamines.31,32 However, the mechanisms through which polyamine production permits tumor growth are unresolved, and other AMPK-dependent mechanisms may also affect medulloblastoma growth. The repertoire of glycolytic genes expressed by medulloblastoma cells changes with differentiation state,7,16,17 and this variation suggest that specific types of tumor cells within medulloblastomas may depend on AMPK for different purposes.
To resolve AMPK function in different tumor cell subpopulations within medulloblastomas, we investigated the consequence of AMPK catalytic domain ablation through Prkaa1 and Prkaa2 co-deletion in medulloblastomas driven by oncogenic, mutant Smo. We used scRNA-seq and IF to examine the impact of AMPK inactivation on different types of cells within medulloblastomas, and compared to Hk2-deleted medulloblastomas to probe the mechanistic role of altered metabolism. Our studies show that Prkaa1/Prkaa2 co-deletion, such as Hk2 deletion, increased intra-tumoral differentiation. Notably, Prkaa1/Prkaa2 co-deletion reduced the populations of OLIG2-expressing stem cells that drive recurrence after conventional therapy. These studies provide new mechanistic insight into how metabolic specialization supports the diverse communities of tumor cells that promote recurrence after therapy.
Results
AMP-activated kinase signaling in Sonic Hedgehog medulloblastoma
To investigate a potential dependence of medulloblastoma on AMPK, we analyzed publicly available data from medulloblastomas resected from patients33 using the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). We did not find a statistically significant effect of PRKAA1 or PRKAA2 expression on overall survival in any subgroup. However, while PRKAA1 and PRKAA2 expression were variable in each subgroup, SHH subgroup medulloblastomas showed significantly higher PRKAA1 expression compared to each of the other tumor types (Figure 1A). The subgroup-specific tendency for increased PRKAA1 expression suggested a role for AMPK activity in the growth of SHH medulloblastomas.
Figure 1.

AMPK inactivation slows the progression of SHH-driven medulloblastoma
(A) PRKAA1 mRNA expression in clinical medulloblastoma samples of indicated subgroups, p values determined by 1-way ANOVA comparing the indicated pairs. In this box and whiskers plot, boxes indicate the upper and lower quartiles, the horizontal line between boxes indicates the mean, and whiskers inidcate 150% of the interquartile range, limited by the total range.
(B and C) Representative sagittal brain sections from G-Smo and G-SmoAMPK mice show similar site of tumor formation and hypercellular tumor pathology.
(D) Increased survival time in G-SmoAMPK mice compared to G-Smo controls with intact AMPK. Survival times in (D) were compared by Log-Rank test.
(E) Representative IF on sagittal sections of G-Smo and G-SmoAMPK medulloblastomas, showing pRB and NEUN expression.
(F) Quantification of the fractions of tumor cells expressing the indicated marker in 6 replicate G-Smo tumors and 5 replicate G-SmoAMPK tumors of each genotype, compared by Student’s t test. Bars = 3 mm in (B) and 50 μm in (C and E).
AMP-activated kinase inactivation in medulloblastomas increase survival of tumor-bearing mice
To test the importance of AMPK function in SHH medulloblastoma experimentally, we generated medulloblastoma-prone mice with CNS-specific Prkaa1/Prkaa2 co-deletion. We intercrossed Gfap-Cre mice that express Cre recombinase in CNS stem cells during development with Prkaa1loxP/loxP/Prkaa2 loxP/loxP29 and SmoM234 mouse lines to generate Gfap-Cre/Prkaa1loxP/loxP/Prkaa2 loxP/loxP/SmoM2 (G-SmoAMPK) mice with brain-wide, Cre-conditional AMPK inactivation, and brain-wide, Cre-conditional SHH hyperactivation.
SHH hyperactivation in Gfap-Cre/SmoM2 (G-Smo) mice produces medulloblastoma with 100% penetrance by postnatal day 10 (P10) and no other brain tumors.35 Similar to G-Smo mice, all G-SmoAMPK mice developed medulloblastoma that was detectable by characteristic change in head shape at P10 and resembled G-Smo medulloblastoma in site and overt pathology, as in representative samples taken at P16 (Figures 1B and 1C). In the absence of treatment, all G-Smo mice die of tumor progression by P22.7,36 In contrast, untreated G-SmoAMPK mice showed significantly longer survival times, with a median survival of 44 days in G-SmoAMPK compared to 16 days in controls (p < 0.0001 by Log-Rank test; Figure 1D). These data show that AMPK inactivation in the G-Smo model did not prevent medulloblastoma formation, but impaired medulloblastoma progression. The reduced medulloblastoma growth is consistent with prior studies in which isolated Prkaa2 deletion reduced tumorigenesis in an SHH medulloblastoma model driven by the SmoA1 allele.31 Considering the consistent tumor-suppressive effect of AMPK inactivation in multiple SHH medulloblastoma models, we studied the changes in growth parameters and cellular heterogeneity in AMPK-inactivated tumors.
We analyzed the populations of replicate G-SmoAMPK mice and G-Smo controls undergoing proliferation, differentiation, and apoptosis. In medulloblastomas from both genotypes harvested at P15, we compared the fractions of cells expressing the proliferation marker phosphorylated RB (pRB), the differentiation marker NEUN, and the apoptosis marker cleaved Capase-3 (cC3; Figure 1E; Figures S1A–S1C). pRB and NEUN showed complementary patterns of regional variation, distinguishing regions with more proliferation from regions with more differentiation (Figures S1A and S1B). G-SmoAMPK medulloblastomas showed significantly lower proliferation index, (defined as pRB+ cells/total cells, p = 3.6x10−5, compared by Student’s t test; fold change (FC) = 0.82) and significantly higher differentiation index (defined as NEUN+ cells/total cells, p = 0.018, compared by Student’s t test; FC = 1.22) (Figure 1F). In contrast, the apoptotic rate trended higher in the G-SmoAMPK tumors with increased variability compared to controls, but was not significantly different when compared by Student’s t test (Figures S1C and S1D). These findings implicate decreased proliferation and increased differentiation rather than increased apoptosis in the slower progression of AMPK-inactivated tumors.
AMP-activated kinase inactivation alters medulloblastoma cellular heterogeneity and induces differentiation
To resolve changes in cellular heterogeneity within the AMPK-inactivated and control tumors, we used scRNA-seq to compare tumors from 4 replicate P15 G-SmoAMPK mice to 6 medulloblastomas from replicate P15 G-Smo controls. We processed tumors using Drop-seq and generated cell-specific, bar-coded sequencing data, using our published methods.11,37,38,39 We filtered putative cells, identified by bead-specific barcodes, to address the common problems of gene drop out, unintentional cell-cell multiplexing and premature cell lysis.40 53% of putative cells from G-Smo mice and 43% of putative G-SmoAMPK cells met QC criteria and were included in the analysis resulting in 11,203 cells across 6 G-Smo replicates and 2426 cells across 4 G-SmoAMPK replicates. To compare the two genotypes at similar sequencing depths, we randomly down-sampled the G-Smo transcript counts to 46.5% of the original depth.41
We used the Harmony algorithm to integrate the scRNA-seq data from G-SmoAMPK and G-Smo tumors in a single analysis, generating a 2-dimensional UMAP projection in which individual cells were clustered with cells of similar gene expression. As in prior studies, this analysis defined both discrete clusters and a group of clusters with shared borders (Figure 2A). Individual replicates from each genotype distributed evenly across the UMAP (Figure S2). We determined the biological relevance of the clusters by generating cluster-specific differential gene expression profiles, comparing the expression by cells within the cluster to the expression by all cells outside the cluster (Data S1). These gene expression profiles identified the cell type of each cluster. The full set of gene expression data with single cell resolution from G-SmoAMPK and G-Smo control tumors can be viewed at: https://malawsky.shinyapps.io/Gerhson_AMPK_analysis.
Figure 2.

AMPK inactivation alters cellular heterogeneity in SHH driven medulloblastomas
(A) UMAP plot of all cells from G-Smo and G-SmoAMPK tumors, color-coded by cluster. Cells are localized according to their proximity in PCA space.
(B) Feature plot showing the expression of Barhl1 and Neurod1, color coded over the UMAP shown in (A).
(C) Heatmap showing the scaled expression of indicated genes in each tumor cell cluster. Hierarchical clustering of genes appropriately grouped the proliferation markers and differentiation markers into two discrete sets. Hierarchical clustering of clusters 0–8 grouped the clusters into two discrete sets that corresponded with the expression of differentiation and proliferation markers.
(D) Comparison between genotypes of the fractions of differentiation tumor cells (Clusters 1, 4 and 6) from each replicate. The blue and red regions show the mean % differentiating and reciprocal % proliferating, indicating the balance between proliferation and differentiation within each genotype.
(E and F) Representative (E) KI67 IF and (F) NEUROD1 IHC, on sagittal sections of G-Smo and G-SmoAMPK medulloblastomas.
(G) Comparison of the fractions of tumor cells from each replicate expressing KI67 and NEUROD1.
(H) Comparison of populations of each stromal cluster in G-Smo and G-SmoAMPK tumors. In panels (D), (G) and (H), horizontal bars indicate the mean values for the conditions, dots represent values for individual replicates, and the number of visible dots may be smaller than the sample size (n) due to multiple replicates showing similar values. In (D) and (G), Student’s t test was used to make pairwise comparisons. In (H), Dirichlet regression was used to make statistical comparisons. In panels (E) and (F), bars = 1 mm in the left panel and 25 μm in the right panel.
As in our prior studies, these methods identified each of the discrete clusters as different types of stromal cells typical of brain tissue. We thus identified astrocytes, oligodendrocytes, ependymal cells, myeloid cells, endothelial cells and fibroblasts (Table 1; Figure 2A). The cluster-specific gene expression patterns identified the multi-cluster aggregate as medulloblastoma cells in a range of states that paralleled CGNP development, from proliferative and undifferentiated to non-proliferative at different stages of neural differentiation (Table 1).
Table 1.
Identification of clusters as specific types of tumor and stromal cells
| Cluster | Cell type designation | distinctive markers | Tumor cell classification |
|---|---|---|---|
| 0 | tumor cells | Barhl1 | proliferative |
| 1 | early differentiating tumor cells | Tubb3, Stmn2 | differentiating |
| 2 | proliferating tumor cells | Top2a, Dek | proliferative |
| 3 | tumor cells | Gas5, Rps20 | proliferative |
| 4 | differentiating tumor cells | Stmn2, Celf4, Nrxn1 | differentiating |
| 5 | mitotic tumor cells | Cenpa, Ube2c | proliferative |
| 6 | late differentiating tumor cells | Cntn2, Gria2, Apc | differentiating |
| 7 | proliferating tumor cells | Top2a, Mki67 | proliferative |
| 8 | proliferating tumor cells | Top2a, Mki67, Ube2c | proliferative |
| 9 | unresolved | Mt1, Mt2 | stromal |
| 10 | immature oligodendrocytes | Olig1/2, Pdgfra | stromal |
| 11 | myeloid cells | C1qc/a, Tyrobp | stromal |
| 12 | myelinating oligodendrocytes | Plp1, Mbp | stromal |
| 13 | endothelial cells | Cldn5 | stromal |
| 14 | fibroblasts | Dcn | stromal |
| 15 | astrocytes | Aqp4, Slc1a3 | stromal |
The expression of the CGNP marker Barhl142 and the CGN differentiation marker NeuroD143 divided the multi-cluster aggregate into two adjacent, mutually exclusive domains, consisting of Barhl1+ clusters 0, 2, 3, 5, 7 expressed and NeuroD1+ clusters 1,4 and 6 (Figure 2B). Similarly, hierarchical clustering using the expression of a panel of genes associated with either cell cycling (Mki67, Pcna, Top2a, Ccnd1 and SHH pathway transcription factor Gli1) or neural differentiation (Rbfox3, Neurod1, Cntn2 and Tubb3) divided clusters 0, 2, 3, 5, 7, and clusters 1,4 and 6 into distinct groups (Figure 2C). Uniformly decreased expression of proliferation markers and increased expression of neural markers identified clusters 1,4 and 6 as non-proliferative and differentiating. In contrast, increased expression of heterogeneous combinations of proliferation markers identified Clusters 0, 2, 3, 5, 7, and 8 as proliferative.
We compared the fractions of differentiating cluster populations (summed populations of clusters 1, 4, and 6) in each replicate G-Smo or G-SmoAMPK animal, using the Student’s t test. G-SmoAMPK tumors showed significantly increased fractions of differentiating cells, and reciprocally decreased proliferating cells. IF studies confirmed significantly fewer cells expressing KI67 protein in G-SmoAMPK tumors (Figures 2D and 2E) and significantly more cells expressing NEUROD1 protein (Figures 2F and 2G) compared to controls. These studies confirm that AMPK inactivation shifted the balance between proliferation and differentiation in G-SmoAMPK tumors toward differentiation, consistent with increased NEUN and decreased pRB.
We disaggregated the cells by genotype to compare the populations and gene expression patterns of the type of cell in G-SmoAMPK and G-Smo control tumors. In view of the inter-relation between populations normalized cluster populations, we used Dirichlet regression to make statistical comparisons of stromal cluster populations in G-SmoAMPK tumors versus controls (Figure 2H).44 Most stromal populations did not show significant differences between genotypes, including astrocytes and myelinating oligodendrocytes which were both within the GFAP lineage and thus subject to Prkaa1/2 deletion. However, immature oligodendrocytes (Cluster 10), which were within the GFAP lineage and thus also subject to Prkaa1/2 deletion, were markedly less numerous in G-SmoAMPK tumors (p = 0.02; FC = 0.33). Additionally, G-SmoAMPK tumors showed a marked increase in endothelial cells (p = 0.02; FC = 3.40), which are not within the GFAP lineage and therefore were not Prkaa1/2-deleted. AMPK inactivation thus increased differentiation in both the tumor and oligodendrocyte lineages and also produced a non-cell autonomous increase in endothelial cells.
AMP-activated kinase-inactivated medulloblastomas show reduced OLIG2+ stem cells
Considering increased differentiation in AMPK-inactivated medulloblastomas and extended survival time, we determined if tumor stem cell populations, which remain undifferentiated and play an essential role in medulloblastoma progression, were reduced. We compared the populations of tumor cells that expressed the stem cell markers Hes1, Olig2, Sox2, and Vim in G-Smo versus G-SmoAMPK tumors. To focus on tumor stem cells, we excluded stromal cells, as astrocytes express Hes1 and Sox2, oligodendrocytes express Sox2 and Olig2, and diverse stromal cells express Vim. G-SmoAMPK medulloblastomas showed significantly fewer Olig2+ cells (FC = 0.43, p = 0.0025 by Student’s t test) and Vim+ cells (FC = 0.52, p = 0.0011 by Student’s t test) (Figures 3A and 3B). Similarly, Sox2+ cells and Hes1+ cells showed trends toward reduced numbers that were not statistically significant (FC = 0.90, p = 0.67 and FC = 0.82 p = 0.15), within the statistical power of the study.
Figure 3.

Decreased stem cell populations in G-SmoAMPK tumors
(A) Expression of indicated stem cell markers in the indicated genotypes, projected onto the disaggregated UMAPs. Hes1, Sox2, Olig2 and Vim were significantly lower in G-SmoAMPK tumors, while Nes was not significantly different.
(B) Dot plot in which the fractions of cells in each genotype that express the marker are represented by dot size. This panel presents the same data as panel (A), visualized using a different method. Hierarchical clustering show relatively similar variation across genotypes for the group of Olig2 and Vim and for the group of Sox2 and Hes1.
(C) Representative OLIG2/SOX10 IF in sagittal sections of tumors of indicated genotype.
(D) Quantification of OLIG2/SOX10 IF as in (C) in replicate samples of each genotype.
(E) Quantification of Yfp+ glial cells in the indicated genotypes. In panels (A), (B), (D) and (E), the p values were determined Student’s t test. In panel (C), bars = 2 mm in the left panel and 100 μm in the right panel.
To compare stem cell populations using an alternative detection method, we analyzed OLIG2 protein expression, detected by IF. As both tumor stem cells and oligodendrocytes express OLIG2, we used the oligodendrocyte marker SOX10 to distinguish between these two cell types, considering OLIG2+/SOX10-cells in medulloblastomas as tumor stem cells. These OLIG2+/SOX10-stem cells were increased in perivascular regions (Figure 3B), consistent with prior descriptions of medulloblastoma stem cells.45,46 The fractions of OLIG2+/SOX10-cells were markedly reduced in G-SmoAMPK tumors (Figures 3C and 3D; p = 0.002; FC = 0.29; Student’s t test), consistent with the differences in Olig2+ populations in the tumor cell populations isolated and analyzed in the scRNA-seq data. The scRNA-seq data on Olig2 mRNA expression and the IF studies of OLIG2 protein consistently demonstrate decreased Olig2-expressing stem-like populations in AMPK-inactivated medulloblastomas.
AMP-activated kinase-inactivation disrupts tumor to glia trans-differentiation
Lineage tracing in mouse models of SHH medulloblastoma show that tumor cells trans-differentiate to generate tumor-derived glial populations,11,13 and that these malignant glial cells play a supportive role in tumor growth.13 Glial trans-differentiation requires stem-like pluripotency, and in light of the reduced stem-like population in G-SmoAMPK tumors, we compared glial trans-differentiation in G-SmoAMPK and G-Smo control tumors.
We traced lineage in the scRNA-seq data using the 3′ Yfp sequence in the Cre-conditional SmoM2 transgene. We previously used this method to show that some astrocytes and oligodendrocytes derive from the tumor lineage in medulloblastomas that form in Math1-Cre/SmoM2 mice.11 In the G-Smo control tumors, we found SmoM2-Yfp+ cells in the astrocytic cluster 15 and in the oligodendrocytic clusters 10 and 12. The detection of the Yfp tag in astrocytes and oligodendrocytes was expected since Gfap-Cre activates SmoM2 in glio-neuronal stem cells of the developing brain that generate both glial and neuronal progeny. However, not all glia were SmoM2-Yfp+, indicating that SmoM2 activation was not uniform in these populations. We therefore considered that tumor-derived glia contributed to the Yfp+ glial populations and used the variation glial Yfp expression to compare the glial cells of tumor lineage between genotypes. G-SmoAMPK tumors showed significantly smaller SmoM2-Yfp+ glial populations, indicating fewer tumor-derived glia (Figure 3E, p = 0.024; FC = 0.18; Student’s t test). This evidence for decreased glial trans-differentiation suggests that the tumor stem cell pluripotency that allows glial differentiation within medulloblastomas was reduced in G-SmoAMPK tumors. However, it is also possible that Prkaa1/2-deleted glia were less able to tolerate SmoM2 expression, and this alternative possibility may also explain the variation in Yfp-tagged glia between genotypes.
Decreased mTORC1 activation in AMP-activated kinase-inactivated medulloblastomas
To identify molecular pathways altered by AMPK inactivation, we defined the set of genes that were differentially expressed in G-SmoAMPK tumors compared to G-Smo control tumors. We analyzed DEGs in the combined set of all tumor cells (Clusters 0–8), and in each separate cluster. Analyzing DEGs within each cluster controlled for differences in the distribution of tumor cells across clusters populations in the two genotypes, and analyzing DEGs that recurred in multiple clusters identified changes in gene expression the were common across cells in multiple states. For these analyses we identified DEGs that showed a corrected p value of <0.05 (Wilcoxon rank-sum test).
Comparing all tumor cells in G-SmoAMPK mice versus G-Smo controls, we identified 64 DEGs, 34 up-regulated in G-SmoAMPK and 30 down-regulated (Data S1). Of these 64 DEGs, 25 were ribosomal genes and 30 were within the broader set of genes ribonucleoprotein complex (p = 2.39x10−37 and 9.84x10−27 by PANTHER Overrepresentation Test using the GO cellular component complete dataset). Consistent with high ribosomal association, gene set enrichment analysis (GSEA) identified translation as the most prominently enriched cellular process (p = 0.00032 by GSEA) (Figure 4A). PANTHER Overrepresentation Test using the GO biologic process complete dataset demonstrated that genes down-regulated in G-SmoAMPK tumors were more strongly associated with translation, with 6/34 up-regulated genes associated with translation (p > 0.05) compared to 20/30 down-regulated genes (p = 5.22 x 10−25). These data implicate decreased translation in G-SmoAMPK tumors.
Figure 4.

Decreased mTORC1 activity in G-SmoAMPK tumors
(A) GSEA showing the pathways significantly enriched in the DEGs in the tumor cells from G-SmoAMPK mice versus G-Smo controls (B) Genes that are down-regulated in 1 or more clusters, ranked by number of clusters with differential expression. Red box marks the 11 genes down-regulated in 7/9 clusters. Blue box marks the 14 genes down regulated in 5/9 clusters.
(C) Venn diagrams of the genes down-regulated in the 5/9 clusters with the highest number of differentially expressed genes, or in the 5/9 clusters with the fewest differentially expressed genes. Cluster 1 is included in both Venn diagrams as a point of consistency, as depicted in the graph. In each Venn diagram, the cluster number is indicated followed by the number of DEGs for that cluster in parentheses. These Venn diagrams depict sets of 12 or 5 genes down-regulated in all included clusters and brackets show the indicated sets of genes commonly down-regulated genes in the list from (B).
(D) Representative p4EBP1 IF in sagittal sections of tumors of indicated genotype. (E) Quantification of p4EBP1 IF as in (D) in replicate samples of each genotype, compared by Student’s t test. In panel (D), bars = 2 mm in the low magnification image and 100 μm in the higher magnification image.
We next analyzed DEGs in each tumor cluster. 36 different genes were significantly down-regulated in any of the 9 tumor cell clusters of G-SmoAMPK tumors and 76 genes significantly up-regulated (Data S1). The DEGs that were decreased in each cluster in G-SmoAMPK tumors showed significant overlap (Figures 4A and 4B; p < 10−15 by extended hypergeometric test47), and the DEGs that were increased also overlapped significantly (p < 0.001 by extended hypergeometric test). AMPK inactivation thus produced changes in gene expression that were common across multiple clusters.
Consistent with pathway enrichment analyses from the comparison of all tumor clusters grouped together, the DEGs that were down-regulated in multiple clusters were associated with translation. We subjected the 14 DEGs down-regulated in 5 or more clusters in G-SmoAMPK tumors to PANTHER overrepresentation analysis. The criterion of differentially expressed in 5 or more clusters was used to limit the analysis to genes that were commonly altered in tumor cells in different states. This analysis identified translation as the primary process statistically implicated (corrected p < 4x10−23), driven by multiple ribosomal proteins and the translation elongation factor Eef1b2 (Figure 4B). In contrast to the pattern detected in the down-regulated genes, GO analysis of the 8 genes up-regulated in 5 or more clusters in G-SmoAMPK tumors did not identify a statistically implicated process. Thus, while the common process underlying the up-regulation of similar genes in multiple clusters remained cryptic, the pattern of genes recurrently down-regulated in multiple clusters indicated reduced translation.
The mTORC1 pathway is known to regulate translation and ribosomal biogenesis,48,49 and we speculated that the decreased expression of translation-related and ribosomal genes in G-SmoAMPK medulloblastomas might reflect decreased mTORC1 activity in the tumors. In support of this proposed connection to mTORC1 activity, we previously found that the down-regulation of translation-related genes in medulloblastomas treated with palbociclib correlated with decreased mTORC1 signaling.50 We therefore investigated whether mTORC1 signaling was reduced in in G-SmoAMPK tumors.
To quantify mTORC1 activation in tumors, we analyzed the phosphorylation of the mTORC1 substrate 4EBP1 (p4EBP1) using IF, comparing G-SmoAMPK and control tumors (Figure 4C). G-SmoAMPK tumors showed significantly fewer p4EBP1+ cells (FC = 0.84; p = 0.0002; Student’s t test) (Figure 4D). Both the recurrent patterns of differential gene expression across clusters and the p4EBP1 studies show reduced mTORC1 activation in G-SmoAMPK tumors compared to G-Smo control tumors. AMPK is known to inhibit mTORC1,51,52,53 and the reduced mTORC1 activity in G-SmoAMPK tumors is opposite of the predicted effect of acute AMPK inhibition. While this unexpected suppression of mTORC1 activity may result from a homeostatic response to the sustained AMPK inactivation in G-SmoAMPK tumors, more information on the timing of the onset of reduced mTORC1 activity will be needed to establish the temporal dynamics of the effect.
Altered glycolytic gene expression in AMP-activated kinase-inactivated medulloblastomas
We previously found that HK2-dependent aerobic glycolysis interacts with AMPK and maintains undifferentiated populations within SHH-driven medulloblastomas. In these prior studies, we conditionally deleted Hk2 in medulloblastomas by crossing G-Smo mice with mice that harbor conditional Hk2 deletion (Hk2 loxP/loxP) to produce the genotype Gfap-Cre/SmoM2/Hk2 loxP/loxP (G-SmoHk2). We found G-SmoHk2 mice developed medulloblastomas with increased differentiation, and that these tumors progressed more slowly, resulting in increased event-free survival of G-SmoHk2 mice compared to G-Smo mice.7 G-SmoHk2 tumors also showed markedly increased AMPK activation, compared to G-Smo tumors.7 The coincident findings of AMPK activation and increased in differentiation in Hk2-deleted tumors suggested a model in which Hk2 deletion causes AMPK activation and AMPK activation causes increased differentiation. However, our current finding of increased differentiation in G-SmoAMPK tumors demonstrates that differentiation does not require AMPK activation, suggesting that the interaction between Hk2 deletion and AMPK activation may not drive reduced tumor growth in the G-SmoHk2 phenotype.
To test directly whether HK2 modulates differentiation through AMPK, we bred mice with medulloblastomas with the deletion of both Hk2 and Prkaa1/2. We crossed Gfap-Cre/Prkaa1loxP/loxP/Prkaa2 loxP/loxP/Hk2 loxP/loxP mice with SmoM2/Prkaa1loxP/loxP/Prkaa2 loxP/loxP/Hk2 loxP/loxP mice to generate the genotype Gfap-Cre/SmoM2/Prkaa1loxP/loxP/Prkaa2 loxP/loxP/Hk2 loxP/loxP (G-SmoAMPK/Hk2). These G-SmoAMPK/Hk2 mice developed medulloblastomas with 100% frequency, and the tumors resembled the previously described G-SmoHk2 medulloblastomas, marked by regions of differentiation (Figure 5A).7 These co-deletion studies show that the increased differentiation phenotype caused by Hk2 deletion does not require AMPK activity, and therefore that AMPK does not operate downstream of HK2-dependent glycolysis to regulate differentiation. In light of these findings, we examined the alternative possibility suggested by AMPK activation in Hk2-deleted tumors, that AMPK may operate upstream to induce HK2-dependent aerobic glycolysis.
Figure 5.

Defining the interaction of AMPK and HK2
(A) Representative H&E sections compare G-Smo control tumors to tumors with the deletion of Hk2 or co-deletion of Hk2, Prkaa1 and Prkaa2. Increased differentiation in Hk2-deleted tumors is not rescued by the co-deletion of Prkaa1 and Prkaa2. IGL regions in G-SmoHk2 and G-SmoAMPK tumors are indicated.
(B) scRNA-seq data showing decreased Hk2 and increased Eno1 in M-SmoAMPK tumors.
(C) Representative OLIG2 immunofluorescence, comparing G-Smo and Hk2-deleted medulloblastomas, with the quantification of replicate samples. p value determined by Student’s t test. Bars = 1 mm in the left panel and 25 μm in the right panel (A). Bars = 1 mm in C.
To determine whether AMPK activity regulates the glycolytic state change that we have previously observed in CGNPs and SHH-driven medulloblastoma,7 we compared the number of cells expressing each gene in the glycolytic pathway in G-SmoAMPK medulloblastomas compared to control tumors. This analysis showed significant differences in the fractions of cells expressing Hk2 and Eno1 in AMPK-deleted tumors. Hk2+ cells were significantly reduced in G-SmoAMPK tumor cells (FC = 0.71; p = 0.008; Student’s t test) (Figure 5B), consistent with the hypothesis that AMPK activity induces HK2 in SHH-driven medulloblastoma, as seen in prior studies of normal muscle cells.54 In contrast, cells expressing Eno1, which acts at the end of glycolysis, were increased in G-SmoAMPK tumors (FC = 2.34; p = 0.003; Student’s t test) (Figure 5B). AMPK inactivation thus induced specific changes in glycolytic gene expression, predicted to reduce the entry of glucose into glycolysis and to increase the processing of glycolytic intermediates toward pyruvate.
Similar stem cell changes in AMP-activated kinase-inactivated and Hk2-deleted medulloblastomas
To determine if alterations in glycolysis may produce specific features of the AMPK-inactivation phenotype, we analyzed OLIG2+ populations in Hk2-deleted medulloblastomas. Comparing G-Smo/Hk2floxed mice to G-Smo controls we found that Hk2-deletion was sufficient to reduce the fraction of OLIG2+ tumor cells (FC = 0.43; p = 0.03; Student’s t test) (Figure 5C). Reduced tumor stem cell populations was thus a common feature of AMPK-inactivated and Hk2-deleted medulloblastomas, suggesting that both genes operate in a common pathway to maintain tumor stem cell populations.
Discussion
Medulloblastomas are highly proliferative tumors that configure energy metabolism to support malignant growth through enhanced aerobic glycolysis7,17 and lipogenesis.18,55 AMPK is an intracellular energy sensor that coordinates anabolic and catabolic processes with nutrient availability and may crucially regulate tumor metabolism in ways that might be predicted either to support or to inhibit tumor growth. In SHH signaling, AMPK has been shown to interact directly with GLI1 to suppress SHH activity in medulloblastoma.56,57 In contrast to the anti-tumor effects of AMPK predicted by its interactions with GLI1, however, a prior study of SHH medulloblastoma showed that the deletion of AMPK subunit Prkaa2 slowed tumor progression in a primary mouse model.31 Understanding the mechanisms through which AMPK inactivation reduces medulloblastoma growth may allow the design of targeted therapies that exploit the role of AMPK in SHH medulloblastoma and potentially in other cancers.
Our data show that AMPK inactivation slowed medulloblastoma growth and altered tumor cell heterogeneity by disproportionately impacting tumor stem cells. We inactivated AMPK by conditionally deleting both catalytic subunits, Prkaa1 and Prkaa2, in mice carrying Gfap-Cre and SmoM2 alleles, generating G-SmoAMPK mice with AMPK inactivation and SHH hyperactivation throughout the brain. In these mice, AMPK-inactivated medulloblastomas formed with 100% penetrance but progressed more slowly than medulloblastomas in controls with SHH hyperactivation but intact AMPK. Like control tumors, AMPK-inactivated tumors comprised medulloblastoma cells with a range of differentiation states. However, AMPK-inactivated medulloblastomas showed a shift in cell populations, with increased differentiated cells compared to controls and specifically reduced populations of OLIG2+ stem cells and malignant glia.
While AMPK inactivation affected different types of tumor cells in different ways, increasing differentiating populations and decreasing tumor stem cells, the effects of gene expression were consistent across multiple cell types. In tumor cells across the differentiation spectrum, AMPK inactivation decreased the expression of multiple genes related to protein translation. This global down-regulation of translation-related genes suggested reduced mTORC1 activity, which was confirmed by the finding of reduced p4EBP1. A decrease in mTORC1 signaling in G-SmoAMPK tumors was unexpected, as acute AMPK activation inhibits mTOR.58,59 We speculate that G-SmoAMPK tumor cells reduce mTORC1 in a homeostatic response to AMPK inactivation.
G-SmoAMPK medulloblastomas also showed altered expression of glycolytic genes, with decreased Hk2 and increased Eno1. By impeding the entry of glucose into the glycolysis through HK2 and enhancing the exit of glycolytic intermediates as phosphoenolpyruvate through ENO1, AMPK inactivation reduced the potential for the diversion of glycolytic intermediates for other purposes.
We noted specific changes in the stromal populations in AMPK-inactivated tumors with important implications. Within the stromal populations subject to the deletion of Prkaa1 and Prkaa2, the less mature oligodendrocyte subset was specifically depleted in G-SmoAMPK medulloblastomas. While additional studies are needed to confirm these findings from scRNA analysis, oligodendrocytes, such as tumor stem cells, express OLIG2, and their apparently altered differentiation suggests an interaction between AMPK and OLIG2 may crucially modulate OLIG2 function.
Within the stromal populations that were not subject to Prkaa1 and Prkaa2 co-deletion, the endothelial cells were significantly increased in G-SmoAMPK medulloblastomas. As these cells were not within the GFAP lineage, their increased population cannot be cell autonomous. Increased endothelial populations may indicated changes in tumor vasculature caused by AMPK inactivation in tumor cells, as we have previously seen in glycolysis deficient medulloblastomas.7 This effect on endothelial cells may thus be another point of similarity between Hk2-deleted and AMPK-inactivated medulloblastomas.
By reducing stem cell self-renewal, glial trans-differentiation, HK2-dependent aerobic glycolysis and mTORC1 activation, AMPK inactivation altered multiple processes important for tumor progression. Each of these processes has been tested in isolation in previous studies. The maintenance of OLIG2+ stem cell pools has been shown to promote both medulloblastoma progression and recurrence after cytotoxic therapy therapy.10 HK2-dependent aerobic glycolysis is similarly required for tumor progression.7 Paracrine signaling between malignant glia and myeloid cells within medulloblastomas also promotes tumor growth.13 The activation of mTORC1 is similarly essential for tumor growth, and mTORC1 inhibition slows progression.60 All of these processes are likely to be interrelated, and to contribute to the anti-tumor effect of AMPK inactivation. AMPK inactivation thus sets in motion a complex set of processes that result in the observed shift from multipotent stem cells to more differentiated tumor cells, with the net effect of slowing tumor growth.
To define causal relationships between processes that we found to be altered in AMPK-inactivated tumors, we compared the phenotypes of AMPK-inactivated and Hk2-deleted tumors. Both Hk2 deletion and Prkaa1/Prkaa2 co-deletion slow tumor growth and extend event-free survival. Importantly, Hk2 deletion impaired stem cell maintenance and increased differentiation, reproducing the altered cellular heterogeneity of the Prkaa1/Prkaa2 co-deleted tumors. These genetic data provide evidence for a linear pathway in which AMPK and HK2 both operate in the same direction to maintain undifferentiated, pluripotent populations within medulloblastomas.
The effect of AMPK inactivation on OLIG2+ stem cells identifies a vulnerability that may be exploited therapeutically for clinical benefit. Tumor stem cells are intrinsically resistant to cytotoxic therapy and drive recurrence. Ways to target these stem cells are urgently needed. Our data show that chronic blockade of AMPK activity disproportionately affects these stem cells, suggesting a way to target these otherwise resistant populations. Follow up studies are needed to test pharmacologic AMPK inhibition in combination with current, cytotoxic therapy. As genetic deletion studies demonstrate that AMPK inactivation can suppress tumor growth, and AMPK deletion in model organisms does not have clear deleterious effects,29 AMPK inhibition may emerge as an important new avenue for cancer therapy.
Limitations of the study
We demonstrate that AMPK inactivation slows tumor progression in SHH medulloblastoma, reduces mTORC1 activity and depletes stem cell populations that express OLIG2. However, the precise mechanisms for the down-regulation of mTORC1 and the disproportionate impact on tumor stem cells are not clear. Additionally, we did not consider sex-specific responses in the study. Additional studies are required to investigate how sustained inactivation of AMPK, which is known to phosphorylate mTORC1, results in net mTORC1 inhibition in tumors in vivo, how AMPK contributes to the maintenance of the stem cell phenotype, and whether there are sex-specific differences in stem cell metabolism. Our scRNA-seq studies identified a large number of differentially expressed genes, and we limited our additional validation studies to a subset of these genes selected to confirm biologically relevant trends in the data. The statistical methods applied to the scRNA-seq data require that the assumptions of the statistical models are satisfied, and these assumptions are difficult to evaluate on the limited set of replicates. For this reason, we have limited our conclusions about changes in cellular diversity induced by AMPK activation to phenotypes that we validated by quantitative studies of protein expression, specifically changes in differentiation, proliferation, OLIG2 expression and mTORC1 activation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| OLIG2 | Cell Marque | Cat.# 387R-14 |
| Sox10 | Cell Signaling Technology | Cat.# 7833S |
| PRB | Cell Signaling Technology | Cat.# 8957 |
| NeuN | Millipore | Cat.# MAB377; RRID: AB_2298772 |
| Ki67 | Cell Signaling Technology | Cat.# 12202S |
| NeuroD1 | Abcam | Cat.# ab213725; RRID: AB_2801303 |
| p4EBP1 | Cell Signaling Technology | Cat.# 2855 |
| Critical commercial assays | ||
| Papain Dissociation System | Worthington Biochemical | Cat.# LK003150 |
| Barcoded Seq B Drop-seq beads | ChemGenes | Cat.# Macosko-2011-10(V+) |
| PDMS device | FlowJEM | Request: “Drop-Seq Device with Aquapel Coating” |
| Deposited data | ||
| scRNA-seq raw and analyzed data | This paper | GEO: GSE150579 |
| scRNA-seq raw and analyzed data | This paper | GEO: GSE190297 |
| Mouse: GFAP-Cre: FVB-Tg(GFAP-cre)25Mes/J | The Jackson Laboratory | RRID:IMSR_JAX:004600 |
| Mouse: Prkaa1loxP: Prkar1atm1.2Lsk/J | The Jackson Laboratory | RRID:IMSR_JAX:036801 |
| Mouse: Prkaa2loxP: Prkaa2tm1.1Sjm/J | The Jackson Laboratory | RRID:IMSR_JAX:014142 |
| Mouse: SmoM2: Gt(ROSA)26Sortm1(Smo/EYFP)Amc/J | The Jackson Laboratory | RRID:IMSR_JAX:005130 |
| Mouse: C57BL/6: | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Software and algorithms | ||
| Seurat R package version 3.1.1 | Satija Lab | https://cran.r-project.org/src/contrib/Archive/Seurat/Seurat_3.1.1.tar.gz |
| Harmony algorithm | Korsunsky et al.61 | https://github.com/immunogenomics/harmony |
| Tissue Studio 4.4.2 | Definiens | Build 60765 x64 |
Resource availability
Lead contact
Further information and and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Timothy R. Gershon, MD, PhD, timothy.gershon@emory.edu.
Materials availability
Identification information for the GFAP-Cre (JAX:004600) Prkaa1loxP (JAX:036801), Prkaa2loxP (JAX:014142), SmoM2 (JAX:005130) and C57BL/6 (JAX:000664) mouse lines are available from Jackson Labs.
Data and code availability
-
•
The scRNA-seq data were deposited in the Gene Expression Omnibus (GEO) database and are publicly available at the time of publication. Accession codes are listed in the key resources table. Microscopy data reported in the paper will be shared by the lead contact upon request.
-
•
This paper did not use or create original code. The scRNA-seq analysis was performed using the publicly available Seurat R package version 3.1.1.
-
•
Any additional information required to reanalyze the data reported in this paper will be made available by the lead contact upon request.
Experimental model and study participant details
Mice
We crossed SmoM2 mice (Jackson Labs, stock # 005130) with GFAP-Cre mice (Jackson Labs, stock # 004600), to generate GFAP-Cre/SmoM2 (G-Smo) mice. We crossed Prkaa1loxP/loxP/Prkaa2 loxP/loxP mice that harbor loxP sites flanking the coding regions of Prkaa1 and Prkaa229 with GFAP-Cre mice to generate Gfap-Cre/Prkaa1loxP/loxP/Prkaa2 loxP/loxP (G-CreAMPK) mice. These mice have been previously published and the effective depletion of PRKAA1 and PRKAA2 proteins in the brains of these mice has been demonstrated.29 We then crossed G-CreAMPK mice with SmoM2 mice generate G-SmoAMPK mice with Prkaa1/2-deleted medulloblastomas. All mice were of species Mus musculus and crossed into the C57BL/6 background through at least five generations. All animal studies were carried out with the approval of the University of North Carolina Institutional Animal Care and Use Committee under protocols (19–098 and 21-011). Male and female animals were randomly selected for each experimental condition.
All mice were immunocompetent and not known to have any confounding health conditions. No animals were previously used in other experiments. All animals were drug naive and no animals were treated with any agents. All mice were housed and maintained by the University of North Carolina Department of Comparative Medicine under standard, humane husbandry conditions. No cell lines were used.
Method details
Histology and immunofluorescence (IF)
Mouse brains were processed, immunostained and quantitatively analyzed as previously described.11,46,62 Briefly, slides were de-paraffinized and stained in an automated processor. Primary antibodies used were: OLIG2 diluted 1:100 (Cell Marque, # 387R-14), SOX10 diluted 1:200 (Cell Signaling Technology, #7833S). Stained images were counterstained with DAPI, digitally imaged using an Aperio Scan Scope XT (Aperio) and imported to Definiens Architect XD 2.7 Build 60765 x64 for analysis with Tissue Studio version 4.4.2.
Tissue preparation for drop-seq
Data from G-Smo and G-SmoAMPK tumors were obtained by Drop-seq in separate batches, using the same methods as described below. The G-Smo data was previously published37 and made publicly available, while the G-SmoAMPK data were newly obtained for this study. In both batches, mice were euthanized by decapitation under isoflurane anesthesia. The brain was divided along the sagittal midline and one-half was processed for histology and a sample of tumor was dissected from the other half and processed for Drop-seq analysis. This sample was dissociated using the Papain Dissociation System (Worthington Biochemical) following the protocol used in previous studies.11,63 Briefly, tumor samples were incubated in papain at 37°C for 15 min, then triturated and the suspended cells were spun through a density gradient of ovomucoid inhibitor.
We resuspended pelleted cells in 1 mL HBSS with 6 g/L glucose and diluted in PBS-BSA solution to a concentration of 95–110 cells/μL. Barcoded Seq B Drop-seq beads (ChemGenes) were diluted in Drop-seq lysis buffer to a concentration between 95 and 110 beads/μL. Tumor cells were co-encapsulated with barcoded beads using FlowJEM brand PDMS devices as previously described.11 All cells were processed within 1 h of tissue dissociation. Droplet breakage and library preparation steps followed Drop-seq protocol V3.1.64 After PCR, amplified cDNA was subjected to Ampure XP cleanup at 0.6x and 1x ratios to eliminate residual PCR primers and debris. If PCR failed to generate adequate cDNA, the PCR was repeated with the 3rd round increased from 11 to 13 cycles.
For quality control (QC) purposes, library pools consisting of the tagmented cDNA from 2,000 beads/run were prepared and sequenced to low depth (∼2.5M reads/2K beads). We used the resulting data to assess library efficiency, including total read losses to PolyA regions, nonsense barcodes and adapter sequences as well as the quality and number of the transcriptomes captured. Passable runs contained 40–60% of reads associated with the top 80–100 barcodes found in 2,000 beads. Drop-seq runs passing QC were then prepared for high-depth sequencing on an Illumina Novaseq S2 flow cell.
Quantification and statistical analysis
Processing of scRNA-seq data
Data analysis was performed using the Seurat R package version 3.1.1.65 Data were subjected to several filtering steps. First, only genes that were detected in at least 30 cells were considered, to prevent misaligned reads appearing as rare transcripts in the data. Cells were then filtered using specific QC criteria to limit the analysis to cells with transcriptomes that were well-characterized and not apoptotic. 4,930 out of 6,743 putative G-SmoAMPK cells, and 8699 out of 16489 G-Smo cells met criteria and were included in the analysis.
We noted that G-Smo cells were sequenced at a greater depth than G-SmoAMPK cells which can introduce unwanted batch effects into the analysis. Consistent with best practices,41 we down-sampled the G-Smo cells to 46.5% of their original depth so as to achieve similar sequencing depth between G-Smo and G-SmoAMPK cells prior to further filtering.
We filtered out putative cells with fewer than 500 detected RNA molecules (nCount) or 200 different genes (nFeature), as likely to represent ambient RNA. We filtered out putative cells with greater than 4 standard deviations above the median nCount or nFeature as likely to be doublets, improperly merged barcodes, or sequencing artifacts. We also filtered out putative cells with more than 10% mitochondrial transcripts which we suspected to be dying cells.
Harmony analysis
To merge the previously published G-Smo control tumor data with the G-SmoAMPK tumor dataset, we used the Harmony algorithm.65 First, data from both tumor types were analyzed in single SCTransform normalization and PCA steps. The Harmony algorithm then used the cells’ PCA coordinates and dataset identity to calculate new coordinates for each cell, to minimize dataset dependence when applying clustering to the cells. This algorithm produced a dimensional reduction that was used as a PCA for the following steps of the analysis.
scRNA-seq data normalization, clustering, differential gene expression, and visualization
We normalized the data using the SCTransform as implemented in Seurat, then selected the top 3,000 most highly variable genes. We performed PCA on these highly variable genes using the RunPCA function. The number of PCs to be used in downstream analysis was chosen based on the elbow plot as implemented by Seurat. We then used the FindNeighbors and FindClusters functions to identify cell clusters in the data.
To identify differential genes between clusters of cells, we used Wilcoxon rank-sum test to compare gene expression of cells within the cluster of interest to all cells outside that cluster as implemented by the FindMarkers function. Cutoffs were set for the absolute value of the log fold change >0.25 between the two compared groups and percent of cells expressing the gene in at least one of the groups set >10%. Uniform Manifold Approximation and Projection was used to reduce the PCs to two dimensions for data visualization using the RunUMAP function.
Cell-type identification
Following PCA and UMAP, we analyzed cluster-specific differential gene expression. Marker genes were identified based on cluster-specific differential gene expression. For this purpose, for each gene we calculated the fraction of cells within the cluster that expressed the gene (referred to in Data S1 as pct1) and the fraction of cells outside the cluster that expressed the gene (referred to in Data S1 as pct2), and ranked genes by the ratio of pct1:pct2. Genes with high pct1:pct2 ratio and high pct1 values were selected as leading cluster-specific markers. The expression patterns of these genes were then examined in publicly available scRNA-seq data describing gene expression during mouse development66 and in our prior published mouse medulloblastoma scRNA-seq studies,11,37,50 to identify cell types.
Statistical analysis of IF data
Counts of total cells in tumor sections of counts of cells expressing each marker under study were analyzed using Microsoft Excel. Statistical tests used are specified in the results section.
Acknowledgments
We thank the UNC CGBID Histology Core, supported by P30 DK 034987 and the UNC Tissue Pathology Laboratory Core supported by NCI CA016086. T.D. was supported by NINDS (F31 NS120459). T.R.G. was supported by NINDS (R01NS088219, R01NS102627, R01NS106227)and by the UNC Department of Neurology Research Fund, and by a TTSA grant from the NCTRACS Institute, which is supported by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through Grant Award Number UL1TR002489.
Author contributions
D.S.M., T.D., J.B., B.D., A.T., and T.R.G. wrote the article. D.S.M., T.D., E.C., and H.L. conducted the experiments and analyzed the data.
Declaration of interests
The authors declare no competing interests. Ethan Castellino is currently a student at Duke University.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. We worked to ensure sex balance in the selection of non-human subjects.
Published: November 14, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108443.
Supplemental information
References
- 1.Northcott P.A., Shih D.J.H., Peacock J., Garzia L., Morrissy A.S., Zichner T., Stütz A.M., Korshunov A., Reimand J., Schumacher S.E., et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kool M., Korshunov A., Remke M., Jones D.T.W., Schlanstein M., Northcott P.A., Cho Y.J., Koster J., Schouten-van Meeteren A., van Vuurden D., et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Northcott P.A., Korshunov A., Witt H., Hielscher T., Eberhart C.G., Mack S., Bouffet E., Clifford S.C., Hawkins C.E., French P., et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalli F.M.G., Remke M., Rampasek L., Peacock J., Shih D.J.H., Luu B., Garzia L., Torchia J., Nor C., Morrissy A.S., et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737–754.e6. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riemondy K.A., Venkataraman S., Willard N., Nellan A., Sanford B., Griesinger A.M., Amani V., Mitra S., Hankinson T.C., Handler M.H., et al. Neoplastic and Immune single cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro Oncol. 2021;24:273–286. doi: 10.1093/neuonc/noab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hovestadt V., Smith K.S., Bihannic L., Filbin M.G., Shaw M.L., Baumgartner A., DeWitt J.C., Groves A., Mayr L., Weisman H.R., et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–79. doi: 10.1038/s41586-019-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gershon T.R., Crowther A.J., Tikunov A., Garcia I., Annis R., Yuan H., Miller C.R., Macdonald J., Olson J., Deshmukh M. Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar neurogenesis and pathogenesis of medulloblastoma. Cancer Metab. 2013;1:2. doi: 10.1186/2049-3002-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliver T.G., Read T.A., Kessler J.D., Mehmeti A., Wells J.F., Huynh T.T.T., Lin S.M., Wechsler-Reya R.J. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 9.Hallahan A.R., Pritchard J.I., Hansen S., Benson M., Stoeck J., Hatton B.A., Russell T.L., Ellenbogen R.G., Bernstein I.D., Beachy P.A., Olson J.M. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L., He X., Liu X., Zhang F., Huang L.F., Potter A.S., Xu L., Zhou W., Zheng T., Luo Z., et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell. 2019;36:302–318.e7. doi: 10.1016/j.ccell.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ocasio J.K., Babcock B., Malawsky D., Weir S.J., Loo L., Simon J.M., Zylka M.J., Hwang D., Dismuke T., Sokolsky M., et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 2019;10:5829. doi: 10.1038/s41467-019-13657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vladoiu M.C., El-Hamamy I., Donovan L.K., Farooq H., Holgado B.L., Sundaravadanam Y., Ramaswamy V., Hendrikse L.D., Kumar S., Mack S.C., et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572:67–73. doi: 10.1038/s41586-019-1158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao M., Ventura P.B., Jiang Y., Rodriguez F.J., Wang L., Perry J.S.A., Yang Y., Wahl K., Crittenden R.B., Bennett M.L., et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell. 2020;180:502–520.e19. doi: 10.1016/j.cell.2019.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardie D.G. Minireview: The AMP-Activated Protein Kinase Cascade: The Key Sensor of Cellular Energy Status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 15.Hardie D.G., Ross F.A., Hawley S.A. AMP-Activated Protein Kinase: A Target for Drugs both Ancient and Modern. Chem. Biol. 2012;19:1222–1236. doi: 10.1016/j.chembiol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tech K., Deshmukh M., Gershon T.R. Adaptations of energy metabolism during cerebellar neurogenesis are co-opted in medulloblastoma. Cancer Lett. 2015;356:268–272. doi: 10.1016/j.canlet.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tech K., Tikunov A.P., Farooq H., Morrissy A.S., Meidinger J., Fish T., Green S.C., Liu H., Li Y., Mungall A.J., et al. Pyruvate Kinase Inhibits Proliferation during Postnatal Cerebellar Neurogenesis and Suppresses Medulloblastoma Formation. Cancer Res. 2017;77:3217–3230. doi: 10.1158/0008-5472.CAN-16-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhatia B., Hsieh M., Kenney A.M., Nahlé Z. Mitogenic Sonic hedgehog signaling drives E2F1-dependent lipogenesis in progenitor cells and medulloblastoma. Oncogene. 2011;30:410–422. doi: 10.1038/onc.2010.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eyrich N.W., Potts C.R., Robinson M.H., Maximov V., Kenney A.M. Reactive Oxygen Species Signaling Promotes Hypoxia-Inducible Factor 1alpha Stabilization in Sonic Hedgehog-Driven Cerebellar Progenitor Cell Proliferation. Mol. Cell Biol. 2019;39 doi: 10.1128/MCB.00268-18. e00268–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malhotra A., Dey A., Prasad N., Kenney A.M. Sonic Hedgehog Signaling Drives Mitochondrial Fragmentation by Suppressing Mitofusins in Cerebellar Granule Neuron Precursors and Medulloblastoma. Mol. Cancer Res. 2016;14:114–124. doi: 10.1158/1541-7786.MCR-15-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng Q., Chen J., Li Y., Werle K.D., Zhao R.X., Quan C.S., Wang Y.S., Zhai Y.X., Wang J.W., Youssef M., et al. LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene. 2017;36:1245–1255. doi: 10.1038/onc.2016.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faubert B., Vincent E.E., Griss T., Samborska B., Izreig S., Svensson R.U., Mamer O.A., Avizonis D., Shackelford D.B., Shaw R.J., Jones R.G. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc. Natl. Acad. Sci. USA. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shackelford D.B., Abt E., Gerken L., Vasquez D.S., Seki A., Leblanc M., Wei L., Fishbein M.C., Czernin J., Mischel P.S., Shaw R.J. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eichner L.J., Brun S.N., Herzig S., Young N.P., Curtis S.D., Shackelford D.B., Shokhirev M.N., Leblanc M., Vera L.I., Hutchins A., et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019;29:285–302.e7. doi: 10.1016/j.cmet.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muraleedharan R., Dasgupta B. AMPK in the brain: its roles in glucose and neural metabolism. FEBS J. 2022;289:2247–2262. doi: 10.1111/febs.16151. [DOI] [PubMed] [Google Scholar]
- 26.Muraleedharan R., Gawali M.V., Tiwari D., Sukumaran A., Oatman N., Anderson J., Nardini D., Bhuiyan M.A.N., Tkáč I., Ward A.L., et al. AMPK-Regulated Astrocytic Lactate Shuttle Plays a Non-Cell-Autonomous Role in Neuronal Survival. Cell Rep. 2020;32:108092. doi: 10.1016/j.celrep.2020.108092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chhipa R.R., Fan Q., Anderson J., Muraleedharan R., Huang Y., Ciraolo G., Chen X., Waclaw R., Chow L.M., Khuchua Z., et al. AMP kinase promotes glioblastoma bioenergetics and tumour growth. Nat. Cell Biol. 2018;20:823–835. doi: 10.1038/s41556-018-0126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams T., Courchet J., Viollet B., Brenman J.E., Polleux F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc. Natl. Acad. Sci. USA. 2011;108:5849–5854. doi: 10.1073/pnas.1013660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dzamko N., van Denderen B.J.W., Hevener A.L., Jørgensen S.B., Honeyman J., Galic S., Chen Z.P., Watt M.J., Campbell D.J., Steinberg G.R., Kemp B.E. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J. Biol. Chem. 2010;285:115–122. doi: 10.1074/jbc.M109.056762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H., Kuick R., Park S.S., Peabody C., Yoon J., Fernández E.C., Wang J., Thomas D., Viollet B., Inoki K., et al. Loss of AMPKalpha2 Impairs Hedgehog-Driven Medulloblastoma Tumorigenesis. Int. J. Mol. Sci. 2018;19:3287. doi: 10.3390/ijms19113287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Amico D., Antonucci L., Di Magno L., Coni S., Sdruscia G., Macone A., Miele E., Infante P., Di Marcotullio L., De Smaele E., et al. Non-canonical Hedgehog/AMPK-Mediated Control of Polyamine Metabolism Supports Neuronal and Medulloblastoma Cell Growth. Dev. Cell. 2015;35:21–35. doi: 10.1016/j.devcel.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kool M., Jones D.T., Jager N., Northcott P.A., Pugh T.J., Hovestadt V., Piro R.M., Esparza L.A., Markant S.L., Remke M., et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao J., Ligon K.L., Rakhlin E.Y., Thayer S.P., Bronson R.T., Rowitch D., McMahon A.P. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schüller U., Heine V.M., Mao J., Kho A.T., Dillon A.K., Han Y.-G., Huillard E., Sun T., Ligon A.H., Qian Y., et al. Acquisition of Granule Neuron Precursor Identity Is a Critical Determinant of Progenitor Cell Competence to Form Shh-Induced Medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malawsky D.S., Weir S., Ocasio J., Babcock B., Dismuke T., Wilhelmsen K., Gershon T.R. Cryptic developmental events determine medulloblastoma radiosensitivity and cellular heterogeneity without altering transcriptomic profile. Commun. Biol. 2020;4:616. doi: 10.1038/s42003-021-02099-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malawsky D.S., Weir S.J., Ocasio J.K., Babcock B., Dismuke T., Cleveland A.H., Donson A.M., Vibhakar R., Wilhelmsen K., Gershon T.R. Cryptic developmental events determine medulloblastoma radiosensitivity and cellular heterogeneity without altering transcriptomic profile. Commun. Biol. 2021;4:616. doi: 10.1038/s42003-021-02099-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swiderska-Syn M., Mir-Pedrol J., Oles A., Schleuger O., Salvador A.D., Greiner S.M., Seward C., Yang F., Babcock B.R., Shen C., et al. Noncanonical activation of GLI signaling in SOX2(+) cells drives medulloblastoma relapse. Sci. Adv. 2022;8:eabj9138. doi: 10.1126/sciadv.abj9138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim C., Dismuke T., Malawsky D., Ramsey J.D., Hwang D., Godfrey V.L., Kabanov A.V., Gershon T.R., Sokolsky-Papkov M. Enhancing CDK4/6 inhibitor therapy for medulloblastoma using nanoparticle delivery and scRNA-seq-guided combination with sapanisertib. Sci. Adv. 2022;8:eabl5838. doi: 10.1126/sciadv.abl5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loo L., Simon J.M., Xing L., McCoy E.S., Niehaus J.K., Guo J., Anton E.S., Zylka M.J. Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun. 2019;10:134. doi: 10.1038/s41467-018-08079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luecken M.D., Theis F.J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019;15:e8746. doi: 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S., Qiu F., Xu A., Price S.M., Xiang M. Barhl1 regulates migration and survival of cerebellar granule cells by controlling expression of the neurotrophin-3 gene. J. Neurosci. 2004;24:3104–3114. doi: 10.1523/JNEUROSCI.4444-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyata T., Maeda T., Lee J.E. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malawsky D., Gershon T.R. scRNA-seq for Microcephaly Research [IV]: Dirichlet Regression for Single-Cell Population Differences. Methods Mol. Biol. 2023;2583:123–125. doi: 10.1007/978-1-0716-2752-5_11. [DOI] [PubMed] [Google Scholar]
- 45.Hambardzumyan D., Becher O.J., Rosenblum M.K., Pandolfi P.P., Manova-Todorova K., Holland E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crowther A.J., Ocasio J.K., Fang F., Meidinger J., Wu J., Deal A.M., Chang S.X., Yuan H., Schmid R., Davis I., Gershon T.R. Radiation Sensitivity in a Preclinical Mouse Model of Medulloblastoma Relies on the Function of the Intrinsic Apoptotic Pathway. Cancer Res. 2016;76:3211–3223. doi: 10.1158/0008-5472.CAN-15-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalinka A. The probability of drawing intersections: extending the hypergeometric distribution. arXiv. 2013 https://arxiv.org/abs/1305.0717 Preprint at. [Google Scholar]
- 48.Morita M., Gravel S.P., Chénard V., Sikström K., Zheng L., Alain T., Gandin V., Avizonis D., Arguello M., Zakaria C., et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18:698–711. doi: 10.1016/j.cmet.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 49.Thoreen C.C., Chantranupong L., Keys H.R., Wang T., Gray N.S., Sabatini D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim C., Dismuke T., Malawsky D., Ramsey J.D., Hwang D., Godfrey V.L., Kabanov A.V., Gershon T.R., Sokolsky-Papkov M. Enhancing CDK4/6 inhibitor therapy for medulloblastoma using nanoparticle delivery and scRNA-seq-guided combination with sapanisertib. Sci. Adv. 2021;8:eabl5838. doi: 10.1126/sciadv.abl5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gwinn D.M., Shackelford D.B., Egan D.F., Mihaylova M.M., Mery A., Vasquez D.S., Turk B.E., Shaw R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corradetti M.N., Inoki K., Bardeesy N., DePinho R.A., Guan K.L. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inoki K., Zhu T., Guan K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 54.Stoppani J., Hildebrandt A.L., Sakamoto K., Cameron-Smith D., Goodyear L.J., Neufer P.D. AMP-activated protein kinase activates transcription of the UCP3 and HKII genes in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2002;283:E1239–E1248. doi: 10.1152/ajpendo.00278.2002. [DOI] [PubMed] [Google Scholar]
- 55.Bhatia B., Potts C.R., Guldal C., Choi S., Korshunov A., Pfister S., Kenney A.M., Nahlé Z.A. Hedgehog-mediated regulation of PPARgamma controls metabolic patterns in neural precursors and shh-driven medulloblastoma. Acta Neuropathol. 2012;123:587–600. doi: 10.1007/s00401-012-0968-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y.H., Luo J., Mosley Y.Y.C., Hedrick V.E., Paul L.N., Chang J., Zhang G., Wang Y.K., Banko M.R., Brunet A., et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015;12:599–609. doi: 10.1016/j.celrep.2015.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang R., Huang S.Y., Ka-Wai Li K., Li Y.H., Hsu W.H., Zhang G.J., Chang C.J., Yang J.Y. Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through beta-TrCP-mediated proteasome degradation. Oncotarget. 2017;8:49869–49881. doi: 10.18632/oncotarget.17769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kimura N., Tokunaga C., Dalal S., Richardson C., Yoshino K.i., Hara K., Kemp B.E., Witters L.A., Mimura O., Yonezawa K. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Gene Cell. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 59.Bolster D.R., Crozier S.J., Kimball S.R., Jefferson L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 60.Wu C.C., Hou S., Orr B.A., Kuo B.R., Youn Y.H., Ong T., Roth F., Eberhart C.G., Robinson G.W., Solecki D.J., et al. mTORC1-Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling-Driven Translation and Medulloblastoma. Dev. Cell. 2017;43:673–688.e5. doi: 10.1016/j.devcel.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ocasio J.K., Bates R.D.P., Rapp C.D., Gershon T.R. GSK-3 modulates SHH-driven proliferation in postnatal cerebellar neurogenesis and medulloblastoma. Development. 2019;146:dev177550. doi: 10.1242/dev.177550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lang P.Y., Nanjangud G.J., Sokolsky-Papkov M., Shaw C., Hwang D., Parker J.S., Kabanov A.V., Gershon T.R. ATR maintains chromosomal integrity during postnatal cerebellar neurogenesis and is required for medulloblastoma formation. Development. 2016;143:4038–4052. doi: 10.1242/dev.139022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M., et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.La Manno G., Siletti K., Furlan A., Gyllborg D., Vinsland E., Mossi Albiach A., Mattsson Langseth C., Khven I., Lederer A.R., Dratva L.M., et al. Molecular architecture of the developing mouse brain. Nature. 2021;596:92–96. doi: 10.1038/s41586-021-03775-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The scRNA-seq data were deposited in the Gene Expression Omnibus (GEO) database and are publicly available at the time of publication. Accession codes are listed in the key resources table. Microscopy data reported in the paper will be shared by the lead contact upon request.
-
•
This paper did not use or create original code. The scRNA-seq analysis was performed using the publicly available Seurat R package version 3.1.1.
-
•
Any additional information required to reanalyze the data reported in this paper will be made available by the lead contact upon request.