

Abstract
The production of a majority of chemicals involves heterogeneous catalysis at some stage, and the rates of many heterogeneously catalyzed processes are governed by transition states for dissociative chemisorption on metals. Accurate values of barrier heights for dissociative chemisorption on metals are therefore important to benchmarking electronic structure theory in general and density functionals in particular. Such accurate barriers can be obtained using the semiempirical specific reaction parameter (SRP) approach to density functional theory. However, this approach has thus far been rather ad hoc in its choice of the generic expression of the SRP functional to be used, and there is a need for better heuristic approaches to determining the mixing parameters contained in such expressions. Here we address these two issues. We investigate the ability of several mixed, parametrized density functional expressions combining exchange at the generalized gradient approximation (GGA) level with either GGA or nonlocal correlation to reproduce barrier heights for dissociative chemisorption on metal surfaces. For this, seven expressions of such mixed density functionals are tested on a database consisting of results for 16 systems taken from a recently published slightly larger database called SBH17. Three expressions are derived that exhibit high tunability and use correlation functionals that are either of the PBE GGA form or of one of two limiting nonlocal forms also describing the attractive van der Waals interaction in an approximate way. We also find that, for mixed density functionals incorporating GGA correlation, the optimum fraction of repulsive RPBE GGA exchange obtained with a specific GGA density functional is correlated with the charge-transfer parameter, which is equal to the difference in the work function of the metal surface and the electron affinity of the molecule. However, the correlation is generally not large and not large enough to obtain accurate guesses of the mixing parameter for the systems considered, suggesting that it does not give rise to a very effective search strategy.
1. Introduction
Transition states formed by the barriers to dissociative chemisorption (DC) can exert a high degree of rate control over the rates of heterogeneously catalyzed reactions proceeding over metal surfaces,1,2 such as ammonia production3,4 and steam reforming.5 It is therefore important to describe such barriers accurately. As the production of the majority of chemicals involves heterogeneous catalysis,6 being able to describe such barriers accurately is important. However, as discussed in several recent papers,7,8 it is not yet possible to use nonempirical present-day electronic structure theory to compute barriers for DC on metal surfaces with guaranteed chemical accuracy (errors ≤1 kcal/mol), although the development of an approach based on diffusion Monte Carlo certainly holds promise in this respect.9 Instead, success with achieving a chemically accurate description of DC on metals has so far been based on a semiempirical approach.7,10 Here, the specific reaction parameter (SRP) approach to density functional theory (DFT) is used to compute a potential energy surface (PES)10−16 or to construct forces used in direct dynamics calculations,17−20 and an empirical parameter in the functional used is tuned to achieve agreement between calculated and measured DC or “sticking” probabilities, as now documented extensively elsewhere.7 The barrier heights extracted from such work have already been collected in databases with barrier heights for DC on transition-metal surfaces.8,21 This endeavor is obviously important to progress with modeling heterogeneous catalysis, to testing existing and new density functionals,8,21 and to test first-principles methods like diffusion Monte Carlo.9 In this sense it is odd that databases used for benchmarking DFT,22−26 which with more than 30,000 papers published annually27 might be called the most important electronic structure method for complex systems, do not yet incorporate a database with barrier heights for DC on transition metals.
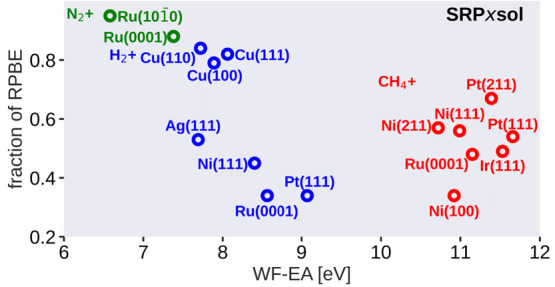
As summarized in a recent review paper7 and in a paper in which an extended database was published,8 the SRP-DFT approach has already been quite successful and has provided considerable physical insight into how DC on transition metals can be modeled with DFT. Accurate barrier heights have now been extracted for 14 systems in which H2, CH4, or N2 dissociate on a metal surface. For systems with a charge transfer parameter ΔECT = WF – EA greater than 7 eV (WF is the work function of the metal surface, and EA is the electron affinity of the molecule) the main challenge to SRP-DFT appears to find a density functional (DF) that generates the correct minimum barrier height for the system.7 Fortunately, and interestingly, for the systems for which the stated condition holds this can be achieved using DFs incorporating exchange from the generalized gradient approximation (GGA), even though a fraction of exact exchange is generally required for reproducing gas-phase reaction barriers.7,8 The success of the SRP-DFT approach for DC on metals strongly suggests,7 and diffusion Monte Carlo calculations on DC of H2 on Al(110) show,28 that nonempirical DFs tend to perform well at reproducing how DC barrier heights vary with geometry in a given system. The validity of minimum barrier heights and their variation with system geometry can be established through comparison of DC probabilities computed with a suitable dynamical model and method with values measured in supersonic molecular beam experiments.7 This statement can be underpinned7 with the so-called hole-model,29 which essentially holds that the DC probability can be computed as the fraction of geometries of the system for which the total energy exceeds the barrier height. Various strategies for developing SRP-DFs have been discussed in the recent review paper, one of which is to rely on the often-found transferability of an SRP-DF among chemically related systems.7
While the SRP-DFT approach has already been highly successful, it is also important to recognize that there have been some inadequacies in the approach used so far. An important shortcoming regarding the strategy of developing new SRP-DFs, which we will address here, has been that the approach to picking an expression for the SRP-DF has been rather ad hoc.10−20 Approaches used so far have been (i) to take a weighted average of two exchange correlation (XC) functionals within the generalized gradient approximation (GGA),10,11 (ii) to take a weighted average of two exchange (X) functionals within the GGA and to combine the resulting X functional with a GGA correlation (C) functional,30 (iii) as in (ii), but use a nonlocal C functional14,15,19,20 also approximately describing the attractive van der Waals interaction,31,32 (iv) to take a GGA exchange functional that was designed to be tunable33 and to combine it with nonlocal van der Waals correlation,12,13 and (v) to use meta-GGA functionals either with semilocal correlation34 or in combination with nonlocal correlation.16
The time is now ripe to address some basic issues in SRP functional construction, such as (i) can we use a generic expression of the density functional (DF) in such a way that the DF will usually work, and (ii) might it be possible to pick the expression in such a way that the tuning parameter (the “specific reaction parameter”) can be made to correlate with a specific property of the system. Two recent and closely related developments ensure that the time is now right. The first is that a new database of dissociative chemisorption barrier heights has recently become available, which has been called the SBH17 database.8 The database holds reference values of barrier heights to DC for 17 systems in which H2, N2, or CH4 dissociates over a metal surface. For 14 of these systems the barrier height was determined using SRP-DFT, while for 3 systems a more ad-hoc semiempirical procedure was used to extract a barrier height from a comparison between theory and experiment.8 In a second development35 it has become clear that the SRP-DFT approach based on GGA exchange functionals has its limits. So far this approach has only been successful for systems in which the charge transfer parameter ΔECT = WF – EA > 7 eV.35 Here WF is the work function of the metal, and EA is the electron affinity of the molecule. The SBH17 database therefore mostly contains systems for which this condition has been obeyed, which is the case for 16 out of the 17 systems.8 For systems with ΔECT < 7 eV the use of even one of the most repulsive GGA X DFs, i.e., RPBE,36 typically leads to underestimated barrier heights.35
Here we test several mixed DF expressions to see if we can derive generic expressions that work for all or most systems in the recently published database,8 to improve the toolbox of strategies aimed at deriving SRP-DFs for DC on metals. We also test the suggestion implicit in ref (35) that the fraction of the RPBE exchange needed in a mixed functional correlates with the value of the charge transfer parameter introduced above. This is also relevant to the strategy of developing new SRP-DFs7 and thereby extending the data available for databases: This should obviously be facilitated if a strong correlation between the fraction of RPBE exchange needed and the charge transfer parameter should exist. Our paper is set up as follows: In Section 2, the Methods Section, we give a short description of the database we use, which is essentially our previously published database with one system removed from it, in Section 2A. In Section 2A we also provide a lengthy discussion showing that high-level first-principles calculations are not yet capable of providing reference values of barrier heights for DC on metals, of how these values are instead extracted with semiempirical SRP-DFT based on experimental measurements, and what the accuracy is of the reference values in SBH17. Section 2B describes the DFs tested, and Section 2C gives computational details. Section 3 presents our results, and conclusions are drawn in Section 4.
2. Methods
2A. The SBH16 Database
The DFs described in Section 2B have been tested on what we here call the SBH16 database, which is the recently described SBH17 database8 with the H2 + Pt(211) system removed from it. The reason that we left out the H2 + Pt(211) system described here is that the results for this system are not that different from those for the H2 + Pt(111) system also contained in the SBH17 system, so that not so much is to be gained by adding results for the H2 + Pt(211) system to the results here presented. The SBH17 database holds results for 8 H2 + metal surface systems (SBH16 for 7 such systems), 2 N2 + metal surface systems, and 7 CH4 + metal systems. The reference values of the barrier heights for these systems and the most important geometrical parameters determining the barrier geometry of the molecule relative to the surface are all presented in Table 2 of ref (8). Ref (8) also provides the references to the papers in which fuller descriptions of the barrier geometries and of how they were derived may be obtained.
A few details regarding the SBH17 database are important to this paper. One is that the barrier heights and geometries are in principle defined best for the 14 out of the 17 systems (13 in SBH16), for which the reference values were obtained with SRP-DFT. Results for three systems (CH4 + Ni(100), CH4 + Ru(0001), and N2 + Ru(101̅0)) were obtained with more ad hoc semiempirical procedures, as discussed in detail in ref (8). The result that, of the three systems for which on average the largest errors were found with the density functionals tested, two systems were among the systems for which more ad hoc semiempirical procedures were used (i.e., CH4 + Ru(0001) and N2 + Ru(101̅0)) is consistent with the lower accuracy anticipated for the ad hoc procedure (the third system for which the DFs tested were least accurate on average was H2 + Ag(111)). Finally, a useful number characterizing the SBH16 database is the average value of the absolute values of the barrier heights contained in it, which is 0.687 eV (15.9 kcal/mol).
For a detailed understanding of our SBH17 database, it is useful to first summarize the state of the art in computing barrier heights for dissociative chemisorption on metals with high-level methods, also comparing them to how the methods concerned perform for databases of gas-phase reactions. For the gas-phase databases we will quote results for the BH76 database (with barrier heights for 38 H atom (HTBH38) and 38 non-H atom (NHTBH38) transfer reactions)23 and the DBH24 database, which is a statistically relevant subset of HTB38 and 44 hydrogen-atom transfer reactions.22 Note that over the years modifications have been made to the reference data, and it is most appropriate to label these databases by the year in which an (updated) database appeared.37
The coupled-cluster with single, double, and perturbative triple particle-hole excitation operators (CCSD(T))38 is typically considered the gold-standard of high-level ab initio methods. This method indeed reproduces reference values of gas-phase reaction barriers with subchemical accuracy, i.e., with a mean absolute error (MAE) of 0.46 eV for the DBH24/08 database.22 To the best of our knowledge, this method has not yet been used in published calculations of barriers for DC on metals. However, a periodic version of CCSD(T) has been used to compute barrier heights for DC of H2 on Si(100), for two reaction paths.39 The computed activation energies (0.70 and 0.75 eV, respectively) were in good agreement with the experimental lower bound quoted for the activation energy (0.6 eV).39 The periodic CCSD(T) method was therefore claimed to exhibit chemical accuracy for this reaction, although it is not quite clear how this can be done on the basis of a lower bound only for the experimental activation energy.
The diffusion Monte Carlo (DMC) method40,41 is likewise a highly accurate first-principles method, with an MAE of 1.2 kcal/mol for the BH76 database.7,42,43 It has been used to compute barrier heights for the H2 + Cu(111),9 H2 + Mg(0001),44 H2 + Al(110),28 and N2 + Cu(111)45 DC reactions. Only for H2 + Cu(111) a comparison based on experiment and semiempirical SRP-DFT calculations has been made, and for this one system it suggested the DMC barrier height to be accurate to within almost chemical accuracy (1.6 ± 1.0 kcal/mol).9
Calculations on DC on metals have also been done with embedded correlated wave function (ECW) methods,46,47 with the correlated wave function methods used including CASPT248,49 (a multireference second order perturbation theory based on a complete-active space reference function) and n-electron valence second-order perturbation theory (NEVPT2).50 The accuracy of these methods should be similar to that of multireference second-order Møller–Plesset perturbation theory (MRMP2), which yields an MAE of 1.4 kcal/mol for the DBH24/08 database.51 A potential energy surface based on embedded CASPT2 allowed DC probabilities of O2 dissociating on a simple metal surface (Al(111)) to be reproduced with near chemical accuracy,52 although the accuracy was also overestimated somewhat by only simulating the reaction of the rotational ground state.35 Calculations with embedded CASPT2 on DC of H2 on Cu(111)53 were less successful, producing a barrier height (0.15 eV) that fails to reproduce the semiempirical SRP-DFT value of 0.63 eV.10 (As explained in ref (8) in ref (53) a mistaken assessment of the quality of embedded CASPT2 was made for H2 + Cu(111) on the basis of an incorrect assumed value of the barrier height of only 0.05 eV.) The embedded NEVPT2 result for the barrier height (0.66 eV)53 is actually in much better agreement with the semiempirical SRP-DFT value (0.63 eV),10 but as the authors stated themselves it is not clear why the embedded CASPT2 and NEVPT2 methods should not agree for H2 + Cu(111).53 It is possible that there is a convergence problem with calculations of embedded CASPT2 and NEVPT2 on DC on transition-metal surfaces as the cost of these methods scales unfavorably with system size.
The random phase approximation (RPA),54−56 which may be viewed as a 5th rung density functional, yields an MAE of 2.3 kcal/mol for the BH76 database,57 which is larger than the DMC value quoted earlier (1.2 kcal/mol). To our knowledge, the RPA has not yet been used for the calculation of barrier heights for DC on metals in published work. The RPA has been used on DC of H2 on Si(100), with the resulting barrier heights showing similar deviations (up to 70 meV) from periodic CCSD(T) values computed in the same work as the DMC barriers computed earlier.39 The RPA has been tested on a reduced version (CE10)58 of a database of 25 chemisorption energies on transition metals (CE25).59 These RPA calculations yielded an MAE of 4.8 kcal/mol for the CE10 database, but there may have been problems with the convergence of these calculations.58 The RPA has also been applied to the calculation of adsorption energies in specific systems,60−63 also with applications to systems relevant to electrochemistry.64,65
It is also worth mentioning a recent ONIOM-type approach.66 In the approach used,67 “high-level” and “low-level” cluster and low-level periodic calculations were used to extract chemisorption energies for a database of 25 molecule-transition metal surface systems and to extract reaction barrier heights for a subset of 5 molecule-transition metal surface systems contained in the SBH10 database. The M06 global hybrid functional68 was used in the high-level cluster calculations, and the dispersion-corrected PBE-D3 GGA functional69−71 was used in the low-level cluster and periodic calculations. The approach gave an MAE of 2.2 kcal/mol for the chemisorption energies and of only 1.1 kcal/mol for the DC barrier heights. The latter number might seem impressive. However, the approach used contains deficiencies. In ref (67) the description of the methodology suggests that they have used transition-state geometries in which the positions of the surface atoms were relaxed in the presence of the molecule, while the reference values quoted in Table 4 of this paper are all for surface geometries relaxed with respect to the vacuum. Furthermore, according to ref (67) a zero-point energy correction was applied to the activation energies, while the reference values in their Table 4, to which they compare their PBE+D3/M06 results, did not contain this correction. This means that the MAE derived is unlikely to be accurate, and we advocate that the PBE+D3/M06 approach also be tested on a larger database such as SBH17,8 in a correct manner. Further objections one might raise to the ONIOM-based approach taken are that clusters with different combinations of numbers of atoms and shape were used for each of the 5 face-centered cubic (fcc) DC systems (although a protocol was followed in setting up the clusters) and that the size of the clusters is subject to restrictions for magnetic metal surfaces. Furthermore, the method will be hard to validate for applications requiring potential energy surfaces, as the approach requires the molecule to be centered on the cluster in the transition state geometry corresponding to one specific impact point of the surface.
In view of the lack of accuracy of high-level first-principles and of DFs on high rungs of Jacob’s ladder of Perdew,72 a different approach has been taken to obtain reference values for DC on transition metals, as already alluded to above. This approach is semiempirical. All reference values in the database are ultimately based on experiment or comparisons of theory with experiment. For most cases in the SBH17 database (see ref (8)), the specific reaction parameter approach to density functional theory (SRP-DFT) was used,7,10 which involves, for each system considered, the development of a functional (the SRP functional or SRP-DF) that is tailored to that specific system. This is done by fitting a parameter in the SRP-DF (the specific reaction parameter or SRP) to reproduce an experimental DC curve (also called sticking curve) for that system, which is given by the DC probability (or sticking probability) as a function of the average incidence energy of the molecule7,10 (see below for expressions used for SRP-DFs). We use sticking probabilities extracted from supersonic molecular beam experiments rather than rate constants because the former reflect the reaction at the well-defined surface geometries on the terraces of the low-index Miller surfaces used in the experiments. Instead, reaction rates often reflect reaction at unknown surface defect geometries.7,73 In the approach adopted, one accepts the DF tried as an SRP-DF if the computed sticking probability curve is shifted relative to the experimental curve along the incidence energy axis by less than 1 kcal/mol, which is generally accepted as the criterion for chemical accuracy. The approach will work if the dynamical model and the dynamics method used are selected to capture the important dynamical effects in the system and if the sticking probability is computed with appropriate thermal averaging over the rovibrational states of the reacting molecule and over the distribution of incidence translational energies. Here, “model” refers to, e.g., inclusion or not of surface atom motion and/or electron–hole pair excitation, and “method” refers to the use of, e.g., quantum or quasi-classical dynamics.7
Because the SRP-DFT approach is not only based on experiment but generally also on a parametrized DF that is adjusted to reproduce a specific experiment, the reference values extracted for the barrier heights for DC on metals have come to be called “semi-empirical”, rather than just “experimental”. Here, semiempirical is meant in a general sense, i.e., as “involving assumptions, approximations, or generalizations designed to simplify calculation or to yield a result in accord with observation” (Merriam-Webster dictionary). Usually computed and measured sticking probabilities exhibit a similar width (or equivalently slope), and this finding7 and DMC and DFT calculations on DC of H2 on Al(110)28 suggest that standard DFs usually get the distribution of the barrier heights (over geometries of the system) right but not the minimum barrier height, which requires tuning of the SRP.7 Because the shapes of the experimental and computed sticking probabilities are usually the same, their discrepancy can be characterized by a single parameter, i.e., the energy shift of the theoretical curve relative to the measured one. This also explains why the SRP-DFT procedure used, in which only one parameter is adapted in the DF being tailored to the experiment, works so well in practice.
As already mentioned above, once the shift is less than 1 kcal/mol the parametrized DF is accepted as an SRP-DF. Particular attention is paid to whether this is true also for the lowest incidence energies, which, in view of the constant energy shift mentioned, is usually the case. The semiempirical minimum barrier height is then extracted by using an appropriate algorithm such as the nudged elastic band74,75 or the dimer76 method, either using the SRP DF directly or using an accurate global fit of the PES computed using SRP DF data and used also in the dynamics calculations. Based on the constraint we put on the energy shift of the sticking probability curves measured and computed for a specific system for the DF to be an SRP-DF, the accuracy of the reference values for the barrier heights in SBH17 that were extracted with SRP-DFT is estimated as 1 kcal/mol. Barrier heights extracted with more approximate semiempirical procedures, as was done for 3 of the 17 systems, are likely less accurate. For a detailed discussion we refer to ref (8). Minimum barrier heights have now been extracted for 14 DC on transition metals with SRP-DFT (see ref (8) for the procedures used for the other 3 systems). The accuracy target of 1 kcal/mol, as also defined some time ago as the target to be set for energies by Pople,77 and usually referred to as “chemical accuracy” (see, e.g., ref (78)), is a useful target to set for the performance of electronic structure methods on DC on metals: it both reflects the accuracy thought to be achieved with SRP-DFT and would seem to be coming within reach with the highest-level first-principles methods now being tested on dissociative chemisorption, as described above.
The procedure used to obtain reference values for databases of gas-phase reaction barriers23,25,79,80 differs from that used for databases of surface reactions in two ways. First of all, in the gas-phase case many reference values come from high-level theoretical methods, and the proportion of the data coming from theoretical methods and the level of these methods have been increasing over the years as more accurate calculations became possible.22,37,81−83 The method adapted may be specific to the system in the database and may involve a specific model chemistry or a so-called multilevel model chemistry.22,77 Here, in this context a model chemistry is usually a combination of a specific high-level ab initio method with a specific basis set.22 Multilevel model chemistries may employ different high-level ab initio methods and/or different basis sets and use these to extrapolate to more accurate results.22 To give an example, at the high end the 2008 version of the DBH24 database, DBH24/08,22 contains some data from so-called Weizmann-4 theory, which uses different basis sets and ab initio methods beyond CCSD(T) and is able to provide atomization energies with an accuracy of 0.1 kcal/mol or better.84
Second, databases of gas-phase reactions, and certainly the older versions of these databases, may contain values that are based on experiments or on experiments and electronic structure calculations. Again, taking the DBH24/08 database as an example, the reference data for some of the reactions were taken from a comparison of rate constants based on measurements and electronic structure calculations, using either quantum dynamics or sophisticated versions of transition state theory, like variational transition state theory.22 For some reasons, these reference values are usually labeled as “experimental”, though arguably there is a semiempirical flavor to the procedure used in the earlier work, as considerations from electronic structure calculations are also taken into account. We therefore conclude that, to a large extent, labeling some of the reference values in gas-phase reaction barrier databases as “experimental” rather than as “semiempirical” is a matter of semantics. We prefer to label the reference data in the SBH17 database as “semiempirical” because in the majority of cases the reference value of the minimum barrier height was derived on the basis of a semiempirical density functional adjusted to reproduce measured sticking coefficients for that reaction.
2B. Mixed Density Functional Expressions
The XC part of DFs used as SRP-DFs has typically been taken as mixtures of the X and C components of standard XC DFs. This has the advantage that constraints enforced in constraint-based X and C DFs can also be enforced in SRP-DFs.85 Based on previous experience, we test the following expressions for the exchange-correlation part of the mixed DFs.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
and
| 6a |
| 6b |
and
| 7a |
| 7b |
The ESRPxXC DF of eq 1 has been
used to arrive at a reparameterized SRP DF for H2 + Cu(111),30 the original version being a weighted average
of the RPBE36 and PW9186 DFs.10 In the limit x = 0 the DF defined by eq 1 corresponds to the PBE69 DF, and in the limit x =
1 it corresponds to the RPBE DF (which has the PBE C DF as the correlation
part of its exchange-correlation functional36). Choosing eq 1 in
attempts to derive an SRP DF for a DC-on-metal-surface system is in
accordance with conventional wisdom that PBE often under-predicts
and RPBE often overpredicts the barrier height for DC on a metal surface.7 In terms of the reduced density gradient 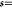
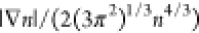 , where n is the total
electron density, the limiting forms of the exchange enhancement factor
of the DF defined by eqs 1–7a for x = 1 (RPBE) and by eqs 1, 3, and 4 for x = 0 (PBE) may be written as follows.
, where n is the total
electron density, the limiting forms of the exchange enhancement factor
of the DF defined by eqs 1–7a for x = 1 (RPBE) and by eqs 1, 3, and 4 for x = 0 (PBE) may be written as follows.
| 8 |
| 9 |
Here, μ = 0.21951, and κ = 0.804.36,69 These exchange enhancement factors are plotted as functions of s in Figure 1.
Figure 1.
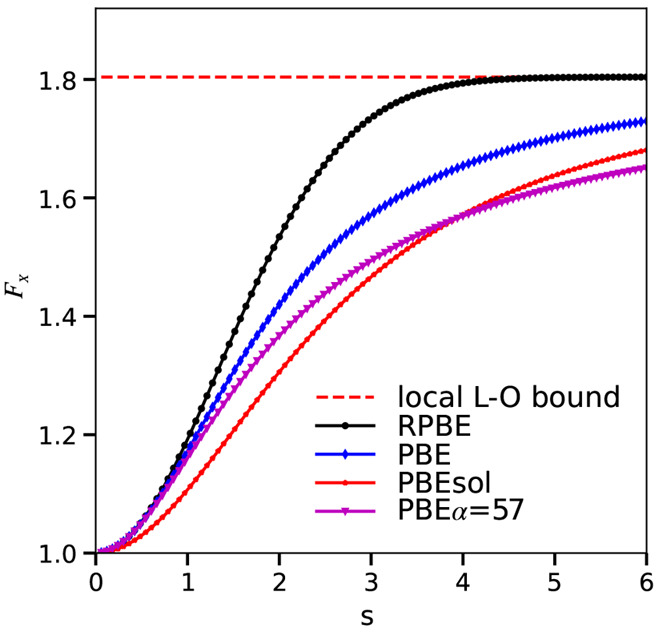
Exchange enhancement factor (Fx) as a function of the reduced density gradient (s) for the RPBE36 (black), PBE69 (blue), PBEα33 (with α = 0.57, magenta), and PBEsol87 (red) functionals. The horizontal red dashed line presents the local Lieb-Oxford bound88 imposed in the construction of these functionals.33,36,69,87
We note that in constructing the PBE DF, the developers of this DF have built69 on the earlier PW91 DF.86 More specifically, PBE was built based on similar, though not identical, nonempirical constraints, with PBE only satisfying those constraints that the developers thought to be energetically significant.69 As a result of this, the exchange-correlation enhancement factors of PW91 and PBE are nearly identical for s ≤ 3.0 (see Figure 1 of ref (69)), and the exchange enhancement factors of these two DFs only start to differ for s > 4.0 (see Figure 1 of ref (89)). The region of s-values for which the two DFs are in good agreement contributes most to the exchange energies of atoms90,91 and covalent molecular systems.90 Perhaps as a result of this, barriers to dissociative chemisorption on metal surfaces computed with PW91 and PBE tend to be in very good agreement with one another (differences smaller than 0.02 eV for barrier heights exceeding 0.6 eV, for instance, cf. Tables 2 (PW91 results) and 3 (PBE results) of ref (92) for CH4 + Ni(111), and see Figure 5 and cf. Figures 7 and 8 of ref (93) for HCl + Ag(111)). Differences between the exchange enhancement factors of PW91 and PBE that occur for large values of the reduced density gradient (s > 4.089) may, however, be relevant to the calculation of the exchange energy in regions where s is large and the density is low.90,94 In these regions exchange and correlation need to be carefully balanced to correctly describe the van der Waals interaction energy of a molecule approaching the surface,90,94 but this obviously also requires the use of a correlation functional that yields at least a qualitative description of the van der Waals energy.31,32 Furthermore, even for systems where the barrier occurs far from the surface like H2 + Ru(0001), differences in PW91 and PBE barrier heights tend to be small (<0.02 eV), as are differences between computed dissociative chemisorption probabilities (see Figures 4 and 9 of ref (14), respectively). Finally, we note that PW91 and PBE gave somewhat different results for monovacancy formation energies of Pt and Al, for reasons that were not well-understood, but the differences are not large.95
A drawback of using eq 1 is that with PBE the barrier height for DC on a metal surface may also be overestimated in specific cases, even though this DF has a negative mean signed error (MSE) of −58 meV for the SBH17 database.8 For this database, overestimated (though often not by much) barrier heights were observed for a few weakly activated or nonactivated H2-metal systems (H2 + Pt(111), Pt(211), and Ru(0001)), for H2 + Ag(111), and for a few CH4 + metal surface systems (CH4 + Ni(100), Pt(211), and Ru(0001)). To avoid this we replaced the X DF of PBE by the X DF of PBEsol,87 which tends to yield lower barriers, this way obtaining the ESRPxsolXC mixed DF of eq 2. It should be noted that for x = 0 ESRPxsolXC does not equal the PBEsol functional, which employs the same expression for the C functional as PBE but uses a different value of a coefficient in it to balance the C part of PBEsol against its X part.87 However, as we will show, the use of ESRPxsolXC comes with the advantage that where necessary it yields lower barrier heights for DC on metals than ESRPxXC, and thus ESRPxsolXC is more tunable than ESRPxXC. Below, we will call the x = 0 limit of ESRPxsolXC PBEsolc, to distinguish it from PBEsol. The limiting x = 0 form of the exchange enhancement factor of the DFs defined by eq 2 and 5 (PBEsol) may be written as
| 10 |
where the form used is identical to the PBE expression (eq 9) but μGE = 0.1235 instead of μ = 0.21951 is used.87 This exchange enhancement factor is also plotted as a function of s in Figure 1.
A drawback of both eqs 1 and 2 is that the attractive van der Waals interaction between molecule and surface is not described with a GGA correlation functional, even though this may be necessary for weakly activated DC of H2 on metals (where the barrier is usually at a fairly large molecule–surface distance so that a proper description of the van der Waals interaction may be important in spite of its weakness12,13) or for CH4 dissociating on a metal surface.19,20 For this reason we also test the DFs of the forms ESRPx-vdW1XC of eq 3 and ESRPx-vdW2XC of eq 4, which contain the vdW-DF1 C functional31 and the vdW-DF2 C functional,32 respectively. The ESRPx-vdW1XC functional has been used successfully to describe supersonic molecular beam experiments on CH4 + Ni(111),19 Pt(111),20 and Pt(211)20 and on H2 + Ru(0001).14 The ESRPx-vdW2XC functional has been used successfully to describe H2 + Ru(0001)14 and Ni(111).15
The DFs described by eq 3 and 4 may have a problem similar to that of the DF described by eq 1, i.e., that the barrier height is already overestimated with x = 0, including PBE exchange only. For instance, the SRP-DF found for H2 + Pt(111)12 and Pt(211)13 is given by EPBEα = 0.57,vdW2XC = EPBEα = 0.57X+EvdW-DF2C, where EPBEα = 0.57X is the inherently tunable PBEα X DF,33 with α = 0.57. For the PBEα exchange DF, the exchange enhancement factor is given by
| 11 |
and is plotted as a function of s in Figure 1 for α = 0.57, which is the lowest value we have used in calculations in this work. (We note that there was a typo in the equation describing FX in the original paper describing the PBEα functional;33 in this equation, x1 should have appeared as x = μs2.)96 As discussed by the developer of the PBEα X functional,33 PBEα = 1 corresponds to the PBE functional, while PBEα = 0.52 is very similar to the X part of the WC functional,97 which like PBEsol87 was developed with a view to a better description of the solid state. The ESRPx-vdW2XC value with x = 0 (only PBE exchange) overestimates the barrier height for almost all systems in the SBH17 database. For this reason, we have also tested the DF ESRPxsol-vdW2XC of eq 5, which for x = 0 consists of PBEsol exchange and the vdW-DF2 correlation functional. In this limit, this DF is expected to yield low barriers like EPBEα = 0.57,vdW2XC.
To increase the tunability of a mixed DF expression as given by eqs 1, 3, and 4, PBE exchange can be replaced by PBEsol exchange, as done in eq 2 to obtain a better tunable DF than the DF of eq 1, and in eq 5 to obtain a better tunable DF than the DF of eq 4. An alternative already implicitly used in the construction of SRP-DFs is to replace PBE exchange by PBEα exchange with α < 1, as done to obtain ESRPx-vdW1-extXC of eq 6a (which should be more tunable than ESRPx-vdW1XC of eq 3) and to obtain ESRPx-vdW2-extXC of eq 7a (which should be more tunable than ESRPx-vdW2XC of eq 4). We have not made use of the possibility of the PBEα functional to interpolate between PBE and RPBE exchange, as the PBEα X functional corresponds to the RPBE X functional only in the limit α →∞, which is a rather awkward limit to work with, and less preferred to a situation where switching from PBE to RPBE exchange can be performed by switching a parameter continuously from 0 to 1, as can be done in eqs 1, 3, and 4.
The DFs of eqs 1–5, 6a, and 7a have been evaluated for x = 0, nΔx with n = 1–9, and 1.0, modifying x by steps Δx equal to 0.1. The DFs of eq 6 and eq 7b have been evaluated for α = 0.57 (x = −0.43), α = 0.70 (x = −0.30), and α = 0.85 (x = −0.15). For each system, the best value of x was defined for the DFs given by eqs 1–7a as described in more detail below. If for the resulting x we have 0.0 ≤ x ≤ 1.0 for a DF defined by one of the eq 1–5 values, the interpolation was successful and the DF expression can be used for the system considered. Similarly, if for the resulting x we have −0.43 ≤ x ≤ 1.0 for a DF defined by one of the eqs 6 or 7 the interpolation was successful and the DF expression can be used for the system considered. Otherwise, extrapolation was used, and the corresponding generic DF was found not to be able to describe the system successfully.
Before closing this section, it is worthwhile to compare the limiting forms of the DFs tested here by inspecting Figure 1 and considering their Taylor expansions. As can be seen, over the range of s considered the exchange enhancement factor of the RPBE DF is consistently larger than that of the PBE DF, which is consistently larger than those of the PBEsol and PBEα = 0.57 DFs. As larger s values tend to correspond to higher barriers,98 we may expect the RPBE barriers to be consistently higher than the PBE barriers, in agreement with conventional wisdom.7 Furthermore, replacing PBE with PBEα or PBEsol should, according to Figure 1, be successful in increasing the tunability of SRP-DFs. Finally, as the exchange enhancement factor of the PBEsol DF is lower than that for PBEα = 0.57 for s up to about 4, one would expect the use of this DF as the lower limit of the SRP-DF to be most successful at increasing its tunability. Finally, one observes that at low s values the RPBE, PBE, and PBEα = 0.57 DFs all behave similarly, while the exchange enhancement factor of the PBEsol DF is clearly lower for small s-values. The enhancement factors of the RPBE, PBE, and PBEα = 0.57 DFs start to diverge only for values of s greater than 1. To understand this, it is useful to consider the Taylor expansions of the four DFs up to the fourth order in s.
| 12a |
| 12b |
| 12c |
| 12d |
The Taylor expansions help us to understand much of the observed behavior. For instance, at low s values the exchange-enhancement factors of the RPBE, PBE, and PBEα = 0.57 DFs are similar because their Taylor expansions are identical up to second order in s. At these small values of s the exchange-enhancement factor of the PBEsol DF is significantly smaller, and it remains smaller up to s ≈ 4 because it uses the gradient expansion form of μ (μGE = 0.1235) that is accurate for slowly varying electron gases.87 As a result, the exchange-enhancement factor ends up being much smaller at small s even though the form of its second-order Taylor expansion is identical to that of the others, which however all use μ = 0.21951. At larger s, where the s4 term kicks in, differences between RPBE, PBE, and PBEα may be understood from the different coefficients in front of the term μ2s4/κ, which equal −0.5, −1, and −1.377 for RPBE, PBE, and PBEα = 0.57, respectively. As noted before, differences between the exchange enhancement factors that occur for large values of the reduced density gradient, as observed in Figure 1, may be relevant to the calculation of the exchange energy in regions where s is large and the density is low.90,94 Finally, we note that in Figure 1 the exchange-enhancement factors all obey the condition that FX(s) ≤ 1.804, which is known as a local Lieb-Oxford bound and which is a sufficient condition36,69 for the global Lieb-Oxford bound on the exchange energy88 being obeyed. As discussed by Marques and co-workers, this is not a necessary condition, and in real systems the local Lieb-Oxford bound may be violated.99
2C. Computational Details
The minimum barrier height is computed as follows.
| 13 |
In eq 13ETS is the energy of the system (molecule + surface) at the minimum barrier geometry, while Easym is the energy of the system with the molecule in its equilibrium geometry at a distance from the surface such that molecule and surface no longer interact. In the so-called medium algorithm that we use, which is defined and explained in detail in ref (8), the surface is set up following DFT geometry optimizations of the bulk lattice (to determine the bulk lattice constant(s) with the DF used) and of the metal slab representing the surface (to determine interlayer spacings in the metal surface slab exposed to vacuum according to the DF used). The geometry of the molecule relative to the surface is taken from earlier SRP-DFT calculations as described in ref (8) (see also Table 2 of ref (8)). In the asymptotic geometry the equilibrium distance of the molecule is likewise computed with the DF tested.8 A crucial point is that the surface is not allowed to relax with respect to the incoming molecule in the calculation of ETS. A minor difference with ref (8) is that in the present work the geometry optimization of the bulk representing the surface was done using the geometry optimization method implemented in VASP. In the earlier calculations of ref (8), a parabola was fitted to the energy of the bulk as a function of the lattice constant, and minimization was used to establish the bulk lattice constant. The new approach led to small differences in the values of the barrier heights (of 10 meV or less) with respect to the early results when they were available for the particular DF tested.
All DFT calculations were performed with a user-modified version of the Vienna ab initio simulation package100−103 (VASP5.4.4). We also used the Atomic Simulation Environment (ASE)104,105 as a convenient interface package. All calculations using the vdW-DF1 or vdW-DF2 C functionals were done with the algorithm of Román-Pérez and Soler106 to speed up their evaluation. All other details regarding the calculations (concerning the pseudopotentials used, the handling of spin-polarization in systems containing Ni, the number of metal layers in the slab representing the surface, the size of the surface unit cell, etc.) are the same as in ref (8), to which we refer for these details.
3. Results and Discussion
3A. Equilibrium Lattice Constants Computed with Mixed Density Functionals
Equilibrium lattice constants computed with the mixed density functional expressions not incorporating the van der Waals interaction are shown in Table S1 of the Supporting Information, stepping through x in SRPx and SRPxsol in steps of 0.1 (results for the other mixed DFs not shown). Comparing with zero-point energy-corrected experimental values we obtain the usual result that the PBE DF somewhat underestimates and that RPBE overestimates lattice constants.107,108 The PBEsolc DF (we recall that PBEsolc is the name we use for the DF with PBEsol exchange and PBE correlation) tends to somewhat underestimate the lattice constant. The PBEsol DF would be expected to do rather well for the lattice constant,108 and we suspect that PBEsolc somewhat underperforms as using PBE correlation with PBEsol exchange should lead to a somewhat unbalanced functional.87 One might of course vary x in the SRPxsol DF to obtain the correct lattice constant, but this is not likely to lead to the correct barrier height, as GGA DFs yielding good molecule–surface interaction energies tend to overestimate metal lattice constants.87,109
3B. Performance of Limiting Forms of the Mixed Density Functionals
To get an impression of how the mixed density functional expressions will perform as generic expressions for fitting SRP functionals, it is a good idea to look at how their limiting forms perform and compare. For this, we first consider the limiting forms of the mixed expressions not using van der Waals correlation functionals, i.e., SRPx (eq 1) and SRPxsol (eq 2), which are PBE and RPBE, and PBEsolc (we recall that this is the name we use for the DF with PBEsol exchange and PBE correlation) and RPBE. Figure 2 shows that for each system in the SBH16 database the barrier height obtained with PBE is lower than that obtained with RPBE, which correlates well with the finding that PBE often underestimates while RPBE often overestimates barrier heights.7 Our results show for all systems investigated here that using RPBE instead of PBE raises the barrier height because the energy of the transition state increases more than the energy of the reactants (of the system with the molecule in the gas phase). This is not completely trivial as the same change in barrier height may also result from the energy of the transition state decreasing less than the energy of the reactants.98 Also, for each system in the SBH16 database, the barrier height obtained with PBEsolc is lower than that obtained with PBE, suggesting that for the purpose of fitting barrier heights, the SRPxsol expression will be tunable over a wider range than the SRPx expression. The barrier heights computed with the PBEsolc, PBE, and RPBE functionals may also be found in Table S2 of Supporting Information.
Figure 2.

Barrier heights Eb computed with the PBEsolc, PBE, and RPBE DFs are shown as a function of the charge transfer parameter for the 16 systems present in the SBH16 database.
Barrier heights obtained for each system in the SBH16 database with the limiting forms of the SRPx (eq 1), SRPxvdW1 (eq 3), and SRPx-vdW2 (eq 4) expressions are shown in Figure 3 for PBE, PBE-vdW1, and PBE-vdW2 and in Figure 4 for RPBE, RPBE-vdW1, and RPBE-vdW2. Whether PBE or PBE-vdW1 yields the lowest barrier height is seen to depend on the value of ΔECT: for ΔECT ≤ 8.055 eV, PBE yields the lowest barrier height, while for ΔECT ≥ 8.395 eV, PBE-vdW1 yields the lowest barrier height. While this might look odd, one should remember that the correlation part of the vdW-DF1 functional is not just a van der Waals term that is added to an energy expression excluding the attractive dispersion interaction (e.g., the PBE energy). Rather, the vdW-DF1 correlation functional is a different correlation functional from the PBE correlation functional. There is thus no a priori reason that the PBE-vdW1 energy should always be lower than the PBE energy or vice versa. Furthermore, the barrier obtained with PBE-vdW2 is almost always higher than that obtained with both PBE-vdW1 and PBE (only for H2 + Ru(0001) is the barrier higher for PBE-vdW1 than for PBE-vdW2). The findings for RPBE, RPBE-vdW1, and RPBE-vdW2 (Figure 4) are analogous to those for PBE, PBE-vdW1, and PBE-vdW2 (Figure 3). The barrier heights computed with the PBE, PBE-vdW1, PBE-vdW2, and RPBE functionals may be found in Table S2 of the Supporting Information, and the barrier heights computed with the RPBE-vdW1 and RPBE-vdW2 functionals may be found in Table S3 of the Supporting Information.
Figure 3.

Barrier heights Eb computed with the PBE, the PBE-vdW1, and the PBE-vdW2 DFs are shown as functions of the charge transfer parameter for the 16 systems present in the SBH16 database.
Figure 4.

Barrier heights Eb computed with the RPBE, the RPBE-vdW1, and the RPBE-vdW2 DFs are shown as a function of the charge transfer parameter for the 16 systems present in the SBH16 database.
Barrier heights obtained for each system in the SBH16 database with the lower-limit forms of the SRPxsol (eq 2) and SRPxsol-vdW2 (eq 5) expressions are shown in Figure 5 for PBEsolc and PBEsol-vdW2. As can be seen, the barriers obtained with PBEsolc-vdW2 are always higher than those obtained with PBEsolc, suggesting that the SRPxsol-vdW2 DF may be slightly less tunable than the SRPxsol DF, which yields very low barriers. The barrier heights computed with the PBEsolc functional are listed in Table S2 of the Supporting Information, and the barrier heights computed with the PBEsol-vdW2 functional are listed in Table S3 of the Supporting Information.
Figure 5.

Barrier heights Eb computed with the PBEsolc and PBEsol-vdW2 DFs are shown as a function of the charge transfer parameter for the 16 systems present in the SBH16 database.
Finally, barrier heights obtained with the PBEα-vdW1 and PBEα-vdW2 DFs are compared in Figure S1 of the Supporting Information for α = 0.57, which is the lowest value of α used here. Figure S1 shows that the PBEα-vdW1 DF consistently yields barrier heights lower than those of the PBEα-vdW2 DF with α = 0.57. This suggests that the PBEα-vdW1 DF is a better tunable mixed DF than the PBEα-vdW2 DF, as the RPBE-vdW1 and RPBE-vdW2 DFs overestimate the barrier height for each system in the SBH16 database (see the discussion of Table 1 below).
Table 1. Performance of the DFs That Represent Limiting Forms of the Mixed Density Functionals Tested on the SBH16 Database Using the Medium Algorithma.
| Med
Algo |
||||
|---|---|---|---|---|
| Functional | MAE | MSE | MAE-SBH17 | MSE-SBH17 |
| PBE | 0.107 | –0.065 | 0.103 | –0.058 |
| RPBE | 0.235 | 0.235 | 0.228 | 0.228 |
| PBEsolc | 0.458 | –0.458 | ||
| PBEsol-vdW-DF2 | 0.269 | –0.265 | ||
| PBE-vdW-DF1 | 0.128 | –0.020 | ||
| PBE-vdW-DF2 | 0.148 | 0.117 | 0.141 | 0.112 |
| PBEα+57-vdW-DF1 | 0.209 | –0.185 | ||
| PBEα57-vdW-DF2 | 0.132 | –0.042 | 0.124 | –0.040 |
| RPBE-vdW-DF1 | 0.278 | 0.278 | ||
| RPBE-vdW-DF2 | 0.424 | 0.424 | ||
| Average | 0.239 | 0.002 | ||
The mean absolute errors (MAEs) and mean signed errors (MSEs) are presented in eV for all density functionals investigated here. For the density functionals for which these results are available we also present MAEs and MSEs computed previously for the closely related SBH17 database.8.
Table 1 shows mean absolute errors (MAEs) and mean signed errors (MSEs) for the SBH16 database, also comparing to the previous SBH17 results for those DFs that have previously been tested on this database.8 Here the error for a specific system is defined as the difference between the barrier height computed here and the reference value tabulated in ref (8) for that system. As can be seen, the MAEs and MSEs computed here for SBH16 differ from previous results known from SBH17 by no more than 10 meV, underscoring the reliability of the results presented here. As previously found, the PBE DF is the best-performing DF in terms of the MAE, the MAE being lowest for the PBE DF. Importantly for this study, the DFs serving as upper limits for mixed DFs here (RPBE for SRPx of eq 1 and SRPxsol of eq 2, RPBE-vdW1 for SRPx-vdW1 of eq 3 and for SRPx-vdW1-ext of eq 6a, and RPBE-vdW2 for SRPxvdW2 of eq 4, SRPxsol-vdW2 of eq 5, and SRPx-vdW2-ext of eq 7a) all have their MSEs equal to their MAEs, suggesting that these DFs all systematically overestimate the barrier height. This is actually a good quality of a functional that is meant to serve as the upper-limit form of a mixed DF. The PBEsolc DF, which is the lower-limit form of the SRPxsol DF of eq 2, shows an MSE that is equal to minus its MAE, suggesting that this DF systematically underestimates the barrier height. This is a good quality of a functional that is meant to serve as the lower-limit form of a mixed DF, and in view of the behavior of the RPBE DF we expect that the SRPxsol DF of eq 2 will perform well as a generic expression for reproducing barrier heights by tuning its x-parameter. Unfortunately PBE (the lower-limit-form of SRPx of eq 1), PBE-vdW1 (the lower limit of SRPx-vdW1 of eq 3), PBE-vdW2 (the lower limit of SRPx-vdW2 of eq 4), PBEsol-vdW2 (the lower limit of SRPxsolvdW2 of eq 5), PBEα57-vdW1 (the lower limit of SRPx-vdW1-ext of eq 6a), and PBEα57-vdW2 (the lower limit of SRPx-vdW2-ext of eq 7a) all have the idea that their MSE is not equal to minus their MAE, meaning that these DFs do not systematically underestimate the barrier height for the systems in SBH17. Of these DFs, on the basis of the correspondence between their MAE and the negative of their MSE, PBEsol-vdW2 and PBEα57-vdW1 are expected to function best as lower-limit forms, and consequently the mixed DFs SRPxsol-vdW2 and SRPx-vdW1-ext are also expected to perform well as tunable mixed DFs.
3C. Performance of Mixed Density Functionals as Tunable SRP DFs
Figure 6 illustrates how we find the optimal value of x for each mixed DF by showing how this was done for the particular examples of the H2 + Cu(111) and CH4 + Pt(111) systems using the mixed DFs SRPx and SRPxsol of eqs 1 and 2. As Figure 6A,B shows, the barrier height obtained with a mixed DF typically depends linearly on x. This means that the optimal value of x can be found using linear interpolation, i.e., from the point where the linearly interpolated barrier height curves (the sloping red and black lines) intersect the horizontal blue line, representing the reference value of the barrier height. If x does not fall between the limits of the mixed DF (0 and 1 for the expressions of eqs 1–5, and −0.43 and 1 for eqs 6 and 7) a value of x can be found by extrapolation. We have not tested whether the DFs that may be obtained by extrapolation lead to reasonable values of the minimum barrier height; we do not recommend their use. However, the values of x obtained in this way may be used in the calculation of the correlation coefficients discussed in the next Section.
Figure 6.
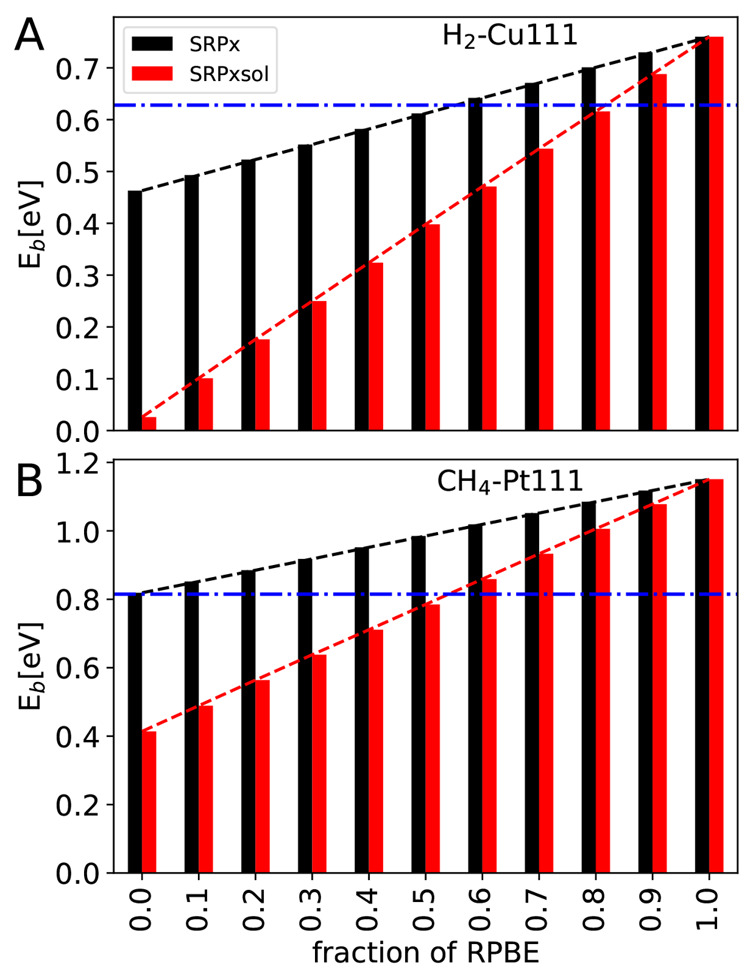
Barrier heights computed with the SRPx DF (black bars) and the SRPxsol DF (red bars) are shown as a function of the fraction of RPBE exchange x, for (A) H2 + Cu(111) (upper panel) and (B) CH4 + Pt(111) (lower panel). Blue horizontal lines indicate the reference value of the barrier height for these systems.8 The black and red dashed lines linearly interpolate the barrier height as a function of x for SRPx and SRPxsol DFs, respectively. The optimal value of x is equal to the value of x for which these lines intersect with the blue lines.
Figures 7 and 8 show the optimal x coefficients computed for the SRPx and SRPxsol DFs of eqs 1 and 2, respectively, as a function of ΔECT. These coefficients are also listed for each DF in Table S4. Figure 7 shows that obtaining the optimum value of x for the SRPx DF required extrapolation to negative values for several H2-metal and CH4-metal surface systems. The use of this mixed DF is therefore not guaranteed to yield a useful SRP DF for systems like the ones investigated here. From the point of view of tunability, the opposite is true for the SRPxsol DF, for which we obtained a value of x falling between 0 and 1 for all systems in the SBH16 database (see Figures 7 & 8).
Figure 7.

Optimum fraction of RPBE exchange x is shown as a function of ΔECT for the SRPx DF (eq 1). Values falling between the two horizontal dot-dashed black lines could be obtained by the interpolation procedure illustrated in Figure 6. The green, blue, and red symbols correspond to N2, H2, and CH4 + metal surface systems, respectively. The black, blue, and red dashed lines provide the linear fits corresponding to the Pearson correlation coefficients computed for all molecules, H2, and CH4 + metal surface systems, respectively, without omitting systems.
Figure 8.

Optimum fraction of RPBE exchange x is shown as a function of ΔECT for the SRPxsol DF (eq 2). Values falling between the two horizontal dot-dashed black lines could be obtained by the interpolation procedure illustrated in Figure 6. The green, blue, and red symbols correspond to N2, H2, and CH4 + metal surface systems, respectively. The black, blue, and red dashed lines provide the linear fits corresponding to the Pearson correlation coefficients computed for all molecules, H2, and CH4 + metal surface systems, respectively, without omitting systems.
Figure 9 shows the optimal x coefficients computed for the SRPxsol-vdW2 DF of eq 5 as a function of ΔECT. These coefficients are also listed for this DF in Table S5. Figure 9 shows that obtaining the optimum value of x for the SRPxsol-vdW2 DF only required extrapolation to a negative value for H2 + Ag(111). This system was classified as problematic in the SBH17 study, with all DFs tested there yielding large MAEs for this system.8 While we conclude that the use of this mixed DF is not guaranteed to yield a useful SRP DF for systems like the ones investigated here, we find that it performs rather well and that it can probably be used if an SRP DF is desired with vdW-DF2 correlation in it. Note that, when coupled to their original partner exchange functionals,31,32 the vdW-DF2 functional32 yields a better description of the S22 database binding energies of gas-phase dimers (MAE of 22 meV)32 than the vdW-DF1 functional32 (MAE of 41 meV).31 However, the vdW-DF1 functional31 generally yields a better description of bulk solids110 than the vdW-DF2 functional.32 The greater tunability of the SRPxsol DF comes from using an exchange enhancement factor that is more appropriate for solids and surfaces than for molecules through the use of μGE = 0.1235 in PBEsol87 instead of μ = 0.21951 in PBE.69 This leads to overall lower values of the gradient enhancement factor as a function of s, which leads to lower barriers.98
Figure 9.

Optimum fraction of RPBE exchange x is shown as a function of ΔECT for the SRPxsol-vdW2 DF (eq 5). Values falling between the two horizontal dot-dashed black lines could be obtained by the interpolation procedure illustrated in Figure 6. The green, blue, and red symbols correspond to N2, H2, and CH4 + metal surface systems, respectively. The black, blue, and red dashed lines provide the linear fits corresponding to the Pearson correlation coefficients computed for all molecules, H2, and CH4 + metal surface systems, respectively, without omitting systems.
Figure 10 shows the optimal x coefficients computed for the SRPxvdW1-ext DF of eq 6a as a function of ΔECT. These coefficients are also listed in Table S5. Figure 10 shows that obtaining the optimum value of x for the SRPx-vdW1-ext DF only required extrapolation to a negative value for H2 + Cu(110) and H2 + Ag(111). The latter system was classified as problematic in the SBH17 study, with all DFs tested there yielding large MAEs for this system.8 The use of the SRPx-vdW1-ext mixed DF is not guaranteed to yield a useful SRP DF for systems like the ones investigated here, but we find that it performs rather well just like SRPxsol-vdW-DF2, and SRPx-vdW1-ext can be used if an SRP-DF is desired with vdW-DF1 correlation in it. As noted above, when partnered with their original exchange functionals vdW-DF1 yields better descriptions of bulk solids, while vdW-DF2 tends to be better for binding energies of gas-phase dimers. Finally, we note that the H2 + Ag(111) system is among the 1 (2) systems for which the optimum fraction of RPBE exchange could not be obtained through interpolation with the SRPxsol-vdW2 DF (the SRPx-vdW1 DF), as can be seen from Figure 9, respectively. This confirms the analysis of ref (8), which already suggested revisiting this system with new experiments and calculations.
Figure 10.

Optimum mixing parameter x is shown as a function of ΔECT for the SRPx-vdW1-ext DF (eq 6a). Values falling between the two horizontal dot-dashed black lines could be obtained by the interpolation procedure illustrated in Figure 6. The green, blue, and red symbols correspond to N2, H2, and CH4 + metal surface systems, respectively. The black, blue, and red dashed lines provide the linear fits corresponding to the Pearson correlation coefficients computed for all molecules, H2, and CH4 + metal surface systems, respectively, without omitting systems.
Figures S2, S3, and S4 show the optimal x coefficients computed for the SRPx-vdW1, SRPx-vdW2, and SRPx-vdW2-ext DFs of eqs 3, 4, and 7a, respectively, as a function of ΔECT. These coefficients are also listed for each DF in Tables S4 and S5. Figures S2–S4 show that obtaining the optimum value of x for these three mixed DFs required extrapolation to negative values for several H2-metal surface and in most cases also for several CH4-metal surface systems, with SRPx-vdW2 performing particularly poorly. The above suggests that these three mixed DFs, and especially SRPx-vdW2, should perhaps not be the first choice for deriving a new SRP-DF for a system like those present in the SBH16 database.
We end by noting that the ability of the SRPx-vdW2 (Figure S3), SRPx-vdW2-ext (Figure S4), and SRPxsol-vdW2 (Figure 9) to interpolate x increases in the order SRPx-vdW2 < SRPx-vdW2-ext < SRPxsol-vdW2. This is related to the tunability of the SRPxsol-vdW2 DF being greatest from the use of μGE = 0.1235 in PBEsol,87 which affects the gradient enhancement factor already to second order in s (see Figure 1). Through the use of PBEα the SRPx-vdW2-ext DF also has a smaller gradient enhancement factor that is smaller than that of SRPx-vdW2 (which uses PBE), but here the decrease comes only from the fourth-order dependence of s, and as Figure 1 shows the gradient enhancement factor of SRPx-vdW2-ext is intermediate between that of PBE and PBEsol. As noted before, the lower the gradient enhancement factor of a DF is as a function of s, the lower the barriers the DF will produce,98 which explains the extent of the tunability of the three mixed DFs using vdW2 correlation that were tested here.
3D. Correlation of the Mixing Parameter with the Charge Transfer Parameter
Table 2 shows correlation coefficients (or Pearson product-moment correlation coefficients)111rxy describing the correlation between the charge transfer parameter taken as independent variable and the mixing coefficient x taken as dependent variable, for the seven mixed DFs tested here. Including all systems, the rxy values are clearly negative for the SRPx and the SRPxsol DFs. The same is true for these DFs if only the H2-metal systems are considered, and for these systems the rxy values get close to the value of −1 indicating a nearly perfect linear relationship if the H2 + Ag(111) system, for which the reference barrier height is somewhat suspect, is not considered. For CH4-metal systems the values of rxy only take on negative values if the CH4 + Ru(0001) and Ni(100) systems, for which the reference barrier heights are also somewhat suspect, are not considered, and these values are small in absolute value. These findings support the analysis of ref (8), which called for more accurate reference data for H2 + Ag(111), CH4 + Ni(100), and CH4 + Ru(0001).
Table 2. Correlation Coefficients Computed for the Dependence of the Optimum Fraction of RPBE Exchange x on the Charge Transfer Parameter for the Mixed DFs Testeda.
| type SRP | All-16 | All-12 | 7 H2-metal | 6 H2-metal | 7 CH4-metal | 5 CH4-metal |
|---|---|---|---|---|---|---|
| SRPx | –0.648 | –0.617 | –0.584 | –0.927 | 0.211 | –0.239 |
| SRPxsol | –0.543 | –0.409 | –0.761 | –0.91 | 0.228 | –0.174 |
| SRPx-vdW-DF1 | 0.264 | 0.528 | 0.752 | 0.684 | 0.362 | 0.003 |
| SRPx-vdW-DF2 | 0.205 | 0.483 | 0.751 | 0.695 | 0.500 | 0.801 |
| SRPxsol-vdW-DF2 | 0.147 | 0.447 | 0.473 | 0.209 | 0.483 | 0.609 |
| SRPx-vdW-DF1-ext | 0.236 | 0.521 | 0.716 | 0.627 | 0.361 | 0.003 |
| SRPx-vdW-DF2-ext | 0.148 | 0.423 | 0.741 | 0.671 | 0.420 | 0.428 |
Computed correlation coefficients are provided for the 7 H2-metal surface systems present in the SBH16 database, the 6 H2-metal surface systems obtained once H2 + Ag(111) is removed, the 7 CH4-metal surface systems in the SBH16 database, the 5 CH4-metal surface systems that remain after CH4 + Ru(0001) and Ni(100) are removed, the 16 systems (All-16) present in the SBH16 database, and the 12 systems (All-12) that remain after the 3 systems already mentioned and N2 + Ru(101̅0) are removed.
The finding of negative correlation coefficients as observed here for the SRPx and SRPxsol DFs is what we expected to see for several reasons. First of all, the MAE of the RPBE DF was previously found to increase from 88 to 167 to 336 meV going from N2-metal systems to H2-metal systems to CH4-metal systems,8 respectively, i.e., going from small values of the charge transfer parameter to large values (see, e.g., Table S2 and Figure 7 for how the charge transfer parameter varies with the type of system). The opposite is true for the PBE DF, where the MAE was found to decrease from 409 to 80 to 45 meV going from N2-metal systems to H2-metal systems to CH4-metal systems,8 respectively. Second, tests on several systems suggest that for systems characterized by charge transfer parameters less than 7 eV even RPBE exchange is not repulsive enough to avoid underestimating the barrier height.35 However, it is also clear that when all three types of systems are considered, the correlation is not that strong, suggesting that when a mixed functional with a fraction of PBE correlation is used, the optimum mixing coefficient also depends on other properties of the system than the charge transfer parameter. In this context we note that rxy for all systems decreases in absolute value if the four systems with suspect reference values (N2 + Ru(101̅0), CH4 + Ru(0001), CH4 + Ni(100), and H2 + Ag(111))8 are excluded from the SBH16 database (see Table 2), which would not be expected if x would only depend on the charge transfer parameter and the relationship would be linear.
The computed values of the correlation coefficients for the DFs incorporating van der Waals correlation are rather different from the values calculated for SRPx and SRPxsol, which incorporate the PBE correlation. Restricting ourselves to the mixed DFs that exhibit high tunability, i.e., SRPxsol-vdW2 and SRPx-vdW1-ext, we see that the former one only exhibits positive correlation coefficients and that the latter one exhibits correlation coefficients that are either positive or close to zero. The reasons for the different values of the correlation coefficients of SRPx and SRPxsol on the one hand (mostly negative) and the other DFs incorporating van der Waals correlation on the other hand (mostly positive) are not clear at this stage; the difference is rather puzzling.
The quality of the description of the mixing coefficient as a linear function of the charge transfer parameter is illustrated in Figures 7–10 by also showing the linear fits corresponding to the computed Pearson correlation coefficients. As can be seen, in no case are good values for x obtained for all systems simultaneously for any of the four mixed functionals described by these figures. The best linear fits were obtained for the SRPxsol DF, and in this case the linear function yields reasonable predictions of x for all H2-metal systems but the H2 + Ag(111) system (Figure 8). In all cases (Figures 7–10) the linear fits of the CH4 + metal surface systems perform poorly at predicting the mixing coefficient for CH4 + Ni(100). The analysis in terms of the linear fits thus provides further evidence that it may be worthwhile to revisit the H2 + Ag(111) and CH4 + Ni(100) systems in order to hopefully obtain better reference values of the barrier heights for these systems.
4. Conclusions and Outlook
We have investigated the tunability of several expressions for mixed density functionals in which mixing parameter x can be tuned to enable the mixed DF to reproduce the reference value of the barrier height to dissociative chemisorption of a molecule on a metal surface. The mixed functionals are tested on the barriers collected in the database we call SBH16, which is equal to the previous SBH17 database in ref (8) with the H2 + Pt(211) system removed from it.
Increasing the fraction of RPBE exchange incorporated into the mixed DFs leads to higher barriers. All mixed DFs tested are well-tunable toward higher barriers, as their limiting forms (RPBE, RPBE-vdW1, and RPBE-vdW2) all systematically overestimate the barrier height for the systems in the SBH16 database. It turns out that the biggest challenge to finding a perfectly tunable mixed DF for describing the SBH16 database is to obtain a mixed DF expression with a good lower-energy form, which consistently underestimates barrier heights for systems such as those present in SBH16. This goal is fully met with the mixed SRPxsol DF that uses PBE correlation and a mixture of PBEsol and RPBE exchange. The mixed SRPxsol-vdW2 DF could describe the minimum barrier height of 15 of the 16 systems using the vdW-DF2 correlation, while the mixed SRPx-vdW1 DF could do so for 14 of the 16 systems using the vdW-DF1 correlation. Being able to use mixed DFs with different correlation functionals may be important to obtaining an SRP DF for a particular system because reproducing the minimum barrier height is a necessary but not a sufficient condition for reproducing measured sticking (or dissociative chemisorption) probabilities, as now used for validating SRP functionals and barrier heights: It is also necessary to provide a description of how the barrier height varies when the molecule’s impact site on the surface and its orientation relative to the surface is changed, and this variation may depend strongly on the correlation functional used.7,14,85
We also tested whether and how the mixing coefficient of the mixed DFs is correlated with the charge transfer parameter describing the system, i.e., the difference between the work function of the metal surface and the electron affinity of the molecule. The answer depends on which mixed DF is used. For the SRPx and SRPxsol DFs, which both use PBE correlation, we found that the optimum fraction of RPBE exchange decreases with the charge transfer parameter, as could be expected on the basis of earlier results. However, the opposite relationship and weaker correlation were found for the mixed DFs using vdW-DF1 or vdW-DF2 correlation. The reason for this difference is not clear.
The results presented here point to several new lines of research. First of all, the results underscore the need to obtain better reference values for the H2 + Ag(111), CH4 + Ru(0001), and CH4 + Ni(100) systems. As noted, the H2 + Ag(111) system is among the few systems for which the optimum fraction of RPBE exchange could not be obtained through interpolation with the SRPxsol-vdW2 and SRPx-vdW1 DFs, which otherwise performed quite well at reproducing minimum barrier heights for the systems in the SBH17 database. Furthermore, the Pearson correlation coefficients describing the relationship between the mixing parameters of the SRPx and SRPxsol DFs and the charge transfer parameter took on values more in line with their expected behavior if the results for H2 + Ag(111), CH4 + Ni(100), and CH4 + Ru(0001) were discarded.
A small improvement over using the SRPxsol mixed DF could be to use a DF that simply mixes the RPBE and PBEsol exchange-correlation functionals. It is also necessary to provide a description of how the barrier height varies when the molecule’s impact site on the surface and its orientation relative to the surface is changed. This usually does not present a problem, as DFT appears to be rather good at describing the variation of barrier height with geometry. This is also attested by the previous success with developing SRP7 and by the comparison of DFT with diffusion Monte Carlo results for H2 + Al(110).28 However, for systems with a deep van der Waals well,85 or systems with a shallow well but an early barrier,14 this variation is best described by including a correlation functional approximately describing the van der Waals interaction.
When it comes to designing mixed functionals incorporating a vdW-DF1 or vdW-DF2 correlation, another idea worth testing might be to investigate mixtures of weakly repulsive GGA exchange DFs that are appropriate matches for the vdW1 and vdW2 correlation functionals with the rather repulsive112 exchange functionals combined with these C functionals in the original vdW-DF131 and vdW-DF232 DFs. Examples of such exchange functionals have been incorporated in the C09113 and CX114 vdW functionals and other exchange functionals mentioned in ref (112). Another idea would be to explore mixtures of repulsive meta-GGA DFs (such as MS-B86bl34) and attractive meta-GGA DFs (such as SCAN115) that tend to overestimate, respectively, underestimate barriers to dissociative chemisorption of molecules on metals.8 It would also be of interest to investigate the performance of mixtures of, or parametrized forms of, screened hybrid functionals such as HSE06116 and screened hybrid functionals incorporating van der Waals correlation.112,117 However, it might be most productive to test such hybrid functionals once a database becomes available that also incorporates good reference values of barrier heights for systems characterized by charge transfer parameters <7 eV, such as O2 + Ag(111)35 and HCl + Au(111).118 Such systems presently defy an accurate description based on DFs incorporating GGA exchange.35,118,119
Acknowledgments
This work was supported financially by a CW-TOP grant (715-017-09), with computer time granted by NWO-EW. We are grateful to Kieron Burke for useful discussions on the PW91 and the PBE functionals and to Donald Truhlar for useful discussions on the benchmarking of gas-phase reaction barrier heights.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpca.3c01932.
Equilibrium lattice constants calculated with the SRPx and the SRPxsol DFs for Ag, Cu, Ir, Pt, and Ru (Table S1). Barrier heights computed with the PBE, the RPBE, the PBEsolc, the PBE-vdW1, the PBE-vdW2, the PBEα-vdW1, the RPBE-vdW1, the RPBE-vdW2, and the PBEsol-vdW2 for 16 molecule-metal surface systems (Tables S2 & S3 and Figures S1–S4). Optimal mixing coefficient x for SRPx, SRPxsol, SRPx-vdW1, SRPx-vdW2, SRPxsol-vdW2, SRPx-vdW1-ext, and SRPx-vdW2-ext (Tables S4 & S5) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Wolcott C. A.; Medford A. J.; Studt F.; Campbell C. T. Degree of rate control approach to computational catalyst screening. J. Catal. 2015, 330, 197–207. 10.1016/j.jcat.2015.07.015. [DOI] [Google Scholar]
- Sabbe M. K.; Reyniers M.-F.; Reuter K. First-principles kinetic modeling in heterogeneous catalysis: an industrial perspective on best-practice, gaps and needs. Catal. Sci. Technol. 2012, 2, 2010–2024. 10.1039/c2cy20261a. [DOI] [Google Scholar]
- Ertl G. Primary steps in catalytic synthesis of ammonia. J. Vac. Sci. Technol., A: Vacuum, Surfaces, and Films 1983, 1, 1247–1253. 10.1116/1.572299. [DOI] [Google Scholar]
- Honkala K.; Hellman A.; Remediakis I.; Logadottir A.; Carlsson A.; Dahl S.; Christensen C. H.; Nørskov J. K. Ammonia synthesis from first-principles calculations. science 2005, 307, 555–558. 10.1126/science.1106435. [DOI] [PubMed] [Google Scholar]
- Chorkendorff I.; Niemantsverdriet J. W.. Concepts of modern catalysis and kinetics ;Wiley Online Library, 2003; Vol. 138. 10.1002/3527602658 [DOI] [Google Scholar]
- Noyori R. Synthesizing our future. Nat. Chem. 2009, 1, 5. 10.1038/nchem.143. [DOI] [PubMed] [Google Scholar]
- Kroes G. J. Computational approaches to dissociative chemisorption on metals: towards chemical accuracy. Phys. Chem. Chem. Phys. 2021, 23, 8962–9048. 10.1039/D1CP00044F. [DOI] [PubMed] [Google Scholar]
- Tchakoua T.; Gerrits N.; Smeets E. W. F.; Kroes G. J. SBH17: Benchmark Database of Barrier Heights for Dissociative Chemisorption on Transition Metal Surfaces. J. Chem. Theory Comput. 2023, 19, 245–270. 10.1021/acs.jctc.2c00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doblhoff-Dier K.; Meyer J.; Hoggan P. E.; Kroes G. J. Quantum Monte Carlo calculations on a benchmark molecule–metal surface reaction: H2+ Cu (111). J. Chem. Theory Comput. 2017, 13, 3208–3219. 10.1021/acs.jctc.7b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz C.; Pijper E.; Olsen R. A.; Busnengo H. F.; Auerbach D. J.; Kroes G. J. Chemically accurate simulation of a prototypical surface reaction: H2 dissociation on Cu(111). Science 2009, 326, 832–834. 10.1126/science.1178722. [DOI] [PubMed] [Google Scholar]
- Sementa L.; Wijzenbroek M.; Van Kolck B. J.; Somers M. F.; Al-Halabi A.; Busnengo H. F.; Olsen R. A.; Kroes G. J.; Rutkowski M.; Thewes C.; et al. Reactive scattering of H2 from Cu (100): Comparison of dynamics calculations based on the specific reaction parameter approach to density functional theory with experiment. J. Chem. Phys. 2013, 138, 044708. 10.1063/1.4776224. [DOI] [PubMed] [Google Scholar]
- Nour Ghassemi E.; Wijzenbroek M.; Somers M. F.; Kroes G.-J. Chemically accurate simulation of dissociative chemisorption of D2 on Pt(111). Chem. Phys. Lett. 2017, 683, 329–335. 10.1016/j.cplett.2016.12.059. [DOI] [Google Scholar]
- Ghassemi E. N.; Smeets E. W.; Somers M. F.; Kroes G. J.; Groot I. M.; Juurlink L. B.; Füchsel G. Transferability of the Specific Reaction Parameter Density Functional for H2+ Pt(111) to H2+ Pt(211). J. Phys. Chem. C 2019, 123, 2973–2986. 10.1021/acs.jpcc.8b11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijzenbroek M.; Kroes G. J. The effect of the exchange-correlation functional on H2 dissociation on Ru(0001). J. Chem. Phys. 2014, 140, 084702. 10.1063/1.4865946. [DOI] [PubMed] [Google Scholar]
- Tchakoua T.; Smeets E. W.; Somers M.; Kroes G.-J. Toward a Specific Reaction Parameter Density Functional for H2+ Ni(111): Comparison of Theory with Molecular Beam Sticking Experiments. J. Phys. Chem. C 2019, 123, 20420–20433. 10.1021/acs.jpcc.9b05928. [DOI] [Google Scholar]
- Smeets E. W.; Kroes G. J. Performance of Made Simple Meta-GGA Functionals with rVV10 Nonlocal Correlation for H2+ Cu(111), D2+Ag(111), H2+Au(111), and D2+Pt(111). J. Phys. Chem. C 2021, 125, 8993–9010. 10.1021/acs.jpcc.0c11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakouri K.; Behler J.; Meyer J.; Kroes G. J. Accurate neural network description of surface phonons in reactive gas–surface dynamics: N2+ Ru (0001). J. Phys. Chem. Lett. 2017, 8, 2131–2136. 10.1021/acs.jpclett.7b00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiering P.; Shakouri K.; Behler J.; Kroes G. J.; Meyer J. Orbital-dependent electronic friction significantly affects the description of reactive scattering of N2 from Ru (0001). J. Phys. Chem. Lett. 2019, 10, 2957–2962. 10.1021/acs.jpclett.9b00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattino F.; Migliorini D.; Kroes G. J.; Dombrowski E.; High E. A.; Killelea D. R.; Utz A. L. Chemically accurate simulation of a polyatomic molecule-metal surface reaction. J. Phys. Chem. Lett. 2016, 7, 2402–2406. 10.1021/acs.jpclett.6b01022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliorini D.; Chadwick H.; Nattino F.; Gutiérrez-González A.; Dombrowski E.; High E. A.; Guo H.; Utz A. L.; Jackson B.; Beck R. D.; Kroes G. J. Surface reaction barriometry: methane dissociation on flat and stepped transition-metal surfaces. J. Phys. Chem. Lett. 2017, 8, 4177–4182. 10.1021/acs.jpclett.7b01905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallikarjun Sharada S.; Bligaard T.; Luntz A. C.; Kroes G. J.; Nørskov J. K. Sbh10: A benchmark database of barrier heights on transition metal surfaces. J. Phys. Chem. C 2017, 121, 19807–19815. 10.1021/acs.jpcc.7b05677. [DOI] [Google Scholar]
- Zheng J.; Zhao Y.; Truhlar D. G. The DBH24/08 database and its use to assess electronic structure model chemistries for chemical reaction barrier heights. J. Chem. Theory Comput. 2009, 5, 808–821. 10.1021/ct800568m. [DOI] [PubMed] [Google Scholar]
- Peverati R.; Truhlar D. G. Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc., A 2014, 372, 20120476. 10.1098/rsta.2012.0476. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Goerigk L.; Hansen A.; Bauer C.; Ehrlich S.; Najibi A.; Grimme S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. 10.1039/C7CP04913G. [DOI] [PubMed] [Google Scholar]
- Morgante P.; Peverati R. Statistically representative databases for density functional theory via data science. Phys. Chem. Chem. Phys. 2019, 21, 19092–19103. 10.1039/C9CP03211H. [DOI] [PubMed] [Google Scholar]
- Pribram-Jones A.; Gross D. A.; Burke K. DFT: A theory full of holes?. Annu. Rev. Phys. Chem. 2015, 66, 283–304. 10.1146/annurev-physchem-040214-121420. [DOI] [PubMed] [Google Scholar]
- Powell A. D.; Kroes G.-J.; Doblhoff-Dier K. Quantum Monte Carlo calculations on dissociative chemisorption of H2+ Al (110): minimum barrier heights and their comparison to DFT values. J. Chem. Phys. 2020, 153, 224701. 10.1063/5.0022919. [DOI] [PubMed] [Google Scholar]
- Karikorpi M.; Holloway S.; Henriksen N.; Nørskov J. K. Dynamics of molecule-surface interactions. Surf. Sci. 1987, 179, L41–L48. 10.1016/0039-6028(87)90111-7. [DOI] [Google Scholar]
- Nattino F.; Díaz C.; Jackson B.; Kroes G. J. Effect of surface motion on the rotational quadrupole alignment parameter of D2 reacting on Cu(111). Phys. Rev. Lett. 2012, 108, 236104. 10.1103/PhysRevLett.108.236104. [DOI] [PubMed] [Google Scholar]
- Dion M.; Rydberg H.; Schröder E.; Langreth D. C.; Lundqvist B. I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. 10.1103/PhysRevLett.92.246401. [DOI] [PubMed] [Google Scholar]
- Lee K.; Murray É. D.; Kong L.; Lundqvist B. I.; Langreth D. C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101. 10.1103/PhysRevB.82.081101. [DOI] [Google Scholar]
- Madsen G. K. H. Functional form of the generalized gradient approximation for exchange: The PBEα functional. Phys. Rev. B 2007, 75, 195108. 10.1103/PhysRevB.75.195108. [DOI] [Google Scholar]
- Smeets E. W.; Voss J.; Kroes G. J. Specific Reaction Parameter Density Functional Based on the Meta-Generalized Gradient Approximation: Application to H2+Cu(111) and H2+Ag(111). J. Phys. Chem. A 2019, 123, 5395–5406. 10.1021/acs.jpca.9b02914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits N.; Smeets E. W.; Vuckovic S.; Powell A. D.; Doblhoff-Dier K.; Kroes G. J. Density functional theory for molecule–metal surface reactions: When does the generalized gradient approximation get it right, and what to do if it does not. J. Phys. Chem. Lett. 2020, 11, 10552–10560. 10.1021/acs.jpclett.0c02452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer B.; Hansen L. B.; Nørskov J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. 10.1103/PhysRevB.59.7413. [DOI] [Google Scholar]
- Verma P.; Wang Y.; Ghosh S.; He X.; Truhlar D. G. Revised M11 exchange-correlation functional for electronic excitation energies and ground-state properties. J. Phys. Chem. A 2019, 123, 2966–2990. 10.1021/acs.jpca.8b11499. [DOI] [PubMed] [Google Scholar]
- Raghavachari K.; Trucks G. W.; Pople J. A.; Head-Gordon M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. 10.1016/S0009-2614(89)87395-6. [DOI] [Google Scholar]
- Tsatsoulis T.; Sakong S.; Groß A.; Grüneis A. Reaction energetics of hydrogen on Si(100) surface: A periodic many-electron theory study. J. Chem. Phys. 2018, 149, 244105 10.1063/1.5055706. [DOI] [PubMed] [Google Scholar]
- Austin B. M.; Zubarev D. Y.; Lester W. A. Quantum Monte Carlo and related approaches. Chem. Rev. 2012, 112, 263–288. 10.1021/cr2001564. [DOI] [PubMed] [Google Scholar]
- Foulkes W.; Mitas L.; Needs R.; Rajagopal G. Quantum Monte Carlo simulations of solids. Rev. Mod. Phys. 2001, 73, 33. 10.1103/RevModPhys.73.33. [DOI] [Google Scholar]
- Zhou X.; Wang F. Barrier heights of hydrogen-transfer reactions with diffusion quantum monte carlo method. J. Comput. Chem. 2017, 38, 798–806. 10.1002/jcc.24750. [DOI] [PubMed] [Google Scholar]
- Krongchon K.; Busemeyer B.; Wagner L. K. Accurate barrier heights using diffusion Monte Carlo. J. Chem. Phys. 2017, 146, 124129 10.1063/1.4979059. [DOI] [PubMed] [Google Scholar]
- Pozzo M.; Alfè D. Hydrogen dissociation on Mg (0001) studied via quantum Monte Carlo calculations. Phys. Rev. B 2008, 78, 245313. 10.1103/PhysRevB.78.245313. [DOI] [Google Scholar]
- Hoggan P. E.; Bouferguène A. Quantum Monte Carlo for activated reactions at solid surfaces: Time well spent on stretched bonds. Int. J. Quantum Chem. 2014, 114, 1150–1156. 10.1002/qua.24676. [DOI] [Google Scholar]
- Huang C.; Pavone M.; Carter E. A. Quantum mechanical embedding theory based on a unique embedding potential. J. Chem. Phys. 2011, 134, 154110 10.1063/1.3577516. [DOI] [PubMed] [Google Scholar]
- Libisch F.; Huang C.; Carter E. A. Embedded correlated wavefunction schemes: Theory and applications. Acc. Chem. Res. 2014, 47, 2768–2775. 10.1021/ar500086h. [DOI] [PubMed] [Google Scholar]
- Andersson K.; Malmqvist P.-Å.; Roos B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. 10.1063/1.462209. [DOI] [Google Scholar]
- Andersson K.; Malmqvist P. A.; Roos B. O.; Sadlej A. J.; Wolinski K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. 10.1021/j100377a012. [DOI] [Google Scholar]
- Angeli C.; Cimiraglia R.; Evangelisti S.; Leininger T.; Malrieu J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. 10.1063/1.1361246. [DOI] [Google Scholar]
- Tishchenko O.; Zheng J.; Truhlar D. G. Multireference model chemistries for thermochemical kinetics. J. Chem. Theory Comput. 2008, 4, 1208–1219. 10.1021/ct800077r. [DOI] [PubMed] [Google Scholar]
- Yin R.; Zhang Y.; Libisch F.; Carter E. A.; Guo H.; Jiang B. Dissociative chemisorption of O2 on Al (111): dynamics on a correlated wave-function-based potential energy surface. J. Phys. Chem. Lett. 2018, 9, 3271–3277. 10.1021/acs.jpclett.8b01470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Zhang X.; Martirez J. M. P.; Carter E. A. Benchmarking an embedded adaptive sampling configuration interaction method for surface reactions: H2 desorption from and CH4 dissociation on Cu (111). J. Chem. Theory Comput. 2020, 16, 7078–7088. 10.1021/acs.jctc.0c00341. [DOI] [PubMed] [Google Scholar]
- Langreth D.; Perdew J. The gradient approximation to the exchange-correlation energy functional: A generalization that works. Solid State Commun. 1979, 31, 567–571. 10.1016/0038-1098(79)90254-0. [DOI] [Google Scholar]
- Langreth D. C.; Perdew J. P. Theory of nonuniform electronic systems. I. Analysis of the gradient approximation and a generalization that works. Phys. Rev. B 1980, 21, 5469. 10.1103/PhysRevB.21.5469. [DOI] [Google Scholar]
- Furche F. Molecular tests of the random phase approximation to the exchange-correlation energy functional. Phys. Rev. B 2001, 64, 195120. 10.1103/PhysRevB.64.195120. [DOI] [Google Scholar]
- Paier J.; Ren X.; Rinke P.; Scuseria G. E.; Grüneis A.; Kresse G.; Scheffler M. Assessment of correlation energies based on the random-phase approximation. New J. Phys. 2012, 14, 043002. 10.1088/1367-2630/14/4/043002. [DOI] [Google Scholar]
- Schmidt P. S.; Thygesen K. S. Benchmark database of transition metal surface and adsorption energies from many-body perturbation theory. J. Phys. Chem. C 2018, 122, 4381–4390. 10.1021/acs.jpcc.7b12258. [DOI] [Google Scholar]
- Wellendorff J.; Silbaugh T. L.; Garcia-Pintos D.; Nørskov J. K.; Bligaard T.; Studt F.; Campbell C. T. A benchmark database for adsorption bond energies to transition metal surfaces and comparison to selected DFT functionals. Surf. Sci. 2015, 640, 36–44. 10.1016/j.susc.2015.03.023. [DOI] [Google Scholar]
- Schimka L.; Harl J.; Stroppa A.; Grüneis A.; Marsman M.; Mittendorfer F.; Kresse G. Accurate surface and adsorption energies from many-body perturbation theory. Nature mater 2010, 9, 741–744. 10.1038/nmat2806. [DOI] [PubMed] [Google Scholar]
- Ren X.; Rinke P.; Joas C.; Scheffler M. Random-phase approximation and its applications in computational chemistry and materials science. J. Mater. Sci. 2012, 47, 7447–7471. 10.1007/s10853-012-6570-4. [DOI] [Google Scholar]
- Yan L.; Sun Y.; Yamamoto Y.; Kasamatsu S.; Hamada I.; Sugino O. Hydrogen adsorption on Pt (111) revisited from random phase approximation. J. Chem. Phys. 2018, 149, 164702 10.1063/1.5050830. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Goltl F.; Sautet P. Diffusion barriers for carbon monoxide on the Cu (001) surface using many-body perturbation theory and various density functionals. J. Chem. Theory Comput. 2021, 17, 7862–7872. 10.1021/acs.jctc.1c00946. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Sautet P. Improving the Accuracy of Modelling CO2 Electroreduction on Copper Using Many-Body Perturbation Theory. Angew. Chem. 2022, 134, e202210060 10.1002/ange.202210060. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Goltl F.; Steinmann S. N.; Sautet P. Modeling electrochemical processes with grand canonical treatment of many-body perturbation theory. J. Phys. Chem. Lett. 2022, 13, 6079–6084. 10.1021/acs.jpclett.2c01376. [DOI] [PubMed] [Google Scholar]
- Svensson M.; Humbel S.; Froese R. D.; Matsubara T.; Sieber S.; Morokuma K. ONIOM: a multilayered integrated MO+ MM method for geometry optimizations and single point energy predictions. A test for Diels- Alder reactions and Pt (P (t-Bu) 3) 2+ H2 oxidative addition. J. Phys. Chem. 1996, 100, 19357–19363. 10.1021/jp962071j. [DOI] [Google Scholar]
- Araujo R. B.; Rodrigues G. L.; Dos Santos E. C.; Pettersson L. G. Adsorption energies on transition metal surfaces: towards an accurate and balanced description. Nature Commun. 2022, 13, 6853. 10.1038/s41467-022-34507-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Allouche A.-R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. 10.1002/jcc.21600. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Climbing the ladder of density functional approximations. MRS bull 2013, 38, 743–750. 10.1557/mrs.2013.178. [DOI] [Google Scholar]
- Klippenstein S. J.; Pande V. S.; Truhlar D. G. Chemical kinetics and mechanisms of complex systems: A perspective on recent theoretical advances. J. Am. Chem. Soc. 2014, 136, 528–546. 10.1021/ja408723a. [DOI] [PubMed] [Google Scholar]
- Henkelman G.; Uberuaga B. P.; Jónsson H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. 10.1063/1.1329672. [DOI] [Google Scholar]
- Henkelman G.; Jónsson H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. 10.1063/1.1323224. [DOI] [Google Scholar]
- Henkelman G.; Jónsson H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. 10.1063/1.480097. [DOI] [Google Scholar]
- Pople J. A. Quantum chemical models (Nobel lecture). Angew. Chem., Int. Ed. 1999, 38, 1894–1902. . [DOI] [PubMed] [Google Scholar]
- Neese F.; Atanasov M.; Bistoni G.; Maganas D.; Ye S. Chemistry and quantum mechanics in 2019: give us insight and numbers. J. Am. Chem. Soc. 2019, 141, 2814–2824. 10.1021/jacs.8b13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Morgante P.; Peverati R. ACCDB: A collection of chemistry databases for broad computational purposes. J. Comput. Chem. 2019, 40, 839–848. 10.1002/jcc.25761. [DOI] [PubMed] [Google Scholar]
- Lynch B. J.; Truhlar D. G. What are the best affordable multi-coefficient strategies for calculating transition state geometries and barrier heights?. J. Phys. Chem. A 2002, 106, 842–846. 10.1021/jp014002x. [DOI] [Google Scholar]
- Zhao Y.; Lynch B. J.; Truhlar D. G. Multi-coefficient extrapolated density functional theory for thermochemistry and thermochemical kinetics. Phys. Chem. Chem. Phys. 2005, 7, 43–52. 10.1039/b416937a. [DOI] [Google Scholar]
- Zhao Y.; González-García N.; Truhlar D. G. Benchmark database of barrier heights for heavy atom transfer, nucleophilic substitution, association, and unimolecular reactions and its use to test theoretical methods. J. Phys. Chem. A 2005, 109, 2012–2018. 10.1021/jp045141s. [DOI] [PubMed] [Google Scholar]
- Karton A.; Rabinovich E.; Martin J. M.; Ruscic B. W4 theory for computational thermochemistry: In pursuit of confident sub-kJ/mol predictions. J. Chem. Phys. 2006, 125, 144108 10.1063/1.2348881. [DOI] [PubMed] [Google Scholar]
- Nattino F.; Migliorini D.; Bonfanti M.; Kroes G. J. Methane dissociation on Pt(111): Searching for a specific reaction parameter density functional. J. Chem. Phys. 2016, 144, 044702. 10.1063/1.4939520. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Chevary J. A.; Vosko S. H.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B: Condens. Matter Mater. Phys. 1992, 46, 6671–6687. 10.1103/PhysRevB.46.6671. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Ruzsinszky A.; Csonka G. I.; Vydrov O. A.; Scuseria G. E.; Constantin L. A.; Zhou X.; Burke K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. 10.1103/PhysRevLett.100.136406. [DOI] [PubMed] [Google Scholar]
- Lieb E. H.; Oxford S. Improved lower bound on the indirect Coulomb energy. Int. J. Quantum Chem. 1981, 19, 427–439. 10.1002/qua.560190306. [DOI] [Google Scholar]
- Pacheco-Kato J. C.; del Campo J. M.; Gázquez J. L.; Trickey S.; Vela A. A PW91-like exchange with a simple analytical form. Chem. Phys. Lett. 2016, 651, 268–273. 10.1016/j.cplett.2016.03.028. [DOI] [Google Scholar]
- Zhang Y.; Pan W.; Yang W. Describing van der Waals Interaction in diatomic molecules with generalized gradient approximations: The role of the exchange functional. J. Chem. Phys. 1997, 107, 7921–7925. 10.1063/1.475105. [DOI] [Google Scholar]
- Zupan A.; Perdew J. P.; Burke K.; Causà M. Density-gradient analysis for density functional theory: Application to atoms. Int. J. Quantum Chem. 1997, 61, 835–845. . [DOI] [Google Scholar]
- Nave S.; Jackson B. Methane dissociation on Ni (111): The effects of lattice motion and relaxation on reactivity. J. Chem. Phys. 2007, 127, 224702 10.1063/1.2800661. [DOI] [PubMed] [Google Scholar]
- Liu T.; Fu B.; Zhang D. H. Six-dimensional potential energy surfaces of the dissociative chemisorption of HCl on Ag (111) with three density functionals. J. Chem. Phys. 2018, 149, 054702 10.1063/1.5036805. [DOI] [PubMed] [Google Scholar]
- Lacks D. J.; Gordon R. G. Pair interactions of rare-gas atoms as a test of exchange-energy-density functionals in regions of large density gradients. Phys. Rev. A 1993, 47, 4681. 10.1103/PhysRevA.47.4681. [DOI] [PubMed] [Google Scholar]
- Mattsson A. E.; Armiento R.; Schultz P. A.; Mattsson T. R. Nonequivalence of the generalized gradient approximations PBE and PW91. Phys. Rev. B 2006, 73, 195123. 10.1103/PhysRevB.73.195123. [DOI] [Google Scholar]
- Madsen G. K. H.personal communication.
- Wu Z.; Cohen R. E. More accurate generalized gradient approximation for solids. Phys. Rev. B 2006, 73, 235116. 10.1103/PhysRevB.73.235116. [DOI] [Google Scholar]
- Zupan A.; Burke K.; Ernzerhof M.; Perdew J. P. Distributions and averages of electron density parameters: Explaining the effects of gradient corrections. J. Chem. Phys. 1997, 106, 10184–10193. 10.1063/1.474101. [DOI] [Google Scholar]
- Vilhena J.; Räsänen E.; Lehtovaara L.; Marques M. Violation of a local form of the lieb-oxford bound. Phys. Rev. A 2012, 85, 052514. 10.1103/PhysRevA.85.052514. [DOI] [Google Scholar]
- Kresse G.; Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Bahn S. R.; Jacobsen K. W. An object-oriented scripting interface to a legacy electronic structure code. Comput. Sci. Eng. 2002, 4, 56–66. 10.1109/5992.998641. [DOI] [Google Scholar]
- Hjorth Larsen A.; Jorgen Mortensen J.; Blomqvist J.; Castelli I. E.; Christensen R.; Dułak M.; Friis J.; Groves M. N.; Hammer B.; Hargus C.; et al. The atomic simulation environment-a Python library for working with atoms. J. Phys.: Condens. Matter 2017, 29, 273002. 10.1088/1361-648X/aa680e. [DOI] [PubMed] [Google Scholar]
- Román-Pérez G.; Soler J. M. Efficient implementation of a van der Waals density functional: application to double-wall carbon nanotubes. Phys. Rev. Lett. 2009, 103, 096102. 10.1103/PhysRevLett.103.096102. [DOI] [PubMed] [Google Scholar]
- Wellendorff J.; Lundgaard K. T.; Møgelhøj A.; Petzold V.; Landis D. D.; Nørskov J. K.; Bligaard T.; Jacobsen K. W. Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation. Phys. Rev. B 2012, 85, 235149. 10.1103/PhysRevB.85.235149. [DOI] [Google Scholar]
- Tran F.; Stelzl J.; Blaha P. Rungs 1 to 4 of DFT Jacob’s ladder: Extensive test on the lattice constant, bulk modulus, and cohesive energy of solids. J. Chem. Phys. 2016, 144, 204120. 10.1063/1.4948636. [DOI] [PubMed] [Google Scholar]
- Schimka L.; Harl J.; Stroppa A.; Grüneis A.; Marsman M.; Mittendorfer F.; Kresse G. Accurate surface and adsorption energies from many-body perturbation theory. Nat. Mater. 2010, 9, 741–744. 10.1038/nmat2806. [DOI] [PubMed] [Google Scholar]
- Klimeš J.; Bowler D. R.; Michaelides A. Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. 10.1103/PhysRevB.83.195131. [DOI] [Google Scholar]
- Hays W. L.Statistics ;Holt–Saunders: Tokyo, Japan, 1981. [Google Scholar]
- Shukla V.; Jiao Y.; Lee J.-H.; Schröder E.; Neaton J. B.; Hyldgaard P. Accurate Nonempirical Range-Separated Hybrid van der Waals Density Functional for Complex Molecular Problems, Solids, and Surfaces. Phys. Rev. X 2022, 12, 041003. 10.1103/PhysRevX.12.041003. [DOI] [Google Scholar]
- Cooper V. R. Van der Waals density functional: An appropriate exchange functional. Phys. Rev. B 2010, 81, 161104. 10.1103/PhysRevB.81.161104. [DOI] [Google Scholar]
- Berland K.; Hyldgaard P. Exchange functional that tests the robustness of the plasmon description of the van der Waals density functional. Phys. Rev. B 2014, 89, 035412. 10.1103/PhysRevB.89.035412. [DOI] [Google Scholar]
- Sun J.; Ruzsinszky A.; Perdew J. P. Strongly constrained and appropriately normed semilocal density functional. Phys. Rev. Lett. 2015, 115, 036402. 10.1103/PhysRevLett.115.036402. [DOI] [PubMed] [Google Scholar]
- Krukau A. V.; Vydrov O. A.; Izmaylov A. F.; Scuseria G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. 10.1063/1.2404663. [DOI] [PubMed] [Google Scholar]
- Shukla V.; Jiao Y.; Frostenson C. M.; Hyldgaard P. vdW-DF-ahcx: a range-separated van der Waals density functional hybrid. J. Phys.: Condens. Matt. 2022, 34, 025902. 10.1088/1361-648X/ac2ad2. [DOI] [PubMed] [Google Scholar]
- Gerrits N.; Geweke J.; Smeets E. W.; Voss J.; Wodtke A. M.; Kroes G. J. Closing the gap between experiment and theory: reactive scattering of HCl from Au (111). J. Phys. Chem. C 2020, 124, 15944–15960. 10.1021/acs.jpcc.0c03756. [DOI] [Google Scholar]
- Behler J.; Delley B.; Lorenz S.; Reuter K.; Scheffler M. Dissociation of O2 at Al(111): The role of spin selection rules. Phys. Rev. Lett. 2005, 94, 036104. 10.1103/PhysRevLett.94.036104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.