

Abstract
CRISPR-Cas9 is a programmable gene editing tool with a promising potential for cancer gene therapy. This therapeutic function is enabled in the present work via the non-covalent delivery of CRISPR ribonucleic protein (RNP) by cationic glucosamine/PEI-derived graphene quantum dots (PEI-GQD) that aid in overcoming physiological barriers and tracking genes of interest. PEI-GQD/RNP complex targeting the TP53 mutation overexpressed in ~50% of cancers successfully produces its double-stranded breaks in solution and in PC3 prostate cancer cells. Restoring this cancer “suicide” gene can promote cellular repair pathways and lead to cancer cell apoptosis. Its repair to the healthy form performed by simultaneous PEI-GQD delivery of CRISPR RNP and a gene repair template leads to a successful therapeutic outcome: 40% apoptotic cancer cell death, while having no effect on non-cancerous HeK293 cells. The translocation of PEI-GQD/RNP complex into PC3 cell cytoplasm is tracked via GQD intrinsic fluorescence, while EGFP-tagged RNP is detected in the cell nucleus, showing the successful detachment of the gene editing tool upon internalization. Using GQDs as non-viral delivery and imaging agents for CRISPR-Cas9 RNP sets the stage for image-guided cancer-specific gene therapy.
Keywords: Graphene Quantum Dots, CRISPR-Cas9, Gene Editing, Cancer, In Vitro
Graphical abstract
Biocompatible PEI-based graphene quantum dots (PEI-GQDs) are utilized for the first time as non-viral delivery platforms and imaging agents for CRISPR-Cas9 complex to correct the mutated TP53 gene in prostate cancer (PC-3) cells. This therapeutic nanoplatform drastically reduces the viability of PC-3 cells down to 60%, propelling gene therapeutics to a new era of personalized medicine.
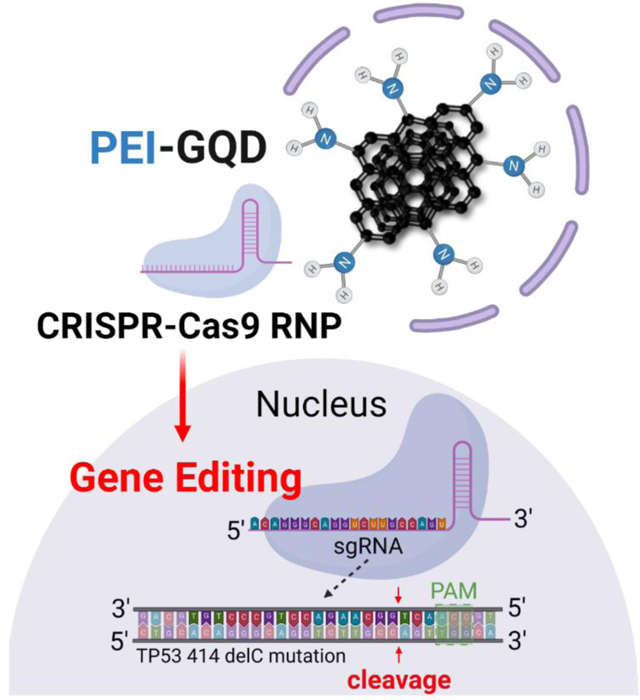
Introduction
Since the release of the first draft of the human genome in 2000 (1, 2), disease-prone genes have been identified through their comparison to the healthy ones (2–4). Until now, at least 291 genes have been associated with cancer, comprising over 1% of the human genome (5). Therefore, the possibility of editing the underlying cancer-driven genes can become critical for successful cancer treatment (6). For instance, almost 50% of cancers possess a mutant form of the tumor protein 53 (TP53) gene, which in its healthy wild-type form leads to cell cycle arrest, DNA repair, and apoptosis (7, 8). Restoring this “guardian of the genome” TP53 gene results in normal cell cycle progression and inhibition of cancer cell proliferation (8–10), leading to cell death (11). Prostate cancer (PC-3) cells appear to be an ideal target for studying the effects of this repair. These cells have a mutation in the TP53 gene, comprising a base pair deletion at codon 138 and generating a frameshift and a new in-frame stop codon at position 169 (exon 5 single base deletion) (12). PC-3 cells are hemizygous for chromosome 17p and have only a single copy of the TP53 gene. For this reason, they require an exogenous DNA repair template to restore the TP53 gene. With prostate cancer being the third most common type of cancer and already having a dysfunctional TP53 gene (13), we find it an appropriate model for gene editing cancer therapy.
With current sequencing methods and the advent of gene editing technologies, genetic sequences are not only read, but can also be written with gene editing tools, such as zinc finger nuclease (ZFN) (14), meganuclease (15) and transcription activator-like effector nuclease (TALEN) (16). These tools are synthetically engineered by fusing individual nucleic acid-targeting proteins with restriction enzymes such as FokI and I-Scel endonucleases (17). Such multi-component synthetic approach, though, is costly and complex, narrowing the practical use of these tools to a specialized audience and upending the field of gene editing. However, in 2012, the discovery of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) method revolutionized this field by overcoming challenges encountered by its predecessors.
CRISPR is derived from the adaptive immune system of prokaryotes and archaea, which is used to fight against viruses by locating and disrupting viral genes (18, 19). Among the family of CRISPR-Cas systems (19–21), CRISPR-Cas9, a type II system, has been chosen and repurposed to work as a gene editing tool because of its single protein effector structure and the ability to make double-stranded breaks (DSBs) in DNA (22). Jinek et al. have synthetically re-engineered a programmable single-guide RNA (sgRNA) bound to the CRISPR-Cas9 ribonucleoprotein (RNP) to target any 20-nucleotide DNA sequence (23). Since then, CRISPR-Cas9 has been rapidly adopted into gene therapeutics for eukaryotic cells (24–26) because of its ease of use (27–29) and efficacy of treatment (30), thus, democratizing the field of gene editing. CRISPR technology has been successfully used for a plethora of therapeutic applications addressing sickle cell anemia, beta-thalassemia, cystic fibrosis, Duchenne muscular dystrophy (DMD), and chimeric antigen receptor (CAR) T-cell therapy (31). However, these treatment approaches are limited to in vitro and ex vivo applications because of CRISPR’s prokaryotic origin, triggering an immunological response and preventing its use in the body (32). Therefore, further advancement of this technology relies on developing safe delivery approaches.
Several viral and non-viral vectors can be successfully used as delivery vehicles for CRISPR-Cas9. For instance, adeno-associated virus (AAV) has recently been established as a promising candidate because of its reduced risk of genomic integration, inherent tissue tropism, and clinically manageable immunogenicity (33, 34). It has been already utilized in clinical trials and FDA-approved gene replacement therapy of spinal muscular atrophy and congenital blindness (34). AAV-mediated CRISPR delivery for DMD gene therapy has been recently demonstrated in vivo (35). Despite these advances, the potential for carcinogenesis, limited loading capacity, restricted scalability and extended presence in the body still hamper wide therapeutic use of viral delivery agents (36–39). Non-viral vectors such as lipid nanoparticles (40), gold nanoparticles (41), single-walled carbon nanotubes (SWCNTs) (42), graphene oxide (GO) (43), and carbon dots (44, 45) can provide an alternative to address some of the aforementioned drawbacks (37, 43, 46–49). For instance, gold nanoparticles have been used to deliver CRISPR-Cas9 RNP along with a repair template enabling homology directed repair (HDR) of CRISPR for the treatment of DMD (41). Lipid nanoparticles can also deliver the RNP for tissue-specific genome editing (50). However, while being effective in delivery, these nanoscale vehicles lack the image-tracking capability. Carbon nanomaterials-based agents can facilitate tracking of the delivery pathways through their intrinsic fluorescence (51–53). Because of the novelty of the field, direct studies involving carbon nanomaterials-assisted CRISPR-Cas9 RNP delivery for cancer therapeutics are yet to be performed (54). Among other nanocarbons, graphene quantum dots (GQDs) exhibit the highest biocompatibility and imaging capabilities suitable for in vitro and in vivo applications (55–57). Owing to their small size, high surface-to-volume ratio, fluorescence in the visible and near-infrared (NIR), as well as the aforementioned high biocompatibility (53, 55, 58), GQDs can serve as an advantageous theragnostic platform (56, 57, 59). They can be utilized as optical pH sensors, visible and NIR imaging agents, and targeted delivery vehicles for drug and gene therapy (55, 60–62). In contrast to other non-viral vectors, GQDs synthesized using the top-down and bottom-up approaches are highly scalable because of their ease of synthesis and widely available precursor materials (57, 63). Their low-cost production can make GQD-delivered therapeutics affordable to the general population. Thus, such a platform can effectively address the needs of CRISPR delivery and imaging for a variety of targets, including cancer, as explored in this work.
In order to ensure the efficient entry of the RNP into biological cells, GQDs in this study are synthesized from glucosamine and polyethylenimine (PEI). PEI is known to induce the proton sponge effect in the endosomes leading to endosomal rupture (64), which can enable the escape of the PEI-GQD-conjugated cargo into the cytoplasm (43, 64). Here, we for the first time synthesize GQDs using the PEI precursor, which is expected to generate cationic nanostructures facilitating the complexation and delivery of negatively charged genes. Using a bottom-up approach to synthesize GQDs from glucosamine and PEI is also expected to reduce the toxicity of PEI, while retaining some of its advantageous properties in the GQD form (45, 65). This is supported by the recent works of Liu et al. in which, similar to the GQDs, carbon dots, synthesized from PEI effectively condense DNA plasmids to deliver them to COS-7 and HepG2 cells (65). Graphene oxide/PEI hybrids conjugated with the RNP have also shown the ability to prevent its enzymatic hydrolysis (43). Thus, it is expected that similar in structure GQDs will also complex and protect the RNP cargo. While carbon-based nanomaterials have been previously used for CRISPR delivery (42–45), to the best of our knowledge, this work describes the first instance of CRISPR cancer therapy delivered by nanocarbons. The immediate goal of this nanotherapeutic approach is to correct the mutant TP53 gene in prostate cancer (PC-3) cells, ultimately, treating a common cancer mutation.
Results and Discussion
Design of the gene editing tool
In this work, we target and edit the TP53 gene at the 414delC null mutation (MUT TP53) in PC-3 cells using CRISPR RNP as the gene-editing tool and a single-stranded oligodeoxynucleotide (ssODN) as the repair template to correct the defective TP53 gene for cancer therapy. The programmable sgRNA bound to the RNP is coded to target the MUT TP53 gene and generate a double-stranded break (DSB) (Figure 1). The sequence of sgRNA is validated with the lowest off-target mismatch score using bioinformatics for minimal risk from off-target binding (66). The score is computed based on the likelihood of creating a stable sgRNA/DNA heteroduplex. This likelihood is lowered with decreasing nucleotide distance of the sgRNA matching region from the RNP’s proto-spacer motif (PAM) recognition sequence site (NGG), which is identified by the RNP to initiate DSB (67–69). The RNP is designed to contain a nuclear localization signal (NLS), short peptides recognized by nuclear transporters such as importins, at both C- and N-terminal ends of the Cas9 protein to promote its entry into the cell nucleus (70, 71). Typically, after a successful double-stranded break via CRISPR-Cas9, cells undergo repair via non-homologous end joining (NHEJ) and homology-directed repair (HDR). The latter mode of repair is preferred as it uses a sister chromatid as the repair template, while decreasing the likelihood of unwanted indels. Since PC-3 cells are not biallelic, they lack a proper copy of the TP53 gene in their chromosome (13). Thus, providing the cell with an exogenous DNA repair template permits for homology-directed repair to occur, converting the mutant gene into its healthy form. The ssODN is selected as a repair sequence instead of a double-stranded DNA to increase the rate of HDR (13, 72). The complete gene editing system developed in this work, therefore, comprises both ssODN and RNP delivered with the PEI-GQD platform.
Figure 1.

Schematic view of a mutant TP53 gene in PC-3 cells being targeted by CRISPR-Cas9 RNP using its sgRNA.
Development and characterization of PEI-GQD/RNP
In order to deliver both RNP and ssODN, the GQD platform was synthesized via a bottom-up approach with pre-selected precursor materials. This one-step microwave-assisted hydrothermal synthesis used glucosamine-HCl and PEI to generate a positively charged nanomaterial. The synthesis involves the dehydration of precursors leading to the polymerization, carbonization, and ultimate formation of the GQDs (63). Among these precursor materials, glucosamine is known to serve as the source for the carbon backbone (63), while PEI is expected to contribute to most of the nitrogen doping. Since GQDs synthesized from solely carbon sources do not have a high net positive charge on their surface (60), PEI was selected to provide positively charged groups that would aid in electrostatic binding to the negatively charged RNP (50). Although 25 kDa PEI by itself is considered the gold standard for transfection of nucleic acids for gene therapy (64), there are still toxicity-related issues observed with its administration in vitro (65). To circumvent this drawback, Hashemzadeh et al. have synthesized carbon dots from lower molecular weight PEI and citric acid aiming to utilize those for the delivery of CRISPR plasmids (45). Similarly, carbon dots produced from 1.8 kDa PEI show biocompatibility up to 160 µg/mL rendering over 80% viability in HeK293 cells. While being substantial compared to that of inorganic nanoparticles, such level of biocompatibility is still not sufficient for gene delivery and imaging in vivo (57). Since our previous studies indicate that GQDs synthesized from glucosamine are biocompatible at over 1 mg/mL in HeK293 cells (55), glucosamine additive is expected to improve the biocompatibility of PEI-GQD platform and retain some of GQDs’ original optical properties which can be utilized for image-tracking (63).
The fluorescence properties of PEI-GQD platform can be affected by synthetic conditions: longer reaction times result in absorption/color changes, red shifts, and variation in fluorescence emission intensities (Figure S1). It is expected that due to electronic confinement-based nature of GQD fluorescence larger GQD structures having electrons confined to larger graphitic regions would exhibit longer wavelength fluorescence. Excitation-emission maps confirm the evolution of optical properties during GQD synthesis (Figure S2), showing that at 1 hr synthetic time at a power of 270 W is optimal for producing highly fluorescing PEI-GQD with an emission peak at 530 nm arising from 400 nm excitation (Figure 2a). PEI-GQDs also exhibit NIR fluorescence emission with a peak at 900 nm excited also in the NIR at 808 nm and attributed to surface defect states (63) (Figure 2b). Unlike their visible fluorescence, NIR excitation and emission wavelengths of PEI-GQDs fall within the biological transparency window, in which tissue absorption, scattering, and autofluorescence are minimized (73). This extends their bioimaging modality into ex vivo and in vivo applications.
Figure 2.

(a) Visible fluorescence spectra of PEI-GQD (black), PEI-GQD/RNP (red), and RNP (blue). (b) NIR emission of PEI-GQD with 808 nm laser excitation. (c) Absorbance spectra of PEI-GQDs (black), PEI-GQD/RNP (red), and RNP (blue). (d) FTIR spectrum of PEI-GQDs.
By simply mixing the PEI-GQDs and RNP aqueous suspensions, we expect the formation of the PEI-GQD/RNP through the electrostatic complexation with the positively charged PEI-GQDs and negatively charged RNP. The quenching of the visible GQD fluorescence upon mixing with the RNP suspension (Figure 2a) can be explained by charge transfer from/to the RNP (74), suggesting its successful complexation with the delivery platform. Functional groups enabling the complexation of PEI-GQD and RNP as well as ensuring high water solubility of the platform are assessed via UV-Vis and Fourier transform infrared (FTIR) spectrophotometry. UV-Vis absorption shows a major peak at 220 nm with a shoulder at 310 nm representing a blue-shifted transition at the bond and an transition at the and bonds, respectively (Figure 2c). The shift in the absorption spectra for can originate from the interaction of the functional groups with GQD sp2 platform. These measurements suggest the presence of oxygen and positively charged nitrogen addends, which is further explored with FTIR. After complexation with the PEI-GQD, RNP having an absorbance shoulder at 280 nm contributes to the spectrum but does not alter the spectral position of the main absorption features of PEI-GQD, suggesting that the complexation is non-covalent.
FTIR further helps to explore the presence of chemical functional groups. In the functional group region, several features represent stretching vibrations of bond (3340 , bond (2925 ) and (1620 ) bonds as well as bond (2846 ), and bond (2000 – 1650 ) bending (Figure 2d). Furthermore, in the fingerprint region, peaks representing the bending vibrations of (1458 ) and stretching vibrations of (1342 ), and (1033 ) are observed. While oxygen functional groups enhance the solubility of PEI-GQD platform, amine groups from PEI can also aid in PEI-GQD complexation with the RNP.
Zeta potential measurements reveal that PEI-GQDs have a net positive charge with a Zeta potential value of 21.4 ± 1.2 mV. Given neutral to negative zeta potentials from solely glucosamine-derived GQDs (60), this net positive value also suggests the abundance of amine groups and the potential for electrostatic RNP attachment (Figure S3). Atomic elemental composition of PEI-GQDs assessed at the nanoscale via EDS shows roughly 81.6% carbon, 10.4% nitrogen, and 4.3% oxygen (Figure S4). PEI-GQDs appear to have a substantially greater nitrogen content than their counterparts created just form glucosamine with only 4.6% nitrogen (60). High-resolution TEM (HRTEM) measurements reveal that PEI-GQDs (Figure 3) have an average size of 5 nm and present lattice fringes with the spacing of 0.21 nm comparable to that of (100) plane of graphene. At the same time, Fast Fourier Transform (FFT) of TEM images also provide the evidence of crystallinity (Inset of Figure 3a). PEI-GQD/RNP complexes form larger nanostructures on the 50 nm scale (Figure 3b). Given the size of the individual RNP of 10 nm (50), these measurements also suggest the complexation of PEI-GQDs with the RNP.
Figure 3.

(a) HRTEM image of PEI-GQD. Inset: FFT image of the selected area. (b) TEM image of PEI-GQD/RNP complex.
The loading capability of the RNP onto PEI-GQDs is further assessed with the gel retardation assay involving varying molar ratios of PEI-GQDs and RNP (Figure S5). At a ratio of 25 pmol of RNP with a PEI-GQD molar excess of 30 pmol, a minimal migration of the PEI-GQD/RNP complex is observed, suggesting a neutral and larger complex in which most PEI-GQDs are bound to the RNP. This result also indicates that more than one PEI-GQD platform can bind to a single RNP. In order to assess whether the cleaving capability of the RNP has been hampered by the complexation with PEI-GQDs, the PEI-GQD/RNP complex is tested for the scission of the MUT TP53 gene amplicon in solution.
Gene editing in solution
The efficacy of the double-stranded breaks by PEI-GQD/RNP on an 1100 bp MUT TP53 gene amplicon and RNP is considered as a critical measure of the therapeutic function retained by the RNP after complexation (43). In order to evaluate that gel electrophoresis performed on MUT TP53 amplicon treated with either RNP, PEI-GQD, or PEI-GQD/RNP at an electrophoretically assessed optimal ratio (Figure 4a). The successful double-stranded break facilitated by the RNP is expected to produce 700 and 400 bp DNA strands from the 1100 bp amplicon as the sgRNA was programmed to target at the 700 bp site. This cleavage site was chosen to ensure enough separation between the bands within the physical constraints of the gel. RNP, as a positive control, exhibited successful cleavage of the MUT TP53 amplicon into separate 700 and above 400 bp bands (Figure 4a). The PEI-GQD/RNP lane also presented the same bands with negligible decrease in intensity attributed to minor steric hindrance from the PEI-GQDs surrounding the RNP. PEI-GQDs alone did not generate distinct bands corresponding to the cleaved MUT TP53 gene amplicon, while smeared bands in the gel was attributed to PEI-GQDs migration only. To assess whether double-stranded breaks from PEI-GQD/RNP can be translated in vitro at the target site, we performed genome editing with PC-3 cells possessing the MUT TP53 gene.
Figure 4.

(a) Cleavage of the mutant TP53 gene amplicon (1100 bp, 160 and 180 ng) with PEI-GQD/RNP (Cas9 (5 pmol) and sgRNA (12 pmol)) complex along with PEI-GQD and RNP alone in the adjacent the gel lanes. The rightmost lane represents intact DNA control. (b) In vitro mismatch cleavage assay of the TP53 gene amplicon (400 bp) performed with the addition of T7E1 restriction enzyme. PC-3 cells were transfected with PEI-GQD/RNP (in 1reaction buffer, 23X reaction buffer, and 33X opti-mem), and Lipofectamine/RNP (4with 0.5 μl of 500 mM of EDTA).
Gene editing in vitro
In vitro CRISPR-Cas9 double-stranded break occurs at the S and G2 phases of the cell cycle, and since PC-3 cells have a doubling time of 33 hr, extraction of edited genomic DNA (gDNA) was considered optimal at 48 hr post-transfection. This time span ensured the cleavage and repair of the targeted DNA sequence via non-homologous end joining, which generally leads to indel formation. The region of the MUT TP53 gene was PCR-amplified to generate a 408 bp amplicon and T7 Endonuclease I (T7E1) assay was employed to check whether a double-stranded break has been achieved at the indel-formed site. The T7 restriction enzyme from this assay recognized and cleaved the heteroduplex DNA at the first, second, or third phosphodiester bond from the 5’ site of the mismatch. These cleaved DNA strands visualized via gel electrophoresis appeared as 236 and 172 bp bands along with the original 408 bp band for PEI-GQD/RNP (Figure 4b). The use of different transfection reagents (reaction buffer and opti-mem) did not have a significant effect on the results of this experiment. Lipofectamine/RNP used as a positive control also shows these cleaved bands, validating the success of generating double-stranded break by PEI-GQD/RNP in vitro. These results demonstrate that PEI-GQD deliver the RNP in cancer cells just like conventional delivery vehicles such as Lipofectamine, while preserving its cleavage efficacy.
Editing the mutant TP53 gene into its wild-type form after the scision requires the additional delivery of the repair template, ssODN, which performs recombination of the cut gene via a homology-directed repair. Exploiting the endogenous cellular pathways suggests that such repair of the TP53 gene is expected to promote a cascade of cellular repair events mediated via caspases, leading to apoptosis and, finally, cancer cell death. To ensure the safe delivery of the ssODN into the cell nucleus, the ssODN is loaded onto the PEI-GQD/RNP via a combination of electrostatic interaction with the positively charged PEI-GQDs and π-π stacking on GQD graphitic surface (60). The gel retardation assay is again used to verify the complexation of ssODN to PEI-GQDs/RNP and to ensure maximum ssODN loading. Among various molar ratios of RNP and ssODN bound to the PEI-GQDs (Figure S6), a 1:1 molar ratio produced a band of least mobility in the electrophoretic gel chamber. Such minimal electrophoretic migration arising from neutralized charge and increased size suggests the maximum electrostatic ssODN loading onto PEI-GQD/RNP complex. In order to assess whether cell death can be achieved by editing the MUT TP53 gene into its healthy wild-type form, PC-3 cell viabilities have been measured via the MTT assay after transfection with PEI-GQD/RNP+ssODN complex.
Since HDR occurs during the S and G2 phases of the cell cycle, the MTT cell viability assay was performed 72 hr of post-transfection. During this time, MTT was reduced by NAD(P)H-dependent cellular oxidoreductase enzymes from metabolically active cells, producing formazan. Absorption of formazan served as an indicator of the number of live cells. These results were further verified via the luminescence assay on healthy HeK293 cells, quantifying the amount of ATP produced from viable cells. PEI-GQDs tested first demonstrate over 80% HeK293 cell viability at concentrations of up to 1.1 mg/mL (Figure 5a), indicating the high biocompatibility of our delivery platform. Moreover, the concentration used for the transfection of PC-3 cells (0.004 mg/mL) produced no toxic response, allowing to assess the gene therapeutic effect without the interference from the delivery vehicle’s cytotoxicity. Cell viabilities above 100% could be in part explained by the degradation and metabolization of the PEI-GQD platform by the cells observed for glucosamine GQDs previously (55). This result also suggests an advantage of the biodegradability of our vehicle. In order to evaluate the cytotoxic effect of the full formulation on healthy cells, MTT assay is performed in HEK 293 cells with the administration of PEI-GQD/RNP+ssODN and Lipofectamine/RNP+ssODN used as a positive control (Figure 5b). Since HeK293 cells already have a wild-type version of the TP53 gene, negligible cell death was observed from their treatment with PEI-GQD/RNP+ssODN and Lipofectamine/RNP+ssODN. However, PC-3 cancer cells with the mutant TP53 gene exhibited drastic cell death with 62% and 15% viable cells left after the transfection with PEI-GQD/RNP+ssODN and Lipofectamine/RNP+ssODN, respectively (Figure 5b). Staining dead cells with trypan blue (Figure 5c, S7) confirms substantial cancer therapeutic effect of the formulation as compared to non-treatment control. Focusing this therapeutic modality to cancer cells only is a critical advantage of this CRISPR-based gene therapy. It is also important that such drastic anticancer effect was not observed without the administration of ssODN, signifying a successful gene repair. To verify that cell death occurred due to cell apoptosis arising from the editing of the MUT TP53 gene into its healthy wild-type form, an apoptotic marker caspase-3/7 assay was employed. This method evaluates caspase-3/7 enzymatic activity, correlated to apoptosis signaling and subsequently leading to the degradation of cellular components. Remarkably, PEI-GQD/RNP+ssODN-treated cells appear to have the same amount of apoptotic cells as those treated with staurosporine, a protein kinase inhibitor known to induce apoptosis (75) (Figure S8). This validates that the administration of PEI-GQD/RNP+ssODN joint formulation leads to apoptosis in PC-3 cancer cells, while having no effect on the non-cancerous ones. Apoptotic cell death after the repair of the cancer suicide TP53 gene is predicted: because of the multiple defects accumulated within the cancer PC-3 cell genome, chromosomal abnormalities and DNA mutations exist. Typically, the p53 protein from a wild-type TP53 gene is activated to induce cell cycle arrest and DNA repair. However, as the DNA damage in PC-3 cells is beyond repair, apoptosis is triggered (75, 76). Although Lipofectamine shows a greater effect than PEI-GQD, the use of PEI-GQD provides the possibility of tailoring the delivery platform structure to improve the therapeutic effect as well as the advantage of using its multifunctionality, where this platform can be also targeted and employed for cancer sensing and imaging (55, 60, 61).
Figure 5.

(a) HeK293 cell viability with the administration of PEI-GQD delivery vehicle (mean ± SE, n = 3). (b) PC-3 and HeK293 cell viability after the treatment with PEI-GQD/RNP+ssODN and Lipofectamine/RNP+ssODN therapeutic formulations (mean ± SE, n = 3, P-values are calculated using one-way ANOVA followed by Tukey post-hoc test, *P<0.05, **P<0.01, ***P<0.001), ****P<0.0001). (c) Trypan blue-stained dead PC-3 cells (encircled).
In vitro imaging
The capability of PEI-GQDs for bioimaging was assessed by tracing the delivery of PEI-GQD/RNP in vitro via confocal fluorescence microscopy in PC-3 cells 24 hr post-transfection. The 24 hr time point was chosen as it ensures maximum internalization of the glucosamine-derived GQDs (55). The RNP was tagged with NLS and EGFP molecules at its N- and C-terminal ends, respectively, to facilitate its entry into the cell nucleus and track its location within the cell independently of the GQDs. This approach aids in determining the stage in which the payload, RNP, separates from PEI-GQD delivery platform. The imaging parameters were offset to render zero autofluorescence while keeping the fluorescence emission of the internalized PEI-GQDs and RNP+EGFP spectrally separated by using 540/605 and 480/535 nm (excitation/emission) filters, respectively.
Confocal image overlays of these channels (Figure 6) indicate that PEI-GQDs fluorescing in red mostly reside in the cytoplasm, while some colocalize with RNP-EGFPs fluorescing in green (overlap shown in yellow). Unlike that of the GQDs, RNP-EGFP emission is also present in the cell nucleus. This suggests the detachment of the therapeutic payload from the nanoplatform, which could occur during the endosomal escape of the complex that may be aided by the PEI’s proton sponge effect (43) and/or partial degradation of the GQD platform within the cells (55). The successful translocation of RNP+EGFP cargo into the cell nucleus further indicates the success of CRISPR-Cas9 RNP delivery for gene editing, opening the avenue for exploration of targeted approaches expected to enhance tumor accumulation and therapeutic efficacy (77–79).
Figure 6.

Confocal imaging of PC-3 cells treated with PEI-GQD/RNP-EGFP formulation: (a) brightfield, (b) fluorescence, and (c) brightfield/fluorescence overlay images. PEI-GQD emission is shown in red and RNP-EGFP emission shown in green.
In this work, for the first time PEI-based graphene quantum dots (PEI-GQDs) were successfully utilized as a non-viral delivery platform and an imaging agent for CRISPR-Cas9 gene therapy correcting the mutated TP53 gene in prostate cancer PC-3 cells. This novel multifunctional platform has substantial advantages over conventional delivery agents, as it enables a variety of GQD-driven potential applications including in vivo NIR imaging, receptor-based cancer targeting, and drug/gene combination therapy. PEI-GQDs demonstrate substantial biocompatibility (at concentrations above 1 mg/mL) superior to many other nanomaterial delivery vehicles. These GQDs were synthesized in the present work using a bottom-up approach via a cost-effective and scalable one-step microwave hydrothermal reaction from glucosamine and PEI precursors. Having a Zeta potential of 21.4 ± 1.2 mV PEI-GQDs were electrostatically bound to the negatively charged CRISPR-Cas9 RNP and ssODN repair template gene editing therapeutic cargo. Gel retardation assay indicates that one or several GQDs at a molar ratio of 1.2:1 (PEI-GQD:RNP) are bound to the RNP in this complex. In solution PEI-GQD/RNP formulation demonstrates active cleavage activity of 1100 bp TP53 amplicon comparable to that of the RNP alone, verifying that the complexation does not inhibit the cleaving capabilities of the RNP. Similar cleavage of the mutant TP53 gene was achieved by the PEI-GQD/RNP in prostate cancer (PC-3) cells. In order to repair the defective TP53 gene for cancer therapy, ssODN donor repair template was further complexed with PEI-GQD/RNP to promote homology directed repair of the mutant gene to its wild-type form. The transfection of PC-3 cells with complete treatment formulation, PEI-GQD/RNP+ssODN, drastically reduced their viability down to 60%, while having negligible effects on healthy HeK293 cells. With the repaired TP53 gene, the caspase cascade pathway is triggered due to the inherent accumulation of DNA mutations in the PC-3 cell genome, leading to cancer cell apoptosis as validated by the caspase 3/7 enzyme activity assay. This critical finding confirms the success of cancer therapeutic outcome of the proposed formulation and suggests that the treatment is cancer-specific with no detrimental effects to non-cancerous cells. Confocal fluorescence microscopy was used to validate the internalization of PEI-GQD/RNP into PC-3 cells showing that RNP+EGFP translocated within the nucleus while PEI-GQDs were mainly distributed in the cytoplasm. This assessment not only elucidates the delivery stages and the destination of the therapeutic payload, but also highlights the potential of the nanoplatform’s image-tracking capabilities. Successful GQD delivery of CRISPR-Cas9 as a gene editing cancer therapeutic described in this work provides a promising avenue for its use in vivo. Overall, given the versatility of this nanoplatform from its modifiable synthetic and imaging modalities, biocompatible GQDs retrofitted with a repertoire of gene editing tools and combination therapies can propel gene therapeutics to the new era of personalized disease-specific medicine.
Experimental Section
CRISPR-Cas9 RNP, sgRNA, and ssODN design
In the design of CRISPR-Cas9 system, Cas9 ribonucleoprotein (RNP) was modified to contain nuclear location signals (NLS) at both its C- and N-terminal ends. This NLS-Cas9-NLS nuclease (Z03469, GenScript, NJ, USA) facilitates the entry of RNP into the cell nucleus. “CCTop-CRISPR/Cas9 target online predictor” and the “RNA-guided endonuclease (RGEN) Tools: Cas-OFF Finder&Cas-Designer” programs were utilized to design the single-guide RNA (sgRNA) (13). The following selected sgRNA sequence 5-ACA GGG CAG GTC TTG CCA GT-3’ was obtained from GenScript, NJ, USA, and bound to the RNP to target the TP53 414delC region.
The cleaved site was repaired via a homology-directed repair mechanism utilizing the HPLC-purified single-stranded oligonucleotide (ssODN, GenScript, NJ, USA) as the repair template with the following sequence: 5′-G*G*G*TGT GGA ATC AAC CCA CAG CTG CAC AGG GCA GGT CTT GGCCAG CTGG CAA AAC ATC TTG TTG AGG GCA GGG GAG TA*C*T*−3’. This ssODN molecule was designed to match the sequence around the cut site with long homology arms promoting high editing efficiency and reducing off-target integration.
Synthesis of PEI-GQDs
4 g of glucosamine HCl (346299, Sigma Aldrich, MO, USA) and 1.5 mL of 2 kDa polyethylenimine (PEI, 408700–250ML, Sigma Aldrich, MO, USA) were mixed with 250 mL of DI water. The mixture underwent a one-step hydrothermal reaction in a commercially available microwave (HB-P90D23AP-ST, Hamilton Beach, NC, USA) at 270 W for 1 hr. The resulting aqueous solution was purified using a 0.5 – 1 kDa dialysis bag (Spectrum Chemical Mfg. Corp., NJ, USA) at a pH of 13.7 to remove Cl- ions bound to the deprotonated amine groups. To remove excess PEI, the sample was passed through a 3 kDa centrifugal filter (Amicon Ultra, UFC500324, Merck Millipore Ltd., Co Cork, Ireland).
Characterization of PEI-GQD and PEI-GQD/RNP
The UV-Vis absorption spectra of all samples were measured in the range of 200 to 1100 nm using Cary 60 spectrophotometer (Agilent Technologies, CA, USA). Visible fluorescence spectra were measured using NanoLog spectrofluorometer (HORIBA Scientific, NJ, USA), while NIR fluorescence spectra were assessed via AvaSpec-HS-TEC spectrofluorometer (Avantes, Apeldoorn, The Netherlands) with a 0.8 W/cm2 808 nm CW laser excitation (808MD-12V-BL, Q-BAIHE, China). Fourier transform infrared spectroscopy (FTIR, Thermo Nicolet Nexus 670, WI, USA) in attenuated total reflection (ATR) mode was utilized to assess the functional groups present in freeze-dried (VirTis Freezemobile, 25ES, SP Scientific, PA, USA) PEI-GQD samples. Zeta potential measurements were performed using a ZetaPALS instrument (Brookhaven Instrument Corporation, NanoBrook, NY, USA). To assess nanoscale structure and composition of the PEI-GQDs and PEI-GQD/RNP, their suspensions were air-dried on a carbon-coated 200 mesh copper grid (Electron Microscopy Sciences, CF300-CU, PA, USA) and imaged using transmission electron microscopy (TEM, JEOL JEM-2100, MA, USA) with consecutive energy-dispersive X-ray analysis (EDS, JEOL, MA, USA).
Gel retardation assay
Gel electrophoresis (EL-100-K4, Walter Products Inc., Ontario, Canada) was performed at 120 V for 1.5 hr using an Enduro power supply (Labnet International, Inc., NJ, USA) in 1X TBE buffer (B52, Thermo Fisher Scientific, MA, USA). 20 µL aliquots of the samples with 2.2 µL of 10X loading buffer solution (Bluejuice, 10816015, Thermo Fisher Scientific, MA, USA) were added into the wells with 1.75% agarose gel (UltraPure Agarose, 16500100, Thermo Fisher Scientific, MA, USA) prepared with 1X TBE buffer. After the electrophoresis experiment the gel was stained on an orbital shaker with 0.5 µg/mL EtBr solution for 20 min and further de-stained in 1X TBE buffer for 5 min.
In solution DNA cleavage assay
CRISPR GenScript protocol was followed for the assembly of CRISPR-Cas9 RNP. First, 5 pmol of sgRNA (0.24 µL of 25 µM) was added to 13.56 µL of DEPC-treated water (AM9915G, Invitrogen, CA, USA) and mixed with 2 µL of 10X nuclease reaction buffer. Next, 0.2 μL of NLS-Cas9-NLS nuclease solution (4 mg/mL) were added into the mixture and incubated at 37 °C for 10 min to form the RNP. 8 µg of PEI-GQDs were added to the mixture and incubated at room temperature for 10 min to generate the PEI-GQD/RNP complex. 80 and 160 ng of the MUT TP53 gene amplicon were introduced into the RNP solutions and incubated at 37 °C for 1 hr to initiate the cleavage. The results of the reaction were evaluated using agarose gel electrophoresis with 1.75% agarose in 1X TBE buffer. 5 µL of a 1:1 mixture of 10 % SDS and 10X loading buffer solution were added to the reaction mixture and incubated at 65 °C for 10 min. After cooling on ice for 5 min, the samples were loaded onto an agarose gel and run in the electrophoretic chamber at 120 V for 1.5 hr. Finally, the gel was stained with EtBr and visualized using a gel imager (FastGene FAS-V, NIPPON Genetics EUROPE, Düren, Germany).
Cell Culture
PC-3 cells (CRL-1435, ATCC, VA, USA) were cultured at 37 °C and 5% CO2 (Midi CO2, Thermo Fisher Scientific, MA, USA) in complete medium containing F-12K nutrient mixture (21127–022, Gibco, Dublin, Ireland). Basal medium was supplemented with 10% FBS (16140–063, Gibco, Dublin, Ireland), 5% L-glutamine (G7513, Sigma-Aldrich, MO, USA), 5% non-essential amino acid solution (M7145, Sigma-Aldrich, MO, USA), and 1% penicillin/streptomycin (P4333, Sigma-Aldrich, MO, USA). HeK293 cells (CRL-1573, ATCC, VA, USA) were cultured using the same incubation parameters and reagents while only utilizing DMEM (D6046, Sigma-Aldrich, MO, USA) as the basal medium.
Genomic DNA extraction
For genomic DNA (gDNA) extraction, the Promega Wizard DNA Extraction Kit (A1120, Promega, WI, USA) was used. At 48 hr post-transfection, the complete medium was first removed from each well. Cells were detached from the well plate by adding 100 µL of trypsin-EDTA to each well and incubating the well plate at 37 °C and 5% CO2 for 2 min. Trypsinized cells were transferred into microcentrifuge tubes and centrifuged (14,000 × g for 10 sec) to pellet the cells. The supernatant was removed and 200 µL of 1X phosphate-buffered saline (PBS, Bioland Scientific LLC, MD, USA) was added to each microcentrifuge tube, and vortexed (20 sec) to wash off and resuspend the cells. The tubes were centrifuged (14,000 × g for 10 sec) again to pellet the cells after which the supernatant was removed. In order to extract the genomic contents of the cells, 600 µL of nuclei lysis solution was added to the microcentrifuge tubes to disrupt the cell membrane. During this process the sample was mixed until no visible cell clumps remained. 3 µL of RNase solution was then added, mixed by inverting the tubes 2 – 5 times, and incubated at 37 °C for 15 – 30 min in a water bath for the enzymatic degradation of RNA contents.
In order to isolate gDNA from the protein contents, the samples were mixed with 200 µL of protein precipitation solution. The mixture was then vortexed (20 sec) and cooled on ice (5 min). The samples were further centrifuged at 16,000 × g for 4 min to pellet the protein content. The DNA-containing supernatant was transferred into microcentrifuge tubes with 600 µL of isopropyl alcohol. Isopropanol in this procedure assisted in removing the hydration shell originating from DNA’s phosphate groups. The solution was mixed by inversion until white, thread-like DNA strands were observed. To remove isopropanol, the contents were again centrifuged (at 16,000 × g for 1 min) and the supernatant was removed. To wash and pellet the DNA, the samples were further mixed with 600 µL of ethanol and centrifuged (16,000 × g for 1 min). Ethanol was carefully aspirated and the DNA contents were air-dried for 10 – 15 min. 15 µL of the DNA rehydration solution was added and incubated overnight at 4 °C to rehydrate the DNA. DNA concentrations were measured using a Nanodrop One spectrophotometer (ND-ONE-W, Thermo Fisher Scientific, MA, USA).
PCR Amplification
PCR amplification was performed to generate 409 bp TP53 gene amplicons from the extracted gDNA of PC-3 cells. 100 ng of gDNA was mixed with 25 µL of 2X Phusion Flash PCR Master Mix (F548S, Thermo Fisher Scientific, MA, USA) containing the DNA polymerase and deoxynucleoside triphosphates (dNTPs). To define the start and the end of the DNA segment amplification, 2.5 µL of 10 µM forward (5’ ACG CCA ACT CTC TCT AGC TC 3’, Thermo Fisher Scientific, MA, USA) and reverse (5’ GCC AGA CCT AAG AGC AAT CAG 3’, Thermo Fisher Scientific, MA, USA) primers were added to the reaction. Binding and amplification of the 409 bp TP53 gene sequence occurred in between the following yellow-color-coded nucleotide segments:
ACGCCAACTCTCTCTAGCTCGCTAGTGGGTTGCAGGAGGTGCTTACGCATGTTTGTTTCTTTGCTGCCGTCTTCCAGTTGCTTTATCTGTTCACTTGTGCCCTGACTTTCAACTCTGTCTCCTTCCTCTTCCTACAGTACTCCCCTGCCCTCAACAAGATGTTTTGCCAACTGGCCAAGACCTGCCCTGTGCAGCTGTGGGTTGATTCCACACCCCCGCCCGGCACCCGCGTCCGCGCCATGGCCATCTACAAGCAGTCACAGCACATGACGGAGGTTGTGAGGCGCTGCCCCCACCATGAGCGCTGCTCAGATAGCGATGGTGAGCAGCTGGGGCTGGAGAGACGACAGGGCTGGTTGCCCAGGGTCCCCAGGCCTCTGATTCCTCACTGATTGCTCTTAGGTCTGGC
PCR amplification reaction was performed using the thermocycling profile shown in Table 1. In short, the thermal cycler (2720, Applied Biosystems, Waltham, MA, USA) was set at 98 °C for 10 sec for the initial denaturation of gDNA. This de-hybridized product underwent 30 cycles of denaturation (98 °C for 1 sec), annealing (63.5 °C for 5 sec), and elongation (72 °C for 10 sec), resulting in 230 targeted DNA region copies. After the final extension at 72 °C for 60 sec, the PCR product was cooled and held at 4 °C.
Table 1:
Thermocycling profile for PCR amplification
| Step | Temperature (°C) | Time (sec) | Cycle |
|---|---|---|---|
| Initial Denaturation | 98.0 | 10 | 1× |
| Denaturation | 98.0 | 1 | 30× |
| Annealing | 63.5 | 5 | |
| Elongation | 72.0 | 10 | |
| Final Extension | 72.0 | 60 | 1× |
| Hold | 4.0 | ∞ | 1× |
In Vitro Mismatch Cleavage Assay
For the in vitro cleavage mismatch assay, 1.8 × 105 cells/well were plated onto a 6-well plate 24 hr prior to transfection. The cells were transfected with PEI-GQD and Lipofectamine (L3000–001, Invitrogen, CA, USA) complexes containing RNP or both RNP and ssODN. Transfected cells were incubated at 37 °C with 5 % CO2 for 48 hr. The PCR-amplified products were placed in a thermal cycler (A48141, Thermo Fisher Scientific, Waltham, MA, USA) with the thermocycling profile depicted in Table 2. In brief, the temperature in the thermal cycler was set at 95 °C for 5 min for the initial denaturation of the PCR products containing the homoduplex DNA. To generate heteroduplex DNA for the T7E1 assay, the products were annealed between 95 and 85 °C at a ramp rate of −2 °C/s and cooled between 85 and 20 °C at a rate of −0.1 °C/s to be further held at 4 °C. The resulting sample was incubated with 1 µL of T7 Endonuclease I (T7E1, Z03396, GenScript, NJ, USA) at 37 °C for 15 min. Since the 408 bp amplicon underwent re-denaturation and re-annealing, heteroduplex and homoduplex forms of DNA were present from the hybridization of edited (indel-formed) and un-edited single strands of DNA. The T7E1 endonuclease enzyme in this reaction recognizes and cleaves heteroduplex DNA 5’ at its first to third phosphodiester bond from the mismatch. 2.2 µL of 10X loading buffer was added to stop the reaction after which the sample was ran in the agarose gel electrophoresis chamber at 120 V for 1 hr. The gel was further stained with EtBr and visualized using a gel imager (FastGene FAS-V, NIPPON Genetics EUROPE, Düren, Germany).
Table 2:
Thermocycling profile for in vitro mismatch cleavage assay
| Thermocycling profile | ||
|---|---|---|
| Step | Temperature (°C) | Time (sec) |
| Initial Denaturation | 95 | 300 |
| Annealing | 95 – 85 | −2°C/s |
| 20 | −0.1°C/s | |
| Hold | 4 | ∞ |
Cell viability assays
The biocompatibility of PEI-GQDs for genome editing applications was evaluated in HEK293 cells via the luminescence-based assay (Cell-Titer Glo, G7572, Promega, WI, USA). Furthermore, the effects of PEI-GQD/RNP+ssODN and Lipofectamine/RNP+ssODN formulations were evaluated via the 3-(4–dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) assay (M6494, Thermo Fisher Scientific, MA, USA) in HeK293 and PC-3 cells, where trypan blue solution, 0.4% (15250061, Thermo Fisher Scientific, MA, USA) was used to stain the cells. Caspase-Glo 3/7 assay (G8090, Promega, WI, USA) was also performed to confirm the successful repair of the TP53 gene and that cell death occurred via apoptosis.
For the luminescence assay, 5,000 cells in 200 µL were seeded into each well of a 96-well plate (701001, Nest Scientific, Wuxi, China) and incubated at 37 °C with 5 % CO2. After 24 hr of incubation, two-fold serial dilutions of PEI-GQD in DMEM were added (starting from 2.2 mg/mL) to different wells and incubated for another 24 hr period. The medium was then replaced with 100 µL of Cell-Titer Glo solution and the luminescence was measured in each well using a microplate reader (FLUOstar OMEGA. BMG LABTECH, NC, USA) after shaking the well plate on an orbital shaker for 5 min at 100 rpm.
For the MTT assay, 10,000 cells in 500 µL were seeded in a 6-well plate with 1.5 mL of complete medium in each well and transfected with PEI-GQD/RNP+ssODN or Lipofectamine/RNP+ssODN after 24 hr of incubation at 37 °C with 5 % CO2. The complete medium was replaced after 72 hr of transfection with 1 mL of MTT (1 mg/mL) and the well plate was incubated again for 4 hr. After removing the MTT solution, 100 µL of DMSO were added and the well plate was placed on an orbital shaker for 5 min at 100 rpm to solubilize the formazan generated by viable cells. Absorbance in the wells was measured using a microplate reader (μQuant, BioTek Instruments, VT, USA) at 570 nm to quantify the amount of formazan.
To evaluate the amount of caspase-3 and −7 enzymes involved in the apoptotic pathway of programmed cell death, 5,000 cells per 200 µL were seeded into each well of a 96-well plate and incubated for 24 hr at 37 °C with 5 % CO2. 100 µL of Caspase-Glo 3/7 solution (G8090, Promega, WI, USA) was added after removing the medium from the treated cells in the well plate 72 hr post-transfection. The well plate was further placed on an orbital shaker at 300 – 500 rpm for 30 sec, incubated at room temperature for 1 hr, and the luminescence from the ATP-dependent luciferase reaction was measured using the FLUOstar OMEGA microplate reader.
Confocal microscopy imaging
A semi-motorized inverted microscope (IX73P2F, Olympus, PA, USA) equipped with an IR-corrected UPLANAPO 60x/0.90na objective (1-UB831, Olympus, Center Valley, PA, USA) was utilized for in vitro confocal fluorescence imaging. Visible fluorescence was collected with a CMOS camera (Prime 95B, Photometrics, AZ, USA) coupled to a confocal disk-spinning unit (DSU, Olympus, PA, USA) with 480 ± 20/535 ± 20 nm and 540 ± 20/605 ± 20 nm (excitation/emission) filters.
To perform cell imaging experiments, 10,000 cells were seeded onto coverslips placed in a 6-well plate. After 24 hr of incubation at 37 °C with 5 % CO2, the cells were transfected with PEI-GQD/RNP and incubated again for 12 hr. The medium from each well was then removed and the cells were washed with 1X PBS. Working as a fixative, 4% formaldehyde (28908, Thermo Fisher Scientific, MA, USA) in 1X PBS was added to each well and left at 4 °C for 30 min. After removing the formaldehyde solution, 1X PBS was added to preserve the cells on the coverslip. Each cover slip received a drop of 1X Fluoromount-G mounting media (00–4958-02, Invitrogen, CA, USA) and was sealed onto a microscope slide with nail polish for imaging. RNP fusion protein with an EGFP protein dye at its C-terminal end (NLS-Cas9-EGFP Nuclease, Z03393, GenScript, NJ, USA) was utilized to visualize and track the delivery pathways of the RNP separately from the GQDs.
Statistical analysis
Statistical analyses were performed by one-way analysis of variance (ANOVA) followed by Tukey post-hoc test using OriginPro (2022b, OriginLab Corp.). A P-value < 0.05 (*) was considered statistically significant.
Supplementary Material
Acknowledgements
Figure 1 is generated using BioRender. The authors would like to thank NIH NIBIB (# 1 R15 EB031528-01) for the grant funding.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Collins FS, Morgan M, Patrinos A, The Human Genome Project: lessons from large-scale biology. Science 300, 286–290 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Olsen UDJGIHTBEPPRPWSSTDNCJ-F et al. , Initial sequencing and analysis of the human genome. nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Collins FS, Medical and societal consequences of the Human Genome Project. New England Journal of Medicine 341, 28–37 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Collins FS, McKusick VA, Implications of the Human Genome Project for medical science. Jama 285, 540–544 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Futreal PA et al. , A census of human cancer genes. Nature reviews cancer 4, 177–183 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian X et al. , CRISPR/Cas9–An evolving biological tool kit for cancer biology and oncology. NPJ precision oncology 3, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olivier M, Hollstein M, Hainaut P, TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor perspectives in biology 2, a001008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teodoro JG, Evans SK, Green MR, Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. Journal of molecular medicine 85, 1175–1186 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Hollstein M, Sidransky D, Vogelstein B, Harris CC, p53 mutations in human cancers. Science 253, 49–53 (1991). [DOI] [PubMed] [Google Scholar]
- 10.Greenblatt M, Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54, 4855–4878 (1994). [PubMed] [Google Scholar]
- 11.Williams AB, Schumacher B, p53 in the DNA-damage-repair process. Cold Spring Harbor perspectives in medicine 6, a026070 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll AG, Voeller HJ, Sugars L, Gelmann EP, p53 oncogene mutations in three human prostate cancer cell lines. The Prostate 23, 123–134 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Batır MB, Şahin E, Çam FS, Evaluation of the CRISPR/Cas9 directed mutant TP53 gene repairing effect in human prostate cancer cell line PC-3. Molecular Biology Reports 46, 6471–6484 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Carroll D, Genome engineering with zinc-finger nucleases. Genetics 188, 773–782 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva G et al. , Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Current gene therapy 11, 11–27 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doudna JA, Charpentier E, The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Church GM, Genome editing and engineering: from TALENs, ZFNs and CRISPRs to molecular surgery. (Cambridge University Press, 2018). [Google Scholar]
- 18.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Soria E, Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of molecular evolution 60, 174–182 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Barrangou R et al. , CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA, Sequence-and structure-specific RNA processing by a CRISPR endonuclease. Science 329, 1355–1358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brouns SJ et al. , Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deltcheva E et al. , CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinek M et al. , A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cong L et al. , Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mali P et al. , RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jinek M et al. , RNA-programmed genome editing in human cells. elife 2, e00471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savić N, Schwank G, Advances in therapeutic CRISPR/Cas9 genome editing. Translational Research 168, 15–21 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Dai W-J et al. , CRISPR-Cas9 for in vivo gene therapy: promise and hurdles. Molecular Therapy-Nucleic Acids 5, e349 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin H, Xue W, Anderson DG, CRISPR–Cas: a tool for cancer research and therapeutics. Nature Reviews Clinical Oncology 16, 281–295 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Lage M, Puig-Serra P, Menendez P, Torres-Ruiz R, Rodriguez-Perales S, CRISPR/Cas9 for cancer therapy: hopes and challenges. Biomedicines 6, 105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Glass Z, Huang M, Chen Z-Y, Xu Q, Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications. Biomaterials 234, 119711 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chew WL, Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 10, e1408 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Senís E et al. , CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnology journal 9, 1402–1412 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Doudna JA, The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y et al. , Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Science advances 6, eaay6812 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang P et al. , Genome editing for cancer therapy: delivery of Cas9 protein/sgRNA plasmid via a gold nanocluster/lipid core–shell nanocarrier. Advanced Science 4, 1700175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin H et al. , Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nature biotechnology 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu CL, Ruan MZ, Mahajan VB, Tsang SH, Viral delivery systems for CRISPR. Viruses 11, 28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L, Hu S, Chen X, Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials 171, 207–218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Q et al. , Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nature nanotechnology 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K et al. , Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature biomedical engineering 1, 889 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demirer GS et al. , High aspect ratio nanomaterials enable delivery of functional genetic material without DNA integration in mature plants. Nature nanotechnology 14, 456–464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue H, Zhou X, Cheng M, Xing D, Graphene oxide-mediated Cas9/sgRNA delivery for efficient genome editing. Nanoscale 10, 1063–1071 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Hasanzadeh A et al. , Photoluminescent functionalized carbon dots for CRISPR delivery: Synthesis, optimization and cellular investigation. Nanotechnology 30, 135101 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Hashemzadeh I et al. , Polyethylenimine-Functionalized Carbon Dots for Delivery of CRISPR/Cas9 Complexes. ACS Applied Bio Materials 4, 7979–7992 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Long C et al. , Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 345, 1184–1188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen J, Zhu Y, Yang X, Li C, Graphene quantum dots: emergent nanolights for bioimaging, sensors, catalysis and photovoltaic devices. Chemical communications 48, 3686–3699 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Cui L, Losic D, Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta biomaterialia 9, 9243–9257 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Eoh J, Gu L, Biomaterials as vectors for the delivery of CRISPR–Cas9. Biomaterials science 7, 1240–1261 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Wei T, Cheng Q, Min Y-L, Olson EN, Siegwart DJ, Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nature communications 11, 1–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gravely M et al. , Aggregation reduces subcellular localization and cytotoxicity of single-walled carbon nanotubes. ACS Applied Materials & Interfaces 14, 19168–19177 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hasan MT et al. , Multi-drug/gene NASH therapy delivery and selective hyperspectral NIR imaging using chirality-sorted single-walled carbon nanotubes. Cancers 11, 1175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell E et al. , Graphene oxide as a multifunctional platform for intracellular delivery, imaging, and cancer sensing. Scientific reports 9, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aghamiri S et al. , Nanoparticles-mediated CRISPR/Cas9 delivery: Applications in cancer treatment and detection. Journal of Drug Delivery Science and Technology, 101533 (2020). [Google Scholar]
- 55.Campbell E, Hasan MT, Gonzalez Rodriguez R, Akkaraju GR, Naumov AV, Doped graphene quantum dots for intracellular multicolor imaging and cancer detection. ACS Biomaterials Science & Engineering 5, 4671–4682 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Hasan MT et al. , Rare-earth metal ions doped graphene quantum dots for near-ir in vitro/in vivo/ex vivo imaging applications. Advanced Optical Materials 8, 2000897 (2020). [Google Scholar]
- 57.Hasan MT et al. , Near-infrared emitting graphene quantum dots synthesized from reduced graphene oxide for in vitro/in vivo/ex vivo bioimaging applications. 2D Materials 8, 035013 (2021). [Google Scholar]
- 58.Cherukuri P, Bachilo SM, Litovsky SH, Weisman RB, Near-infrared fluorescence microscopy of single-walled carbon nanotubes in phagocytic cells. Journal of the American Chemical Society 126, 15638–15639 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Hong G, Diao S, Antaris AL, Dai H, Carbon nanomaterials for biological imaging and nanomedicinal therapy. Chemical reviews 115, 10816–10906 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Ajgaonkar R et al. , Detection of Pancreatic Cancer miRNA with Biocompatible Nitrogen-Doped Graphene Quantum Dots. Materials 15, 5760 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Campbell E et al. , Graphene quantum dot formulation for cancer imaging and redox-based drug delivery. Nanomedicine: Nanotechnology, Biology and Medicine 37, 102408 (2021). [DOI] [PubMed] [Google Scholar]
- 62.Frieler M et al. , Effects of doxorubicin delivery by nitrogen-doped graphene quantum dots on cancer cell growth: experimental study and mathematical modeling. Nanomaterials 11, 140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hasan MT et al. , Photo-and Electroluminescence from Nitrogen-Doped and Nitrogen–Sulfur Codoped Graphene Quantum Dots. Advanced Functional Materials 28, 1804337 (2018). [Google Scholar]
- 64.Akinc A, Thomas M, Klibanov AM, Langer R, Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. The Journal of Gene Medicine: A cross-disciplinary journal for research on the science of gene transfer and its clinical applications 7, 657–663 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Liu C et al. , Nano-carrier for gene delivery and bioimaging based on carbon dots with PEI-passivation enhanced fluorescence. Biomaterials 33, 3604–3613 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Stemmer M, Thumberger T, del Sol Keyer M, Wittbrodt J, Mateo JL, CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PloS one 10, e0124633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodríguez‑Rodríguez DR, Ramírez‑Solís R, Garza‑Elizondo MA, Garza‑Rodríguez MDL, Barrera‑Saldaña HA, Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases. International journal of molecular medicine 43, 1559–1574 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Labun K et al. , CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic acids research 47, W171–W174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Görlich D, Jäkel S, Nucleocytoplasmic transport. Protein Targeting, Transport, and Translocation, 293–321 (2002). [Google Scholar]
- 71.Lu J et al. , Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell communication and signaling 19, 1–10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Biagioni A et al. , Type II CRISPR/Cas9 approach in the oncological therapy. Journal of Experimental & Clinical Cancer Research 36, 80 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith AM, Mancini MC, Nie S, Second window for in vivo imaging. Nature nanotechnology 4, 710–711 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khakbaz F, Mahani M, Micro-RNA detection based on fluorescence resonance energy transfer of DNA-carbon quantum dots probes. Analytical biochemistry 523, 32–38 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Yang ES, Huh YJ, Park J-W, RNA interference targeting sensitive-to-apoptosis gene potentiates doxorubicin-and staurosporine-induced apoptosis of PC3 cells. Anticancer Research 33, 847–855 (2013). [PubMed] [Google Scholar]
- 76.Nachshon-Kedmi M, Yannai S, Fares F, Induction of apoptosis in human prostate cancer cell line, PC3, by 3, 3′-diindolylmethane through the mitochondrial pathway. British journal of cancer 91, 1358–1363 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suriamoorthy P et al. , Folic acid-CdTe quantum dot conjugates and their applications for cancer cell targeting. Cancer nanotechnology 1, 19–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shrestha S et al. , X-ray induced photodynamic therapy with copper-cysteamine nanoparticles in mice tumors. Proceedings of the National Academy of Sciences 116, 16823–16828 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Menon I, Zaroudi M, Zhang Y, Aisenbrey E, Hui L, Fabrication of active targeting lipid nanoparticles: Challenges and perspectives. Materials Today Advances 16, 100299 (2022). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.