

Abstract
Background
Anthracycline‐induced cardiomyopathy is a leading cause of premature death in childhood cancer survivors, presenting a need to understand the underlying pathogenesis. We sought to examine differential blood‐based mRNA expression profiles in anthracycline‐exposed childhood cancer survivors with and without cardiomyopathy.
Methods and Results
We designed a matched case‐control study (Children's Oncology Group‐ALTE03N1) with mRNA sequencing on total RNA from peripheral blood in 40 anthracycline‐exposed survivors with cardiomyopathy (cases) and 64 matched survivors without (controls). DESeq2 identified differentially expressed genes. Ingenuity Pathway Analyses (IPA) and Gene Set Enrichment Analyses determined the potential roles of altered genes in biological pathways. Functional validation was performed by gene knockout in human‐induced pluripotent stem cell‐derived cardiomyocytes using CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein 9) technology. Median age at primary cancer diagnosis for cases and controls was 8.2 and 9.7 years, respectively. Thirty‐six differentially expressed genes with fold change ≥±2 were identified; 35 were upregulated. IPA identified “hepatic fibrosis” and “iron homeostasis” pathways to be significantly modulated by differentially expressed genes, including toxicology functions of myocardial infarction, cardiac damage, and cardiac dilation. Leading edge analysis from Gene Set Enrichment Analyses identified lactate dehydrogenase A (LDHA) and cluster of differentiation 36 (CD36) genes to be significantly upregulated in cases. Interleukin 1 receptor type 1, 2 (IL1R1, IL1R2), and matrix metalloproteinase 8, 9 (MMP8, MMP9) appeared in multiple canonical pathways. LDHA‐knockout human‐induced pluripotent stem cell‐derived cardiomyocytes showed increased sensitivity to doxorubicin.
Conclusions
We identified differential mRNA expression profiles in peripheral blood of anthracycline‐exposed childhood cancer survivors with and without cardiomyopathy. Upregulation of LDHA and CD36 genes suggests metabolic perturbations in a failing heart. Dysregulation of proinflammatory cytokine receptors IL1R1 and IL1R2 and matrix metalloproteinases, MMP8 and MMP9 indicates structural remodeling that accompanies the clinical manifestation of symptomatic cardiotoxicity.
Keywords: anthracyclines, cardiomyopathy, childhood cancer survivors, gene expression, peripheral blood, transcriptome
Nonstandard Abbreviations and Acronyms
- CRISPR/Cas9
clustered regularly interspaced short palindromic repeats/CRISPR‐associated protein 9
- CVRFs
cardiovascular risk factors
- DEGs
differentially expressed genes
- GSEA
gene set enrichment analysis
- hiPSC‐CMs
human induced pluripotent stem cell‐derived cardiomyocytes
- IPA
Ingenuity Pathway Analysis
Clinical Perspective.
What Is New?
A gene expression profile of peripheral blood can distinguish anthracycline‐exposed childhood cancer survivors with cardiomyopathy from those without.
LDHA, CD36, IL1R1, IL1R2, MMP8 and MMP9 genes were significantly upregulated in childhood cancer survivors with cardiomyopathy when compared with those without, suggesting metabolic and structural perturbations in a failing heart.
What Are the Clinical Implications?
This study suggests that transcriptional dysregulation is associated with anthracycline‐induced cardiomyopathy and can be inferred from the blood transcriptome.
Anthracyclines are a highly effective class of chemotherapy used in the treatment of childhood lymphoma, leukemia, and several solid tumors 1 , 2 ; 60% of children with cancer are treated with anthracyclines. 3 However, the clinical utility of anthracyclines is limited because of cumulative and irreversible cardiomyopathy, leading to congestive heart failure. 4 The mechanisms underlying anthracycline‐induced cardiotoxicity are multifactorial and include mitochondrial injury, oxidative stress, apoptosis, ferroptosis, and dysregulation of autophagy. 5 Demographic characteristics such as young age at anthracycline exposure, female sex, chest radiation, and presence of cardiovascular risk factors (CVRFs: diabetes, hypertension, dyslipidemia) modify the anthracycline‐cardiomyopathy association. 6 The 5‐year survival rate for anthracycline‐induced congestive heart failure is estimated to be <50%. 7 While the cardiomyopathy risk is dose‐dependent, there is considerable interpatient variability at any anthracycline dose, suggesting a need to understand the underlying genetic basis. 8 , 9 We and others have identified genomic variants associated with cardiomyopathy, but these explain only a modest proportion of cardiomyopathy risk. 10 , 11
Comparing mRNA transcript levels between 2 contrasting phenotypes allows for the identification of differentially expressed genes (DEGs) that can shed mechanistic insights into the disease. RNA sequencing allows the quantification of mRNA levels using an unbiased approach. Differential constitutive gene expression in peripheral blood of anthracycline‐exposed cancer survivors with and without cardiomyopathy could enhance our knowledge of biological functions impacted by DEGs and the pathogenesis of anthracycline‐induced cardiomyopathy. While obtaining heart biopsies from cancer survivors is logistically difficult, peripheral blood is easily obtained, and gene expression levels in blood correlate with the cardiac transcriptome. 12 , 13 , 14 We tested the hypothesis that assessment of differential gene expression in peripheral blood followed by an examination of mechanistically plausible DEGs in human‐induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs) can help elucidate molecular mechanisms underlying anthracycline‐induced cardiotoxicity.
Methods
Data Availability
The data discussed in this publication have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus and are accessible through GEO Series accession number GSE218276 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218276).
Study Design
Participants were drawn from a COG (Children's Oncology Group) study (COG‐ALTE03N1, PI: S. Bhatia) that uses a matched case–control design to understand the pathogenesis of cardiomyopathy in childhood cancer survivors. COG member institutions enrolled patients after obtaining approval from local institutional review boards. Written informed consent/assent was obtained from patients or parents/legal guardians. Cases consisted of childhood cancer survivors who developed cardiomyopathy after exposure to anthracyclines. For each case, up to 3 anthracycline‐exposed survivors with no signs or symptoms of cardiomyopathy were randomly selected as controls from the same COG cohort, matched on primary cancer diagnosis, year of diagnosis (±5 years), and race or ethnicity. The selected controls also needed a longer duration of cardiomyopathy‐free follow‐up compared with the time from cancer diagnosis to cardiomyopathy for the corresponding case. Participants provided peripheral blood samples in PAXgene blood RNA tubes for germline RNA at the time of enrollment to the study (single time point).
Cases fulfilled the American Heart Association criteria for cardiac compromise by presenting with signs or symptoms (dyspnea, orthopnea, fatigue, edema, hepatomegaly, or rales); or, in the absence of signs or symptoms, had echocardiographic features of left ventricular dysfunction (ejection fraction ≤40% or fractional shortening ≤28%). Lifetime anthracycline exposure was calculated by multiplying the cumulative dose (mg/m2) of individual anthracyclines (doxorubicin [n=90], daunorubicin [n=26], mitoxantrone [n=4], and idarubicin [n=3]) by a factor that reflects the drug's cardiotoxic potential 15 and then summing the results. Radiation to the chest with the heart in the field was captured as a yes/no variable. CVRFs were captured as a yes/no variable.
RNA Isolation, Library Construction, and Sequencing
RNA was isolated from whole blood using the PAXgene blood RNA kit (Qiagen Inc., CA). RNA concentration was measured using a Nanodrop ND‐1000 Spectrophotometer (ThermoFisher Scientific Inc., MA). RNA quality was checked on Bioanalyzer Nanochip (Agilent Technologies, CA), and samples with RNA integrity number (RIN) >7 were submitted to the Genomic Services Laboratory at HudsonAlpha Institute for Biotechnology, Huntsville, AL. Poly‐adenylated RNAs were isolated using NEBNext Magnetic Oligo d(T)25 beads. Libraries were prepared using the TruSeq RNA Sample Preparation Kit (Illumina). Each library was pair‐end sequenced (100 bp) using the TruSeq SBS Kit v4‐HS (Illumina), on a NovaSeq 6000 platform. Raw reads were de‐multiplexed using bcl2fastq Conversion Software (Illumina Inc., CA) with default settings.
Differential Gene Expression Analysis
TrimGalore! 16 was used to trim off primer adapter sequences found in raw FASTQ files. STAR was used to align trimmed RNA‐Sequencing FASTQ reads to the human reference genome from Gencode (GRCh38 p7 Release 25). 17 HTSeq‐count was used to count the reads mapping to each gene from the STAR alignments. 18 Normalization and differential expression were then applied to the count files using DESeq2. 19
Ingenuity Pathway Analysis
DEGs with a fold change of ±1.5 and P value <0.05 were analyzed by the Ingenuity Pathway Analysis (IPA) software (Qiagen Inc., CA) to identify canonical pathways and toxicological functions. Canonical signaling pathways enriched by DEGs were identified and ranked according to P values. The ratios of significantly involved canonical signaling pathways were calculated by dividing the numbers of DEGs in the canonical signaling pathway by the number of total genes in the pathway and indicated as a percentage. IPA‐Tox was used to examine toxicological functions and identify subsets of DEGs predictive of toxicity end points.
Gene Set Enrichment Analysis and Leading‐Edge Analysis
Gene Set Enrichment Analysis (GSEA) uses prior gene sets that have been grouped by their involvement in the same biological pathway and searches for sets of genes significantly over‐represented in a given list of genes. The unranked list of DEGs from DESeq2 was exported and analyzed with GSEA (v.4.1.0) applying the hallmark (50 gene set) and cardiomyopathy (19 gene set) collections from MSigDB (Tables S1 and S2). False discovery rate and nominal P value estimates determined the statistical significance of the enrichment score. A gene set with a normalized enrichment score of >1.5, false discovery rate <0.25, and a nominal P value <0.01 was considered significantly enriched. We also examined genes that were enriched in the hallmark and cardiomyopathy gene sets using the leading edge analysis tool in GSEA.
Functional Analyses
Criteria for prioritizing candidate genes for functional analyses
We used the following criteria to prioritize genes for functional studies: (1) “protein‐coding” genes obtained by the “biotype” feature from DESeq2 output (P adj≤0.05 and fold‐change ≥±2); (2) genes with ≥50% of matched sets showing differential expression between the case and their matched control(s); (3) leading edge genes of an enriched gene set; (4) robust expression in the adult human heart and hiPSC‐CMs 20 , 21 ; and (5) mechanistic plausibility and association with cardiac dysfunction informed by literature review (Table S3).
We used existing hiPSC line 19c3 22 previously generated from peripheral blood mononuclear cells from a healthy individual using the CytoTune‐iPS 2.0 Sendai Reprogramming Kit (Invitrogen, A16518) (Data S1). To generate gene knockout gRNA expression vectors, 1 to 2 gRNAs targeting all splicing variants of the targeted genes were designed using an online CRISPR design tool (Integrated DNA Technologies) with a high predicted on‐target score and minimal predicted off‐target effect. Table S4 includes primers for sgRNA expression vector generation and sequencing primers.
CRISPR/Cas9‐mediated knockout of candidate genes
Details of the CRISPR/Cas9‐mediated knockout of candidate genes are summarized in Data S1. We used quantitative reverse transcription‐polymerase chain reaction to examine the success of the candidate gene knockout. All polymerase chain reactions were performed in triplicates in a 384‐well plate format using TaqMan Fast Advanced Master Mix (Applied Biosystems, 4444557) in a QuantStudio 5 Real‐Time polymerase chain reaction System (Applied Biosystems, A28140). Table S5 summarizes the TaqMan probes. Differentiation into cardiomyocytes was completed following our previously described protocol 23 using a hiPSC line expressing an exogenous TNNT2 promoter‐driven Zeocin antibiotic selection resistance cassette for cardiomyocyte purification (Data S1). hiPSC‐CMs at day 30 after initiation of differentiation were treated for 72 hours with doxorubicin (0.01–100 μM) diluted in RPMI 1640 medium (Corning) supplemented with 500 μg/mL fatty acid‐free bovine serum albumin (GenDEPOT, A0100). Cell viability was assessed after the 72‐hour period using a resazurin assay. Fluorescence was measured using a VarioSkan Lux Multi‐Mode Reader (Thermo Scientific) using top read, an excitation wavelength of 560 nm, and emission wavelength of 590 nm. Data were presented as mean±SEM. Comparisons used 1‐way ANOVA test, an unpaired 2‐tailed Student t‐test, or F‐test. Data were analyzed using Excel and graphed using Prism 7.0 software (GraphPad), depicting standard dose–response guidelines.
Results
Patient Characteristics
The median age at primary cancer diagnosis for the 40 cases and 64 matched controls was 8.2 and 9.7 years, respectively (Table 1). Cases received a higher cumulative anthracycline exposure than the controls (≥250 mg/m2: 62.5% versus 35.9%, P=0.008). Cases were more likely to have received chest radiation (47.5% versus 20.3%; P=0.003) and more likely to have had a CVRF (35% versus 3.1%; P<0.0001). The median time between cancer diagnosis and cardiomyopathy for cases was 5.3 years; controls were followed for a significantly longer period (median, 10.1 years; P=0.0006).
Table 1.
Characteristics of Anthracycline‐Exposed Childhood Cancer Survivors
| Variables | Cases (n=40) | Controls (n=64) | P value* |
|---|---|---|---|
| Age at primary cancer diagnosis, y | |||
| Median (IQR) | 8.2 (3.6–13.9) | 9.7 (3.3–14.4) | 0.9 |
| Sex, n (%) | |||
| Female | 24 (60.0) | 34 (53.1) | 0.5 |
| Male | 16 (40.0) | 30 (46.9) | |
| Cumulative anthracycline exposure, n (%) | |||
| <250 mg/m2 | 15 (37.5) | 41 (64.1) | 0.008 † |
| ≥250 mg/m2 | 25 (62.5) | 23 (35.9) | |
| Chest radiation | |||
| Yes (n, %) | 19 (47.5) | 13 (20.3) | 0.003 † |
| Dose in cGy (Mean±SD) | 1417.3 ± 284.1 | 674.1 ± 226.4 | 0.003 † |
| Race or ethnicity (n, %) | |||
| Non‐Hispanic White | 23 (57.5) | 37 (57.8) | Matched |
| Hispanic | 9 (22.5) | 16 (25.0) | |
| Black | 5 (12.5) | 7 (10.9) | |
| Asian | 3 (7.5) | 3 (4.7) | |
| Mixed race ethnicity | 0 (0.0) | 1 (1.6) | |
| Primary diagnosis, n (%) | |||
| Acute lymphoblastic leukemia | 9 (22.5) | 16 (25.0) | Matched |
| Acute myeloid leukemia | 2 (5.0) | 3 (4.7) | |
| Ewing sarcoma | 4 (10.0) | 8 (12.5) | |
| Hodgkin lymphoma | 7 (17.5) | 10 (15.6) | |
| Kidney tumors | 2 (5.0) | 2 (3.1) | |
| Neuroblastoma | 5 (12.5) | 8 (12.5) | |
| Non‐Hodgkin lymphoma | 5 (12.5) | 8 (12.5) | |
| Osteosarcoma | 4 (10.0) | 7 (10.9) | |
| Soft tissue sarcoma | 2 (5.0) | 2 (3.1) | |
| CVRF, n (%) | |||
| No | 24 (60.0) | 62 (96.9) | <0.0001 † |
| Yes | 14 (35.0) | 2 (3.1) | |
| Missing | 2 (5.0) | 0 (0.0) | |
| Time from cancer diagnosis to study enrollment, y | |||
| Median (IQR) | 11.1 (4.3–18.8) | 10.1 (7.1–14.5) | 0.8 |
| Time from cancer diagnosis to cardiac event for cases, y | |||
| Median (IQR) | 5.3 (0.8–12.8) | ‐ | |
cGy indicates centiGray; CVRF, cardiovascular risk factors; and IQR, interquartile range.
P values were estimated using either chi‐square or Fisher exact test for categorical variables and the Wilcoxon/Kruskal–Wallis test for continuous variables.
Indicates statistical significance at P < 0.05.
Gene Expression and Anthracycline‐Induced Cardiomyopathy
Of the 43198 genes expressed in the peripheral blood of the study participants, we filtered 28026 low‐expressing genes (RNA counts <10 in ≥70% of samples). The remaining 15172 genes comprised protein‐coding genes (82%) and lncRNA/pseudogenes/immunoglobulin genes (18%). Using DESeq2, we identified 36 DEGs with an adjusted P value cutoff of <0.05 and a fold‐change cutoff of ±2, when comparing all cases to all controls (DESeq2 does not allow matched case–control comparisons) (Table 2). Thirty‐five of the 36 DEGs were upregulated among the cases (IFI27, RAP1GAP, HBG2, HBD, ARG1, CD177, GYPB, ORM1, AHSP, MCEMP1, FAM20A, IFIT1B, CA1, TSPO2, SLC6A9, ALAS2, KLF1, MMP8, OLFM4, ENSG00000274173, FCGR1CP, HP, VSIG4, S100A12, SPTA1, HLA‐DRB6, ANKRD9, ITLN1, KLHDC8A, KRT1, ZDHHC19, DAAM2, KLC3, CEACAM8 and IGKV1‐27); 1 gene (NKX3.1) was downregulated. We plotted the normalized RNA counts of each case with their corresponding matched control(s) for the 36 genes (Figure S1).
Table 2.
List of Differentially Expressed Genes in Cases Versus Controls
| Ensembl ID | Gene name | Gene biotype | Fold change | P value | P adj |
|---|---|---|---|---|---|
| ENSG00000165949 | IFI27 | protein_coding | 5.93 | 1.32E‐09 | 6.55E‐06 |
| ENSG00000076864 | RAP1GAP | protein_coding | 4.09 | 1.48E‐06 | 6.89E‐04 |
| ENSG00000196565 | HBG2 | protein_coding | 4.00 | 4.36E‐06 | 1.20E‐03 |
| ENSG00000223609 | HBD | protein_coding | 3.43 | 2.06E‐07 | 2.36E‐04 |
| ENSG00000118520 | ARG1 | protein_coding | 3.23 | 3.82E‐09 | 1.42E‐05 |
| ENSG00000204936 | CD177 | protein_coding | 2.70 | 1.43E‐04 | 7.67E‐03 |
| ENSG00000250361 | GYPB | protein_coding | 2.69 | 1.56E‐04 | 7.92E‐03 |
| ENSG00000229314 | ORM1 | protein_coding | 2.61 | 1.64E‐06 | 7.17E‐04 |
| ENSG00000169877 | AHSP | protein_coding | 2.56 | 1.57E‐05 | 2.19E‐03 |
| ENSG00000183019 | MCEMP1 | protein_coding | 2.50 | 8.53E‐10 | 6.35E‐06 |
| ENSG00000108950 | FAM20A | protein_coding | 2.48 | 3.46E‐10 | 5.15E‐06 |
| ENSG00000204010 | IFIT1B | protein_coding | 2.36 | 5.24E‐05 | 4.40E‐03 |
| ENSG00000133742 | CA1 | protein_coding | 2.34 | 1.57E‐03 | 2.78E‐02 |
| ENSG00000112212 | TSPO2 | protein_coding | 2.31 | 4.06E‐05 | 3.78E‐03 |
| ENSG00000196517 | SLC6A9 | protein_coding | 2.30 | 4.03E‐05 | 3.78E‐03 |
| ENSG00000158578 | ALAS2 | protein_coding | 2.29 | 1.31E‐04 | 7.42E‐03 |
| ENSG00000105610 | KLF1 | protein_coding | 2.29 | 3.41E‐05 | 3.41E‐03 |
| ENSG00000118113 | MMP8 | protein_coding | 2.24 | 6.60E‐04 | 1.75E‐02 |
| ENSG00000102837 | OLFM4 | protein_coding | 2.19 | 3.49E‐03 | 4.32E‐02 |
| ENSG00000274173 | ‐ | lncRNA | 2.19 | 7.01E‐05 | 5.02E‐03 |
| ENSG00000265531 | FCGR1CP | Unprocessed pseudogene | 2.17 | 4.07E‐07 | 3.57E‐04 |
| ENSG00000257017 | HP | protein_coding | 2.17 | 8.99E‐06 | 1.67E‐03 |
| ENSG00000155659 | VSIG4 | protein_coding | 2.16 | 8.09E‐07 | 5.24E‐04 |
| ENSG00000163221 | S100A12 | protein_coding | 2.14 | 7.26E‐07 | 4.91E‐04 |
| ENSG00000163554 | SPTA1 | protein_coding | 2.14 | 5.33E‐05 | 4.40E‐03 |
| ENSG00000229391 | HLA‐DRB6 | Transcribed unprocessed pseudogene | 2.14 | 2.40E‐03 | 3.57E‐02 |
| ENSG00000156381 | ANKRD9 | protein_coding | 2.11 | 9.11E‐06 | 1.68E‐03 |
| ENSG00000179914 | ITLN1 | protein_coding | 2.09 | 1.41E‐04 | 7.61E‐03 |
| ENSG00000162873 | KLHDC8A | protein_coding | 2.06 | 2.76E‐05 | 3.09E‐03 |
| ENSG00000167768 | KRT1 | protein_coding | 2.04 | 1.35E‐03 | 2.60E‐02 |
| ENSG00000163958 | ZDHHC19 | protein_coding | 2.04 | 4.41E‐06 | 1.20E‐03 |
| ENSG00000146122 | DAAM2 | protein_coding | 2.03 | 1.72E‐03 | 2.93E‐02 |
| ENSG00000104892 | KLC3 | protein_coding | 2.01 | 3.25E‐04 | 1.25E‐02 |
| ENSG00000124469 | CEACAM8 | protein_coding | 2.01 | 4.69E‐03 | 5.12E‐02 |
| ENSG00000244575 | IGKV1‐27 | IG_V_gene | 2.01 | 9.16E‐05 | 6.04E‐03 |
| ENSG00000167034 | NKX3.1 | protein_coding | −2.04 | 0.00204 | 3.25E‐02 |
P adj ≤0.05 and fold change ≥±1.5 were considered significant, and the list with fold change ≥±2 is shown. A positive fold change means that the affected gene is overexpressed in CASES. A negative fold change means that the affected gene is overexpressed in CONTROLS. P adj indicates adjusted P value corrected for multiple testing using the Benjamini and Hochberg method in DESeq2.
IPA for Biological Interpretation of Differentially Expressed Genes
IPA was used to identify canonical pathways and toxicological processes of biological importance among the DEGs. After sorting the canonical signaling pathways from large to small by −log (P value), the top 25 canonical signaling pathways with P<0.05 (−log=1.3) were identified and are listed in Table 3. The ratios of DEGs to total genes in these signaling pathways are also listed. The top enriched canonical signaling pathways included: Hepatic fibrosis/Hepatic stellate cell activation, Iron homeostasis signaling pathway, LXR/RXR activation pathway, Osteoarthritis pathway and Heme biosynthesis II pathway. Two cytokine receptor genes (IL1R1 and IL1R2) and 2 matrix metalloproteinases (MMP8 and MMP9) overlapped in several canonical pathways as shown in Table 3. Using IPA‐Tox, the toxicological effects were classified as cardiotoxicity, hepatotoxicity, and nephrotoxicity, along with various subcategories. The analysis revealed that the probability of myocardial infarction, cardiac enlargement, cardiac damage, cardiac arrhythmia, cardiac inflammation, cardiac degeneration, and heart failure was above the significance threshold, which is consistent with the clinical manifestation of cardiomyopathy (Table 4).
Table 3.
Canonical Pathways Generated by IPA Analysis
| Ingenuity canonical pathways | ‐log (P value) | Ratio | Gene Symbol |
|---|---|---|---|
| Hepatic fibrosis/Hepatic stellate cell activation | 6.05 | 0.1 | COL13A1, COL4A3, COL5A3, COL9A2, IL18RAP, IL1R1* , IL1R2* , LY96, MMP9* , MYL4, MYL9 |
| Iron homeostasis signaling pathway | 4.85 | 0.1 | ALAS2, CD163, FECH, GDF15, HBB, HBD, HBG2, HBQ1, HP, TFR2 |
| LXR/RXR activation | 4.18 | 0.1 | IL18RAP, IL1R1* , IL1R2* , LY96, MMP9* , ORM1, S100A8 |
| Osteoarthritis pathway | 3.41 | 0.1 | ALPL, FZD5, IL18RAP, IL1R1* , IL1R2* , ITLN1, MMP9* , S100A8, S100A9 |
| Heme biosynthesis II | 3.36 | 0.3 | ALAS2, FECH, HMBS |
| Airway pathology in chronic obstructive pulmonary disease | 3.26 | 0.1 | ELANE, LCN2, MMP8* , MMP9* , ORM1 |
| Granulocyte adhesion and diapedesis | 2.68 | 0.1 | IL18RAP, IL1R1* , IL1R2* , MMP8* , MMP9* , SELP |
| IL‐10 signaling | 2.64 | 0.1 | BLVRB, IL18RAP, IL1R1 *, IL1R2 *, SOCS3 |
| Agranulocyte adhesion and diapedesis | 2.54 | 0.1 | IL1R1 *, MMP8 *, MMP9 *, MYL4, MYL9, SELP |
| Tetrapyrrole biosynthesis II | 2.51 | 0.4 | ALAS2, HMBS |
| Atherosclerosis signaling | 2.39 | 0.1 | COL5A3, MMP9 *, ORM1, S100A8, SELP |
| GP6 signaling pathway | 2.13 | 0.1 | COL13A1, COL4A3, COL5A3, COL9A2, ITGA2B |
| Role of IL‐17A in psoriasis | 2.08 | 0.3 | S100A8, S100A9 |
| Role of osteoblasts, osteoclasts, and chondrocytes in rheumatoid arthritis | 1.92 | 0.1 | ALPL, FZD5, IL18RAP, IL1R1 *, IL1R2 *, MMP8 *, NAIP |
| Role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis | 1.79 | 0.0 | FCGR1A, FZD5, IGHG1, IL18RAP, IL1R1 *, IL1R2 *, IRAK3, SOCS3, TLR5 |
| Glucocorticoid receptor signaling | 1.6 | 0.0 | BCL2L1, CD163, FCGR1A, FKBP5, IL1R2 *, KRT1, KRT73, POLR2I, SLPI |
| Phagosome formation | 1.59 | 0.1 | FCGR1A, FCGR1B, IGHG1, MARCO, TLR5 |
| Creatine‐phosphate biosynthesis | 1.45 | 0.5 | CKB |
| Urea cycle | 1.45 | 0.5 | ARG1 |
| GDP‐L‐fucose biosynthesis I (from GDP‐D‐mannose) | 1.45 | 0.5 | TSTA3 |
| L‐serine degradation | 1.45 | 0.5 | SDSL |
| Pyrimidine deoxyribonucleotides de novo biosynthesis I | 1.44 | 0.1 | NME2, RRM2 |
| Vitamin‐C transport | 1.44 | 0.1 | SLC2A1, TXN |
| Hepatic cholestasis | 1.42 | 0.0 | IL18RAP, IL1R1 *, IL1R2 *, IRAK3, LY96 |
| Inhibition of matrix metalloproteases | 1.39 | 0.1 | MMP8 *, MMP9 * |
IPA indicates Ingenuity Pathway Analysis. The P value for each pathway is expressed as ‐log (P value). The ratio represents the number of differentially expressed genes from our data set in a given pathway that meets the cutoff criteria, divided by the total number of genes that comprise that canonical pathway.
*Indicates overlapping genes.
Table 4.
Toxicity List Generated by the IPA Analysis
| Categories | Diseases or functions | P value | No.of genes | Gene symbols |
|---|---|---|---|---|
| Myocardial infarction* | Infarction of heart | 1.54E‐07 | 16 | BCL2L1, CA1, CA4, CD163, CREG1, FCGR1A, FCGR1B, GDF15, IL1R1, ITGA2B, KCNE1, LGALS2, MMP9, SELP, TGM2, TUBB2A |
| Myocardial infarction | 6.11E‐07 | 15 | CA1, CA4, CD163, CREG1, FCGR1A, FCGR1B, GDF15, IL1R1, ITGA2B, KCNE1, LGALS2, MMP9, SELP, TGM2, TUBB2A | |
| Acute myocardial infarction | 3.22E‐04 | 7 | FCGR1A, FCGR1B, IL1R1, ITGA2B, KCNE1, SELP, TUBB2A | |
| ST‐segment–elevation myocardial infarction | 2.43E‐03 | 4 | IL1R1, ITGA2B, KCNE1, TUBB2A | |
| Reperfusion injury of myocardium | 1.79E‐02 | 1 | SERPING1 | |
| Cardiac dilation, cardiac enlargement* | Dilated cardiomyopathy | 1.59E‐03 | 11 | BCL2L1, GLRX5, GPER1, IL1R1, KAZN, KCNE1, MMP8, MMP9, SLC4A1, SLC6A8, STAB1 |
| Enlargement of heart | 3.56E‐03 | 18 | BCL2L1, BIRC5, BMX, COL9A2, FKBP1B, GDF15, GLRX5, GPER1, IL1R1, KAZN, KCNE1, MMP8, MMP9, MYL9, SLC4A1, SLC6A8, STAB1, TXN | |
| Enlargement of heart ventricle | 9.25E‐03 | 7 | BIRC5, BMX, FKBP1B, MMP9, MYL9, SLC4A1, TXN | |
| Cardiac damage* | Rupture of heart | 1.87E‐03 | 2 | GDF15, MMP9 |
| Damage of cardiac muscle | 2.68E‐03 | 3 | GDF15, SERPING1, TXN | |
| Damage of myocardium | 6.33E‐03 | 2 | GDF15, SERPING1 | |
| Damage of heart | 7.78E‐03 | 4 | GDF15, MMP9, SERPING1, TXN | |
| Liver inflammation/hepatitis | Inflammation of liver | 1.45E‐03 | 14 | ABCG2, ACKR1, ALPL, BCL2L1, FCGR1A, FCGR1B, HP, IL1R1, KAZN, LGALS3, MMP9, SOCS3, STAB1, TK1 |
| Acute hepatitis | 1.47E‐02 | 3 | ALPL, IL1R1, MMP9 | |
| Acute alcoholic hepatitis | 1.87E‐02 | 2 | ALPL, IL1R1 | |
| Pulmonary hypertension | Pulmonary hypertension | 3.49E‐03 | 6 | ARG1, BCL2L1, BIRC5, CA1, CA4, SLC2A1 |
| Renal atrophy | Atrophy of kidney | 5.17E‐03 | 3 | COL4A3, MXI1, SLC4A1 |
| Glomerular injury, renal fibrosis | Fibrosis of renal glomerulus | 8.34E‐03 | 2 | COL4A3, STAB1 |
| Hepatocellular carcinoma, liver hyperplasia/hyperproliferation | Hepatocellular carcinoma | 4.37E‐03 | 25 | ARG1, ASPH, BCL2L1, CD163, FLT3, GPER1, HBB, HLA‐G, HP, KAZN, LGALS3, MARCO, MMP9, MT1E, RRM2, SERPING1, SLC4A1, SOCS3, STAB1, TNNT1, TP53I3, TPX2, TUBB2A, TXN, VSIG4 |
| Growth of hepatocellular carcinoma | 8.34E‐03 | 2 | RRM2, TPX2 | |
| Cardiac arrhythmia* | Cardiac fibrillation | 1.20E‐02 | 6 | FKBP1B, KCNE1, MMP9, MYL4, TGM2, TUBB2A |
| Arrhythmia of heart ventricle | 1.58E‐02 | 4 | ASPH, FKBP1B, KCNE1, TGM2 | |
| Familial atrial fibrillation type 18 | 1.79E‐02 | 1 | MYL4 | |
| Arrhythmia | 1.84E‐02 | 8 | ASPH, FKBP1B, KCNE1, MMP9, MYL4, TGM2, TUBB2A, TXN | |
| Liver hyperplasia/hyperproliferation | Growth of liver tumor | 1.34E‐02 | 3 | RRM2, SOCS3, TPX2 |
| Cardiac inflammation* | Acute myocarditis | 1.79E‐02 | 1 | IL1R1 |
| Experimental autoimmune myocarditis | 1.87E‐02 | 2 | IL1R1, MMP9 | |
| Cardiac degeneration* | Myocytolysis of heart | 1.79E‐02 | 1 | MMP9 |
| Cardiac arrhythmia, congenital heart anomaly* | Jervell and Lange‐Nielsen syndrome type 2 | 1.79E‐02 | 1 | KCNE1 |
| Susceptibility to acquired long QT syndrome type 5 | 1.79E‐02 | 1 | KCNE1 | |
| Heart failure* | Progressive heart failure | 1.79E‐02 | 1 | BIRC5 |
| Cardiorenal syndrome | 2.19E‐02 | 2 | CA1, CA4 | |
| Glomerular injury, renal inflammation, renal nephritis | Severe glomerulonephritis | 1.79E‐02 | 1 | COL4A3 |
| Kidney failure | Septic acute kidney injury | 1.79E‐02 | 1 | LCN2 |
| Renal dilation | Vasodilation of renal artery | 1.79E‐02 | 1 | MMP9 |
| Glomerular injury | Glomerulosclerosis | 1.60E‐02 | 6 | AQP1, COL4A3, IRAK3, STAB1, TK1, TNS1 |
| Renal necrosis/cell death | Apoptosis of tubular cells | 1.61E‐02 | 3 | BIRC5, CA4, LCN2 |
| Nephrosis | Steroid dependent nephrotic syndrome | 1.87E‐02 | 2 | FCGR1A, FCGR1B |
IPA indicates Ingenuity Pathway Analysis.
*Indicates cardiac‐related categories.
Gene Set Enrichment Analysis
The unranked expressed gene list obtained from DESeq2 analysis was uploaded to GSEA. The heat map with the top‐50 upregulated and top‐50 downregulated DEGs in all cases compared with all controls is displayed in Figure 1 and listed in Table S6. GSEA results using the “hallmark” collection showed that “adipogenesis” and “oxidative phosphorylation” gene sets were enriched (false discovery rate <0.1, normalized enrichment score >1.5). GSEA results using the “cardiomyopathy” gene set showed that “KAAB failed heart atrium dn” and “KAAB failed heart ventricle dn” were also enriched (false discovery rate <0.1, normalized enrichment score >1.5) (Tables S7 through S10). Leading edge analysis identified lactate dehydrogenase A (LDHA) to be the top‐ranking gene that contributed most to the enrichment signal in the “oxidative phosphorylation,” “KAAB failed heart atrium dn” and “KAAB failed heart ventricle dn” gene sets, and CD36 in the “adipogenesis” gene set (Figure 2).
Figure 1. Gene Set Enrichment Analysis generated heat map of the top‐100 differentially expressed genes in cases vs controls (50 upregulated genes and 50 downregulated genes).
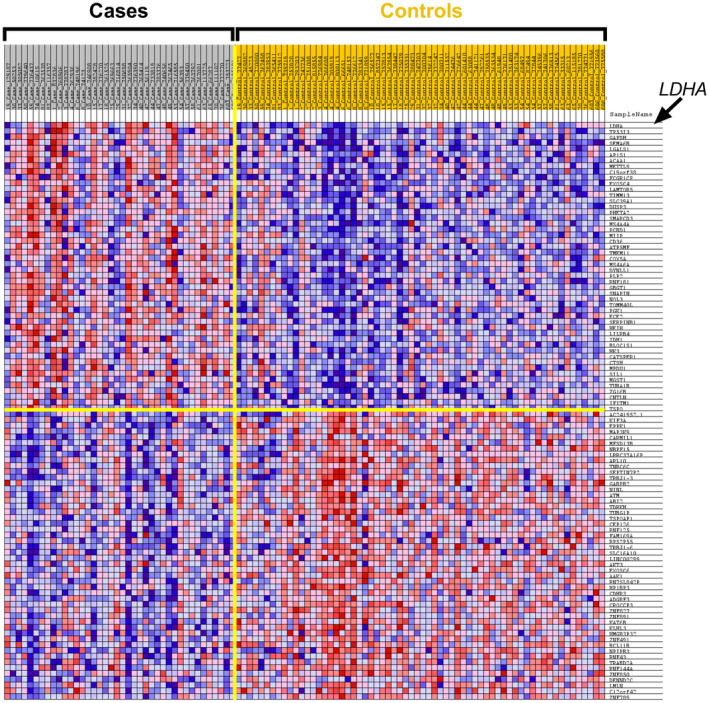
Rows: genes; Columns: samples; Colors range from dark red to dark blue representing respectively the highest and lowest expression of a gene. LDHA indicates lactate dehydrogenase A.
Figure 2. Enrichment plots of top 4 gene sets generated by Gene Set Enrichment Analysis (normalized enrichment score >1.5; P <0.01 and false discovery rate q value <0.25).
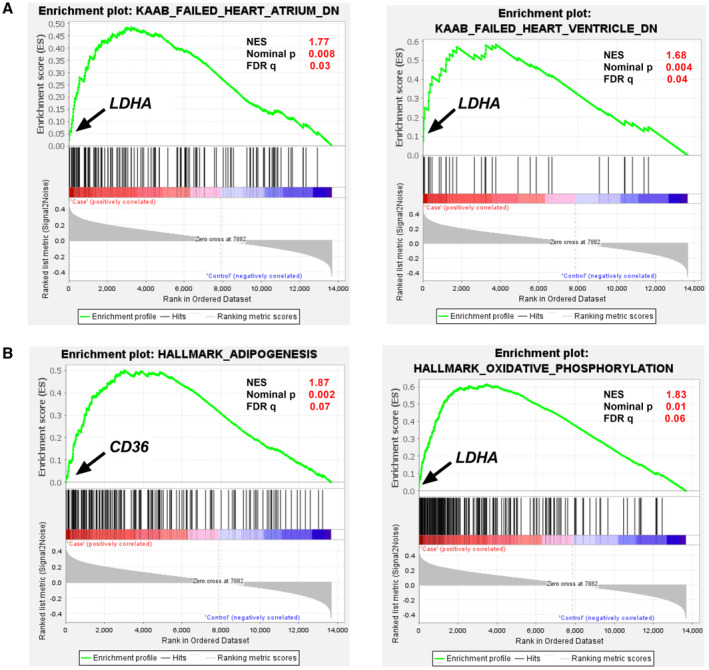
Green line shows the running enrichment scores for the gene set as the analysis walks along the ranked list, and the bottom portion shows the ranked genes as vertical black bars. Y‐axis: ranking metric, X‐axis: individual ranks for all genes. Upregulated (red); down‐regulated (blue). Normalized Enrichment score, Nominal P value and false discovery rate q‐values are shown for each gene set. Top hit genes are shown for each gene set. A, Enrichment plot of KAAB_FAILED_HEART_ATRIUM_DN and KAAB_FAILED_HEART_VENTRICLE_DN for Cardiomyopathy gene sets from MSigDB; B, Enrichment plot of HALLMARK_ADIPOGENSIS and HALLMARK_OXIDATIVE_PHOSPHORYLATION for Hallmark gene sets from MSigDB. CD36 indicates cluster of differentiation 36; and LDHA, lactate dehydrogenase A.
Genes for Functional Analyses
Using our predetermined prioritization strategies (Table S3) and supported by the leading edge GSEA findings, LDHA was shortlisted for functional analysis. We examined whether the loss of function of LDHA altered the viability of cardiomyocytes derived from an isogenic hiPSC line (ISO) upon exposure to doxorubicin. LDHA knockout (ISO‐KO) line was generated via a CRISPR/Cas9–mediated approach. 7‐bp (Exon 2) deletion in LDHA was confirmed by Sanger sequencing (Figure S2), and decreased gene expression in the cell line was confirmed by reverse transcription‐polymerase chain react (Figure 3A; Table S5). The cell viability assay showed that the ISO‐LDHA hiPSC‐CMs (LD50=0.67μM) was 6.7‐fold more sensitive to doxorubicin (72 hours) as compared with ISO control (LD50=4.49 μM, P<0.0001) (Figure 3B).
Figure 3. Assessment of in vitro doxorubicin‐induced cardiotoxicity in patient–specific human‐induced pluripotent stem cell–derived cardiomyocytes.
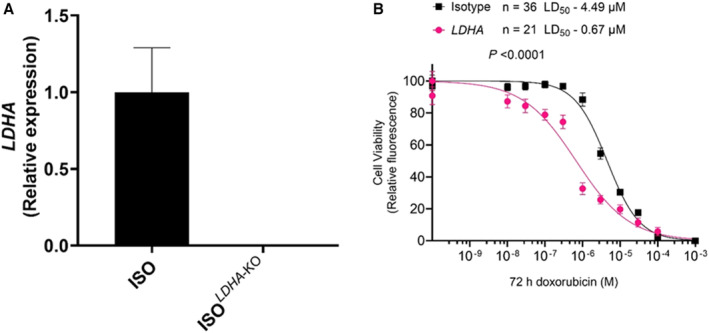
A, Validation of KO by reverse transcription‐polymerase chain reaction for LDHA; B, Effect of Doxorubicin (72 hours) on human‐induced pluripotent stem cell–derived cardiomyocytes viability in ISO (Control) (n=36) and LDHA–KO (n=21). ISO indicates isotype; LDHA, lactate dehydrogenase A; and KO, knock out.
Given the previous observations that LDHA plays a role in the adaptive response to hemodynamic stress 24 and is significantly upregulated by pressure overload in the heart, 24 , 25 we examined the LDHA expression levels in patients with cardiomyopathy who did or did not have CVRFs. We found that the case–control difference in the LDHA expression was larger among those with cardiomyopathy/congestive heart failure and CVRFs (3177 versus 2440, P=0.0004) compared with those without CVRFs (2697 versus 2320, P=0.005).
Discussion
We found distinct differential gene expression in the peripheral blood mRNA from childhood cancer survivors with and without anthracycline‐induced cardiomyopathy. Thirty‐five genes were upregulated, and 1 gene was downregulated in cases versus controls with an absolute fold‐change of >2. IPA of genes with absolute fold‐change >1.5 implicated “hepatic fibrosis/hepatic stellate cell activation” and the “iron homeostasis signaling” canonical pathways. Identification of the hepatic fibrosis/hepatic stellate cell activation pathway may be supported by the fact that doxorubicin is metabolized in the liver via microsomal enzymes; alteration of doxorubicin metabolism may allow accumulation of toxic anthracycline metabolites. 26 The “iron homeostasis signaling pathway” is supported by prior evidence showing that iron overload exacerbates the cardiotoxic effects of anthracyclines. 27 , 28 In addition, “myocardial infarction,” “cardiac damage,” “cardiac dilation,” “cardiac inflammation,” “cardiac enlargement,” and “heart failure” were identified as significantly activated toxicological pathways. The GSEA analysis showed significant enrichment of LDHA and CD36 genes in cardiomyopathy cases. The heart predominantly uses 2 fuel sources concomitantly: fatty acids and glucose, while use of a single energy source elicits heart disease. 29 LDHA and CD36 are key genes in the glycolytic and fatty acid uptake pathways in the heart. 30
Lactate dehydrogenase (LDH) is a key enzyme in the regulation of glycolysis. It has 2 isoforms, LDHA and LDHB. LDHA has a higher affinity for pyruvate, preferentially converting pyruvate to lactate. Administration of anthracyclines is associated with reduction in LDHA expression. 31 In our hiPSC‐CM model, knockout of LDHA showed that loss of this gene results in increased sensitivity to doxorubicin and fits the observation that anthracyclines have an inhibitory impact on LDHA expression. 31 This downregulation of LDHA likely does not persist after anthracycline exposure. Anthracycline exposure triggers cardiac injury metabolic reprogramming and myocardial remodeling, progressing to overt cardiotoxicity over time. 4 Hypoxia is a prominent feature of cardiac hypertrophy, and metabolic remodeling precedes and plays a role in cardiac hypertrophic growth. Hypoxia also induces hypoxia‐inducible factor 1‐alpha, which triggers LDHA in the hypertrophic heart and results in a second metabolic switch from oxidative phosphorylation to glycolysis in the myocardium, resulting in overt anthracycline‐related cardiomyopathy in cancer survivors. 32 , 33 , 34 This is further exacerbated because of CVRF‐induced pressure overload and hypoxia, 35 , 36 , 37 resulting in a metabolic transition from oxidative phosphorylation to glycolysis and induction of LDHA. Hypertrophied hearts show increased production and efflux of lactate from the myocardium, 38 , 39 , 40 supporting the well observed clinical finding that the abnormal extracellular appearance of LDH signifies tissue damage. Indeed, LDH was first proposed as a diagnostic aid for myocardial infarction in the year 1957 41 ; elevated serum LDH is associated with cardiovascular disease risk and heart failure. 25 , 42 These findings suggest the role of the upregulation of LDHA in anthracycline‐induced cardiomyopathy.
We also observed a significant upregulation of CD36 gene in cases with anthracycline‐induced cardiomyopathy. CD36/FAT (cluster of differentiation 36/fatty acid translocase) is a transmembrane protein that regulates cellular lipid metabolism. It is estimated that 70% of heart muscle fatty acid uptake is regulated by CD36. In an enlarged heart, the main fuel source switches from fatty acids to glucose. This fuel shift is also associated with contractile dysfunction. Fatty acid or lipid metabolism dysfunction triggered by CD36 dysregulation plays a key role in the development of obesity‐induced cardiac dysfunction and diabetic cardiomyopathy. 43 Myocardial CD36 expression is upregulated in aging mice, and there is a concomitant increase in intramyocardial lipid content associated with energy compromise. 44 Further, cardiomyocyte‐specific deletion of Cd36 in mice accelerates the progression of pressure overload‐induced cardiac hypertrophy to cardiac dysfunction. 45 These findings suggest the role of the upregulation of CD36 in anthracycline‐induced cardiomyopathy observed in our study.
Interleukin receptors (IL1R1 and IL1R2) and matrix metalloproteinase‐8 and ‐9 genes (MMP8 and MMP9) appeared in 9 out of 25 canonical pathways. Inflammatory process is a key component in the mechanism of different cardiac pathologies. The interleukin‐1 (IL1) family of ligands and receptors is the main cytokine family associated with acute and chronic inflammation. IL1 mediates inflammation by binding its receptor, termed type 1 (IL1R1), whereas IL1R2 preferentially binds IL1β and, in doing so, sequesters IL1β from binding to IL1R1. 46 The cytokine hypothesis of heart failure suggests that a precipitating event triggers activation of pro‐inflammatory cytokines, which leads to detrimental effects on left ventricular function and accelerates the progression of heart failure. 47 , 48 A study in mice showed that IL1 mediates doxorubicin cardiotoxicity, and a sequential study showed that blocking IL1 with Anakinra in such mice diminished doxorubicin‐induced microstructural damages of cardiac tissue and rescued doxorubicin‐caused reduction of cardiac functions exemplified by left ventricle ejection fraction and fractional shortening. 49 IL1 was also found to play a role in radiation‐induced cardiomyopathy in IL1R1 knock‐out mice exposed to thoracic X‐ray therapy. 50 Patients with coronary artery disease have higher levels of IL1RI compared with normal people. 51 Genetic loss of IL1R1 in mice decreases dilation of the infarcted heart, reducing collagen deposition and attenuating matrix metalloproteinases expression. 52 , 53 MMP9 regulates pathological remodeling processes that involve inflammation and fibrosis in cardiovascular disease. MMP9 directly degrades extracellular matrix proteins and activates cytokines and chemokines to regulate tissue remodeling. 54 MMP9 can cleave collagen and is found to increase during several cardiovascular diseases. 54 MMP9 deletion or inhibition is beneficial in several animal models of cardiovascular disease. 55
The study approach of using differential single gene expression analysis with GSEA and leading‐edge analyses has several advantages over single‐gene methods. We identified pathways, processes, and gene sets to elucidate key players in relevant biological processes. Nonetheless, this study needs to be considered in the context of its limitations. Ideally, gene expression should be measured in the affected tissue (ie, cardiac tissue). Obtaining heart biopsies from cancer survivors is logistically challenging and not without risk. On the other hand, peripheral blood is easily obtained, and gene expression levels in peripheral whole blood samples correlate with the cardiac transcriptome. 12 , 13 , 14 , 56 Furthermore, the molecular fingerprints of whole blood and peripheral blood mononuclear cells have a good overlap and concordance in their gene expression with common pathways and mechanisms represented by these genes, providing the rationale for using whole blood rather than cellular subtypes, given the logistic issues with obtaining the latter. 57 Though the hiPSC‐CM model offers considerable advantages over animal models, the immaturity of hiPSC‐CMs compared with adult human cardiomyocytes and having a heterogeneous cardiac cell population in the culture must be taken into account before extrapolating results to adult human cardiac physiology.
Limitations notwithstanding, to our knowledge, this is the first study to show DEGs and pathways in anthracycline‐exposed childhood cancer survivors with cardiomyopathy when compared with those without. These findings shed light on the molecular mechanism underlying anthracycline‐related cardiomyopathy and should be explored further to identify potential therapeutic targets for the treatment of anthracycline‐related cardiomyopathy.
Conclusions
The present study demonstrates that dysregulated expression of LDHA, CD36, IL1R1, IL1R2, MMP8, and MMP9 in blood is associated with anthracycline‐related cardiomyopathy in childhood cancer survivors. These findings provide evidence of a possible explanation for the role of metabolic and structural remodeling in the heart following anthracycline exposure (Figure 4).
Figure 4. Schematic figure and timeline proposing the role of altered gene expression in anthracycline‐induced cardiomyopathy.
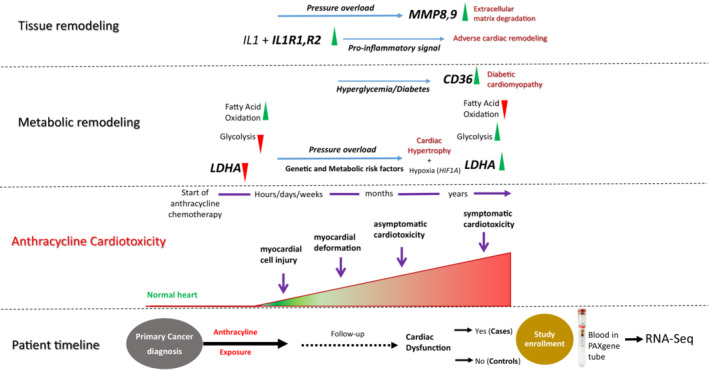
Progression of anthracycline‐induced cardiotoxicity involves a spectrum of remodeling at various levels, including structural, electrophysiological, metabolic, and functional events in the heart. Anthracycline exposure significantly reduces the expression of LDHA. Inhibition of LDHA obstructs aerobic glycolysis and activates fatty acid oxidation, which exacerbates cardiomyopathy under pressure overload. Hypoxia is a key regulator of cardiac hypertrophy and hypoxia‐inducible factor 1‐alpha activates transcription of LDHA, resulting in a second metabolic shift to aerobic glycolysis. Increase in CD36 facilitates the uptake of fatty acids and accumulation of lipids in cardiac muscle. Cardiac tissue remodeling involves activation of proinflammatory cytokines IL1R1 and IL1R2 that accelerate the progression of heart failure. MMP8 and MMP9 directly degrade extracellular matrix proteins, resulting in cardiomyocyte death and fibrosis. CD36 indicates cluster of differentiation 36; IL1R (1 and 2) interleukin 1 receptor type 1 and 2; LDHA, lactate dehydrogenase A; and MMP (8 and 9) matrix metalloproteinase 8 and 9.
Sources of Funding
National Cancer Institute (NCI) (R35CA220502; Principal Investigator [PI]: S. Bhatia), Leukemia and Lymphoma Society (6563‐19; PI: S. Bhatia), The V Foundation for Cancer Research (DT2019‐010; PI: S. Bhatia), NCI (R01CA220002 and R01CA261898; PI: P.W. Burridge), The Children's Oncology Group study reported here is supported by the National Clinical Trials Network Operations Center Grant (U10CA180886; PI: D.S. Hawkins); the National Clinical Trials Network Statistics & Data Center Grant (U10CA180899; PI: Alonzo); the Children's Oncology Group Chair's Grant (U10CA098543; PI: Adamson); The Children's Oncology Group Statistics & Data Center Grant (U10CA098413; PI: Anderson); the NCI Community Oncology Research Program Grant (UG1CA189955; PI: Pollock); and the Community Clinical Oncology Program Grant (U10CA095861; PI: Pollock), and the St Baldrick's Foundation through an unrestricted grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
Supporting information
Data S1
Tables S1–S10
Figures S1–S2
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.029954
P. Singh and D. A. Shah contributed equally to this article.
For Sources of Funding and Disclosures, see page 13.
This manuscript was sent to Tochukwu M. Okwuosa, DO, Associate Editor, for review by expert referees, editorial decision, and final disposition.
References
- 1. Lipshultz SE, Miller TL, Lipsitz SR, Neuberg DS, Dahlberg SE, Colan SD, Silverman LB, Henkel JM, Franco VI, Cushman LL, et al. Continuous versus bolus infusion of doxorubicin in children with ALL: long‐term cardiac outcomes. Pediatrics. 2012;130:1003–1011. doi: 10.1542/peds.2012-0727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6 [DOI] [PubMed] [Google Scholar]
- 3. Bhatia S. Genetics of anthracycline cardiomyopathy in cancer survivors: JACC: cardiooncology state‐of‐the‐art review. JACC CardioOncol. 2020;2:539–552. doi: 10.1016/j.jaccao.2020.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cardinale D, Iacopo F, Cipolla CM. Cardiotoxicity of anthracyclines. Front Cardiovasc Med. 2020;7:26. doi: 10.3389/fcvm.2020.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abdullah CS, Alam S, Aishwarya R, Miriyala S, Bhuiyan MAN, Panchatcharam M, Pattillo CB, Orr AW, Sadoshima J, Hill JA, et al. Doxorubicin‐induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci Rep. 2019;9:2002. doi: 10.1038/s41598-018-37862-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alagona P Jr, Ahmad TA. Cardiovascular disease risk assessment and prevention: current guidelines and limitations. Med Clin North Am. 2015;99:711–731. doi: 10.1016/j.mcna.2015.02.003 [DOI] [PubMed] [Google Scholar]
- 7. Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying causes and long‐term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–1084. doi: 10.1056/NEJM200004133421502 [DOI] [PubMed] [Google Scholar]
- 8. Bhatia S. Role of genetic susceptibility in development of treatment‐related adverse outcomes in cancer survivors. Cancer Epidemiol Biomark Prev. 2011;20:2048–2067. doi: 10.1158/1055-9965.EPI-11-0659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gramatges MM, Bhatia S. Evidence for genetic risk contributing to long‐term adverse treatment effects in childhood cancer survivors. Annu Rev Med. 2018;69:247–262. doi: 10.1146/annurev-med-041916-124328 [DOI] [PubMed] [Google Scholar]
- 10. Al‐Otaibi TK, Weitzman B, Tahir UA, Asnani A. Genetics of anthracycline‐associated cardiotoxicity. Front Cardiovasc Med. 2022;9:867873. doi: 10.3389/fcvm.2022.867873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim Y, Seidman JG, Seidman CE. Genetics of cancer therapy‐associated cardiotoxicity. J Mol Cell Cardiol. 2022;167:85–91. doi: 10.1016/j.yjmcc.2022.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wingrove JA, Daniels SE, Sehnert AJ, Tingley W, Elashoff MR, Rosenberg S, Buellesfeld L, Grube E, Newby LK, Ginsburg GS, et al. Correlation of peripheral‐blood gene expression with the extent of coronary artery stenosis. Circ Cardiovasc Genet. 2008;1:31–38. doi: 10.1161/CIRCGENETICS.108.782730 [DOI] [PubMed] [Google Scholar]
- 13. Sinnaeve PR, Donahue MP, Grass P, Seo D, Vonderscher J, Chibout SD, Kraus WE, Sketch M Jr, Nelson C, Ginsburg GS, et al. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS One. 2009;4:e7037. doi: 10.1371/journal.pone.0007037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Devaux Y. Transcriptome of blood cells as a reservoir of cardiovascular biomarkers. Biochim Biophys Acta, Mol Cell Res. 2017;1864:209–216. doi: 10.1016/j.bbamcr.2016.11.005 [DOI] [PubMed] [Google Scholar]
- 15. Feijen EAM, Leisenring WM, Stratton KL, Ness KK, van der Pal HJH, van Dalen EC, Armstrong GT, Aune GJ, Green DM, Hudson MM, et al. Derivation of anthracycline and anthraquinone equivalence ratios to doxorubicin for late‐onset cardiotoxicity. JAMA Oncol. 2019;5:864–871. doi: 10.1001/jamaoncol.2018.6634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krueger F, James F, Ewels P, Afyounian E, Weinstein M, Schuster‐Boeckler B, Hulselmans G, Sclamons . FelixKrueger/TrimGalore: v0.6.10. Zenodo. 2023. doi: 10.5281/zenodo.7598955. [DOI]
- 17. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmstrom A, Chang AC, Coronado MJ, Ebert AD, et al. Human induced pluripotent stem cell‐derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin‐induced cardiotoxicity. Nat Med. 2016;22:547–556. doi: 10.1038/nm.4087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Magdy T, Jiang Z, Jouni M, Fonoudi H, Lyra‐Leite D, Jung G, Romero‐Tejeda M, Kuo HH, Fetterman KA, Gharib M, et al. RARG variant predictive of doxorubicin‐induced cardiotoxicity identifies a cardioprotective therapy. Cell Stem Cell. 2021;28:2076–2089.e2077. doi: 10.1016/j.stem.2021.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuo HH, Gao X, DeKeyser JM, Fetterman KA, Pinheiro EA, Weddle CJ, Fonoudi H, Orman MV, Romero‐Tejeda M, Jouni M, et al. Negligible‐cost and weekend‐free chemically defined human iPSC culture. Stem Cell Reports. 2020;14:256–270. doi: 10.1016/j.stemcr.2019.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Magdy T, Jouni M, Kuo HH, Weddle CJ, Lyra‐Leite D, Fonoudi H, Romero‐Tejeda M, Gharib M, Javed H, Fajardo G, et al. Identification of drug transporter genomic variants and inhibitors that protect against doxorubicin‐induced cardiotoxicity. Circulation. 2022;145:279–294. doi: 10.1161/CIRCULATIONAHA.121.055801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dai C, Li Q, May HI, Li C, Zhang G, Sharma G, Sherry AD, Malloy CR, Khemtong C, Zhang Y, et al. Lactate dehydrogenase A governs cardiac hypertrophic growth in response to hemodynamic stress. Cell Rep. 2020;32:108087. doi: 10.1016/j.celrep.2020.108087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu W, Ma Y, Guo W, Lu J, Li X, Wu J, Qin P, Zhu C, Zhang Q. Serum level of lactate dehydrogenase is associated with cardiovascular disease risk as determined by the Framingham risk score and arterial stiffness in a health‐examined population in China. Int J Gen Med. 2022;15:11–17. doi: 10.2147/IJGM.S337517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. LiverTox: Clinical and Research Information on Drug‐Induced Liver Injury. Bethesda (MD): NCBI Bookshelf; 2012. [Google Scholar]
- 27. Gammella E, Maccarinelli F, Buratti P, Recalcati S, Cairo G. The role of iron in anthracycline cardiotoxicity. Front Pharmacol. 2014;5:25. doi: 10.3389/fphar.2014.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qin Y, Guo T, Wang Z, Zhao Y. The role of iron in doxorubicin‐induced cardiotoxicity: recent advances and implication for drug delivery. J Mater Chem B. 2021;9:4793–4803. doi: 10.1039/d1tb00551k [DOI] [PubMed] [Google Scholar]
- 29. Glatz JFC, Wang F, Nabben M, Luiken J. CD36 as a target for metabolic modulation therapy in cardiac disease. Expert Opin Ther Targets. 2021;25:393–400. doi: 10.1080/14728222.2021.1941865 [DOI] [PubMed] [Google Scholar]
- 30. Niken Puspa Kuspriyanti EFA, Mas RA, Syamsunarno A. Role of Warburg effect in cardiovascular diseases: a potential treatment option. The Open Cardiovascular Medicine Journal. 2021;15:6–17. doi: 10.2174/1874192402115010006 [DOI] [Google Scholar]
- 31. Korga A, Ostrowska M, Iwan M, Herbet M, Dudka J. Inhibition of glycolysis disrupts cellular antioxidant defense and sensitizes HepG2 cells to doxorubicin treatment. FEBS Open Bio. 2019;9:959–972. doi: 10.1002/2211-5463.12628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu D, Linders A, Yamak A, Correia C, Kijlstra JD, Garakani A, Xiao L, Milan DJ, van der Meer P, Serra M, et al. Metabolic maturation of human pluripotent stem cell‐derived cardiomyocytes by inhibition of HIF1alpha and LDHA. Circ Res. 2018;123:1066–1079. doi: 10.1161/CIRCRESAHA.118.313249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ordono J, Perez‐Amodio S, Ball K, Aguirre A, Engel E. The generation of a lactate‐rich environment stimulates cell cycle progression and modulates gene expression on neonatal and hiPSC‐derived cardiomyocytes. Biomater Adv. 2022;139:213035. doi: 10.1016/j.bioadv.2022.213035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yuan X, Braun T. Multimodal regulation of cardiac myocyte proliferation. Circ Res. 2017;121:293–309. doi: 10.1161/CIRCRESAHA.117.308428 [DOI] [PubMed] [Google Scholar]
- 35. Ainscow EK, Zhao C, Rutter GA. Acute overexpression of lactate dehydrogenase‐A perturbs beta‐cell mitochondrial metabolism and insulin secretion. Diabetes. 2000;49:1149–1155. doi: 10.2337/diabetes.49.7.1149 [DOI] [PubMed] [Google Scholar]
- 36. Cai X, Wang T, Ye C, Xu G, Xie L. Relationship between lactate dehydrogenase and albuminuria in Chinese hypertensive patients. J Clin Hypertens (Greenwich). 2021;23:128–136. doi: 10.1111/jch.14118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia‐inducible factor 1 and cAMP response elements. J Biol Chem. 1995;270:21021–21027. doi: 10.1074/jbc.270.36.21021 [DOI] [PubMed] [Google Scholar]
- 38. Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. 2019;8:e012673. doi: 10.1161/JAHA.119.012673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolwicz SC Jr, Tian R. Glucose metabolism and cardiac hypertrophy. Cardiovasc Res. 2011;90:194–201. doi: 10.1093/cvr/cvr071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. 2015;6:331–351. doi: 10.1002/cphy.c150016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Macdonald RP, Simpson JR, Nossal E. Serum lactic dehydrogenase; a diagnostic aid in myocardial infarction. J Am Med Assoc. 1957;165:35–40. doi: 10.1001/jama.1957.02980190037009 [DOI] [PubMed] [Google Scholar]
- 42. Wu Y, Lu C, Pan N, Zhang M, An Y, Xu M, Zhang L, Guo Y, Tan L. Serum lactate dehydrogenase activities as systems biomarkers for 48 types of human diseases. Sci Rep. 2021;11:12997. doi: 10.1038/s41598-021-92430-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glatz JFC, Luiken J. Dynamic role of the transmembrane glycoprotein CD36 (SR‐B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59:1084–1093. doi: 10.1194/jlr.R082933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koonen DP, Febbraio M, Bonnet S, Nagendran J, Young ME, Michelakis ED, Dyck JR. CD36 expression contributes to age‐induced cardiomyopathy in mice. Circulation. 2007;116:2139–2147. doi: 10.1161/CIRCULATIONAHA.107.712901 [DOI] [PubMed] [Google Scholar]
- 45. Sung MM, Byrne NJ, Kim TT, Levasseur J, Masson G, Boisvenue JJ, Febbraio M, Dyck JR. Cardiomyocyte‐specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H552–H560. doi: 10.1152/ajpheart.00626.2016 [DOI] [PubMed] [Google Scholar]
- 46. Szekely Y, Arbel Y. A review of interleukin‐1 in heart disease: where do we stand today? Cardiol Ther. 2018;7:25–44. doi: 10.1007/s40119-018-0104-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin‐1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ Res. 2020;126:1260–1280. doi: 10.1161/CIRCRESAHA.120.315937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL‐1 pathway in atherosclerosis. Circ Res. 2018;122:1722–1740. doi: 10.1161/CIRCRESAHA.118.311362 [DOI] [PubMed] [Google Scholar]
- 49. Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, Zhu S, Zhang R, Han W. Recombinant human interleukin‐1 receptor antagonist protects mice against acute doxorubicin‐induced cardiotoxicity. Eur J Pharmacol. 2010;643:247–253. doi: 10.1016/j.ejphar.2010.06.024 [DOI] [PubMed] [Google Scholar]
- 50. Mezzaroma E, Mikkelsen RB, Toldo S, Mauro AG, Sharma K, Marchetti C, Alam A, Van Tassell BW, Gewirtz DA, Abbate A. Role of interleukin‐1 in radiation‐induced cardiomyopathy. Mol Med. 2015;21:210–218. doi: 10.2119/molmed.2014.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Z, Zhang M, Wu J, Zhou P, Liu Y, Wu Y, Yang Y, Lu X. Serum CD121a (interleukin 1 receptor, type I): a potential novel inflammatory marker for coronary heart disease. PLoS One. 2015;10:e0131086. doi: 10.1371/journal.pone.0131086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG. Interleukin‐1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. 2008;173:57–67. doi: 10.2353/ajpath.2008.070974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bageghni SA, Hemmings KE, Yuldasheva NY, Maqbool A, Gamboa‐Esteves FO, Humphreys NE, Jackson MS, Denton CP, Francis S, Porter KE, et al. Fibroblast‐specific deletion of interleukin‐1 receptor‐1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight. 2019;5:e125074. doi: 10.1172/jci.insight.125074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase‐9: many shades of function in cardiovascular disease. Physiology (Bethesda). 2013;28:391–403. doi: 10.1152/physiol.00029.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. DeLeon‐Pennell KY, Meschiari CA, Jung M, Lindsey ML. Matrix metalloproteinases in myocardial infarction and heart failure. Prog Mol Biol Transl Sci. 2017;147:75–100. doi: 10.1016/bs.pmbts.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Basu M, Wang K, Ruppin E, Hannenhalli S. Predicting tissue‐specific gene expression from whole blood transcriptome. Sci Adv. 2021;7:7. doi: 10.1126/sciadv.abd6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bondar G, Cadeiras M, Wisniewski N, Maque J, Chittoor J, Chang E, Bakir M, Starling C, Shahzad K, Ping P, et al. Comparison of whole blood and peripheral blood mononuclear cell gene expression for evaluation of the perioperative inflammatory response in patients with advanced heart failure. PLoS One. 2014;9:e115097. doi: 10.1371/journal.pone.0115097 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S10
Figures S1–S2
Data Availability Statement
The data discussed in this publication have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus and are accessible through GEO Series accession number GSE218276 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218276).