

Abstract
5-Demethylnobiletin (5DN) is a unique flavonoid mainly found in citrus fruits. In this study, we determined the chemopreventive effects of 5DN and its major colonic metabolites on both a colitis-driven colon carcinogenesis mouse model and a human colon cancer cell model. In azoxymethane/dextran sulfate sodium-treated mice, dietary 5DN (0.05% w/w in the diet) significantly decreased the tumor incidence, multiplicity and burden, and showed potent anti-proliferative, proapoptotic, and anti-inflammatory activities in mouse colon tissue. Three major metabolites of 5DN, named 5,3ʹ-didemethylnobiletin (M1), 5,4ʹ-didemethylnobiletin (M2) and 5,3ʹ,4ʹ-tridemethylnobiletin (M3) were found in the colonic mucosa of 5DN-treated mice, and the combined level of these metabolites in mouse colonic mucosa was 1.56-fold higher than that of 5DN. Cell culture studies revealed that 5DN and its colonic metabolites profoundly inhibited the growth of human colon cancer cells by inducing cell cycle arrest, triggering apoptosis and modulating key signaling proteins related to cell proliferation and apoptosis. Importantly, the colonic metabolites, especially M1 showed much stronger effects than those produced by 5DN itself. Overall, our results demonstrated that dietary 5DN significantly inhibited colitis-driven colon carcinogenesis in mice, and this chemopreventive effect was associated with its metabolites in colon.
Keywords: 5-demethylnobiletin, Colitis, Colorectal carcinogenesis, Bioactive compounds, Chemoprevention
Graphical Abstract
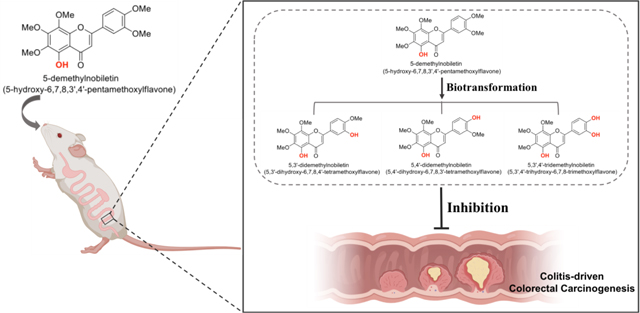
The inhibitory effect of dietary 5-demethylnobiletin on colitis-driven colorectal carcinogenesis and the potential roles of its colonic metabolites were reported.
1. Introduction
Colorectal cancer (CRC) is a major health problem with the third highest rate of morbidity and mortality in the United States.1,2 Chronic inflammation is implicated as a risk factor for colorectal carcinogenesis as it involves the interaction of various immune and inflammatory cells, chemokines, cytokines and pro-inflammatory mediators which can enhance the growth and invasion of malignant cells, promote angiogenesis, boost tumor metastasis, and alter tumor response to chemotherapeutic agents,3–7 therefore is considered to be a direct cause of colitis-driven cancer in numerous experimental models and humans.8–10 The correlation between colitis and CRC has been broadly accepted. Indeed, clinical research had showed that patients with inflammatory bowel disease (IBD) such as Crohn’s disease and ulcerative colitis had 2- to 3-fold higher risk of developing CRC compared to the general population,11–13 creating an urgent need for more efficacious chemoprevention strategies targeting colorectal carcinogenesis.14,15
Epidemiological studies have indicated an inverse relationship between fruits, vegetables and medicinal herbs intake and human colon cancer, which may be at least partially attributed to the bioactive components existed in these bioactive foods.16–21 Citrus fruit contains several chemopreventive compounds against cancers. Among them, 5-demethylated polymethoxyflavones are a unique subclass of polymethoxyflavones (PMFs) that have been recently isolated and documented to have numerous health-beneficial activities, including anti-cancer, anti-oxidation and anti-inflammation ones.22–24 For example, 5-demethylnobiletin (5-hydroxy-6,7,8,3’,4’-pentamethoxyflavone, 5DN), which is one of the most abundant 5-demethylated PMFs in citrus fruits, has shown potent inhibitory effects against lipid accumulation, multiple cancer cells and lung tumorigenesis.25–27
Biotransformation plays a critical role in the biological effects of orally administered compounds. Because the metabolites generated in the body through biotransformation have different chemical structures, which may result in stronger bioactivities in comparison with their parent compounds.26,28–31 Therefore, to better understand the in vivo efficacy of dietary compounds, it is important to investigate the biological activities of their metabolites generated in body. Previously, we have demonstrated the inhibitory effect of 5DN on NNK-induced lung tumorigenesis in mice, and this effect was associated with its two major metabolites in lung tissue.32 We first reported the inhibitory effect of 5DN on colitis-driven colon carcinogenesis in azoxymethane (AOM)/dextran sulfate sodium (DSS)-treated mice,33 then other group confirmed our findings that 5DN indeed showed potent inhibition on colorectal carcinogenesis.34 However, the potential contribution of biotransformation to the reported inhibitory effect of 5DN on colon cancer still remains unclear. In this paper, we systematically investigated the inhibitory effect of dietary 5DN on colitis-driven colon carcinogenesis in male CD-1 mice, identified and quantified the major colonic metabolites of 5DN in mice, and demonstrated the superior inhibitory activities of these metabolites on human colon cancer cells.
2. Materials and methods
2.1. Animals, diets, and experimental design
This experimental protocol was approved by Institutional Animal Care and Use Committee of University of Massachusetts Amherst (#2014–0079). Approximately 5-week old male CD-1 mice were obtained from Charles River Laboratory (Wilmington, MA). After one-week of acclimation, mice were randomly assigned to three experimental groups (negative control group, positive control group, and 5DN group, 20 mice each) and placed on an AIN-93G diet. Then the animals in positive control group and 5DN group received a single intraperitoneal injection of AOM (12 mg kg−1 body weight) in saline, and animals in negative group received same volume of saline. One week after AOM injection, 1.5% DSS (molecular weight: 36 000–50 000, MP Biomedicals, Solon, OH) was administered in the drinking water for 4 days followed by one week of regular water for recovery, and this cycle was repeated four times (negative control group received regular drinking water). Starting one week after AOM injection, negative and positive control groups were fed with AIN-93G diet, while 5DN group was fed with AIN-93G diet containing 5DN (0.05% in diet, w/w) until the end of the experiment. The body weight was recorded weekly. All mice were humanely sacrificed via CO2 asphyxiation 20 weeks after AOM injection. The liver and spleen were removed, rinsed with PBS (pH 7.4) and weighted. At necropsy, after measuring the length, the colons were opened longitudinally, flushed with PBS (pH 7.4) and weighted. The number of tumors was counted under a dissection microscope, and the size of the tumor was measured using a caliper. The tumor volume was determined using the formula V (mm3) = 0.5 × (length × width × width) as reported35–37. Then the colons were cut along the main axis. Half of the colon was fixed in 4% buffered formalin (pH 7.4) for 24 h for histopathological and immunohistochemical analysis. The other half was stored at −80 °C for ELISA, qRT-PCR, and HPLC analysis.
2.2. Histopathological and immunohistochemical analysis
The fixed colon tissue was dehydrated, embedded in paraffin, sectioned (5 μm), mounted on glass slides and stained with hematoxylin and eosin (H&E) as we previously described.38 The histological alterations such as mucosal dysplasia, and carcinoma were evaluated under a microscope according to the criteria previously described by a trained pathologist blinded to the study design.39,40 In brief, colonic mucosal dysplasia is characterized by elongated, crowded and pseudostratified nuclei. Carcinoma was defined as a high-grade dysplasia of colonic mucosa that had invaded beyond the muscularis mucosa and into the submucosa. Histological scores were assigned following the chronic colitis scoring system by Chinen et al.41 Immunohistochemisty staining was performed on the colon tissue sections as we previously described.38,42,43 Cell proliferation in the colon tissue was determined by positive staining of proliferating cell nuclear antigen (PCNA) and Ki-67 (1:1000, Dako, Denmark). Cellular apoptosis was determined by staining with antibodies against cleaved caspase-3 (1:1000, Cell Signaling Technology, Beverly, MA). Colonic inflammation was measured by staining with antibodies against inducible nitric oxide synthase (iNOS, 1:200, Cell Signaling Technology, Danvers, MA, USA). Briefly, colon tissue sections were deparaffinized in serial xylene, rehydrated through graded ethanol solutions. Antigen retrieval was performed by heating the sections in 0.01 mol/L citrate buffer (pH 6.0) for 20 minutes in a PT Module antigen retrieval device (Thermo Fisher Scientific, Agawam, MA, USA). Endogenous peroxidase was quenched in 0.3% hydrogen peroxide. Nonspecific binding was blocked by incubating the sections with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA) for 30 minutes. Then, primary antibodies were applied to the sections and incubated overnight at 4 °C. After thorough washed with PBS, sections were incubated with a biotinylated secondary antibody for 30 minutes at the room temperature and subsequently incubated with the chromogen 3-diaminobenzidine (DAB). Sections were then counterstained with hematoxylin for 3 minutes. Positive staining was observed using a Nikon E400 microscope. Digital images were captured with a SPOT Idea 1.3 Mp camera.
2.3. ELISA and real-time qRT-PCR analysis
Colonic mucosa was scraped and homogenized in a phosphate buffer solution containing 0.4 M NaCl, 0.05% Tween-20, 0.5% BSA, 0.1 mM benzethonium, and 1% protease inhibitor cocktail (Boston Bioproducts, Ashland, MA). Then the homogenates were centrifuged at 10000 g for 30 min at 0 °C. The supernatant was collected and used for quantification of cytokines, i.e. IL-1β, IL-6, and tumor necrosis factor-α (TNF-α) by ELISA kits (R&D System, Minneapolis, MN) according to the manufacturer’s instructions. Real Time qRT-PCR analysis was performed as previously described.44 The primer pairs were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA) with the following primers: IL-1β F: 5’-ACCTGCTGGTGTGTGACGTT-3’, R: 5’-TCGTTGCTTGGTTCTCCTTG-3’; IL-6 F: 5’-GAGGATACCACTCCCAACAGACC-3’, R: 5’-AAGTGCATCATCGTT GTTCATACA-3’; TNF-α F: 5’-AGCACAGAAAGCATGATC CG-3’, R: 5’-CTGATGAGAGGGAGGCCATT-3’; β-actin F: 5’-AAGAGAGGCATCCTCACCCT-3’, R: 5’-TACATGGCTGGGGTGTTGAA-3’.45 The copy number of each transcript was calculated with respect to the β-actin copy number, using the 2-ΔΔCt method.46
2.4. Quantification of colonic 5DN and its metabolites by HPLC
Colonic mucosa samples were homogenized in methanol (50% in phosphate buffered saline, pH=5.00) and then extracted with ethyl acetate for three times. Pooled ethyl acetate fractions were dried under vacuum and reconstituted in 50% methanol. Identification and quantification of 5DN and its metabolites were performed using HPLC method as we previously described.47,48 5DN, M1, M2, and M3, with purity greater than 98%, were used as external standards and tangeretin (>98%) was used as an internal standard. Tangeretin was purchased from Sigma-Aldrich (St. Louis, Mo). 5DN, M1, M2, and M3 were synthesized as we described previously.47,48
2.5. Cell viability, cell cycle and cellular apoptosis analysis
The analysis of cell viability, cell cycle and apoptosis were conducted as we previously described.25,49 In brief, HCT116 (ATCC, Manassas, VA) human colorectal cancer cells were seeded at a density of 2500 cells/well in 96-well plates. After 24 h incubation, cells were treated with treatments in serum complete media for 72 h. The cell viability was then determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. For cell cycle and apoptosis assay, cells were seeded at a density of 5×104 cells/well in 6-well plates. After 24 h of incubation for cell attachment, cells were treated with different treatments in serum complete media. After 24h or 48h, floating cells in media and adherent cells were harvest and subjected to cell cycle and apoptosis analysis by flow cytometry method.
2.6. Immunoblotting
Cells were seeded in 150mm culture dishes. After 24h of incubation for cell attachment, cells were treated with different treatments. After another 24 or 48 h, cells were harvested, combined with floating cells, if any. Whole cell lysates were prepared and then subjected to Western blotting analysis as we previously described.25,49 For colon tissue, samples were homogenized with RIPA lysis buffer (Tris-Hcl pH7.2, 25mM; SDS 0.1%; Triton X-100 1%; sodium deoxycholate 1%; NaCl 0.15% M; ethylenediaminetetraacetic acid (EDTA) 1mM (Boston Bioproducts, Ashland, MA) containing 1% protease inhibitor cocktail, then subjected to western blotting analysis.
2.7. Statistical analysis
All data were presented as mean ± SD or mean ± SEM. Student’s t-test was used to test the mean difference between two groups, whereas analysis of variance (ANOVA) followed by Tukey’s HSD test was used for the comparison of difference among three or more groups. Tumor incidence was analyzed by Fisher’s exact probability test. Differences were considered statistically significant when p < 0.05.
3. Results and Discussion
3.1. General observation
AOM/DSS-treated mice model was used in this study to determine the chemopreventive effect of 5DN on colitis-driven colorectal carcinogenesis. In this model, a single injection of a colon carcinogen AOM in combination with cyclic administration of DSS in drinking water induced the development of colitis, colorectal dysplasia, and cancer.50,51 The dose of 5DN (0.05%) in mice treatment was equivalent to 250 mg per day in human approximately according to formula emanated from Reagan-Shaw et al,52 which can be conveniently achievable through dietary supplementation. All animals survived the experimental period. Bloody and soft stool was observed in a few mice that received DSS treatment. In positive control group, anal prolapse due to severe inflammation and tumor development in the distal colon was observed as well. As shown in Figure 1, the mean body weight of mice in positive group was lower compared to other groups at the end of the experiment (p=0.29). This might be caused by severe inflammation induced by DSS treatment. Dietary 5DN, however, alleviated the body weight loss by DSS treatment. Colonic weight/length ratio is correlated with the severity of colitis and therefore is a indicative measurement of colonic wall thickening, severity of inflammation and neoplasia development.53,54 Our results showed that dietary 5DN significantly prevented the shortening of colon length and decreased the elevated weight/length ratio caused by AOM/DSS treatment when compared to the positive control group (Table 1). These results together suggested the protective effect of dietary 5DN against severe inflammation induced by AOM/DSS treatment.
Figure 1.

Effect of 5DN on the weight changes during colon carcinogenesis.
Table 1.
Final liver, spleen weights, and colon assessment of mice
| Group | Negative control | Positive control | 5DN treated |
|---|---|---|---|
| Liver Weight (mg) | 2376.34 ± 68.52 | 2262.86 ± 92.74 | 2278.04 ± 105.40 |
| Spleen Weight (mg) | 236.65 ± 33.56 | 230.60 ± 30.12 | 212.78 ± 26.20 |
| Colon Length (mm) | 96.82 ± 2.64a | 86.14 ± 3.72b | 95.98 ± 2.92a |
| Colon W/L ratio (mg/mm) | 3.68 ± 0.30a | 4.32 ± 0.50b | 3.76 ± 0.28a |
| Tumor incidence | 0a | 100% b (20/20) | 65%c (13/20) |
| Tumor multiplicity | 0a | 5.70 ± 1.10b | 2.50 ± 0.50c |
| Tumor burden (mm3) | 0a | 13.48 ± 2.32b | 6.69 ± 1.82c |
All values are represented as mean ± SEM. Different notations indicate statistically significant difference (p < 0.05, n=20) according to ANOVA analysis followed by Tukey’s HSD test. Tumor incidence is analyzed by Fisher’s exact probability test.
We also examined the main organ weight of the animals, which have been widely accepted as important markers and sensitive indicators of potential toxicities of test agents, as well as treatment-related effects.55,56 As shown in Table 1, there was no significant difference in the weight of liver and spleen among the groups. Histologically, no pathological alterations or lesions in the main organs (liver, kidney and spleen) of mice were found (data not shown), suggesting that dietary administration of 5DN did not cause noticeable toxicity in male CD-1 mice.
3.2. Dietary 5DN suppressed AOM/DSS-induced colonic tumorigenesis
At the end of the experiment (Table 1), all the mice in positive control group developed colonic tumors with a multiplicity of 5.70 ± 1.10 (mice in negative control group showed no tumor). Notably, the dietary administration of 0.05% 5DN for 20 weeks significantly decreased the tumor incidence and multiplicity by 35% (100 versus 65%) and 56.14% (5.7 ± 1.1 versus 2.5 ± 0.5), respectively. Moreover, tumor burden was decreased from 13.48 ± 2.32 mm3 (positive control group) to 6.69 ± 1.82 mm3 (mice fed 0.05% 5DN).
A trained histopathologist then examined and characterized the histological alterations of colon samples by H&E staining. As shown in Figure 2, AOM/DSS treatment resulted in significant alterations of colonic mucosa, including loss of crypts, surface erosion with exuberant inflammatory exudate, infiltration of inflammatory cells in to the lamina propria, formation of dysplasia, and tumor development. In contrast, dietary 5DN significantly attenuate histologic damage (with maintenance of crypt organization in normal mucosa), decrease the chronic colitis histologic score (from 12.38 ± 0.57 to 5.38 ± 0.60) and the occurrence of AOM/DSS-induced tumor compared to positive control. Specifically, the colon of 5DN-treated mice appeared to largely maintain the normal colon morphology or dysplasia with inflammation, the mucosa had tightly packed glands with normal architecture of goblet cells. Together these findings demonstrated that dietary administration of 5DN (0.05% in diet) effectively alleviate colitis-driven colon carcinogenesis in male CD-1 mice.
Figure 2.

Histological characterization of colonic mucosa and tumors of AOM/DSS-treated mice. Representative H&E staining histological sections were showed. Overall histology scores were calculated according the chronic colitis scoring system mentioned in materials and methods. Data are presented as mean ± SD. Asterisk indicates statistical significance in comparison with control (p < 0.01, n=8) by Student’s-t test.
3.3. Dietary 5DN inhibited cell proliferation, induced apoptosis, and decreased the levels of proinflammatory cytokines in the colon of AOM/DSS-treated mice
Colorectal cancer initiation and progression are strong associated with enhanced cell proliferation and evasion of apoptosis.57 Therefore, if one could inhibit proliferation and induce apoptosis, it could suppress the development of colorectal cancer.58–60 One of the most reliable method to examine colorectal cell proliferation is the evaluation of Ki-67 and PCNA expression through immunostaining61. Ki-67 and PCNA are closely correlated with somatic cell proliferation. Thus, increased proliferation of colon epithelial cell, which was characterized as hyperplasia, can be detected with the Ki-67 and PCNA proliferation markers.62,63 As shown in Figure 3A and 3B, colonic sections from the positive control mice showed intense staining of Ki-67 and PCNA, indicating a high cell proliferation rate. In contrast, the positive staining of Ki-67 and PCNA was markedly decreased by 35.86% (41.13 ± 2.90% versus 26.38 ± 1.31%) and 61.81% (47.13 ± 3.53% versus 18.00 ± 1.60%) respectively in 5DN treatment group, suggesting a significant inhibition in cell proliferation. It is well accepted that a reduction in tumor incidence is generally correlated to a decrease in cellular proliferation and/or increase in apoptosis.64 Thus, the induction of apoptosis is an effective strategy in the chemoprevention of caner.65 By using cleaved caspase-3, an activated regulator of apoptosis as a marker, we found that the number of apoptotic cells in the colonic tumors was 10.00 ± 1.05% in 5DN treatment group. When compared with the positive group (3.38 ± 0.89 %), it was a 2.96-fold increase (Figure 3C), indicating an intense apoptosis triggered by 5DN treatment.
Figure 3.

Effect of dietary 5DN on colonic immunohistochemical staining of (A) Ki-67, (B) PCNA, (C) Cleaved caspase-3 and (D) inducible nitric oxide synthase (iNOS) in AOM/DSS-treated mice. Representative colon sections from control and 5DN treatment groups are shown (Magnification 150× or 300×). Positive staining is brown colored. Data are presented as mean ± SD and asterisks indicate statistical significance in comparison with control (p < 0.01, n=8) by Student’s-t test.
It is well known that over-expression of pro-inflammatory cytokines and/or enzymes amplifies inflammatory cascade signaling, causes intestinal tissue damage, and increases the risk of colorectal carcinogenesis.6,66–68 Thus, management of cytokine equilibrium was considered to be a promising strategy for both prevention and treatment of various malignancies including colorectal cancer.69,70 Herein, we investigated the effects of dietary 5DN on the expression of pro-inflammatory factors in colon by immunohistochemistry and ELISA analysis. As shown in Figure 3D, an intense staining of iNOS was observed in the colon tissue of positive control mice, indicating a high level of inflammation. Remarkably, dietary 5DN significantly decreased the expression of iNOS by 39.95% (50.38 ± 2.75% versus 30.25 ± 1.88%). To further confirm the effect of 5DN on AOM/DSS induced inflammation, ELISA analysis of colon mucosa samples was performed. Our results (Figure 4A) showed that 5DN treatment significantly decreased the levels of IL-1β, IL-6 and TNF-α by 86.24% (53.79 ± 6.43 versus 7.40 ± 1.74%), 73.69% (112.53 ± 32.09 versus 29.61 ± 7.57) and 57.23% (9.47 ± 1.66 versus 4.05 ± 0.80), respectively, when compared to those of the positive control group. Then we determined the effect of dietary 5DN on the mRNA expression of pro-inflammatory cytokines by real-time qRT-PCR analysis (Figure 4B). Our results revealed that the mRNA expressions of IL-1β, IL-6 and TNF-α in the colon mucosa of 5DN-treated mice were dramatically reduced by 93.79%, 95.95% and 63.63%, respectively, compared with those found in the positive control group mice.
Figure 4.

Effects of 5DN treatment on (A) protein levels and (B) mRNA levels of IL-1β, IL-6 and TNF-α in colonic mucosa of AOM/DSS-treated mice. Samples were randomly collected from the middle and distal colon, and then subjected to ELISA or qRT-PCR analysis. Data are presented as mean ± SD of three independent experiments. The level of IL-1β and IL-6, and TNF-α mRNA expression was normalized to that of β-actin. Asterisks indicate statistically significance in comparison with control (p < 0.01, n=3) by Student’s-t test.
Together, these results demonstrated that dietary administration of 0.05% 5DN significantly inhibited the colitis-driven colon carcinogenesis. And this effect was further evidenced by reduced abnormal cell proliferation, elevated cellular apoptosis, and attenuated the mRNA and protein expression of pro-inflammatory cytokines in the colonic mucosa of AOM/DSS-treated mice.
3.4. Identification and quantification of colonic metabolites of 5DN in mice
The metabolic fate of dietary component is critical for their biological activities. We and others have documented that orally administration of PMFs, including NBT, tangeretin, and 5DN, resulted in the production of various metabolites via extensive biotransformation.47,48,71,72 Particularly, we have identified three major metabolites of 5DN in the urine of 5DN-fed mice as: 5,3ʹ-didemethylnobiletin (M1), 5,4ʹ-didemethylnobiletin (M2) and 5,3ʹ,4ʹ-tridemethylnobiletin (M3) (Figure 5A).47 Interestingly, all these metabolites exhibited similar, even stronger inhibition against the growth of human lung cancer cells in comparison with their parent compound.26 In this study, to determine the role of biotransformation in the chemopreventive effect of 5DN against colitis-driven colon carcinogenesis, the identity and abundance of the metabolites of 5DN in the colonic mucosa need to be investigated. This is because the metabolites, rather than 5DN itself, may be responsible for the biological activities we observed due to their potentially stronger activities and greater abundance. We expected that the urinary metabolites of 5DN we identified previously would be found in colonic mucosa of 5DN treated mice. By conducting HPLC followed by mass spectroscopy analysis, we confirmed that oral administration of 5DN resulted in the formation of three major metabolites (M1, M2, and M3) in colonic mucosa of mice (Figure 5). The mechanism of the transformation from 5DN to these metabolites is not clear yet. However, it is likely that phases I and II metabolism play an important role in their formation as well as the biotransformation by gut microbiome.73
Figure 5.

(A) Chemical structure of 5DN and its metabolites M1, M2, and M3. (B) Representative HPLC profile of colonic mucosa samples from the 0.05% 5DN treated group. Samples were detected by using an electrochemical detector at 300 mV. Four major peaks in the chromatogram were identified as 5DN (retention time at 20.2 min), M1 (retention time at 17.5 min), M2 (retention time at 19.2 min) and M4 (retention time at 16.7 min).
The levels of a specific component and its metabolites in body after oral administration greatly influence the overall bioactivity in vivo. Thus, we further quantified the levels of 5DN and its metabolites in the mucosa of 5DN-fed mice by HPLC method we established previously.47 As shown in table 2, the colonic levels of 5DN, M1, M2, and M3 were 13.58 ± 2.68, 8.09 ± 2.27, 10.16 ± 2.50, and 2.92 ± 0.59 nmol/g, respectively. Specifically, the level of M1 and M2 was similar to that of 5DN itself, and the level of M3 was relatively lower than others. Importantly, the combined level of metabolites was 1.56–fold higher than that of 5DN in the colonic mucosa. Considering the potential stronger activity of these metabolites, this information suggested that the inhibitory effect of 5DN against colitis-driven colon carcinogenesis we observed in the animal study might be at least partially attributed to its colonic metabolites. Together for the first time, we successfully identified M1, M2, and M3 as three major colonic metabolites of 5DN in mice after long-term oral administration of 5DN. Most interestingly, the level of metabolites combined was much higher than 5DN itself in the colonic mucosa, suggesting the importance of biotransformation in the biological effects of orally administered 5DN.
Table 2.
Quantification of 5DN and its metabolites in the colonic mucosa of 5DN treated mice
| Compound | Concentration (nmol per gram tissue) | Percentage among compounds (%) |
|---|---|---|
| 5DN | 13.58 ± 2.68 | 39.1 |
| M1 | 8.09 ± 2.27 | 23.3 |
| M2 | 10.16 ± 2.50 | 29.2 |
| M3 | 2.92 ± 0.59 | 8.4 |
5DN and its metabolites were extracted three times with ethyl acetate from the colonic mucosa homogenate of 5DN treated mice, and then quantified by HPLC. Values are presented as mean ± SD (n=6).
3.5. Colonic metabolites of 5DN showed stronger effects than 5DN on inhibiting the growth, inducing cell cycle arrest and apoptosis of human colon cancer cells
Due to the fact that long-term administration of 5DN resulted in the presence of 5DN and abundant levels of its metabolites in the colonic mucosa, we hypothesized that these metabolites might play an important role in the inhibitory effect of dietary 5DN on colitis-driven colon carcinogenesis. To confirm our hypothesis, we investigated the effects of 5DN and its metabolites (M1, M2, and M3) on the growth of human HCT116 colon cancer cells. HCT116 cells were treated with serial concentrations of 5DN (4–20 μM), M1 (0.1–0.5 μM), M2 (4–20 μM), and M3 (4–20 μM). As shown in figure 6A, all the compounds significantly inhibited the growth of HCT116 cells in a dose-dependent manner after 72 h of incubation. Furthermore, all three metabolites of 5DN showed more potent inhibition than their parent compound, 5DN. Markedly, M1 showed the strongest inhibitory effect among all compounds. Specifically, M1 at only 0.5 μM inhibited cell growth by 82%, which is much stronger than those produced by 5DN at much higher concentration (20 μM). The estimated IC50 values of 5DN, M1, M2, and M3 were 13.5, 0.22, 11.5 and 7.0 μM, respectively. It is noteworthy that the IC50 values of all the metabolites were lower than 5DN. Especially, the IC50 value of M1 was about 61-fold lower than that of 5DN. To better understand the contribution of the metabolites to the inhibitory effect of orally administrated 5DN, we defined a parameter as “inhibitory index” which equals to the level of a specific compound found in colon mucosa (showed in Table 2) divided by its IC50 value. According to this, the inhibitory index of 5DN, M1, M2, and M3 was 1.006, 36.770, 0.884, and 0.417, respectively. Considering that 5DN and its three major metabolites were co-existing in the colon mucosa as a mixture, the combined inhibitory index of the mixture was 38.9-fold higher than that of 5DN alone (Figure 6B), suggesting that the biotransformation of 5DN, which results in the formation of M1, M2, and M3 in the mice, indeed elevates the overall inhibitory effect on colon carcinogenesis by oral administration of 5DN. Together, our results suggested that the higher potency of M1, M2, and M3 than 5DN might contribute to the inhibition of colon carcinogenesis.
Figure 6.

(A) Inhibitory effects of 5DN and its metabolite (M1, M2, and M3) on the growth of HCT116 human colon cancer cells. Cells were seeded in 96-well plates and treated with serial concentrations of 5DN, M1, M2, and M3. After 72 hours of incubation, cell viability was quantified by the MTT assay as described in the Materials and methods section. Data are represented as mean ± SD (n=6), and the asterisks indicate the statistical significance in comparison with the control cells (p < 0.01). (B) Inhibitory Index of 5DN and the mixture of 5DN and its three metabolites. The inhibitory index equals to the level of a compound in colon mucosa divided by its IC50 value.
To further illustrate the mechanisms by which 5DN and its metabolites inhibit the cancer cell growth, we studied their effects on cell cycle progression and apoptosis by flow cytometry. As showed in Figure 7A, all the compounds were able to modulate cell population distribution but in different manners and to different extents. Specifically, 5DN at 20 μM and M2 at 10 μM significantly increased cell population in G2/M phase, and decreased cell population in S phase. Interestingly, M1 at a much lower dose (0.25 μM) caused similar effect when compared to 5DN and M2. Furthermore, M3 at 10 μM showed same manner of cell cycle arrest as those caused by 5DN, M1 and M2, however with much more potency. Overall, these results revealed that 5DN, M1, M2, and M3 caused G2/M phase cell cycle arrest with different potency on human HCT-116 colon cancer cells. Interestingly, the pattern and potency of cell cycle arrest caused by 5DN and its metabolites on human lung and colon cancer cells were not always consistent. For example, M2 significantly arrested human A549, H460 and H1299 lung cancer cells in G0/G1 phase, however M1 and M3 caused G2/M phase arrest in those cells26,32, which was consistent with the results in this study. Together, these findings suggested that their difference in chemical structures may lead to different molecular mechanisms that be involved in their modulation on cell cycle progression.
Figure 7.

Effects of 5DN (20 μM) and its colonic metabolites M1 (0.25μM), M2 (10 μM) and M3 (10 μM) on the cell cycle progression and apoptosis of HCT116 human colon cancer cells. Cells were seeded in 6-well plates and then treated with 5DN and its metabolites. After 24 or 48 h of treatments, cells were harvested and subject to cell cycle and apoptosis analysis as described in the Materials and methods section. All data are represented as mean ± SD (n=3). Different notations in the bar charts indicate statistical significance (p < 0.01) according to ANOVA analysis followed by Tukeys’s HSD test.
Inducing apoptosis in cancer cells is one of the effective strategies in cancer chemoprevention. To determine if triggering apoptosis contributed to the growth inhibition of 5DN and its metabolites on colon cells, the Annexin-V/PI double staining assay was conducted by flow cytometry. As shown in Figure 7B, after 48 h of treatment, compared to control, both early and late apoptotic cell populations were significantly increased by all the compounds on HCT116 cells. Importantly, all three metabolites, especially M1 and M3 showed much stronger effect than 5DN. For instance, treatment with 5DN at 20 μM increased early apoptotic cell population by 3.1-fold compared to the control. However, M1 at only 0.25 μM increased early apoptotic cell population by 7.6-fold, and M3 at 10 μM, which is half dose of 5DN, increased 11.8-fold of early apoptotic cell population compared to control. These results demonstrated that 5DN and its metabolites induced significant apoptosis in human colon cancer cells. More importantly, the metabolites, especially M1 and M3 showed much stronger effect, indicating their important roles in inhibiting colon carcinogenesis.
3.6. 5DN and its colonic metabolites modulated key signaling proteins related to cell proliferation and apoptosis
To illustrate the molecular mechanisms of the inhibition produced by 5DN and its colonic metabolites, we investigated their effects on the expression of key signaling proteins related to cell cycle progression and apoptosis pathways in HCT116 human colon cancer cells. Cell cycle related proteins were analyzed after 24 h of treatment, and apoptosis related proteins were analyzed after 48 h of treatment. We found that (Figure 8) 5DN and its three colonic metabolites significantly increased p21Cip1/Waf1 and p27kip1 expression. Moreover, all the metabolites, especially M3 at 10 μM decreased the expression levels of cyclin A2 and cyclin B1. The loss of normal cell proliferation caused by abnormal regulation of cell cycle is one of the hallmarks of cancer. Cyclins, cyclin dependent kinases (CDKs), and CDK inhibitors play important roles in regulating cell cycle progression. The formation of cyclin/CDK complexes drives the cell cycle transition. The G2/M phase transition is driven by cyclin B-CDK1 complex. These cyclin-CDK complexes often bind to the endogenous inhibitor proteins (CKIs) p21Cip1/Waf1 and p27kip1, which inhibit their kinase activities and prevent cell cycle progression.74–76 Our results showed that the metabolites of 5DN significantly decreased the expressions of cyclin B1, which at least in part, down-regulated the level of cyclin B1-CDK1 complex, and led to cell accumulation in G2/M phase. Cyclin A is thought be involved in the activation and stabilization of cyclin B/CDK1 complex,77,78 the down-regulated cyclin A2 expression caused by the metabolites of 5DN might decrease the stability and activation of cyclin B/CDK1 complex, which further arrested cells in G2/M phase. It is also well known that the activation of cyclin B1-CDK1 complex by the phosphorylation of cyclin B1 are mandatory for a cell to enter into mitosis at G2/M transition.79 Interestingly, 5DN and its metabolites showed different effects on the phosphorylation of cyclin B1Ser147. Specifically, 5DN and M2 significantly increased the expression of phosphorylated cyclin B1Ser147. In contrast, M3 at 10 μM showed the opposite effect, which might decrease the activation of cyclin B1-CDK1 complex. These findings might partially explain why M3 had the strongest effect in inducing cell cycle arrest at G2/M phase in comparison with other compounds. P21Cip1/Waf1 is a negative CDK regulator that can directly bind to the cyclin B1/CDK1 complex and inhibit its activity, which further block cells in G2/M phase.80,81 In addition, p21Cip1/Waf1 is able to diminish CDK1 protein level by decreasing Cdc2 mRNA transcriptions and its promoter activity.82 According to these, the increased p21/p27 expression could be another possible mechanism by which HCT116 human colon cancer cells undergo cell cycle arrest during exposure to 5DN and its metabolites.
Figure 8.

Effects of 5DN (20 μM), M1 (0.25μM), M2 (10μM) and M3 (10μM) on cell cycle and apoptosis related key proteins in HCT116 human cancer cells. Cells were seeded in 15 cm culture dishes for 24 hours and then treated with 5DN and its metabolites at different concentrations. After another 24 or 48 hours of incubation, cells were collected for immunoblotting as described in the Materials and methods section. The number underneath the blots represents the band intensity (normalized to β-actin, means of three independent experiments) measured by Image Studio software. The SDs (all within ±15% of the means) are not shown. β-Actin was served as an equal loading control. Asterisks indicate statistical significance in comparison with the control (p < 0.05, n = 3).
The evasion of apoptosis is considered to facilitate the development of various cancer.83 The central engine of apoptosis is the caspases cascade that implement cell death by cleaving a variety of intracellular substrate. The activation (cleavage) of caspase-9 results in the activation its downstream effector caspase-3, which will trigger cellular apoptosis. The activation (cleavage) of caspase-3 also leads to the activation of other key effectors, such like poly ADP ribose polymerase (PARP), which ultimately promote apoptosis by interfering chromatin condensation and DNA fragmentation.84 We found that 5DN and its colonic metabolites were able to active the caspase cascade by cleavage of caspase-9, caspase-7, caspase-3, and their final protein target PARP in HCT116 cells. And these actions might be driven by the upregulation of p53. Markedly, the metabolites, especially M1 at only 0.25 μM and M3 at 10 μM, showed stronger effects than that of 5DN on the modulation of these apoptosis-related proteins, which is consistent with previous annexin-V/propidium iodine double staining assay (Figure 7B). Together our results revealed that the major colonic metabolites of 5DN had stronger effects on the activation of caspase cascade for apoptosis in comparison with 5DN itself.
Markedly, the effects of 5DN and its major metabolites on p21Cip1/Waf1, p53 and caspase cascade were consistent with our previous findings in lung tumorigenesis, suggesting that these proteins and related signal pathways might be the potential molecular targets for 5DN and its major metabolites to modulate cell cycle progression and apoptosis of human cancer cells.
4. Concluding remarks
This study demonstrated that dietary 5DN (0.05% in diet, w/w) significantly inhibited colitis-driven colon carcinogenesis in AOM/DSS-treated CD-1 mice. Specifically, 5DN decreased the tumor incidence, multiplicity and tumor burden by 35%, 54.16% and 50.37% in mice, respectively. For the first time, we identified and quantified three major metabolites of 5DN in the colonic mucosa of 5DN-fed tumor-bearing mice, namely 5,3ʹ-didemethylnobiletin (M1), 5,4ʹ-didemethylnobiletin (M2) and 5,3ʹ,4ʹ-tridemethylnobiletin (M3). The level of each metabolite was similar to that of 5DN in the colonic mucosa, however the combined level of these metabolites was about 1.5-fold higher than that of 5DN itself. We further demonstrated that the colonic metabolites of 5DN had more potent anticancer activities than 5DN, which was evidenced by their superior effects in inhibiting human colon cancer cell growth, inducing cell cycle arrest, triggering apoptosis, and modulating key signaling proteins related to cell proliferation and apoptosis, including cyclin A2, cyclin B1, p21Cip1/Waf1, p27kip1, cleaved caspase-3, caspase-7, caspase-9, PARP and p53. In conclusion, our results suggested that the chemopreventive effect of dietary 5DN against colitis-driven colon carcinogenesis were closely associated with its colonic metabolites.
Acknowledgements
This work was partially supported by a NIH grant (R01AT010229 to H. X.), an USDA Special Grant on bioactive food components (to H. X.), a National Natural Science Foundation of China (31801505 to M. S.), a Natural Science Foundation of Guangdong Province (2018A0303130008 to M.S.) and a Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2019ZT08N291 to J. X.)
Abbreviations
- 5DN
5-demethylnobiletin
- AOM
azoxymethane
- DSS
dextran sulfate sodium
- H&E
hematoxylin and eosin
- PMFs
polymethoxyflavones
- iNOS
inducible nitric oxide synthase
- PCNA
proliferating cell nuclear antigen
- CDKs
cyclin dependent kinases
- PARP
poly ADP ribose polymerase
Footnotes
Conflict of interest
The authors have declared no conflict of interest.
References
- 1.Half E. and Arber N, Expert Opin. Pharmacother, 2009, 10, 211–219. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD and Jemal A, CA. Cancer J. Clin, 2019, 69, 7–34. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani A, Mantovani A, Allavena P, Allavena P, Sica A, Sica A, Balkwill F. and Balkwill F, Nature, 2008, 454, 436–44. [DOI] [PubMed] [Google Scholar]
- 4.Janakiram NB and Rao CV, Adv. Exp. Med. Biol, 2014, 816, 25–52. [DOI] [PubMed] [Google Scholar]
- 5.Korniluk A, Koper O, Kemona H. and Dymicka-Piekarska V, Ir. J. Med. Sci, 2017, 186, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klampfer L, Curr. Cancer Drug Targets, 2011, 11, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beaugerie L. and Itzkowitz SH, N. Engl. J. Med, 2015, 372, 1441–1452. [DOI] [PubMed] [Google Scholar]
- 8.Mizoguchi E, Kanneganti M. and Mino-Kenudson M, J. Biomed. Biotechnol, 2011, 2011. [DOI] [PMC free article] [PubMed]
- 9.Azer SA, Eur. J. Gastroenterol. Hepatol, 2013, 25, 271–281. [DOI] [PubMed] [Google Scholar]
- 10.Rubin DC, Shaker A. and Levin MS, Front. Immunol, 2012, 3, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein CN, Blanchard JF, Kliewer E. and Wajda A, Cancer, 2001, 91, 854–862. [DOI] [PubMed] [Google Scholar]
- 12.Von Roon AC, Reese G, Teare J, Constantinides V, Darzi AW and Tekkis PP, Dis. Colon Rectum, 2007, 50, 839–855. [DOI] [PubMed] [Google Scholar]
- 13.Harmon BE, Wirth MD, Boushey CJ, Wilkens LR, Draluck E, Shivappa N, Steck SE, Hofseth L, Haiman CA, Le Marchand L. and Hébert JR, J. Nutr, 2017, 147, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall JR, Gastroenterol. Clin. North Am, 2008, 37, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohammed A, Yarla NS, Madka V. and Rao CV, Int. J. Mol. Sci, 2018, 19. [DOI] [PMC free article] [PubMed]
- 16.Mehta RG, Murillo G, Naithani R. and Peng X, Pharm. Res, 2010, 27, 950–961. [DOI] [PubMed] [Google Scholar]
- 17.Han Y. and Xiao H, Annu. Rev. Food Sci. Technol, 2020, 11, 1–25. [DOI] [PubMed] [Google Scholar]
- 18.Dang H, Zhang T, Yi F, Ye S, Liu J, Li Q, Li H. and Li R, Aquac. Fish, 2019, 4, 114–121. [Google Scholar]
- 19.Lee J, Shin A, Oh JH and Kim J, World J. Gastroenterol, 2017, 23, 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu M, Chen YM, Huang J, Fang YJ, Huang WQ, Yan B, Lu MS, Pan ZZ and Zhang CX, Br. J. Nutr, 2016, 116, 1275–1287. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H, Hao J, Niu Y, Liu D, Chen D. and Wu X, Sci. Rep, 2018, 8, 7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohno H, Suzuki R, Curini M, Epifano F, Maltese F, Gonzales SP and Tanaka T, Int. J. Cancer, 2006, 118, 2936–42. [DOI] [PubMed] [Google Scholar]
- 23.Li S, Pan M-H, Lo C-Y, Tan D, Wang Y, Shahidi F. and Ho C-T, J. Funct. Foods, 2009, 1, 2–12. [Google Scholar]
- 24.Wang M, Meng D, Zhang P, Wang X, Du G, Brennan C, Li S, Ho CT and Zhao H, J. Agric. Food Chem, 2018, 66, 3155–3160. [DOI] [PubMed] [Google Scholar]
- 25.Qiu P, Dong P, Guan H, Li S, Ho C-T, Pan M-H, McClements DJ and Xiao H, Mol. Nutr. Food Res, 2010, 54 Suppl 2, S244–52. [DOI] [PubMed] [Google Scholar]
- 26.Song M, Charoensinphon N, Wu X, Zheng J, Gao Z, Xu F, Wang M. and Xiao H, J. Agric. Food Chem, 2016, 64, 4943–4949. [DOI] [PubMed] [Google Scholar]
- 27.Tung YC, Li S, Huang Q, Hung WL, Ho CT, Wei GJ and Pan MH, J. Agric. Food Chem, 2016, 64, 3196–3205. [DOI] [PubMed] [Google Scholar]
- 28.Wu X, Song M, Rakariyatham K, Zheng J, Wang M, Xu F, Gao Z. and Xiao H, J. Agric. Food Chem, 2015, 63, 10921–7. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Song M, Rakariyatham K, Zheng J, Guo S, Tang Z, Zhou S. and Xiao H, J. Funct. Foods, 2015, 19, 278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Song M, Gao Z, Sun Y, Wang M, Li F, Zheng J. and Xiao H, J. Nutr. Biochem, 2017, 42, 17–25. [DOI] [PubMed] [Google Scholar]
- 31.Rupasinghe HPPV, Parmar I. and Neir SV, Oxid. Med. Cell. Longev, 2019, 2019, 4750795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song M, Wu X, Charoensinphon N, Wang M, Zheng J, Gao Z, Xu F, Li Z, Li F, Zhou J. and Xiao H, Food Funct., 2017, 8, 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song M, Wu X, Zheng J. and Xiao H, FASEB J., 2014, 28, 123.3. [Google Scholar]
- 34.Wu J-C, Tung Y-C, Zheng Y-N, Tsai M-L, Lai C-S, Ho C-T and Pan M-H, J. Food Bioact, 2018, 2, 98–103. [Google Scholar]
- 35.Jensen MM, Jørgensen JT, Binderup T. and Kjær A, BMC Med. Imaging, 2008, 8, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, Da Costa RG, Colaço B, Pires MJ, Colaço J, Ferreira R. and Ginja M, Lab Anim. (NY), 2013, 42, 217–224. [DOI] [PubMed] [Google Scholar]
- 37.Guo Y, Liu Y, Zhang C, Su Z-Y, Li W, Huang M-T and Kong A-N, Carcinogenesis, 2016, 37, 616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao H, Hao X, Simi B, Ju J, Jiang H, Reddy BS and Yang CS, Carcinogenesis, 2008, 29, 113–119. [DOI] [PubMed] [Google Scholar]
- 39.Pascal RR, Hum. Pathol, 1994, 25, 1160–1171. [DOI] [PubMed] [Google Scholar]
- 40.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, Pitot HC, Halberg RB, Itzkowltz SH, Groden J. and Coffey RJ, Gastroenterology, 2003, 124, 762–777. [DOI] [PubMed] [Google Scholar]
- 41.Chinen T, Komai K, Muto G, Morita R, Inoue N, Yoshida H, Sekiya T, Yoshida R, Nakamura K, Takayanagi R. and Yoshimura A, Nat. Commun, 2011, 2, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu G, Xiao H, You H, Lin Y, Jin H, Snagaski B. and Yang CS, Clin. Cancer Res, 2008, 14, 4981. [DOI] [PubMed] [Google Scholar]
- 43.Wu X, Song M, Wang M, Zheng J, Gao Z, Xu F, Zhang G. and Xiao H, Mol. Nutr. Food Res, 2015, 59, 2383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo S, Qiu P, Xu G, Wu X, Dong P, Yang G, Zheng J, McClements DJ and Xiao H, J. Agric. Food Chem, 2012, 60, 2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiou YS, Ma NJL, Sang S, Ho CT, Wang YJ and Pan MH, J. Agric. Food Chem, 2012, 60, 3441–3451. [DOI] [PubMed] [Google Scholar]
- 46.Livak KJ and Schmittgen TD, Methods, 2001, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 47.Zheng J, Song M, Dong P, Qiu P, Guo S, Zhong Z, Li S, Ho C-T and Xiao H, Mol. Nutr. Food Res, 2013, 57, 1999–2007. [DOI] [PubMed] [Google Scholar]
- 48.Zheng J, Bi J, Johnson D, Sun Y, Song M, Qiu P, Dong P, Decker E. and Xiao H, J. Agric. Food Chem, 2015, 63, 509–516. [DOI] [PubMed] [Google Scholar]
- 49.Charoensinphon N, Qiu P, Dong P, Zheng J, Ngauv P, Cao Y, Li S, Ho C-T and Xiao H, Mol. Nutr. Food Res, 2013, 57, 2103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clapper ML, Cooper HS and Chang WCL, Acta Pharmacol. Sin, 2007, 28, 1450–1459. [DOI] [PubMed] [Google Scholar]
- 51.Neufert C, Becker C. and Neurath MF, Nat. Protoc, 2007, 2, 1998–2004. [DOI] [PubMed] [Google Scholar]
- 52.Reagan-Shaw S, Nihal M. and Ahmad N, FASEB J., 2008, 22, 659–661. [DOI] [PubMed] [Google Scholar]
- 53.Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O. and Sartor RB, J. Clin. Invest, 2000, 105, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Robertis M, Massi E, Poeta ML, Carotti S, Morini S, Cecchetelli L, Signori E. and Fazio VM, J. Carcinog, 2011, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Black HE, Handbook of Toxicologic Pathology, Elsevier, 2002, vol. 1. [Google Scholar]
- 56.Sellers RS, Morton D, Michael B, Roome N, Johnson JK, Yano BL, Perry R. and Schafer K, Toxicol. Pathol, 2007, 35, 751–5. [DOI] [PubMed] [Google Scholar]
- 57.Rosenberg DW, Giardina C. and Tanaka T, Carcinogenesis, 2009, 30, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Afrin S, Giampieri F, Gasparrini M, Forbes-Hernández TY, Cianciosi D, Reboredo-Rodriguez P, Amici A, Quiles JL and Battino M, Food Funct., 2018, 9, 2145–2157. [DOI] [PubMed] [Google Scholar]
- 59.Ekbatan SS, Li XQ, Ghorbani M, Azadi B. and Kubow S, Int. J. Mol. Sci, 2018, 19, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Otto T. and Sicinski P, Nat. Rev. Cancer, 2017, 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo ZW, Zhu MG, Zhang ZQ, Ye FJ, Huang WH and Luo XZ, BMC Cancer, 2019, 19, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKay JA, Douglas JJ, Ross VG, Curran S, Loane JF, Ahmed FY, Cassidy J, McLeod HL and Murray GI, J. Pathol, 2002, 196, 386–393. [DOI] [PubMed] [Google Scholar]
- 63.Kikuchi Y, Dinjens WN and Bosman FT, Virchows Arch., 1997, 431, 111–7. [DOI] [PubMed] [Google Scholar]
- 64.Barnes LD, Garrison PN, Siprashvilia Z, Guranowskia, Robinson K, Ingram SW, Croce CM, Ohta M. and Huebner K, Biochemistry, 1996, 35, 11529–11535. [DOI] [PubMed] [Google Scholar]
- 65.Khan N, Afaq F. and Mukhtar H, Carcinogenesis, 2007, 28, 233–239. [DOI] [PubMed] [Google Scholar]
- 66.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C. and Mukaida N, J. Clin. Invest, 2008, 118, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Terzić J, Grivennikov S, Karin E. and Karin M, Gastroenterology, 2010, 138, 2101–2114. [DOI] [PubMed] [Google Scholar]
- 68.Zhao X, Si R, He M, Jiang X. and Zou S, Aquac. Fish, 2019, 4, 98–104. [Google Scholar]
- 69.Dranoff G, Nat. Rev. Cancer, 2004, 4, 11–22. [DOI] [PubMed] [Google Scholar]
- 70.Mager LF, Wasmer MH, Rau TT and Krebs P, Front. Oncol, 2016, 6, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li S, Sang S, Pan MH, Lai CS, Lo CY, Yang CS and Ho CT, Bioorganic Med. Chem. Lett, 2007, 17, 5177–5181. [DOI] [PubMed] [Google Scholar]
- 72.Nielsen SE, Breinholt V, Cornett C. and Dragsted LO, Food Chem. Toxicol, 2000, 38, 739–746. [DOI] [PubMed] [Google Scholar]
- 73.Wang M, Song M, Wu X, Gao Z, Xu F, Cao Y. and Xiao H, Abstr. Pap. Am. Chem. Soc, 2015, 250, Meeting abstract: 306. [Google Scholar]
- 74.Grana X. and Reddy EP, Oncogene, 1995, 11, 211–219. [PubMed] [Google Scholar]
- 75.Eymin B. and Gazzeri S, Cell Adh. Migr, 2009, 4, 114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karimian A, Ahmadi Y. and Yousefi B, DNA Repair (Amst)., 2016, 42, 63–71. [DOI] [PubMed] [Google Scholar]
- 77.Yam CH, Fung TK and Poon RYC, Cell. Mol. Life Sci, 2002, 59, 1317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sherr CJ and Bartek J, Annu. Rev. Cancer Biol, 2017, 1, 41–57. [Google Scholar]
- 79.Molinari M, Cell Prolif., 2000, 33, 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor WR and Stark GR, Oncogene, 2001, 20, 1803–1815. [DOI] [PubMed] [Google Scholar]
- 81.Charrier-Savournin FB, Château M-T, Gire V, Sedivy J, Piette J. and Dulic V, Mol. Biol. Cell, 2004, 15, 3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taylor WR, Schönthal AH, Galante J. and Stark GR, J. Biol. Chem, 2001, 276, 1998–2006. [DOI] [PubMed] [Google Scholar]
- 83.Fesik SW, Nat. Rev. Cancer, 2005, 5, 876–885. [DOI] [PubMed] [Google Scholar]
- 84.Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G. and Murcia JM, J. Biol. Chem, 1998, 273, 33533–33539. [DOI] [PubMed] [Google Scholar]