

ABSTRACT
Matrix remodeling outcomes largely dictate patient survival post myocardial infarction. Moreover, human-restricted noncoding regulatory elements have been shown to worsen fibrosis, but their mechanism of action remains elusive. Here, we demonstrate, using induced pluripotent stem cell-derived cardiac fibroblasts (iCFs), that inflammatory ligands abundant in the remodeling heart after infarction activate AP-1 transcription factor signaling pathways resulting in fibrotic responses. This observed signaling induces deposition of fibronectin matrix and is further capable of supporting immune cell adhesion; pathway inhibition blocks iCF matrix production and cell adhesion. Polymorphisms in the noncoding regulatory elements within the 9p21 locus (also referred to as ANRIL) redirect stress programs, and in iCFs, they transcriptionally silence the AP-1 inducible transcription factor GATA5. The presence of these polymorphisms modulate iCF matrix production and assembly and reduce cell–cell signaling. These data suggest that this signaling axis is a critical modulator of cardiac disease models and might be influenced by noncoding regulatory elements.
Keywords: Inflammation, Fibrosis, Myocardial regeneration, Gene expression, Gene regulation, Myocardial infarction
Summary: A novel AP-1 signaling axis modulates cardiac fibroblast stress responses to agonists post-myocardial infarction. Polymorphisms in human non-coding regulatory elements suppress this signaling axis.
INTRODUCTION
Heart disease is the leading cause of death worldwide, causing 18.6 million fatalities annually (Virani et al., 2021). After acute myocardial infarction (MI), a scar is formed to maintain tissue integrity and prevent cardiac rupture; however, the formation of a rigid scar impairs both mechanical and electrical coupling of the tissue, leading to worsened heart function. Although fibrosis occurs in many organ systems in various states of disease (Wynn, 2004), mechanisms are not always conserved, and thus need to be investigated in a context-dependent manner. Additionally, tissue-level organization can impact fibrotic models – as an extreme example, the heart lacks a commensal mucosal barrier, which is known to drive inflammation in idiopathic pulmonary fibrosis and Crohn's disease (Glass et al., 2020; Stange and Schroeder, 2019). Similarly, niche composition and organization differ between the heart and liver, with cells from the same developmental origin (yolk sac macrophages) driving seemingly opposite fibrotic responses (Bennett et al., 2020; Dick et al., 2019). Therefore, tissue-restricted environmental cues can drastically alter matrix processes. Moreover, although many molecular and genetic tools are available for study of disease in mice, they lack many of the regulatory regions of the genome that exist in higher primates, and thus do not always yield the same phenotype (Jarinova et al., 2009; Stange and Schroeder, 2019; Visel et al., 2010). This is evident from the difficulty of inducing coronary artery disease in mice (Liao et al., 2015), and is why some groups turn to other animal models. The wealth of experiments performed in mice, however, provide an excellent starting point for computational predictions, assuming they are ultimately confirmed in orthogonal models. Taken together, this suggests that a human cardiac-specific model is most likely to serve as a litmus test for murine observations.
Many studies from the past few decades have identified single receptors, ligands or biophysical stimuli that modulate fibrosis following MI. We previously sought to organize these signaling ‘nodes’ into a network by analysis of several RNA-seq datasets, cell-type knockout models, and biochemical studies from existing literature, and found that many inflammatory agonists have been reported to activate AP-1 transcription factor stress pathways, including transforming growth factor β (TGFβ), angiotensin II (AngII) and low molecular weight hyaluronic acid (LMW-HA) (Whitehead and Engler, 2021). Furthermore, in regenerative models, these processes are either inhibited or are only weakly induced by models of infarction. Given these previous observations, we hypothesize that convergent signaling through these pathways ultimately determines matrix outcomes, as this would explain why single molecule, ligand or receptor inhibition only partially rescues heart fibrosis in vivo.
Through the use of our recent induced pluripotent stem cell (iPSC)-derived cardiac fibroblast protocol, we are also able to investigate how these stress responses and pathways might be altered in the presence of disease-causing long non-coding RNA (lncRNA) single nucleotide polymorphisms (SNPs). Our previous work has demonstrated that lncRNA SNPs at the 9p21 locus act through JNK, a family of kinases upstream of AP-1, to cause gap junction decoupling in cardiomyocytes (Kumar et al., 2019). Recent evidence demonstrates that AP-1 family proteins cooperatively interact with these lncRNAs to promote activation of smooth muscle cells (Lo Sardo et al., 2018; Zhao et al., 2019). We hypothesize that this mechanism might be conserved across cardiac cellular populations and manifest in matrix differences in cardiac fibroblasts. At a population level, an estimated 25% of humans are homozygous for these SNPs, which clinically present with increased arrhythmias, sudden cardiac death and poor post-MI remodeling – maladies typically governed by fibroblasts. For these reasons, we generated human cardiac-specific epicardial fibroblasts (iCFs; Whitehead et al., 2022) from an H9 cell line as well as pluripotent stem cells with lncRNA SNPs at the 9p21 locus (Lo Sardo et al., 2018) to test our murine-derived computational model of sterile inflammation resulting in fibrosis (Whitehead and Engler, 2021), and used haplotype-dependent differences to investigate abnormal stress responses in the context of 9p21 SNPs.
In this study, we computationally and experimentally identify AP-1 signaling pathways as critical modulators of fibroblast stress responses and matrix production. Specifically, we find that AP-1 promotes cardiac fibroblast fibrotic responses and also enhances GATA5 transcription factor expression. The observed signaling pathway is shown to enhance the expression and assembly of fibronectin, which promotes the adhesion of monocytes to the resulting extracellular matrix (ECM). Finally, we find that lncRNA SNPs at the 9p21 locus redirect AP-1 downstream signaling and disrupt gap junction production and remodeling. Given the highly coordinated and complex nature of wound healing, disruption of ECM formation and immune cell adhesion is likely to lead to detrimental outcomes following MI.
RESULTS
MI-mediated inflammation suppresses regeneration in adult hearts
The regenerative potential of hearts is largely linked with age, where older hearts are considered nonregenerative compared to their younger counterparts (Aurora et al., 2014; Giudice et al., 2014; Wang et al., 2020). To better understand the molecular pathways involved in age-dependent heart regeneration, we analyzed a single-cell cardiac ATAC sequencing dataset produced by Wang et al. (2020) (GSE153481) to discern fibroblast-specific subpopulation changes with age and following infarction. Initial clustering of fibroblasts demonstrated that non-regenerative mice at 3 days following an MI procedure [postnatal day (P)8 3d MI] largely contribute to a unique population (Fig. 1A, cluster 4) that is enriched in collagen 1 and fibronectin peaks (Fig. 1B), suggesting that these cells are responsible for matrix responses post-MI. Moreover, cluster 4 was highly enriched in AP-1 family motifs (Fig. 1C,D) relative to the other clusters, suggesting that AP-1 differences are driving the transcriptional programs. AP-1 motifs, such as MA144-1 and MA0476-1, are also uniquely enriched in nonregenerative infarcted hearts as a whole (Fig. 1E). Other potential pathways have also been proposed to help explain the age-related decrease in heart regeneration. For example, using pre-plated cardiac fibroblasts from P1, P28 and P60 mice sequenced by Giudice et al. (2014), we are able to see a drastic increase in an inflammatory phenotype, through upregulation of nuclear factor κB (NFκB) and TNF signaling pathways, in the first four weeks of age (Fig. S1A–D). This correlates with the loss of regenerative ability after the first week of life (Wang et al., 2019), and raises the possibility of age-dependent inflammatory priming driving altered healing responses in vivo. Together, these data suggest an important role for AP-1 signaling and inflammation in modulating fibroblast stress responses following MI, which could ultimately influence regenerative outcomes.
Fig. 1.

Enrichment of AP-1 family motifs in infarcted hearts. (A) Clustering of cardiac fibroblasts colored by sample (left) and non-biased cluster (right) from neonatal day 1 (P1), neonatal day 8 (P8) and 3 days post-MI (3d MI) or sham control (3d Sham) treated mice. (B) Score of collagen and fibronectin accessibility using the add_score_module function of Signac. (C) Top motif results from ChromVar when contrasting cluster 4 with 1. Similar results were obtained when comparing against each other cluster. (D) Violin plot of AP-1 motif enrichment in accessible regions for each cluster. (E) Feature plot of the AP-1 motif, split by sample origin. The arrow highlights previously identified cluster 4. Data adapted from Wang et al. (2020), GSE153479.
Post-MI stress agonists upregulate matrix production and lead to AP-1 phosphorylation
To determine the role of AP-1 in driving matrix responses, we first employed H9 embryonic stem cell (ESC)-derived cardiac fibroblasts and challenged them with agonists predicted to generate large matrix responses in adult hearts (Whitehead and Engler, 2021). TGF-β, AngII, and LMW-HA were found to induce an upregulation of the cellular isoform of fibronectin (as measured by antibodies specific to the EDA domain), but not of collagen I after 3 days of treatment (Fig. 2A). Given that 2D matrices do not support BMP-1 cleavage of pro-collagen 1 preventing collagen fibril assembly in shorter timeframes (<2 weeks) (Lareu et al., 2007; Puerta Cavanzo et al., 2020), and artificially inducing BMP stabilization through crowding agents yields varied matrix responses (Chen et al., 2009; Rønnow et al., 2020), we instead decided to assay fibronectin. Fibronectin is generally expressed in response to the agonists used and within the same timeframe as type 1 and 3 fibrillar collagens (Whitehead and Engler, 2021); therefore, the resulting protein can be used in downstream functional assays. Next, we measured phosphorylation of the AP-1 pathway (via c-Jun) we previously predicted to drive matrix formation. We found that all three agonists drove a peak in phosphorylation of c-Jun after 30 min of stimulation, while total transcription factor expression remained constant (Fig. 2B). Similarly, immunofluorescence analysis following 30 min post-agonist treatment revealed enhanced expression of phosphorylated c-Jun (Fig. 2C). Additionally, ESC-derived cardiac fibroblasts treated with both an agonist as well as T-5224, a pharmacological AP-1 inhibitor (AP-1i; Tsuchida et al., 2004), were observed to reduce phosphorylation of NFκB, suggesting possible AP-1 involvement in NFκB activation (Fig. S2A), as has been previously reported (Ji et al., 2019). To further probe the interactions between the two pathways, mass spectrometry was performed on lysates obtained following a phosphorylated NFκB immunoprecipitation (Table S1). The proteins identified were then compared against known NFκB and AP-1 pathway molecules using the STRING database (Szklarczyk et al., 2019). The data presented show complex protein associations between samples, thus suggesting potential interconnectivity among pathways in response to agonist stimulation (Fig. S2B). Independently, we also found that phosphorylation of NF-κB (via its p65 subunit, also known as RelA) was enhanced 60 min post agonist stimulation (Fig. S3A,B). However, the full scope of interactions is beyond the scope of this work. Nonetheless, AP-1 is found to activate in response to agonists.
Fig. 2.

Stress response agonists enhance matrix expression and phosphorylate key transcription factors. (A) Representative western blot (left) and quantification (right) of cellular fibronectin, collagen, and actin as a loading control after the indicated treatment of iCF cells for 72 h. The bars denote mean for three independent experiments. **P<0.01; N.S., not significant (one-way ANOVA with Dunnett's test). (B) Representative western blots of three biological replicates for phosphorylated AP-1 (p-AP-1) and total AP-1 following 0, 15, 30, 60 and 120 min of agonist treatment. The arrowhead highlights the brightest band with highest p-AP-1 expression. (C) Representative immunofluorescence images (left) and quantification (right) of phospho-c-Jun (p-AP-1) levels (30 min post-agonist treatment). Error bars denote mean (n=3). **P<0.01 (one-way ANOVA with Dunnett's).
AP-1 activation promotes matrix expression and assembly
To elucidate the role of AP-1 in modulating fibroblast matrix responses, we pharmacologically inhibited the binding of AP-1 factors to DNA (Tsuchida et al., 2004). We found that inhibition of AP-1 reduced the expression of fibronectin in response to TGFβ, LMW-HA, AngII or a combination of all agonists (Fig. 3A). Furthermore, agonist treatment enhanced the assembly and deposition of fibronectin on to glass coverslips (Fig. 3B). This in turn allowed U937 monocytes, a human myeloid leukemia cell line that is traditionally grown in suspension but adheres to RGD adhesion domains, to adhere to the assembled matrix (Fig. 3C; Fig. S4). We observed that decellularized matrices from cardiac fibroblasts (CFs) treated with both AP-1i and agonists had reduced monocyte adhesion (Fig. 3C). These data suggest that AP-1 activation not only increases cellular expression of fibronectin, but also enhances the matrix assembly allowing for the deposition of functional ECM. This, in turn, promotes monocyte adhesion, demonstrating that AP-1 activation in fibroblasts can functionally recruit immune cells to the site of injury. NFκB inhibition yielded similar results with reduced fibronectin expression and monocyte adhesion (Fig. S5), further demonstrating the interconnectivity between the two pathways. These data confirm the ability of AP-1 to influence fibroblast stress responses. However, additional downstream targets and the relevancy of the elucidated pathway to cardiovascular disease remain unclear.
Fig. 3.
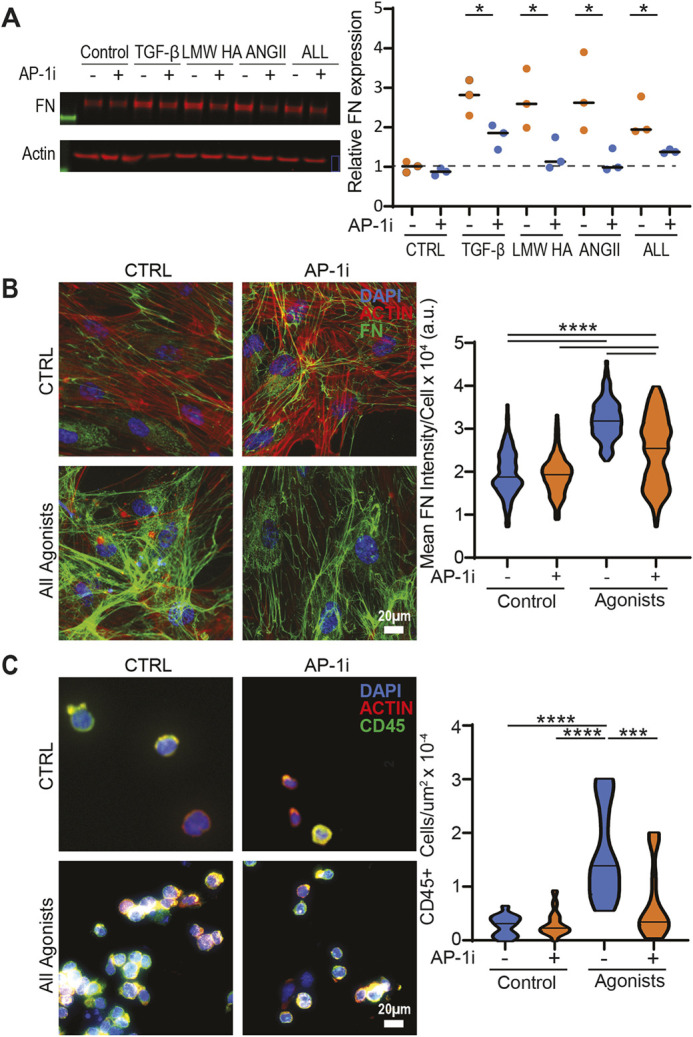
AP-1 mediates agonist matrix responses. (A) Representative western blot (left) and quantification (right) of cellular fibronectin (FN) in control versus AP-1 inhibitor (T-5224)-treated iCFs exposed to TGFβ, LMW HA, AngII or a combination of all agonists for 72 h. Error bars denote mean for three independent experiments, *P<0.05 (unpaired two-tailed Student's t-test). (B) Representative immunofluorescent images of iCFs treated with TGFβ, LMW HA and ANGII for 72 h (left) and quantification of mean FN signal per cell (right) presented as a violin plot with median marked. ****P<0.0001 (one-way ANOVA with Dunnett's test). a.u., arbitrary units. (C) Representative immunofluorescent images of U937 monocytes adhered to decellularized matrices obtained from control versus AP-1 inhibitor exposed iCFs treated with TGFβ, LMW HA and AngII for 72 h. The quantification shows number of CD45+ monocytes adhered per µm2, with results are presented as a violin plot with median marked (n=15). **P<0.01; ***P<0.001 (nonparametric Kruskal–Wallis test).
ANRIL modulates stress responses via the AP-1 and GATA5 signaling pathway
Given that AP-1 coordinates fibroblast responses to physiological events like myocardial infarction, we wanted to investigate how AP-1 might interact with regions of the genome associated with cardiovascular disease, specifically the 9p21 locus, also referred to as ANRIL. This non-coding region of the genome contains ∼61 SNPs in high linkage disequilibrium and with a Mendelian distribution across the global population; SNPs within ANRIL are also correlated with high clinical incidence of recurrent MI, arrythmias and sudden cardiac death, pathologies associated with matrix remodeling (Helgadottir et al., 2007; Jarinova et al., 2009; Lo Sardo et al., 2018; Newton-Cheh et al., 2009; Yamagishi et al., 2009). Our group has previously demonstrated that the presence of these SNPs alters cardiomyocyte responses to stiffness, resulting in differences in gap junction assembly and suggesting that ANRIL non-coding RNA might be induced by stress (Kumar et al., 2019). To probe this hypothesis, we generated six iCF lines in triplicate, spanning two individuals with ANRIL SNPs. For each homozygous risk (RR) line, which contains SNPs within ANRIL, a TALEN knockout (RKO) of the SNP-containing region was performed, generating three isogenic clonal comparisons (Fig. 4A). Moreover, HOMER motif analysis predicted two AP-1-binding sites at the promoter region of ANRIL, a region in which ENCODE UCSC genome browser tracks highlight as a probable super enhancer (Fig. 4B). To further understand how ANRIL SNPs alter genomic stress responses, and given that the many permutations of AP-1 complexes eliminate the feasibility of single transcription factor CHIP-seq, we performed ATAC-sequencing on RR and RKO cell lines. After excluding lowly accessible regions (concentration <3 in Diffbind), 3569 sites were found to be differentially accessible [log2 fold change±2, false discovery rate (FDR)<0.01] between risk and knockout lines (Fig. 4C). HOMER motif finding identified enrichment in AP-1-binding motifs in both risk and knockout lines. Interestingly, we found that 581 regions with AP-1 motifs are unique to RR lines and 94 regions are unique to RKO lines (Table S2). However, only knockout lines contained GATA motifs (Fig. 4D). Regions containing either motif were intersected, generating a list of regions that contain both GATA and AP-1 motifs (Table S3). Connexin 43 (GJA1) was predicted to be co-regulated by both transcription factor families, and accessibility at the promoter was found to be suppressed in RR lines (Fig. 4E). Additionally, the promoter region of GATA5 was inaccessible in RR cells, suggesting that ANRIL SNPs silence GATA5 epigenetically. The ANRIL promoter region was also found to be equally accessible between genotypes (Fig. 4B), further suggesting that SNPs, not lncRNA accessibility, are driving CF differences. Together, these data identify differential binding sites for AP-1 between RR and RKO cell lines.
Fig. 4.

ANRIL is AP-1 inducible and transcriptionally silences GATA5. (A) Schematic of cell lines used. Peripheral blood mononuclear cells (PBMCs) from RR individuals are isolated and converted into iPSCs. These cells are then differentiated into CFs. (B) UCSC Genome track with ENCODE tracks labeling H3K27Ac (top) and representative IGV track demonstrating open chromatin at the promoter region. (C) Heatmap of differentially accessible regions between haplotypes. (D) Top 12 motifs, in order, for regions unique to risk and risk knockout haplotypes, respectively. AP-1 family motifs are colored blue and GATA family motifs are colored green. (E) IGV screenshots of all 18 lines, colored by haplotype (red, risk; green, risk knockout).
Given the complexity of the AP-1 signaling pathway, we next looked to identify specific AP-1 factors and their overall ability to modulate ANRIL-mediated cardiac fibroblast activation. To accomplish this, we first performed RNA-seq on RR and RKO cell lines (Fig. 5A). Through analysis of known AP-1 factors (Table S4), JunD was found to be the most highly expressed and could therefore be responsible for downstream AP-1 activation (Fig. 5B,C). Interestingly, JunD is also known to be involved in connexin 43 (GJA1) gene expression (Mitchell and Lye, 2005) and has been demonstrated to be able to bind within the gene body of GJA1 as demonstrated by H1 ESC ENCODE CHIP data (Fig. 5D). Experimentally, connexin 43 gene expression was also suppressed with AP-1 inhibition, and RR lines were observed to have reduced expression when compared to RKO lines (Fig. 6A). RKO lines had enhanced GATA5 expression; this was reduced in the presence of AP-1 inhibitor (Fig. 6B). Functionally, RKO cells had enhanced fibronectin expression, which was also dampened in the presence of AP-1 inhibitor (Fig. 6C). Consistent with this, RKO cells produce functional matrix allowing for enhanced monocyte adhesion, both of which are inhibited in the presence of AP-1i (Fig. 6D). These data suggest that AP-1 and GATA5 synergistically enhance CF matrix responses, and that ANRIL can disrupt this process. Overall, ANRIL-mediated AP-1 misdirection in the genome, combined with reduced transcription factor accessibility and expression of GATA5 suppresses CF matrix responses and connexin 43 expression (Fig. 6E). Given that healing following myocardial infarction is a highly coordinated and complex process, the suppression of ECM production and gap junction expression could disrupt remodeling, thus resulting in detrimental outcomes.
Fig. 5.

AP-1 factors are differentially expressed to RR and RKO cell lines. (A) Pearson correlation plots for RR (1–5, ED70 and 2–3) and RKO (1–9, ED65 and WB46) cell lines. (B) Heatmap displaying expression of AP-1 factors within RR and RKO cell lines. (C) Relative expression in transcripts per million (TPM) of JunD (n=3). The box represents the 25–75th percentiles, and the median is indicated. The whiskers show the range. (D) UCSC genome track with ENCODE tracks showing JunD binding at GJA1.
Fig. 6.

ANRIL mediates gap junction expression as well as matrix expression and assembly. (A) qPCR data of connexin 43 (Cx43) gene expression in RR and RKO iCFs with or without pharmacological AP-1 inhibition. Data normalized to RR control. Error bars denote mean (n=3). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001 (one-way ANOVA with Dunnett's test). (B) Representative western blot (left) and quantification (right) of GATA5 expression in RR and RKO iCFs with or without AP-1 inhibitor (T-5224). Error bars denote mean for three independent experiments. *P<0.05; **P<0.01 (one-way ANOVA with Dunnett's test). (C) Representative immunofluorescent images of 1–5 RR and 1–9 RKO iCFs treated with TGFβ, LMW HA and AngII for 72 h (left) and quantification of mean fibronectin signal per cell presented as a violin plot with median marked (right). **P<0.01; ****P<0.0001 (nonparametric Kruskal–Wallis test). (D) Quantification of the number of U937 monocytes adhered to decellularized matrices obtained from RR or RKO iCFs treated with TGFβ, LMW HA, and ANGII for 72 h presented as a violin plot with median marked. ****P<0.0001 (nonparametric Kruskal–Wallis test). (E) Agonists associated with stress responses following MI activate AP-1 resulting in enhanced fibronectin expression and monocyte adhesion. Long non-coding RNA SNPs at the 9p21 locus (also referred to as ANRIL) alter AP-1-suppressed activity through possible genomic redirection.
DISCUSSION
Fibrosis is a major detrimental outcome following MI as it impairs both mechanical and electrical coupling of the tissue eventually resulting in impaired heart function. In this study, we looked to better understand the molecular mechanisms associated with this process. Using recently established protocols, we demonstrated that iCFs can be used to model post-MI inflammation and found that AP-1 is a primary initiator of matrix responses to stress. AP-1 consists of a family of transcription factors that are composed of homodimers and heterodimers of Jun proteins (v-Jun, c-Jun, JunB and JunD), Fos proteins (v-Fos, c-Fos, FosB, Fral, and Fra2), ATF [ATF2, ATF3 (also known as LRF1), B-ATF, JDP1 and JDP2] and MAF proteins (c-Maf, MafB, MafA, MafG, MafF and MafK, and Nrl) (Angel and Karin, 1991; Hai et al., 1988; Ye et al., 2014). Activation of this pathway in response to stress stimuli resulted in enhanced matrix production, which was abrogated with pharmacological inhibition. Matrix production was not only enhanced intracellularly but we also observed an increase in extracellular assembled and functional matrix (Fig. 3B). This ECM allowed for the adhesion of U937 monocytes, which are non-adherent cells that would otherwise be grown in suspension, through integrin-mediated interactions (Fig. 3C; Fig. S4) (Chung and Kao, 2009; Gao et al., 2021; Harris and Ralph, 1985). Although other pathways have also been reported to regulate fibroblast function and fibrotic responses and monocyte adhesion, including SMAD signaling pathways (Hinz, 2007; Szeto et al., 2016; Verrecchia and Mauviel, 2002), our data show that stress responses through AP-1-related pathways are also critical and can result in the production of functional matrix as well as the adhesion of immune cells. The ability of multiple pathways to modulate the fibrotic response highlights their interconnectivity, as we have also demonstrated with the activation of NFκB and its role in enhancing fibronectin deposition in response to agonist stimulation (Figs S2 and S5). Further analysis, however, is required to fully uncover the network of pathways involved and their interactions with one another within the fibrosis.
Interestingly, we also demonstrate that SNPs in the lncRNA at the 9p21 locus, also referred to as ANRIL, produce a silencing effect on GATA5, a critical transcription factor for cardiac development (Bonachea et al., 2014; Reiter et al., 1999; Singh et al., 2010). These genetic abnormalities are associated with a number of different pathological states, including cardiovascular disease as well as various forms of cancer. Within cardiovascular diseases, ANRIL SNPs are correlated with recurrent MI, arrythmias and sudden cardiac death, all pathologies associated with matrix remodeling (Helgadottir et al., 2007; Jarinova et al., 2009; Lo Sardo et al., 2018; Newton-Cheh et al., 2009; Yamagishi et al., 2009). However, the mechanisms associated with disease progression are not clearly understood. In this study, we find that AP-1 works in conjunction with ANRIL SNPs to dictate stress responses. More specifically, we find that ANRIL SNPs transcriptionally silence GATA5, reduce the expression of connexin 43, and suppress fibronectin expression and assembly (Fig. 6A–D). These data suggest a novel pathway by which ANRIL mediates iCF activation. Although ANRIL is clinically associated with exacerbated cardiovascular disease post-MI, our results indicate that fibronectin matrix production might not be solely responsible for pathology. Instead, the stress programs that are canonically activated after MI are rewired in the presence of 9p21 SNPs – with 94 and 581 unique AP-1 binding motifs in RR and RKO lines, respectively – resulting in a five-fold net increase in unique AP-1 motifs, the physiological function of which remains unclear. When the RR AP-1 regions were submitted to GREAT GO, no ontological terms were found to be enriched (data not shown). The function of the silenced RKO AP-1 sites, however, is more easily deciphered. Connexin-43, a gap junction-forming protein, was found to be both less accessible and expressed at lower levels in RR cells, which could result in electrical decoupling and poor conductivity in heart tissues. Nevertheless, our study validates the use of iCFs in studying human specific pathology, as it has revealed that ANRIL SNPs present unique pathological challenges that directly influence cardiac fibroblast function and might play a pivotal role in post-MI outcomes.
Although we propose a novel pathway to further the understanding of myocardial fibrosis, there are several key questions that remain. For example, more biomimetic culture substrates similar in mechanical properties to physiological or pathological cardiac tissues could provide better insight into the mechanobiology involved in vitro. Although tissue culture plastic is a common first step, use of other three-dimensional substrates might also further validate the proposed mechanism. In addition, crosstalk with additional cell types that are commonly present within cardiac tissue, such as macrophages (Whitehead and Engler, 2021), could provide feedback mechanisms associated with the proposed signaling. Macrophages are key producers of TGFβ, and other inflammatory and healing cytokines associated with MI, which are also known to influence fibroblast activation and function (Biswas et al., 2012). Therefore, co-culture models might also provide a better understanding of the feedback and molecular mechanisms involved in fibroblast as well as macrophage function. Nevertheless, the data presented provide key steps into better understanding cardiac fibrosis.
MATERIALS AND METHODS
Ethical compliance and cell lines
The authors have complied with all ethical regulations approved by UCSD (IRB #141315) for all RR iPSCs, which were derived originally by the Scripps Research Institute (IRB #11-5676). Characterization of these lines has been previously reported elsewhere (Lo Sardo et al., 2018).
Differentiation and cell culture methods
RR iPSCs and H9 ESCs were cultured using mTeSR1 and passaged using Versene (15040066, Thermo Fisher Scientific), 5 μM ROCK inhibitor Y27632 (Y-5301, LC Labs) and cell scrapers prior to differentiation. Differentiations into cardiac fibroblasts were carried out as previously described (Whitehead et al., 2022). Briefly, iPSCs were seeded at 250,000–500,000 cells per well of 12-well tissue culture Matrigel-treated plates and converted into cardiac progenitor cells (CPCs) using 5–6 μM CHIR (SML1046, Sigma-Aldrich) and 0.25 μM IWP2 (S7085, Sellechem). Cells in these cultures were then converted into epicardial cells using 3 μM CHIR, and into cardiac fibroblasts over 20 days with 10 ng/ml bFGF (233-FB, RnD Systems). CFs were expanded and maintained in Fibroblast Growth Medium 3 (C-23025, Promocell) and passaged using Accutase for 5–10 min. All cells used for experiments were passage 5 or less. Any assays requiring matrix deposition were carried out in RPMI1640 plus 10% FBS and 100 mg/l ascorbic acid (36237, Alfa Aesar). For assays measuring protein phosphorylation, cells were serum starved overnight using basal Fibroblast Growth Medium 3 prior to agonist dosing. AP-1 was inhibited using 80 μM T-5224 (22904, Cayman Chemical), added concurrently with agonists. AngII (10 nM, 1158, Tocris 1158), LMW-HA (8–15 kDa, 30 ng/ml, 40583-10MG, Millipore Sigma) and TGF-β1 (10 ng/ml, 240-B-002, RnD Systems) were used as agonists, and were added daily for 3 days with cells harvested 72 h after the first dose.
RNA-seq mining
Sequencing files were obtained from the GEO database under accessions GSE49906 (Giudice et al., 2014) and GSE153481 (Wang et al., 2020). Bulk FASTQ files were aligned to the mm10 genome using STAR with the following settings: --readFilesCommand zcat --genomeLoad LoadAndRemove --outFilterType BySJout --outFilterMultimapNmax 10 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 4 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 100000 (Dobin et al., 2013). BAM files were sorted and indexed using samtools (Li et al., 2009). Raw and transcripts per kilobase million (TPM) normalized tag directories were generated using HOMER command makeTagDirectory and analyzeRepeats scripts (Heinz et al., 2010). Statistical significance for Giudice et al. raw counts was determined using EdgeR (Robinson et al., 2010) in the getDiffExpression HOMER script. Counts were read in using DGEList while library sizes and normalization factors were calculated from Tag Directory sizes. Reads were counted using DGEList, with each sample constituting a treatment in the design matrix. Common dispersion was estimated at 0.05 as recommended. P-values were generated using an Exact Test and corrected using the Benjamini–Hochberg method. Only genes with an adjusted P-value of 0.05, minimum fold change of ±2 and minimum 32 tags in one dataset per gene were considered. Biological process and molecular function gene ontologies were generated using Metascape (Zhou et al., 2019). Heatmaps and PCA plots of TPM-normalized values were generated using R and ggplot (https://cran.r-project.org/web/packages/ggplot2/index.html) and pheatmap (https://cran.r-project.org/web/packages/pheatmap/index.html) packages. Single-cell ATAC was processed using Signac (Stuart et al., 2021; version 1.4.0), Seurat (Stuart et al., 2021; version 4.0.5), and ChromVar (Schep et al., 2017; version 1.16.0) according to methods from Wang et al. (2020). ECM gene signature was compiled using the AddModuleScore function of Seurat using Fn1, Col1a1 and Col1a2 genes, and significance was calculated using stat_compare_means function in ggpubr (https://cran.r-project.org/web/packages/ggpubr/index.html) with a Kruskal–Wallis test for overall P-value generation and Wilcox signed-rank test to compare individual groups. The single-cell accessibility of motif MA1144-1 is shown, but similar results were obtained using other AP-1 motifs such as MA0476-1.
Western blotting
Samples were prepared as previously described (Whitehead et al., 2022). Briefly, cells were lysed then further agitated and amassed using cell scrapers, and vortexed every 5 min for 30 min total. Samples were then centrifuged at 23,000 g for 15 min and the supernatant was transferred to a new tube to remove DNA. Protein concentrations were calculated using a bicinchoninic acid assay (23225, Thermo Fisher Scientific), and after denaturing at 95°C for 5 min, 10 μg of protein in 30 μl of RIPA buffer was loaded per lane on a 4–12% Bis-Tris Plus gel (for phospho-proteins, NW04122BOX, Thermo Fisher Scientific) or 25 μl in 3–8% Tris-Acetate gel (for fibronectin and collagen 1, EA0375BOX, Themo Fisher Scientific) in reducing conditions. Gels were run at 80 V for 15 min, then 140 V for 1 h, and transferred using an iBlot nitrocellulose transfer membrane (IB301001, Thermo Fisher Scientific). Membranes were blocked using Azure Blot blocking buffer (AC2190, Azure Biosystems) for 1 h, incubated with primary antibodies (collagen 1, 14695-1-AP, Proteintech, 1:1000; fibronectin-EDA, NBP1-51723, Novus Bio, 1:2500; β-actin, ab-8226, Abcam, 1:5000; phospho-c-Jun, 9261S, Stem Cell Technologies, 1:1000; phospho-p65, 3033S, Stem Cell Technologies, 1:1000) overnight at 4°C and secondary antibodies (A11374 and A10038, Invitrogen, 1:5000) for 1 h at room temperature, and imaged using a LI-COR Odyssey (LI-COR, Lincoln, NE). Raw intensities were calculated using LI-COR ImageStudio Lite and significance was determined using a one-way ANOVA with a Kruskal–Wallis test.
Mass spectrometry and STRING analysis
Cells lysates were collected as described in the previous section. Lysates were treated with an anti-phospho-p65 (3033S, Stem Cell Technologies, 1:100) antibody prior to being incubated overnight at 4°C on a tube rotator. Pierce Protein A/G Magnetic Beads (Thermo Fisher Scientific, 88802) were then washed, added to the lysate and incubated overnight at 4°C. Beads were washed three times and proteins eluted using Lamaelli sample buffer (Bio-Rad, 1610747) with 50 mM DTT. The samples were boiled and then run on a gel followed by isolation of samples within the gel for mass spectrometry, which was performed by the biomolecular and proteomics mass spectrometry facility at UCSD. Proteins identified were then input the STRING database (Szklarczyk et al., 2019) along with known AP-1 and NFκB pathway signaling molecules to generate associations maps.
Immunofluorescent staining and analysis
Cells were fixed using 3.7% methanol-stabilized formaldehyde for 15 min and rinsed three times with PBS. Samples were blocked in 10% donkey serum, 0.3 M glycine and 1% bovine serum albumin for 1 h, permeabilized in blocking buffer with 0.1% Triton X-100 for 15 min, stained with primary antibodies (Gata5, AF2170, RnD Systems; Cx43, 71-0700, Invitrogen, both 1:100) for 2 h or overnight at 4°C, and then incubated with secondary antibodies (A21202, A10042, and A21447, Invitrogen) for another 2 h. Nuclei were stained with DAPI for 15 min at 1:10,000 dilution in deionized water, and three washes were performed between each incubation for 5 min each. Samples were imaged using a Keyence BZ-X microscope, keeping the exposure for each channel constant between sample groups. All image analysis was performed using FIJI (Schindelin et al., 2012). For transcription factor analysis, the intensity for each color was calculated and normalized to the number of nuclei in each image; a total of three randomized fields of view were analyzed across three or four biological replicates. For monocyte adhesion, the number of nuclei was normalized to the area imaged; five randomized fields of view were analyzed across three biological replicates. All other image analyses involved calculating the mean fluorescence intensity per cell. Briefly, a minimum of 150 cells were outlined and analyzed across five randomized fields of view and three biological replicates.
Decellularization and adhesion assay
CFs were grown for 3 days on sterile glass coverslips in RPMI1640 plus 10% FBS and 100 mg/l ascorbic acid for 3 days to allow for matrix deposition. Samples were decellularized using NP-40 according to a previously published protocol (Mao and Schwarzbauer, 2005), without the use of wash buffer 1 as it caused monolayer contraction. U937 human monocytes (CRL-1593.2, ATCC) were grown in RPMI1640 plus 10% FBS and seeded onto decellularized matrix at 1×106 cells per coverslip and allowed to attach for 2 h. Coverslips were then fixed and stained as described above.
ATAC-Seq
ATAC-sequencing was performed on RR (1–5, ED70 and 2–3) and RKO (1–9, ED65 and WB46) iPSC clones in triplicate. Library preparation and sequencing was performed by the UCSD Center for Epigenomics. ATAC-seq was performed on 50,000 nuclei per sample. Samples were permeabilized in cold permeabilization buffer [0.2% IGEPAL-CA630 (I8896, Sigma), 1 mM DTT (D9779, Sigma), protease inhibitor (05056489001, Roche), and 5% BSA (A7906, Sigma) in PBS (10010-23, Thermo Fisher Scientific)] for 10 min on a rotator at 4°C followed by centrifugation for 5 min at 500 g at 4°C. The pellet was resuspended in cold tagmentation buffer [33 mM Tris-acetate pH 7.8 (BP-152, Thermo Fisher Scientific), 66 mM K-acetate (P5708, Sigma), 11 mM Mg-acetate (M2545, Sigma), 16% DMF (DX1730, EMD Millipore)] in molecular biology grade water (46000-CM, Corning) followed by incubation with Tagmentation enzyme (FC-121-1030; Illumina) at 37°C with shaking at 500 rpm for 30 min. Tagmented DNA was purified using MinElute PCR purification kit (28004, Qiagen). The resulting libraries were amplified using NEBNext High-Fidelity 2X PCR Master Mix (M0541, NEB) with primer extension at 72°C for 5 min, denaturation at 98°C for 30 s, followed by 8 cycles of denaturation at 98°C for 10 s, annealing at 63°C for 30 s and extension at 72°C for 60 s. After purification of amplified libraries using MinElute PCR purification kit (28004, QIAGEN), double-sided size selection was performed using SPRIselect beads (B23317, Beckman Coulter) with 0.55× beads and 1.5× to sample volume. Libraries were sequenced on a NextSeq500 (Illumina).
Adaptor-trimmed fastq files were aligned to hg38 by Bowtie2 (Langmead and Salzberg, 2012) using parameters ‘-X2000 –local’ and converted into bams and sorted using samtools (Li et al., 2009). Files were then filtered to remove improper mapped, unmapped, not primary, failing platform, and poorly mapping reads using samtools with the following parameters: ‘view -F 1804 -f 2 -q 30’ and then sorted with sambamba (Tarasov et al., 2015). Mate coordinates were then filled using samtools fixmate, and then samtools filtering and sambamba sorting was repeated. Next, Picard was used to mark and remove duplicates. Bigwig files were producted from resultant bam files using deepTools (Ramírez et al., 2016) bamCoverage with the following settings: ‘--binSize 10 -normalizeUsing RPGC -effectiveGenomeSize 2150570000 -irgnoareForNormalization chrX -extendReads.’ Bams were also sorted by name using ‘samtools sort -n’ and then converted into bedpe using bamtools (Barnett et al., 2011). Mitochondrial reads were then removed using grep and a Tn5 shift was reads were shifted to remove Tn5 adaptors. Peaks were then called using MACS2 (Zhang et al., 2008) using the following parameters: -q 0.01 -nomodel -nolambda -shift 100 -extsize 200 -B -keep-dup all -call-summits.
Peaks and bam files were then read into Diffbind (Ross-Innes et al., 2012). Low read-count regions were then filtered using a cutoff sum of 15 in dba.count, normalized by sequencing depth, and then a contrast was defined by haplotype. Differential accessibility was calculated using the dba.analyze wrapper for DESEQ2 (Love et al., 2014), and peaks were annotated using the HOMER annotatepeaks.pl script (Heinz et al., 2010). Peaks were then filtered with a cutoff of FDR <0.01, log2 fold change of ±2, and average concentration of >2. The top 3000 peaks that were unique to each haplotype were then fed into the HOMER findMotifsGenome.pl script (Heinz et al., 2010) using a size of 200. Following the identification of haplotype-dependent AP-1 and GATA motifs, similar motifs were merged for each transcription factor family [AP-1: Fra1, Fos, Atf3, JunB, BATF, Fra2, AP-1 (from GSE21512), and GATA: Gata1–Gata6] and DARS were annotated for each group using annotatepeaks.pl. GATA motifs were identified as being enriched in RKO AP-1 containing DARS (vs RR DARS) using a Chi-squared test (P<0.001). Additional transcription factors (i.e. TEAD family proteins) were present in line pair-wise comparison, but not significant when tested for haplotype-dependent enrichment using a Chi-squared test. Regions containing predicted motifs for both AP-1 and GATA are provided in Table S1.
RNA-Seq
RNA-sequencing was performed on RR (1–5, ED70 and 2–3) and RKO (1–9, ED65 and WB46) iPSC clones in triplicate. RNA was isolated using Qiagen mini-RNA isolation kits and paired-end poly-A RNA library preparation and sequencing was performed by Novogene. FASTQ files were aligned against the hg38 genome using STAR (Dobin et al., 2013) with the following parameters: --outFilterType BySJout --outFilterMultimapNmax 10 --alignSJoverhangMin 8 --alignSJDBoverhangMin 1 --outFilterMismatchNmax 4 --alignIntronMin 20 --alignIntronMax 1000000 --alignMatesGapMax 100000. Strand-specific tag directories were generated with the HOMER (Heinz et al., 2010) makeTagDirectories.pl script, and raw and TPM-normalized count matrices were generated using the analyzeRepeats.pl script to count exons and condense genes. The HOMER getDiffExpression wrapper of DESEQ2 was used to compare gene expression between genotypes for each clonal pair of fibroblasts. Plots were generated in R using ggplot2 and pheatmap packages.
qPCR
Cells were washed with 1× PBS twice, and RNA was isolated using TRIzol (15596026, Thermo Fisher Scientific). After 5 min of incubation, cells were scraped, transferred to 1.5 ml Eppendorf tubes, and 0.2 ml of molecular grade chloroform was added. Tubes were shaken and left to equilibrate for 3 min, after which tubes were centrifuged for 30 min at 4°C and 3700 g. Aqueous phases were transferred to new tubes and 0.5 ml of isopropanol was added. Tubes were inverted and left to sit for 10 min. Tubes were centrifuged again using the same parameters as before, but for 20 min. The supernatant was removed and 1 ml of cold 75% molecular grade ethanol in DEPC water was added and vortexed. Tubes were then centrifuged at 3700 g for 12 min, decanted and allowed to air dry. RNA was then resuspended in DEPC water and purified using the RNeasy Mini Kit (74104, Qiagen). The RNA concentration was measured by use of Nanodrop (ND-2000, Thermo Fisher Scientific), and 1 μg of RNA was used per reverse transcription reaction using Superscript IV and oligo(dT) (18091050, Thermo Fisher Scientific). cDNA was stored at −80°C prior to amplification. 10 μl reactions were performed using 5 ng of cDNA, 1 μM forward and reverse primers in DEPC water, and 5 μl of Sybr Green (4309155, Thermo Fisher Scientific). Each primer pair was optimized for melt temperature, and efficiency was validated to be between 80 and 120%. Primer sequences used include (forward, reverse): GJA1: 5′-GGTGACTGGAGCGCCTTAG-3′, 5′-GCGCACATGAGAGATTGGGA-3′; ACTB: 5′-CACCAACTGGGACGACAT-3′, 5′-ACAGCCTGGATAGCAACG-3′. Cycles were performed using a two-step protocol with 10 s denaturing at 95°C and 30 s of annealing and extension. All amplicons were less than 400 base pairs. Expression was calculated using 2−ΔΔCT method using ACTB as the housekeeping gene.
Supplementary Material
Acknowledgements
The authors would like to thank Dr Tatiana Kisseleva for helpful discussions.
Footnotes
Author contributions
Conceptualization: A.J.W., H.A., B.R., A.J.E.; Methodology: H.A., J.D.H., A.J.E.; Validation: A.J.W., H.A., A.J.E.; Formal analysis: A.J.W., H.A., J.D.H.; Investigation: A.J.W., H.A., A.J.E.; Resources: A.J.E.; Data curation: A.J.W., H.A., J.D.H.; Writing - original draft: A.J.W., H.A.; Writing - review & editing: A.J.W., H.A., B.R., A.J.E.; Visualization: A.J.W., H.A.; Supervision: B.R., A.J.E.; Funding acquisition: A.J.E.
Funding
The authors acknowledge funding and equipment support from the National Institutes of Health (R01AG045428 to A.J.E. and S10OD026929 to UCSD IGM Genomics Center). Fellowship support was provided by the National Science Foundation (to A.J.W.), the ARCS Foundation (to A.J.W.), and the National Institutes of Health (T32GM008666 to J.D.H. and T32HL007444 to H.A.). Work at the Center for Epigenomics was supported in part by the UC San Diego School of Medicine. Deposited in PMC for release after 12 months.
Data availability
ATAC-Seq and RNA-seq data are deposited at Gene Omnibus Express (GEO) under accession no. GSE225736. Source data files are provided and all supporting data from this study are available from the corresponding author upon reasonable request.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/lookup/doi/10.1242/jcs.261152.reviewer-comments.pdf
References
- Angel, P. and Karin, M. (1991). The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta Rev. Cancer 1072, 129-157. 10.1016/0304-419X(91)90011-9 [DOI] [PubMed] [Google Scholar]
- Aurora, A. B., Porrello, E. R., Tan, W., Mahmoud, A. I., Hill, J. A., Bassel-Duby, R., Sadek, H. A. and Olson, E. N. (2014). Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382-1392. 10.1172/JCI72181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P. and Marth, G. T. (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691-1692. 10.1093/bioinformatics/btr174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, H., Troutman, T. D., Sakai, M. and Glass, C. K. (2020). Epigenetic regulation of kupffer cell function in health and disease. Front. Immunol. 11, 609618. 10.3389/fimmu.2020.609618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas, S. K., Chittezhath, M., Shalova, I. N. and Lim, J.-Y. (2012). Macrophage polarization and plasticity in health and disease. Immunol. Res. 53, 11-24. 10.1007/s12026-012-8291-9 [DOI] [PubMed] [Google Scholar]
- Bonachea, E. M., Chang, S.-W., Zender, G., Lahaye, S., Fitzgerald-Butt, S., Mcbride, K. L. and Garg, V. (2014). Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr. Res. 76, 211-216. 10.1038/pr.2014.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. Z. C., Peng, Y. X., Wang, Z. B., Fish, P. V., Kaar, J. L., Koepsel, R. R., Russell, A. J., Lareu, R. R. and Raghunath, M. (2009). The Scar-in-a-Jar: studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br. J. Pharmacol. 158, 1196-1209. 10.1111/j.1476-5381.2009.00387.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, A. S. and Kao, W. J. (2009). Fibroblasts regulate monocyte response to ECM-derived matrix: the effects on monocyte adhesion and the production of inflammatory, matrix remodeling, and growth factor proteins. J. Biomed. Mater. Res. A 89A, 841-853. 10.1002/jbm.a.32431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick, S. A., Macklin, J. A., Nejat, S., Momen, A., Clemente-Casares, X., Althagafi, M. G., Chen, J., Kantores, C., Hosseinzadeh, S., Aronoff, L.et al. (2019). Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29-39. 10.1038/s41590-018-0272-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J., Wang, S., Dong, X. and Wang, Z. (2021). RGD-expressed bacterial membrane-derived nanovesicles enhance cancer therapy via multiple tumorous targeting. Theranostics 11, 3301-3316. 10.7150/thno.51988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice, J., Xia, Z., Wang, E. T., Scavuzzo, M. A., Ward, A. J., Kalsotra, A., Wang, W., Wehrens, X. H. T., Burge, C. B., Li, W.et al. (2014). Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun. 5, 3603. 10.1038/ncomms4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, D. S., Grossfeld, D., Renna, H. A., Agarwala, P., Spiegler, P., Kasselman, L. J., Glass, A. D., Deleon, J. and Reiss, A. B. (2020). Idiopathic pulmonary fibrosis: molecular mechanisms and potential treatment approaches. Respir. Investig. 58, 320-335. 10.1016/j.resinv.2020.04.002 [DOI] [PubMed] [Google Scholar]
- Hai, T. W., Liu, F., Allegretto, E. A., Karin, M. and Green, M. R. (1988). A family of immunologically related transcription factors that includes multiple forms of ATF and AP-1. Genes Dev. 2, 1216-1226. 10.1101/gad.2.10.1216 [DOI] [PubMed] [Google Scholar]
- Harris, P. and Ralph, P. (1985). Human Leukemic Models of Myelomonocytic Development: A Review of the HL-60 and U937 Cell Lines. J. Leukoc. Biol. 37, 407-422. 10.1002/jlb.37.4.407 [DOI] [PubMed] [Google Scholar]
- Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., Cheng, J. X., Murre, C., Singh, H. and Glass, C. K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576-589. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgadottir, A., Thorleifsson, G., Manolescu, A., Gretarsdottir, S., Blondal, T., Jonasdottir, A., Jonasdottir, A., Sigurdsson, A., Baker, A., Palsson, A.et al. (2007). A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 316, 1491-1493. 10.1126/science.1142842 [DOI] [PubMed] [Google Scholar]
- Hinz, B. (2007). Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 127, 526-537. 10.1038/sj.jid.5700613 [DOI] [PubMed] [Google Scholar]
- Jarinova, O., Stewart, A. F. R., Roberts, R., Wells, G., Lau, P., Naing, T., Buerki, C., Mclean, B. W., Cook, R. C., Parker, J. S.et al. (2009). Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler. Thromb. Vasc. Biol. 29, 1671-1677. 10.1161/ATVBAHA.109.189522 [DOI] [PubMed] [Google Scholar]
- Ji, Z., He, L., Regev, A. and Struhl, K. (2019). Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. USA 116, 9453-9462. 10.1073/pnas.1821068116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A., Thomas, S. K., Wong, K. C., Lo Sardo, V., Cheah, D. S., Hou, Y.-H., Placone, J. K., Tenerelli, K. P., Ferguson, W. C., Torkamani, A.et al. (2019). Mechanical activation of noncoding-RNA-mediated regulation of disease-associated phenotypes in human cardiomyocytes. Nat. Biomed. Eng. 3, 137-146. 10.1038/s41551-018-0344-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareu, R. R., Subramhanya, K. H., Peng, Y., Benny, P., Chen, C., Wang, Z., Rajagopalan, R. and Raghunath, M. (2007). Collagen matrix deposition is dramatically enhanced in vitro when crowded with charged macromolecules: the biological relevance of the excluded volume effect. FEBS Lett. 581, 2709-2714. 10.1016/j.febslet.2007.05.020 [DOI] [PubMed] [Google Scholar]
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R., and 1000 Genome Project Data Processing Subgroup. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078-2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, J., Huang, W. and Liu, G. (2015). Animal models of coronary heart disease. J. Biomed. Res. 30, 3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Sardo, V., Chubukov, P., Ferguson, W., Kumar, A., Teng, E. L., Duran, M., Zhang, L., Cost, G., Engler, A. J., Urnov, F.et al. (2018). Unveiling the role of the most impactful cardiovascular risk locus through haplotype editing. Cell 175, 1796-1810.e20. 10.1016/j.cell.2018.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, Y. and Schwarzbauer, J. E. (2005). Stimulatory effects of a three-dimensional microenvironment on cell-mediated fibronectin fibrillogenesis. J. Cell Sci. 118, 4427-4436. 10.1242/jcs.02566 [DOI] [PubMed] [Google Scholar]
- Mitchell, J. A. and Lye, S. J. (2005). Differential activation of the connexin 43 promoter by dimers of activator protein-1 transcription factors in myometrial cells. Endocrinology 146, 2048-2054. 10.1210/en.2004-1066 [DOI] [PubMed] [Google Scholar]
- Newton-Cheh, C., Cook, N. R., Vandenburgh, M., Rimm, E. B., Ridker, P. M. and Albert, C. M. (2009). A common variant at 9p21 is associated with sudden and arrhythmic cardiac death. Circulation 120, 2062-2068. 10.1161/CIRCULATIONAHA.109.879049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puerta Cavanzo, N., Bigaeva, E., Boersema, M., Olinga, P. and Bank, R. A. (2020). Macromolecular crowding as a tool to screen anti-fibrotic drugs: the Scar-in-a-Jar system revisited. Front. Med. (Lausanne) 7, 615774. 10.3389/fmed.2020.615774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez, F., Ryan, D. P., Grüning, B., Bhardwaj, V., Kilpert, F., Richter, A. S., Heyne, S., Dündar, F. and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160-W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter, J. F., Alexander, J., Rodaway, A., Yelon, D., Patient, R., Holder, N. and Stainier, D. Y. R. (1999). Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 13, 2983-2995. 10.1101/gad.13.22.2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M. D., Mccarthy, D. J. and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139-140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rønnow, S. R., Dabbagh, R. Q., Genovese, F., Nanthakumar, C. B., Barrett, V. J., Good, R. B., Brockbank, S., Cruwys, S., Jessen, H., Sorensen, G. L.et al. (2020). Prolonged Scar-in-a-Jar: an in vitro screening tool for anti-fibrotic therapies using biomarkers of extracellular matrix synthesis. Respir. Res. 21, 108. 10.1186/s12931-020-01369-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes, C. S., Stark, R., Teschendorff, A. E., Holmes, K. A., Ali, H. R., Dunning, M. J., Brown, G. D., Gojis, O., Ellis, I. O., Green, A. R.et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389-393. 10.1038/nature10730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep, A. N., Wu, B., Buenrostro, J. D. and Greenleaf, W. J. (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975-978. 10.1038/nmeth.4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B.et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, M. K., Li, Y., Li, S., Cobb, R. M., Zhou, D., Lu, M. M., Epstein, J. A., Morrisey, E. E. and Gruber, P. J. (2010). Gata4 and Gata5 cooperatively regulate cardiac myocyte proliferation in mice. J. Biol. Chem. 285, 1765-1772. 10.1074/jbc.M109.038539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange, E. F. and Schroeder, B. O. (2019). Microbiota and mucosal defense in IBD: an update. Expert. Rev. Gastroenterol. Hepatol. 13, 963-976. 10.1080/17474124.2019.1671822 [DOI] [PubMed] [Google Scholar]
- Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. and Satija, R. (2021). Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333-1341. 10.1038/s41592-021-01282-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto, S. G., Narimatsu, M., Lu, M., He, X., Sidiqi, A. M., Tolosa, M. F., Chan, L., De Freitas, K., Bialik, J. F., Majumder, S.et al. (2016). YAP/TAZ are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 27, 3117-3128. 10.1681/ASN.2015050499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., Simonovic, M., Doncheva, N. T., Morris, J. H., Bork, P.et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607-D613. 10.1093/nar/gky1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. and Prins, P. (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032-2034. 10.1093/bioinformatics/btv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida, K., Chaki, H., Takakura, T., Yokotani, J., Aikawa, Y., Shiozawa, S., Gouda, H. and Hirono, S. (2004). Design, synthesis, and biological evaluation of new cyclic disulfide decapeptides that inhibit the binding of AP-1 to DNA. J. Med. Chem. 47, 4239-4246. 10.1021/jm049890+ [DOI] [PubMed] [Google Scholar]
- Virani, S. S., Alonso, A., Aparicio, H. J., Benjamin, E. J., Bittencourt, M. S., Callaway, C. W., Carson, A. P., Chamberlain, A. M., Cheng, S., Delling, F. N.et al. (2021). Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation 143, e254-e743. 10.1161/CIR.0000000000000950 [DOI] [PubMed] [Google Scholar]
- Verrecchia, F. and Mauviel, A. (2002). Transforming growth factor-β signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J. Investig. Dermatol. 118, 211-215. 10.1046/j.1523-1747.2002.01641.x [DOI] [PubMed] [Google Scholar]
- Visel, A., Zhu, Y., May, D., Afzal, V., Gong, E., Attanasio, C., Blow, M. J., Cohen, J. C., Rubin, E. M. and Pennacchio, L. A. (2010). Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 464, 409-412. 10.1038/nature08801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Cui, M., Shah, A. M., Ye, W., Tan, W., Min, Y.-L., Botten, G. A., Shelton, J. M., Liu, N., Bassel-Duby, R.et al. (2019). Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc. Natl. Acad. Sci. USA 116, 18455-18465. 10.1073/pnas.1905824116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Cui, M., Shah, A. M., Tan, W., Liu, N., Bassel-Duby, R. and Olson, E. N. (2020). Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. 33, 108472. 10.1016/j.celrep.2020.108472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead, A. J. and Engler, A. J. (2021). Regenerative cross talk between cardiac cells and macrophages. Am. J. Physiol. Heart Circ. Physiol. 320, H2211-H2221. 10.1152/ajpheart.00056.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead, A. J., Hocker, J. D., Ren, B. and Engler, A. J. (2022). Improved epicardial cardiac fibroblast generation from iPSCs. J. Mol. Cell. Cardiol. 164, 58-68. 10.1016/j.yjmcc.2021.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn, T. A. (2004). Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4, 583-594. 10.1038/nri1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi, K., Folsom, A. R., Rosamond, W. D., Boerwinkle, E. and ARIC Investigators. (2009). A genetic variant on chromosome 9p21 and incident heart failure in the ARIC study. Eur. Heart J. 30, 1222-1228. 10.1093/eurheartj/ehp087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, N., Ding, Y., Wild, C., Shen, Q. and Zhou, J. (2014). Small molecule inhibitors targeting Activator Protein 1 (AP-1). J. Med. Chem. 57, 6930-6948. 10.1021/jm5004733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W.et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q., Wirka, R., Nguyen, T., Nagao, M., Cheng, P., Miller, C. L., Kim, J. B., Pjanic, M. and Quertermous, T. (2019). TCF21 and AP-1 interact through epigenetic modifications to regulate coronary artery disease gene expression. Genome Med. 11, 23. 10.1186/s13073-019-0635-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Zhou, B., Pache, L., Chang, M., Khodabakhshi, A. H., Tanaseichuk, O., Benner, C. and Chanda, S. K. (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.