

Abstract
Oral squamous cell carcinoma of the gingivo‐buccal region (OSCC‐GB) is the most common cancer among men in India, and is associated with poor prognosis and frequent recurrence. Cellular heterogeneity in OSCC‐GB was investigated by single‐cell RNA sequencing of tumors derived from the oral cavity of 12 OSCC‐GB patients, 3 of whom had concomitant presence of a precancerous lesion (oral submucous fibrosis [OSMF]). Unique malignant cell types, features, and phenotypic shifts in the stromal cell population were identified in oral tumors with associated submucous fibrosis. Expression levels of FOS, ATP1A, and DUSP1 provided robust discrimination between tumors with or without the concomitant presence of OSMF. Malignant cell populations shared between tumors with and without OSMF were enriched with the expression of partial epithelial–mesenchymal transition (pEMT) or fetal cell type signatures indicative of two dominant cellular programs in OSCC‐GB—pEMT and fetal cellular reprogramming. Malignant cells exhibiting fetal cellular and pEMT programs were enriched with the expression of immune‐related pathway genes known to be involved in antitumor immune response. In the tumor microenvironment, higher infiltration of immune cells than the stromal cells was observed. The T cell population was large in tumors and diverse subtypes of T cells with varying levels of infiltration were found. We also detected double‐negative PLCG2 + T cells and cells with intermediate M1–M2 macrophage polarization. Our findings shed light on unique aspects of cellular heterogeneity and cell states in OSCC‐GB.
Keywords: cellular diversity, fetal cell‐type signature, partial epithelial–mesenchymal transition, single‐cell RNA sequencing, tumor ecosystem
This is the first report of intratumor cellular heterogeneity at the single‐cell level in gingivo‐buccal oral cancer (OSCC‐GB). Some unique malignant cell types were found when a precancerous lesion, oral submucous fibrosis, was concomitantly present with frank cancer (OSCC‐GB). Malignant cell populations shared between tumors exhibited partial epithelial–mesenchymal transition or were enriched with fetal cell‐type signatures. Double negative PLCG2 + T cells and intermediate M1–M2 macrophage polarization were also detected.
Abbreviations
- CAF
cancer‐associated fibroblast
- CNV
copy number variation
- DN
double negative
- DP
distance profiling
- EMT
epithelial–mesenchymal transition
- HNSCC
head and neck squamous cell carcinoma
- myCAF
cancer‐associated myofibroblast
- NK
natural killer
- OSCC‐GB
oral squamous cell carcinoma of the gingivo buccal region
- OSMF
oral tumor with associated submucous fibrosis
- pEMT
partial epithelial–mesenchymal transition
- scRNA‐seq
single‐cell RNA sequencing
- TCR
T cell receptor
- TME
tumor microenvironment
- Treg
regulatory T cell
- UMAP
uniform manifold approximation and projection
1. INTRODUCTION
Oral squamous cell carcinoma is the 16th most common malignancy in the world and is associated with high mortality. Oral squamous cell carcinoma of the gingivo‐buccal region, commonly referred to as gingivo‐buccal oral cancer, ranks the highest among men, and sixth among women in India in terms of incidence and mortality. 1 It is associated with tobacco chewing, presentation at advanced stages (stages III and IV), and a high rate of loco‐regional failure, leading to poor prognosis. 2 Oral cavity tumors contain diverse cell types that show considerable interindividual variation and distinct genomic, epigenomic, and phenotypic features. 3 Such cellular heterogeneity is a key determinant of differences in tumor progression, treatment failure/recurrence, and overall survival of a cancer patient. 4 , 5 Single‐cell genomics has provided unparalleled opportunities to explore tumor heterogeneity at cellular resolution. 6 Recent single‐cell transcriptomic analysis of HNSCCs has revealed considerable intra‐ and intertumor cellular diversity. 7 , 8 , 9 However, these studies were undertaken on patients with tongue cancer or involved a comparison of oral cancer patients with and without HPV infection. The intratumor cellular heterogeneity of gingivo‐buccal oral cancer remains to be characterized. We have previously shown from bulk‐sequencing studies that there are notable differences in gene‐expression profiles between tongue cancer and OSCC‐GB patients. 10 We, therefore, undertook this study to characterize the landscape of cellular diversity in OSCC‐GB tumors using scRNA‐seq, and obtained novel information on the heterogeneity of cell types and cell states in this form of cancer.
2. MATERIALS AND METHODS
2.1. Sample collection
Sections of freshly resected, treatment‐naïve OSCC‐GB tumors were collected from 12 patients (Table S1), with written informed consent, who presented themselves for treatment at the R. Ahmed Dental College and Hospital, Kolkata. Resected tissues were collected in prechilled MACS tissue storage solution (Miltenyi Biotec) and transported on ice immediately after surgical procurement.
2.2. Preparation of single‐cell suspension
Tumor tissue was dissociated within 2 h after collection. Each tissue was minced into ≤1–2 mm pieces in 1 mL enzyme mix (enzymes H, R, and A from Miltenyi Biotech in RPMI‐1640 medium). The minced tissue suspension and the remaining enzyme mix were transferred to a gentleMACS C tube and then placed onto the gentleMACS Octo dissociator (Miltenyi Biotech). The gentleMACS program 37C_h_TDK_3 was run for dissociation after attaching the heating elements. The homogenate was centrifuged briefly and filtered through a 70 μm cell strainer (BD Biosciences). The cell suspension was then centrifuged at 300 g for 7 min, the supernatant was aspirated, and the pellet was resuspended in RPMI‐1640 medium (Gibco). Red blood cells were lysed by incubating the pellet in RBC lysis buffer (Miltenyi Biotec) for 10 min at 4°C, after which 10 mL chilled RPMI‐1640 medium was added, centrifuged at 300 g for 10 min at 4°C, and the cell pellet was resuspended in RPMI‐1640. The number of cells and fractions of live cells were counted using Trypan blue and Live/Dead Viability/Cytotoxicity Kit (Thermo Fisher Scientific) in a Countess II FL Automated Cell Counter (Thermo Fisher Scientific). Cell suspensions with 75% or more viable cells were then diluted to a final concentration of ~1000 cells/μL.
2.3. Single‐cell RNA sequencing library construction and sequencing
Single‐cell gel bead‐in emulsion and scRNA‐seq library were generated using the Chromium controller (10X Genomics), Chromium Single Cell 3′ Library and Gel Bead Kit and Chromium Single Cell 3′ Chip Kit (10X Genomics) according to the manufacturer's instructions. The quality of each final library was evaluated using a High Sensitivity chip in TapeStation (Agilent) and quantitated by real‐time PCR in QuantStudio‐7 (Thermo Fisher Scientific). The final libraries were then pooled for paired‐end sequencing (2 × 100 bp) in NovaSeq‐6000 (Illumina) at an average depth of 50,000 raw reads per cell and 200 million paired‐end reads per sample.
2.4. Single‐cell RNA sequencing data processing
The generated base call files were converted into FASTQ files using CellRanger version 4.0.0 (10X Genomics), then the reads were aligned to the GRCh38 reference transcriptome, and a filtered gene‐barcode matrix was generated. Seurat version 4.0.5 R toolkit 11 was used for quality control and downstream analysis of our scRNA‐seq data taking the gene‐barcode matrix as input. Quality control, clustering, visualization, and cell type annotation were carried out as described in Supplementary Methods.
2.5. Identification of malignant cells
We devised a new method—the DP method—to identify malignant cells. As oral squamous cell carcinoma is epithelial cancer, it is expected that the malignant cells will reside solely within the epithelial cell portion and their gene expression pattern will be distinct from nonepithelial cells. Therefore, we calculated the Euclidean distance between the average expression levels of all genes expressed in nonepithelial cells of the tumor (which was used as the reference vector of expression level) and that of each epithelial cell of the tumor. The frequency distribution of the distance values was plotted, which invariably had an antimode possibly indicating two cell subsets—epithelial malignant cells (with larger distances from the reference nonepithelial gene‐expression vector) and the remaining epithelial cells. The non‐epithelial cells of the tumor are normal nonmalignant cells, even if they comprise of heterogeneous cell types. It is, therefore, expected that the gene expression levels of malignant cells, all of which are epithelial, will be grossly different from normal cells, and the remaining epithelial cells will be closer to normal cells. The presence of an antimode in the frequency distribution of distances among cells indicated the fullfilment of these expectations, identifying malignant and nonmalignant epithelial cell subsets. To safeguard against possible misclassification, we discarded the malignant cells that belonged to the bottom 5% of the distribution of distances. Further details are provided in Figure S1A.
We have also used the previously published method, InferCNV, 12 to identify malignant cells and compare the results with those obtained with the DP method. Nonepithelial cells with minimum CNV scores were identified as a reference by the hierarchical clustering method. These reference cells were used to identify the CNV pattern of epithelial cells. A cell with a CNV score >0.03 and CNV correlation >0.4 was classified as malignant (Figure S1B,C).
2.6. Analysis of subclusters within malignant and nonmalignant cell clusters
After cell type identification, the malignant and nonmalignant cells (nonepithelial cells) were extracted using the “SubsetData” function of Seurat and clustered separately for further detailed analysis, using 15 PCs and resolution ranging from 0.5 to 1.0. Cell types were annotated as described earlier. We identified differentially expressed genes and undertook gene set enrichment analysis using methods as described in Supplementary Methods. Putative single‐cell trajectories were constructed using the R package Monocle (version 2.22.0). 13 We also undertook a comparative analysis (see Supplementary Methods) between our dataset and previously published HNSCC datasets on patients from Western countries.
3. RESULTS
3.1. Characteristics of scRNA‐seq data
We profiled 52,393 cells from primary tumors of 12 treatment‐naïve OSCC‐GB patients, of which 28,186 cells were retained after quality control (mean number of cells per patient = 2557; Table S2). We evaluated the quality of our scRNA‐seq data by comparing these data with bulk RNA‐seq data previously published by us, 10 which revealed that the total number of expressed genes in each of the two datasets was nearly equal (Figure S2). The mean expression levels of genes commonly expressed (n = 14,716) in scRNA‐seq and bulk RNA‐seq data were highly correlated (r = 0.83, p < 2.2 × 10−16), reflective of high reliability of our methods of single‐cell capture and analyses.
3.2. Cell type classification and broad characteristics of classified cells
Cell types were identified by annotation of 16 cell clusters as described earlier (Figures 1B,C and S3, Tables S3 and S4). Among the epithelial cells, we identified malignant cells separately by DP and InferCNV methods (Figure S1A–C, Table S5). Malignant cells identified exclusively by the DP method had lower levels of expression of DNA repair and apoptotic genes than those identified as malignant exclusively by the InferCNV method (Figure S1D,E). These malignant cells also clustered separately from the nonmalignant epithelial cells (Figure S4) Hence, for further analysis, we used the 7386 cells classified as malignant by the DP method, the results of which were more robust than the ones obtained by InferCNV.
FIGURE 1.
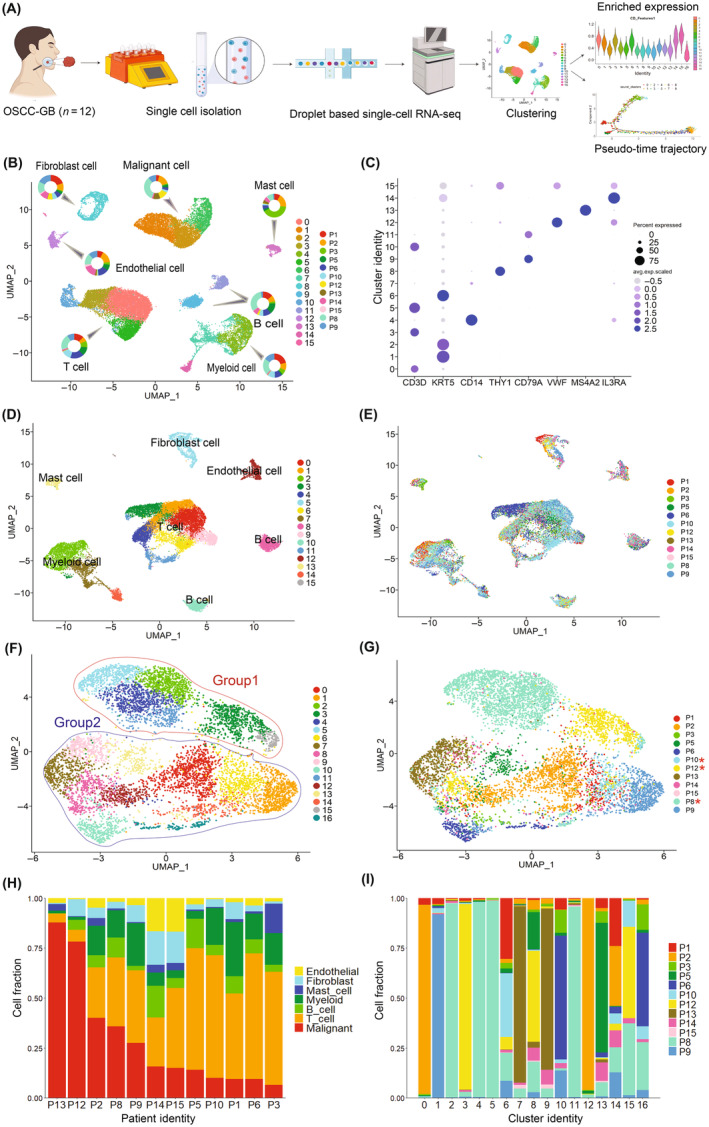
Cell type classification in oral squamous cell carcinoma of the gingivo‐buccal region (OSCC‐GB). (A) Workflow for generating single‐cell RNA sequencing profiles of OSCC‐GB. (B) Clustering of 28,186 cells from 12 OSCC‐GB patients identified 16 clusters. UMAP plot showing seven major cell types identified (T cells, B cells, myeloid, endothelial cells, epithelial malignant cells, fibroblasts, and mast cells). Pie chart showing the proportion of cells from each patient for each cell types. Each dot represents a cell. UMAP is colored by cell types and pie charts are colored by patient identity. (C) Dot plot showing expression of cell type representative marker genes. Cluster identity is on the y‐axis and gene names are on the x‐axis; color intensity represents the average gene expression level. (D–G) Clustering of (D, E) 20,800 nonmalignant cells and (F, G) 7386 malignant cells of OSCC‐GB patients. UMAP plot showing (D) nonmalignant cells and (F) malignant cells, colored by clusters. UMAP plot showing identified (E) nonmalignant cells and (G) malignant cells, colored by patients. *The patient has oral submucous fibrosis. (H) Proportions of cell types across OSCC‐GB patients are arranged by the decreasing proportion of malignant cells. (I) Proportions of malignant cells belonging to the various clusters derived from different patients.
Nonmalignant nonepithelial cell clusters were cell type‐specific and with low interindividual variation in gene expression pattern, although the proportions of specific cell types varied across patients (Figure 1D,E,H). Significantly (p < 0.00001) higher infiltration of immune cells (B, T, myeloid, and mast) compared to stromal cells (endothelial and fibroblast) was observed; this pattern was similar among patients with or without OSMF (Table S6). The major cell populations in the TME were T cells and myeloid cells (Figure 1H). In contrast, subclusters of malignant cells were composed of cells from tumors either of multiple patients (Clusters 6, 8, 14, 15, 16; henceforth denoted as C6, C8, etc.), or predominantly of specific patients (Clusters 0, 1, 2, 3, 4, 5, 7, 9, 10, 11, 12, 13) (Figure 1F,G,I).
3.3. Patients with prior OSMF showed distinctive gene expression and cell profiles
We noted that some clusters of malignant cells were specific to patients with OSMF. These clusters also seemed to be affine in the space of the first two UMAPs (Figure 1F). We called these clusters of cells Group 1 and the remaining as Group 2; in all, 17 clusters of malignant cells were identified. Clusters of cells in Group 1 were almost exclusively from three OSCC‐GB patients (P8, P10, and P12) who had a concomitant presence of OSMF (Figure 1G, Table S1). Cell clusters of Group 2 comprised cells from all OSCC‐GB patients, but predominantly from the nine patients in whom OSMF was absent. Mean expression levels of three genes (FOS, ATP1A1, and DUSP1) were significantly (p < 0.05) different between the two groups (Table 1), which also provided perfect discrimination between the two groups, as revealed by the probability of correct classification of patients (100% for each of the two groups) in stepwise discriminant analysis. These three genes are predominantly expressed (level of expression and proportion of cells expressing) in cells belonging to Group 2. However, cells of patients belonging to Group 1 also express these genes, although at a lower level and proportion than cells in Group 2 (Figure S5, Table S7). It is noteworthy that most clusters of malignant cells were patient‐specific. Only 5 of the 17 clusters—C6, C8, C14, C15, and C16—comprised cells from multiple patients.
TABLE 1.
Discriminant analysis of clusters of malignant cells, designated Group1 and Group 2.
| (A) Tests of equality of group means between Group 1 and Group 2 | |||||
|---|---|---|---|---|---|
| Wilks' L | F | df1 | df2 | p value | |
| SNHG25 | 0.904 | 1.065 | 1 | 10 | 0.3260 |
| FOS | 0.262 | 28.123 | 1 | 10 | <0.0001 |
| RPS23 | 0.893 | 1.193 | 1 | 10 | 0.3000 |
| ATP1A1 | 0.592 | 6.901 | 1 | 10 | 0.0250 |
| DUSP1 | 0.671 | 4.908 | 1 | 10 | 0.0490 |
| PLCG2 | 0.841 | 1.895 | 1 | 10 | 0.1990 |
| KRT1 | 0.801 | 2.485 | 1 | 10 | 0.1460 |
| (B) Classification results a between Group 1 and Group 2 | ||||
|---|---|---|---|---|
| Group | Predicted group membership | Total | ||
| 1 | 2 | |||
| Original group membership | ||||
| Count | 1 | 3.0 | 0.0 | 3.0 |
| 2 | 0.0 | 9.0 | 9.0 | |
| % | 1 | 100.0 | 0.0 | 100.0 |
| 2 | 0.0 | 100.0 | 100.0 | |
100.0% of original grouped cases are correctly classified.
3.4. Epithelial–mesenchymal transition in malignant cells
Cells in C6, C14, and C15 (multiple patient clusters) and C1 (patient‐specific cluster) were significantly enriched with the expression of genes in pathways known to be involved in EMT (Figure 2A, Tables [Link], [Link]), including ECM–receptor interaction, focal adhesion, adherence junction, and regulation of actin cytoskeletons pathways. Cells in these clusters (40.61% of all malignant cells) were also enriched with the expression of EMT, invasion, and metastasis gene signatures (Figure 2A, Table S13), suggesting that these cells undergo EMT leading to tumor invasion and metastasis.
FIGURE 2.
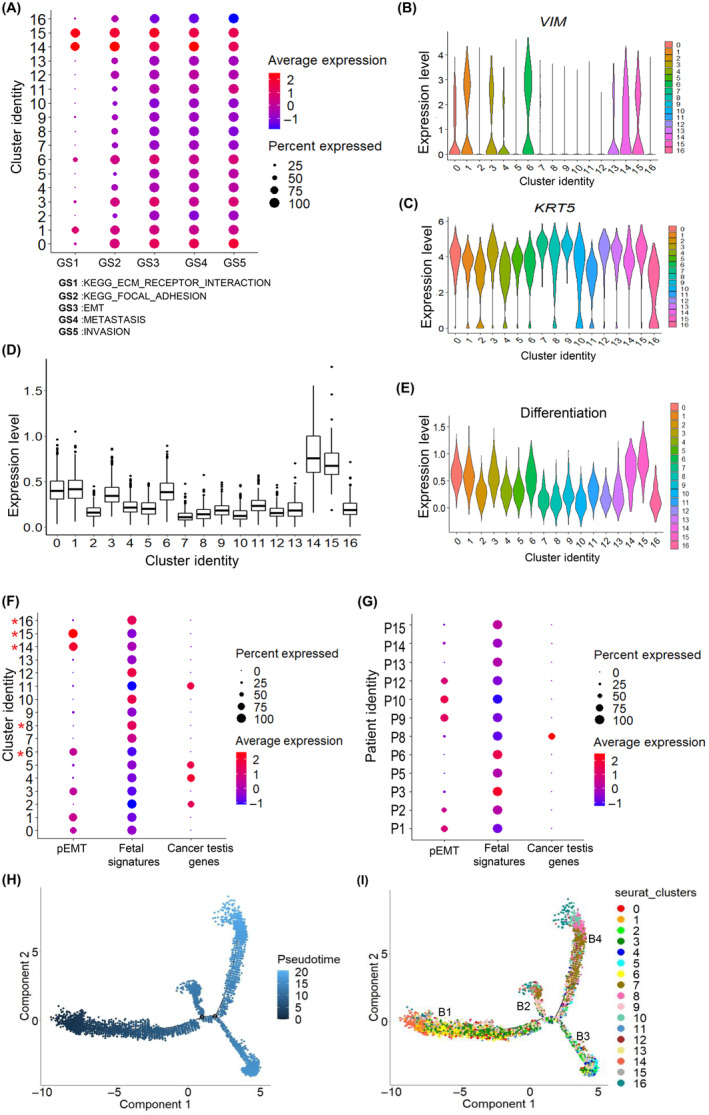
Malignant cell state heterogeneity in oral squamous cell carcinoma of the gingivo‐buccal region (OSCC‐GB): partial epithelial–mesenchymal transition (pEMT) and fetal and germ‐like cell state in OSCC‐GB tumors. (A) Dot plot of the average expression of Kyoto Encyclopedia of Genes and Genomes (KEGG) ECM receptor interaction, KEGG focal adhesion, epithelial–mesenchymal transition (EMT), metastasis, and invasion gene signatures across clusters (gene signatures on the x‐axis; cluster identity on the y‐axis); color represents the average gene expression level. (B, C) Violin plots showing expression of (B) VIM and (C) KRT5 across clusters. Cluster identity on the x‐axis; expression level on the y‐axis. (D) The box plot shows the average expression of pEMT‐associated genes across clusters. Cluster identity are on the x‐axis; average expression levels are on the y‐axis. (E) Violin plot showing expression of differentiation gene signature across clusters. Cluster identity on the x‐axis; expression level on the y‐axis. (F, G) Dot plots of the average expression of pEMT and fetal cell type signatures and cancer testis genes across (F) clusters and (G) patients (gene signatures on the x‐axis; cluster/patient identity on the y‐axis); color represents the average gene expression level. *Clusters containing cells from multiple patients. (H, I) Pseudotime analysis exploring the development trajectories of malignant cells. Each dot corresponds to a single cell, colored by (H) pseudotime and (I) cluster.
In addition to the cells in the four clusters with enriched expressions of EMT genes, cells in two other clusters, C0 and C3, were enriched with expression of SNAI2 (Figure S6), epithelial and mesenchymal markers and pEMT‐related genes (Figure 2B–D, Table S13) indicative of pEMT state. 7
3.5. Fetal and germ‐like epithelial cells are abundant in OSCC‐GB tumors
Multiple clusters of malignant cells, viz., C8 and C16 (contributed by cells from multiple patients; 6.9% of malignant cells) and C7, C9, C10, C12, and C13 (patient‐specific; 31.7% of cells) expressed genes that are fetal epithelial cell‐specific in nature, including fetal squamous epithelial cells, fetal lung squamous epithelial cells and fetal stomach squamous epithelial cells (Figure 2F, Table S14). The expressions of cancer/testis genes are usually restricted to the germ cells. However, in many cancers, these genes are reactivated and expressed in tumors. 14 Cells in C2, C4, C5, and C11 (29.2% of malignant cells)—not enriched with EMT/pEMT or fetal epithelial signature—showed expression of cancer‐testis genes, including CTAG2, MAGEA4, MAGEA1, MAGEA10, and MAGEB2, indicative of germ‐like epithelial cells (Figure 2F, Tables [Link], [Link]).
3.6. Relationship between different malignant cell states
Pseudotime trajectory analysis revealed four branches (denoted as B1, B2, B3, and B4). B1 (at the beginning of the trajectory) was populated by cells with enriched expression of pEMT signature while B4 (at one end of the trajectory) was by cells with enriched expression of fetal cell type signature (Figure 2H,I). Cells enriched with expression of fetal epithelial cell type signatures and cancer‐testis genes were found to show lower expression of differentiation signature than the remaining malignant cells (Figure 2E). Of the 12 patients, 6 patients (P3, P5, P6, P13, P14, and P15) were enriched with cells expressing fetal cell type signatures and 5 patients (P1, P2, P9, P10, and P12) with cells expressing pEMT signatures; P8 was enriched with cells expressing of cancer testis genes (Figure 2G). The mean expression levels of fetal cell type signature and pEMT signature genes in cells across patients were negatively correlated except in P8 (range of correlation coefficient = −0.11 to −0.40) (Table S15). Such enrichment of cells expressing fetal cell type signature was also observed in previous HNSCC single‐cell studies 7 , 9 (Figure S7). The mean expression levels of fetal cell type signature and pEMT signature genes in cells across patients were also negatively correlated in 80% and 75% of patients in GSE103322 and GSE164690 datasets, respectively (Table S15).
3.7. Malignant cells abundantly express genes of immune‐related pathways
Malignant cells, enriched with expression of immune‐related pathway genes (31.7% of malignant cells) known to be involved in antitumor immune response, were abundant in C6, C14, C15, and C16 (cells from multiple patients) and C1 and C10 (patient‐specific clusters). In particular, we detected cells enriched with expression of antigen processing and presentation pathway genes (C1, C6, C10, C14, C15, and C16), cytosolic DNA sensing pathway (C6), cytokine–cytokine receptor interaction (C6), NOD‐like receptor signaling pathway (C6, C16), Toll‐like receptor signaling pathway (C1, C6), and chemokine signaling pathway (C6) (Tables [Link], [Link]).
3.8. T cells are abundant in tumors, with diverse subtypes and levels of infiltration
T cells accounted for approximately 36.4% of all cells in the OSCC‐GB tumors. Variability in the proportion of infiltrated T cell population was observed across patients (Figure 3A). The proportion of T cells was very small (approximately 5%) in two patients, P12 and P13, relative to the total number of cells sampled from the tumor, indicative of low T cell infiltration. Furthermore, the T cell population was transcriptionally heterogeneous as indicated by the formation of 10 subclusters (Figure 3B,C,H,I). The major clusters consisted of Tregs, CD4 + T cells, CD8 + T cells, DN T cells, and NK T cells. Cells in C4 were predominantly Tregs, as evidenced by high levels of expression of Treg‐specific markers, FOXP3 and IL2RA. C6 was enriched with cells expressing high levels of NK cell marker (NCAM1) and killer cell lectin‐like receptor D1 (KLRD1) (Figure 3C), thus representing NK T cells. Clusters C2 and C9 were enriched with CD4 − and CD8 − T cells (Figure 3B,C,H). In concordance with previous reports, 15 this T cell subpopulation lacked expression of FOXP3 as well as natural killer cell marker (NCAM1), thus representing DN T cells (CD4 − CD8 − NCAM1 −). These DN T cells also showed high expression of PLCG2 which is known to play an important role in T cell receptor signal transduction and T cell selection 16 (Figure 3D). Pseudotime trajectory analysis revealed six branches, and branch B1 (at the beginning of the trajectory) was populated by DN T cells (Figure 3F,G), thus suggesting that these DN T cells differentiate into CD4+ and CD8+ T cells in concordance with the role of DN T cells in T cell development and differentiation. 17 , 18 A higher proportion of CD4 − CD8 − PLCG2 + T cells was observed in our study compared to previous HNSCC studies (Figure S8A–F). Clustering the T cells from all three datasets revealed a distinct cluster (C5) with enriched DN PLCG2 + T cells. This cluster was dominated by DN PLCG2 + T cells from our data (Figure S8G–L), thus suggesting a distinct T cell subtype abundance in OSCC‐GB.
FIGURE 3.
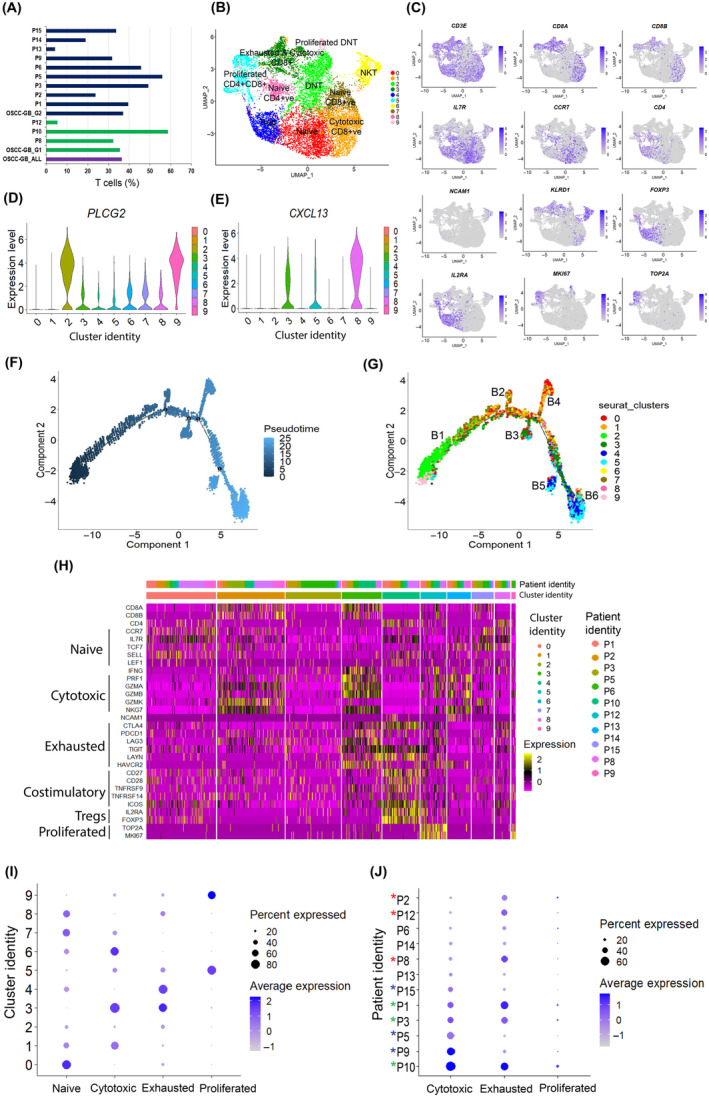
T cell state heterogeneity in oral squamous cell carcinoma of the gingivo‐buccal region (OSCC‐GB) tumors. (A) Fraction of T cells among OSCC‐GB patients. Patient identity on the y‐axis. Colors indicate groups (purple, all OSCC‐GB; green, Group 1; blue, Group 2). (B) UMAP plot of expression profiles of T cells from 12 patients, UMAP is colored by cluster. (C) Feature plot showing expression patterns of T cell marker genes. Each cell is denoted by a dot. (D, E) Violin plots showing expression of (D) PLCG2 and (E) CXCL13 across clusters. Cluster identity on the x‐axis; expression levels on the y‐axis. (F, G) Pseudotime analysis exploring the development trajectories of T cells. Each dot corresponds to a single cell, colored by (F) pseudotime and (G) cluster. (H) Heatmap showing expression levels of T cell state‐specific genes across clusters. Patients and cluster identity are on the x‐axis; gene names on the y‐axis. (I, J) Dot plots showing the average expression level of T cell state‐specific genes across (I) clusters and (J) patients. Cluster/patient identity are on the y‐axis. Color represents the average gene expression level.
Cells in C5 (CD8 + and CD4 + T cells) and C9 (DN T cells) expressed high levels of cell proliferation markers MKI67 and TOP2A, representing proliferative T cells. C0, C7, and C8 showed enrichment of cells expressing high levels of naïve T cell markers ILR7 and CCR7, representing naïve T cells. C1 showed enrichment of cells expressing high levels of cytotoxicity‐related genes (GZMA, GZMB, GZMK, PRF1, and NKG7), representing CD8 + cytotoxic T cells. Cells in C3 (CD8 + T cells) expressed cytotoxicity‐related genes (GZMA, GZMB, GZMK, PRF1, and NKG7) and CTLA4, LAG3, PDCD1 (PD1), HAVCR2 (TIM3), TIGIT, and LAYN, suggestive of cytotoxic and exhausted CD8 + T cells 19 (Figure 3B,H,I). C3 and C8 showed enrichment of cells expressing high levels of CXCL13 (Figure 3E). P9, P5, and P15 were enriched with cytotoxic T cells whereas P8, P1, and P2 were enriched with exhausted T cells (Figure 3J, Table S16). P1, P3, and P10 were enriched with both cytotoxic and exhausted T cells.
3.9. Plasma and memory cells are dominant B cell subtypes
B cells accounted for approximately 7.5% of all the cell populations and consisted of nine subclusters (Figure S9). Variation in the proportion of B cells across tumors was observed. Plasma B cells were represented by C0, C3, C6, and C8, which expressed CD38 and CD27 but did not express MS4A1/CD20 or CD24. C1, C2, C4, C5, and C7 comprised memory B cells that expressed MS4A1/CD20, CD19, and CD27 but did not express CD38. Additionally, C6 was found to be enriched with cells expressing high levels of CCL5, interleukins including IL32 and IL7R, and GNLY, indicating their cytotoxic potential. Cells in C0 showed high expression of immunoglobulin genes including IGKC, IGHG2, IGLC3, IGLC2, and JCHAIN. C8 was enriched with cells expressing MZB1, suggestive of a marginal zone (MZ) B cell subpopulation. Cells expressing FOXP1 and YBX3, critical for early B cell development and differentiation, were enriched in C7.
3.10. Among myeloid cell subtypes, M1 macrophages are the most common
The myeloid cell population (14% of the total cell population) was represented by 13 major clusters mostly consisting of macrophages (Clusters 0, 3, 4, 5, 6, and 10) expressing CD14, CD68, and FCGR3A (CD16) (Figure 4A,B,E). C1 was enriched with cells expressing CD1C and CLEC10A, indicative of myeloid dendritic cells, whereas C2 and C12 were enriched with cells expressing CLEC4C and IL3RA, suggestive of plasmacytoid dendritic cells. 20 Macrophages showed elevated expression of pro‐inflammatory‐related genes (IL1A, IL1B, IL6, and TNF) compared to anti‐inflammatory‐related genes (IL10 and TGFB1) (Figure 4C,D). Cells expressing APOBEC3A, which is involved in promoting pro‐inflammatory M1 macrophage polarization, 21 were enriched in C5. Cells in C0 and C4 were enriched with the expression of LGMN that was earlier reported to be involved in inducing M2 phenotype. 22 Cells expressing FABP5, a marker for lipid‐rich foamy macrophages, 23 were enriched in C3, suggesting that they could be lipid‐laden foam macrophages. In concordance with a previous report 24 there was a distinct subcluster (C6) with enrichment of cells expressing the T cell marker gene (CD3D) and TCR marker genes TRAC (Figure 4A,E), suggesting that they could be CD3 + TCR+ macrophage cell populations.
FIGURE 4.
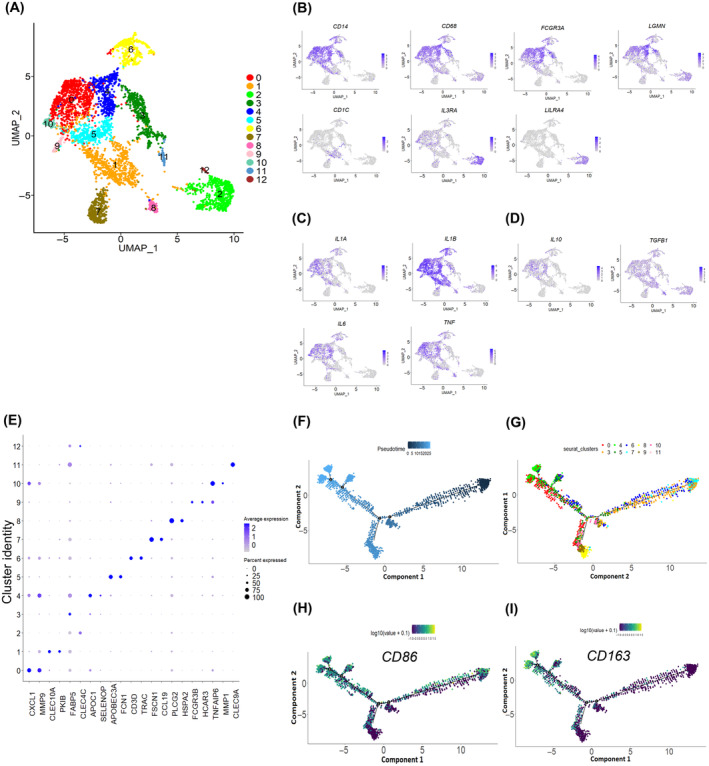
Myeloid cell population in oral squamous cell carcinoma of the gingivo‐buccal region. (A) UMAP plot of expression profiles of myeloid cells. Each cell is denoted by a dot. UMAP is colored by cluster. (B‐D) Feature plots showing expression levels of (B) macrophages and dendritic cell marker genes, (C) pro‐inflammatory genes, and (D) anti‐inflammatory genes across clusters. Each cell is denoted by a dot and the color intensity represents the gene expression level. (E) Dot plot showing distribution and expression of top two differentially expressed genes across clusters; color intensity represents the average gene expression level. Genes are on the x‐axis; cluster identity on the y‐axis. (F–I) Pseudotime analysis of macrophages shows four decision points. Each dot corresponds to a single cell, colored by (F) pseudotime and (G) cluster and expression of (H) M1 marker (CD86) and (I) M2 marker (CD163) genes.
3.11. Macrophages in tumors exhibit an intermediate M1–M2 polarization state
Lineage relationships among the macrophage subsets were inferred using pseudotime trajectory analysis. The trajectory consisted of four decision points (Figure 4F,G). Cells started with an M1 phenotype expressing M1 marker genes (CD86) and traveled through branch points one to four whereas cells with an M2 phenotype expressing M2 marker genes (CD163) traveled through branch points two to four (Figure 4H,I). These features indicate that the macrophages in OSCC‐GB tumors analyzed in our study showed an intermediate M1–M2 macrophage polarization state. We also observed that macrophages showed elevated expression of pro‐ and anti‐inflammatory‐related genes and M1 and M2 marker genes in previous HNSCC datasets (Figures S10 and S11).
3.12. Endothelial cell subtypes that perform diverse cellular functions are present
Endothelial cells accounted for 3% of the total cell population and consisted of nine major clusters (Figure S12). C2 was enriched with cells expressing ESM1, an endothelial tip cell‐related gene. 25 These cells were also enriched with CA2 and COL13A1 expression. CA2 expression supports endothelial cell survival under lactic acidosis in the tumor ecosystem 26 while COL13A1 expression is involved in cell–matrix, and cell–cell adhesion. C4 was a distinct cluster enriched with cells expressing PDPN, PROX1, and IGF1. PDPN was earlier reported to be expressed in lymphatic endothelium 27 and PROX1 is widely used as a marker for lymphatic endothelial cells. 28 C5 was enriched with cells expressing SEMA3G and SULF1, previously reported as aiding angiogenesis. 29 C0 and C6 were enriched with cells expressing ACKR1, 30 suggestive of being enriched with venous endothelial cells. These cells also showed high expression of the vascular cell adhesion molecule 1 (VCAM1) and chemokine CCL14. C7 was enriched with cells expressing PDGFRB, a pericyte marker, 31 suggesting that these cells were pericytes. As reported earlier, 32 these cells expressed genes encoding collagens I, III, and VI (COL1A1, COL3A1, and COL6A3, respectively). C8 was enriched with cells expressing proliferation markers MKI67 and TOP2A, representing proliferative endothelial cells.
3.13. Two major groups of fibroblasts found in OSCC‐GB
Subclustering of the fibroblast cell population (5% of total cells) revealed two major groups, with specific characteristics as reported earlier 7 , 33 (Figures 5 and S13). The first group consisted of cells (C0, C3, and C7) with an enriched expression of ECM‐related genes, including the fibroblast activation protein (FAP), podoplanin (PDPN), and MMP‐related genes (MMP2, MMP1, and MMP3) (Figure 5A,B,D–G), thus, representing CAFs. The second group consists of cells (C1, C4, C5, and C6) with an enriched expression of alpha‐smooth muscle actin (ACTA2) and myosin light chain proteins (MYLK) (Figure 5A,C,E,F,H), thus representing myCAFs. We also observed these two major fibroblast populations (CAF and myCAFs) in GSE103322 and GSE164690 datasets (Figures S14 and S15). Interestingly, we found that in C0, a large proportion (>60%) of fibroblasts were from OSMF‐associated tumors and OSCC‐associated OSMF tumors show a higher proportion of CAF than non‐OSMF‐associated tumors (Figure 5A,I,J), thus suggestive of the distinct role of CAF in OSMF‐associated tumors. In concordance with a previous report, 9 C7 cells were characterized by the expression of elastic fibroblasts markers (MFAP4, MFAP5, and FBLN1), representing the elastic fibroblasts (Figure 5D).
FIGURE 5.
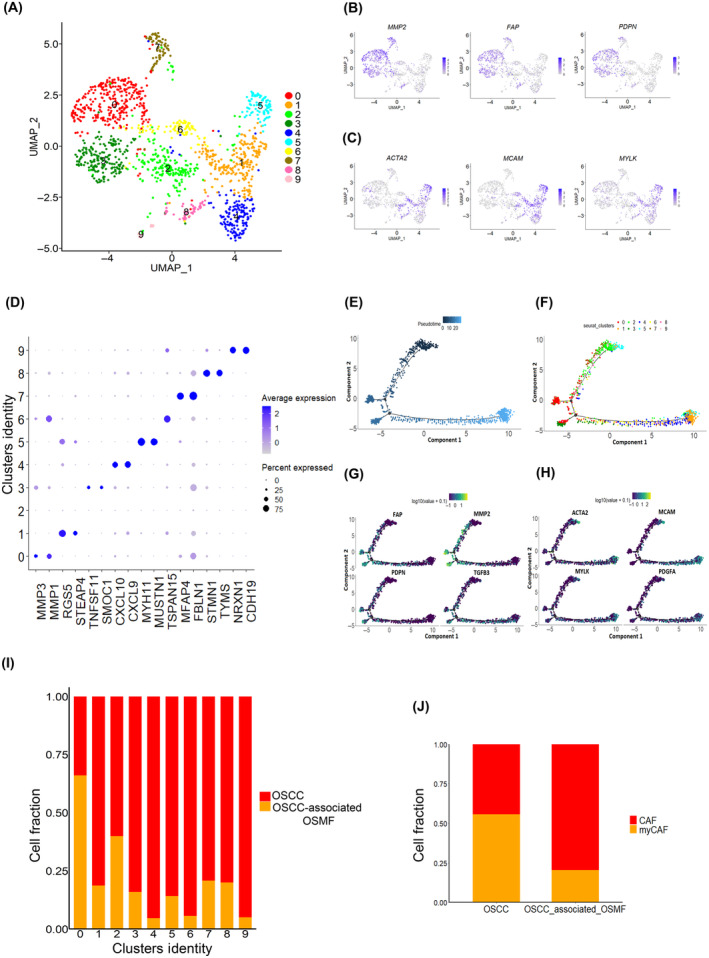
Cancer‐associated fibroblast (CAF) and myofibroblast (myCAF) in oral squamous cell carcinoma of the gingivo‐buccal region (OSCC‐GB). (A) UMAP plot of the expression profiles of fibroblast cells from 12 OSCC‐GB tumors. Each cell is denoted by a dot. UMAP is colored by cluster. (B, C) Feature plots showing distribution and expression of fibroblast marker genes across clusters. Each cell is denoted by a dot and color represents the average gene expression level. (D) Dot plot showing distribution and expression of top two differentially expressed genes across clusters. Color represents the average gene expression level. Genes are on the x‐axis; cluster identity on the y‐axis. (E–H) Pseudotime analysis of fibroblasts. Each dot corresponds to a single cell, colored by (E) pseudotime and (F) cluster and expression of (G) CAF and (H) myCAF marker genes. (I, J) Column graphs showing fractions of (I) fibroblast cells from OSCC‐GB and oral submucous fibrosis (OSMF)‐associated OSCC‐GB tumors across clusters and (J) CAF and myCAF populations within OSCC‐GB and OSMF‐associated OSCC‐GB tumors. Cluster identity/groups are on the x‐axis; cell fraction on the y‐axis.
3.14. Coexpression states in cells expressing driver genes of OSCC‐GB
We earlier identified genes that are significantly and frequently mutated specifically in OSCC‐GB tumors (ARID2, MLL4, KMT2B, USP9X, UNC13C, and TRPM3), genes with recurrent amplification (DROSHA, MECOM, YAP1, and NFIB) and DDX3X, a gene with homozygous deletion. 34 We found significant upregulation of VMP1 and NEAT1 in cells that express ARID2, KMT2B, USP9X, YAP1, NFIB, and DDX3X compared to the nonexpressing cells (Table S17) in our samples. Significant upregulation of MALAT1 in cells expressing ARID2, KMT2B, YAP1, and NFIB as well as upregulation of F3 in cells expressing ARID2, USP9X, YAP1, and NFIB, compared to the respective nonexpressing cells, were also observed. LAMA3 was significantly upregulated in cells that express ARID2, YAP1, and NFIB, while PLCG2 was significantly upregulated in cells expressing KMT2B and DDX3X. MACF1 was significantly upregulated in cells expressing ARID2 and KMT2B while CDK6 was found to be significantly upregulated in cells that expressed DROSHA and NFIB compared to the nonexpressing cells.
3.15. Comparison of scRNA‐seq data with bulk transcriptomic data
Our scRNA‐seq analysis revealed that the upregulation of immune checkpoint genes previously identified 10 in our bulk RNA sequencing data, CD274, CD80 and IDO1, were predominately present in specific myeloid cell populations (Figure S16). Of 13 clusters of the myeloid cell population, C5, C7, C10, and C11 showed prominent expression of these genes, highlighting their multifaceted functions within the myeloid cell compartments. Additionally, consistent with our bulk data, we observed significant enrichments of pathways such as ECM‐receptor interaction and focal adhesion (Figure 2A) in OSCC‐GB in specific malignant cell clusters C0, C1, C3, C6, C14, and C15.
4. DISCUSSION
In the oral cavity, OSCC‐GB is the most common form of cancer. It shows distinctive gene expression profiles compared to that of other oral cavity cancers, for example, tongue cancer. Patients display considerable clinical heterogeneity and variable response to treatment, which is likely due to the heterogeneity of cell types and gene expression states within the tumors. 10 Such heterogeneity is difficult, if not impossible, to decipher from bulk genomic analyses of tumor. 35 We, therefore, used scRNA‐seq and novel statistical methods of data analysis (notably, to identify malignant cells that performed more accurately than the commonly used InferCNV method) for an unbiased characterization—globally, the first report on OSCC‐GB—of the cell type and cell state diversity in the tumor of individual OSCC‐GB patients.
Consistent with previous studies 7 , 36 we found that nonmalignant cells clustered by cell type. Most, but not all, of the malignant cell clusters (12 of 17 clusters of malignant cells) were, however, patient‐specific. This feature reflects that some transcriptional programs of malignant cells are shared while others are unique to individual patients, possibly because sets of cancer‐associated somatic alterations are heterogeneous across patients. Our finding differs from earlier reports where all malignant cell clusters were patient‐specific. 7 , 35
We found two major subgroups of malignant cells; one subgroup was specific to tumors that were associated with OSMF, whereas the other was not associated with this precancerous lesion. Expression of FOS, ATP1A1, and DUSP1 provided perfect discrimination between these two groups. This finding is suggestive of the existence of malignant cells unique to oral tumors with associated submucous fibrosis, indicating that tumors in these patients might have developed from their precancerous lesions.
An interesting finding in our study was the presence of fetal and germ‐like epithelial malignant cells. This agrees with earlier reports of tumor cells recapitulating signatures of early developmental programs including embryonic and fetal development. 37 Epithelial tissues underwent fetal‐like cellular reprogramming and adopted an undifferentiated fetal‐like state. 38 In our samples, the subpopulation of malignant cells expressing these fetal gene signatures and cancer‐testis genes also exhibited reduced expression of differentiation signatures indicative of undifferentiated fetal‐like and germ‐like states. An earlier report suggests that in hepatocellular carcinoma, a population of monocyte‐derived macrophages and PLVAP endothelial cells are reprogrammed to acquire a fetal‐like phenotype. 39 However, this is the first report of such fetal‐like states in malignant cells, which seems to be a dominant cell state in OSCC‐GB.
Epithelial–mesenchymal transition, an important hallmark of cancer, is a continuous process by which cells with epithelial phenotype acquire the mesenchymal phenotype. In concordance with the role of EMT in tumor invasion and metastasis, 40 a subpopulation of malignant cells in OSCC‐GB with enriched EMT, invasion, and metastasis signatures could contribute to tumor invasion and metastasis through EMT. Recent evidence indicates that malignant cells can acquire a hybrid and transient state called pEMT. 41 Cells expressing EMT signatures also expressed both the epithelial and mesenchymal markers but did not express most of the EMT transcription factors except SNAI2 in concordance with previous HNSCC scRNA‐seq studies. 7 , 42 SNAI2 overexpression was found to be correlated with pEMT in non‐small‐cell lung cancer and HNSCC, 7 , 42 thus indicative of the existence of a subpopulation of malignant cells being in pEMT state. In this subpopulation, genes of the ECM‐receptor and focal adhesion pathways were highly expressed and were also highly correlated with the levels of expression of the pEMT genes. These findings suggest the involvement of ECM‐receptor and focal adhesion pathways in pEMT in OSCC‐GB, as previously reported in some other cancers. 43 , 44 , 45
Even though cell populations of patients with prior OSMF clustered separately from those without, we identified four small cell populations that were shared between the two patient types. Of these, two were in the pEMT state while the remaining two were in the fetal‐like state, indicative of pEMT and fetal cellular reprogramming, respectively. The pEMT and fetal‐like states were mutually exclusive in our dataset. Pseudotime trajectory analysis suggested that the cells in the pEMT state are tumor‐initiating and transitioning to cells in the fetal‐like state. Thus, these findings indicate the importance of pEMT and fetal cellular reprogramming in OSCC‐GB. We found evidence of the existence of such a fetal cell type signature in HNSCC datasets from other regions of the world, 7 , 9 suggesting that these cell states are present and largely mutually exclusive in malignant cells in HNSCC.
Double negative T cell infiltration in other solid tumor types such as non‐small‐cell lung cancer, liver, glioma, and pancreatic tumors has been reported. 46 , 47 , 48 Double negative T cells have strong antitumor effects 49 known to inhibit cell proliferation and invasion in human cancer cells. 50 The DN T cells observed in our patients likely play an important role in antitumor activity in OSCC‐GB. Higher abundance of CD4 − CD8 − PLCG2 + (DN PLCG2 + ) T cells in our data compared to previous HNSCC studies, 7 , 9 suggest a predominance of this T cell subtype in OSCC‐GB.
We found that the myeloid cells were the second largest immune cell population in the OSCC‐GB tumor ecosystem, underlining the importance of this cell‐type in OSCC‐GB pathogenesis. The coexistence of both M1 and M2 macrophages observed in our patients suggested an intermediate M1–M2 macrophage polarization, which has been recently found in other cancer types. 51 , 52 These macrophages exhibited both pro‐inflammatory and pro‐tumorigenic characteristics as evidenced by the expression of both cytotoxic and anti‐inflammatory cytokines. This simultaneous expression of M1 and M2 markers in the macrophage population is indicative of a transition in which the TME becomes more conducive to the proliferation of malignant cells. A unique population of macrophages (CD3+ TCR+ macrophages) was found to be present in almost all patients, underscoring its importance in OSCC‐GB tumorigenesis.
Our data show two major groups of fibroblasts of OSCC‐GB patients: CAFs and myCAFs. As reported earlier, 53 the myofibroblast population is high in OSMF leading to severe fibrosis. However, we found ~80% of fibroblasts in OSMF‐associated OSCC‐GB were CAFs. These CAFs were enriched with the expression of ECM‐related genes indicative of ECM remodeling phenotype. This suggests a phenotype shift in the stromal population, which might have a link with OSMF‐associated OSCC‐GB pathogenesis.
The reliability of our scRNA‐seq data is underscored by the high concordance between the single‐cell data and our previously published bulk RNA sequencing data. Additionally, we found that genes that predominate in the bulk data are mostly contributed by specific subsets of the cell population. In particular, cells of myeloid origin are instrumental in immune evasion in OSCC‐GB. This further highlights the dynamic nature of the TME that regulates immune response within the TME, facilitating tumor growth and survival. Our single‐cell analysis has provided enhanced insights over those obtained from the results of bulk sequencing analysis. From scRNA‐seq data, we identified genes whose expressions in the malignant cells are associated with tumor progression and poor prognosis (NEAT1, MALT1, etc.) which express some of the driver genes of OSCC‐GB identified by us earlier using the bulk sequencing data.
In summary, OSCC‐GB‐associated OSMF tumors show an expression profile distinct from tumors not associated with OSMF. Oral squamous cell carcinoma of the gingivo‐buccal region with concomitant presence of OSMF is likely to have developed from the precancerous lesion. This inference is also supported by the fact that the mean age at onset of OSCC‐GB in patients with OSMF (44.7 years; range, 36–60 years) is over 10 years earlier than patients without OSMF (59.2 years; range, 46–76 years). Fetal cellular reprogramming and pEMT appear to play major roles in OSCC‐GB tumorigenesis. Double negative PLCG2 + T cells and intermediate M1–M2 macrophage polarization were also detected in this cancer type. Our study presents the first comprehensive analysis of the tumor ecosystem of OSCC‐GB. The results of the single‐cell transcriptomic analysis presented here, in addition to being consistent with our earlier findings obtained from bulk RNA sequencing data, have provided deep insights into the nature and extent of cellular heterogeneity, evidenced by the diversity of cell types and cell states, as well as helped identify unique gene expression programs that could not be obtained from bulk‐cell analysis.
AUTHOR CONTRIBUTIONS
Sillarine Kurkalang: Data curation; formal analysis; investigation; methodology; validation; visualization; writing – original draft; writing – review and editing. Sumitava Roy: Data curation; formal analysis; investigation; methodology; software; validation; visualization; writing – original draft; writing – review and editing. Arunima Acharya: Data curation; methodology; software; validation; visualization; writing – review and editing. Paramita Mazumder: Methodology; resources. Somnath Mazumder: Methodology; resources. Subrata Patra: Methodology; resources. Shekhar Ghosh: Methodology; resources. Sumanta Sarkar: Methodology; resources. Sudip Kundu: Methodology; resources. Nidhan Kumar Biswas: Investigation; supervision. Sandip Ghose: Conceptualization; investigation; methodology; resources; supervision. Partha P Majumder: Conceptualization; formal analysis; funding acquisition; investigation; supervision; writing – original draft; writing – review and editing. Arindam Maitra: Conceptualization; formal analysis; funding acquisition; investigation; project administration; supervision; writing – original draft; writing – review and editing.
FUNDING INFORMATION
This study was funded by the Department of Biotechnology, Ministry of Science and Technology, Government of India in the Systems Medicine Cluster (SyMeC) (Grant # BT/Med II/NIBMG/SyMeC/2014/Vol.II).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
Approval of the research protocol by an institutional review board: The study was approved by the Institutional Ethics Committees of Dr. R Ahmed Dental College and Hospital and the National Institute of Biomedical Genomics, India.
Informed consent: Written informed consent was obtained from every patient.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
Supporting information
Data S1
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Figure S10
Figure S11
Figure S12
Figure S13
Figure S14
Figure S15
Figure S16
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Table S11
Table S12
Table S13
Table S14
Table S15
Table S16
Table S17
ACKNOWLEDGMENTS
We are grateful to Professor Aviv Regev, Professor Alex Shalek of the Broad Institute of MIT and Harvard, and Dr. Matthew D. Young of The Wellcome Sanger Institute for their advice, support, and guidance. The National Genomics Core of NIBMG is acknowledged for providing support in sequencing data generation. Partha P. Majumder acknowledges the support of his J. C. Bose Fellowship and National Science Chair fellowship from SERB, Government of India. Sumitava Roy acknowledges the support of his Senior Research Fellowship (NET) from the University Grants Commission of India (UGC India). Arunima Acharya acknowledges support from NIBMG. Sillarine Kurkalang acknowledges the support of her DBT‐Research associateship by the Department of Biotechnology, Government of India. We acknowledge the intramural support provided by NIBMG for this study.
Kurkalang S, Roy S, Acharya A, et al. Single‐cell transcriptomic analysis of gingivo‐buccal oral cancer reveals two dominant cellular programs. Cancer Sci. 2023;114:4732‐4746. doi: 10.1111/cas.15979
Sillarine Kurkalang and Sumitava Roy contributed equally to this study.
Contributor Information
Partha P. Majumder, Email: parmaj2023@gmail.com.
Arindam Maitra, Email: am1@nibmg.ac.in.
DATA AVAILABILITY STATEMENT
The scRNA‐seq data generated in this study is available in the GEO database under the accession number GSE215403. Other data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209‐249. doi: 10.3322/caac.21660 [DOI] [PubMed] [Google Scholar]
- 2. de Camargo CM, Voti L, Guerra‐Yi M, Chapuis F, Mazuir M, Curado MP. Oral cavity cancer in developed and in developing countries: population‐based incidence. Head Neck. 2010;32(3):357‐367. doi: 10.1002/hed.21193 [DOI] [PubMed] [Google Scholar]
- 3. Hua X, Zhao W, Pesatori AC, et al. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat Commun. 2020;11(1):2459. doi: 10.1038/s41467-020-16295-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park SY, Gönen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Investig. 2010;120(2):636‐644. doi: 10.1172/JCI40724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sottoriva A, Spiteri I, Piccirillo SGM, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009‐4014. doi: 10.1073/pnas.1219747110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanay A, Regev A. Scaling single‐cell genomics from phenomenology to mechanism. Nature. 2017;541(7637):331‐338. doi: 10.1038/nature21350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puram SV, Tirosh I, Parikh AS, et al. Single‐cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611‐1624.e24. doi: 10.1016/j.cell.2017.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cillo AR, Kürten CHL, Tabib T, et al. Immune landscape of viral‐ and carcinogen‐driven head and neck cancer. Immunity. 2020;52(1):183‐199.e9. doi: 10.1016/j.immuni.2019.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kürten CHL, Kulkarni A, Cillo AR, et al. Investigating immune and non‐immune cell interactions in head and neck tumors by single‐cell RNA sequencing. Nat Commun. 2021;12(1):7338. doi: 10.1038/s41467-021-27619-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das D, Maitra A, Panda CK, et al. Genes and pathways monotonically dysregulated during progression from normal through leukoplakia to gingivo‐buccal oral cancer. NPJ Genom Med. 2021;6(1):32. doi: 10.1038/s41525-021-00195-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hao Y, Hao S, Andersen‐Nissen E, et al. Integrated analysis of multimodal single‐cell data. Cell. 2021;184(13):3573‐3587.e29. doi: 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tickle TI, Georgescu C, Brown M, Haas B. inferCNV of the Trinity CTAT Project .
- 13. Qiu X, Mao Q, Tang Y, et al. Reversed graph embedding resolves complex single‐cell trajectories. Nat Methods. 2017;14(10):979‐982. doi: 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang C, Gu Y, Zhang K, et al. Systematic identification of genes with a cancer‐testis expression pattern in 19 cancer types. Nat Commun. 2016;7(1):10499. doi: 10.1038/ncomms10499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hillhouse EE, Lesage S. A comprehensive review of the phenotype and function of antigen‐specific immunoregulatory double negative T cells. J Autoimmun. 2013;40(1):58‐65. doi: 10.1016/j.jaut.2012.07.010 [DOI] [PubMed] [Google Scholar]
- 16. Fu G, Chen Y, Schuman J, Wang D, Wen R. Phospholipase Cγ2 plays a role in TCR signal transduction and T cell selection. J Immunol. 2012;189(5):2326‐2332. doi: 10.4049/jimmunol.1103458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anderson G, Jenkinson EJ. Lymphostromal interactions in thymic development and function. Nat Rev Immunol. 2001;1(1):31‐40. doi: 10.1038/35095500 [DOI] [PubMed] [Google Scholar]
- 18. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. 2018;48(2):202‐213. doi: 10.1016/j.immuni.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crawford A, Angelosanto JM, Kao C, et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity. 2014;40(2):289‐302. doi: 10.1016/j.immuni.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heger L, Balk S, Lühr JJ, et al. CLEC10A is a specific marker for human CD1c+ dendritic cells and enhances their toll‐like receptor 7/8‐induced cytokine secretion. Front Immunol. 2018;9:744. doi: 10.3389/fimmu.2018.00744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alqassim EY, Sharma S, Khan ANMNH, et al. RNA editing enzyme APOBEC3A promotes pro‐inflammatory M1 macrophage polarization. Commun Biol. 2021;4(1):102. doi: 10.1038/s42003-020-01620-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun SG, Guo JJ, Qu XY, et al. The extracellular vesicular pseudogene LGMNP1 induces M2‐like macrophage polarization by upregulating LGMN and serves as a novel promising predictive biomarker for ovarian endometriosis recurrence. Hum Reprod. 2022;37(3):447‐465. doi: 10.1093/humrep/deab266 [DOI] [PubMed] [Google Scholar]
- 23. Umbarawan Y, Enoura A, Ogura H, et al. Fabp5 is a sensitive marker for lipid‐rich macrophages in the luminal side of atherosclerotic lesions. Int Heart J. 2021;62(3):666‐676. doi: 10.1536/ihj.20-676 [DOI] [PubMed] [Google Scholar]
- 24. Jiang Y, Zhang S, Tang L, et al. Single‐cell RNA sequencing reveals TCR+ macrophages in HPV‐related head and neck squamous cell carcinoma. Front Immunol. 2022;13:1030222. doi: 10.3389/fimmu.2022.1030222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. del Toro R, Prahst C, Mathivet T, et al. Identification and functional analysis of endothelial tip cell‐enriched genes. Blood. 2010;116(19):4025‐4033. doi: 10.1182/blood-2010-02-270819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Annan DA, Maishi N, Soga T, et al. Carbonic anhydrase 2 (CAII) supports tumor blood endothelial cell survival under lactic acidosis in the tumor microenvironment. Cell Commun Signal. 2019;17(1):169. doi: 10.1186/s12964-019-0478-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jha SK, Rauniyar K, Jeltsch M. Key molecules in lymphatic development, function, and identification. Ann Anat. 2018;219:25‐34. doi: 10.1016/j.aanat.2018.05.003 [DOI] [PubMed] [Google Scholar]
- 28. Pan Y, di Wang W, Yago T. Transcriptional regulation of podoplanin expression by Prox1 in lymphatic endothelial cells. Microvasc Res. 2014;94:96‐102. doi: 10.1016/j.mvr.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 29. Chen DY, Sun NH, Chen X, et al. Endothelium‐derived semaphorin 3G attenuates ischemic retinopathy by coordinating β‐catenin‐dependent vascular remodeling. J Clin Investig. 2021;131(4):e135296. doi: 10.1172/JCI135296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schupp JC, Adams TS, Cosme C, et al. Integrated single‐cell atlas of endothelial cells of the human lung. Circulation. 2021;144(4):286‐302. doi: 10.1161/CIRCULATIONAHA.120.052318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miners JS, Schulz I, Love S. Differing associations between Aβ accumulation, hypoperfusion, blood–brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer's disease. J Cereb Blood Flow Metab. 2018;38(1):103‐115. doi: 10.1177/0271678X17690761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sakhneny L, Epshtein A, Landsman L. Pericytes contribute to the islet basement membranes to promote beta‐cell gene expression. Sci Rep. 2021;11(1):2378. doi: 10.1038/s41598-021-81774-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H, Courtois ET, Sengupta D, et al. Reference component analysis of single‐cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017;49(5):708‐718. doi: 10.1038/ng.3818 [DOI] [PubMed] [Google Scholar]
- 34. India Project Team of ICGC . Mutational landscape of gingivo‐buccal oral squamous cell carcinoma reveals new recurrently‐mutated genes and molecular subgroups. Nat Commun. 2013;4(1):2873. doi: 10.1038/ncomms3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu F, Fan J, He Y, et al. Single‐cell profiling of tumor heterogeneity and the microenvironment in advanced non‐small cell lung cancer. Nat Commun. 2021;12(1):2540. doi: 10.1038/s41467-021-22801-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science (1979). 2016;352(6282):189‐196. doi: 10.1126/science.aad0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yong KJ, Gao C, Lim JSJ, et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med. 2013;368(24):2266‐2276. doi: 10.1056/nejmoa1300297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nusse YM, Savage AK, Marangoni P, et al. Parasitic helminths induce fetal‐like reversion in the intestinal stem cell niche. Nature. 2018;559(7712):109‐113. doi: 10.1038/s41586-018-0257-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sharma A, Seow JJW, Dutertre CA, et al. Onco‐fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183(2):377‐394.e21. doi: 10.1016/j.cell.2020.08.040 [DOI] [PubMed] [Google Scholar]
- 40. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105‐111. doi: 10.1038/35102167 [DOI] [PubMed] [Google Scholar]
- 41. Sha Y, Haensel D, Gutierrez G, Du H, Dai X, Nie Q. Intermediate cell states in epithelial‐to‐mesenchymal transition. Phys Biol. 2019;16(2):021001. doi: 10.1088/1478-3975/aaf928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emprou C, le van Quyen P, Jégu J, et al. SNAI2 and TWIST1 in lymph node progression in early stages of NSCLC patients. Cancer Med. 2018;7(7):3278‐3291. doi: 10.1002/cam4.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rahbari NN, Kedrin D, Incio J, et al. Anti‐VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med. 2016;8(360):360ra135. doi: 10.1126/scitranslmed.aaf5219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shah PP, Fong MY, Kakar SS. PTTG induces EMT through integrin αVβ3‐focal adhesion kinase signaling in lung cancer cells. Oncogene. 2012;31(26):3124‐3135. doi: 10.1038/onc.2011.488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deng B, Yang X, Liu J, He F, Zhu Z, Zhang C. Focal adhesion kinase mediates TGF‐β1‐induced renal tubular epithelial‐to‐mesenchymal transition in vitro. Mol Cell Biochem. 2010;340(1–2):21‐29. doi: 10.1007/s11010-010-0396-7 [DOI] [PubMed] [Google Scholar]
- 46. Fang L, Ly D, Wang S, et al. Targeting late‐stage non‐small cell lung cancer with a combination of DNT cellular therapy and PD‐1 checkpoint blockade. J Exp Clin Cancer Res. 2019;38(1):123. doi: 10.1186/s13046-019-1126-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. di Blasi D, Boldanova T, Mori L, Terracciano L, Heim MH, de Libero G. Unique T‐cell populations define immune‐inflamed hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2020;9(2):195‐218. doi: 10.1016/j.jcmgh.2019.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hall M, Liu H, Malafa M, et al. Expansion of tumor‐infiltrating lymphocytes (TIL) from human pancreatic tumors. J Immunother Cancer. 2016;4(1):61. doi: 10.1186/s40425-016-0164-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li Y, Dong K, Fan X, et al. DNT cell‐based immunotherapy: progress and applications. J Cancer. 2020;11(13):3717‐3724. doi: 10.7150/jca.39717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu Y, Hu P, Zhou H, et al. Double‐negative T cells inhibit proliferation and invasion of human pancreatic cancer cells in co‐culture. Anticancer Res. 2019;39(11):5911‐5918. doi: 10.21873/anticanres.13795 [DOI] [PubMed] [Google Scholar]
- 51. Jin S, Li R, Chen MY, et al. Single‐cell transcriptomic analysis defines the interplay between tumor cells, viral infection, and the microenvironment in nasopharyngeal carcinoma. Cell Res. 2020;30(11):950‐965. doi: 10.1038/s41422-020-00402-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peng J, Sun BF, Chen CY, et al. Single‐cell RNA‐seq highlights intra‐tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019;29(9):725‐738. doi: 10.1038/s41422-019-0195-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Angadi PV, Kale AD, Hallikerimath S. Evaluation of myofibroblasts in oral submucous fibrosis: correlation with disease severity. J Oral Pathol Med. 2011;40(3):208‐213. doi: 10.1111/j.1600-0714.2010.00995.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Figure S8
Figure S9
Figure S10
Figure S11
Figure S12
Figure S13
Figure S14
Figure S15
Figure S16
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Table S10
Table S11
Table S12
Table S13
Table S14
Table S15
Table S16
Table S17
Data Availability Statement
The scRNA‐seq data generated in this study is available in the GEO database under the accession number GSE215403. Other data that support the findings of this study are available from the corresponding author upon request.