

Abstract

An efficient, microwave/ultrasound-irradiated synthesis of novel chromenopyrimidines has been established. 2-Amino-5-oxo-4-(thiophen-2-yl)-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (1) underwent cyclization reactions with various assorted reagents under sustainable conditions to afford a family of fused pyrimidine derivatives. The proposed structures of the designed fused pyrimidines were confirmed by several spectral techniques. Moreover, the targeted pyrimidines were estimated for their in vitro cytotoxic activities toward three carcinoma cell lines: breast (MCF7), hepatocyte (HepG2), and lung (A549) cancer cell lines, as well as one noncancerous cell line (MCF-10A). Structure–activity relationship (SAR) analyses revealed that derivatives 3 and 7 exhibited the highest potency in inhibiting the growth of cancer cells tested in vitro. Particularly, 3-amino-4-imino-5-(thiophen-2-yl)-3,4,5,7,8,9-hexahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3) displayed a robust impact with IC50 values ranging from 2.02 to 1.61 μM. Interestingly, compound 3 was observed to have low cytotoxicity toward noncancerous cell (MCF-10A) compared to the standard drug (Doxorubicin). Further, quantum chemical computations of the designed molecules utilizing density functional theory (DFT) were conducted and shown to be compatible with the observed antiproliferative properties. Thorough docking investigations revealed that the assembled compounds possess exceptionally low binding energies toward our three selected proteins: 4b3z-Lung, HepG2–2JW2, and 6ENV-MCV-7. Based on these intriguing results, compound 3 could be further evaluated for preclinical screening, potentially paving the way for its utilization as a promising cancer treatment.
Introduction
Fused heterocyclic frameworks have plenty of merits due to their remarkable applicability as synthones in organic conversions as well as their possible pharmacological activities.1 Furthermore, the structural variety and biochemical significance of oxygen- and nitrogen-comprising heterocycles have rendered them appealing synthesis candidates for several decades.2 Considering their significance as precursors for several therapeutically active molecules, there has been extensive work on discovering protocols to design such systems.3,4 The design of nitrogen heterocyclic molecules and their analogues have a significant role in the field of synthetic and natural chemistry owing to their potential medicinal and pharmaceutical attributes.5 In the same context, pyrimidine-based heterocycles are one of the most robust anticancer agents.6 There are many approved and marketed anticancer drugs that contain pyrimidine pharmacophores in their structure. Pyrimidine analogue rugs, such as Adrucil (also known as 5-fluorouracil), Gemcitabine, and Floxuridine, are widely used as cytotoxic chemotherapies in the treatment of various vicious ailments, like colorectal cancer, pancreatic cancer, and breast cancer7 (Figure 1).
Figure 1.

Commercially available pyrimidine-based anticancer drugs.
Likewise, chromenes represent a substantial group of molecules that are commonly found in a wide range of plants, particularly in edible fruits and vegetables.8 The existence of chromene motifs in several natural products has been attributed to their ability to inhibit diseases.9 Over the years, chromene alternatives, such as antimicrobial,10,11 molluscidial,12 antianaphylactic, spasmolytic, anticoagulant, and diuretic properties,13 have been designed, several of which have demonstrated outstanding pharmacological benefits.14 Fused chromenopyrimidines are commonly found in numerous naturally occurring substances.15 These compounds are known for their significant biological activity and promising pharmacological characteristics.16 They additionally have been widely investigated in medical applications as useful building blocks in diverse biological and pharmaceutical activities such as anticancer,17 antitumor,18 and antiproliferative.19 Their therapeutic effectiveness is attributed to their selectivity for biomolecules such as deoxyribonucleic acid (DNA).19 However, recent advancements have led to the development of improved approaches for assembling these compounds.1,2,20 The majority of these methodologies often involve the employment of hazardous solvents, prolonged times at elevated temperatures, and costly reagents.16 As carcinoma is a potentially fatal disease worldwide21 and the fundamental challenge in current therapy strategies, such as radiotherapy and chemotherapy, is the occurrence of multidrug resistance (MDR),22 designing remarkably effective protocols that deliver greater structural diversity and complexity with the lowest number of steps to establish products with remarkable features is a significant issue in contemporary drug development.23 Based on the above-mentioned studies, it is an interesting challenge to design robust and straightforward synthetic protocols for the effective synthesis of heterocycles comprising a chromenopyrimidine motif. Herein, we conducted the synthesis of a chromenopyrimidine series employing sustainable techniques as part of our objective to establish alternative environmentally friendly and selective protocols for the development of such heterocycles. The synthesized chromenopyrimidines were then tested against three anticancer cell lines and one nonmalignant cell line. Additionally, DFT analyses were also conducted to investigate the electronic structural properties associated with their antiproliferative activities. Finally, docking investigations were carried out to gain insights into the compounds’ interactions with three selected proteins.
Results and Discussion
Driven by the high functionality of compound 1 and the aforementioned pyrimidines merits as potent cytotoxic agents,6,7 2-aminochromene-3-carbonitrile derivative 1 was utilized as a platform to construct diverse fused pyrimidines (Schemes 1–3). Thiophene-2-carbaldehyde easily underwent a Knoevenagel reaction with malononitrile, and the resulting product underwent a Michael addition reaction with cyclohexane-1,3-dione to yield essential intermediate 1, which was then used for synthesizing the chromenopyrimidine library. Various eco-friendly approaches, such as microwave and ultrasonic irradiation, will be exploited to design the targeted chromenopyrimidines (Schemes 1–3).
Scheme 1. Assembly of Chromenopyrimidine Derivatives (3–7) from 1.
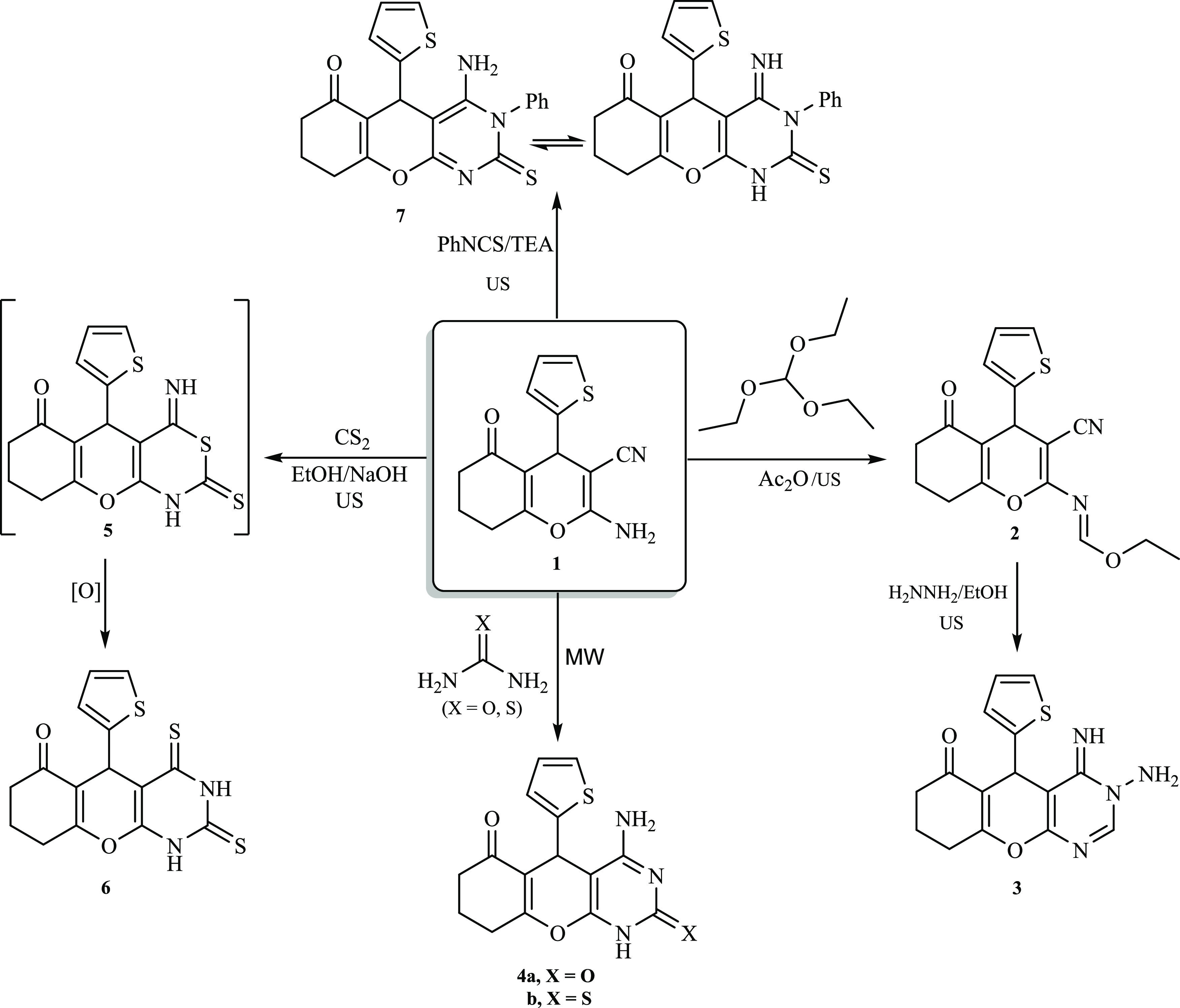
Scheme 3. Assembly of Chromenopyrimidine Derivatives (17–19) from 1.
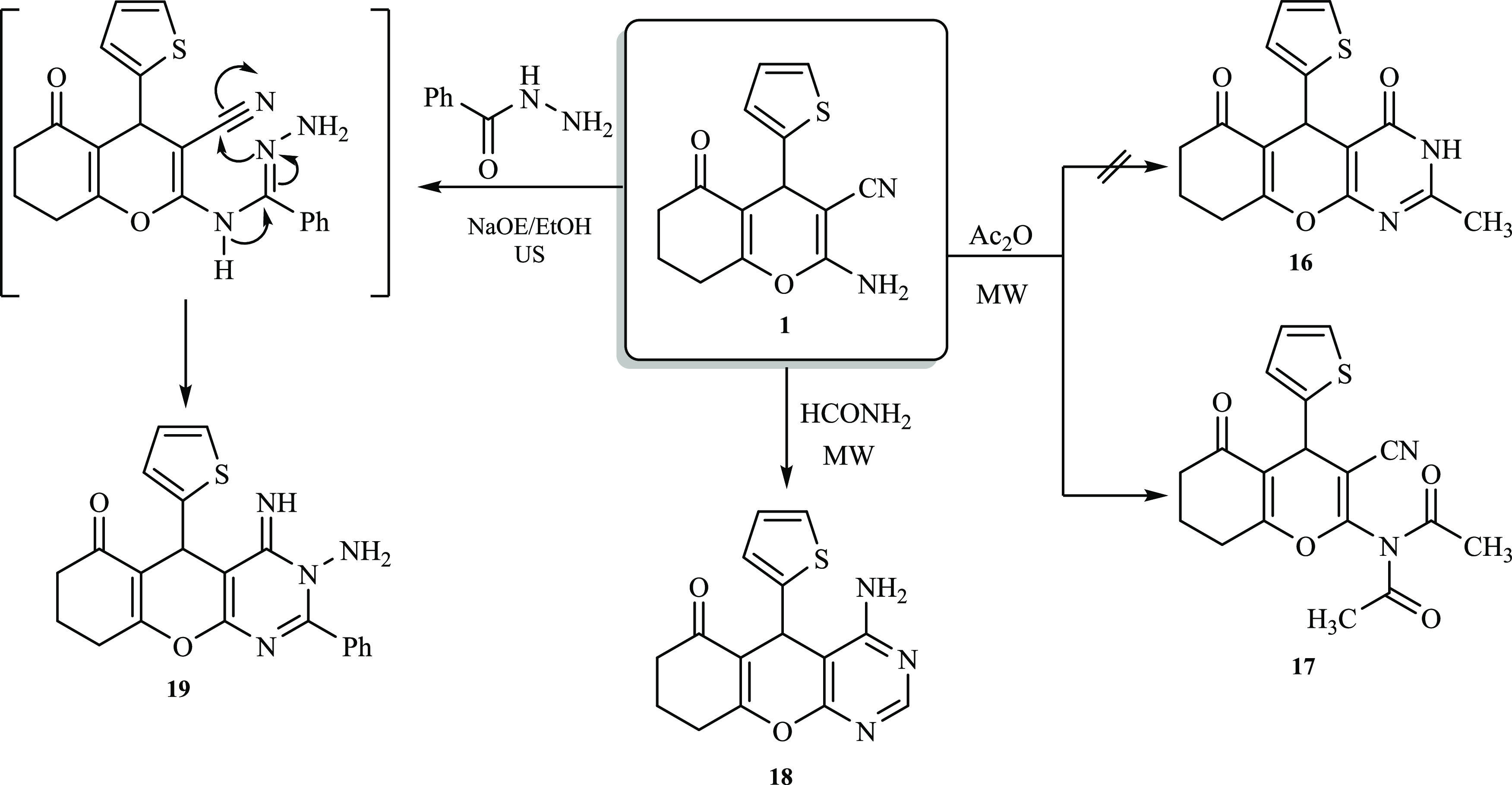
As depicted in Scheme 1, condensation of 2-amino-tetrahydro-4H-chromene-3-carbonitrile derivative 1 with triethyl orthoformate under ultrasonic irradiation afforded formimidate derivative 2. The 1H NMR spectrum of compound 2 exhibited a triplet-quartet pattern belonging to the ethyl motif at δ 4.28 and 1.26 ppm, while the methine proton (N=CH) was observed at δ 8.52 ppm. Furthermore, the 13C NMR spectrum of derivative 2 displayed a distinctive signal at δ 157 ppm attributed to the methine carbon (CH=N), in addition to two signals at 64 and 14 ppm assigned to the carbons of the ethyl motif. Subsequently, formimidate derivative 2 underwent a ring closure reaction upon treatment with hydrazine hydrate to afford isolable chromeno[2,3-d] pyrimidin-6-one derivative 3 in a good yield (76%, Scheme 1). The Fourier transform infrared (FT-IR) spectrum of pyrimidine derivative 3 was devoid of any stretching frequencies for the C≡N group, but a new stretching band attributed to the C=N was detected at 1597 cm–1. Additionally, the 1H NMR spectrum exhibited three distinguished singlet signals at δ 11.70, 8.30, and 4.71 ppm ascribed to the NH group, a pyrimidine proton (C=CH), and an amino proton (NH2), respectively. Furthermore, the mass spectrum of derivative 3 disclosed the prospective molecular ion peak at m/z = 314, corresponding to C15H14N4O2S. To improve the reaction yield and rate, the above-mentioned reaction (Scheme 1) was sequentially repeated under ultrasound irritation. The reactants (1 and triethyl orthoformate) were irradiated at 60 °C for 30 min (TLC). After the removal of the excess reagent under vacuum, a solution of hydrazine hydrate in ethanol was added to the residue, and irradiation was recommenced at ambient temperature for 1 h (TLC). The target product 3 was obtained in 98% yield without employing any purification steps. In order to have access to pyrimidine derivatives 4a,b, different synthetic strategies were explored (Scheme 1). Initially, by employing basic alumina as a solid support, aminocarbonitrile 1 underwent a cyclization reaction with either urea or thiourea. This resulted in the successful synthesis of the anticipated pyrimidine derivative 4a,b in approximately 73% yield. The use of basic alumina as a mineral support eliminates the necessity for an external base or acid to obtain desired products 4a,b in an acceptable purity, as confirmed by TLC analysis. Interestingly, conducting the same reaction under solvent-free and catalyst-free conditions diminished the yield remarkably, and only a 20% yield was accessible. In contrast, performing the same reaction in DMF afforded the product in 67% yield. This enhanced yield might be attributed to the unique properties of DMF (energy transfer solvent, high dielectric constant, and a homogenizer to elevate the temperature of the reaction).24 In comparison to the solid-supported approach, the latter approach afforded the product in a relatively lower purity and yield. Therefore, a novel, cost-effective, convenient, and eco-friendly one-pot strategy for the synthesis of 4a,b is urgent. Accordingly, by subjecting equimolar amounts of compound 1 and either urea or thiourea to microwave irradiation, using either basic alumina or DMF, the desired compounds (4a,b) were obtained in significantly high yields (88 and 90%, respectively) within 45 min. The 1H NMR spectrum of derivative 4a revealed two distinguishing singlets at δ 11.20 and 7.91 ppm attributed to the NH and NH2 moieties, respectively. In addition, the 13C NMR spectrum of derivative 4a demonstrated two carbonyl carbons at δ 194.9 and 168.1 ppm together with four distinctive signals assigned to the thiophene moiety. The existence of a C=S stretching band at approximately 1346 cm–1 and the absence of an S–H motif absorption band around 2550 cm–1 in the FT-IR spectrum of derivative 4b indicated that this derivative was in the thione form. This was further supported by the absence of any thiol-related signals in 1H NMR. Furthermore, thiazinethione intermediate 5 was prepared using different techniques. Boiling aminocarbonitrile derivative 1 with an excess amount of carbon disulfide in pyridine for 2 h gave rise to intermediate 5. Driven by the basic conditions utilized, intermediate 5 underwent a subsequent rearrangement reaction to afford isolable pyrimidinedithione 6 in a good yield (73%, Scheme 1).
In another trial to obtain a better yield, the reaction was repeated under ultrasonication (30 °C, 40 min, 88%), notably finding that both the reaction rate and yield were improved. Noteworthy, when the aforementioned reaction (Scheme 1) was performed under ultrasonication utilizing 15% alcoholic sodium hydroxide solution (10.0 mL) rather than pyridine, the desired product 6 was acquired in an outstanding yield (93%) within a short time (30 min). The dithione compound 6 showed accurate values in the spectral data and elemental analyses. Accordingly, NH signals could be observed in the FT-IR spectrum in the region of 3201–3137 cm–1, while no absorption bands for the cyano motif were detected. Additionally, the 1H NMR spectrum of compound 6 displayed two singlets attributed to NH motifs at δ = 11.09 and 12.19 ppm. Further, a cyclization reaction took place upon protracted boiling of aminocarbonitrile derivative 1 with phenylisothiocyanate in DMF, comprising a catalytic portion of TEA, and 4-aminothiopyrimidine derivative 7 was isolable in good yield (83%, Scheme 2). A considerable enhancement in both the reaction rate and yield was acquired by conducting the same reaction (Scheme 1) under ultrasonic irradiation conditions (90 °C, 3 h, 97%). The FT-IR spectrum of 7 demonstrated absorption bands at 3350–3310 and 1310 cm–1 attributed to NH2 and C=S moieties, respectively. Moreover, the 1H NMR spectrum of compound 7, along with its MS data, provided additional supportive results for the proposed structure.
Scheme 2. Assembly of Chromenopyrimidine Derivatives (11–15) from 1.
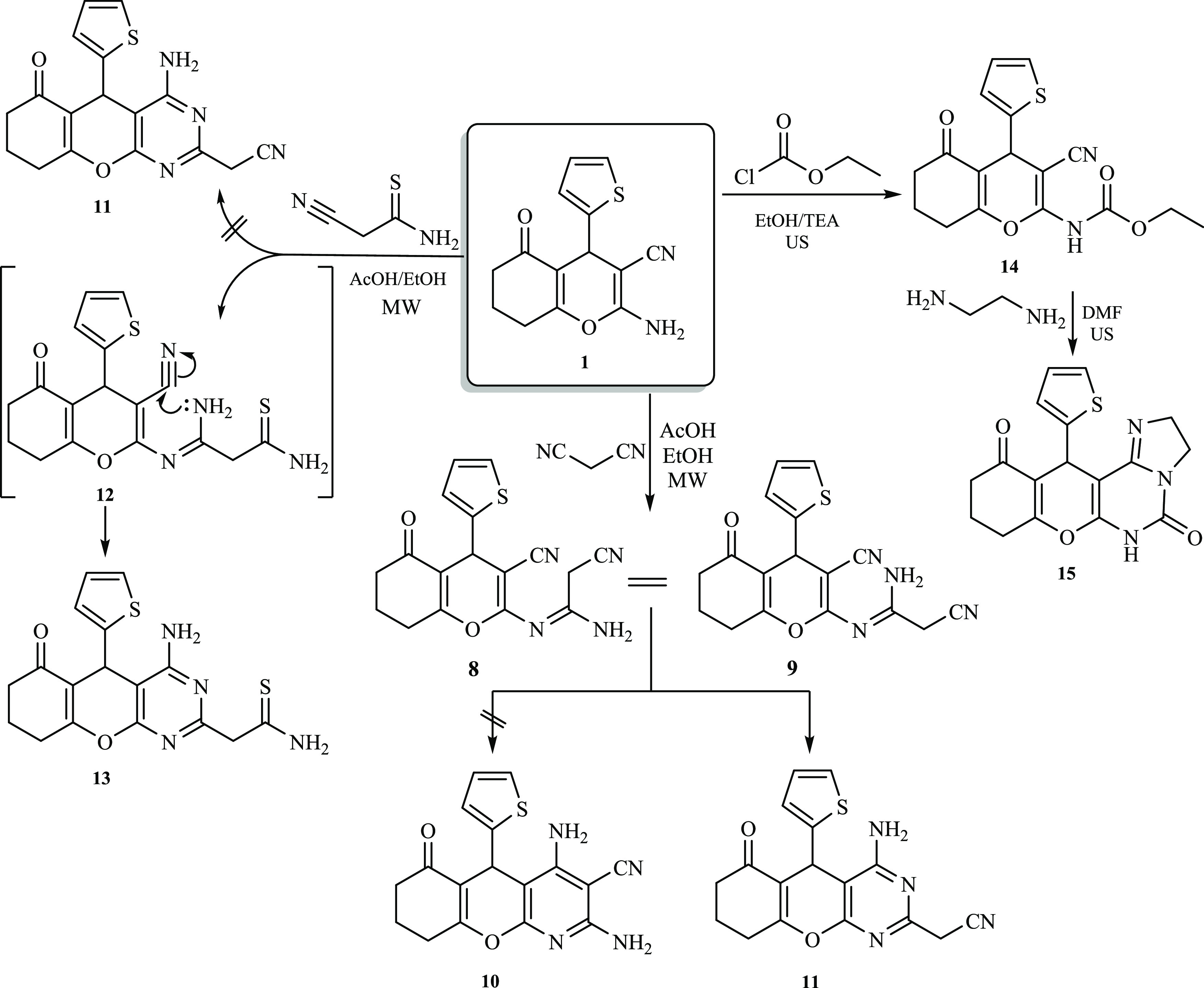
Moreover, under acidic reaction conditions, derivative 1 underwent a reaction with malononitrile to afford a brown solid. It was proposed that the reaction would submit pyridine derivative 10 through intermediate 8 (Scheme 2). Surprisingly, a methylene motif was recorded by 1H NMR as a characteristic singlet signal at δ 3.69 ppm. This result was also supported by the 13C NMR spectrum which manifested a distinctive signal at δ 24.8 ppm assigned to the methylene carbon (CH2CN). Therefore, the cyclization most probably occurred via a nucleophilic attack of the intermediate 9 NH2 group on the C≡N originating from 1 to afford the pyrimidine derivative 11 (Scheme 2). Such a reaction pathway was previously reported by Metwally and co-workers.25 Interestingly, performing the latter reaction under microwave irradiation (MW) conditions in ethanol comprising a catalytic amount of AcOH gave a comparable yield, but a notable improvement in the reaction rate was observed (from 8 to 1 h). Likewise, derivative 1 underwent a similar reaction with cyanothioacetamide in an acidic medium under microwave irradiation. In this respect, the reaction had two possibilities: either the formation of intermediate 9 via the elimination of hydrogen sulfide, followed by a cyclization process to afford pyrimidine derivative 11, or the formation of intermediate 12 via Michael’s addition of the NH2 motif of compound 1 to the C≡N group of cyanothioacetamide, followed by cyclization to pyrimidine derivative 13. Nevertheless, the melting point and spectral data of the end product of the reaction did not match compound 11. Accordingly, it was anticipated that the reaction of cyanothioacetamide would occur through intermediate 12, as depicted in Scheme 2. The 1H NMR spectrum of produced derivative 13 demonstrated three distinguishing singlets at δ 6.82 (NH2), 6.66 (NH2), and 3.74 ppm (CH2). In addition, the MS of compound 13 revealed a molecular ion peak at m/z = 372 [M], corresponding to C17H16N4O2S2 and in agreement with the molar mass of the suggested structure. Furthermore, derivative 1 effectively reacted with ethyl chloroformate at ambient temperature under ultrasonication conditions to yield a substantial amount of carbamate derivative 14 (Scheme 2). A subsequent cyclization reaction took place, and compound 15 was accessible via the sonication of an equimolar quantity of ethylenediamine with carbamate derivative 14 for 2 h at 60 °C. The acquired product was identified as 12-(thiophen-2-yl)-2,6,8,9,10,12-hexahydro-5H-chromeno[3,2-e]imidazo[1,2-c]pyrimidine-5,11(3H)-dione (15) based on the elemental analyses and spectral data. The 1H NMR spectrum of derivative 15 exhibited a singlet signal at δ 10.12 ppm assigned to the NH moiety, in addition to two distinguishing multiplets at δ 4.21 and 3.70 ppm corresponding to the imidazole protons. Also, high-resolution mass spectrometry (HRMS) revealed a molecular ion peak at m/z 341.0831 [M], corresponding to C17H15N3O3S, which is in agreement with the molar mass of the suggested structure. Interestingly, pyrimidine derivative 16 was expected to be the sole product of reacting 2-aminochromene-3-carbonitrile (1) with acetic anhydride under microwave irradiation conditions. However, in stark contrast to the proposed structure, diacetylated derivative 17 was obtained instead (Scheme 3).26
The FT-IR spectrum of compound 17 showed the existence of absorption bands at 2212 and 1674 cm–1 that are distinctive to C≡N and C=O functions, respectively. Further, the 1H NMR spectrum of compound 17 exhibited a unique singlet signal assigned to two CH3 groups at δ = 2.98 ppm. In addition, the molecular ion peak at m/z = 356 was equivalent to that of C18H16N2O4S and was consistent with the molar mass of the presented structure. Moreover, 2-aminochromene-3-carbonitrile (1) reacted with an excess quantity of formamide under microwave irradiation conditions to afford the corresponding pyrimidine derivative 18 in good yield (Scheme 3). The structure of the obtained compound was elucidated using several spectroscopic techniques. For instance, a distinctive singlet signal was detected at δ 6.45 ppm that was assigned to the amino moiety, and a singlet signal was observed at δ 8.13 ppm that was attributed to the pyrimidine proton. Also, the HRMS spectrum of acquired compound 18 disclosed a molecular ion peak at m/z 322.0624 [M + Na], which is equivalent to C15H13N3O2S. Moreover, a cyclocondensation reaction was observed upon sonication of aminocarbonitrile derivative 1 with benzohydrazide in the presence of a catalytic amount of NaOEt, and pyrimidine derivative 19 was isolated in excellent yield (Scheme 3). In the latter transformation, various bases, such as NaOH, TEA, and piperidine, were explored; however, sodium ethoxide provided the desired product at a better rate and yield. This suggests that the development of a robust hydrogen bond with the anionic oxygen of the ethoxide strengthened the amino group’s nucleophilicity.27 The chemical structure of compound 19 was confirmed from its spectral and analytical results. In the 1H NMR spectrum, two multiplet signals attributed to the phenyl ring appeared at δ = 7.51–7.35 ppm. Besides, the spectrum exhibited two singlet signals due to NH2 and NH protons at δ 8.68 and 11.61 ppm, respectively. The 13C NMR spectrum displayed 19 signals, in agreement with the proposed structure. The FT-IR absorption bands observed at 3346–3127 cm–1 were attributed to the stretching vibrations of the NH and NH2 groups. Also, the HRMS spectrum of 19 revealed a molecular ion peak at m/z = 413.1047 [M + Na], equivalent to C21H18N4O2S.
Antiproliferative Activity
Pyrimidines play a crucial role as building blocks in a diverse array of naturally occurring compounds such as pterins, nucleotides, purines, vitamins, coenzymes, and nucleic acids. The fact that pyrimidine is a significant constituent of RNA and DNA could be clarified by its wide range of medical applications. The various mechanisms through which pyrimidine-based frameworks successfully destroy cells indicate that they might interact with a wide range of receptors and enzymes. Notably, cancer has emerged as a prominent cause of mortality in the modern era. Cancer is often regarded as a leading cause of mortality worldwide. According to the World Health Organization (WHO),28 lung cancer is the leading cause of mortality among all cancer types, followed by hepatocyte cancer. On the other hand, breast cancer is the leading cause of cancer-related deaths among women worldwide.28 Based on the aforementioned facts, the antiproliferative activities of newly synthesized compounds were examined using the MTT approach against three cancerous human cell lines: breast (MCF7), hepatocyte (HepG2), and lung (A549), as well as a noncancerous cell line (MCF-10A). Table 1 and Figure 2 display the obtained results as the IC50 values.
Table 1. IC50 Values for Evaluated Molecules Employing Doxorubicin as a Standard Druga.
| IC50 values (μM) |
|||||
|---|---|---|---|---|---|
| entry | compound | HepG2 | MCF7 | A549 | MCF-10A |
| 1 | 1 | 33.87 ± 3.34 | 39.76 ± 3.35 | 36.27 ± 3.37 | NT |
| 2 | 2 | 27.33 ± 3.87 | 29.39 ± 2.35 | 29.87 ± 3.05 | NT |
| 3 | 3 | 2.02 ± 0.15 | 1.61 ± 0.12 | 1.87 ± 0.32 | 77.29 ± 3.22 |
| 4 | 4a | 13.74 ± 3.81 | 14.27 ± 2.05 | 14.87 ± 3.12 | 60.12 ± 2.31 |
| 5 | 4b | 17.82 ± 2.04 | 18.76 ± 3.19 | 17.11 ± 3.17 | 58.12 ± 2.34 |
| 6 | 6 | 15.39 ± 3.12 | 15.21 ± 2.16 | 15.57 ± 2.94 | 57.02 ± 1.28 |
| 7 | 7 | 6.26 ± 0.32 | 5.60 ± 1.12 | 7.28 ± 1.34 | 78.54 ± 2.05 |
| 8 | 11 | 50.48 ± 3.16 | 60.10 ± 3.45 | 50.67 ± 3.23 | NT |
| 9 | 13 | 19.26 ± 2.07 | 18.09 ± 2.05 | 19.15 ± 3.88 | 51.57 ± 1.98 |
| 10 | 14 | 37.22 ± 1.45 | 39.33 ± 3.12 | 40.39 ± 3.15 | NT |
| 11 | 15 | 19.27 ± 2.06 | 19.27 ± 3.37 | 18.99 ± 3.45 | 52.33 ± 2.18 |
| 12 | 17 | 25.29 ± 1.34 | 29.02 ± 2.38 | 24.28 ± 3.88 | NT |
| 13 | 18 | 39.67 ± 1.22 | 45.62 ± 3.28 | 45.97 ± 3.65 | NT |
| 14 | 19 | 13.33 ± 1.96 | 15.21 ± 1.65 | 14.33 ± 1.96 | 60.45 ± 1.94 |
| 15 | doxorubicin | 2.71 ± 0.12 | 1.68 ± 0.11 | 2.22 ± 0.22 | 87.34 ± 1.47 |
The percentage inhibition (IC50) was determined from three successive tests utilizing an MTT procedure after 72 h of treatment. The statistics are reported as the mean standard deviation (S.D.). A standard drug is Doxorubicin. NT = Not Tested.
Figure 2.

Antiproliferative activities of the synthesized compounds against doxorubicin (Dox.) [x-axis: compounds under investigation; y-axis: IC50 values (μM)].
In the beginning, the antiproliferative potential of 2-aminochromene-3-carbonitrile derivative 1 was evaluated, and a moderate impact on the cell lines studied was observed (Table 1, entry 1 and Figure 2). As depicted in Table 1 and Figure 2, the cyclization of platform compound 1 to the targeted fused pyrimidines showed an immense improvement in cytotoxicity in almost all derivatives. For example, derivatives 3, 4a, 7, and 19 exhibited the highest antiproliferative effects (IC50 < 15.21 μM) toward MCF–7, A549, and HepG2. In particular, pyrimidine derivative 3 exhibited very potent antiproliferative effects, which were slightly better than those of Doxorubicin (Table 1 and Figure 2).
Structure–Activity Relationship
The improvement of the anticancer activities toward the tested cell lines was achieved by the cyclization of the 2-aminochromene-3-carbonitrile derivative 1 to fused pyrimidines, emphasizing the importance of the pyrimidine motif for the antiproliferative activities. The condensation reaction of 1 with triethyl orthoformate afforded formimidate derivative 2 with improved activity toward all tested cells (entry 2, Table 1). Interestingly, cyclization of compound 2 with hydrazine afforded amino-iminopyrimidine derivative 3 that exhibited the most potent anticancer activities against the three tested cells (IC50 values ranged from 1.61 to 2.02 μM, entry 3, Table 1). This observation might be attributed to the presence of ortho amino-imino groups.29 Furthermore, cyclization of derivative 1 to 4-aminopyrimidines 4a,b improved the activities toward the examined cell lines. Pyrimidinone derivative 4a showed superior activity to thione derivative 4b; this may be attributed to the hydrophilic impact of the carbonyl group.30 Additionally, the antiproliferative activities of the dithione compound 6 were improved over derivative 1 but decreased compared with the thione derivative 4b. Moreover, 4-amino-3-phenylpyrimidine 7 exhibited promising cytotoxic effects. We speculated that the toxicity of approximately 6 μM might be derived from the phenyl moiety connecting the pyrimidine ring. 4-(Aminopyrimidin-2-yl)acetonitrile derivative 11 exhibited marginal effects on the tested cancerous cell lines (IC50 > 50 μM, entry 8, Table 1). The presence of an electron-withdrawing group (C≡N) might be the reason for the diminished activity of derivative 11. The replacement of the acetonitrile motif in compound 11 with the more electron-rich ethanethioamide motif 13 causes an improvement in the cytotoxic impact on all of the examined cancer cells (IC50 ranged between 18.09 and 19.26 μM). Furthermore, the amidation of derivative 1 with ethyl chloroformate produced ethyl carbamate derivative 14, which exhibited a slight decrease in all of the tested cell line activities. Notably, a considerable improvement in the antiproliferative activities was observed with the cyclization of derivative 14 to imidazopyrimidine derivative 15. However, the N-acetyl acetamide derivative 17, which comprises two electron-withdrawing groups (COCH3), demonstrated a remarkable increase in anticancer activities (IC50, 24.28–29.02 μM) in comparison with starting compound 1. Besides, the cyclization of 1 to 4-aminopyrimidine 18 resulted in diminished activity, especially for the A-549 cell line. Also, the cyclization of derivative 1 to 3-amino-4-imino-2-phenylpyrimidine (19) led to increased activity for all tested cancer cells in comparison with compound 1 (entry 14, Table 1). Finally, we can deduce that the incorporation of a pyrimidine nucleus with a hydrophobic group such as (NH, −NH2) or (−NAc2) is favorable and greatly enhances the activity. Furthermore, compounds 3 and 7 demonstrated acceptable efficacy in preventing the proliferation of all cancer cells examined when compared to the standard drug, Dox. (Figure 2).
Quantum Chemical Calculations
The quantum chemistry calculations of the constructed compounds were performed utilizing the DFT/B3LYP/6-311 G(2d,2p) approach (Table 2).31 Several computationally advantageous parameters, such as ELUMO, EHOMO, ΔE (ELUMO – EHOMO), electronegativity (χ), electrophilicity index (ω), hardness (η), and softness (S or σ), were computed to evaluate the reactivity of the obtained derivatives (Table 2). Additionally, the effects of these computational parameters on the biological activity of the prepared compounds were studied.
Table 2. Computed Quantum Chemical Variables Received from B3LYP/6-311G++(d, p) of the Assembled Molecules.
| comp. | ELUMO | EHOMO | ΔE | A | I | X | η | S or σ | ω |
|---|---|---|---|---|---|---|---|---|---|
| 1 | –0.15044 | –0.24997 | 0.09953 | 0.15044 | 0.24997 | 0.200205 | 0.049765 | 20.09444 | 0.805426 |
| 2 | –0.15212 | –0.25029 | 0.09817 | 0.15212 | 0.25029 | 0.201205 | 0.049085 | 20.37282 | 0.824762 |
| 3 | –0.15936 | –0.17032 | 0.01096 | 0.15936 | 0.17032 | 0.16484 | 0.00548 | 182.4818 | 4.958435 |
| 4a | –0.16162 | –0.23822 | 0.0766 | 0.16162 | 0.23822 | 0.19992 | 0.0383 | 26.10966 | 1.043551 |
| 4b | –0.15177 | –0.23501 | 0.08324 | 0.15177 | 0.23501 | 0.19339 | 0.04162 | 24.02691 | 0.898599 |
| 6 | –0.15168 | –0.23109 | 0.07941 | 0.15168 | 0.23109 | 0.191385 | 0.039705 | 25.18574 | 0.922509 |
| 7 | –0.15128 | –0.21043 | 0.05915 | 0.15128 | 0.21043 | 0.180855 | 0.029575 | 33.81234 | 1.105952 |
| 11 | –0.14965 | –0.25212 | 0.10247 | 0.14965 | 0.25212 | 0.200885 | 0.051235 | 19.51791 | 0.787641 |
| 13 | –0.14948 | –0.23671 | 0.08723 | 0.14948 | 0.23671 | 0.193095 | 0.043615 | 22.92789 | 0.854882 |
| 14 | –0.15048 | –0.25091 | 0.10043 | 0.15048 | 0.25091 | 0.200695 | 0.050215 | 19.91437 | 0.802121 |
| 15 | –0.15277 | –0.25192 | 0.09915 | 0.15277 | 0.25192 | 0.202345 | 0.049575 | 20.17146 | 0.82589 |
| 17 | –0.15416 | –0.25136 | 0.0972 | 0.15416 | 0.25136 | 0.20276 | 0.0486 | 20.57613 | 0.845918 |
| 18 | –0.15197 | –0.25243 | 0.10046 | 0.15197 | 0.25243 | 0.2022 | 0.05023 | 19.90842 | 0.813953 |
| 19 | –0.17259 | –0.24379 | 0.0712 | 0.17259 | 0.24379 | 0.20819 | 0.0356 | 28.08989 | 1.217502 |
| Dox. | –0.20501 | –0.29094 | 0.08593 | 0.20501 | 0.29094 | 0.247975 | 0.042965 | 23.27476 | 1.431202 |
Table 2 shows the optimized compounds’ optimal molecular structures with the lowest energy. The outcomes of the biological activity assessments revealed that compounds 3, 4a, 4b, 6, 7, 13, 15, and 19 are the most potent derivatives, which may also be analyzed by computational estimations. The significantly low values of gap energy (ΔE) have predominantly been attributed to higher activities and possibly enhanced inhibitory efficiency.32 Compound 1 has a relatively high value of ΔE (0.09953 eV), which might explain its low cytotoxic behavior toward the examined cell lines. The conversion of compound 1 into amino-iminopyrimidine derivative 3 dramatically diminishes the ΔE from 0.09953 to 0.01096 eV (Table 2 and Figure 3), which was anticipated to enhance the biological activities of this molecule 3 (ΔE for Doxorubicin is 0.08593). However, the cyclization of derivative 1 to aminopyrimidines 4a and 4b decreases the ΔE values by about 0.02293 and 0.01629 eV, respectively, which is likely to improve these compounds’ biological behaviors in comparison with 1. The same trend could also be observed for compounds 6, 7, 13, 15, 17, and 19 (Table 2). Besides, derivatives 11, 14, and 18, which were obtained from the cyclization of compound 1, showed a remarkable increase in the ΔE values (Table 2). These results might explain the lower anticancer performance of these derivatives (11, 14, and 18) in comparison with that of compound 1. Furthermore, the amount of ω (global electrophilicity indicator) is the explicative concept of reactivity and is essential to describe the compounds’ toxicity.32 As demonstrated in Table 2, compound 3 achieved the highest value of ω (4.958435), affirming its superior anticancer activity against the investigated cell lines. Ionization potential (I) is another crucial measure of chemical reactivity, a compound with a smaller value of (I) possessed superior biological activity (was less stable) and conversely.32 As observed in Table 2, compound 3 possessed the lowest ionization potential value (I = 0.17032 eV). Thus, derivative 3 was expected to be the most active derivative among all of the derivatives (I for Doxorubicin is 0.29094). As compound 18 exhibited the highest ionization potential (I = 0.25243 eV, Table 2), it was expected to behave as a weak anticancer drug. Softness (σ) and hardness (η) are the other two parameters that could be used to predict the stability and biological activity of the designed compounds.32 The most active compound exhibited the greatest softness (σ) and smallest hardness (η) values. Based on the computed parameters (η and σ), the biological activity patterns for the most active compounds could be specified as 3 > 7 > 19 (Table 2). The chemical properties of the prepared compounds could also be predicted from the calculation of the chemical potential (μ = −χ) parameter. The susceptibility of inhibitor compounds to accepting electrons could be anticipated by estimating the electronegativity (χ).32 According to the calculated values of χ (Table 2), compounds 3 and 7 had the smallest values of χ (0.16484 and 0.180855, respectively), which indicates that these derivatives assist in the transfer of charge and, consequently, enhance their anticancer performance. According to the obtained data from quantum chemical calculations and the experimental results of anticancer activity, we can confirm that there is an excellent agreement between the practical and predicted results (Table 2 and Figure 3).
Figure 3.

DFT-optimized structures and HOMO/LUMO of compounds 1, 3, and 7.a The optimal molecular structures of fused pyrimidines in Table 2 are presented in Table S1 in the Supporting Information.
Electrostatic Potential (Esp)
The electrostatic potential (ESP) is another essential parameter for elucidating the atomic structure and chemical reactivity of molecules. The application of Density Functional Theory (DFT) allows for systematic and efficient investigation of the electrostatic potential (ESP) distribution of molecules.33 The nucleophilic and electrophilic properties of a molecule were investigated using ESP. This also serves as one of the key research approaches for understanding molecular reactivity. Accordingly, the maximal quantity of the positive area, wherein the nucleophilic reaction occurs, is indicated by the shaded blue in the ESP charts. The negative area, in which the electrophilic reaction occurs, is distinguished by the red color, and the zero potential is shown by the shade of green.34 As displayed in Figure 4 (Table S2), the electron densities of the designed fused pyrimidines increased around nitrogen, oxygen, and sulfur atoms, with a negative electrostatic potential of the compounds that might be due to the lone pair of electrons of these atoms.34 The present study suggests that such atoms might be more effectively incorporated into electrophilic processes. Subsequently, ESP mapping of the developed molecules revealed the active sites. Owing to the diverse substituents, their impacts on the reaction activity are varied. In particular, compound 3 (Figure 4) demonstrated an electrostatic potential area concentrated within a broad zone (strong binding interaction), suggesting that compound 3 has lower stability than the others. Accordingly, its biological activities are comparatively stronger, which is in agreement with the observed biological activities and quantum calculations. On the other hand, compound 11 (Figure 4) showed that the electrostatic potential is mainly concentrated in a narrow zone, confirming that compound 11 has greater stability than the others. As a result, compound 11 possessed low biological activities, which was also in agreement with the obtained data from biological tests and quantum calculations (Tables 1 and 2).
Figure 4.
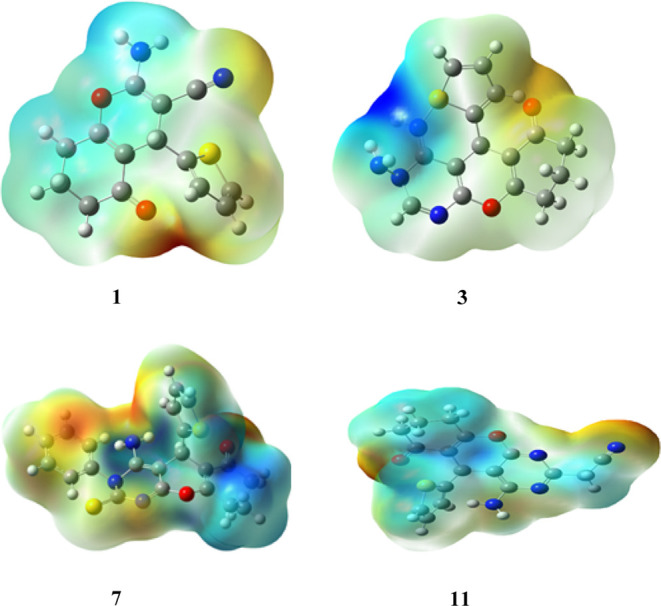
Computed electrostatic potential (ESP) surface of compounds 1, 3, 7, and 11.a The computed electrostatic potential (ESP) surface for other assembled molecules is presented in Table S2 in the Supporting Information.
Docking Studies
To obtain a better overview of the estimated binding interactions between the designed fused pyrimidines and various proteins, the ligand-protein docking of the assembled pyrimidine derivatives was studied. The Molecular Operating Environment (MOE) model 2014.09 was used for this investigation. Three proteins have been selected in this study: 6ENV, 4b3z, and 2JW2. These proteins were selected to represent different biological systems and provide a comprehensive understanding of the compounds’ interactions and potential modes of action. The first is 6ENV, a protein that is often found in intestinal mucous membranes. The potential of 6ENV to store and bind with iron when coupled with a phosphate-ferric hydroxide complex to produce ferritin is essential for biological functions. On the other hand, it has been observed that in breast cancer, ferritin is liberated by invading macrophages and thereby promotes carcinogenesis.35 Thus, we expected that the affinity of the designed compounds with 6ENV might be caused by apoptosis in MCF7 cell lines. The second protein is 4b3z, which was the first recognized candidate of the Collapsin Response Mediator Protein (CRMP) family and is important for both mediating the differentiation of neurons and reducing lung cancer growth.36 Because such protein has been demonstrated to be downregulated in various spreading tumors with excellent binding energies with several pyrimidine compounds,37 we employed it as a potential approach for managing cancer metastasis, particularly lung cancer. Zülbiye and his co-workers38 reported that the pyrimidine derivatives have an excellent binding energy with 2JW2 protein, especially in inhibiting the growth of hepatocellular carcinoma. Accordingly, such a protein was chosen to investigate its potential to bind to assembled pyrimidines. Figures 5–7 demonstrate that the active conformations of every molecule bind to the active region and overlap with one another. The results indicated that the compounds exhibited favorable receptor–ligand binding interactions, suggesting their potential as effective agents in the context of the studied cancer types.
Figure 5.
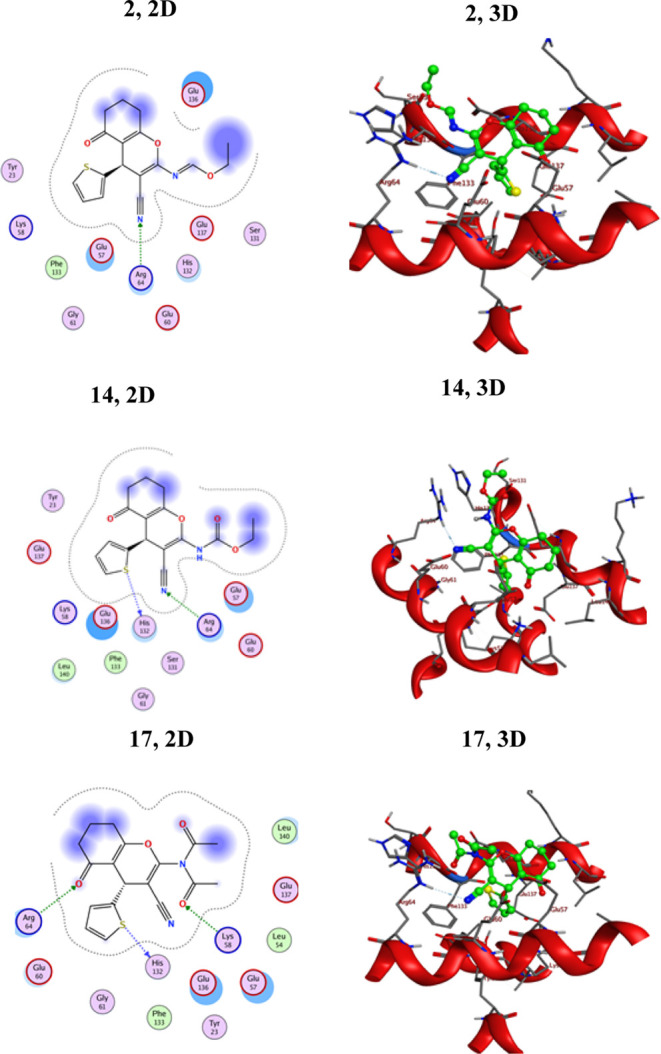
2D and 3D interactions of ligands 2, 14, and 17 against the active site of 6ENV-MCV-7.
Figure 7.
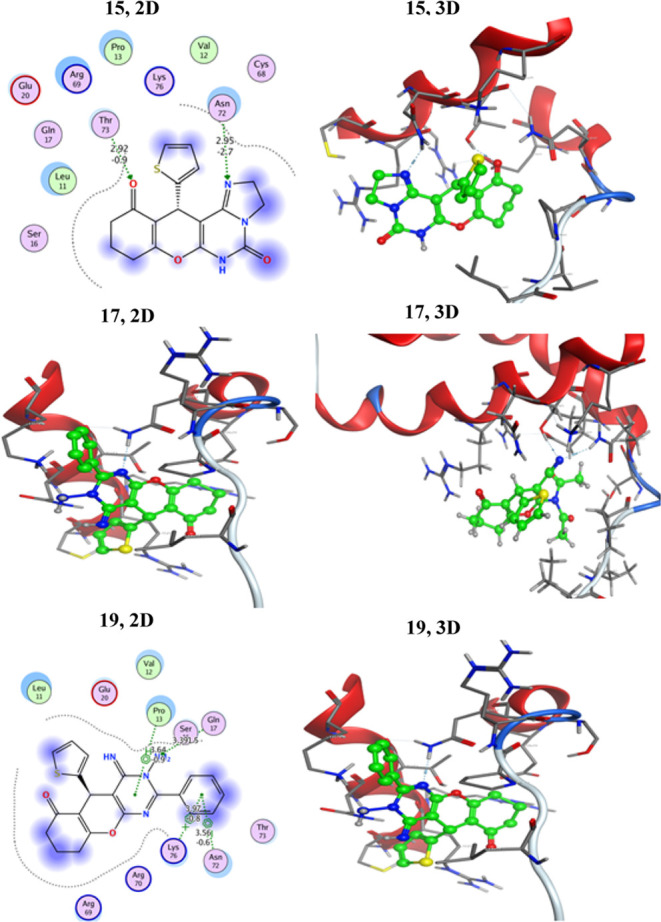
2D and 3D interactions of ligands 15, 17, and 19 against the active site of HepG2–2JW2.
To evaluate the obtained results, we utilized doxorubicin, a widely prescribed anticancer therapy, as a reference drug. After analysis of the docking interactions, the conformation with a slightly lower docking energy score was selected. This choice was made because this particular conformation signifies the ligand’s strongest binding potential within the target. As depicted in Table 3, the binding energy scores imply that the majority of the assembled fused pyrimidines have superior or equivalent susceptibility to 6ENV-MCV-7 and 4b3z-Lung. Moreover, almost all of the assembled pyrimidines exhibited promising binding energies, ranging from −5.11563 to −5.95474 kcal/mol, when docked against the HepG2–2JW2 protein. It was also observed that the assembled compounds exhibited superior inhibition activities with the HepG2–2JW2 protein in comparison with the standard drug Doxorubicin.
Table 3. Binding Scores of the Newly Prepared Compounds and Dox to 6ENV-MCV-7, 4b3z-Lung, and HepG2-2JW2.
| binding
score (kcal/mol) |
|||
|---|---|---|---|
| comp. | 6ENV-MCV-7 | 4b3z-lung | HepG2–2JW2 |
| 1 | –5.47951 | –5.19771 | –4.89862 |
| 2 | –6.25608 | –5.88317 | –5.49264 |
| 3 | –5.8625 | –5.50277 | –5.5669 |
| 4a | –5.83893 | –5.40306 | –5.16215 |
| 4b | –5.77245 | –5.42736 | –5.11563 |
| 6 | –5.82066 | –5.69825 | –5.14694 |
| 7 | –5.94382 | –6.17619 | –5.43515 |
| 11 | –6.13866 | –5.80001 | –5.29499 |
| 13 | –6.0665 | –6.03983 | –5.4955 |
| 14 | –6.22066 | –6.21129 | –5.43210 |
| 15 | –5.69644 | –5.6829 | –5.59437 |
| 17 | –6.5531 | –6.12329 | –5.59266 |
| 18 | –5.72812 | –5.17581 | –5.26642 |
| 19 | –6.11341 | –6.33115 | –5.95474 |
| Dox. | –8.1865 | –7.2391 | –6.61660 |
Binding Score of the Assembled Ligands to 6ENV-MCV-7
According to the docking profile, most of the synthesized compounds had a defined significant binding affinity for the selected protein, as evidenced by their docking score results. The reference drug Doxorubicin showed three hydrogen bonding interactions with Glu253, Lys273, and Asn282; its docking score was −8.1865 kcal/mol. The designed molecules fit best in the allosteric site of the protein. According to the obtained results (Table 3), compound 17 is the most potent inhibitor for the 6ENV-MCV-7 protein, with the lowest binding score value (−6.5531 kcal/mol). Compound 17 displayed strong hydrogen bonding with the corresponding amino acid residues, as demonstrated in Figure 5. This derivative (17) formed three binding interactions via hydrogen bonds: carbonyl oxygen bound with Arg64, amide carbonyl oxygen bound with Lys58, and thiophene-S bound with His132. Consequently, the presence of two acetyl groups in compound 17 enhanced its opportunity for hydrogen bonding interactions with the electron-rich sites of the 6ENV-MCV-7 protein. Compounds 2 and 14, comprising a nitrile group, demonstrated slightly high docking scores of −6.25608 and −6.22066 kcal/mol, respectively. Further, derivatives 2, 11, 13, 14, and 19 exhibited superior susceptibility for the 6ENV-MCV-7 protein with binding score ranges of −6.25608 to −6.0665 kcal/mol. The other derivatives (3, 4a, 4b, 6, 7, 15, and 18) demonstrated moderate susceptibilities (−5.94382 to −5.69644 kcal/mol) toward the 6ENV-MCV-7 protein in comparison with Doxorubicin (Table 3). The lower docking score of compound 15 compared to those of the other compounds was due to the presence of an imidazole motif fused with the pyrimidine ring. As could be observed, the geometry of the assembled ligands influenced their binding properties as well as their inhibitory activities. Also, all of the assembled compounds showed different types of interactions with the evaluated protein (Figure 5 and Table S3 [Supporting Information]).
Binding Score of the Assembled Ligands to 4b3z-Lung
The results revealed that most of the pyrimidine derivatives had superior or equivalent affinity for 4b3z-Lung, as determined by binding energy scores (Table 3). Compound 19 containing a phenyl ring attached to the second position of the pyrimidine ring had a docking score (−6.33115 kcal/mol) comparable to that of the standard drug. Derivative 19 was the top candidate with a binding energy docking score of −6.33115 kcal/mol. This derivative formed three binding interactions via hydrogen bonds: the amino group bound with both Glu353 and Ser80, and thiophene-S bound with Gln81. Additionally, compound 19 exhibited a hydrophobic interaction between the pyrimidine motif and Sr80. Moreover, the results indicate that derivative 18 is the weakest inhibitor of 4b3z-Lung, with a total intermolecular interaction energy of −5.17581 kcal/mol. Compound 4a showed five hydrogen bonding interactions with Gln81, Glu353, 2Pro79, and Tyr174 and recorded a binding energy score of −5.40306 kcal/mol, which was lower than that of the standard drug. Compound 4b showed six hydrogen bonding interactions with Gln81, Glu353, 2Pro79, Tyr168, and Tyr174 and recorded a binding energy score of −5.42736 kcal/mol. The only difference between compounds 4a and 4b was the presence of carbonyl and thiocarbonyl motifs in these compounds, respectively. The other compounds demonstrated moderate to good inhibition activities in comparison with those of doxorubicin (Table 3 and Figure 6). The 2D and 3D docking data of the newly assembled compounds to 4b3z-Lung are presented in Table S4 [Supporting Information].
Figure 6.
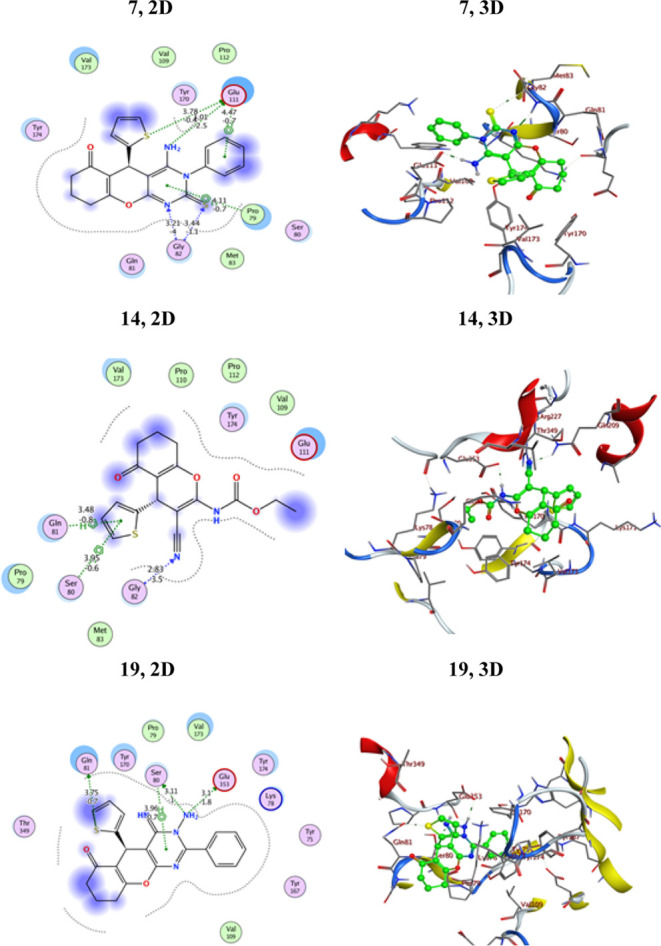
2D and 3D interactions of ligands 7, 14, and 19 against the active site of 4b3z-Lung.
Binding Score of the Assembled Ligands to HepG2–2JW2
As displayed in Table 3, all of the assembled derivatives exhibited moderate inhibitory activities toward HepG2–2JW2 in comparison with Doxorubicin (Table 3). In addition, they recorded binding scores lower than those of the starting reactant 1, affirming the potential role of the pyrimidine motif in inhibiting the HepG2–2JW2 protein. It is noteworthy that out of all of the synthesized compounds, derivative 19 had the best binding score to the 2JW2 protein. Compound 19 demonstrated three binding interactions via hydrophobic interactions: a pyrimidine ring bent with Pro13 and a phenyl ring bent with both Lys76 and Asn72, as well as one hydrogen bond of the amino group with Gln17, with a total intermolecular interaction energy of −5.95474 kcal/mol (Figure 7). Further, derivatives 15 and 17 displayed similar inhibitory activities with bending scores of approximately 5.59 kcal/mol (Figure 7). Also, the pyrimidine 3 modeling configuration with the HepG2–2JW2 protein showed hydrogen bonding interactions of Thr73, Gln17, Ser16, and Asn72 with NH, NH2, and C=O groups, respectively, with a binding energy score of −5.5669 kcal/mol. Moreover, derivatives 2, 7, 11, 13, 14, and 18 exhibited moderate activities with binding scores ranging between −5.5669 and −5.26642 kcal/mol (Table 3), whereas derivatives 4a, 4b, and 6 displayed lower activities in comparison with Doxorubicin. The pyrimidinone derivative 4a modeling configuration with HepG2–2JW2 showed two hydrogen bonding interactions with Arg69 and Gln and recorded a binding energy score of −5.16215 kcal/mol. Whereas, the pyrimidinethione derivative 4b (the thione derivative of 4a) modeling configuration with HepG2–2JW2 showed two hydrogen bonding interactions with Gln17 and Thr73, in addition to one hydrophobic interaction with Pro13. It recorded a binding energy score of −5.11563 kcal/mol. Moreover, the compound 6 modeling configuration with the target protein showed three hydrogen bonding interactions with Thr73, Gln17, and Sr16. In addition, it exhibited two hydrophobic interactions with Thr73 and Pro13 and recorded a binding energy score of −5.14694 kcal/mol. Thus, we can conclude that the presence of carbonyl (4a) or thiocarbonyl (4b and 6) motif resulted in diminishing binding energy score. On the other hand, 3-phenyl pyrimidinethione derivative 7 showed a slight improvement in its bending energy (−5.43515 kcal/mol) in comparison to compounds 4a, 4b, or 6. This might be due to the presence of a phenyl moiety attached to the third position of the pyrimidine ring. Moreover, derivatives 2, 11, 13, 14, and 18 exhibited moderate activities with binding scores ranging between −5.5669 and −5.26642 kcal/mol (Table 3). 2D and 3D docking data as well as the types of interactions of the newly assembled compounds with HepG2–2JW2 are presented in Table S5 [Supporting Information].
Conclusions
In summary, the present work demonstrated a versatile approach for designing a series of chromenopyrimidine derivatives using ultrasonic or microwave irritation conditions. The structures of the obtained fused pyrimidines were established by using several spectral tools. Further, the synthesized chromenopyrimidine derivatives were evaluated against three cancerous cells (A549, HepG2, and MCF7) as well as a nontumorigenic cell line (MCF-10A, representing breast cells). The comprehensive findings highlighted the significant activity of compounds 3 and 7 against cancer cells, with derivative 3 surpassing the performance of the reference drug Doxorubicin. Compound 3 demonstrated the highest cytotoxic activity, indicating its superior selective killing ability toward malignant cells while maintaining favorable selectivity over nonmalignant cells and the standard drug. The quantum chemical parameters of the assembled molecules were calculated and thoroughly discussed, with ΔE, I, χ, η, σ, and ω values representing key indicators. Notably, chromenopyrimidine derivative 3 emerged as the most active candidate, and the outcomes of the antiproliferative activity aligned with the outcomes of the quantum investigations. Moreover, molecular docking studies of the assembled molecules were conducted to assess their interaction with three specific proteins (6ENV-MCV-7, 4b3z-Lung, and HepG2–2JW2). The results of the binding energy scores indicated that the prepared chromenopyrimidines possess significant inhibitory potential against the activity of the evaluated proteins, further confirming their efficacy.
Experimental Section
The melting points were determined by using an electrothermal IA9100 melting point device (U.K.) and were uncorrected. Infrared spectra were recorded by using a Shimadzu DR-8001 spectrometer. For NMR data collection, a Jeol 600 MHz NMR analyzer was utilized, with TMS serving as the internal reference and DMSO-d6 as the solvent. The MS data were collected at the Microanalytical Center using a Shimadzu Qp-2010 Plus instrument. Analytical thin-layer chromatography (TLC) was performed by using an EM silica gel F254 sheet (0.2 mm). Ultrasonication was carried out using an SY5200DH-T ultrasound cleaner. Various solvents and fine materials were obtained from suppliers, such as Merck, Fluka, and other commercial sources, and they were used without prior purification.
Synthesis of Ethyl (E)-N-(3-Cyano-5-oxo-4-(thiophen-2-yl)-5,6,7,8-tetrahydro-4H-chromen-2-yl)formimidate (2) and 3-Amino-4-imino-5-(thiophen-2-yl)-3,4,5,7,8,9-hexahydro-6H-chromeno[2,3-d]pyrimidin-6-one (3)
A mixture of triethyl orthoformate (10.0 mL) and compound 1 (2.0 mmol) was sonicated for 30 min at 60 °C (verified by TLC). The extra reagent was then pumped out. A solution of hydrazine hydrate (5.0 mmol) in 10.0 mL of EtOH was added to resultant product 2, and sonication was kept at room temperature for an extra hour. The crystalline product 3 obtained was filtered and washed with EtOH (2 × 5.0 mL).
Compound 2
Yellow crystals (dioxane); yield: 93%; mp 197–200 °C. FT-IR νmax 2234 (C≡N), 1663 (C=O), 1598 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 8.52 (s, 1H, N=CH–O), 7.35 (s, 1H, Ar–H), 6.91 (s, 2H, Ar–H), 4.72 (s, 1H, CH), 4.28 (q, J = 7.1 Hz, 2H, CH2CH3), 2.61 (br, 2H, CH2), 2.31 (br, 2H, CH2), 1.97–1.85 (m, 2H, CH2), 1.26 (t, J = 7.2 Hz, 3H, CH2CH3). 13C NMR (DMSO-d6), δC (ppm): 196.3 (C=O), 165.2, 162.5, 157.2, 156.7, 147.1, 127.5, 125.7, 125.6, 117.6, 113.2, 82.7, 64.5, 36.7, 32.6, 26.9, 20.5, 14.2. Analysis calcd for C17H16N2O3S: C, 62.18; H, 4.91; N, 8.53%. Found: C, 62.22; H, 4.87; N, 8.50%.
Compound 3
Brown crystals (dioxane); yield: 91%; mp 227–228 °C. FT-IR νmax 3275–3114 (NH), 1675 (C=O), 1597 cm–1 (C=N). 1H NMR (DMSO-d6), δH (ppm): 11.70 (s, 1H, NH), 8.67 (s, 1H, Ar–H), 8.30 (s, 2H, NH2), 7.30 (s, 1H, ArH), 6.99 (s, 2H, Ar–H), 4.71 (s, 1H, CH), 2.61 (br, 2H, CH2), 2.30 (br, 2H, CH2), 1.99–1.81 (m, 2H, CH2). MS (EI) m/z: calcd for C15H14N4O2S (M+) 314, found 314. Analysis calcd for C15H14N4O2S: C, 57.31; H, 4.49; N, 17.82%. Found: C, 57.18; H, 4.53; N, 17.57%.
Synthesis of 4-Amino-5-(thiophen-2-yl)-5,7,8,9-tetrahydro-2H-chromeno[2,3-d]pyrimidine-2,6(1H)-dione (4a) and 4-Amino-5-(thiophen-2-yl)-2-thioxo-1,2,5,7,8,9-hexahydro-6H-chromeno[2,3-d]pyrimidin-6-one (4b)
Employing DMF
An equimolar mixture of compound 1 (1.0 mmol) and urea or thiourea (1.0 mmol) in DMF (5.0 mL) was subjected to microwave irradiation for a suitable period (validated by TLC). The precipitate formed upon cooling was recrystallized from dioxane to obtain the desired product.
Employing Basic Alumina as Solid Support
A mixture containing urea or thiourea (1.0 mmol) and compound 1 (1.0 mmol) adsorbed on basic alumina was irradiated within a microwave apparatus for an appropriate period of time (as verified by TLC). Following elution of the solids with MeOH, the recoverable basic alumina was isolated through filtration, resulting in the isolation of yellow products (4a,b) in a pure form.
Compound 4a
Yellow crystals (dioxane); yield: 88%; mp 167–168 °C. FT-IR νmax 3319–3187 (NH), 1663 and 1657 (C=O), 1591 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 11.20 (s, 1H, NH), 7.91 (s, 2H, NH2), 7.24 (s, 1H, Ar–H), 6.89–9.81 (s, 2H, Ar–H), 4.52 (s, 1H, CH), 2.59–2.51 (m, 2H, CH2), 2.32–2.25 (m, 2H, CH2), 1.96–1.79 (m, 2H, CH2). 13C NMR (DMSO-d6), δC (ppm): 194.9, 168.1 (C=O), 163.0, 153.1, 149.0, 135.9, 128.7, 126.0, 125.9, 116.1, 75.6, 37.4, 31.5, 27.6, 21.0. HRMS (EI) m/z: calcd for C15H13N3O3S (M + Na) 338.0575, found 338.0579. Analysis calcd for C15H13N3O3S: C, 57.13; H, 4.16; N, 13.33%. Found: C, 57.18; H, 4.10; N, 13.28%.
Compound 4b
Yellow crystals (dioxane); yield: 90%; mp 178–179 °C. FT-IR νmax 3278–3109 (NH), 1679 (C=O), 1590 (C=C), 1346 cm–1 (C=S). 1H NMR (DMSO-d6), δH (ppm): 11.90 (s, 1H, NH), 8.27 (s, 2H, NH2), 7.35 (s, 1H, Ar–H), 6.90 (s, 2H, Ar–H), 4.60 (s, 1H, CH), 2.54 (br, 2H, CH2), 2.29 (br, 2H, CH2), 2.00–1.86 (m, 2H, CH2). HRMS (EI) m/z: calcd for C15H13N3O2S2 (M + Na) 354.0346, found 354.0348. Analysis calcd for C15H13N3O3S2: C, 54.36; H, 3.95; N, 12.68%. Found: C, 54.39; H, 3.90; N, 12.62%.
Synthesis of 5-(Thiophen-2-yl)-2,4-dithioxo-1,2,3,4,5,7,8,9-octahydro-6H-hromeno[2,3-d]pyrimidin-6-one (6)
A solution of compound 1 (1.0 mmol) and carbon disulfide (4.0 mL) in a 15% alcoholic sodium hydroxide solution (10.0 mL) was sonicated at 70 °C for 30 min (confirmed by TLC). The resulting solution was cooled to ambient temperature, leading to the formation of a solid product. The product was subsequently collected, dissolved in water (20.0 mL), and acidified using HCl. The resulting crystals were recrystallized from dioxane, yielding compound 6.
Brown crystals (dioxane); yield: 93%; mp 246–245 °C. FT-IR νmax 3201–3137 (NH), 1683 (C=O), 1607 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 12.19 (s, 1H, NH), 11.09 (s, 1H, NH), 7.28 (s, 1H, Ar–H), 6.89–6.80 (s, 2H, Ar–H), 4.50 (s, 1H, CH), 2.55–2.53 (m, 2H, CH2), 2.30–2.28 (m, 2H, CH2), 1.96–1.79 (m, 2H, CH2). MS (EI) m/z: calcd for C15H12N2O2S3 (M) 348, found 347 (M – 1). Analysis calcd for C15H12N2O2S3: C, 51.70; H, 3.47; N, 8.04%. Found: C, 51.76; H, 3.40; N, 7.96%.
Synthesis of 4-Amino-3-phenyl-5-(thiophen-2-yl)-2-thioxo-2,3,5,7,8,9-hexahydro-6H-chromeno[2,3-d]pyrimidin-6-one (7)
A mixture consisting of compound 1 (1.0 mmol) and phenylisothiocyanate (1.0 mmol) in DMF (15.0 mL) was sonicated at 90 °C for 3 h in the presence of TEA. After cooling, the reaction mixture was filtered, diluted with water (15.0 mL), and neutralized using 2 M HCl. The resulting crystals were filtered and washed with EtOH (3 × 5 mL).
Yellow crystals (DMF/EtOH); yield: 97%; mp 287–289 °C. FT-IR νmax 3350–3310 (NH), 1672 (C=O), 1618 (C=C), 1310 cm–1 (C=S). 1H NMR (DMSO-d6), δH (ppm): 7.49–7.32 (m, 5H, Ar–H), 7.20 (s, 1H, Ar–H), 6.93–6.86 (s, 2H, Ar–H), 6.42 (s, 2H, NH2), 4.35 (s, 1H, CH), 2.62 (br, 2H, CH2), 2.38 (br, 2H, CH2), 2.20–1.98 (m, 2H, CH2). MS (EI) m/z: calcd for C21H17N3O2S2 (M) 407, found 407 (M). Analysis calcd for C15H12N2O2S3: C, 61.90; H, 4.21; N, 10.31%. Found: C, 61.87; H, 4.29; N, 10.48%.
Synthesis of 2-(4-Amino-6-oxo-5-(thiophen-2-yl)-6,7,8,9-tetrahydro-5H-chromeno[2,3-d]pyrimidin-2-yl)acetonitrile (11) and 2-(4-Amino-6-oxo-5-(thiophen-2-yl)-6,7,8,9-tetrahydro-5H-chromeno[2,3-d]pyrimidin-2-yl)ethanethioamide (13)
A mixture containing compound 1 (1.0 mmol) and malononitrile (1.0 mmol) or cyanothioacetamide (1.0 mmol) in ethanol (5.0 mL) with a catalytic amount of acetic acid was microwave irradiated for 1 h at 90 °C. After cooling to ambient temperature, the formed solid was filtered and recrystallized to yield compounds 11 and 13, respectively.
Compound 11
Brown crystals (dioxane); yield: 89%; mp 298–300 °C. FT-IR νmax 3287–3274 (NH2), 2257 (C≡N), 1675 (C=O), 1601 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 7.29 (s, 1H, Ar–H), 6.92 (s, 2H, Ar–H), 6.30 (br, 2H, NH2), 4.68 (s, 1H, CH), 3.69 (s, 2H, CH2CN), 2.56–2.52 (br, 2H, CH2), 2.31–2.28 (br, 2H, CH2), 2.00–1.82 (m, 2H, CH2). 13C NMR (DMSO-d6), δC (ppm): 192.4 (C=O), 173.1, 165.2, 159.8, 152.9, 143.0, 127.9, 125.8, 123.9, 117.4, 113.0, 109.9, 37.0, 33.9, 27.0, 24.8, 20.1. HRMS (EI) m/z: calcd for C17H14N4O2S (M) 338.0837, found 338.0834. Analysis calcd for C17H14N4O2S: C, 60.34; H, 4.17; N, 16.56%. Found: C, 60.39; H, 4.11; N, 16.50%.
Compound 13
Brown crystals (EtOH/dioxane); yield: 91%; mp 253–254 °C. FT-IR νmax 3387–3098 (NH2), 1679 (C=O), 1599 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 7.30 (s, 1H, Ar–H), 7.01–6.70 (m, 4H, NH2 + Ar–H), 6.66 (s, 2H, NH2), 4.68 (s, 1H, CH), 3.74 (s, 2H, CH2C=S), 2.84–2.69 (m, 4H, 2CH2), 1.71 (br, 2H, CH2). HRMS (EI) m/z: calcd for C17H16N4O2S2 (M + H) 373.0793, found 373.0795. MS (EI) m/z: calcd 372, found 372 (M). Analysis calcd for C17H16N4O2S2: C, 54.82; H, 4.33; N, 15.04%. Found: C, 54.89; H, 4.27; N, 14.96%.
Synthesis of Ethyl (3-Cyano-5-oxo-4-(thiophen-2-yl)-5,6,7,8-tetrahydro-4H-chromen-2-yl)carbamate (14) and 12-(Thiophen-2-yl)-2,6,8,9,10,12-hexahydro-5H-chromeno[3,2-e]imidazo[1,2-c]pyrimidine-5,11(3H)-dione (15)
A mixture of compound 1 (1.0 mmol) and ethyl chloroformate (1.0 mmol) in pyridine (5.0 mL) was sonicated at ambient temperature for 1 h. After solvent removal under reduced pressure, the obtained solid 14 (a few milligrams were crystallized from ethanol for analysis purposes) was washed with water (2 × 3.0 mL) and redissolved in DMF (5.0 mL). Ethylenediamine (2.0 mL) was added dropwise to this solution, and the resulting mixture was sonicated for 2 h at 60 °C. After the mixture was cooled, 5.0 mL of water was added, and the resulting solid was filtered and crystallized from dioxane, resulting in compound 15.
Compound 14
Yellow crystals (dioxane); yield: 98%; mp 167–168 °C. FT-IR νmax 3328 (NH), 1711, 1666 (C=O), 1597 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 10.48 (s. 1H, NH), 7.38–7.34 (m, 1H, Ar–H), 6.93–6.88 (m, 2H, Ar–H), 4.58 (s, 1H, CH), 4.31–4.28 (q, J = 7.3 Hz, 2H, CH2), 2.64–2.59 (m, 2H, CH2), 2.34–2.29 (m, 2H, CH2), 2.02–1.81 (m, 2H, CH2), 1.30–1.23 (t, 2H, CH3). HRMS (EI) m/z: calcd for C17H16N2O4S (M + H) 345.0909, found 345.0907. Analysis calcd for C17H16N2O4S: C, 59.29; H, 4.68; N, 8.13%. Found: C, 59.33; H, 4.60; N, 8.11%.
Compound 15
Yellow crystals (dioxane); yield: 94%; mp 256–259 °C. FT-IR νmax 3237 (NH), 1669, 1652 (C=O), 1586 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 10.12 (s. 1H, NH), 7.28 (s, 1H, Ar–H), 6.90–6.81 (m, 2H, Ar–H), 4.49 (s, 1H, CH), 4.21–4.19 (m, 2H, CH2), 3.70–3.68 (m, 2H, CH2), 2.58–2.51 (m, 2H, CH2), 2.31–2.28 (m, 2H, CH2), 1.97–1.97 (m, 2H, CH2). HRMS (EI) m/z: calcd for C17H15N3O3S (M) 341.0834, found 341.0831. MS (EI) m/z: calcd 341, found 340 (M – 1). Analysis calcd for C17H15N3O3S: C, 59.81; H, 4.43; N, 12.31%. Found: C, 59.77; H, 4.46; N, 12.26%.
Synthesis of N-Acetyl-N-(3-cyano-5-oxo-4-(thiophen-2-yl)-5,6,7,8-tetrahydro-4H-chromen-2-yl)acetamide (17)
Derivative 1 (1.0 mmol) was mixed with 10.0 mL of acetic anhydride and heated under microwave irradiation conditions for 1 h at 80 °C. After cooling to ambient temperature, the obtained solution was poured onto a water/ice mixture (40.0 mL). Upon stirring for 2 h, a yellow precipitate formed, which was subsequently crystallized from dioxane, yielding compound 17.
Yellow crystals (dioxane); yield: 83%; mp 231–233 °C. FT-IR νmax 2212 (C≡N), 1674, 1661 (C=O), 1602 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 7.29 (s, 1H, Ar–H), 6.98–6.91 (m, 2H, Ar–H), 4.56 (s, 1H, CH), 2.98 (s, 6H, 2CH3), 2.59–2.56 (m, 2H, CH2), 2.40–2.37 (m, 2H, CH2), 2.16–2.05 (m, 2H, CH2). MS (EI) m/z: calcd 356, found 356 (M). Analysis calcd for C18H16N2O4S: C, 60.66; H, 4.53; N, 7.86%. Found: C, 60.61; H, 4.59; N, 7.79%.
Synthesis of 4-Amino-5-(thiophen-2-yl)-5,7,8,9-tetrahydro-6H-chromeno[2,3 d]pyrimidin-6-one (18)
A mixture of derivative 1 (1.0 mmol) and formamide (7.0 mL) was heated under microwave irradiation conditions at 120 °C for 1 h. After cooling to room temperature, the resulting solid was filtered and crystallized from dioxane to afford compound 18.
Yellow crystals (dioxane); yield: 93%; mp 269–271 °C. FT-IR νmax 3312–3289 (NH), 1667 (C=O), 1612 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 8.13 (s, 1H, Ar–H), 7.28 (s, 1H, Ar–H), 6.89–6.81 (m, 2H, Ar–H), 6.45 (s, 2H, NH2), 4.61 (s, 1H, CH), 2.71–2.67 (m, 2H, CH2), 2.48–2.39 (m, 2H, CH2), 2.10–1.92 (m, 2H, CH2). HRMS (EI) m/z: calcd for C15H13N3O2S (M + Na) 322.0624, found 322.0624. Analysis calcd for C15H13N3O2S: C, 60.19; H, 4.38; N, 14.04%. Found: C, 60.23; H, 4.31; N, 13.96%.
Synthesis of 3-Amino-4-imino-2-phenyl-5-(thiophen-2-yl)-3,4,5,7,8,9-hexahydro-6H-chromeno[2,3-d]pyrimidin-6-one (19)
To a solution of sodium ethoxide (15.0 mL), prepared by mixing 1.0 mmol of metallic sodium with 15.0 mL of absolute ethanol, was added 1.0 mmol of derivative 1 was added. Subsequently, benzohydrazide (1.0 mmol) was added dropwise to the resulting solution, which was then sonicated at 60 °C for 90 min. The obtained solution was cooled to ambient temperature and neutralized with AcOH (0.5 M). The resulting solid was filtered and recrystallized from ethanol containing a few drops of dioxane, yielding compound 19.
Reddish brown crystals (EtOH/dioxane); yield: 88%; mp 229–232 °C. FT-IR νmax 3346-3127 (NH2), 1659 (C=O), 1621 cm–1 (C=C). 1H NMR (DMSO-d6), δH (ppm): 11.61 (s, 1H, NH), 8.68 (s, 1H, NH2), 7.51–7.35 (m, 5H, Ar–H), 7.20 (s, 1H, Ar–H), 6.99–6.91 (m, 2H, Ar–H), 4.24 (s, 1H, CH), 2.62–2.53 (br, 2H, CH2), 2.31–2.18 (br, 2H, CH2), 1.97–1.82 (m, 2H, CH2). 13C NMR (DMSO-d6), δC (ppm): 196.1 (C=O), 165.0, 159.2, 158.0, 157.1, 148.9, 147.3, 145.4, 130.2, 129.1, 127.8, 127.0, 126.1, 120.1, 114.2, 37.0, 35.9, 26.8, 20.1. HRMS (EI) m/z: calcd for C21H18N4O2S (M + Na) 413.1049, found 413.1047. Analysis calculated for C21H18N4O2S: C, 64.60; H, 4.65; N, 14.35%. Found: C, 64.57; H, 4.69; N, 14.28%.
Antiproliferative Activity Assay
Cell Lines
The cell lines MCF7, A549, HepG2 (tumorigenic cells), and MCF-10A (nontumorigenic cell) were obtained from ATCC via holding company for biological products and vaccines (VACSERA), Cairo, Egypt.
MTT Assay
The potential carcinogenic activities of the obtained molecules have been assessed using the MTT approach. Tumor cell lines were plated in 96 multiwell plates (104 cells/well) for 24 h before treatment with compounds to allow cell attachment to the wall of the plate. Then, several concentrations of each compound (0, 2.5, 5, 10, and 15 mg/mL) were added to the cell monolayer triplicate wells after preparation for each individual dose. The final concentration of dimethyl sulfoxide (DMSO) in each sample did not exceed 1% v/v. Dox. was employed as a standard antitumor drug for comparison. Monolayer cells were incubated with the compounds for 48 h at 37 °C in an atmosphere of 5% CO2. After 48 h, cells were fixed, washed, and stained with MTT stain. The excess stain was washed with acetic acid, and the attached stain was recovered with a tris-EDTA buffer. Cytotoxicity was evaluated as the concentration that caused approximately 50% loss of monolayer. The assay was utilized to examine the newly prepared derivatives.39
Molecular Docking
Ligand-Proteins docking has been studied in order to estimate the binding interactions between the designed pyrimidines with three selected proteins that have already been collected from the Protein Data Bank (PDB: 6ENV, 4b3z, and 2JW2) for (MCV-7) breast, (A549) lung, and (HepG2) liver cancer cell lines, respectively. The Molecular Operating Environment (MOE) version 2014.09 was used for this investigation.40 The two-dimensional (2D) structure of the ligands was generated by ChemDraw. MOE subsequently utilized its WASH module to add hydrogen atoms and partial electrical charges to the ligands, converting them into three dimensions (3D).41 The 3D structure of every molecule further diminished, until the gradient decreased to 0.1 RMS kcal/mol. Upon the generation of protein and ligand files, docking was carried out using the Triangle Matcher docking approach and the London G scoring function. Thirty docked conformations have been retained for each ligand, and the most optimized docked orientations were identified based on the docking score and binding interactions.
Density Functional Theory (DFT) Analysis
All Density Functional Theory (DFT) analyses are made utilizing B3LYP, which involves DFT exchange terms correlated with the gradient-corrected exchange-correlation functional of Lee, Yang, and Parr (LYP). It has fewer integration issues than the pure DFT methods. In this paper, therefore, B3LYP has been used to perform quantum calculations. Full geometry optimizations of all additives were carried out with the standard B3LYP/6- 311G++(d,p) basis set using Gaussian-09.31 LUMO–HOMO calculations and their other parameters, such as energy gap (ΔE), global electrophilicity (ω), softness (σ), electronegativity (χ), hardness (η), and ionization potential (I), were also calculated.32
Molecular Electrostatic Potential
Molecular electrostatic potential (MEP) that visualizes both yellow and blue regions to indicate the electrophilic and nucleophilic regions, respectively, was computed at B3LYP/6-31G on the basis of the optimized structure.34
Acknowledgments
The authors extend their thanks to the Central Laboratory at Jouf University, KSA, for performing the spectral analyses.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c07434.
1H, 13C NMR, and MS spectra of the synthesized compounds; HOMO, LUMO, and 2D and 3D docking of the assembled molecules (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Eicher T.; Hauptmann S.. The Chemistry of Heterocycles: Structures, Reactions. Synthesis, and Applications, 2nd ed.; Wiley-VCH: Weinheim, 2003. [Google Scholar]; b Pozharskii A. F.; Soldatenkov A. T.; Katritzky A. R.. Heterocycles in Life and Society; John Wiley & Sons, Ltd: West Sussex, 1997. [Google Scholar]
- a Taylor A. P.; Robinson R. P.; Fobian Y. M.; Blakemore D. C.; Jones L. H.; Fadeyi O. Modern advances in heterocyclic chemistry in drug discovery. Org. Biomol. Chem. 2016, 14, 6611–6637. 10.1039/C6OB00936K. [DOI] [PubMed] [Google Scholar]; b Kumar D.; Jain S. K. A Comprehensive Review of N-Heterocycles as Cytotoxic Agents. Curr. Med. Chem. 2016, 23, 4338–4394. 10.2174/0929867323666160809093930. [DOI] [PubMed] [Google Scholar]
- Pandey S. M.; Pandey J.; Saraswat K.; Kant R. A binuclear dihalodioxido molybdenum compounds for bio medicinal use. J. Indian Chem. Soc. 2022, 99, 100759 10.1016/j.jics.2022.100759. [DOI] [Google Scholar]
- Pandey J.; Jha A. K.; Hajela K. Synthesis and biological activities of some new dibenzopyranones and dibenzopyrans: search for potential estrogen receptor agonists and antagonists. Bioorg. Med. Chem. 2004, 12, 2239–2249. 10.1016/j.bmc.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Kate A.; Sahu L. K.; Pandey J.; Mishra M.; Sharma P. K. Green catalysis for chemical transformation: the need for the sustainable development. Curr. Res. Green Sustainable Chem. 2022, 5, 100248 10.1016/j.crgsc.2021.100248. [DOI] [Google Scholar]
- Ramandeep K.; Prabhkirat K.; Sahil S.; Gurpreet S.; Samir M.; Preet B. M. S.; Kunal N. Anti-Cancer Pyrimidines in Diverse Scaffolds: A Review of Patent Literature. Recent Pat. Anti-Cancer Drug Discovery 2015, 10, 23–71. 10.2174/1574892809666140917104502. [DOI] [PubMed] [Google Scholar]
- Albratty M.; Alhazmi H. A. Novel pyridine and pyrimidine derivatives as promising anticancer agents: A review. Arab. J. Chem. 2022, 15, 103846 10.1016/j.arabjc.2022.103846. [DOI] [Google Scholar]
- Curini M.; Cravotto G.; Epifano F.; Giannone G. Chemistry and Biological Activity of Natural and Synthetic Prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. 10.2174/092986706775197890. [DOI] [PubMed] [Google Scholar]
- O’Kennedy P.; Thornes R. D.. Coumarins: Biology, Applications and Mode of Action; J. Wiley & Sons, Chichester, U.K, 1997. [Google Scholar]
- Tangmouo J. G.; Meli A. L.; Komguem J.; Kuete V.; Ngounou F. N.; Lontsi D.; Beng V. P.; Choudhary M. I.; Sondengam B. L. Crassiflorone, a new naphthoquinone from Diospyros crassiflora (Hien). Tetrahedron Lett. 2006, 47, 3067–3070. 10.1016/j.tetlet.2006.03.006. [DOI] [Google Scholar]
- Kitamura R. O.; Romoff S. P.; Young M. C. M.; Kato M. J.; Lago J. H. G. Chromenes from Peperomia serpens (Sw.) Loudon (Piperaceae). Phytochemistry 2006, 67, 2398–2402. 10.1016/j.phytochem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Abdelrazek F. M.; Metz P.; Kataeva O.; Jäger A.; El-Mahrouky S. F. Synthesis and Molluscidial Activity of New Chromene and Pyrano[2,3-c]pyrazole Derivatives. Arch. Pharm. 2007, 340, 543–548. 10.1002/ardp.200700157. [DOI] [PubMed] [Google Scholar]
- Singh K.; Singh J.; Singh H. A synthetic entry into fused pyran derivatives through carbon transfer reactions of 1,3-oxazinanes and oxazolidines with carbon nucleophiles. Tetrahedron 1996, 52, 14273–14280. 10.1016/0040-4020(96)00879-4. [DOI] [Google Scholar]
- Borges F.; Roleira F.; Milhazes N.; Santana L.; Uriarte E. Simple coumarins and analogues in medicinal chemistry: occurrence, synthesis and biological activity. Curr. Med. Chem. 2005, 12, 887–916. 10.2174/0929867053507315. [DOI] [PubMed] [Google Scholar]
- Andrianasolo E. H.; Haramaty L.; Rosario-Passapera R.; Vetriani C.; Falkowski P.; White E.; Lutz R. Hydroxyethylamine Chromene Derivatives from a Cultured Marine Hydrothermal Vent Bacterium, Thermovibrio ammonificans. Mar. Drugs 2012, 10, 2300–2311. 10.3390/md10102300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curini M.; Cravotto G.; Epifano F.; Giannone G. Chemistry and biological activity of natural and synthetic prenyloxycoumarins. Curr. Med. Chem. 2006, 13, 199–222. 10.2174/092986706775197890. [DOI] [PubMed] [Google Scholar]
- Bhosle M. R.; Wahul D. B.; Bondle G. M.; Sarkate A.; Tiwari S. V. An efficient multicomponent synthesis and in vitro anticancer activity of dihydropyranochromene and chromenopyrimidine-2,5-diones. Synth. Commun. 2018, 48, 2046–2060. 10.1080/00397911.2018.1480042. [DOI] [Google Scholar]
- El-Agrody A. M.; Fouda A. M.; Al-Dies A. A. M. Studies on the Synthesis in Vitro Antitumor Activity of 4H-Benzo[h]Chromene, 7H-Benzo[h]Chromene[2,3-d]Pyrimidine Derivatives and Structure-Activity Relationships of the 2,3- and 2,3-Positions. Med. Chem. Res. 2014, 23, 3187–3199. 10.1007/s00044-013-0904-x. [DOI] [Google Scholar]
- El-Agrody A. M.; Halawa A. H.; Fouda A. M.; Al-Anood M. A. D. The anti-Proliferative Activity of Novel 4H-Benzo[h]Chromenes, 7H-Benzo[h]-Chromeno[2,3-d]Pyrimidines and the Structure–Activity Relationships of the 2-, 3-Positions and Fused Rings at the 2, 3-Positions. J. Saudi Chem. Soc. 2017, 21, 82–90. 10.1016/j.jscs.2016.03.002. [DOI] [Google Scholar]
- Malviya J.; Singh R. K. P. One- pot three-component synthesis of chromeno[2,3-d]pyrimidine derivatives: Novel, simple, and efficient electrochemical approach. J. Heterocycl. Chem. 2020, 57, 39–49. 10.1002/jhet.3741. [DOI] [Google Scholar]
- Jadidi K.; Ghahremanzadeh R.; Bazgir A. Efficient Synthesis of Spiro[chromeno[2,3-d]pyrimidine-5,3′indoline]-tetraones by a One-Pot and Three-Component Reaction. J. Comb. Chem. 2009, 11, 341–344. 10.1021/cc800167h. [DOI] [PubMed] [Google Scholar]
- Andrade C. B. V.; Lopes L. V. A.; Ortiga-Carvalho T. M.; Matthews S. G.; Bloise E. Infection and disruption of placental multidrug resistance (MDR) transporters: Implications for fetal drug exposure. Toxicol. Appl. Pharmacol. 2023, 459, 116344 10.1016/j.taap.2022.116344. [DOI] [PubMed] [Google Scholar]
- Dömling A. Recent advances in isocyanide-based multicomponent chemistry. Curr. Opin. Chem. Biol. 2002, 6, 303–313. 10.1016/S1367-5931(02)00328-9. [DOI] [PubMed] [Google Scholar]
- Dandia A.; Arya K.; Khaturia S.; Yadav P. Facile one pot microwave enhanced multistep synthesis of novel biologically important scaffold spiro[indole-pyridopyrimidines]. Arkivoc 2005, 2005 (xiii), 80–88. 10.3998/ark.5550190.0006.d07. [DOI] [Google Scholar]
- Metwally N. H.; Abdelrazek F. M. Reaction of Anthranilonitrile with Some Active Methylene Reagents: Synthesis of Some New Quinoline and Quinazoline Derivatives. Synth. Commun. 2005, 35, 2481–2487. 10.1080/00397910500192068. [DOI] [Google Scholar]
- Litvinov Y. M.; Shestopalov A. M. Convenient selective synthesis of pyrano[2,3-d]pyrimidines. Russ. Chem. Bull. 2008, 57, 2223–2226. 10.1007/s11172-008-0308-0. [DOI] [Google Scholar]
- Beyzaei H.; Aryan R.; Moghaddam-Manesh M.; Ghasemi B.; Karimi P.; Delarami H. S.; Sanchooli M. Evaluation and structure-activity relationship analysis of a new series of 4-imino-5H-pyrazolo[3,4-d]pyrimidin-5-amines as potential antibacterial agents. J. Mol. Struct. 2017, 1144, 273–279. 10.1016/j.molstruc.2017.05.050. [DOI] [Google Scholar]
- WHO Cancer Reports, 2020https://apps.who.int/iris/rest/bitstreams/1267643/retrieve.
- Stolarczyk M.; Wolska A.; Mikołajczyk A.; Bryndal I.; Cieplik J.; Lis T.; Matera-Witkiewicz A. A New Pyrimidine Schiff Base with Selective Activities against Enterococcus faecalis and Gastric Adenocarcinoma. Molecules 2021, 26, 2296 10.3390/molecules26082296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y. D. L.; Xing Z.; Xu Y.; Yuhong W. Synthesis and Anticancer Activity of Mono-Carbonyl Analogues of Curcumin. Sci. Res. 2013, 4, 113–123. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian; Gaussian, Inc.: Wallingford CT, 2016.
- Arafa W. A. A.; Hussein M. F. Design, Sonosynthesis, Quantum-Chemical Calculations, and Evaluation of New Mono- and Bis-pyridine Dicarbonitriles as Antiproliferative Agents. Chin. J. Chem. 2020, 38, 501–508. 10.1002/cjoc.201900494. [DOI] [Google Scholar]
- Kökbudak Z.; Saracoglu M.; Akkoç S.; Çimen Z.; Yilmazer M. I.; Kandemirli F. Synthesis, cytotoxic activity and quantum chemical calculations of new 7-thioxopyrazolo[1,5-f]pyrimidin-2-one derivatives. J. Mol. Struct. 2020, 1202, 127261 10.1016/j.molstruc.2019.127261. [DOI] [Google Scholar]
- Wu S.; Zhang W.; Qi L.; Ren Y.; Ma H. Investigation on 4-amino-5-substituent-1,2,4-triazole-3-thione Schiff bases an antifungal drug by characterization (spectroscopic, XRD), biological activities, molecular docking studies and electrostatic potential (ESP). J. Mol. Struct. 2019, 1197, 171–182. 10.1016/j.molstruc.2019.07.013. [DOI] [Google Scholar]
- Monti D. M.; Ferraro G.; Petruk G.; Maiore L.; Pane F.; Amoresano A.; Cinellu M. A.; Merlino A. Ferritin nanocages loaded with gold ions induce oxidative stress and apoptosis in MCF-7 human breast cancer cells. Dalton Trans. 2017, 46, 15354–15362. 10.1039/C7DT02370G. [DOI] [PubMed] [Google Scholar]
- Pan S.-H.; Chao Y.-C.; Chen H.-Y.; Hung P.-F.; Lin P.-Y.; Lin C.-W.; Chang Y.-L.; Wu C.-T.; Lee Y.-C.; Yang S.-C.; Hong T.-M.; Yang P.-C. Long form collapsin response mediator protein-1 (LCRMP-1) expression is associated with clinical outcome and lymph node metastasis in non-small cell lung cancer patients. Lung Cancer 2010, 67, 93–100. 10.1016/j.lungcan.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Nakamura F.; Ohshima T.; Goshima Y. Collapsin Response Mediator Proteins: Their Biological Functions and Pathophysiology in Neuronal Development and Regeneration. Front. Cell. Neurosci. 2020, 14, 188 10.3389/fncel.2020.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zülbiye K.; Senem A.; Halis K.; Burak T.; Güzin A. In Silico and In Vitro Antiproliferative Activity Assessment of New Schiff Bases. ChemistrySelect 2022, 7, e202103679 10.1002/slct.202103679. [DOI] [Google Scholar]
- Aljuhr S. A.; Abdelaziz G.; Selim A. A.; Zaghary W. A.; Sakr T. M. Anti-cancer, anti-inflammatory and antioxidant effects of Vit-A/CSeNPs in mutual diethylnitrosamine and carbon tetrachloride induced hepatocellular damage in albino rats. J. Drug Delivery Sci. Technol. 2022, 75, 103723 10.1016/j.jddst.2022.103723. [DOI] [Google Scholar]
- Swain B.; Singh P.; Angeli A.; Aashritha K.; Nagesh N.; Supuran C. T.; Arifuddin M. 3-Functionalised benzenesulphonamide based 1,3,4-oxadiazoles as selective carbonic anhydrase XIII inhibitors: Design, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2021, 37, 127856 10.1016/j.bmcl.2021.127856. [DOI] [PubMed] [Google Scholar]
- Manjunatha K.; Poojary B.; Lobo P. L.; Fernandes J.; Kumari N. S. Synthesis and biological evaluation of some 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5225–5233. 10.1016/j.ejmech.2010.08.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.