

Abstract
The combination of ArB(OH)2 transmetallation with cationic gold(I) [LAu]+ and electrochemical anodic oxidation (EAO) approach was successfully developed for the preparation of AuIII-Ar intermediate for the first time. This in-situ generated aryl gold intermediate gave rapid and controllable transmetallation with ArB(OH)2 or alkyne followed by reductive elimination to generate either di-aryl coupling or sp2-sp Sonogashira-type coupling products under mild conditions with no need of external oxidants, which significantly extended the versatility of electrochemical approach in promoting gold redox catalysis.
Keywords: Electrochemistry, Gold redox catalysis, Oxidative coupling, Arylation
Graphical Abstract
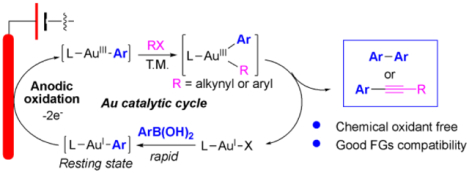
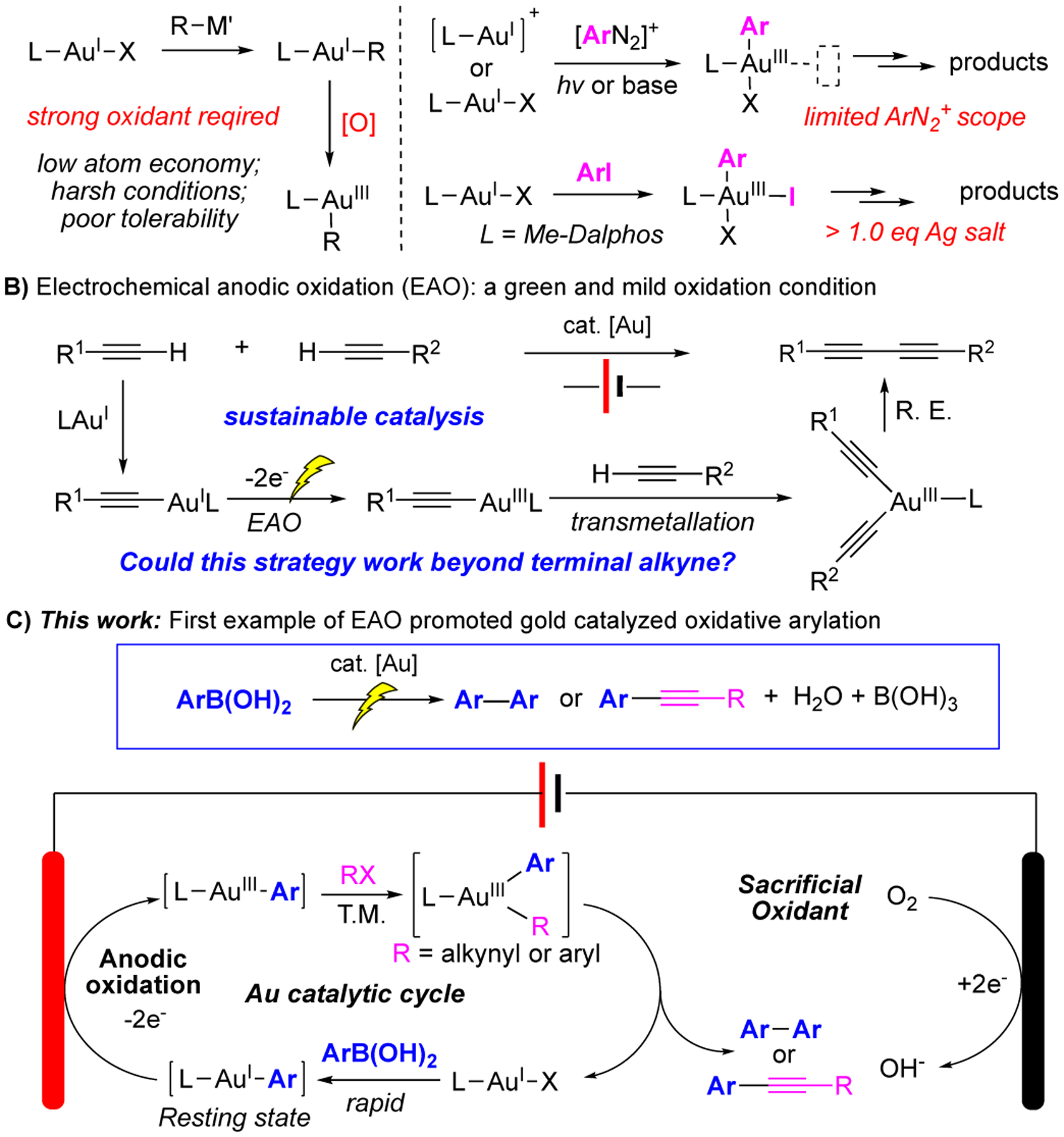
Gold redox catalysis has attracted much attention during the past decades owing to the unique reactivity of gold as both a strong π-acid for C-C multiple bond activation and rapid reductive elimination for C-C or C-X bond formation [1]. While offering the versatile reactivity and high efficiency, gold redox catalysis suffered from one major obstacle: how to achieve effective AuI oxidation conditions while compatible with general reaction substrates. The high redox potential between AuI and AuIII (1.4 eV) makes this process challenging since typical strong “oxidative environment” is likely needed to achieve this process [2]. As shown in Scheme 1A, a typical approach to achieve AuI oxidation is the application of strong oxidants like Selectfluor or PhI(OAc)2 etc. [3]. The need of stoichiometric amount of highly reactive reagents resulted in low atom economy and poor substrate tolerability. To overcome this problem, alternative oxidation methods have been developed with the focus on both milder oxidation condition and alternative substrates that could produce AuIII-C intermediates to improve the overall atom economy [4]. Two representative examples that have been reported are (A) photoactivation of diazonium to achieve AuIII-Ar [5]; (B) ligand promoted and silver assisted aryl-iodide oxidative addition to L-AuI-X. Both methods successfully achieved the goal to have the oxidants (diazonium or aryl iodide) serve as part of the reactants, which therefore improved the overall atom economy. However, they still suffer from major limitations.
Scheme 1.
Gold redox catalysis with electrochemical oxidation.
For the photocatalytic activation of diazonium approach, the overall reaction performance is highly depending on the reaction conditions due to the formation of radical intermediates [6]. Typical oxygen-free and water-free conditions are needed to ensure optimal reactivity. To overcome this problem, our group developed the base assisted diazonium activation at a slightly elevated temperature (50 °C) [7]. This optimization allowed reaction to proceed under more practical and robust conditions with no need for exhausted gassing and further extend the reaction scopes to substrate that are sensitive to photo-catalysis. However, substituted groups greatly influenced the reaction, where EDG modified diazonium did not work due to a low oxidation potential [8].
Please provide all schemes or figures drew in software of Chemdraw for modifying easily.
Recently, Bourissou and co-workers reported a ligand assisted oxidative addition of gold(I) to aryl iodide under mild conditions [9]. The key for the success of this method is the application of P-N bidentate ligands (Me-Dalphos), which help to promote aryl iodide oxidative addition and stabilize the resulting AuIII intermediate. With this method, electron rich aryl precursors were successfully applied as valid coupling partners via gold redox catalysis [10]. However, the major drawback of this method is the requirement of stoichiometric amount of silver salts, which greatly reduced the overall atom economy and limited the reaction utility.
Synthetic organic electrochemistry has witnessed tremendous growth during the past decade [11]. As a sustainable and environmentally friendly oxidation strategy, electrochemical anodic oxidation (EAO) has been applied to achieve challenging oxidation of various transition metal mediated catalysis. Recently, our group reported the first example of EAO promoted gold redox catalysis with the application of terminal alkyne coupling for the synthesis of conjugated 1,3-diyne (Scheme 1B) [12]. The major concern of conducting gold redox chemistry under electrochemical conditions is the rapid cation reduction (formation of Au0) on cathode. In that study, practical conditions were developed through the rapid formation of gold acetylide (Au-alkyne) to prevent gold(I) from reduction. As a result, effective alkyne homo- and cross-couplings were achieved with no external oxidants. One intriguing question is whether this EAO-gold catalysis could be applied into other functional groups beyond terminal alkyne. Herein, we report the successful development of EAO gold redox catalysis in arylation coupling reactions (Scheme 1C). Based on mechanistic investigations, optimal conditions are developed for rapid boronic acid transmetallation. Oxygen gas was identified as the necessary sacrificial oxidants, which was getting reduced on cathode. Both diaryl and aryl-alkyne oxidative cross coupling were achieved with a large group of substrates and good overall yields, which highlighted the mild conditions and high efficiency of this new oxidative coupling strategy.
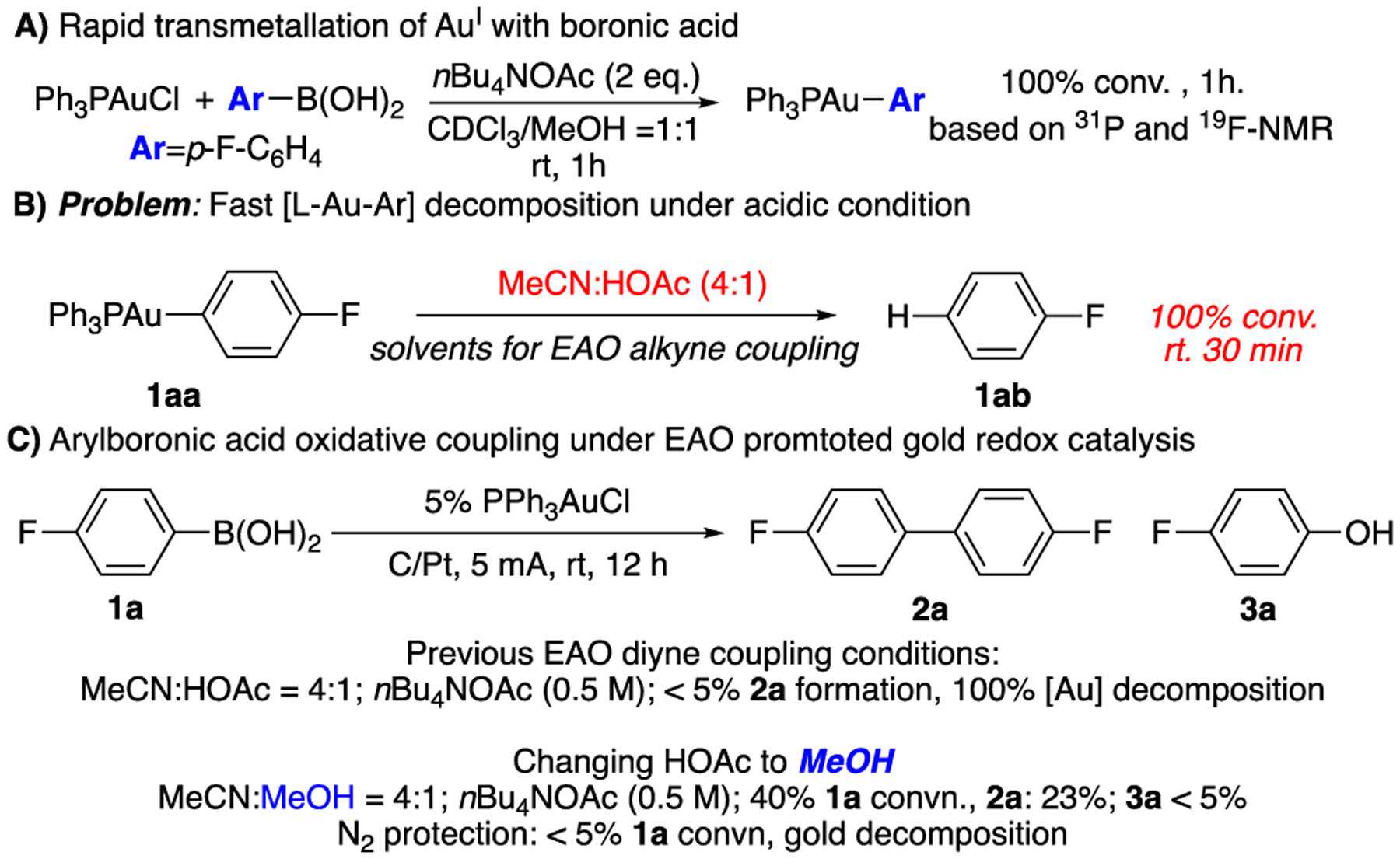
As reported in our previous diyne coupling, one of the crucial factors to achieve EAO gold redox catalysis is the application of protic solvents (1:1 mixture of MeCN and HOAc). Clearly, the main concern for electrochemically promoted gold catalysis is quenching of gold cation upon cathode reduction. Increase the reaction mixture acidity helps to reduce gold decomposition with proton oxidation (H2 formation). However, this strategy will NOT work for the di-aryl oxidative coupling. First, aryl C-H is significantly less reactive than terminal alkyne C-H and leads to the slow formation of Au-Ar. As a result, the gold cations will be “exposed” to the E-chem reaction environment, causing rapid decomposition (reduction). To avoid this problem, aryl-boronic acids derivatives, ArB(OH)2 is likely the suitable source with the expectation of rapid transmetallation to form [AuI-Ar] intermediate as the key gold resting state in the catalytic cycle. To evaluate this idea, reaction between PPh3AuCl and p-F-phenylboronic acid 1a was conducted. The reaction was monitored using 31P, 19F and 1H NMR (details in Supporting informaiton). According to NMR, [PPh3-Au-Ar] 1aa was formed within 1 h at room temperature as shown in Fig. 1A.
Fig. 1.
EAO promoted oxidative diaryl coupling.
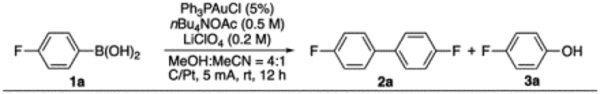
This result is encouraging since it confirmed that boronic acid could give rapid transmetallation with gold cation and form [L-Au-Ar]+ as the potential resting state for sequential redox coupling as we expected. However, treating gold complex 1aa under previous optimal EAO condition (diyne coupling) gave only fluorobenzene 1ab (from protodeauration) with no diaryl 2a observed. Monitoring the reaction using NMR confirmed the rapid aryl-gold decomposition under acidic conditions (Fig. 1B). Similar reactivity was obtained under EAO condition, where low conversion of 1a was obtained associated with rapid gold decomposition (reduction on cathode, Fig. 1C). To avoid protodeauration, we screened various alternative solvents. Finally, mixture of MeOH and MeCN were identified as plausible solution, giving the desired coupling product 2a in 23% yield with 40% 1a conversion. Notably, in this reaction, molecular O2 was suggested as the sacrificial oxidants. Conducting the reaction under oxygen-free conditions gave no coupling product 2a with gold decomposition over time. The use of molecular oxygen O2 as the oxidant makes this transformation more attractive and practical. Encouraged by these initial results, we conducted a comprehensive condition screening (details in Supporting information). To our delight, with the combination of MeOH and MeCN (4:1) as solvents, nBu4NOAc (0.5 M) and LiClO4 (0.2 mol/L) as electrolyte, under 5 mA constant current (initial potential 2.5 V), the desired diaryl coupling product 2a was obtained in 78% isolated yields. Some alternative conditions are summarized in Table 1.
Table 1.
| |||
|---|---|---|---|
| Entry | Variation from “standard conditions” | Yield (%)b | |
| 2a | 3a | ||
| 1 | none | 78 | <5 |
| 2 | No LiClO4 | 23 | 10 |
| 3 | 0.2 mol/L nBu4NOAc instead | 36 | 16 |
| 4 | 0.5 mol/L LiClO4 instead | trace | <5 |
| 5 | 0.5 mol/L LiOAc 2H2O as only electrolyte | 36 | <5 |
| 6 | 5 mL MeOH, no MeCN | 15 | 10 |
| 7 | MeOH:MeCN = 1:4 | 23 | 8 |
| 8 | Under Ar | trace | 0 |
| 9 | Under O2 | 68 | 8 |
| 10 | “water-free” operation | 38 | <5 |
| 11 | No [Au] | 0 | <5 |
Conditions: 1a (0.5 mmol), Ph3PAuCl (0.025 mmol), nBu4NOAc (2.5 mmol), LiClO4 (1 mmol), MeOH (4 mL), MeCN (1 mL), air, r.t., 12 h.
1H NMR yields using 1,3,5-trimethoxybenzene as an internal standard.
Notably, although the yield of 2a is not excellent, under the optimal condition, the only byproduct observed is the phenol 3a, which highlighted the mild conditions of this electrochemical process. Conducting reaction without LiClO4 significantly reduced the reaction rate, giving 2a at only 23% yield along with 10% phenol 3a (entry 2). Reducing the loading of nBu4NOAc caused slower reaction with increasing amount of phenol formation (entry 3). Interestingly, further increasing LiClO4 loading to 0.5 mol/L quenched the reaction, suggesting the needed balance for conductivity and overall catalyst reactivity (entry 4). Switching electrolyte to LiOAc also led to the yield decreasing of 2a (entry 5), suggesting the critical balanced ratio between Li+ and OAc− for optimal performance. The ratio of MeOH and MeCN could greatly impact the reaction due to the reduced solubility of substrates (entry 6,7). As expected, conducting the reaction under argon gave rapid gold catalyst decomposition, which is consistent with the proposed molecular oxygen oxidation on cathode for this transformation (entry 8, see Supporting information for detailed study on the “oxygen effect”). Interestingly, conducting the reaction in anhydrous condition gave poor yield, suggesting that presence of water could help this reaction, likely due to improved solubility and conductivity (entry 9). Finally, the control experiment confirmed that gold is necessary for this transformation (entry 10).
With the optimal conditions in hand, we explore the reaction scope. The results are summarized in Scheme 2 (Scheme is more suitable here).
Scheme 2.
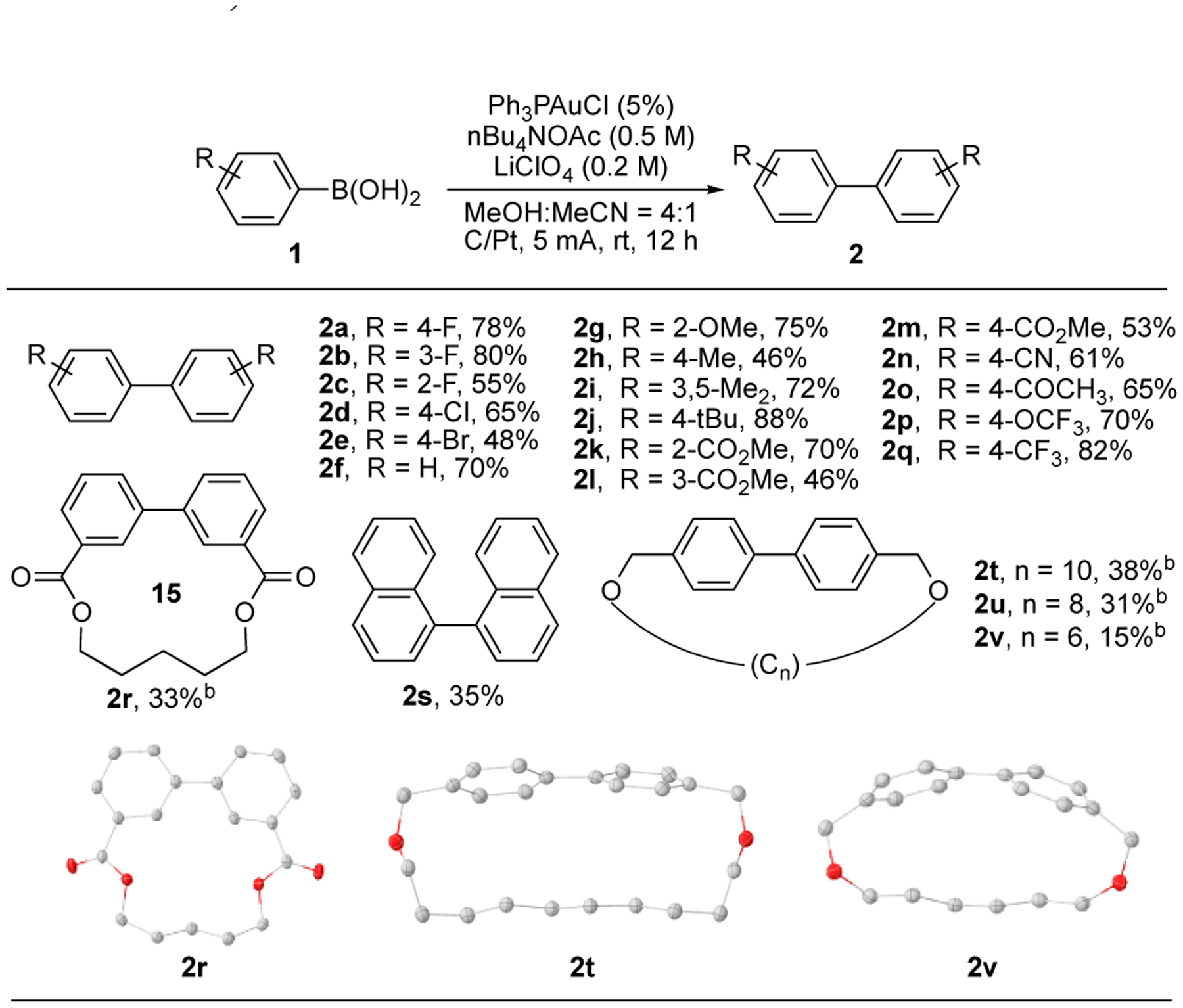
Substrate scope of aryl homo-coupling. Conditions: 1 (0.5 mmol), Ph3PAuCl (0.025 mmol), nBu4NOAc (2.5 mmol), LiClO4 (1 mmol), MeOH (4 mL), MeCN (1 mL), r.t., 12 h, Isolated yield.
b Reaction was performed at a lower concentration of 1 (0.05 mmol), Ph3PAuCl (0.005 mmol), 4 h.
Various aryl boronic acids homo-coupling products were achieved under this electrochemical condition. Both EWG and EDG modified arenes worked well, giving corresponding product with moderate to good yields (2a-2q). The ortho substituted arene performed well, suggesting the good tolerability of steric hinderance with this EAO promoted gold redox catalysis (2c, 2g, 2k). Interestingly, intramolecular cyclization was successfully achieved using this method, forming macrocycles, though at modest yields (2r, 2t-2v). It is important to note that the macrocyclization requires low concentration to prevent undesired polymerization. The success in achieving this macrocyclization highlighted the good efficiency of this electrochemical promoted gold redox chemistry under mild conditions.
Comparing with Pd chemistry, the gold catalyzed oxidative coupling has a different mechanism with metal oxidation as turnover limiting step (TLS) and a much faster reductive elimination step (AuIII vs. PdII). Therefore, the identification of electrochemical oxidation strategy could be potentially applied into other coupling partners. One immediate question is whether this strategy could be applied into the cross-coupling for the synthesis of hetereo-diaryl compounds. In this regard, the electron density of aryl group and substituents on boronic ester are clear the two important factors that will influence the overall reactivity and selectivity. Treating 1:1 mixture of 1j and 1m under the optimal conditions gave the homocoupling product 2j as the dominant product (60% yield) with only trace amount cross-coupling product observed (Fig. 2A). This result suggested that the electron rich aryl boronic acid not only helps the transmetalation, but also gives faster AuI-Ar oxidation comparing with EWG modified substrate. While tunning aryl electronic density is not a valid approach for cross-coupling, we put our attention to different boron derivatives. As shown in Fig. 2B, monitoring the reaction with NMR confirmed that ArB(OH)2 gave faster transmetalation than ArBPin. In addition, significantly lower conversion was observed with ArBF3K and no L-Au-Ar formation was observed with Ar-B(MIDA). These results revealed the different transmetalation rate with various boronic acid derivatives. Reaction of p-tBuC6H4BF3K and L-Au-Ar gave 18% cross coupling product, while no homocoupling product 2a was observed (Fig. 2C). This result suggested that ArBF3K could be oxidized prior to AuI-Ar. However, the ArBF3K oxidation is slow, which led to gold decomposition over time. Notably, although the diaryl cross-coupling seems not practical using this EAO catalysis, this study provides the important mechanistic insight for future development of coupling reactions under gold-redox catalysis, which is currently under investigation in our lab.
Fig. 2.
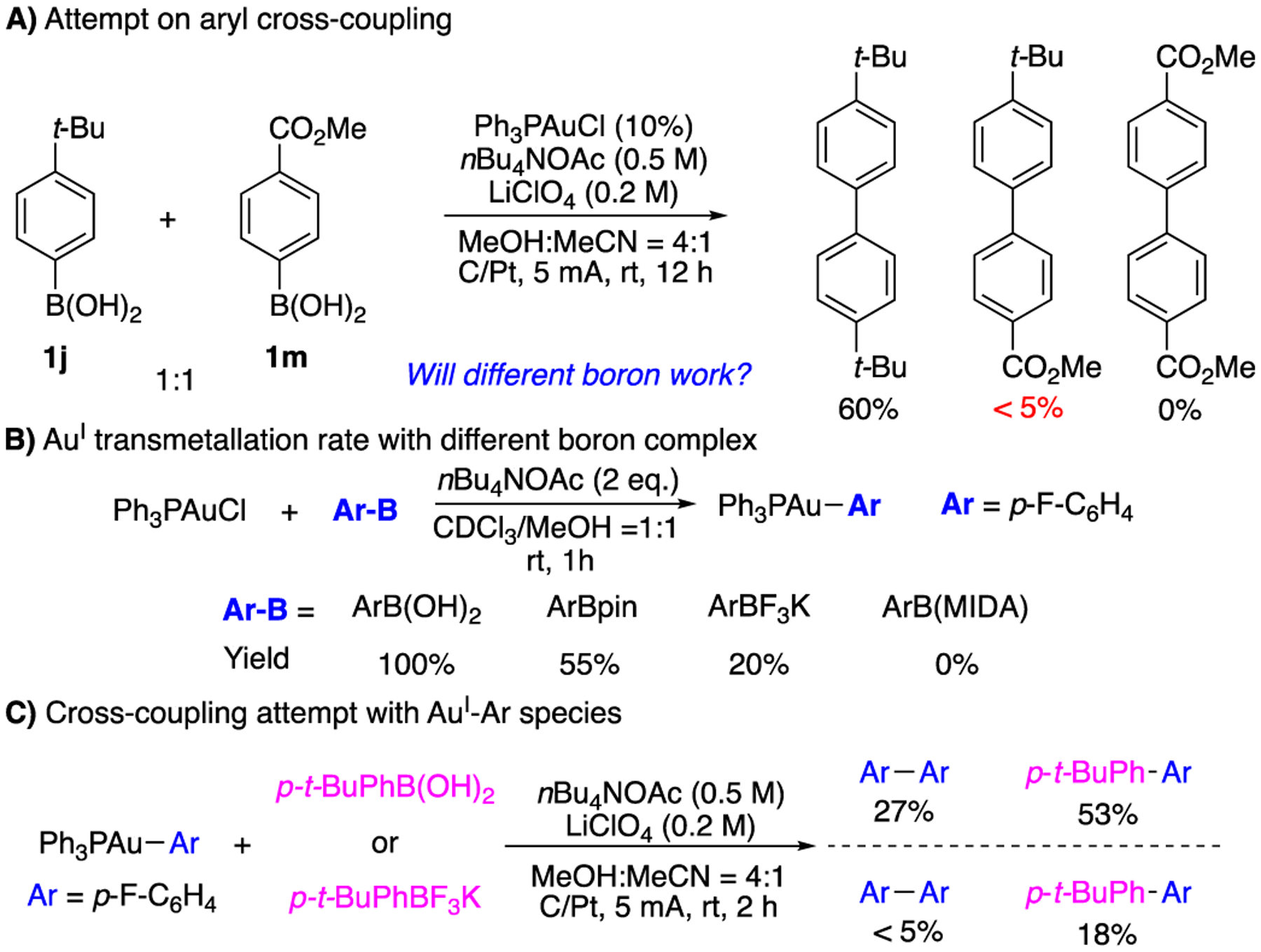
Investigating gold catalyzed oxidative aryl cross-coupling.
While diaryl cross-coupling was limited by the reaction scope, it is plausible to achieve coupling with alternative coupling partner such as the terminal alkynes. As shown in Fig. 3A, both ArB(OH)2 and terminal alkyne could react with LAuCl to form corresponding L-Au-Ar and L-Au-Alkyne within 1 h. NMR investigations revealed that L-Au-Ar is favored over alkyne in a 2:1 ratio. Charging the 1:1 mixture of ArB(OH)2 1a and alkyne 4a under EAO conditions gave the desired aryl-alkyne coupling product 5a in 32%, along with diaryl product 2a (51%) and phenol 3a (10%). Notably, no diyne 6 was observed. This result suggested that (A) L-Au-Ar was easier to be oxidized over L-Au-alkyne under this EAO condition and (B) L-Au-alkyne gave faster transmetalation to [AuIII-Ar], which facilitated the desired aryl-alkyne cross-coupling.
Fig. 3.
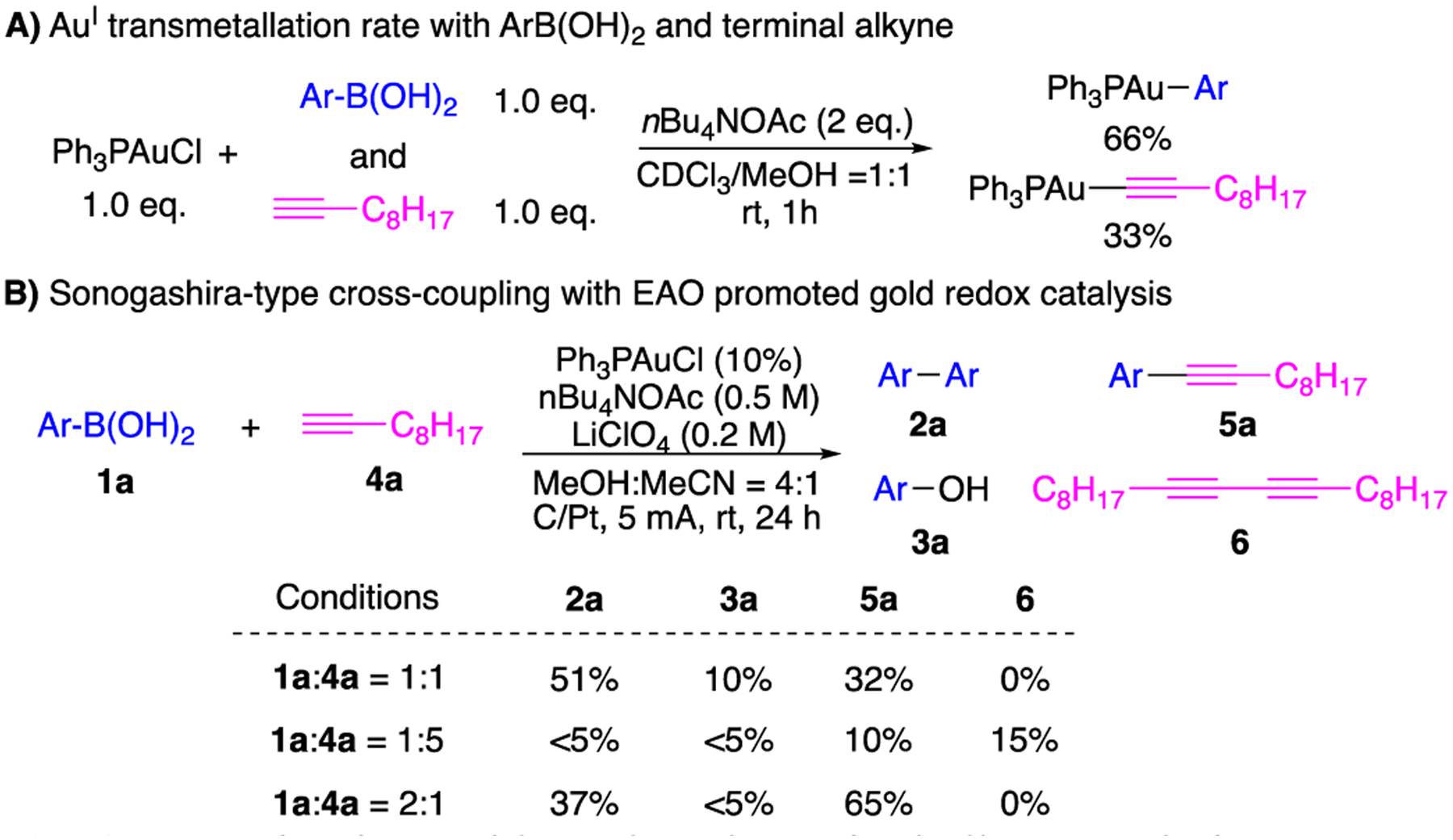
Investigating gold catalyzed terminal alkyne arylation.
Technically, the yields of cross coupling product 5a could be further improved with the addition of excess alkyne. However, with the application of 5 equiv. 4a, reduced yields of all arylation products (2a, 3a, and 5a) were observed, suggesting the importance of L-Au-Ar formation (with certain concentration) for effective transformation. Based on this mechanistic insight, increasing amount of aryl boronic acid 4a (2 equiv.) was applied, which improved the cross-coupling product 5a to 65% yield (detailed screening conditions in Supporting information). With the optimal conditions in hand, reaction scope was evaluated of this oxidative alkyne arylation. The result is summarized in Scheme 3
Scheme 3.
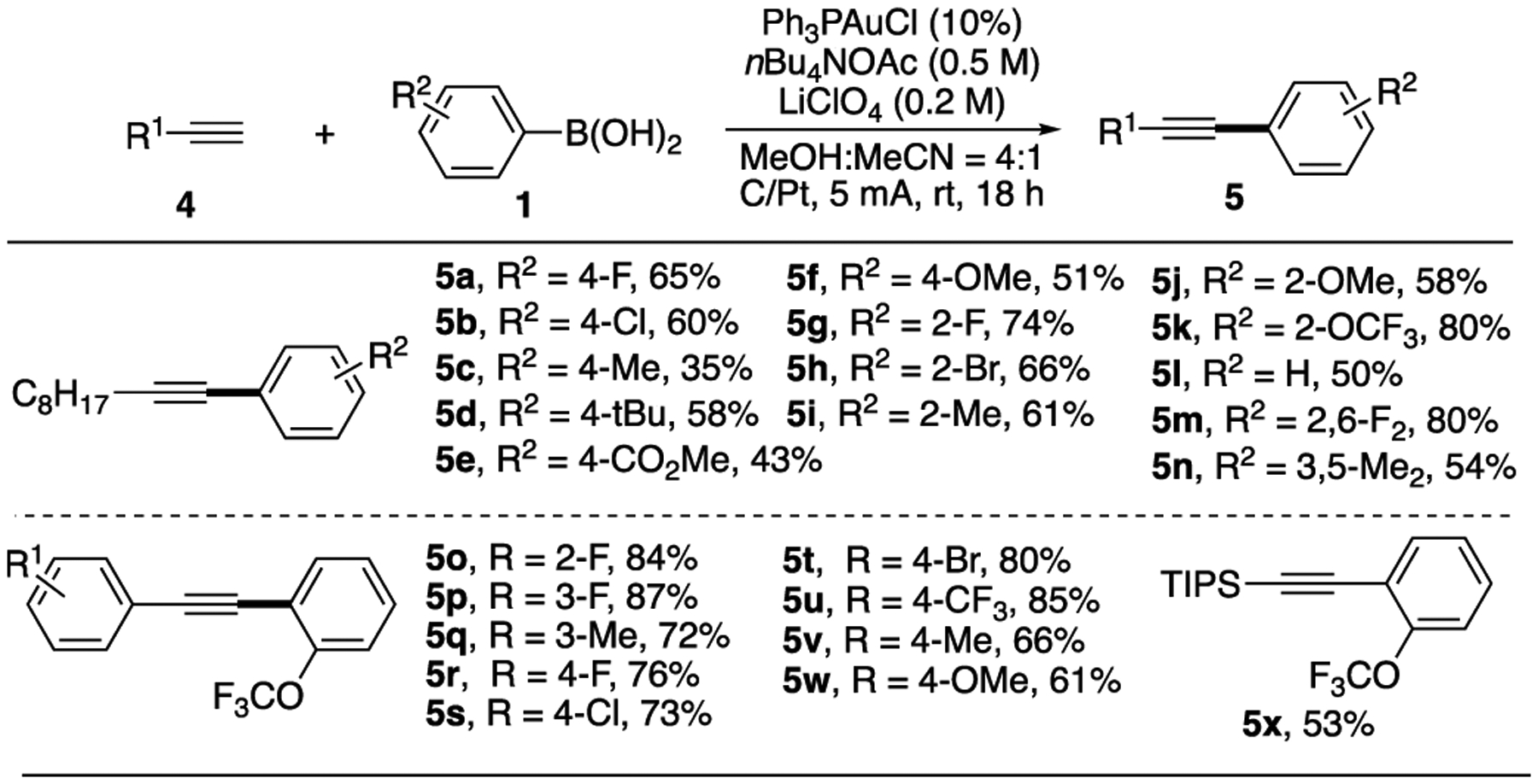
Substrate scope of Sonogashira-type coupling. Conditions: 1 (0.5 mmol), 4 (0.25 mmol), Ph3PAuCl (0.025 mmol), nBu4NOAc (2.5 mmol), LiClO4 (1 mmol), MeOH (4 mL), MeCN (1 mL), r.t., 18 h. isolated yield.
In the reaction with 1-decyne 4a, various EDGs modified arene works well under this EAO condition, giving the desired coupling products in good yields (5a-5n). Electron deficient aryl substrates gave significantly reduced yields (such as 5e), suggesting slow transmetallation with gold cation as discussed above. Interestingly, ortho substituted substrates performed better than the para substituted ones (5g-5k, 5m), implying the influence of steric hindrance on the transmetalation step. Various terminal alkynes were all suitable for this reaction, giving the coupling products in good yields (5o-5w). TIPS alkyne also tolerated in this reaction (5x), which not only highlighted the mild conditions of this EAO strategy, but also provide a practical synthesis of terminal alkyne through simple TIPS deprotection.
In conclusion, we reported the first example of the preparation and utilization of AuIII-Ar intermediate under electrochemical anodic oxidation conditions. Both di-aryl homocoupling and Sonogashira-type sp2-sp coupling were achieved under this EAO promoted gold catalysis. Besides the value in practical synthesis (mild conditions and no external oxidants), the detailed mechanistic studies revealed key reactivity of gold complexes under electrochemistry conditions (rate of transmetallation and catalyst resting state). These mechanistic insights set up the solid foundation for extending this EAO-gold catalysis system into other challenging transformations, which is currently under investigation in our lab.
Supplementary Material
Acknowledgments
We are grateful to the National Science Foundation (NSF) (No. CHE-1665122) and National Institutes of Health (NIH) (1R01GM120240–01) for financial support. Full name
References
- [1].(a) Huang BR, Hu MY, Toste FD, Trends Chem 2 (2020) 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kar A, Mangu N, Kaiser HM, Tse MK, J. Organomet. Chem 694 (2009) 524–537. [Google Scholar]; (c) Kramer S, Synthesis 52 (2020) 2017–2030. [Google Scholar]; (d) Nijamudheen A, Datta A, Chem. Eur. J 26 (2020) 1442–1487. [DOI] [PubMed] [Google Scholar]
- [2].(a) Bratsch SG, J. Phys. Chem. Ref. Data 18 (1989) 1–21. [Google Scholar]; (b) Joost M, Amgoune A, Bourissou D, Angew. Chem. Int. Ed 54 (2015) 15022–15045. [DOI] [PubMed] [Google Scholar]
- [3].(a) Miró J, del Pozo C, Chem. Rev 116 (2016) 11924–11966. [DOI] [PubMed] [Google Scholar]; (b) Fricke C, Reid WB, Schoenebeck F, Eur. J. Org. Chem (2020) 10.1002/ejoc.202000856. [DOI] [Google Scholar]; (c) Kramer S, Chem. Eur. J 22 (2016) 15584–15598. [DOI] [PubMed] [Google Scholar]; (d) Zhang G, Peng Y, Cui L, Zhang L, Angew. Chem. Int. Ed 48 (2009) 3112–3115. [DOI] [PubMed] [Google Scholar]; (e) de Haro T, Nevado C, J. Am. Chem. Soc 132 (2010) 1512–1513. [DOI] [PubMed] [Google Scholar]; (f) Ball LT, Lloyd-Jones GC, Russell CA, J. Am. Chem. Soc 136 (2014) 254–264. [DOI] [PubMed] [Google Scholar]; (g) Tkatchouk E, Mankad NP, Benitez D, Goddard WA, Toste FD, J. Am. Chem. Soc 133 (2011) 14293–14300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Peng HH, Xi YM, Ronaghi N, Dong BL, Akhmedov NG, Shi XD, J. Am. Chem. Soc 136 (2014) 13174–13177. [DOI] [PubMed] [Google Scholar]; (i) Ye XH, Peng HH, Wei CY, Yuan T, Wojtas L, Shi XD, Chem 4 (2018) 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Liu K, Li N, Ning Y, Zhu C, Xie J, Chem 5 (2019) 2718–2730. [Google Scholar]
- [4].(a) Akram MO, Banerjee S, Saswade SS, Bedi V, Patil NT, Chem. Commun 54 (2018) 11069–11083. [DOI] [PubMed] [Google Scholar]; (b) Levin MD, Toste FD, Angew. Chem. Int. Ed 53 (2014) 6211–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang J, Zhang S, Xu C, Wojtas L, Akhmedov NG, Chen H, Shi X, Angew. Chem. Int. Ed 57 (2018) 6915–6920. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang J, Wei C, Li X, Zhao P, Shan C, Wojtas L, Chen H, Shi X, Chem. Eur. J 26 (2020) 5946–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].(a) Hopkinson MN, Tlahuext-Aca A, Glorius F, Acc. Chem. Res 49 (2016) 2261–2272. [DOI] [PubMed] [Google Scholar]; (b) Medina-Mercado I, Porcel S, Chem. Eur. J (2020) 10.1002/chem.202000884, [DOI] [PubMed] [Google Scholar]; (c) Sahoo B, Hopkinson MN, Glorius F, J. Am. Chem. Soc 135 (2013) 5505–5508. [DOI] [PubMed] [Google Scholar]; (d) Shu XZ, Zhang M, He Y, Frei H, Toste FD, J. Am. Chem. Soc 136 (2014) 5844–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huang L, Rudolph M, Rominger F, Hashmi ASK, Angew. Chem. Int. Ed 55 (2016) 4808–4813. [DOI] [PubMed] [Google Scholar]
- [6].Huang L, Rominger F, Rudolph M, Hashmi ASK, Chem. Commun 52 (2016) 6435–6438. [DOI] [PubMed] [Google Scholar]
- [7].(a) Cai R, Lu M, Aguilera EY, Xi YM, Akhmedov NG, Petersen JL, Chen H, Shi XD, Angew. Chem. Int. Ed 54 (2015) 8772–8776. [DOI] [PubMed] [Google Scholar]; (b) Peng HH, Cai R, Xu C, Chen H, Shi XD, Chem. Sci 7 (2016) 6190–6196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dong BL, Peng HH, Motika SE, Shi XD, Chem. Eur. J 23 (2017) 11093–11099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jimoh AA, Hosseyni S, Ye XH, Wojtas L, Hu Y, Shi XD, Chem. Commun 55 (2019) 8150–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sherborne GJ, Gevondian AG, Funes-Ardoiz I, Dahiya A, Fricke C, Schoenebeck F, Angew. Chem. Int. Ed 59 (2020) 15543–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zeineddine A, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D, Nat. Coummun 8 (2017) 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].(a) Rodriguez J, Zeineddine A, Sosa Carrizo ED, Miqueu K, Saffon-Merceron N, Amgoune A, Bourissou D, Chem. Sci 10 (2019) 7183–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rigoulet M, du Boullay OT, Amgoune A, Bourissou D, Angew. Chem. Int. Ed 59 (2020) 16625–16630. [DOI] [PubMed] [Google Scholar]; (c) Rodriguez J, Adet N, Saffon-Merceron N, Bourissou D, Chem. Commun 56 (2020) 94–97. [DOI] [PubMed] [Google Scholar]; (d) Zhang SY, Wang CH, Ye XH, Shi XD, Angew. Chem. Int. Ed 59 (2020) 20470–20474. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Messina MS, Stauber JM, Waddington MA, Rheingold AL, Maynard HD, Spokoyny AM, J. Am. Chem. Soc 140 (2018) 7065–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Akram MO, Das A, Chakrabarty I, Patil NT, Org. Lett 21 (2019) 8101–8105. [DOI] [PubMed] [Google Scholar]
- [11].(a) Moeller KD, Tetrahedron 56 (2000) 9527–9554. [Google Scholar]; (b) Yan M, Kawamata Y, Baran PS, Chem. Rev 117 (2017) 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ma C, Fang P, Mei TS, Acs Catalysis 8 (2018) 7179–7189. [Google Scholar]; (d) Mohle S, Zirbes M, Rodrigo E, Gieshoff T, Wiebe A, Waldvogel SR, Angew. Chem. Int. Ed 57 (2018) 6018–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sauermann N, Meyer TH, Qiu YA, Ackermann L, Acs Catalysis 8 (2018) 7086–7103. [Google Scholar]; (f) Wiebe A, Gieshoff T, Mohle S, Rodrigo E, Zirbes M, Waldvogel SR, Angew. Chem. Int. Ed 57 (2018) 5594–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ghosh M, Shinde VS, Rueping M, Beilstein J Org. Chem 15 (2019) 2710–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang HM, Gao XL, Lv ZC, Abdelilah T, Lei AW, Chem. Rev, 119 (2019) 6769–6787. [DOI] [PubMed] [Google Scholar]; (i) Barham JP, Konig B, Angew. Chem. Int. Ed 59 (2020) 11732–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Siu JC, Fu N, Lin S, Acc. Chem. Res 53 (2020) 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Xiong P, Xu HC, Acc. Chem. Res 52 (2019) 3339–3350. [DOI] [PubMed] [Google Scholar]
- [12].Ye X, Zhao P, Zhang S, Zhang Y, Wang Q, Shan C, Wojtas L, Guo H, Chen H, Shi X, Angew. Chem. Int. Ed 58 (2019) 17226–17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.