

Summary
Stem cells regulate their self-renewal and differentiation fate outcomes through both symmetric and asymmetric divisions. m6A RNA methylation controls symmetric commitment and inflammation of hematopoietic stem cells (HSCs) through unknown mechanisms. Here, we demonstrate that the nuclear speckle protein SON is an essential m6A target required for murine HSC self-renewal, symmetric commitment, and inflammation control. Global profiling of m6A identified that m6A mRNA methylation of Son increases during HSC commitment. Upon m6A depletion, Son mRNA increases, but its protein is depleted. Reintroduction of SON rescues defects in HSC symmetric commitment divisions and engraftment. Conversely, Son deletion results in a loss of HSC fitness, while overexpression of SON improves mouse and human HSC engraftment potential by increasing quiescence. Mechanistically, we found that SON rescues MYC and suppresses the METTL3-HSC inflammatory gene expression program through CCL5 transcriptional regulation. Thus, our findings define a m6A-SON-CCL5 axis that controls inflammation and HSC fate.
Graphical Abstract
eTOC Blurb
Kharas and colleagues report SON, a central component of nuclear speckles as a critical m6A target required for murine HSC self-renewal, symmetric commitment, and inflammation control. SON suppresses inflammatory programs including double stranded DNA and chemokines in HSCs downstream of RNA methylation.
Introduction
The balance between self-renewal and differentiation fate in hematopoietic stem cells (HSCs) is crucial for maintaining blood homeostasis1–4. Upon activation, HSCs divide in either a symmetric or asymmetric manner, with the mode of division predicting the fates of their direct progenies5–10. Asymmetric division ensures both self-renewal and commitment fates (differentiation to multipotent progenitors (MPPs) or a direct megakaryocyte fate11–13), while symmetric divisions maximize a single fate outcome. Therefore, symmetric divisions play a critical role in the in vitro expansion of HSCs during transplantation, as well as in the rapid differentiation of blood cells during bone marrow recovery after stress14–17.
N6-methyladenosine (m6A) RNA methylation is a crucial regulator of stem cell fate determination18–22. m6A is the most abundant post-transcriptional RNA modification–observed on mRNAs and long non-coding RNAs23. The m6A mark is deposited on mRNAs by METTL3, the catalytic subunit of the METTL3-METTL14-WTAP-VIRMA-ZC3H13-RBM15 protein complex24, and is recognized by m6A erasers and readers, which play critical roles in controlling cell fate and myeloid differentiation25–33. Work from our lab showed that m6A RNA methylation controls HSC fate by maintaining normal symmetric commitment divisions18. Despite its demonstrated role in HSC maintenance, conflicting hypotheses exist regarding m6A’s mode of action in HSCs. We and others proposed that m6A stabilizes Myc transcripts to control HSC symmetric commitment18,19. Others suggest an indirect role through maintenance of mRNA structure that prevents dsRNA formation resulting in an innate immune response34. However, studies with the m6A reader YTHDF2 argues against this indirect role as YTHDF2 suppresses inflammation through destabilizing m6A-modified inflammatory transcripts in HSCs30. It therefore remains unclear how inflammatory responses are regulated in HSCs. This question is of clinical importance since inflammatory activation sensitizes cancer to immune checkpoint blockade. Moreover, small molecules targeting m6A RNA methylation have been developed in solid tumor and leukemia and proposed to be combined with immune checkpoint blockade in clinical trials35,36. Thus, we investigated how m6A controls both HSC fate and activation of the inflammatory program.
Results
DART-seq identifies SON as a m6A target in mouse and human HSCs
To understand how RNA methylation changes during HSC commitment, we sought a method that could accurately map m6A sites in rare cell types. Several approaches have combined RNA-binding protein (RBP) fusions with editing enzymes to assess direct mRNA targets in HSCs and other cell types37,38. One such approach, DART-seq, has been developed to measure m6A sites by fusing the YTH domain of m6A reader protein (YTHDF2) to the RNA editing protein APOBEC139 (Figure 1A). This method permits identification of direct YTH domain binding transcripts (i.e., m6A targets), as well as the C to U editing sites that reflect direct binding, as identified by RNA-seq.
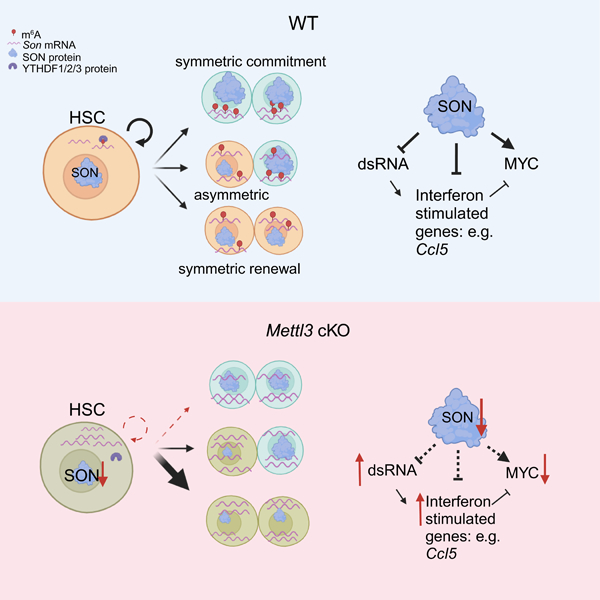
Figure 1: DART-seq identifies SON as a m6A target in mouse HSCs. See also Figure S1 and Table S1–S3.
(A) DART-seq experimental scheme in sorted mouse HSC, MPP1, MPP2, and MPP4s.
(B) DART-seq identified significant edit sites from n=3 independent experiments. (sites with padj<0.1, differential editing > 0.1 were defined as significant sites).
(C) DART-seq identified significant target genes from n=3. independent experiments. (cut-off for significance: padj<0.1, differential editing > 0.1).
(D) DART-seq targets in mouse HSCs mainly localized to 3’UTR.
(E) De novo motif search analysis using edit sites in mouse HSCs and MPPs.
(F) Majority of HSC DART-seq targets overlap with MPP DART-seq targets.
(G) Scheme of genes whose m6A modifications increase from HSC to MPP commitment.
(H) Overlap of sites that have unique/increased m6A modification during HSC to MPP commitment with two other published datasets (m6A-seq31 and SLIMseq43).
(I) Representative track showing the C to U edit site on Son transcript and its adjacency to a DRACH motif. Editing frequencies in each sample was annotated. Yellow boxed ‘C’s were the edit sites and the sequences boxed with dotted black line were the adjacent DRACH motif. n=3 independent experiments. EV: empty vector.
To determine whether DART-seq could be effectively employed to monitor m6A in hematopoietic stem and progenitor cells (HSPCs), we mapped m6A sites in purified human cord blood CD34+ HSPCs treated with vehicle or with the METTL3 inhibitor STM245735 (Figure S1A). In the untreated control group, we identified 230 m6A sites and 187 m6A target genes (Table S1), with the majority (~90%) of m6A sites located in the 3’UTR (Figure S1B–D). The m6A target genes were substantially enriched for transcription factors essential for hematopoietic differentiation, including MYC, EKLF, and RUNX2 (Figure S1E, Table S1). STM2457 treatment produced a greater than 50% reduction in both the number of m6A sites and genes detected, as well as a decrease in APOBEC-YTH editing frequency (Figure S1B and Figure S1F). We also observed an enrichment of the canonical m6A DRACH motif within the significant sites compared to non-significant sites (Figure S1G). Although we identified fewer m6A sites compared to other mapping approaches, such as m6A-SAC-seq40, we were able to unambiguously distinguish target genes that were sensitive to METTL3 inhibition, thus demonstrating the utility of DART-seq to study dynamic regulation of m6A by METTL3 in HSPCs.
To compare the regulation of m6A in different HSPC populations, we next conducted DART-seq on purified HSCs, MPP1s, which are considered unbiased; MPP2s, which have a bias toward megakaryocytic and erythroid lineages; and MPP4s, with lymphoid lineage bias12,41,42 from wildtype (WT) mice (Figure 1A and Figure S1H). Our analysis revealed that each population had close to 300 m6A sites and 250–300 gene targets (Figure 1B and Figure 1C, Table S2). The MPP2 population showed the most editing with 1128 sites and 880 genes detected (Figure 1B and Figure 1C). Although Apobec-Yth transcripts in the MPP2 population are modestly increased (~1.3 fold increase in Apobec-Yth read counts in MPP2 vs other populations), we found elevated YTH-binding in the MPP2. Interestingly, the MPP2 population has also the greatest expansion in frequency upon m6A loss18. Consistent with human CD34+ cells, the vast majority of m6A sites were mainly localized to the 3’UTR and enriched for the m6A DRACH motif (Figure 1D–E, Figure S1I–J).
We found that a significant proportion of m6A targets in HSCs (76%) were also present in MPPs, but that only a small fraction of the MPP targets were shared with HSCs (20%) (Figure 1F). Our analysis of the m6A targets in HSCs revealed enrichment in three key biological processes: innate immune response pathways, including the NFκB pathway and the Toll-like receptor cascades; cellular signaling processes, such as the RhoGTPase and ERK/MAPK signaling targets; and metabolic processes, such as amino acid transport and fatty acid oxidation pathways (Figure S1K). In contrast, the m6A targets unique to MPPs were enriched for lineage differentiation and myeloid programs, such as MLL, GATA1, PU.1, and CEBPD (Figure S1L, Table S3). Taken together, these findings suggest that DART-seq is capable of mapping m6A in a cell type-specific manner.
To further explore the significance of m6A modification in the early stages of hematopoiesis, we focused on the targets with significantly increased m6A modification from HSCs to MPPs (Figure 1G), hypothesizing that these modifications may play a crucial role in the commitment of HSCs. To expand our analysis, we integrated two additional published datasets (m6A-seq31 and SLIMseq43) that employ distinct methodologies to identify m6A targets in murine hematopoiesis. By intersecting the genes with increased m6A modification in MPPs compared to HSCs across all three datasets, we identified Son as a common target with increased m6A modification in MPPs (Figure 1H and Figure 1I). Of note, despite the larger number of sites detected by SLIM-seq43, Myc was undetected even though it was an m6A methylated target identified by DART-seq, m6A-seq31 and other studies18,19,34,44–46. This finding suggests a potential functional importance of SON in the regulation of hematopoietic stem cell differentiation.
The SON RNA binding protein is a central component of nuclear speckles that has been implicated in a variety of cellular processes, including splicing control and transcriptional repression47,48. Of clinical relevance, de novo heterozygous loss-of-function variants in the SON gene (Zhu-Tokita-Takenouchi-Kim Syndrome; ZTTK) have been associated with hematological symptoms, as well as neurological and developmental delays49,50. Our previous miCLIP analysis in human leukemia cells revealed that SON RNA transcript has one of the highest numbers of m6A sites51. Furthermore, we observed that three out of the four m6A sites on the SON transcript were sensitive to METTL3 inhibition in human CD34+ cells (Figure S1M). Collectively, these findings suggest that SON is a conserved m6A target in both mouse and human HSCs.
Depletion of m6A reduces SON protein abundance
To investigate whether the m6A modification controls SON expression, we used the Mx1-cre Mettl3 conditional knockout mice (Mettl3 cKO) as a genetic model to deplete m6A in HSCs and examined the effect on SON. Our results showed that genetic depletion of METTL3 led to an increase in Son transcripts in both HSCs and MPP populations (Figure 2A and Figure S2A). In contrast to its RNA expression, SON protein abundance was reduced in Mettl3 cKO HSCs and HSPCs (Figure 2B–C and Figure S2B). Similarly, in the more committed MPP2 and MPP4 populations, but not in MPP1 cells, SON protein levels were decreased upon METTL3 depletion (Figure S2C). Additionally, we found that METTL3 enzymatic activity was necessary to maintain SON protein levels, as METTL3 inhibition by STM2457 resulted in a similar reduction of SON in HSCs (Figure 2D–E).
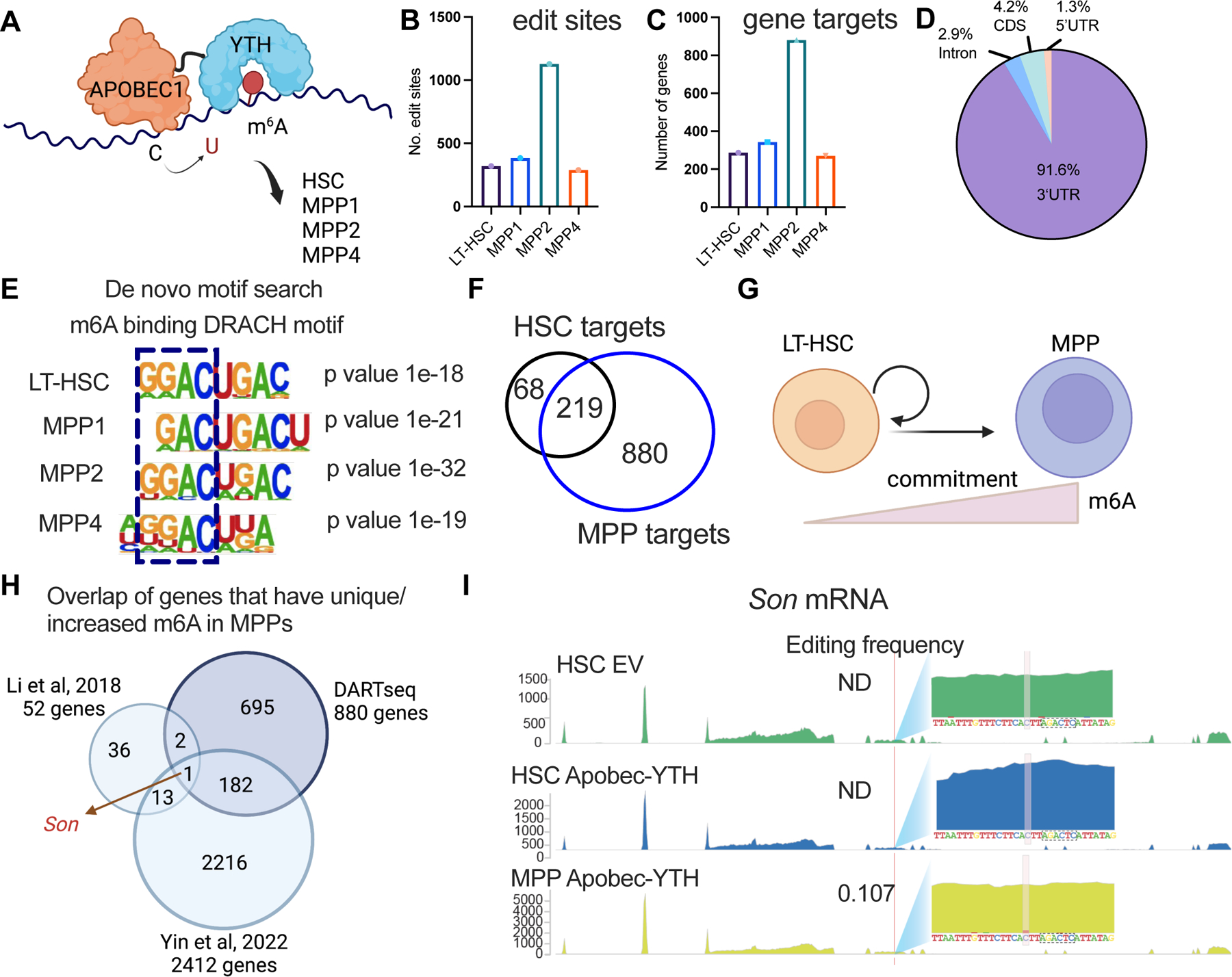
Figure 2: Depletion of m6A reduces SON protein abundance. See also Figure S2.
(A) Increased Son mRNA level in Mettl3 cKO HSC and MPPs. n=3, n represents number of mice.
(B) Representative immunofluorescence images of SON protein abundance in WT and Mettl3 cKO HSCs. Scale bar: 10μm.
(C) Reduced SON protein abundance by immunofluorescence in Mettl3 cKO HSCs. n=3 independent experiments.
(D) Representative immunofluorescence images of SON protein abundance in WT HSCs with DMSO or STM2457 treatment (20μM 40hr). Scale bar: 10μm.
(E) Reduced SON protein abundance in WT HSCs with STM2457 treatment (20μM 40hr) measured by immunofluorescence.
(F) Targeted demethylation of Son mRNA reduced SON protein abundance in WT HSCs.
(G) Representative confocal immunofluorescence images of HSC paired daughter cell assays. Paired daughter cells were stained with DAPI (blue), NUMB (green), SON (red). Scale bar: 10μm.
(H) Percentages of doublet cells in each type of cell division in WT HSCs with STM2457 treatment (20μM 40hr) measured by immunofluorescence. Number of daughter pairs assessed: n=111 (WT HSC quantified using NUMB IF); n=115 (WT HSC STM2457 quantified using NUMB IF); n=60 (WT HSC quantified using SON IF); n=71 (WT HSC STM2457 quantified using SON IF). P value was calculated using Chi-square test. n=2 independent experiments. Data in (A), (C), (E), (F) represent means ± s.e.m. , * represents p < 0.05. ** represents p < 0.01. *** represents p < 0.001. **** represents p<0.0001. ns represents p > 0.05.
To examine whether SON protein abundance is controlled by the m6A modification, we performed targeted RNA demethylation of the Son mRNA in WT HSCs using engineered m6A eraser (dCas9-FTO) and assessed SON protein abundance. The dCas9-FTO fusion was designed previously to achieve site-specific demethylation of RNAs52. We found that recruiting dCas9-FTO to the m6A sites on Son using both sgRNAs combinations targeting either the 5’ or 3’ of the site reduced SON protein abundance (Figure 2F). These data support that the m6A modification of Son mRNA controls its protein abundance.
We then sought to understand which m6A cytoplasmic reader is responsible for this effect on SON. We examined CLIP53 and TRIBE-STAMP54 datasets and found that YTHDF1/2/3 bind the SON transcript. These binding sites match the m6A sites identified by our DART-seq data in human CD34+ HSPCs (Figure S2D). Furthermore, depletion of the cytosolic m6A readers YTHDF1 or YTHDF3, but not YTHDF2, reduced SON protein abundance in cKit+-enriched WT bone marrow cells (Figure S2E–F). Depletion of YTHDF2 increased Son transcripts, suggesting the effect of YTHDF2 binding on Son transcript controls its mRNA levels (Figure S2G–H). Together, these results suggest that m6A’s control of SON protein abundance likely occurs through cytosolic m6A reader proteins YTHDF1 or YTHDF3.
Segregation of key cell fate determinants such as NUMB and MYC proteins, as well as active mitochondria and lysosomes, can effectively predict self-renewal or commitment outcomes during HSC division18,55–59. As we previously reported, loss of m6A results in a symmetric commitment defect18. Consistent with these observations, we found that STM2457 treatment in WT HSCs mimicked the effect of METTL3 loss, leading to an increase in symmetric NUMB-low (self-renewal) divisions and a loss of symmetric NUMB-high divisions (symmetric commitment) (Figure 2G–H). Unexpectedly, we found that SON could be segregated into asymmetrically dividing HSCs as well as symmetrically dividing “low” and “high” cells correlating with NUMB expression (Figure 2G and Figure S2I, see movie 1). These data suggest that increased SON abundance serves as a marker for cellular commitment and that it may play a functional role in HSC fate determination.
Forced SON expression rescues the function of m6A deficient HSCs
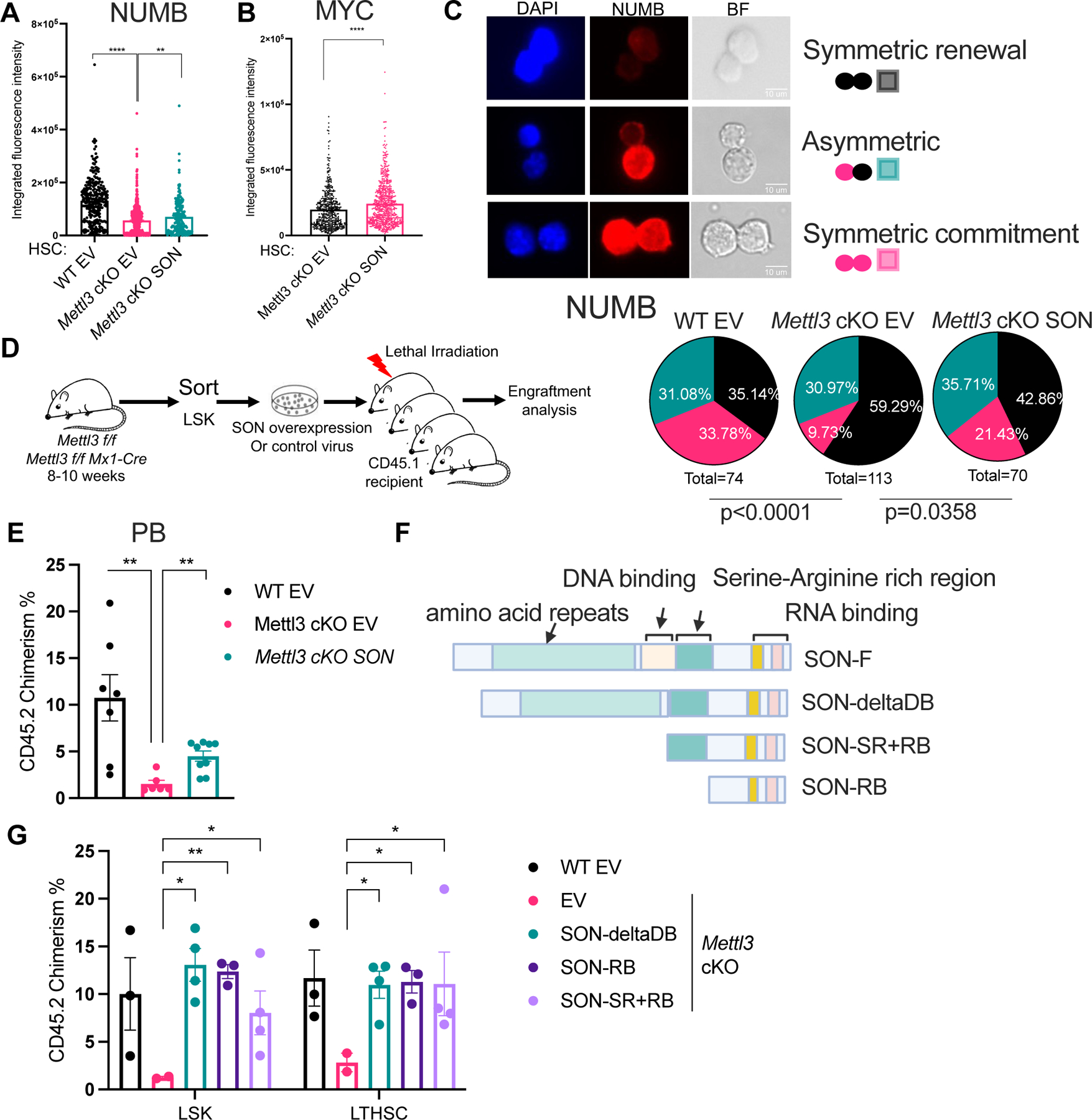
We next examined whether SON could rescue HSC function in METTL3-depleted cells. SON overexpression in Mettl3 cKO HSCs increased the overall abundance of NUMB and MYC proteins in HSCs (Figure 3A and Figure 3B) and increased the symmetric NUMB-hi or MYC-hi paired divisions (Figure 3C, Figure S3A–B). Importantly, we found that NUMB abundance was not increased in SON overexpressing cells that underwent a symmetric commitment fate (Figure S3C). Thus, the modest increase in NUMB abundance was likely due to the rescue of the percentage of cells undergoing symmetric commitment. In contrast, MYC showed a statistically significant increase in all HSC fate categories, with the highest increase observed in the asymmetric committed cells and symmetric commitment daughter pairs (Figure S3D). These results suggest that SON controls MYC protein abundance while also rescuing the percentage of cells undergoing MYC symmetric commitment. Furthermore, we found that overexpression of SON in Mettl3 cKO LSKs partially rescues multilineage engraftment (Figure 3D–E, S3E–H). Together, these results suggest that SON can recover the reduced HSC symmetric commitment and engraftment defects arising from METTL3 depletion.
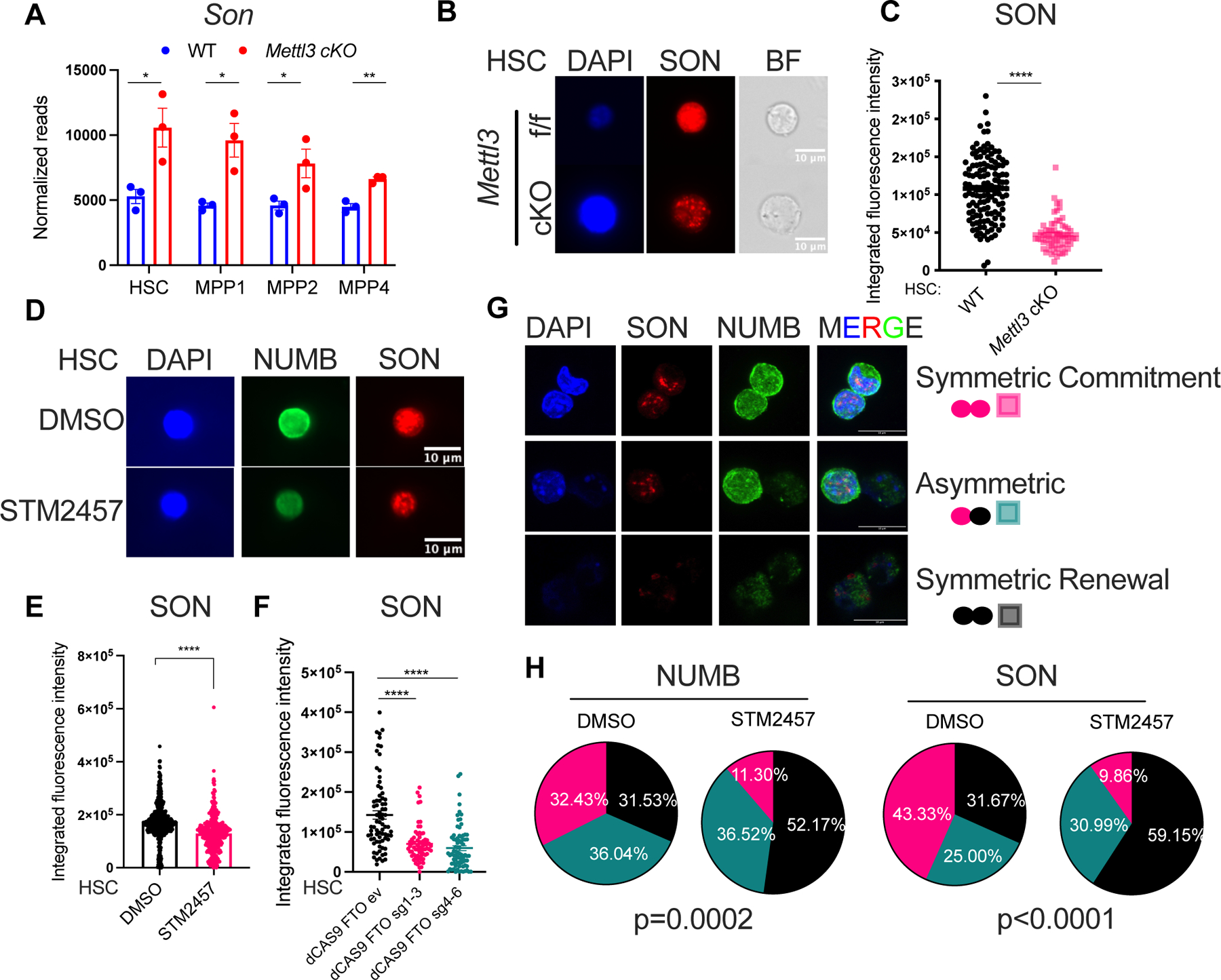
Figure 3: Forced SON expression rescues the function of m6A deficient HSCs. See also Figure S3.
(A) NUMB expression in Mettl3 cKO HSCs with SON overexpression compared to Mettl3 cKO HSCs and WT (Mettl3 f/f) HSCs as quantified by immunofluorescence. EV: empty vector controls; n=3 independent experiments.
(B) MYC expression in Mettl3 cKO HSCs with SON overexpression compared to Mettl3 cKO HSCs as quantified by immunofluorescence. n=2 independent experiments.
(C) SON overexpression in Mettl3 cKO HSC rescues its symmetric commitment defect measured by NUMB symmetric high divisions. Above: representative immunofluorescence images of paired daughter cells stained with DAPI (blue), NUMB (red), and brightfield. Scale bar: 10μm. Below: percentages of doublet cells in each type of cell division. n=74 (WT HSC EV); n=113 (Mettl3 cKO EV); n=70 (Mettl3 cKO SON); P value was calculated using Chi-square test.
(D) Scheme of transplant strategy in (E).
(E) Quantification of the frequency of donor-derived cells was shown in the peripheral blood at 4-week post transplantation, n = 6–9. n represents number of mice. Representative of three independent experiments.
(F) Scheme of SON truncation mutants.
(G) Quantification of the frequency of donor-derived cells was shown in the bone marrow LSK population 16 weeks post transplantation. n=2–4, n represents number of mice. Representative of three independent experiments. Data in (A), (B), (E), (G) represent means ± s.e.m. , * represents p < 0.05. ** represents p < 0.01. *** represents p < 0.001. **** represents p<0.0001. ns represents p > 0.05.
To gain insight into how SON mediates its restorative effects from METTL3 loss, we characterized the relative contributions of its conserved domains. SON’s N-terminus consists of amino acid repeats, while the C-terminus includes both its DNA- and RNA-binding domains60. We first determined that the C-terminal SON construct was necessary and sufficient to rescue the engraftment within the HSPC compartment (Figure S3I–J). To further probe the precise C-terminal domains responsible for mediating these rescue phenotypes, we generated three additional SON truncation constructs harboring individual deletions of either the DNA-binding domain (SON-deletion-DB), a fragment containing the serine-arginine rich region and two RNA-binding domains (SON-SR+RB), or a fragment containing the two RNA-binding domains (SON-RB) (Figure 3F). We found that the SON-RB were sufficient to rescue the engraftment at Mettl3 cKO HSCs (Figure 3G). Notably, we confirmed by genotyping that Mettl3 remains deleted in the engrafted cells from the SON rescue groups (Figure S3K). Taken together, these results indicate that SON rescues the Mettl3 cKO HSC and engraftment through its C-terminal region and requires its RNA-binding ability.
SON controls hematopoietic stem cell fate
Given our finding that SON can rescue the downstream defects resulting from METTL3 depletion, we postulated that SON may play an independent functional role in maintaining HSC homeostasis. To assess the requirement of SON for HSC engraftment, we employed a genetic approach to delete Son by CRISPR Cas9 resulting in reduced expression in HSPCs (Figure 4A and Figure S4A). We also observed a marked decrease in long-term multi-lineage donor engraftment upon Son deletion (Figure 4B, S4B–D). These data further suggest that SON is indispensable for HSC engraftment.
Figure 4: SON controls hematopoietic stem cell fate. See also Figure S4.
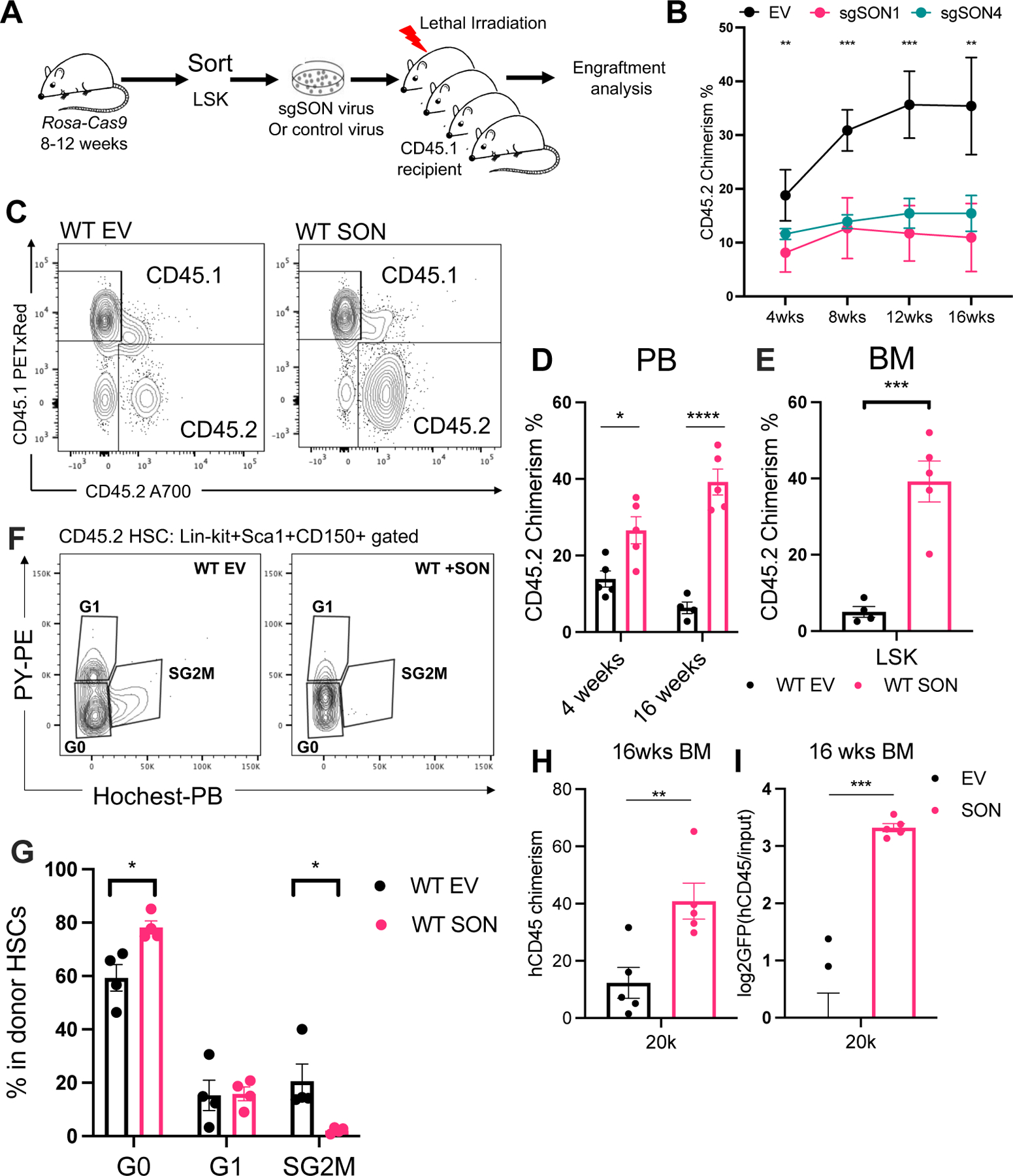
(A) Scheme of the transplantation experiment in (B).
(B) Donor CD45.2 chimerism over 16 weeks in the peripheral blood. n=2–5, n represents number of mice. Representative of two independent experiments.
(C) Peripheral blood donor engraftment in 4- and 16-weeks post transplantation. n=4–5, n represents number of mice. Representative of two independent experiments.
(D) Representative flow cytometry plots of the CD45.1 and CD45.2 engraftment in the peripheral blood in experiments shown in (C).
(E) SON overexpression in WT LSKs resulted in increased donor engraftment in the LSK population 16 weeks post transplantation. n=4–5, n represents number of mice.
(F) Representative flow cytometry plots of cell cycle analysis using Hoechst and Pyronin Y staining in the CD45.2+ Lin-Ckit+Sca1+CD150+ population.
(G) Quantification of cell cycle status of the CD45.2 Lin-Ckit+Sca1+CD150+ population. n=4, n represents number of mice.
(H) Human CD45 engraftment 16 weeks post transplantation were plotted. n=5, n represents number of mice. Representative of three independent experiments.
(I) GFP frequency in the hCD45 engrafted population were measured and the relative enrichment of GFP+ donor cells were plotted at 16 weeks post transplantation. n=5, n represents number of mice. Data in (B), (D), (E), (G), (H), (I) represent means ± s.e.m. , * represents p < 0.05. ** represents p < 0.01. *** represents p < 0.001. **** represents p<0.0001. ns represents p > 0.05.
SON could improve HSC function in the absence of perturbed METTL3 activity as it significantly enhanced multi-lineage engraftment of donor cells (Figure 4C–D, S4E) and within the HSC compartment (Figure 4E and Figure S4F). Consistent with prior studies that linked increased quiescence and reduced cell cycle activation to improved stem cell function61,62, we found that SON-overexpressing cells were more quiescent than controls (Figure 4F–G). Importantly, the engraftment advantage of SON-overexpressing cells persisted even after secondary transplantation (Figure S4G). Furthermore, SON’s ability to enhance self-renewal in vivo was also conserved in human cord blood derived CD34+ cells, as SON overexpression improved human engraftment (Figure 4H–I and Figure S4H–I). Together, our results demonstrate that SON is a positive regulator of stem cell engraftment and function.
SON partially rescues the inflammatory program and enhances stem cell programs in Mettl3 cKO HSCs
Our previous studies have demonstrated that m6A deficiency in HSCs leads to the expansion of two single cell RNA-seq derived clusters (KO-specific clusters, KOsp1 and Kosp2)18. KOsp1 represents an HSC/MPP-like population with reduced self-renewal potential, while KOsp2 is enriched for an HSC/MPP cluster with a skewed megakaryocyte gene expression signature18. To explore how SON overexpression affects these altered HSC/MPP populations, we performed single-cell RNA-seq analysis on WT and Mettl3 cKO LSKs transduced with empty vector control (EV) or SON-overexpressing lentivirus (Figure 5A). We found that SON overexpression expanded the HSC cluster in WT LSKs (Figure S5A–B) and decreased the frequency of several dysregulated clusters (KOsp1–1 and CCL5+) in Mettl3 cKO LSKs by 60% and 50%, respectively (Figure 5B–C). These findings reveal that SON can partially restore the frequency of HSPC populations following METTL3 loss.
Figure 5: SON partially rescues the inflammatory program in Mettl3 cKO HSCs. See also Figure S5 and Table S4–9.
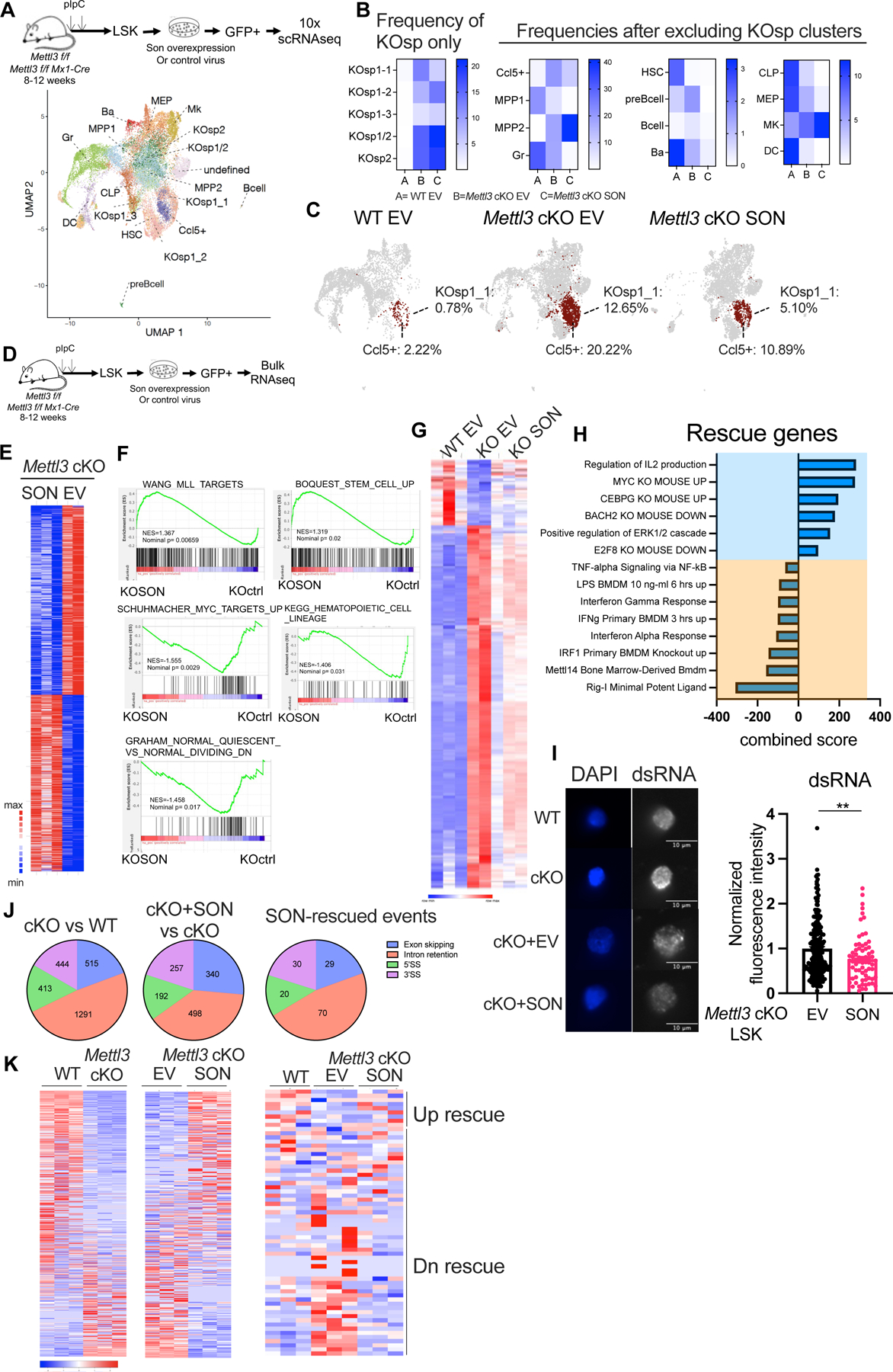
(A) Cell-type cluster assignments in Mettl3 f/f and Mettl3 cKO LSK cells transduced with EV (empty vector) or SON overexpressing lentivirus, overlaid on a uniform manifold approximation and projection (UMAP) of the single-cell RNA sequencing (scRNA-seq).
(B) Quantification of cell frequencies of different clusters in WT EV, Mettl3 cKO EV and Mettl3 cKO SON LSK cells based on scRNA-seq.
(C) SON overexpression rescues the frequency of KOsp1–1 and CCL5+ clusters, labeled as red.(D) Scheme of the bulk RNA-seq experiment. n=3 WT ctrl; n=2 Mettl3 cKO ctrl; n=3 Mettl3 cKO SON; n represents independent experiments.
(E) Heatmap showing the differential expressed genes between the Mettl3 cKO EV and Mettl3 cKO SON overexpressing LSK cells. (cut-off for differential expressed genes: padj <0.1)
(F) GSEA analysis showing the pathways enriched in the Mettl3 cKO SON overexpressing cells using the gene expression rank-list comparing Mettl3 cKO EV vs Mettl3 cKO SON in LSKs.
(G) Heatmap showing the differential expressed genes between the WT EV, Mettl3 cKO EV and Mettl3 cKO SON overexpressing LSK cells. (cut-off for differential expressed genes: padj <0.1)
(H) Enrichr pathway enrichment of the genes that were differentially expressed between Mettl3 f/f and Mettl3 cKO and were rescued by SON overexpressing in Mettl3 cKO LSKs. (cut-off for differential expressed genes: padj < 0.1)
(I) Left: Representative immunofluorescence images of dsRNA abundance in WT, Mettl3 cKO LSKs, Mettl3 cKO empty vector and Mettl3 cKO SON LSKs. Scale bar: 10μm. Right: Quantification of dsRNA abundance by immunofluorescence. Data were pooled and normalized to EV group. n=3 independent experiments.
(J) Percentage of alternative splicing events compared between Mettl3 cKO EV versus WT EV LSKs, Mettl3 cKO SON versus Mettl3 cKO EV LSK and SON rescue alternative splicing events were shown as pie charts. 3’SS: alternative 3’ splice site; 5’SS: alternative 5’ splice site. (cut-off for differential splicing events: padj <= 0.05, abs(diff_mean) >= 0.05)
(K) Left: differential intron retention events between WT EV and Mettl3 cKO EV LSKs. Middle: differential intron retention events between Mettl3 cKO EV and Mettl3 cKO SON LSKs. Right: differential intron retention events between WT and Mettl3 cKO LSKs that were rescued by SON overexpression. (cut-off for differential intron retention events: padj <= 0.05, abs(diff_mean) >= 0.05)。 Data in (I) represent means ± s.e.m. , * represents p <0.05. ** represents p < 0.01. *** represents p < 0.001. **** represents p<0.0001. ns represents p > 0.05.
To understand how SON alters the identity and marker gene expression of the KOsp1–1 and CCL5+ clusters, we performed a maximum likelihood projection onto annotated HSPC clusters in WT control mice (Figure S5C). In the Mettl3 cKO EV sample, the CCL5+ cluster predominantly clustered with MPP2 (60%) and to a lesser extent with HSC (30%) and MPP1 (5.5%). However, SON overexpression shifted the clustering towards HSC (37%) and MPP1 (12.4%) at the expense of MPP2 (48%). The KOsp1–1 cluster in the Mettl3 cKO ctrl sample mainly clustered with MPP2 (94%) and HSC (4.7%), but SON overexpression doubled the contribution from the HSC cluster (9.8%). These findings suggest that SON overexpression imparts a more HSC-like program compared to the clusters with loss of METTL3. Additionally, we noticed that these two clusters share a core set of innate immune-related genes, including Oasl2, Ifitm3, Ccl5, Irf7, Oasl1, and Isg15 (Table S4). SON overexpression downregulated Type-1 interferon response genes Irf7 and Isg15 in both clusters (Figure S5D–E, Table S5). SON overexpression also upregulated MYC target genes, such as Tuba1a, Tuba1c, and Slc38a2 (Figure S5D–E). These results suggest that SON rescues METTL3 loss by enhancing the HSC signature and reducing the inflammatory program.
To examine the molecular changes induced by SON, we conducted RNA sequencing analysis of WT and Mettl3 cKO LSKs with EV or SON overexpression (Figure 5D). First, we investigated the impact of SON overexpression in WT LSKs. Our analysis revealed an increase in the expression of genes associated with dormant and low-output HSC signatures42,63–66 (Figure S5F–G). Moreover, we found that genes upregulated by SON overexpression are enriched for targets of key pluripotency factors such as NANOG, SOX2, and OCT4 (Figure S5H). These findings support our scRNA-seq results (Figure S5A–B) indicating that SON promotes a stem cell program.
To investigate the molecular mechanisms underlying SON-mediated rescue of Mettl3 cKO HSCs, we analyzed the differential gene expression profiles between WT EV vs Mettl3 cKO EV groups. Consistent with prior studies18,34, we found an enrichment of the METTL3 KO embryonic stem cell signature21, as well as a significant upregulation of inflammatory genes30, including members of the TNF and innate immune pathways (such as Irf7, Oasl2, Tnfaip2, Tnfaip3, Cxcl10, and Ccl5) (Figure S5I–J, Table S6).
We identified 257 upregulated genes and 227 downregulated genes between the Mettl3 cKO EV vs Mettl3 cKO SON groups (Figure 5E, Table S7). Gene set enrichment analysis revealed that MLL target genes, stem cell and quiescence programs were positively enriched in the Mettl3 cKO SON group, along with MYC targets and hematopoietic lineage genes (Figure 5F). Furthermore, SON overexpression rescued a substantial set of gene expression profiles that were dysregulated in the Mettl3 cKO LSKs (Figure 5G). Specifically, SON overexpression significantly upregulated pathways involved in cellular response to IL2, while downregulating pro-inflammatory pathways, including RIGI-mediated innate immune signaling, TNF alpha signaling, and IRF1 targets (Figure 5H). These results indicate that SON rescues the altered inflammatory pathways in Mettl3 cKO LSKs and enhances stem cell programs.
SON reduces dsRNA formation by decreasing intron retention
We next sought to understand the molecular mechanism behind the enhanced inflammatory program following METTL3 depletion and SON’s inhibitory effect on it. One major activator of the innate inflammatory response is cellular accumulation of double stranded RNAs (dsRNA). As previously reported in m6A deficient fetal liver LSKs34, we also found that adult LSKs have increased dsRNA levels in Mettl3 cKO (Figure S5K). Furthermore, overexpression of both SON and SON-RB rescued this increase in dsRNA levels (Figure 5I, Figure S5L), suggesting that loss of SON contributes to the aberrant dsRNA formation upon m6A loss.
To identify the potential source of dsRNAs, we examined the role of endogenous retroviruses (ERVs), as prior studies have suggested that METTL3 loss increases the expression of these elements in embryonic stem cells67,68. Although we observed a modest increase of ERVs in Mettl3 cKO EV (Figure S5M), SON overexpression did not significantly repress them (Figure S5M, Table S8). We next explored intron retention as an alternative potential source of dsRNA69,70 by testing whether overexpressing SON could rescue intron retention events in Mettl3 cKO LSKs. We analyzed the alternative splicing differences between WT EV vs Mettl3 cKO EV LSKs, as well as Mettl3 cKO SON vs Mettl3 cKO EV LSKs. We observed a total of 2663 and 1287 different types of alternative splicing events in each comparison respectively (Figure 5J). We focused on alternative splicing events that were differentially regulated between WT and Mettl3 cKO LSKs and were rescued by SON overexpression in Mettl3 cKO LSKs. Almost 50% of the SON-rescued alternative splicing events (70 out of 149 events) belonged to differential intron retention events (Figure 5J). Remarkably, 85% of the rescued intron retention events accounted for the downregulation of intron retention in Mettl3 cKO LSKs upon SON overexpression (Figure 5K, Table S9). Our findings suggest that SON reduces dsRNA formation by reducing aberrantly retained introns.
SON binds to and suppresses pro-inflammatory chemokine CCL5 expression
We found that the SON-rescued alternative splicing gene targets and SON-rescued differential expressed genes mostly did not overlap, suggesting that SON controls its downstream target expression likely through a splicing-independent mechanism (Figure S5N). Furthermore, SON reversed the expression of the RIG-I signature and Oasl1, both of which are sensors of dsRNA (Figure 5H). When we analyzed innate immune response genes affected by m6A loss and rescued by SON overexpression, we found that pro-inflammatory chemokines Ccl5 and Cxcl10 were the top genes downregulated by SON overexpression (Figure 6A). Both the Ccl5 and Cxcl10 RNA expression in Mettl3 cKO LSKs were upregulated by greater than two-fold and were almost completely rescued by SON overexpression (Figure S6A, Table S6). SON-RB also reduced both Ccl5 and Cxcl10 RNA expression in Mettl3 cKO LSKs (Figure S6B). However, SON overexpression in the WT LSK cells reduced Ccl5 but not Cxcl10 (Figure S6C). CCL5, also known as RANTES, is known to be highly expressed in aged bone marrow microenvironment and contributes to HSC myeloid skewing71. Therefore, we focused our studies on the role of CCL5 downstream of SON.
Figure 6: CCL5 is a downstream of SON that controls HSC symmetric commitment fate. See also Figure S6.
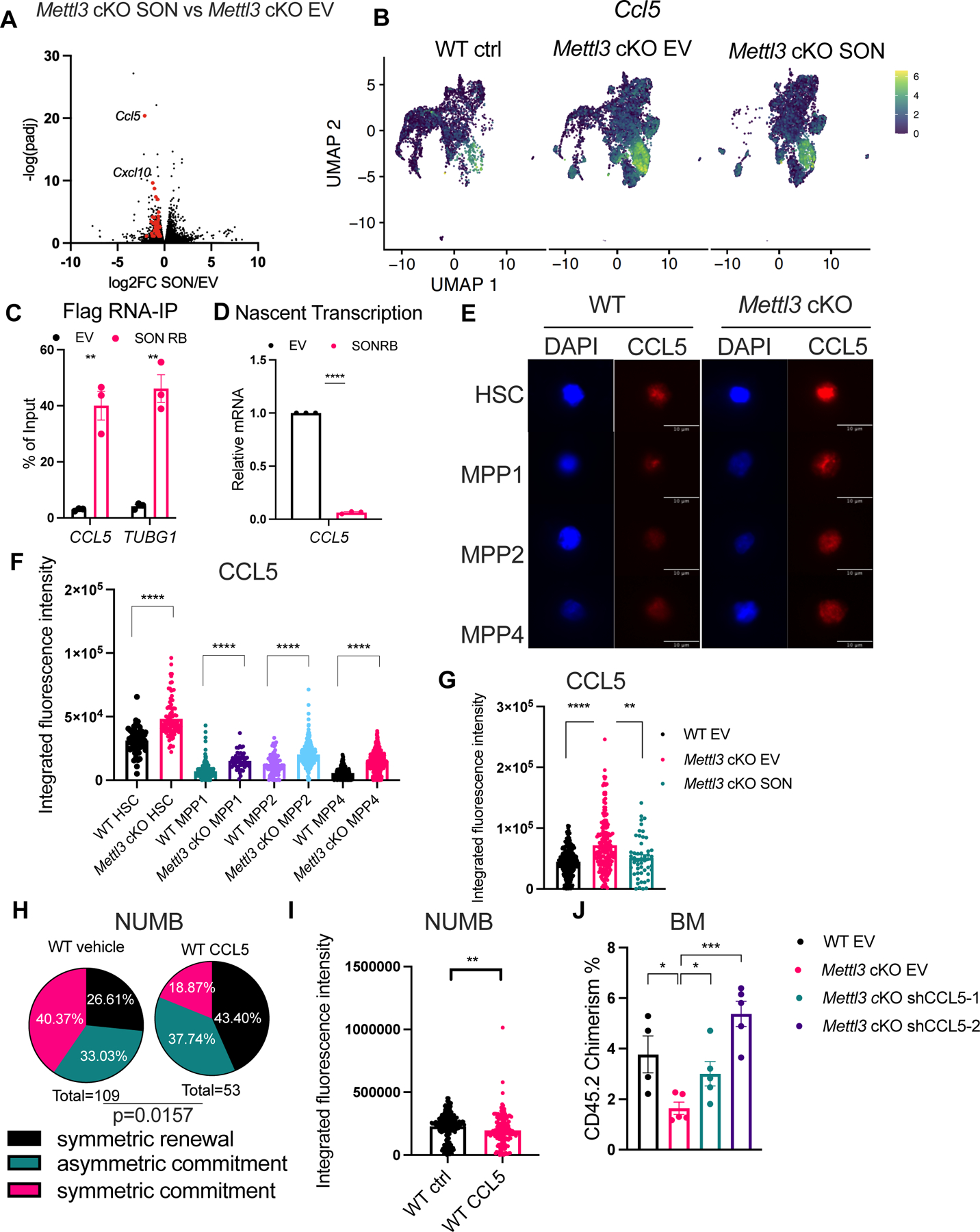
(A) Ccl5 and Cxcl10 are among the most downregulated innate immune genes by SON overexpression in Mettl3 cKO LSKs shown by Volcano plot. Genes labeled in red are shown in Table S6.
(B) Ccl5 expression in Mettl3 cKO LSKs in the scRNA-seq data projected in a UMAP, similar to (A).
(C) RNA-IP in THP1 cells expressing empty vector (MIG) or SON-RB-Flag retrovirus followed by RNA extraction and qPCR of target genes. n=3–4, n represents independent experiments.
(D) Nascent transcription of the CCL5 transcript in THP1 cells. n=3 independent experiments.
(E) Representative immunofluorescence images of CCL5 protein abundance in WT and Mettl3 cKO HSCs and MPPs. Scale bar: 10μm.
(F) Increased CCL5 protein abundance in Mettl3 cKO HSCs and MPPs. n=2 independent experiments.
(G) CCL5 protein abundance in Mettl3 cKO LSKs. n=3 independent experiments.
(H) Percentages of doublet cells in each type of cell division in WT HSCs with or without CCL5 treatment. n=109 (WT vehicle using NUMB IF); n=53 (WT CCL5 using NUMB IF); P value was calculated using Chi-square test.
(I) CCL5 treatment in WT HSCs reduced NUMB protein abundance. n=2 independent experiments.
(J) Quantification of the frequency of donor-derived cells was shown in the bone marrow 7 weeks post transplantation. n=4–5, n represents number of mice. Data in (C), (D), (F), (G), (I), (J) represent means ± s.e.m. , * represents p < 0.05. ** represents p < 0.01. *** represents p < 0.001. **** represents p<0.0001. ns represents p > 0.05.
The expanded CCL5-specific cluster in the Mettl3 cKO EV sample was partially normalized by SON overexpression (Figure 5C). Moreover, we also observed that overall Ccl5 expression levels were rescued by SON overexpression (Figure 6B). Collectively, our results suggest that SON targets pro-inflammatory chemokines CCL5, which is the likely functional effector of the inflammatory response in Mettl3 cKO LSKs.
SON-RB was capable of binding CCL5 mRNA, as well as TUBG160, which is a known SON binding target in human leukemia cells (THP1) (Figure 6C and Figure S6D–E). We also observed binding of the SON-RB to the MYC transcript, suggesting that it may be another direct SON target (Figure S6F). We found a significant reduction in CCL5 nascent transcription, as well as the CXCL10 and CEBPB targets in SON-RB expressing cells compared to the controls (Figure 6D and Figure S6G).
To understand how the SON-RB controls nascent transcription of CCL5, we examined the localization of SON-RB in cells. We overexpressed Flag-tagged SON, SON-RB, SON-SR+RB , SON-DB (DNA-binding domain containing fragment alone) in 293T cells and assessed the colocalization of Flag with endogenous SON. We found that ~ 40% of SON-RB also colocalizes to nuclear speckles, suggesting that the RNA-binding domain-containing fragment of SON shares a reduced but similar nuclear localization to full-length SON (Figure S6H and Movie 2–5). These results suggest that the RNA-binding domain is sufficient to recapitulate the rescue function of SON. Interestingly, the SON-DB displayed the least colocalization to endogenous SON (~20%). Nuclear speckles that contain RNA-processing proteins, transcription factors and RNAs act as a hub for transcriptional regulation72–74. Given SON’s known role in transcriptional repression47, these results suggest that SON-RB’s role in nascent transcription is closely associated with nuclear speckles.
CCL5 is a functional downstream target of SON in HSC fate control
We investigated the role of CCL5 in HSC cell fate, based on its direct link to the METTL3-SON pathway and its critical role in HSC and inflammation71. We confirmed that CCL5 protein abundance was increased in Mettl3 cKO HSCs and MPPs (Figure 6E–F). Interestingly, WT HSCs expressed higher levels of CCL5 when compared to MPPs (Figure 6E–F), and SON overexpression reduced CCL5 protein levels in Mettl3 cKO LSKs (Figure 6G). Next, we examined whether CCL5 was sufficient to induce a block in symmetric commitment in HSCs. We found that CCL5 treatment reduced NUMB abundance and significantly increased the symmetric NUMB low divisions while reducing the symmetric NUMB high divisions, thus mimicking the effects of m6A depletion (Figure 6H–I).
To determine whether SON’s suppression of CCL5 drives HSC commitment, we treated Mettl3 cKO SON-overexpressing HSCs with exogenous CCL5, anticipating that reduced HSC commitment would be reflected by reduced MYC protein abundance. As we previously found, SON overexpression increased MYC in Mettl3 cKO HSCs (Figure 3C and Figure S6I). Furthermore, CCL5 addition to Mettl3 cKO SON-overexpressing HSCs abolished the MYC rescue effect (Figure S6I). We then assessed how depletion of CCL5 impacts Mettl3 cKO LSKs function and found that it also can significantly improve engraftment (Figure 6J and Figure S6J–L). Thus, these results define a m6A-SON-CCL5 axis that controls both HSC commitment and MYC abundance. Importantly, increased CCL5 and CXCL10 levels after METTL3 depletion is validated by other datasets obtained from mouse or human hematopoietic cells19,34,75,76 (Figure S6M). This axis may also be relevant in the leukemic context, as we observed a negative correlation between METTL3 and CCL5 in AML patients77 and in leukemia stem cell enriched patient samples78 (Figure S6N–O). Additionally, we found a negative correlation between SON and CCL5 expression among METTL3 low AML patients78 (Figure S6P). Taken together, these results suggest that the m6A-SON-CCL5 axis may play a role in both normal and malignant contexts.
Discussion
Our study combines global m6A mapping, functional assays, and single-cell analysis to identify SON as a downstream effector of m6A RNA methylation, controlling early stem cell fate decisions and inflammation. We demonstrated that SON enhances the functional in human CD34+ HSPCs, suggesting that targeting SON and its downstream program may be a promising strategy for expanding HSC function similar to a prior study that found the SON-E isoform improves in vitro replating47. Importantly, our data on SON’s essential role in normal hematopoiesis phenocopies patients with the ZTTK syndrome that have bone marrows failures and leukopenia79. Some of these clinical features are likely due to defects in stem cell self-renewal and maintenance.
Given that SON forms the core component of nuclear speckle enriched for pre-mRNA splicing factors, the asymmetric segregation of SON may imply a close association of the splicing machinery with HSC fate determination. Interestingly, asymmetric segregation of nuclear speckles was reported to play a role in the post-transcriptional regulation essential for cell fate determination during spermatogenesis in M. vestita80. Further studies are needed to address the functional relevance of this observation in adult stem cells.
We and others have previously identified MYC as a functional m6A target18,19. Although MYC overexpression rescued HSPC engraftment in m6A-deficient HSCs, it also resulted in a fatal myeloproliferative and AML-like disease, indicating that additional mechanisms may be at play to fine-tune this program18. Unlike MYC, whose RNA stability and protein abundance are both reduced upon m6A loss, the abundance of SON RNA and protein is differentially regulated by m6A.
We found that SON directly binds Myc transcripts and enhances MYC protein abundance. SON and MYC may also interact on the common downstream targets of m6A in HSCs. Furthermore, SON’s regulation of MYC could be blocked by CCL5 administration, supporting a m6A-SON-CCL5 axis that also controls MYC. These data suggest a multistep regulatory control of symmetric commitment through MYC.
SON controls the inflammatory program through both indirect and direct mechanisms. Prior studies found that loss of m6A leads to abnormal formation of dsRNA that triggers an excessive innate interferon response34. We demonstrate that SON indirectly controls dsRNA formation through decreasing intron retention. The inflammatory program is further regulated directly by SON’s suppression of the expression of proinflammatory chemokine CCL5.
Previous studies have shown that CCL5 controls myeloid-biased HSCs and that elevated levels of CCL5 in the aging bone marrow microenvironment contributes to the HSC myeloid skewing phenotype71,81. Our findings demonstrate that exposure to CCL5 alters early HSC fate choices, resulting in reduced HSC symmetric commitment divisions and increased symmetric renewal divisions. This may provide a mechanism for the phenotypic expansion of aged HSCs with poor regenerative properties. Moreover, CCL5, which is targeted by SON, functions to attract T cells and is a promising marker for immunotherapy response in various cancers82–84. Overall, our findings illustrate how the SON RNA-binding protein, which is regulated by m6A, plays a critical role in both HSC cell fate determination and innate immune response.
Limitations of the study
In this study, we have identified SON as a critical m6A target using DART-seq. Although DART-seq offers significant advantages, such as antibody-free detection and compatibility with rare cells, it does have limitations. One limitation is its reliance on transgene overexpression levels and the cellular localization of the APOBEC1-YTH fusion protein. Our co-expression analysis suggests that the m6A-SON-CCL5 axis may also operate in a leukemia setting; however, further experimental testing is necessary to validate its functionality in various cellular contexts.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael G. Kharas (kharasm@mskcc.org).
Materials availability
There are no restrictions to the availability of all materials mentioned in the manuscript.
Data and code availability
DART-seq data (mouse and human), Single-cell RNA-seq data, and bulk RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOUCRCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| CD45.1-PE TexasRed | BD Bioscience | Cat# 562452, RRID:AB_11152958 | |
| CD45.2-A700 | Invitrogen | Cat# 56-0454-82, RRID:AB_657752 | |
| CD3-PECy5 | Invitrogen | Cat# 15-0031-83, RRID:AB_468691 | |
| CD8-PECy5 | Invitrogen | Cat# 15-0081-83, RRID:AB_468707 | |
| Gr1-PECy5 | Invitrogen | Cat# 15-5931-82, RRID:AB_468813 | |
| B220-PECy5 | Invitrogen | Cat# 15-0452-83, RRID:AB_468756 | |
| CD19-PECy5 | Invitrogen | Cat# 15-0193-83, RRID:AB_657673 | |
| Ter119-PECy5 | Invitrogen | Cat# 15-5921-82, RRID:AB_468810 | |
| cKit-APC-Cy7 | BioLegend | Cat# 105826, RRID:AB_1626278 | |
| Sca-1-Pacific Blue | BioLegend | Cat# 122520, RRID:AB_2143237 | |
| CD150-APC | BioLegend | Cat# 115910, RRID:AB_493460 | |
| CD48-PE | BD Bioscience | Cat# 557485, RRID:AB_396725 | |
| Gr1-APC | Invitrogen | Cat# 17-5931-82, RRID:AB_469476 | |
| Mac1-Pacific Blue | BioLegend | Cat# 101224, RRID:AB_755986 | |
| Ter119-PE-Cy5 | Invitrogen | Cat# 15-5921-82, RRID:AB_468810 | |
| CD41-PE | BD Bioscience | Cat# 558040, RRID:AB_397004 | |
| CD3-Pacific Blue | eBioscience | Cat# 48-0031-80, RRID:AB_10733280 | |
| CD4-PE | BD Bioscience | Cat# 557308, RRID:AB_396634 | |
| B220-PE-Cy7 | BD Bioscience | Cat# 552772, RRID:AB_394458 | |
| CD19-PE-Cy5 | Invitrogen | Cat# 15-0193-83, RRID:AB_657673 | |
| IgM-APC | BioLegend | Cat# 406509, RRID:AB_315059 | |
| Sca-1-PECy7 | BioLegend | Cat# 122514, RRID:AB_756199 | |
| CD48-FITC | BioLegend | Cat# 103404, RRID:AB_313019 | |
| human CD45 PerCP Cy5.5 | eBioscience | Cat# 45-0459-42, RRID:AB_10717530 | |
| mouse CD45 APC | BD Bioscience | Cat# 559864, RRID:AB_398672 | |
| anti-SON | Abcam | Cat# ab121759, RRID:AB_11132447 | |
| anti-CCL5 | thermo | Cat# 710001, RRID:AB_2532515 | |
| Anti-METTL3 | Proteintech | Cat# 150731-1-AP, RRID: AB_2142033 | |
| Anti-ACTIN | Sigma | Cat# A3854, RRID: AB_262011 | |
| Anti-YTHDF1 | Abcam | Cat# ab220162, RRID: AB_2892231 | |
| Anti-YTHDF2 | Proteintech | Cat# 24744-1-AP, RRID: AB_2687435 | |
| Anti-YTHDF3 | Abcam | Cat# A3854, RRID: AB_2868574 | |
| Anti-VINCULIN | NovusBio | Cat# NBP2-20859, RRID: N/A | |
| donkey anti-mouse Alexa fluor 568 | Invitrogen | Cat# A-10037, RRID: AB_2534013 | |
| donkey anti-rabbit Alexa fluor 647 | Invitrogen | Cat# A-31573, RRID:AB_2536183 | |
| dsRNA-specific antibody J2 | Abcam | Cat# ab288755 | |
| streptavidin-Cy5 | Invitrogen | Cat# 43-4316 | |
| anti-NUMB | Abcam | Cat# ab4147, RRID:AB_304320 | |
| anti-MYC | Cell Signaling | Cat# 5605, RRID:AB_1903938 | |
| donkey anti-goat Alexa fluor 488 | Invitrogen | Cat# A-11055, RRID:AB_2534102 | |
| donkey anti-goat Alexa fluor 647 | Invitrogen | Cat# A-21447, RRID:AB_141844 | |
| donkey anti-rabbit Alexa fluor 568 | Invitrogen | Cat# A10042, RRID:AB_2534017 | |
| Anti-mouse IgG | Sigma | Cat# 12-371, RRID: AB_145840 | |
| anti-Flag antibody | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 | |
| Bacterial and virus strains | |||
| N/A | N/A | N/A | |
| Biological samples | |||
| Cord blood | National Cord Blood Program, NY Blood Center | N/A | |
| Chemicals, peptides, and recombinant proteins | |||
| pIpC HMW | InVivogen | Cat# Vac-Pic | |
| Pyronin Y | Sigma | Cat# 213519 | |
| Polybrene | Millipore | Cat# TR-1003-G | |
| Murine SCF | Peprotech | Cat# 250-03 | |
| Murine IL-3 | Peprotech | Cat# 213-13 | |
| Murine IL-6 | Peprotech | Cat# 216-16 | |
| Mouse TPO | Peprotech | Cat# 315-14 | |
| Mouse Flt3L | Peprotech | Cat# 250-31L | |
| SFEM medium | Stem Cell Technologies | Cat# NC9753895 | |
| X-vivo 10 medium | Lonza | Cat# BE04-380Q | |
| Human SCF | Peprotech | Cat# 300-07 | |
| Human Flt3L | Peprotech | Cat# 300-19 | |
| Human TPO | Peprotech | Cat# 300-18 | |
| Human IL7 | Peprotech | Cat# 200-07 | |
| STM2457 | MedChemExpress | Cat# 134836 | |
| Recombinant mouse CCL5 | R&D Systems | Cat# 478-MR-025 | |
| Critical commercial assays | |||
| Murine CD117 MicroBeads Kit | Miltenyi Biotec | Cat# 130-091-224 | |
| Verso cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# AB1453-B | |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4367659 | |
| Human CD34 MicroBeads Kit | Miltenyi Biotec | Cat# 130-046-702 | |
| Dynabeads Protein A/G | Invitrogen | Cat# 10015D | |
| M.O.M (Mouse on Mouse) Immunodetection kit | Vector Laboratories | Cat# BMK-2202 | |
| Neon transfection Kit | Invitrogen | Cat# N-1096 | |
| Click-iT Nascent RNA Capture Kit | Thermo Fisher Scientific | Cat# C10365 | |
| Deposited data | |||
| Mouse HSC and MPP DART-seq data | This paper | GSE231862 | |
| Human CD34 HSPC DART-seq data | This paper | GSE231861 | |
| Single cell RNA-seq data | This paper | GSE231863 | |
| Bulk RNA-seq data | This paper | GSE231637 | |
| Experimental models: Cell lines | |||
| Human: THP1 leukemia cell line | ATCC | TIB-202 | |
| Experimental models: Organisms/strains | |||
| Mouse: Mx1-cre Mettl3 flox/flox | Cheng et al18 | N/A | |
| Mouse: Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (RosaCAS9) | The Jackson Laboratory | Strain #024858 | |
| Mouse: RosaCAS9 Mx1-cre Mettl3 flox/flox | This paper | N/A | |
| Mouse: C57BL/6J | The Jackson Laboratory | Strain # 000664 | |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ | The Jackson Laboratory | Strain # 013062 | |
| Mouse: B6.SJL-Ptprc <a> /Boy | Taconic Biosciences | Strain # 4007 | |
| Oligonucleotides | |||
| shCCL5-1 oligo F: CCGGTCTTGATTCTGACCCTGTATACTCGAGTATACAGGGTCAGAATCAAGATTTTTG |
This paper | N/A | |
| shCCL5-1 oligo R: AATTCAAAAATCTTGATTCTGACCCTGTATACTCGAGTATACAGGGTCAGAATCAAGA |
This paper | N/A | |
| shCCL5-2 oligo F: CCGGCCACGTCAAGGAGTATTTCTACTCGAGTAGAAATACTCCTTGACGTGGTTTTTG |
This paper | N/A | |
| shCCL5-2 oligo R: AATTCAAAAACCACGTCAAGGAGTATTTCTACTCGAGTAGAAATACTCCTTGACGTGG |
This paper | N/A | |
| sgSON-1: cctgaaaacctgctcgatgt |
This paper | N/A | |
| sgSON-4: gggtcttagatacggagctg |
This paper | N/A | |
| sgCCL5-3: tgcagagggcggctgcagtg |
This paper | N/A | |
| sgCCL5-4: gtgaactaagtacctaccgt |
This paper | N/A | |
| Primers, see Supplementary Table S10 | This paper | N/A | |
| Targeted Son demethylation oligoes, see Supplementary Table S10 | This paper | N/A | |
| sgRNAs against mus YTHDF1/2/3, see Supplementary Table S10 | This paper | N/A | |
| Recombinant DNA | |||
| Lenti-EGFP-Blast | This paper | N/A | |
| Lenti-SON-EGFP-Blast | This paper | N/A | |
| MSCV-IRES-GFP | Addgene | # 20672 | |
| MSCV-APOBEC-YTH | This paper | N/A | |
| MSCV-SON-N-GFP | This paper | N/A | |
| MSCV-SON-C-GFP | This paper | N/A | |
| MSCV-SON-delDB-GFP | This paper | N/A | |
| MSCV-SON-RB-GFP | This paper | N/A | |
| MSCV-SON-DB-GFP | This paper | N/A | |
| MSCV-SON-SRRB-GFP | This paper | N/A | |
| FTO-dCas9 | Addgene | # 134781 | |
| PLKO.1.GFP | This paper | N/A | |
| PLKO5.sgRNA.EFS.GFP | Addgene | # 57822 | |
| Software and algorithms | |||
| Image J version 2.1.0 | Image J | N/A | |
| FlowJo software (version10.2) | FlowJo | N/A | |
| GraphPad Prism 9 | GraphPad Software | N/A | |
| Imaris | BITPLANE | N/A | |
| STAR | Dobrin et al.87 | https://github.com/alexdobin/STAR | |
| GATK | Broad institute | https://gatk.broadinstitute.org/hc/en-us/articles/360035531192 | |
| HOMER (version 4.11) | Heinz et al.88 | http://homer.ucsd.edu/homer/ | |
| HISAT3N (version 2.2.1-3n) | Zhang et al.78 | http://daehwankimlab.github.io/hisat2/hisat-3n/ | |
| Bullseye | Tegowski et al.89 | https://github.com/mflamand/Bullseye | |
| MEME suite (version 1.4.1) | Bailey et al.79 | https://meme-suite.org/meme/ | |
| Cutadapt (version 4.1) | Marcel Martin | https://github.com/marcelm/cutadapt | |
| HTSeq (version 2.0.2) | Putri et al.90 | https://github.com/htseq/htseq | |
| DESeq (version 1.34.0) | Love et al.91 | https://github.com/mikelove/DESeq2 | |
| EnhancedVolcano (version 1.12.0) | Blighe et al | https://github.com/kevinblighe/EnhancedVolcano | |
| VAST-TOOLS (version 2.5.1) | Irimia et al92 | https://github.com/vastgroup/vast-tools | |
| gplots (version 3.1.3) | T. Galili | https://github.com/talgalili/gplots | |
| Cell Ranger (version 7.1.0) | Zheng et al.93 | N/A | |
| Seurat (version 4.3.0) | Stuart et al.94 | https://github.com/satijalab/seurat | |
| Harmony (version 0.1.1) | Korsunsky et al.82 | https://github.com/immunogenomics/harmony | |
| clustree (version 0.5.0) | Oshlack et al.83 | https://github.com/lazappi/clustree | |
| SCINA (version 1.2.0) | Zhang et al.95 | https://github.com/jcao89757/SCINA | |
| scDissector | Martin et al.86 | https://github.com/effiken/scDissector | |
Experimental model and study participant details
Mice
Mx1-cre Mettl3 flox/flox mice were previously published18. We crossed the Mx1-cre Mettl3 flox/flox mice to the RosaCAS9 mice to generate the RosaCAS9 Mx1-cre Mettl3 flox/flox mice. Deletion of Mettl3 was initiated using pIpC HMW (InVivogen, Vac-Pic) by intraperitoneal injections at a dose of 10 mg/kg on 2 consecutive days as showed previously18. Littermates from both the Mx1-cre Mettl3 flox/flox mice and the RosaCAS9 Mx1-cre Mettl3 flox/flox mice were used for experiments. C57BL/6J mice were used as WT mice in experiments in Figure 1, Figure 2D–H, Figure 4C–G, Figure 6H–I, Figure S5E–G. RosaCAS9 mice were used in experiments in Figure 4A–B, Figure S3E–H and Figure S5A–D. NSG-SGM3 mice were used as recipients for human xenograft studies. B6-CD45.1 mice were used as recipients for all other transplantation experiments. Mice used for this experiment were 8 to 12 weeks of age. Both male and female mice were used. All mice were housed on a 12:12 hr light:dark cycle at 25C and received water and chow ad libitum. All the animal experiments were approved under the Institutional Animal Care and Use Committee.
HSPC Cell Sorting, Cell Culture, Viral Infection, and Transplant
LSK or HSC cells were sorted following protocol described above. For each experiment at least three mice were pooled for sorting per genotype. Sorted LSK cells were cultured in the SFEM medium (STEM CELL Technologies, NC9753895) supplemented with murine cytokines (50ng/mlSCF,10ng/mlIL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L, PeproTec). Cells were then transduced with high titer concentrated retroviral or lentiviral suspensions in the presence of 8mg/ml polybrene and followed with spin infection for 1.5hr. Next day, second round of transduction was performed as described. For transplant, 10,000 LSK cells plus 500K CD45.1 whole bone marrow cells were injected into lethally irradiated B6SJL congenic CD45.1 recipients. For chimerism, either peripheral blood or bone marrow cells was extracted and subjected to flow cytometry at different time points.
Human Cord Blood CD34+ Cells Purification and Xenotransplantation
Purification of human CD34+ HSPCs was performed as previously described. Human CD34+ HSPCs were purified from at least 6 mixed CB units (each unit from one healthy donor). Freshly purified CD34+ cells were cultured overnight in X-Vivo 10 media containing 1% BSA, 100ng/ml hSCF, 100ng/ml hFlt3L, 50ng/ml hTPO, and 10ng/ml hIL7, PeproTec. CD34+ cells were transduced with lentivirus expressing either control empty vector or SON. 2 days post infection, initial GFP+% were measured in transduced CD34+ cells. Equal amount of CD34+ cells (20k) were transplanted into NSG-SGM3 mice that were irradiated at 200rad a day before.
METHOD DETAILS
Plasmid Constructs and Transfection
For plasmid transfection, 293T cells were transfected with certain constructs using lipofectamine 2000 or lipofectamine 3000 (Invitrogen) following instruction. EGFP or SON-EGFP were cloned in lentiviral backbone with blasticidin selection. SON deletion mutants, including SON-delta DB (amino acids 1,263 – 1,818 deleted, based on SON full length sequence), SON-RB (aa 2054 ~ 2426, based on SON full length sequence), SON SR+RB (aa 1819 ~ 2426, based on SON full length sequence) and SON DB (aa 1263 – 1818, based on SON full length sequence), were cloned in MIGR1 retroviral backbone. SON-N and SON-C mutants were cloned in MIGR1 retroviral backbone and gifted by Dr. Erin Ahn’s laboratory. For sgRNA knockout experiment, sgRNAs were cloned in PLKO.5 backbone with GFP except otherwise noted. sgYTHDF1 sequences were: sgDF1–2: CCCGAGACGCCCCTACCTGG; sgDF1–3: GGTAAGGATCACTCATCGAG; sgDF1–4: GTGCCACCAAGGGAGCTAGG; sgYTHDF2 sequences were (sgYTHDF2 backbone were in ilentiguide-GFP): sgDF2–1: CATGGCTCTCGGATCCTCAC; sgDF2–2: GGCATGAATACTATAGACCA; sgDF2–3: CACCTCGGAACCGTGGCAGT; sgDF2–4: gaatactatagaccaaggga; sgYTHDF3 sequences were sgDF3–2: cctcacctgatccacgctag; sgDF3–4: attcttcaccgtaacccgag; sgSON sequences were: sg1: cctgaaaacctgctcgatgt; sg4: gggtcttagatacggagctg. sgCCL5 sequences were: sg3: tgcagagggcggctgcagtg; sg4: gtgaactaagtacctaccgt. For shRNA knockdown experiment, shRNAs were cloned in PLKO.1 backbone with GFP. shCCL5 sequences were:
shCCL5–1 oligo F: CCGGTCTTGATTCTGACCCTGTATACTCGAGTATACAGGGTCAGAATCAAGATTTTTG
shCCL5–1 oligo R: AATTCAAAAATCTTGATTCTGACCCTGTATACTCGAGTATACAGGGTCAGAATCAAGA
shCCL5–2 oligo F: CCGGCCACGTCAAGGAGTATTTCTACTCGAGTAGAAATACTCCTTGACGTGGTTTTTG
shCCL5–2 oligo R: AATTCAAAAACCACGTCAAGGAGTATTTCTACTCGAGTAGAAATACTCCTTGACGTGG
PCR and Quantitative RT-PCR
Genomic DNA from engrafted donor whole bone marrow cells was isolated by using genomic DNA extraction kit (QIAGEN) and used as template for PCR to genotype. For Mettl3 genotyping, primers are Forward: ACCACAACAGCCAAGGAACA; Reverse: CCGGAGCTCTGAAACCTTGT. For Mx1-cre genotyping, primers are Forward: TCCCAACCTCAGTACCAAGCCAAG; Reverse: ACGACCGGCAAACGGACAGAAGCA. For qRT-PCR, total RNA was extracted from cells using TRIZOL (Life Technologies) following the standard manual. Equal amount of RNA from samples was reverse transcribed into cDNA with Verso cDNA Synthesis Kit (Thermo), and qPCR was performed using an ABI7500 sequence detection system using primers together with SYBR green master mix (ABIsystems). Primers are listed below:
hMYC Forward: TTCGGGTAGTGGAAAACCAG
hMYC Reverse: CAGCAGCTCGAATTTCTTCC
hCCL5 Forward: GGTACCATGAAGGTCTCCGC
hCCL5 Reverse: CTTGACCTGTGGACGACTGC
hCXCL10 Forward: CCACGTGTTGAGATCATTGCT
hCXCL10 Reverse: TGCATCGATTTTGCTCCCCT
hCEBPB Forward: TTTGTCCAAACCAACCGCAC
hCEBPB Reverse: GCATCAACTTCGAAACCGGC
hTUBG1 Forward: CGGCTACACCCCTCTCACTA
hTUBG1 Reverse: CTGTGGACACCATCACGTTC
hACTIN Forward: GGACTTCGAGCAAGAGATGG
hACTIN Reverse: AGCACTGTGTTGGCGTACAG
mSon Forward: GGAGAGAAGGAGAAGGATTAGGA
mSon Reverse: CTGGTAAGGGCTTCCATTTCT
mCcl5 Forward: TGCTGCTTTGCCTACCTCTC
mCcl5 Reverse: TCCTTCGAGTGACAAACACGA
mCxcl10 Forward: CCACGTGTTGAGATCATTGCC
mCxcl10 Reverse: GAGGCTCTCTGCTGTCCATC
mActin Forward: ACCAACTGGGACGACATGGAGAAG
mActin Reverse: TACGACCAGAGGCATACAGGGACA
Flow Cytometry and Cell Sorting
Bone marrow cells were harvested and subjected to red blood cell lysis. For transplanted mice, we used CD45.1-PE TexasRed (BDBioscience,562452) and CD45.2-A700 (Invitrogen,56-0454-82) to distinguish donor and recipient cells. To measure HSPC compartments, cells were further stained with following cocktail: Lineage marker85 (CD3 (15-0031-83), CD8(15-0081-83), Gr1(15-5931-82), B220(15-0452-83), CD19(15-0193-83) and Ter119(15-5921-82))- PE-Cy5(Invitrogen, eBioscience)), cKit-APC-Cy7 (BioLegend, 105826), Sca-1-Pacific Blue (BioLegend, 122520), CD150-APC (BioLegend, 115910), CD48-PE (BD Bioscience, 557485). To monitor myeloid linage differentiation, cells were stained with cocktail including Gr1-APC (Invitrogen, 17-5931-82), Mac1-Pacific Blue (BioLegend, 101224), Ter119-PE-Cy5 (Invitrogen, 155921–82), CD41-PE (BD Bioscience, 558040). To monitor lymphoid linage differentiation, cells were stained with cocktail containing CD3-Pacific Blue (eBioscience, 48-0031-80), CD4-PE (BD Bioscience, 557308), B220-PE-Cy7 (BD Bioscience, 552772), CD19-PE-Cy5 (Invitrogen, 15-0193-83), IgM-APC (BioLegend, 406509). For cell cycle analyses PY/Hoechst staining, bone marrow cells stained with HSC surface markers (Linage marker (CD3, CD8, Gr1, B220, CD19 and Ter119)PE-Cy5, cKit-APC-Cy7, Sca-1-PECy7 (BioLegend, 122514), CD150-APC, CD48-FITC (BioLegend, 103404)) were resuspended in 500ul NASS buffer (1 phosphate citrate tablet (sigma), 0.9g EDTA, 0.45 g NaCl, 0.25 g BSA in 50ml ddH2O), 3.3ul diluted Hoechst (1:10 in NASS) was added followed by 30mins in room temperature. Samples were then incubated on ice for 5 mins to stop reaction. 2.5ul of diluted PY (1:100 of 10mg/ml PY stock in NASS) was added to samples. Incubate for 10mins at 4 degrees. Samples were washed with PBS before analysis. Cells were analyzed on a BD FACS LSR or Fortessa instrument. For HSPC (LSK), HSC (LSK, CD150+CD48-) or MPPs (MPP1: LSK, CD150-CD48-; MPP2: LSK, CD150+CD48+; MPP4: LSK, CD150-CD48+) cell sorting, bone marrow cells were harvested and incubated with MACS beads (CD117, Miltenyi Biotec, 130-091-224). Then enriched c-Kit+ cells were collected by running AutoMACS (Miltenyi Biotec) according to the manufacturer’s instructions. The cells were then stained with cocktail: Lineage marker (CD3, CD8, Gr1, B220, CD19 and Ter119)- PE-Cy5, cKit-APC-Cy7, Sca-1-Pacific Blue, CD150-APC, CD48-PE. Specific cell populations (HSCs and MPPs) were sorted on BD Aria machine. For human xenograft analysis in NSGS mice, human CD45 PerCP Cy5.5 (eBioscience 45-0459-42) and mouse CD45 APC (BD 559864) were used to distinguish donor and recipient cells.
Chemical Treatment
HSCs were sorted following protocol shown before. For each experiment at least 4 mice were pooled for sorting per genotype. Sorted HSCs were cultured with SFEM media (STEMCELL Technologies, NC9753895) containing 50ng/ml SCF,10ng/ml IL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L (PeproTec) in 96 round-bottom wells. For CCL5 treatment, recombinant murine CCL5 protein was added into the culture media at 30ng/ml for 16hr followed by the paired daughter cell assay. For METTL3 inhibitor treatment in murine HSCs, STM2457 was added in the culture media at a concentration of 20μM for 16hr followed by the paired daughter cell assay. The procedures of the paired daughter cell assay were described in the Immunofluorescence section.
Peripheral Blood Analysis
Peripheral blood was collected from the retro-orbital cavity using a heparinized glass capillary tube. For flow cytometry, peripheral blood was treated by red blood cell lysis to remove red blood cells and then applied to flow as described earlier.
Paired Daughter Cell Assay
Sorted HSCs were cultured with SFEM media (STEMCELL Technologies, NC9753895) containing 50ng/ml SCF,10ng/ml IL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L, PeproTec in 96 round-bottom wells for 16 h with or without the chemical treatment (STM2457 20μM or CCL5 30ng/ml). HSCs were then treated with 10nM Nocodazole for 24 h. Cells were then fixed with 1.6% paraformaldehyde 15 mins at room temperature and permeabilized with cold methanol followed by immunofluorescence staining described below. For SON overexpression, sorted HSCs were cultured with SFEM media (STEMCELL Technologies, NC9753895) containing 50ng/ml SCF,10ng/ml IL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L, PeproTec in 96 round-bottom wells for 16 h and performed electroporation as described below. We then treated cells with 10nM Nocodazole for 24hr the next morning. After incubation, cells were fixed with 1.6% paraformaldehyde 15 mins at room temperature and permeabilized with cold methanol followed by immunofluorescence.
Immunofluorescence
HSCs or MPP1, MPP2 and MPP4 populations were sorted following protocol shown before. For each experiment at least 4 mice were pooled for sorting per genotype. Fresh sorted cells were fixed with 1.6% paraformaldehyde 15 mins at room temperature and permeabilized with cold methanol. Fixed HSCs or MPPs were then cytospun onto glass slides and were blocked for 1h followed with staining on slides with anti-SON (Abcam 121759), or anti-CCL5 (thermo 710001) and secondary Ab (donkey anti-rabbit Alexa fluor 647, Invitrogen) with DAPI counterstaining.
For dsRNA staining, LSKs were purified from RosaCAS9 Mettl3 f/f and Mettl3 cKO mice, then fixed and cytospun onto glass slides as described. Cells were stained following the M.O.M (Mouse on Mouse) Immunodetection kit protocol. Briefly, slides were incubated for 1hr in M.O.M Mouse IgG blocking reagent, followed by two washes with PBS. Slides were incubated for 5 mins in working solution of prepared M.O.M diluent and 30 mins in dsRNA-specific antibody J2 (Abcam, ab288755) at 1:200 dilution. After two washes with PBS, slides were incubated with M.O.M Biotinylated anti-Mouse IgG reagent for 10mins. With another two washes with PBS, slides were incubated with streptavidin-Cy5 antibody at 1:200 dilution for 20mins with DAPI counterstaining.
For immunofluorescence in the paired daughter cell assays, fixed HSCs were then cytospun onto glass slides and were blocked for 1h followed with staining on slides with anti-NUMB (Abcam, ab4147), anti-MYC (Cell Signaling, 5605) and secondary Ab (donkey anti–goat Alexa fluor 488 or 647, Invitrogen, A11055 or A21447; donkey antirabbit Alexa fluor 568, Invitrogen, A10042; Molecular Probes) with DAPI counterstaining. We evaluate symmetric and asymmetric percentages based on the fluorescence signal intensity of each cell acquired by Axio Imager M2 microscope (Carl Zeiss) or confocal microscope and quantified by FIJI. Thresholds to determine NUMB high/low/asymmetric were set for experimental replicates. Briefly, both of daughter cells with high NUMB staining or average staining intensity is above NUMB high threshold and there is less than 2-fold difference in the daughter pairs, this condition was counted as symmetric commitment. Daughter pair cells were scored as a symmetric renewal division when both were low, or no staining or average staining intensity is below NUMB high threshold, and there is less than 2-fold difference in the daughter pairs. If else, the division was considered an asymmetric division55.
For immunofluorescence in the colocalization analysis, 293T cells were seeded in an 8-well chamber slide at a concentration of 100k/500ul media. The chamber slides were pre-coated with 200ul poly D lysine per well for 3–4 hours at room temperature. 293T cells were then transfected with SON-full length, SON-RB, SON-SR+RB, SON-DB constructs (1ug each) as previously described. 2 days post transfection, 293T cells were fixed with 4% PFA for 15 mins at room temperature and permeabilized with cold methanol. Fixed 293T cells were blocked for 1hr followed with staining on slides with anti-FLAG (Sigma, F1804), anti-SON (Abcam 121759) and secondary antibody (donkey anti–mouse Alexa fluor 568, Invitrogen, A10037; donkey antirabbit Alexa fluor 647, Invitrogen, A10042; Molecular Probes) with DAPI counterstaining. We evaluate colocalization based on the overlap between Flag (all the constructs are flag tagged) and endogenous SON signal using FIJI. It is worth noting that the SON antibody recognizes the SON’s DNA binding domain. Thus, SON-RB and SON-SR+RB will only be recognized by FLAG antibody due to lack of DNA binding domain, The colocalization analysis was then performed using confocal microscope and quantified by FIJI.
Western Blotting
For SON protein abundance detection, fresh LSK cells were sorted from Mettl3 f/f and Mettl3 cKO bone marrow cells following protocol described before. Sorted cells were then lysed in XT protein sample buffer and boiled for 5min. Whole-cell lysates were run on a 3%−8% gradient Tris-Acetate and transferred to a nitrocellulose membrane. Membranes were probed with the SON (Abcam 121759), METTL3 (Proteintech,150731-1-AP) and ACTIN (Sigma, A3854) antibody.
For YTHDF1/2/3 protein abundance detection, fresh RosaCAS9 WT bone marrow cells were harvested and incubated with MACS beads (CD117, Miltenyi Biotec, 130-091-224). Then enriched c-Kit+ cells were collected by running AutoMACS (Miltenyi Biotec) according to the manufacturer’s instructions. Ckit+ enriched bone marrow cells were then transduced with respective lentivirus including empty vector (EV) and sgYTHDF1/2/3. GFP+ cells were then sorted and lysed in Laemmli protein loading buffer and boiled for 5min. Whole-cell lysates were run on a 4%–15% gradient SDS–PAGE and transferred to a nitrocellulose membrane. Membranes were probed with the YTHDF1 (abcam, ab220162), YTHDF2 (Proteintech, 24744-1-AP), or YTHDF3 (abcam, ab220161) and ACTIN (Sigma, A3854) or VINCULIN (NovusBio, NBP2–20859) antibody.
Electroporation Transfection
Neon nucleofection of HSPCs was performed as described86. HSCs were sorted and cultured in the SFEM medium for 1–3 hours before transfection. 1ug Plasmids were electroporated into 50k-100k HSCs or LSKs by using 10ul Neon transfection system (Thermo Fisher Scientific) under a condition:1700V, 20ms, 1 pulse. If higher numbers of cells were needed, multiple electroporations were performed (e.g., 6 electroporations of 100,000 LSKs for 600,000 total cells).
Targeted demethylation on Son mRNA
HSCs were sorted following protocol shown before. For each experiment at least 5 mice were pooled for sorting. Sorted HSCs were cultured with SFEM media (STEMCELL Technologies, NC9753895) containing 50ng/ml SCF,10ng/ml IL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L (PeproTec) in 96 round-bottom wells overnight. Next day, HSCs were collected and targeted demethylation was performed as previously described52. Briefly, HSCs were electroporated with dCas9-FTO, and pLKO5 empty vector, sgRNA1–3, or sgRNA4–6, as well as the corresponding PAMer sequences. In each condition, 0.75ug dCas9-FTO, 0.25ug of each sgRNAs (1–3 or 4–6 combination, sgRNAs were designed to target surrounding the detected DART-seq target site on Son mRNA (Chr 16, position 91663413), with sgRNAs1–3 target the 5’ of the site and sgRNAs 4–6 target the 3’ of the site), and 1pmol of corresponding PAMer sequences to each sgRNAs (total of 3pmol) were added. Electroporations were performed as described above. HSCs were cultured for 2 days and fixed for immunofluorescence. sgRNA sequences and PAMer sequences were listed below (PAMer sequence were HPLC-purified and consist of mixed DNA and 2’OMe RNA bases, commercially synthesized by Integrated DNA Technologies):
sgRNA1: taaaagattttaaacatttg; PAMer1: aacatttgtggcagggtaaatagtaac
sgRNA2: gaatggaaaataaggcatga; PAMer2: aggcatgatggagagggcacaattga
sgRNA3: gttttttttaaatttatttc; PAMer3: tttatttcaggactgtaacttaagaat
sgRNA4: agcaatagaggctttttgct; PAMer4: tttttgctaggtctttttgaagttata
sgRNA5: ttagagaaagagcttgcatt; PAMer5: cttgcatttggtaagtaagcaatagag
sgRNA6: agttgggagagacttgtcag; PAMer6: cttgtcagagggacaagaacgctgaag
RNA-IP
THP1 cells overexpressing MIG empty vector or MIG-SON-RB-Flag were collected (20 × 106 cells were used per IP reaction) and washed twice with ice-cold PBS. Cells were lysed in ice-cold IP lysis buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl and 0.5% NP40) for 30 min on ice and frozen at −80 °C immediately to aid the lysis. On the day of IP, the lysate was centrifuged to precipitate the debris. Supernatant was collected and incubated with 7.5 μg of anti-mouse IgG (Sigma, 12–371) or anti-Flag antibody (Sigma-Aldrich, clone M2-F1804) overnight at 4 °C. RNA–SON-RB-Flag antibody complexes were pulled down using Dynabeads Protein A/G (Millipore) and washed five times in 100% IP lysis buffer, 70% IP lysis buffer and 30% PBS, 50% IP lysis buffer and 50% PBS, 30% IP lysis buffer and 70% PBS and 100% PBS. RNA was extracted using the phenol–chroloroform method and quantified by qRT–PCR.
Nascent Transcription
Empty vector control (MIG) and Flag SON-RB overexpressing THP1 cells were incubated with EU (5-ethynyl uridine) for 1hr and harvested. Total RNA was purified by Trizol and newly synthesized RNA transcripts were isolated using a Click-iT Nascent RNA Capture kit (Invitrogen). cDNA was synthesized from RNA using a SuperScript VILO cDNA Synthesis kit (Invitrogen). qRT–PCR was performed as described above.
Sample Preparation for DART-seq
HSC, MPP1, MPP2, and MPP4 were sorted from C57BL/6J mice based on cell surface marker. For each experiment at least 5 mice were pooled for sorting. Cells were then cultured in SFEM media (STEMCELL Technologies, NC9753895) containing 50ng/ml SCF,10ng/ml IL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L, PeproTec in 96 round-bottom wells and infected with high-titer MIG empty vector, or Apobec-YTH retrovirus. GFP+ cells were sorted two days post infection. 3 replicates were performed. For DARTseq in human cord blood CD34+ cells, human cord blood CD34+ cells were purified as previously described. Freshly purified CD34+ cells were cultured overnight in X-Vivo 10 media containing 1% BSA, 100ng/ml hSCF, 100ng/ml hFlt3L, 50ng/ml hTPO, and 10ng/ml hIL7, PeproTec and then transduced with retrovirus expressing either control empty vector or APOBEC-YTH fusion protein. 24hr post infection, CD34+ cells were treated with STM2457 at a concentration of 20μM for 2 days. GFP+ cells were sorted the next day. 3 replicates were performed.
Identification of RNA Editing Events in mouse and human DART-seq Data
RNA editing frequency in mouse DART-seq data was calculated as previously described37. Briefly, we aligned the paired-end RNA-seq reads to mouse (mm10) genome using STAR aligner55. Next, we followed the GATK56 workflow for calling variants in RNA-seq (https://software.broadinstitute.org/gatk/documentation/article?id=3891) to identify all the mutations in each RNA-seq library. We then restricted to the mutations within annotated mRNA transcripts, as well as restricting to C-to-T mutations in transcripts encoded by the forward strand and G-to-A mutations in transcripts encoded by the reverse strand. We also filtered out mutations found in the dbSNP database since they are most likely DNA-level mutations. We then combined the filtered sets of RNA editing events from all RNA-seq libraries of the same experiment and counted the number of reads containing reference (C/G) and alternative (T/A) alleles from each library at each site. For de novo motif discovery, we first extracted sequences extending 100 bp from both sides of each edit site and considered all these windows as the target sequence pool for the HOMER program. Overlapping sequences were merged into a single sequence. Background sequences with length 201 bp were randomly selected from 3′UTRs in the genome that did not overlap with the target sequence pool. We used the HOMER software to search for enriched motifs of length 6, 7, or 8, and regional oligomer auto-normalization of up to length 3. To identify the RNA editing events in the human CD34+ HSPC DART-seq data with STM2457 treatment, we aligned the paired-end RNA-seq reads to the human (hg38) genomes using HISAT3N87 (v2.2.1–3n) with –base-change C,T and –no-repeat-index as parameters. The Bullseye pipeline (https://github.com/mflamand/Bullseye) was employed to identify C-to-T mutations in each RNA-seq library. A minimal coverage of 10 reads and duplicate removal were required to construct the variant quantification matrices using the parseBAM.pl script, provided by the Bullseye pipeline. The Bullseye/Find_edit_site.pl was applied with parameters --ControlMinCoverage 10, --EditMinCoverage 10, --minEdit 2.5, --maxEdit 95, --editFoldThreshold 1, --bedt, --intron, and --extUTR. Additionally, --minEditSites 1 was used to account for edited sites that may appear as single positions instead of clusters. The MIG empty vector sample without the YTH construct served as the control sample for comparisons. Edited sites identified were collapsed per replicate and condition. A beta binomial distribution was fitted to each site to distinguish editing signal from background noise. Benjamin-Hochberg corrected P values were reported, and sites with a log2Fold edit greater than 1.5 and a adjusted P value < 0.05 were defined as significantly edited sites. Motif analysis of the DART-seq CD34+ data was performed using the memes package v1.4.1 (https://github.com/snystrom/memes), wrapper of the MEME suite v5.4.188. Briefly, we extracted 50 nt sequences around the edited site (edited site in the center) for significant sites (as defined previously) as well as non-significant sites. For motif discovery, we use the DREME (Discriminative Regular Expression Motif Elicitation89) algorithm, comparing the significant edited sites sequences against shuffled input sequences as control. Both analyses were performed independently between conditions.
Transposable Elements (TEs) Analysis
The pipeline used to analyze transposable elements (TEs) expression was adapted from67. Adapters were trimmed using Cutadapt v4.1. Paired-end read alignment onto the Mouse reference genome (mm10) and the LENTI-SON construct was performed with STAR v2.7.10b using the same parameters described in67. Precomputed RepeatMasker annotations v4.0.5 for all TEs were downloaded from RepeatMasker (https://www.repeatmasker.org/species/mm.html), which were then used to reconstruct full-length LTR copies with ‘one code to find them all v1.0’ with the default parameters. The reconstructed TEs annotation and the mm10 basic genes annotation from GENCODE v25 were merged and was used as the reference database for gene quantification in HTSeq v2.0.2. Next, we extracted the read counts for non-ambiguous, non-DNA TEs. P values for differential expression were computed with DESeq2 v1.34.0 and adjusted with the Benjamini-Hochberg Procedure. ERVs were classified as significantly deregulated if its absolute log2FC >= 1 and false discovery rate (FDR) <= 0.05. Volcano plots were generated using EnhancedVolcano v1.12.0.
Differential Alternative Splicing Analysis
Adapters were trimmed using Cutadapt v4.1. Differential alternative splicing analysis was performed on the trimmed reads with VAST-TOOLS v2.5.1 with the database Mm2, using the default parameters. Alternative splicing events were classified as significantly deregulated if its absolute change in mean frequency >= 0.05 and false discovery rate (FDR) <= 0.05 (percent spliced in for exon, percent 5’ splice site usage for 5’SS, percent 3’ splice site usage for 3’SS, percent intron retention for INT). Alternative splicing events were classified as being rescued by SON overexpression if it is either significantly upregulated in Mettl3 cKO with respect to WT and significantly downregulated in Mettl3 cKO SON overexpression with respect to that without overexpression (Dn rescue), or vice versa (Up rescue). To generate the heatmaps, Z-scores for each event were computed across all samples and plotted with gplots v3.1.3.
Bulk RNA-Seq Analysis
LSKs were sorted from Mettl3 f/f and Mettl3 cKO mice based on cell surface marker. Cells were then cultured in SFEM media (STEMCELL Technologies, NC9753895) containing 50ng/ml SCF,10ng/ml IL-3, and 10 ng/ml IL-6, 10ng/ml TPO and 20 ng/ml FLT3L, PeproTec) in 96 round-bottom wells and infected with high-titer empty vector, or SON overexpressing lentivirus. GFP+ cells were sorted post two days of infection. 3 replicates were performed. RNA was extracted from bulk cells using the SMARTer RNA extraction method for minimal cells RNA extraction. After amplification, cDNA was subjected to automated Illumina paired-end library construction. Libraries were sequenced on Illumina HiSeq2000 instruments with paired reads of 100 bp in length per sample. The samples were sequenced at 90–100 million reads per sample. Sequence data were aligned using the tophat software to the mouse (mm10) reference genome. Fragments Per Kilobase of transcript per Million mapped reads were calculated using the cufflink software.
Gene Set Enrichment and Enrichr Analysis
Gene set enrichment analysis were performed using the GSEA software (https://www.gsea-msigdb.org/gsea/index.jsp). Briefly, differentially expressed gene rank lists were generated based on gene expression profiles in Mettl3 cKO EV vs Mettl3 SON OE, or WT EV vs WT SON OE groups (cutoff padj<0.05, rank by log2FC). Annotated gene sets were obtained from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) or curated from66.
Enrichr analysis were performed using the Enrichr website (https://maayanlab.cloud/Enrichr/) 90–92. Gene sets (including m6A targets or differential expressed genes as shown in supplementary tables) generated with cutoff noted in figure legends were used. The combined score is computed by taking the log of the p-value from the Fisher exact test and multiplying that by the z-score of the deviation from the expected rank.
c = log(p) * z, where c = the combined score, p = Fisher exact test p-value, and z = z-score for deviation from expected rank.
Single-Cell RNA-Seq
Single-cell RNA-seq data processing, alignment, cell type classification and clustering: 10x data were processed using Cell Ranger 7.1.0 with default parameters. Reads were aligned to a custom-built reference that included the cDNA sequence from the Son Overexpression construct in addition to the mm10 reference. Downstream analysis of Cell Ranger results was performed using the Seurat package v4.3.093. Briefly, cells with < 300 detected genes or percentage of mitochondrial reads > 20% were filtered out. In addition, cells that showed lower number of genes than expected from the number of UMIs (< 1000) detected were filtered out as low-quality cells. The data was scaled, and normalization was performed using the SCTransform function, regressing out potential confounders, including the proportion of mitochondrial and ribosomal genes as well as the difference between the G2M and S phase scores. Principal component analysis was performed using variable genes. Normalized SCT values were integrating with Harmony v0.1.194 using the Son Overexpression and Mettl3 cKO as grouping variables. The 3000 most variable features were used to conduct a principal component analysis (PCA). Subsequently, clustering options were inspected using clustree v0.5.095, selecting a cluster resolution of 0.9. For cluster visualization, the Uniform Manifold Approximation and Projection (UMAP) algorithm was fed with the initial 40 principal components that demonstrated statistical significance. Clusters were annotated with a combination of manual and semi-supervised methods like SCINA v1.2.0, using as a reference the gene markers reported in our Mettl3 cKO study18 as well as previously reported marker genes96,97. Differential gene expression analysis was conducted using Seurat’s FindMarkers function with a log2-transformed fold change threshold of ≥ 0.25 to compare between cell types. Likewise, for the comparison of cell clusters across genotypes, the default Wilcox-Rank sum test in FindMarkers was used to identify DEGs present in at least 25% of the cells in either group (min.pct = 0.25). The final set of DEGs was determined by filtering for a P value adjusted < 0.05.
Maximum Likelihood Analysis
We calculated a gene expression estimation for each cluster defined in our WT LSK scRNA-seq data to perform maximum likelihood projection across genotypes. The cluster gene expression estimation was created by determining the fraction of unique molecular identifiers (UMIs) that mapped to each gene, relative to the total UMIs detected for that cluster. We then calculated the log-likelihood of each cell in our KOsp and Ccl5+ clusters to map to each of the gene expression models, separately for each genotype, using the getLikelihood function from the scDissector package98. Based on the log-likelihood values, we assigned the cells in the special clusters of the Mettl3 cKO and Mettl3 cKO + Son Overexpression conditions to their respective cell identities.
Co-expression Analysis Using Patient Data
Normalized expression data (RPKM), for n = 707 patient samples, was obtained from the BeatAML2 portal (http://www.vizome.org/aml2/)99. ggstatsplot (v0.11.1) package was use to visualize and calculate the Pearson correlation with confidence intervals of 95. Normalized expression data from sorted fractions of K Ng.’s AML cohort78. The data comprises BeadArray Illumina probes and was obtained using the package GEOquery (v2.64.2) and ggpmisc (v0.5.3) was used to generate the plots and add the Pearson correlation with confidence intervals of 95.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were processed using GraphPad Prism v.9. All analyses were performed using two-tailed Student’s t tests, except were stated otherwise. Graphs and error bars reflect means ± s.e.m., except were stated otherwise. In all corresponding figures, * represents p < 0.05. ** represents p < 0.01. *** represents p < 0.001. ns represents p > 0.05. Replicate information is indicated in the figures.
Supplementary Material
Highlights.
DART-seq identified SON as a m6A target critical for HSC commitment
SON rescues m6A HSC defect and is asymmetrically segregated during HSC division
SON enhances mouse and human HSC function
The m6A-SON-CCL5 axis controls HSC inflammation and fate
Acknowledgements
We would like to thank members of the Kharas laboratory for their discussions and advice. We thank the MSKCC Single-cell Analytics Innovation Lab, Integrated Genomics Operation (IGO), Flow Cytometry Core and Molecular Cytology Core. We acknowledge MSK cancer center grant NCI P30 CA 008748. H.L is supported by NYSTEM training award contract C32599GG, and K99 DK128602–01. This work was supported in part by a grant to Memorial Sloan Kettering Institute from the Howard Hughes Medical Institute through the Gilliam Fellows Program to I.W. M. G. K. is a Scholar of the Leukemia and Lymphoma Society and supported by the US NIH National Institute of Diabetes Digestive and Kidney Diseases Career Development Award; NIDDK NIH R01-DK101989–01A1; NCI 1R01CA193842–01, R01HL135564, R01 CA274249–01A1, and R01CA225231–01; the Starr Cancer Consortium; the Alex’s Lemonade Stand A Award; the LLS Translation Research Program; the Susan and Peter Solomon Fund; and D.A.L and M.G.K are supported by the Tri-Institutional Stem Cell Initiative 2016–014. D.A.L is supported by the National Heart Lung and Blood Institute (R01HL157387–01A1) and the MacMillan Family and the MacMillan Center for the Study of the Non-Coding Cancer Genome at the New York Genome Center. E.E.A is supported by R01 HL168659, R01 CA236911. Q.D.M and C.L.T is supported by R01 HG013328–01A1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
MGK is a SAB member of 858 Therapeutics and received honorarium from Kumquat, AstraZeneca and consulting at Transition Bio. D.A.L. has served as a consultant for AbbVie, AstraZeneca, and Illumina and is on the Scientific Advisory Board of Mission Bio, Pangea, Alethiomics, and C2i Genomics; D.A.L. has received prior research funding from BMS, 10x Genomics, Ultima Genomics, and Illumina unrelated to the current manuscript. Remaining authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Supplementary table:
Table S1: List of DARTseq sites and targets in mouse HSC and MPPs. Related to Figure 1.
Table S2: Enrichr analysis of HSC and MPP DART-seq targets. Related to Figure 1.
Table S3: List of human CD34+ HSPC DARTseq targets. Related to Figure 1.
Table S4: List of Top 50 genes per cluster in scRNA-seq. Related to Figure 5.
Table S5: List of differentially expressed genes in KOsp1–1 and ccl5 cluster between mettl3 cKO ctrl and mettl3 cKO SON overexpression. Related to Figure 5.
Table S6: List of differentially expressed genes among WT and Mettl3 cKO ctrl and SON overexpression bulkRNA-seq. Related to Figure 5.
Table S7: List of SON rescue genes, inflammatory genes and pathway enrichment. Related to Figure 5.
Table S8: Differential retrotransposon analysis. Related to Figure 5.
Table S9: List of genes with Differential alternative splicing events in WT ctrl Mettl3 KO ctrl and SON overexpression groups. Related to Figure 5.
Table S10: Oligoes used in this study.
Supplementary Movie 1: SON asymmetrically segregate in dividing HSCs. Blue: DAPI; Red: SON; Green: NUMB. Sorted HSCs were cultured for 16hrs and then treated with 10nM Nocodazole for 24h. HSC doublets were then fixed, cytospun on to slide followed by immunofluorescence. Z-stack confocal images of a representative SON asymmetric division HSC daughter pair were acquired. Scale bar was shown in the movie. Related to Figure 2.
Supplementary Movie 2: SON full length colocalize with endogenous nuclear speckle. Blue: DAPI; Red: SON; Green: Flag. 293T cells were transfected with Flag tagged SON-FL (full length). Z-stack confocal images of SON and Flag immunofluorescence were acquired. Scale bar was shown in the movie. Related to Figure 6.
Supplementary Movie 3: 40% of SON RB colocalize with endogenous nuclear speckle. Blue: DAPI; Red: SON; Green: Flag. 293T cells were transfected with Flag tagged SON-RB (RNA binding domain containing fragment). Z-stack confocal images of SON and Flag immunofluorescence were acquired. Scale bar was shown in the movie. Related to Figure 6.
Supplementary Movie 4: 60% of SON SR+RB colocalize with endogenous nuclear speckle. Blue: DAPI; Red: SON; Green: Flag. 293T cells were transfected with Flag tagged SON-SR+RB (serine arginine rich and RNA binding domain containing fragment). Z-stack confocal images of SON and Flag immunofluorescence were acquired. Scale bar was shown in the movie. Related to Figure 6.
Supplementary Movie 5: ~20% of SON DB colocalize with endogenous nuclear speckle. Blue: DAPI; Red: SON; Green: Flag. 293T cells were transfected with Flag tagged SON-DB (DNA binding domain containing fragment). Z-stack confocal images of SON and Flag immunofluorescence were acquired. Scale bar was shown in the movie. Related to Figure 6.
Reference:
- 1.Laurenti E, and Gottgens B (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seita J, and Weissman IL (2010). Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2, 640–653. 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metcalf D (2007). On hematopoietic stem cell fate. Immunity 26, 669–673. 10.1016/j.immuni.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Haas S, Trumpp A, and Milsom MD (2018). Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 22, 627–638. 10.1016/j.stem.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Brummendorf TH, Dragowska W, Zijlmans J, Thornbury G, and Lansdorp PM (1998). Asymmetric cell divisions sustain long-term hematopoiesis from single-sorted human fetal liver cells. J Exp Med 188, 1117–1124. 10.1084/jem.188.6.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrison SJ, and Kimble J (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068–1074. 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 7.Takano H, Ema H, Sudo K, and Nakauchi H (2004). Asymmetric division and lineage commitment at the level of hematopoietic stem cells: inference from differentiation in daughter cell and granddaughter cell pairs. J Exp Med 199, 295–302. 10.1084/jem.20030929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ho AD (2005). Kinetics and symmetry of divisions of hematopoietic stem cells. Exp Hematol 33, 1–8. 10.1016/j.exphem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Loeffler D, and Schroeder T (2021). Symmetric and asymmetric activation of hematopoietic stem cells. Curr Opin Hematol 28, 262–268. 10.1097/MOH.0000000000000644. [DOI] [PubMed] [Google Scholar]
- 10.Jassinskaja M, Gonka M, and Kent D (2023). Resolving the hematopoietic stem cell state by linking functional and molecular assays. Blood 10.1182/blood.2022017864. [DOI] [PMC free article] [PubMed]
- 11.Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. (2016). Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116. 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 13.Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, and Passegue E (2015). Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell 17, 35–46. 10.1016/j.stem.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, et al. (2014). Chronic variable stress activates hematopoietic stem cells. Nat Med 20, 754–758. 10.1038/nm.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cellot S, and Sauvageau G (2006). In vitro stem cell expansion: stepping closer towards self-renewal. Gene Ther 13, 1617–1618. 10.1038/sj.gt.3302804. [DOI] [PubMed] [Google Scholar]
- 16.Walasek MA, van Os R, and de Haan G (2012). Hematopoietic stem cell expansion: challenges and opportunities. Ann N Y Acad Sci 1266, 138–150. 10.1111/j.1749-6632.2012.06549.x. [DOI] [PubMed] [Google Scholar]
- 17.Cheshier SH, Prohaska SS, and Weissman IL (2007). The effect of bleeding on hematopoietic stem cell cycling and self-renewal. Stem Cells Dev 16, 707–717. 10.1089/scd.2007.0017. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Bruning JC, Vu LP, et al. (2019). m(6)A RNA Methylation Maintains Hematopoietic Stem Cell Identity and Symmetric Commitment. Cell Rep 28, 1703–1716 e1706. 10.1016/j.celrep.2019.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, and Ding L (2019). Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol 21, 700–709. 10.1038/s41556-019-0318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16, 191–198. 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer KD, and Jaffrey SR (2017). Rethinking m(6)A Readers, Writers, and Erasers. Annu Rev Cell Dev Biol 33, 319–342. 10.1146/annurev-cellbio-100616-060758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang P, Doxtader KA, and Nam Y (2016). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell 63, 306–317. 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. (2017). FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 31, 127–141. 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, Chen H, Su R, Yin Z, Li W, et al. (2020). RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 27, 64–80 e69. 10.1016/j.stem.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, Yin R, Shan Y, Wen J, Xie X, et al. (2020). Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 27, 81–97 e88. 10.1016/j.stem.2020.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y, Xie W, Pickering BF, Chu KL, Savino AM, Yang X, Luo H, Nguyen DT, Mo S, Barin E, et al. (2021). N(6)-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39, 958–972 e958. 10.1016/j.ccell.2021.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheng Y, Wei J, Yu F, Xu H, Yu C, Wu Q, Liu Y, Li L, Cui XL, Gu X, et al. (2021). A critical role of nuclear m6A reader YTHDC1 in leukemogenesis by regulating MCM complex-mediated DNA replication. Blood 138, 2838–2852. 10.1182/blood.2021011707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mapperley C, van de Lagemaat LN, Lawson H, Tavosanis A, Paris J, Campos J, Wotherspoon D, Durko J, Sarapuu A, Choe J, et al. (2021). The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J Exp Med 218. 10.1084/jem.20200829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. (2018). Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res 28, 904–917. 10.1038/s41422-018-0072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA, Sepulveda C, et al. (2019). Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 25, 137–148 e136. 10.1016/j.stem.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaccara S, and Jaffrey SR (2020). A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell 181, 1582–1595 e1518. 10.1016/j.cell.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y, Vasic R, Song Y, Teng R, Liu C, Gbyli R, Biancon G, Nelakanti R, Lobben K, Kudo E, et al. (2020). m(6)A Modification Prevents Formation of Endogenous Double-Stranded RNAs and Deleterious Innate Immune Responses during Hematopoietic Development. Immunity 52, 1007–1021 e1008. 10.1016/j.immuni.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG, et al. (2021). Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601. 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guirguis AA, Ofir-Rosenfeld Y, Knezevic K, Blackaby W, Hardick D, Chan YC, Motazedian A, Gillespie A, Vassiliadis D, Lam EY, et al. (2023). Inhibition of METTL3 results in a cell-intrinsic interferon response that enhances anti-tumour immunity. Cancer Discov 10.1158/2159-8290.CD-23-0007. [DOI] [PubMed]
- 37.Nguyen DTT, Lu Y, Chu KL, Yang X, Park SM, Choo ZN, Chin CR, Prieto C, Schurer A, Barin E, et al. (2020). HyperTRIBE uncovers increased MUSASHI-2 RNA binding activity and differential regulation in leukemic stem cells. Nat Commun 11, 2026. 10.1038/s41467-020-15814-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brannan KW, Chaim IA, Marina RJ, Yee BA, Kofman ER, Lorenz DA, Jagannatha P, Dong KD, Madrigal AA, Underwood JG, and Yeo GW (2021). Robust single-cell discovery of RNA targets of RNA-binding proteins and ribosomes. Nat Methods 18, 507–519. 10.1038/s41592-021-01128-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer KD (2019). DART-seq: an antibody-free method for global m(6)A detection. Nat Methods 16, 1275–1280. 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu L, Liu S, Peng Y, Ge R, Su R, Senevirathne C, Harada BT, Dai Q, Wei J, Zhang L, et al. (2022). m(6)A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol 40, 1210–1219. 10.1038/s41587-022-01243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oguro H, Ding L, and Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116. 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J, Calogero RA, Klein AM, and Camargo FD (2018). Clonal analysis of lineage fate in native haematopoiesis. Nature 553, 212–216. 10.1038/nature25168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin R, Chang J, Li Y, Gao Z, Qiu Q, Wang Q, Han G, Chai J, Feng M, Wang P, et al. (2022). Differential m(6)A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell 29, 149–159 e147. 10.1016/j.stem.2021.09.014. [DOI] [PubMed] [Google Scholar]
- 44.Vu LP, Cheng Y, and Kharas MG (2019). The Biology of m(6)A RNA Methylation in Normal and Malignant Hematopoiesis. Cancer Discov 9, 25–33. 10.1158/2159-8290.CD-18-0959. [DOI] [PubMed] [Google Scholar]
- 45.Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123. 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. (2018). METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 22, 191–205 e199. 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW, Huang G, Yan X, Pauli-Behn F, Myers RM, et al. (2016). SON and Its Alternatively Spliced Isoforms Control MLL Complex-Mediated H3K4me3 and Transcription of Leukemia-Associated Genes. Mol Cell 61, 859–873. 10.1016/j.molcel.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu X, Ng HH, and Bubulya PA (2014). The role of SON in splicing, development, and disease. Wiley Interdiscip Rev RNA 5, 637–646. 10.1002/wrna.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, and Yang F (2020). A de novo heterozygous variant in the SON gene is associated with Zhu-Tokita-Takenouchi-Kim syndrome. Mol Genet Genomic Med 8, e1496. 10.1002/mgg3.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR, Bosch DGM, Bubulya PA, Shashi V, Petrovski S, et al. (2016). De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome. Am J Hum Genet 99, 711–719. 10.1016/j.ajhg.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376. 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu XM, Zhou J, Mao Y, Ji Q, and Qian SB (2019). Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol 15, 865–871. 10.1038/s41589-019-0327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, and Jaffrey SR (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Flamand MN, Ke K, Tamming R, and Meyer KD (2022). Single-molecule identification of the target RNAs of different RNA binding proteins simultaneously in cells. Genes Dev 36, 1002–1015. 10.1101/gad.349983.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, Morgan K, Tam W, Paktinat M, Okabe R, et al. (2010). Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med 16, 903–908. 10.1038/nm.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu M, Kwon HY, Rattis F, Blum J, Zhao C, Ashkenazi R, Jackson TL, Gaiano N, Oliver T, and Reya T (2007). Imaging hematopoietic precursor division in real time. Cell Stem Cell 1, 541–554. 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Florian MC, Dorr K, Niebel A, Daria D, Schrezenmeier H, Rojewski M, Filippi MD, Hasenberg A, Gunzer M, Scharffetter-Kochanek K, et al. (2012). Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 10, 520–530. 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, Sesaki H, Li T, Yu J, Wunderlich M, et al. (2020). Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 26, 420–430 e426. 10.1016/j.stem.2020.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loeffler D, Wehling A, Schneiter F, Zhang Y, Muller-Botticher N, Hoppe PS, Hilsenbeck O, Kokkaliaris KD, Endele M, and Schroeder T (2019). Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573, 426–429. 10.1038/s41586-019-1531-6. [DOI] [PubMed] [Google Scholar]
- 60.Ahn EY, DeKelver RC, Lo MC, Nguyen TA, Matsuura S, Boyapati A, Pandit S, Fu XD, and Zhang DE (2011). SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol Cell 42, 185–198. 10.1016/j.molcel.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Morree A, and Rando TA (2023). Regulation of adult stem cell quiescence and its functions in the maintenance of tissue integrity. Nat Rev Mol Cell Biol 10.1038/s41580-022-00568-6. [DOI] [PMC free article] [PubMed]
- 62.Brunet A, Goodell MA, and Rando TA (2023). Ageing and rejuvenation of tissue stem cells and their niches. Nat Rev Mol Cell Biol 24, 45–62. 10.1038/s41580-022-00510-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB, Pastor-Flores D, Roma LP, Renders S, Zeisberger P, et al. (2017). Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 169, 807–823 e819. 10.1016/j.cell.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 64.Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 65.Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sanchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, et al. (2015). Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 16, 712–724. 10.1016/j.stem.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodriguez-Fraticelli AE, Weinreb C, Wang SW, Migueles RP, Jankovic M, Usart M, Klein AM, Lowell S, and Camargo FD (2020). Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature 583, 585–589. 10.1038/s41586-020-2503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, Dossin F, Fouassier C, Lameiras S, and Bourc’his D (2021). m(6)A RNA methylation regulates the fate of endogenous retroviruses. Nature 591, 312–316. 10.1038/s41586-020-03135-1. [DOI] [PubMed] [Google Scholar]
- 68.Xu W, Li J, He C, Wen J, Ma H, Rong B, Diao J, Wang L, Wang J, Wu F, et al. (2021). METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591, 317–321. 10.1038/s41586-021-03210-1. [DOI] [PubMed] [Google Scholar]
- 69.Chen YG, and Hur S (2022). Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol 23, 286–301. 10.1038/s41580-021-00430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bowling EA, Wang JH, Gong F, Wu W, Neill NJ, Kim IS, Tyagi S, Orellana M, Kurley SJ, Dominguez-Vidana R, et al. (2021). Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 184, 384–403 e321. 10.1016/j.cell.2020.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ergen AV, Boles NC, and Goodell MA (2012). Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 119, 2500–2509. 10.1182/blood-2011-11-391730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim J, Venkata NC, Hernandez Gonzalez GA, Khanna N, and Belmont AS (2020). Gene expression amplification by nuclear speckle association. J Cell Biol 219. 10.1083/jcb.201904046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, and Belmont AS (2018). Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217, 4025–4048. 10.1083/jcb.201807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spector DL, and Lamond AI (2011). Nuclear speckles. Cold Spring Harb Perspect Biol 3. 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, Bailis W, Cao G, Kroehling L, Chen Y, et al. (2017). m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342. 10.1038/nature23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131. 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. (2018). Functional genomic landscape of acute myeloid leukaemia. Nature 562, 526–531. 10.1038/s41586-018-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, Arruda A, Popescu A, Gupta V, Schimmer AD, et al. (2016). A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437. 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 79.Vukadin L, Kim JH, Hackwelder TB, Ungerleider N, Flemington E, and Ahn EYE (2019). Hematological Disorders in Human Patients with Son Mutations. Blood 134. 10.1182/blood-2019-130616. [DOI] [Google Scholar]
- 80.Boothby TC, and Wolniak SM (2011). Masked mRNA is stored with aggregated nuclear speckles and its asymmetric redistribution requires a homolog of Mago nashi. BMC Cell Biol 12, 45. 10.1186/1471-2121-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Piryani SO, Kam AYF, Vu UT, Chao NJ, and Doan PL (2019). CCR5 Signaling Promotes Murine and Human Hematopoietic Regeneration following Ionizing Radiation. Stem Cell Reports 13, 76–90. 10.1016/j.stemcr.2019.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mowat C, Mosley SR, Namdar A, Schiller D, and Baker K (2021). Anti-tumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10. J Exp Med 218. 10.1084/jem.20210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Huffman AP, Lin JH, Kim SI, Byrne KT, and Vonderheide RH (2020). CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight 5. 10.1172/jci.insight.137263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reschke R, Yu J, Flood B, Higgs EF, Hatogai K, and Gajewski TF (2021). Immune cell and tumor cell-derived CXCL10 is indicative of immunotherapy response in metastatic melanoma. J Immunother Cancer 9. 10.1136/jitc-2021-003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Challen GA, Pietras EM, Wallscheid NC, and Signer RAJ (2021). Simplified murine multipotent progenitor isolation scheme: Establishing a consensus approach for multipotent progenitor identification. Exp Hematol 104, 55–63. 10.1016/j.exphem.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 86.Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, Hsu JI, Hoegenauer KA, Rooney CM, Goodell MA, and Nakada D (2016). Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep 17, 1453–1461. 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang Y, Park C, Bennett C, Thornton M, and Kim D (2021). Rapid and accurate alignment of nucleotide conversion sequencing reads with HISAT-3N. Genome Res 31, 1290–1295. 10.1101/gr.275193.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res 43, W39–49. 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bailey TL (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene Set Knowledge Discovery with Enrichr. Curr Protoc 1, e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zappia L, and Oshlack A (2018). Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7. 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang R, Zhang P, Wang J, Ma L, E W, Suo S, Jiang M, Li J, Chen H, Sun H, et al. (2023). Construction of a cross-species cell landscape at single-cell level. Nucleic Acids Res 51, 501–516. 10.1093/nar/gkac633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. (2015). Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677. 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 98.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, Gettler K, Chuang LS, Nayar S, Greenstein AJ, et al. (2019). Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell 178, 1493–1508 e1420. 10.1016/j.cell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bottomly D, Long N, Schultz AR, Kurtz SE, Tognon CE, Johnson K, Abel M, Agarwal A, Avaylon S, Benton E, et al. (2022). Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell 40, 850–864 e859. 10.1016/j.ccell.2022.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DART-seq data (mouse and human), Single-cell RNA-seq data, and bulk RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper does not report original code. Microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOUCRCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| CD45.1-PE TexasRed | BD Bioscience | Cat# 562452, RRID:AB_11152958 | |
| CD45.2-A700 | Invitrogen | Cat# 56-0454-82, RRID:AB_657752 | |
| CD3-PECy5 | Invitrogen | Cat# 15-0031-83, RRID:AB_468691 | |
| CD8-PECy5 | Invitrogen | Cat# 15-0081-83, RRID:AB_468707 | |
| Gr1-PECy5 | Invitrogen | Cat# 15-5931-82, RRID:AB_468813 | |
| B220-PECy5 | Invitrogen | Cat# 15-0452-83, RRID:AB_468756 | |
| CD19-PECy5 | Invitrogen | Cat# 15-0193-83, RRID:AB_657673 | |
| Ter119-PECy5 | Invitrogen | Cat# 15-5921-82, RRID:AB_468810 | |
| cKit-APC-Cy7 | BioLegend | Cat# 105826, RRID:AB_1626278 | |
| Sca-1-Pacific Blue | BioLegend | Cat# 122520, RRID:AB_2143237 | |
| CD150-APC | BioLegend | Cat# 115910, RRID:AB_493460 | |
| CD48-PE | BD Bioscience | Cat# 557485, RRID:AB_396725 | |
| Gr1-APC | Invitrogen | Cat# 17-5931-82, RRID:AB_469476 | |
| Mac1-Pacific Blue | BioLegend | Cat# 101224, RRID:AB_755986 | |
| Ter119-PE-Cy5 | Invitrogen | Cat# 15-5921-82, RRID:AB_468810 | |
| CD41-PE | BD Bioscience | Cat# 558040, RRID:AB_397004 | |
| CD3-Pacific Blue | eBioscience | Cat# 48-0031-80, RRID:AB_10733280 | |
| CD4-PE | BD Bioscience | Cat# 557308, RRID:AB_396634 | |
| B220-PE-Cy7 | BD Bioscience | Cat# 552772, RRID:AB_394458 | |
| CD19-PE-Cy5 | Invitrogen | Cat# 15-0193-83, RRID:AB_657673 | |
| IgM-APC | BioLegend | Cat# 406509, RRID:AB_315059 | |
| Sca-1-PECy7 | BioLegend | Cat# 122514, RRID:AB_756199 | |
| CD48-FITC | BioLegend | Cat# 103404, RRID:AB_313019 | |
| human CD45 PerCP Cy5.5 | eBioscience | Cat# 45-0459-42, RRID:AB_10717530 | |
| mouse CD45 APC | BD Bioscience | Cat# 559864, RRID:AB_398672 | |
| anti-SON | Abcam | Cat# ab121759, RRID:AB_11132447 | |
| anti-CCL5 | thermo | Cat# 710001, RRID:AB_2532515 | |
| Anti-METTL3 | Proteintech | Cat# 150731-1-AP, RRID: AB_2142033 | |
| Anti-ACTIN | Sigma | Cat# A3854, RRID: AB_262011 | |
| Anti-YTHDF1 | Abcam | Cat# ab220162, RRID: AB_2892231 | |
| Anti-YTHDF2 | Proteintech | Cat# 24744-1-AP, RRID: AB_2687435 | |
| Anti-YTHDF3 | Abcam | Cat# A3854, RRID: AB_2868574 | |
| Anti-VINCULIN | NovusBio | Cat# NBP2-20859, RRID: N/A | |
| donkey anti-mouse Alexa fluor 568 | Invitrogen | Cat# A-10037, RRID: AB_2534013 | |
| donkey anti-rabbit Alexa fluor 647 | Invitrogen | Cat# A-31573, RRID:AB_2536183 | |
| dsRNA-specific antibody J2 | Abcam | Cat# ab288755 | |
| streptavidin-Cy5 | Invitrogen | Cat# 43-4316 | |
| anti-NUMB | Abcam | Cat# ab4147, RRID:AB_304320 | |
| anti-MYC | Cell Signaling | Cat# 5605, RRID:AB_1903938 | |
| donkey anti-goat Alexa fluor 488 | Invitrogen | Cat# A-11055, RRID:AB_2534102 | |
| donkey anti-goat Alexa fluor 647 | Invitrogen | Cat# A-21447, RRID:AB_141844 | |
| donkey anti-rabbit Alexa fluor 568 | Invitrogen | Cat# A10042, RRID:AB_2534017 | |
| Anti-mouse IgG | Sigma | Cat# 12-371, RRID: AB_145840 | |
| anti-Flag antibody | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 | |
| Bacterial and virus strains | |||
| N/A | N/A | N/A | |
| Biological samples | |||
| Cord blood | National Cord Blood Program, NY Blood Center | N/A | |
| Chemicals, peptides, and recombinant proteins | |||
| pIpC HMW | InVivogen | Cat# Vac-Pic | |
| Pyronin Y | Sigma | Cat# 213519 | |
| Polybrene | Millipore | Cat# TR-1003-G | |
| Murine SCF | Peprotech | Cat# 250-03 | |
| Murine IL-3 | Peprotech | Cat# 213-13 | |
| Murine IL-6 | Peprotech | Cat# 216-16 | |
| Mouse TPO | Peprotech | Cat# 315-14 | |
| Mouse Flt3L | Peprotech | Cat# 250-31L | |
| SFEM medium | Stem Cell Technologies | Cat# NC9753895 | |
| X-vivo 10 medium | Lonza | Cat# BE04-380Q | |
| Human SCF | Peprotech | Cat# 300-07 | |
| Human Flt3L | Peprotech | Cat# 300-19 | |
| Human TPO | Peprotech | Cat# 300-18 | |
| Human IL7 | Peprotech | Cat# 200-07 | |
| STM2457 | MedChemExpress | Cat# 134836 | |
| Recombinant mouse CCL5 | R&D Systems | Cat# 478-MR-025 | |
| Critical commercial assays | |||
| Murine CD117 MicroBeads Kit | Miltenyi Biotec | Cat# 130-091-224 | |
| Verso cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# AB1453-B | |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4367659 | |
| Human CD34 MicroBeads Kit | Miltenyi Biotec | Cat# 130-046-702 | |
| Dynabeads Protein A/G | Invitrogen | Cat# 10015D | |
| M.O.M (Mouse on Mouse) Immunodetection kit | Vector Laboratories | Cat# BMK-2202 | |
| Neon transfection Kit | Invitrogen | Cat# N-1096 | |
| Click-iT Nascent RNA Capture Kit | Thermo Fisher Scientific | Cat# C10365 | |
| Deposited data | |||
| Mouse HSC and MPP DART-seq data | This paper | GSE231862 | |
| Human CD34 HSPC DART-seq data | This paper | GSE231861 | |
| Single cell RNA-seq data | This paper | GSE231863 | |
| Bulk RNA-seq data | This paper | GSE231637 | |
| Experimental models: Cell lines | |||
| Human: THP1 leukemia cell line | ATCC | TIB-202 | |
| Experimental models: Organisms/strains | |||
| Mouse: Mx1-cre Mettl3 flox/flox | Cheng et al18 | N/A | |
| Mouse: Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (RosaCAS9) | The Jackson Laboratory | Strain #024858 | |
| Mouse: RosaCAS9 Mx1-cre Mettl3 flox/flox | This paper | N/A | |
| Mouse: C57BL/6J | The Jackson Laboratory | Strain # 000664 | |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ | The Jackson Laboratory | Strain # 013062 | |
| Mouse: B6.SJL-Ptprc <a> /Boy | Taconic Biosciences | Strain # 4007 | |
| Oligonucleotides | |||
| shCCL5-1 oligo F: CCGGTCTTGATTCTGACCCTGTATACTCGAGTATACAGGGTCAGAATCAAGATTTTTG |
This paper | N/A | |
| shCCL5-1 oligo R: AATTCAAAAATCTTGATTCTGACCCTGTATACTCGAGTATACAGGGTCAGAATCAAGA |
This paper | N/A | |
| shCCL5-2 oligo F: CCGGCCACGTCAAGGAGTATTTCTACTCGAGTAGAAATACTCCTTGACGTGGTTTTTG |
This paper | N/A | |
| shCCL5-2 oligo R: AATTCAAAAACCACGTCAAGGAGTATTTCTACTCGAGTAGAAATACTCCTTGACGTGG |
This paper | N/A | |
| sgSON-1: cctgaaaacctgctcgatgt |
This paper | N/A | |
| sgSON-4: gggtcttagatacggagctg |
This paper | N/A | |
| sgCCL5-3: tgcagagggcggctgcagtg |
This paper | N/A | |
| sgCCL5-4: gtgaactaagtacctaccgt |
This paper | N/A | |
| Primers, see Supplementary Table S10 | This paper | N/A | |
| Targeted Son demethylation oligoes, see Supplementary Table S10 | This paper | N/A | |
| sgRNAs against mus YTHDF1/2/3, see Supplementary Table S10 | This paper | N/A | |
| Recombinant DNA | |||
| Lenti-EGFP-Blast | This paper | N/A | |
| Lenti-SON-EGFP-Blast | This paper | N/A | |
| MSCV-IRES-GFP | Addgene | # 20672 | |
| MSCV-APOBEC-YTH | This paper | N/A | |
| MSCV-SON-N-GFP | This paper | N/A | |
| MSCV-SON-C-GFP | This paper | N/A | |
| MSCV-SON-delDB-GFP | This paper | N/A | |
| MSCV-SON-RB-GFP | This paper | N/A | |
| MSCV-SON-DB-GFP | This paper | N/A | |
| MSCV-SON-SRRB-GFP | This paper | N/A | |
| FTO-dCas9 | Addgene | # 134781 | |
| PLKO.1.GFP | This paper | N/A | |
| PLKO5.sgRNA.EFS.GFP | Addgene | # 57822 | |
| Software and algorithms | |||
| Image J version 2.1.0 | Image J | N/A | |
| FlowJo software (version10.2) | FlowJo | N/A | |
| GraphPad Prism 9 | GraphPad Software | N/A | |
| Imaris | BITPLANE | N/A | |
| STAR | Dobrin et al.87 | https://github.com/alexdobin/STAR | |
| GATK | Broad institute | https://gatk.broadinstitute.org/hc/en-us/articles/360035531192 | |
| HOMER (version 4.11) | Heinz et al.88 | http://homer.ucsd.edu/homer/ | |
| HISAT3N (version 2.2.1-3n) | Zhang et al.78 | http://daehwankimlab.github.io/hisat2/hisat-3n/ | |
| Bullseye | Tegowski et al.89 | https://github.com/mflamand/Bullseye | |
| MEME suite (version 1.4.1) | Bailey et al.79 | https://meme-suite.org/meme/ | |
| Cutadapt (version 4.1) | Marcel Martin | https://github.com/marcelm/cutadapt | |
| HTSeq (version 2.0.2) | Putri et al.90 | https://github.com/htseq/htseq | |
| DESeq (version 1.34.0) | Love et al.91 | https://github.com/mikelove/DESeq2 | |
| EnhancedVolcano (version 1.12.0) | Blighe et al | https://github.com/kevinblighe/EnhancedVolcano | |
| VAST-TOOLS (version 2.5.1) | Irimia et al92 | https://github.com/vastgroup/vast-tools | |
| gplots (version 3.1.3) | T. Galili | https://github.com/talgalili/gplots | |
| Cell Ranger (version 7.1.0) | Zheng et al.93 | N/A | |
| Seurat (version 4.3.0) | Stuart et al.94 | https://github.com/satijalab/seurat | |
| Harmony (version 0.1.1) | Korsunsky et al.82 | https://github.com/immunogenomics/harmony | |
| clustree (version 0.5.0) | Oshlack et al.83 | https://github.com/lazappi/clustree | |
| SCINA (version 1.2.0) | Zhang et al.95 | https://github.com/jcao89757/SCINA | |
| scDissector | Martin et al.86 | https://github.com/effiken/scDissector | |