

Abstract
Many advanced human cancers contain regions of intratumoral hypoxia, with O2 gradients extending to anoxia. Hypoxia-inducible factors (HIFs) are activated in hypoxic cancer cells and drive metabolic reprogramming, vascularization, invasion, and metastasis. Hypoxia induces breast cancer stem cell (BCSC) specification by inducing the expression and/or activity of the pluripotency factors KLF4, NANOG, OCT4, and SOX2. Recent studies have identified HIF-1-dependent expression of PLXNB3, NARF, and TERT in hypoxic breast cancer cells. PLXNB3 binds to and activates the MET receptor tyrosine kinase, leading to activation of the SRC non-receptor tyrosine kinase and subsequently focal adhesion kinase, which promotes cancer cell migration and invasion. PLXNB3-MET-SRC signaling also activates STAT3, a transcription factor that mediates increased NANOG gene expression. Hypoxia-induced NARF binds to OCT4 and serves as a coactivator by stabilizing OCT4 binding to the KLF4, NANOG, and SOX2 genes and by stabilizing the interaction of OCT4 with KDM6A, a histone demethylase that erases repressive trimethylation of histone H3 at lysine 27, thereby increasing KLF4, NANOG, and SOX2 gene expression. In addition to increasing pluripotency factor expression by these mechanisms, HIF-1 directly activates expression of the TERT gene encoding telomerase, the enzyme required for maintenance of telomeres, which is required for the unlimited self-renewal of BCSCs. HIF-1 binds to the TERT gene and recruits NANOG, which serves as a coactivator by promoting the subsequent recruitment of USP9X, a deubiquitinase that inhibits HIF-1α degradation, and p300, a histone acetyltransferase that mediates acetylation of H3K27, which is required for transcriptional activation.
Keywords: breast cancer stem cells, hypoxia, pluripotency factors, self-renewal, telomerase
Graphical Abstract
Graphical Abstract.
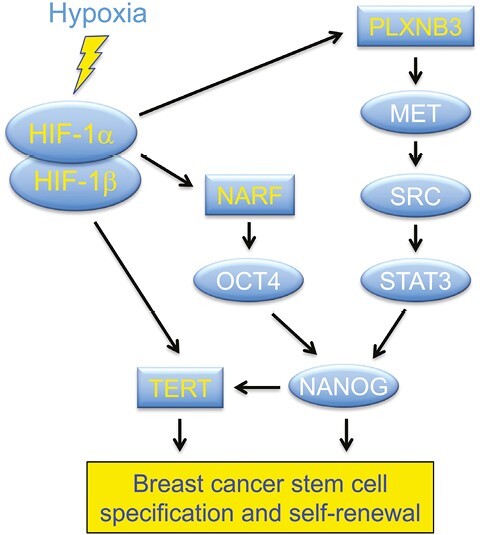
Significance Statement.
Cancer stem cells are a small subpopulation of cells that are capable of tumor initiation and infinite self-renewal. Intratumoral hypoxia induces HIF-1, which activates PLXNB3, NARF, and TERT gene expression, leading to increased expression and activity of pluripotency factors and telomerase, which mediate breast cancer stem cell specification and self-renewal, respectively. Development of HIF-1 inhibitors may provide a novel therapeutic strategy for the eradication of breast cancer stem cells.
Intratumoral Hypoxia
The pioneering work of Peter Vaupel and his colleagues established that one of the unifying features of advanced human cancers is the presence of intratumoral hypoxia. Using Eppendorf microelectrodes, pO2 was directly measured in accessible tumors, such as those of the breast, head/neck, and uterine cervix. Based on 1009 measurements from 16 subjects, Vaupel’s group found a broad range of pO2 values in normal breast tissue from 15 to 100 mmHg, with median pO2 = 65 mmHg; by contrast, in 851 measurements from 15 breast cancers, the median pO2 was 10 mmHg, with the most common measurement values = 0-5 mmHg.1 This profound intratumoral hypoxia reflects a severe mismatch between O2 supply and demand, with supply impaired by the presence of blood vessels that are structurally and functionally abnormal, and demand driven by dysregulated cell proliferation.1,2 In the uterine cervix, a large difference in pO2 between normal and cancer tissue (42 and 9 mmHg, respectively) was also reported and pO2 < 10 mmHg in cervical cancer was associated with decreased disease-free and overall survival.3
Within each tumor, pO2 levels vary according to the distance from the nearest functional blood vessel. O2 undergoes radial diffusion from blood vessels and is consumed by cells for respiration. In many tumors, adequate O2 is available to cancer cells that are less than 100 µm from the nearest blood vessel and these cells are said to be normoxic, whereas when cancer cells are located greater than 100 µm from a vessel, O2 becomes limiting and the cells are said to be hypoxic; and, in many tumors, when cells are located greater than 200 µm, O2 has become depleted and the cells are said to be anoxic (and dead). In their landmark 1955 paper, Thomlinson and Gray concluded that O2 depletion was the primary factor leading to tumor necrosis.4
Hypoxia-Inducible Factors
Decreased O2 availability triggers the accumulation of hypoxia-inducible factors (HIFs). HIFs are heterodimeric proteins that consist of an O2-regulated HIF-α subunit (HIF-1α, HIF-2α, or HIF-3α)and a constitutively expressed HIF-1β subunit, as originally determined for HIF-1.5,6 Under normoxic conditions, the HIF-α subunits are subjected to hydroxylation on 2 proline residues (P402 and P564 in human HIF-1α), leading to binding of the von-Hippel-Lindau (VHL) protein, which targets HIF-α proteins for ubiquitination and proteasomal degradation; under hypoxic conditions, the hydroxylation reaction (which uses O2 as a substrate) is inhibited, and the non-hydroxylated HIF-α subunits rapidly accumulate, dimerize with HIF-1β, bind to the consensus sequence 5ʹ-RCGTG-3ʹ (R = A or G) at target genes and activate their transcription.7-9
Immunohistochemistry using antibodies against HIF-1α to analyze primary tumor biopsies has demonstrated that increased HIF-1α expression is associated with increased patient mortality in bladder, brain, breast, cervical, colorectal, endometrial, esophageal, gastric, head/neck/ oropharyngeal, hepatocellular, lung, ovarian, and pancreatic cancer as well as acute leukemias and melanoma.9,10 In many cancers, HIF-1α expression is observed in a peri-necrotic pattern, representing the viable cells that are furthest away from a blood vessel and therefore the most hypoxic. However, HIF-1α can also be activated by oncogenic mutations in an O2-independent manner (most notably in the clear cell type of renal cell carcinoma which is characterized by VHL loss-of-function), leading to HIF-1α expression throughout the tumor biopsy.11 Focal, hypoxia-induced vs diffuse, O2-independent HIF-1α expression patterns were observed in two-thirds and one-third of oropharyngeal cancers, respectively.12 Exposure of SUM159 human breast cancer cells (see Table 1 for characterization of breast cancer cell lines mentioned in this review) to hypoxia (1% O2 for 24 h) led to increased expression of hundreds of RNAs and decreased expression of hundreds of RNAs in a HIF-dependent manner.13 Similar results have been observed in every other type of cancer, with each cancer cell line showing a unique pattern of altered gene expression in response to hypoxia.14 These HIF-dependent changes in gene expression promote vascularization, metabolic reprogramming, migration, invasion, metastasis, and immune evasion.7,8,10,15-17
Table 1.
Expression of estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2) and BRCA1 tumor suppressor protein in breast cancer cell lines described in this review.
| Cell line | Species | ER | PR | HER2 | BRCA1 |
|---|---|---|---|---|---|
| 4T1 | Mouse | − | − | − | + |
| HCC1954 | Human | − | − | + | + |
| MCF−7 | Human | + | + | - | + |
| MDA-MB-231 | Human | − | − | − | + |
| SUM149 | Human | − | − | − | − |
| SUM159 | Human | − | − | − | + |
| T47D | Human | + | + | − | + |
Breast Cancer Stem Cells
Cancer stem cells (CSCs), which were first discovered in acute myeloid leukemia,18,19 have been identified in many types of cancer, including brain tumors20; breast,21 colon,22 head/neck,23 pancreatic,24 and prostate25 cancer; melanoma26; and neuroblastoma.27 CSCs are defined by their ability to self-renew by asymmetrically dividing into one CSC, which is capable of infinite cell divisions, and one transient amplifying cell, which is capable of rapid cell division but only for a finite number of divisions.28 CSCs are the only cells within a tumor that are capable of giving rise to a secondary (recurrent or metastatic) tumor.29-31 CSCs are specified and maintained by the expression of core pluripotency factors, which were originally identified in embryonic stem cells (ESCs), including octamer binding factor 4 (OCT4), SRY-box 2 (SOX2), Kruppel-like factor 4 (KLF4), and NANOG.32-35 Cell populations that are enriched for breast CSCs (BCSCs) can be identified by flow cytometry as CD44hiCD24lo,21 except in many triple-negative breast cancer (TNBC) cell lines, which constitutively express CD44.36
BCSCs express aldehyde dehydrogenase 1 (ALDH1), which oxidizes retinaldehyde to retinoic acid, which induces gene expression by binding to retinoic acid receptors (RARs) and licensing their transcriptional activity.37 ALDH1 is directly linked to the pluripotency factors, as KLF4 and SOX2 have been reported to activate expression of one or more ALDH1 genes (ALDH1A1, ALDH1A2, ALDH1A3) in breast cancer.38,39 High ALDH1 expression (as measured by ALDH enzyme activity detected by use of a fluorogenic substrate and flow cytometry) is correlated with increased risk of metastasis and patient mortality.37,40,41 BCSCs propagate as multicellular spheroids (mammospheres) when placed in suspension culture using ultra-non-adherent plates.42 The primary mammospheres can be harvested, dissociated into single cells, and replated to assay for secondary spheroid formation as a measure of cell renewal.43 In addition to self-renewal and limitless cell division, BCSCs have tumor-initiating properties: 500 ALDH1+ human breast cancer cells will reliably form tumors when injected into the mammary fat pad of immunodeficient mice, whereas 50 000 ALDH1− cells from the same breast cancer will not.37 Selection of CD44hiCD24lo cells from human breast cancers also enriches for tumor-initiating cells.21
Hypoxia, HIF-1, and BCSCs
Exposure of human breast cancer cells to 1% O2 for 3 days increases the percentage of BCSCs by several fold and this increase in BCSC specification is dependent on the induction of multiple HIF-1 target genes but is independent of HIF-2.44,45 In contrast, the enrichment of BCSCs among cells surviving chemotherapy is dependent on both HIF-1 and HIF-2.45 In 4T1 mouse mammary carcinoma cells, HIF-2α expression was found to be enriched among ALDH1+ cells, treatment with an ALDH1 inhibitor led to decreased HIF-2α mRNA expression, and HIF-2α knockdown by RNA interference led to decreased OCT4 mRNA expression in these cells under non-hypoxic conditions.46 Knockdown of ALDH1A1 expression in MCF-7 cells impaired mammosphere formation, whereas ALDH1A1 overexpression increased mammosphere formation and increased the expression of the stemness markers CD133 and KLF4.47 Furthermore, changes in ALDH1A1 expression were associated with corresponding changes in the expression of HIF-1α and VEGFA, which is the product of a HIF-1 target gene.48 Remarkably, exposure of ALDH1A1-knockdown cells to exogenous retinoic acid restored HIF-1α and VEGFA expression, whereas exposure of cells overexpressing ALDH1A1 to the RAR inhibitor AGN-193109 led to decreased HIF-1α and VEGFA expression.47 HIF-1α knockdown inhibited expression of both VEGFA and CD133, thereby placing HIF-1 both upstream and downstream of stemness markers in MCF-7 cells. ALDH1A1 → HIF-1α → VEGFA signaling triggered angiogenesis in MCF-7 tumor xenografts.47 This result, like many others, demonstrates that BCSC specification is simply a component (along with angiogenesis, migration/invasion, metabolic reprogramming, and chemotherapy resistance) of the “high HIF” phenotype, also known as the lethal cancer phenotype.
In contrast to BCSCs, exposure of human ESCs to hypoxia and reoxygenation was shown to induce the expression of pluripotency factors in a HIF-2-dependent manner49 and hypoxia induced the binding of HIF-2 to the promoters of the genes encoding NANOG, OCT4, and SOX2.50 Hypoxia also induced pluripotency factor expression in many cancer cell lines in a HIF-dependent manner.51 In hypoxic breast cancer cells, the mechanism of induction appears to be indirect: multiple HIF-1 target genes encode proteins that activate signal transduction pathways leading to increased expression or activity of NANOG, OCT4, and other pluripotency factors (Fig. 1). This principle is illustrated by several recent studies that are described below, which have implicated PLXNB3 and NARF as novel mediators of breast cancer stem cell specification and elucidated a novel mechanism by which telomerase activity is reactivated in breast cancer stem cells.
Figure 1.
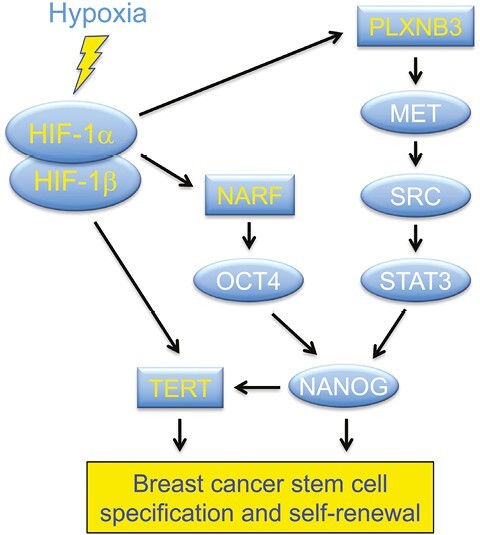
Hypoxia induces HIF-1-dependent transcriptional activation of the PLXNB3, NARF, and TERT genes, leading to increased breast cancer stem cell specification and self-renewal.
PLXNB3
Plexin-B3 (PLXNB3) is a large transmembrane protein that interacts with semaphorin 5A to mediate axon guidance during the development of the nervous system.52 PLXNB3 mRNA is expressed in many cancers and is associated with increased pan-cancer patient mortality.53 However, PLXNB3 was not previously implicated in cancer stem cell specification. PLXNB3 mRNA expression was induced by hypoxia in SUM159 human TNBC cells as part of a battery of RNAs associated with axon guidance13 and was correlated with the expression of a HIF metagene signature in 1218 human breast cancers in The Cancer Genome Atlas (TCGA BRCA dataset).54 Immunohistochemical analysis revealed that HIF-1α and PLXNB3 protein expression were highly correlated in human breast cancers and that PLXNB3 protein expression greater than the median was associated with decreased overall survival of women with breast cancer.54
Expression of short hairpin RNA (shRNA) targeting HIF-1α in triple-negative SUM159 or human epidermal growth factor receptor 2-positive (HER2+) HCC1954 breast cancer cells blocked the hypoxic induction of PLXNB3 mRNA and protein, whereas expression of shRNA targeting HIF-2α had no effect. Chromatin immunoprecipitation (ChIP) assays revealed the hypoxia-induced binding of HIF-1α and HIF-1β, but not HIF-2α, at 2 sites located approximately 6 and 10 kb downstream of the transcription start site of the PLXNB3 gene in SUM159 and HCC1954 cells (Fig. 2A), demonstrating that PLXNB3 is direct HIF-1 target gene.54
Figure 2.
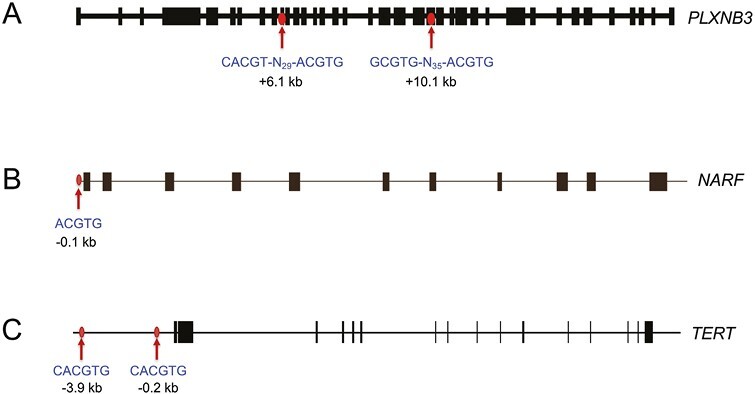
Localization of HIF-1 binding sites in the PLXNB351 (A), NARF55 (B), and TERT56 (C) genes. Chromatin immunoprecipitation identified hypoxia-induced binding of HIF-1α and HIF-1β to genomic regions containing sequences (shown beneath the arrows) that match the HIF consensus binding site 5ʹ-RCGTG-3ʹ (R = A or G) or its complement 5ʹ-CACGY-3ʹ (Y = C or T). Note that these genes contain from 1 to 4 HIF consensus sequences in various orientations at locations (denoted by arrows) which are either upstream or downstream of the transcription start site (at the 5ʹ end of the first exon). Exon-intron structures of the genes are shown extending from left to right in the same orientation as the sequences.
Exposure of HCC1954 and SUM159 cells to hypoxia induced: increased migration and invasion; increased ALDH1 and NANOG expression; and enrichment of CD24−CD44+ and mammosphere-forming cells, all of which were lost in subclones expressing shRNA targeting PLXNB3.54 Since migration, invasion, and CSC properties are all fundamental to metastatic breast cancer cells, we injected MDA-MB-231 subclones with altered PLXNB3 expression into the mammary fat pad of immunodeficient female mice. The parental MDA-MB-231 TNBC cells form primary tumors that spontaneously metastasize to the lungs. Primary tumor growth was not significantly affected by the expression of either of the 2 different shRNAs targeting PLXNB3. However, metastasis to the lungs was virtually eliminated in the absence of PLXNB3 expression. When limiting numbers of MDA-MB-231 cells were injected into the mammary fat pad (1 × 103 rather than 2 × 106 used in the prior experiment), tumors formed in 10 out of 10 mice injected with cells expressing a non-targeting control (NTC) shRNA but in only 3 or 4 of out of 10 mice injected with cells expressing either of 2 different shRNAs targeting PLXNB3, demonstrating a significant impairment of tumor formation by PLXNB3 loss of function.54
The studies described above provide molecular, cellular, and in vivo data indicating the PLXNB3 is essential for BCSC specification. But what is the mechanism? First, in SUM159 cells, the knockdown of SEMA5A, which is the specific ligand for PLXNB3,52 impaired hypoxia-induced migration, invasion, and enrichment of ALDH1+ and mammosphere-forming cells, similar to the effect of PLXNB3 knockdown.54 Second, activation of PLXNB3 by SEMA5A is known to enable PLXNB3 to interact with, and activate, the MET receptor tyrosine kinase, even in the absence of hepatocyte growth factor, which is the cognate ligand for MET.52 In many cancers, MET signals to the non-receptor tyrosine kinase SRC,57 which activates focal adhesion kinase (FAK), which in turn is essential for BCSC specification (Fig. 1) as well as breast cancer invasion and metastasis.55,58,59 In SUM159 cells, both hypoxia-induced invasion of Matrigel (a tumor basement membrane preparation) and enrichment of ALDH1+ BCSCs was blocked by treatment with an inhibitor of MET, SRC, or FAK.54 SRC also signaled to STAT3, which activates NANOG transcription, and hypoxia-induced NANOG mRNA and protein expression were blocked by treatment with an inhibitor of MET, SRC, or STAT3.54 Thus, oncogenic signal transduction pathways leading to BCSC specification and metastasis are activated, in the absence of genetic alterations, within the hypoxic tumor microenvironment.
NARF
RNA-seq data from SUM159 cells revealed that hypoxia-induced the expression of NARF mRNA,13 which encodes nuclear prelamin A recognition factor, a poorly characterized nuclear protein that is also known as iron-only hydrogenase-like protein 2 (despite its name, it has no hydrogenase activity).60,61 NARF has not been previously implicated in breast cancer pathogenesis or cancer stem cell biology. The reader should be aware that the acronym NARF has also been applied to the nemo-like kinase ring finger protein, which is the product of the RNF138 gene. NARF expression was induced by hypoxia in all breast cancer cell lines analyzed, including hormone (estrogen and/or progesterone) receptor-positive (HR+) MCF-7 and T47D cells, HER2+ HCC1954 cells, and triple-negative (HR− and HER2−) MDA-MB-231, SUM149, and SUM159 cells.62 Knockdown experiments in HR+ MCF-7 and HR− MDA-MB-231 cells revealed that hypoxia-induced NARF expression was dependent on HIF-1α and independent of HIF-2α. This conclusion was supported by ChIP assays demonstrating hypoxia-induced binding of HIF-1α and HIF-1β, but not HIF-2α, at a consensus HIF binding site (5ʹ-ACGTG-3ʹ) located 95 bp 5ʹ to the transcription start site of the NARF gene (Fig. 2B). A 55-bp sequence encompassing the site functioned as a hypoxia response element when inserted into a luciferase reporter plasmid, whereas a 3-bp mutation in the HIF binding site (ACGTG to AAAAG) eliminated hypoxia-induced luciferase expression.62 These studies demonstrated that NARF was a direct HIF-1 target gene.
In 1218 breast cancers (TCGA BRCA dataset), NARF mRNA expression was significantly correlated with a 10-gene HIF signature (R = 0.42; P < .0001) and a 20-gene BCSC signature (R = 0.50; P < .0001). Hypoxia-induced enrichment of ALDH1+ cells and mammosphere-forming cells was impaired in MDA-MB-231 and MCF-7 subclones with NARF knockdown.62 When MDA-MB-231 subclones were injected into the mammary fat pad of immunodeficient mice (2 × 106 cells, such that BCSCs were not limiting for tumor initiation), there was no difference in primary tumor growth rate between cells expressing a non-targeting control shRNA (NTC) and cells expressing either of 2 different shRNAs targeting NARF, but lung metastasis of NARF-knockdown cells was markedly impaired. When only 1000 cells were injected (such that BCSCs were limiting), tumors formed in 10/10 mice injected with NTC cells, but only 3/10 and 2/9 mice injected with either of 2 NARF-knockdown subclones, indicating that NARF plays a critical role in BCSC specification and tumor initiation, as described above for PLXNB3. Analysis of human breast cancer biopsy tissues by immunohistochemistry revealed that high NARF protein expression was associated with decreased overall survival of breast cancer patients.62
Exposure of HR+ MCF-7 and HR− MDA-MB-231 cells to hypoxia induced the NARF-dependent expression of KLF4, NANOG, and SOX2 mRNA and protein, whereas OCT4 (encoded by the POU5F1 gene) was constitutively expressed and was not affected by NARF knockdown.62 In MDA-MB-231 cells, OCT4 bound to, and was required for expression of, the KLF4, NANOG, and SOX2 genes. NARF was also required for hypoxia-induced expression of KLF4, NANOG, and SOX2.62 NARF was recruited to OCT4 binding sites in the KLF4, NANOG, and SOX2 genes in an OCT4-dependent manner, and NARF, in turn, increased the occupancy of OCT4 at these sites. In contrast, OCT4 binding to the POU5F1 gene was neither hypoxia-induced nor NARF-dependent. Hypoxia-induced recruitment of the histone-lysine demethylase KDM6A by NARF and OCT4 to the KLF4, NANOG, and SOX2 genes led to decreased trimethylation of lysine-27 of histone H3 (H3K27me3, a repressive histone mark) and increased gene expression.62 In contrast, KDM6A recruitment to the OCT4 site in the POU5F1 gene was neither hypoxia-induced nor NARF-dependent. These data indicate that whereas OCT4 autoregulates its own expression in a constitutive manner, the ability of OCT4 to increase the expression of KLF4, NANOG, and SOX2 in response to hypoxia is dependent on the induction of NARF and its association with both OCT4 and KDM6A. Thus, NARF functions as a coactivator for OCT4, stabilizing its binding to DNA and promoting the recruitment of KDM6A, which erases the repressive histone mark H3K27me3, thereby facilitating transcription of the KLF4, NANOG, and SOX2 genes.
TERT
While the ability of pluripotency factors such as NANOG and OCT4 to regulate the expression of one another is well established, the targets of these transcription factors that mediate self-renewal are less well studied. Telomeres are several-kilobase-long repeats of the sequence 5ʹ-TTAGGG-3ʹ at the tips of each human chromosome.63 The maintenance of telomere length by the enzyme telomerase is required for the infinite self-renewal of ESCs and CSCs.28,64 Telomerase consists of the telomerase reverse transcriptase (TERT) and an RNA that serves as a template for reverse transcription.65 TERT is expressed during embryogenesis, repressed in adult cells, and reactivated in BCSCs, although TERT gene amplification and promoter mutations are rare in common forms of breast cancer, thereby leaving the mechanism of reactivation unexplained.66-68 Hypoxia-induced TERT expression mediated by HIF-1 has been reported in several types of cancer56,69,70 as well as ESCs,71 but the mechanism that restricts HIF-1-dependent TERT expression to stem cells has not been determined.
NANOG knockdown in MDA-MB-231 cells significantly decreased the percentage of ALDH1+ cells and mammosphere-forming cells under both hypoxic and non-hypoxic culture conditions.72 NANOG overexpression increased the expression of KLF4, OCT4, and SOX2 but did not increase the percentage of ALDH1+ cells or mammosphere-forming cells under non-hypoxic conditions. In contrast, NANOG overexpression did increase the percentage of BCSCs under hypoxic culture conditions and this effect was lost in HIF-1α-knockdown cells. Overexpression of NANOG with HIF-1α, but not with HIF-2α, increased the percentage of BCSCs under non-hypoxic conditions.72 These results indicated that NANOG is necessary but not sufficient for BCSC specification and suggested that both NANOG and HIF-1α were required for BCSC specification.
TERT knockdown in MDA-MB-231 cells resulted in progressive telomere shortening and loss of proliferative ability after 6 passages.72 The percentage of ALDH1+ cells also decreased progressively as TERT-knockdown subclones were passaged. Exposure of ER+ MCF-7 and ER- MDA-MB-231 cells to hypoxia increased TERT mRNA and protein expression, telomerase activity, and telomere length, all of which were lost by knockdown of HIF-1α but not HIF-2α.72 ChIP assays identified hypoxia-induced binding of HIF-1α and HIF-1β at 2 sites located approximately 0.2 and 3.9 kb 5ʹ to the TERT gene (Fig. 2C). TCGA BRCA dataset analysis revealed that the 10-gene HIF signature was significantly correlated with a 43-gene telomerase signature,73 suggesting that HIF-1 plays a key role in regulating telomerase activity in primary human breast cancers.72
NANOG knockdown blocked hypoxia-induced TERT expression, similar to the effect of HIF-1α knockdown. NANOG knockdown also blocked hypoxia-induced telomerase activity and telomere lengthening, similar to the effect of HIF-1α or TERT knockdown. ChIP assays revealed that NANOG was recruited to HIF-1 binding sites in the TERT gene and that NANOG knockdown impaired the hypoxia-induced binding of HIF-1α and HIF-1β to the TERT promoter. NANOG interacted directly with the transactivation domain of HIF-1α and recruited USP9X, a deubiquitinase, to HIF-1α, thereby blocking its ubiquitination and proteasomal degradation. In addition, NANOG interacted with the histone acetyltransferase p300 to stabilize its interaction with HIF-1α, thereby increasing acetylation of histone H3 at lysine residue 9 (H3K9ac) and H3K27ac, which are histone marks associated with transcriptional activation, at the TERT promoter.72 Thus, NANOG functions as a coactivator that increases HIF-1α protein stability and transactivation function in breast cancer cells.
Conclusions
BCSCs are the only cells within a primary tumor capable of giving rise to a secondary (recurrent and/or metastatic) tumor, based on their ability for infinite self-renewal, which is controlled by the expression of pluripotency factors that were first described in ESCs. However, unlike ESCs, BCSCs are not pluripotent and rather than representing a developmental state, they are better characterized as representing a reversible physiological state that is induced by intratumoral hypoxia. Indeed, the studies described above indicate that an intimate, cooperative relationship between HIF-1 and NANOG is required for BCSC specification and for self-renewal, which is dependent upon TERT expression. Moreover, BCSCs are inherently invasive and metastatic, as demonstrated in the PLXNB3 study. HIFs also activate the expression of genes encoding proteins that mediate immune evasion by cancer cells.15,17 Thus, high HIF activity drives the lethal cancer phenotype. To effectively treat breast cancer one must eliminate BCSCs and inhibition of HIF-1 activity may aid in achieving this goal. Recently, the HIF-2-selective inhibitor Belzutifan has been approved for the treatment of renal cell carcinoma in patients with von Hippel-Lindau syndrome,74 but the studies summarized here suggest this drug will not be effective in blocking hypoxia-induced BCSC specification, which appears to be controlled exclusively by HIF-1. Thus, the development of dual HIF-1/2 inhibitors may provide a more effective therapeutic strategy for eradicating BCSCs.
Funding
G.L.S. is the C. Michael Armstrong Professor at the Johns Hopkins University School of Medicine and research in the author’s laboratory is supported by the Armstrong Family Foundation.
Conflict of Interest
GLS is an inventor on patent application PCT/US2022/039883 and is a founder of, and holds equity in, HIF Therapeutics Inc. This arrangement has been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies.
Data Availability
No new data were generated or analyzed in support of this review.
References
- 1. Vaupel P, Höckel M, Mayer A.. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007;9(8):1221-1235. 10.1089/ars.2007.1628 [DOI] [PubMed] [Google Scholar]
- 2. Carmeliet P, Jain RK.. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417-427. 10.1038/nrd3455 [DOI] [PubMed] [Google Scholar]
- 3. Höckel M, Schlenger K, Aral B, et al. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996;56(19):4509-4515. [PubMed] [Google Scholar]
- 4. Thomlinson RH, Gray LH.. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9(4):539-549. 10.1038/bjc.1955.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang GL, Jiang BH, Rue EA, Semenza GL.. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92(12):5510-5514. 10.1073/pnas.92.12.5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang GL, Semenza GL.. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270(3):1230-1237. 10.1074/jbc.270.3.1230 [DOI] [PubMed] [Google Scholar]
- 7. Liao C, Liu X, Zhang C, Zhang Q.. Tumor hypoxia: from basic knowledge to therapeutic implications. Semin Cancer Biol. 2023;88:172-186. 10.1016/j.semcancer.2022.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magliulo D, Bernardi R.. Hypoxic stress and hypoxia-inducible factors in leukemias. Front Oncol. 2022;12:973978. 10.3389/fonc.2022.973978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol Mech Dis. 2014;9:47-71. 10.1146/annurev-pathol-012513-104720 [DOI] [PubMed] [Google Scholar]
- 10. Wicks EE, Semenza GL.. Hypoxia-inducible factors: cancer progression and clinical translation. J Clin Invest. 2022;132(11):e159839. 10.1172/JCI159839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625-634. 10.1038/onc.2009.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aebersold DM, Burri P, Beer KT, et al. Expression of hypoxia-inducible factor-1α: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001;61(7):2911-2916. [PubMed] [Google Scholar]
- 13. Wang Y, Lyu Y, Tu K, et al. Histone citrullination by PADI4 is required for HIF-dependent transcriptional responses to hypoxia and tumor vascularization. Sci Adv. 2021;7(35):eabe3771. 10.1126/sciadv.abe3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lombardi O, Li R, Halim S, Choudhry H, Ratcliffe PJ, Mole DR.. Pan-cancer analysis of tissue and single-cell HIF-pathway activation using a conserved gene signature. Cell Rep. 2022;41(7):111652. 10.1016/j.celrep.2022.111652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Noman MZ, Hasmim M, Lequeux A, et al. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: new opportunities and challenges. Cells. 2019;8(9):1083. 10.3390/cells8091083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rankin EB, Giaccia AJ.. Hypoxic control of metastasis. Science. 2016;352(6282):175-180. 10.1126/science.aaf4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Semenza GL. Intratumoral hypoxia and mechanisms of immune evasion mediated by hypoxia-inducible factors. Physiology (Bethesda). 2021;36(2):73-83. 10.1152/physiol.00034.2020 [DOI] [PubMed] [Google Scholar]
- 18. Bonnet D, Dick JE.. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730-737. 10.1038/nm0797-730 [DOI] [PubMed] [Google Scholar]
- 19. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukemia after transplantation into SCID mice. Nature. 1994;367(6464):645-648. 10.1038/367645a0 [DOI] [PubMed] [Google Scholar]
- 20. Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821-5828. [PubMed] [Google Scholar]
- 21. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF.. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983-3988. 10.1073/pnas.0530291100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O’Brien CA, Pollett A, Gallinger S, Dick JE.. A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. Nature. 2007;445(7123):106-110. 10.1038/nature05372 [DOI] [PubMed] [Google Scholar]
- 23. Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104(3):973-978. 10.1073/pnas.0610117104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313-323. 10.1016/j.stem.2007.06.002 [DOI] [PubMed] [Google Scholar]
- 25. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ.. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946-10951. 10.1158/0008-5472.CAN-05-2018 [DOI] [PubMed] [Google Scholar]
- 26. Fang D, Nguyen TK, Leishear K, et al. A tumorigenic subpopulation with stem cell properties in melanoma. Cancer Res. 2005;65(20):9328-9337. 10.1158/0008-5472.CAN-05-1343 [DOI] [PubMed] [Google Scholar]
- 27. Ross RA, Spengler BA.. Human neuroblastoma stem cells. Semin Cancer Biol. 2007;17(3):241-247. 10.1016/j.semcancer.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 28. Shay JW, Wright WE.. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010;584(17):3819-3825. 10.1016/j.febslet.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Charafe-Jauffret E, Ginestier C, Iovino F, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69(4):1302-1313. 10.1158/0008-5472.CAN-08-2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oskarsson T, Batlle E, Massague J.. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14(3):306-321. 10.1016/j.stem.2014.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140(1):62-73. 10.1016/j.cell.2009.12.007 [DOI] [PubMed] [Google Scholar]
- 32. Hu T, Liu S, Breiter DR, et al. Octamer 4 small interfering RNA results in cancer stem cell-like apoptosis. Cancer Res. 2008;68(16):6533-6540. 10.1158/0008-5472.CAN-07-6642 [DOI] [PubMed] [Google Scholar]
- 33. Leis QY, Eguirara A, Lopez-Arribillaga E, et al. Sox2 expression in breast tumors and activation in breast cancer stem cells. Oncogene. 2012;31(11):1354-1365. 10.1038/onc.2011.338 [DOI] [PubMed] [Google Scholar]
- 34. Santaliz-Ruiz LE IV, Xie X, Old M, et al. Emerging role of Nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014;135(12):2741-2748. 10.1002/ijc.28690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu F, Li J, Chen H, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30(18):2161-2172. 10.1038/onc.2010.591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kao J, Salari K, Bocanegra M, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4(7):e6146. 10.1371/journal.pone.0006146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555-567. 10.1016/j.stem.2007.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Domenici G, Aurrekoetxea-Rodriguez I, Simoes BM, et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene. 2019;38(17):3151-3169. 10.1038/s41388-018-0656-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shi L, Tang X, Qian M, et al. A SIRT1-centered circuitry regulates breast cancer stemness and metastasis. Oncogene. 2018;37(49):6299-6315. 10.1038/s41388-018-0370-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marcato P, Dean CA, Pan D, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29(1):32-45. 10.1002/stem.563 [DOI] [PubMed] [Google Scholar]
- 41. Ozaki A, Motomura H, Tamori S, et al. High expression of p62 and ALDH1A3 is associated with poor prognosis in luminal B breast cancer. Anticancer Res. 2022;42(7):3299-3312. 10.21873/anticanres.15818 [DOI] [PubMed] [Google Scholar]
- 42. Dontu G, Abdallah WM, Foley JM, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17(10):1253-1270. 10.1101/gad.1061803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506-5511. 10.1158/0008-5472.CAN-05-0626 [DOI] [PubMed] [Google Scholar]
- 44. Conley SJ, Gheordunescu E, Kakarala P, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA. 2012;109(8):2784-2789. 10.1073/pnas.1018866109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xiang L, Semenza GL.. Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Adv Cancer Res. 2019;141:175-212. 10.1016/bs.acr.2018.11.001 [DOI] [PubMed] [Google Scholar]
- 46. Kim RJ, Park JR, Roh KJ, et al. High aldehyde dehydrogenase activity enhances stem cell features in breast cancer cells by activating hypoxia-inducible factor-2α. Cancer Lett. 2013;333(1):18-31. 10.1016/j.canlet.2012.11.026 [DOI] [PubMed] [Google Scholar]
- 47. Ciccone V, Terzuoli E, Donnini S, et al. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J Exp Clin Cancer Res. 2018;37(1):311. 10.1186/s13046-018-0975-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forsythe JA, Jiang BH, Iyer NV, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16(9):4604-4613. 10.1128/MCB.16.9.4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Forristal CE, Wright KL, Hanley NA, Oreffo ROC, Houghton FD.. Hypoxia-inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. Reproduction. 2010;139(1):85-97. 10.1530/REP-09-0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petruzzelli R, Christensen DR, Perry KL, et al. HIF-2α regulates NANOG expression in human embryonic stem cells following hypoxia and reoxygenation through the interaction with an Oct-Sox cis regulatory element. PLoS One. 2014;9(10):e108309. 10.1371/journal.pone.0108309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mathieu J, Zhang Z, Zhou W, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71(13):4640-4652. 10.1158/0008-5472.CAN-10-3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Artigiani S, Conrotto P, Fazzari P, et al. Plexin-B3 is a functional receptor for semaphorin 5A. EMBO Rep. 2004;5(7):710-714. 10.1038/sj.embor.7400189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang X, Shao S, Li L.. Characterization of class-3 semaphorin receptors, neuropilins and plexins, as therapeutic targets in a pan-cancer study. Cancers. 2020;12(7):1816. 10.3390/cancers12071816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zuo Q, Yang Y, Lyu Y, et al. Plexin-B3 expression stimulates MET signaling, breast cancer stem cell specification and lung metastasis. Cell Rep. 2023;42(3):112164. 10.1016/j.celrep.2023.112164 [DOI] [PubMed] [Google Scholar]
- 55. Mitra SK, Lim ST, Chi A, Schlaepfer DD.. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene. 2006;25(32):4429-4440. 10.1038/sj.onc.1209482 [DOI] [PubMed] [Google Scholar]
- 56. Nishi H, Nakada T, Kyo S, et al. Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT). Mol Cell Biol. 2004;24(13):6076-6083. 10.1128/MCB.24.13.6076-6083.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bradley CA, Salto-Tellez M, Laurent-Puig P, et al. ; MErCuRIC consortium. Targeting c-MET in gastrointestinal tumors: rationale, opportunities and challenges. Nat Rev Clin Oncol. 2017;14(9):562-576. 10.1038/nrclinonc.2017.40 [DOI] [PubMed] [Google Scholar]
- 58. Luo M, Fan H, Nagy T, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69(2):466-474. 10.1158/0008-5472.CAN-08-3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pylayeva Y, Gillen KM, Gerald W, et al. Ras- and PI3K-dependent breast tumorigenesis inn mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119(2):252-266. 10.1172/JCI37160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barton RM, Worman HJ.. Prenylated prelamin A interacts with NARF, a novel nuclear protein. J Biol Chem. 1999;274(42):30008-30018. 10.1074/jbc.274.42.30008 [DOI] [PubMed] [Google Scholar]
- 61. Song D, Lee FS.. A role for IOP1 in mammalian cytosolic iron-sulfur protein biogenesis. J Biol Chem. 2008;283(14):9231-9238. 10.1074/jbc.M708077200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang Y, Chen C, Zuo Q, et al. NARF is a hypoxia-induced coactivator for OCT4-mediated breast cancer stem cell specification. Sci Adv. 2022;8(49):eabo5000. 10.1126/sciadv.abo5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Greider CW. Telomere length regulation. Annu Rev Biochem. 1996;65:337-365. 10.1146/annurev.bi.65.070196.002005 [DOI] [PubMed] [Google Scholar]
- 64. Hannen R, Bartsch JW.. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018;592(12):2023-2031. 10.1002/1873-3468.13084 [DOI] [PubMed] [Google Scholar]
- 65. Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E.. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat Struct Mol Biol. 2010;17(4):513-518. 10.1038/nsmb.1777 [DOI] [PubMed] [Google Scholar]
- 66. Da Silva E, Selenica P, Vahdatinia M, et al. TERT promoter hotspot mutations and gene amplification in metaplastic breast cancer. npj Breast Cancer. 2021;7(1):43. 10.1038/s41523-021-00250-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gay-Bellile M, Veronese L, Combes P, et al. TERT promoter status and gene copy number gains: effect on TERT expression and association with prognosis in breast cancer. Oncotarget. 2017;8(44):77540-77551. 10.18632/oncotarget.20560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lin T, Meng L, Li Y, Tsai RYL.. Tumor-initiating function of nucleostemin-enriched mammary tumor cells. Cancer Res. 2010;70(22):9444-9452. 10.1158/0008-5472.CAN-10-2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Song H, Chen X, Jiao Q, et al. HIF-1α-mediated telomerase reverse transcriptase activation inducing autophagy through mammalian target of rapamycin promotes papillary thyroid carcinoma progression during hypoxia stress. Thyroid. 2021;31(2):233-246. 10.1089/thy.2020.0023 [DOI] [PubMed] [Google Scholar]
- 70. Yatabe N, Kyo S, Maida Y, et al. HIF-1-mediated activation of telomerase in cervical cancer cells. Oncogene. 2004;23(20):3708-3715. 10.1038/sj.onc.1207460 [DOI] [PubMed] [Google Scholar]
- 71. Coussens M, Davy P, Brown L, et al. RNAi screen for telomerase reverse transcriptase transcriptional regulators identifies HIF1α as critical for telomerase function in murine embryonic stem cells. Proc Natl Acad Sci USA. 2010;107(31):13842-13847. 10.1073/pnas.0913834107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu H, Lyu Y, Tran L, et al. HIF-1 recruits NANOG as a coactivator for TERT gene transcription in hypoxic breast cancer cells. Cell Rep. 2021;36(13):109757. 10.1016/j.celrep.2021.109757 [DOI] [PubMed] [Google Scholar]
- 73. Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49(3):349-357. 10.1038/ng.3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel-Lindau disease. N Engl J Med. 2021;385(22):2036-2046. 10.1056/NEJMoa2103425 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were generated or analyzed in support of this review.