

Abstract
Anaplastic lymphoma kinase (ALK) is a potent oncogenic driver in lung cancer. ALK tyrosine kinase inhibitors yield significant benefit in patients with ALK fusion-positive (ALK+) lung cancers; yet the durability of response is limited by drug resistance. Elucidation of on-target resistance mechanisms has facilitated the development of next-generation ALK inhibitors, but overcoming ALK-independent resistance mechanisms remains a challenge. In this Review, we discuss the molecular underpinnings of acquired resistance to ALK-directed therapy and highlight new treatment approaches aimed at inducing long-term remission in ALK+ disease.
Over the past few decades, advances in lung cancer diagnostics and treatments have transformed patient outcomes1,2. Translational research, clinical genotyping and drug discovery have enabled the molecular stratification of lung cancers, namely adenocarcinomas, based on the presence of oncogenic drivers and the development of targeted therapies matched to the respective oncogenes2. The ALK gene fusion defines one molecular subtype of non-small cell cancer (NSCLC), comprising 4–6% of lung adenocarcinomas3. A chromosomal rearrangement involving the ALK gene on chromosome 2 leads to ectopic expression of the tyrosine kinase-containing portion of ALK and its constitutive activation. ALK+ lung cancers exhibit ALK dependency and are typically sensitive to ALK inhibition using tyrosine kinase inhibitors (TKIs). So far, five ALK TKIs have received approval by the US Food and Drug Administration (FDA) for treatment of advanced ALK+ NSCLC, with more in clinical development.
Despite their remarkable responses to ALK TKIs4–9, almost all patients with advanced ALK+ lung cancers ultimately experience disease relapse through on-target and off-target resistance mechanisms10. Tumor cells with on-target resistance retain their dependence on ALK, whereas those with off-target mechanisms activate ALK-independent pathways to support proliferation and survival. Re-biopsies and genotyping of resistant clinical samples are key in elucidating the mechanisms of resistance and guiding sequential therapeutic approaches. Yet, challenges remain in addressing the heterogeneity of resistance mechanisms and preventing disease relapse.
In this Review, we provide an overview of the underlying biology of oncogenic ALK fusions, discuss the current understanding of acquired resistance to ALK-directed therapy and highlight the latest therapeutic strategies aimed at inducing long-term remission in advanced ALK+ lung cancers, centered on the hypothetical, yet imperative, question of what it would take to cure metastatic ALK+ lung cancer.
Physiological role of ALK
The ALK gene was first cloned in 1994 when the nucleophosmin (NPM1)–ALK fusion protein was identified in anaplastic large cell lymphoma (ALCL)11. ALK encodes a highly conserved receptor tyrosine kinase (RTK) in the insulin receptor superfamily12. The native ALK protein is thought to be essential for the development and functioning of the nervous system13. Structurally, ALK is composed of an N-terminal extracellular domain, a hydrophobic single-pass transmembrane region and an intracellular kinase domain (Fig. 1). ALK is activated when ALKAL proteins (endogenous ligands of ALK) bind to its extracellular domain, resulting in dimerization and autophosphorylation and activation of downstream signaling pathways critical for cell proliferation, survival and differentiation14–16.
Fig. 1 |. Oncogenic ALK signaling.
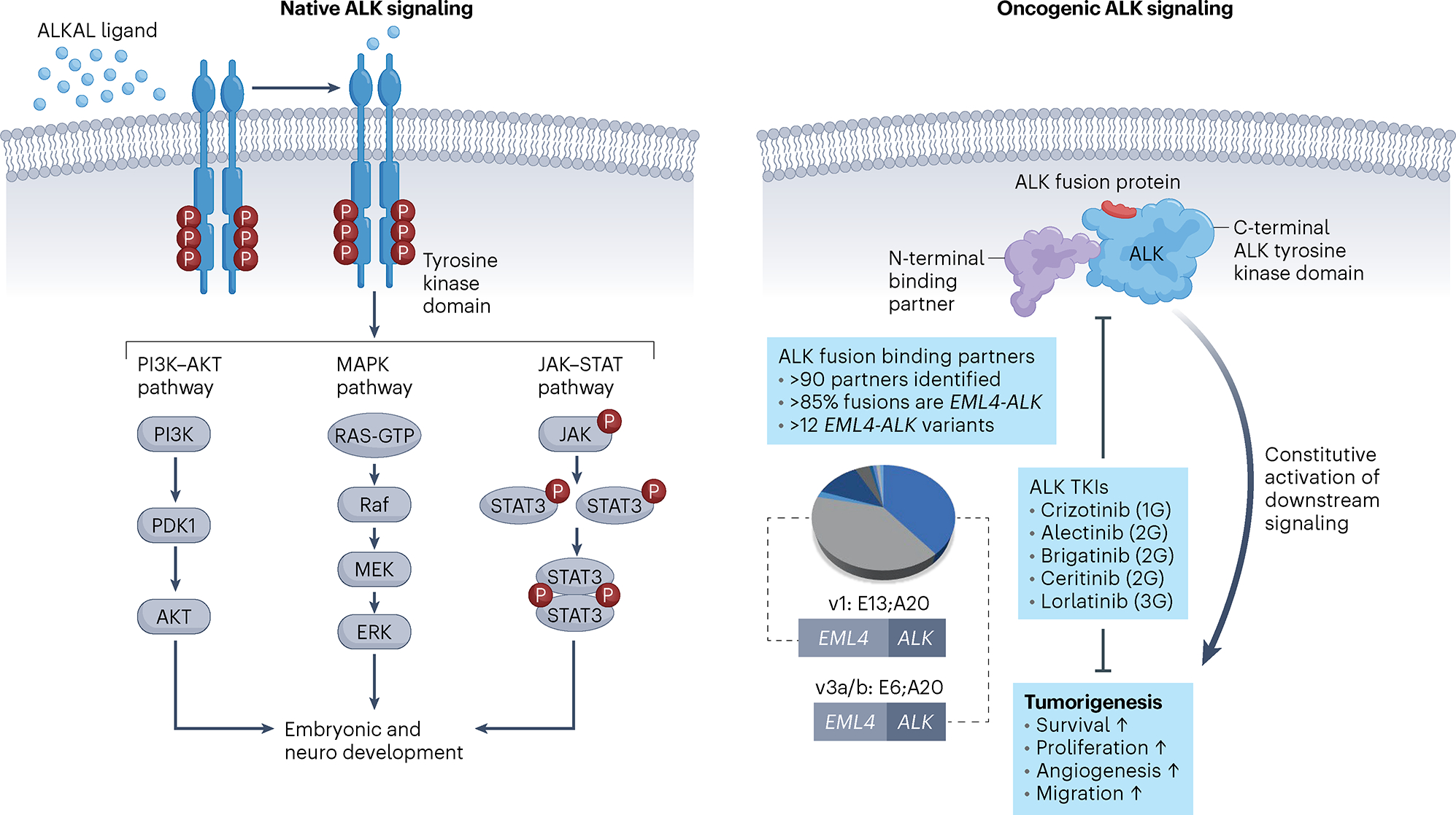
Left, wild-type ALK is a plasma membrane-bound RTK that undergoes autophosphorylation upon ligand binding and receptor oligomerization. ALK activates downstream signaling pathways that contribute to organ development and homeostasis. Right, a chromosomal translocation leads to formation of an ALK fusion gene and translation of an ALK chimeric oncoprotein that is composed of the C-terminal kinase domain of ALK joined with various N-terminal, non-kinase fusion partners. Constitutive activation of ALK promotes cell survival pathways and tumorigenesis. Although many ALK binding partners have been elucidated across all tumor types, the EML4–ALK fusion is the most common, of which EML4-ALK variant 1 (E13;A20) and variant 3 (E6;A20) are the most prevalent in lung cancer. FDA-approved ALK TKIs and their generation are depicted. PDK1, pyruvate dehydrogenase kinase 1; v, variant; 1G, first generation; 2G, second generation; 3G, third generation.
Oncogenic ALK fusions
In malignancies, ALK point mutations or chromosomal rearrangements result in aberrant activation of ALK and downstream signaling cascades17. In NSCLC and cancer types that include ALCL, diffuse large B cell lymphoma, inflammatory myofibroblastic tumors, glioma and colon cancer14,18–20, the oncogenic driver is a structural ALK rearrangement whereby the kinase domain-encoding region of ALK at the 3′ end is fused to various partner genes at the 5′ end. Echinoderm microtubule-associated protein-like 4 (EML4)-ALK is the most common ALK fusion in NSCLC, identified in approximately 85% of cases21,22. Delineation of ALK fusion partners continues, with more than 90 distinct 5′ partner genes reported to date in NSCLC22. ALK fusions result in ligand-independent dimerization and hyperactivation of pro-mitogenic and anti-apoptotic signaling including the RAS–mitogen-activated protein kinase (MAPK)23, PI3K–AKT24 and JAK–STAT25 cascades (Fig. 1). In the case of EML4–ALK, the MAPK pathway is a critical downstream effector, the activation of which is mediated by the HELP domain of EML4 (ref. 23).
Within EML4-ALK alone, at least 12 distinct variants exist as determined by breakpoints in EML4. Variant 1 (E13;A20) and variant 3 (E6a/b;A20) are the most common and are associated with differences in protein stability, drug sensitivity26 and distinct ALK resistance mutations27. The identity of the 5′ partner can influence the intrinsic properties of the fusion protein by altering kinase activity, protein stability, transformative potential and drug sensitivities in vitro28. However, selection of ALK inhibitors currently remains agnostic of fusion type. Sequencing analysis showed that 5′ ALK is retained in the genome in some cases, in addition to 3′ ALK. Whether these reciprocal or nonreciprocal translocations are independent predictors of poorer outcomes in patients treated with next-generation ALK TKIs remains to be seen29.
Most ALK fusions lack a transmembrane region and are not anchored to the plasma membrane, unlike native ALK. Early studies of ALK fusions in ALCL showed differential cytoplasmic, nuclear and granular subcellular localization depending on the ALK binding partner identity30, likely representing an important feature in the modulation of protein–protein interactions, activation of distinct signaling cascades and stability and/or degradation of the fusion product. Recent studies have revealed that certain RTK fusion oncoproteins including ALK assemble de novo in their own subcellular compartment that can phase separate based on coalescence of cytoplasmic membraneless protein granules31–33. These discrete structures concentrate oncogenic ALK with components of MAPK, PLC-γ, PI3K and JAK–STAT cascades to coordinate RTK signaling, suggesting that disruption of protein granule assembly and function could be therapeutically targeted. However, more work is needed to identify factors that regulate these subcellular condensates.
ALK has served as a framework for investigating other fusion oncoproteins in lung cancer such as ROS1, RET, TRKA–TRKC and NRG1. Although ALK-driven NSCLC is an archetypal example of oncogene addiction, whether genomic heterogeneity in ALK+ NSCLC, in terms of binding partners, reciprocal translocations and breakpoint variants, accounts for differences in treatment responses to TKIs remains unclear34. Furthermore, the etiology of ALK fusions in cancers is largely unknown, as is its propensity to affect young patients35,36.
ALK as a therapeutic target
The field of ALK+ NSCLC has been transformed by the development of successive generations of increasingly selective, potent and brain-penetrant ALK TKIs, serving as a paradigm of a successful bench-to-bedside approach and highlighting the clinical benefit of understanding molecular dependencies of cancer. The success in targeting ALK+ lung cancer is also a model of reverse translation, as insights from molecular analysis of patient samples have directly guided basic science discoveries and shaped drug development.
Preclinical modeling of ALK+ NSCLC has proven critical in recapitulating ALK pathophysiology in vitro and in vivo and facilitating the development of ALK inhibitors. For instance, overexpression of fusion ALK in untransformed cells28,37,38 facilitated in vitro studies in isogenic backgrounds and enabled mutagenesis screening, drug testing and tracking of resistant clones37. Early germline transgenic mouse models and tissue-specific expression of EML4–ALK in alveolar epithelial cells established the transforming role of ALK fusion in vivo39. Tissue-specific Cre-inducible transgenic models and CRISPR–Cas9-based, viral-mediated delivery of EML4-ALK were developed to study drug resistance in vivo40,41. Although patient-derived cell lines (PDCs) and xenografts lack certain advantages of orthotopic ALK-driven NSCLC murine models, they have been invaluable in elucidating clinically relevant mechanisms of resistance in ALK- and other oncogene-driven cancers42,43.
Targeting ALK in the clinic
Preclinical modeling of ALK+ lung cancers spurred the development of numerous ALK TKIs (summarized in Table 1). Crizotinib, the first-generation ALK inhibitor, is a multitargeted TKI that was originally developed as an inhibitor of mesenchymal–epithelial transition (MET)44. In 2011, crizotinib was granted accelerated FDA approval based on phase 1–2 studies showing clinical activity in advanced ALK+ NSCLC45, followed by two large phase 3 trials demonstrating its superiority to cytotoxic chemotherapy in this patient population4,5. However, the median progression-free survival (PFS) of patients treated with crizotinib was limited to 8–11 months, with relapse observed in the central nervous system (CNS) due to poor drug penetration of the blood–brain barrier4. Several second-generation ALK TKIs that were developed (of which ceritinib, alectinib and brigatinib have received FDA approval) showed clinical activity also in the CNS in the post-crizotinib setting and overcame some common crizotinib-refractory ALK resistance mutations46. Second-generation ALK TKIs were also effective in the absence of crizotinib-resistant ALK mutations, suggesting incomplete ALK inhibition by crizotinib.
Table 1 |.
Summary of ALK inhibitors approved by the FDA or in clinical testing
| ALK TKI | TKI generation | Status | Trial namea (phase) | Comparator | n | Median PFSb, months (HR, 95% CI) | ORR, % (95% CI) | IC-ORRc, % (95% CI) | ALK mutations refractory to TKI | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Crizotinib | 1G | Approved for 1L and beyond | PROFILE 1014 (phase 3) | Chemotherapy (platinum-based doublet) | 343 | 10.9 versus 7.0 (0.45, 0.35–0.60) |
74% (67–81%) versus 45% (37–53%) |
N/A | L1196M G1202R I1171T/N/S G1269A/S S1206Y I1151Tins L1152P/R C1156Y/T F1174C/L/V V1180L S1206C/Y E1210K |
4,5,43,60 |
| Ceritinib | 2G | Approved for 1L and beyond | ASCEND-4 (phase 3) | Chemotherapy (platinum-based doublet) | 376 | 16.6 versus 8.1 (0.55, 0.42–0.73) |
73% (66–79%) versus 27% (21–34%) |
73% (50–89%) versus 27% (11–50%) |
G1202R/del I1151Tins L1152P/R C1156Y/T F1174C/L/V D1203K G1269A |
6,45 |
| Alectinib | 2G | Approved for 1L and beyond | ALEX (phase 3) | Crizotinib | 303 | 25.7 versus 10.4 (0.50, 0.3–0.70) |
83% (76–89%) versus 76% (68–82%) (INV) |
81% (58–95%) versus 50% (28–72%) (INV) |
G1202R/del V1180L I1171T/N/S L1196M |
7,45,49-51 |
| Brigatinib | 2G | Approved for 1L and beyond | ALTA-1L (phase 3) | Crizotinib | 275 | 24.0 versus 11.1 (0.48, 0.35–0.66) |
74% (66–82%) versus 62% (54–70%) |
78% (52–94%) versus 26% (10–48%) |
G1202R/del E1210K |
8,45-47,90 |
| Ensartinib | 2G | Phase 3 | eXalt3 (phase 3) | Crizotinib | 290 | 25.8 versus 12.7 (0.51, 0.35–0.72) |
74% (66–81%) versus 67% (58–74%) |
64% versus 21% |
G1269A G1202R/del E1210K | 48,90 |
| Lorlatinib | 3G | Approved for 1L and beyond | CROWN (phase 3) | Crizotinib | 296 | NR versus 9.3 (0.27, 0.18–0.39) |
76% (68–83%) versus 58% (49–66%) |
82% (57–96%) versus 23% (5–54%) |
C1156Y + L1198F I1171N + L1198F G1202R + F1174L G1202R + L1196M D1203N + L1196M G1202R + S1206Y G1202R + C1156Y G1202R + G1269A C1156Y + G1269A I1171N/T + D1203N G1202R + G1269A G1202R + L1204V + G1269A G1202R + S1206F + G1269A D1203N + E1210K + G1269A |
9,36,52-54,65,68 |
| TPX-0131 | 4G | Phases 1–2 | FORGE-1 (NCT04849273) |
None | N/A | N/A | N/A | N/A | N/A | 97 |
| NVL-655 | 4G | Phases 1–2 | ALKOVE-1 (NCT05384626) |
None | N/A | N/A | N/A | N/A | N/A | 98 |
Seminal global randomized phase 3 trials are listed for the FDA-approved agents and for ensartinib, which is approved as first-line treatment in China.
Median PFS according to blinded independent review committee assessment is shown.
Intracranial response rates in patients with baseline measurable brain metastases are shown, according to blinded independent review committee assessment unless indicated otherwise. CI, confidence interval; HR, hazard ratio; IC-ORR, intracranial ORR; INV, per-investigator assessment; NR, not reached; N/A, not available.
Although they were initially evaluated in the post-crizotinib setting, second-generation ALK TKIs have since supplanted crizotinib as the preferred initial therapy in advanced ALK+ lung cancer. Alectinib7, brigatinib8,47,48 and ensartinib49 were compared directly to crizotinib in the first-line setting, demonstrating superior efficacy of second-generation TKIs across the board (Table 1). The global randomized phase 3 ALEX study7,50–52 established alectinib as a preferred first-line therapy with mature data showing significantly prolonged PFS with alectinib compared to crizotinib (median PFS of 25.7 versus 10.4 months, respectively; hazard ratio (HR) for disease progression or death of 0.50).
Lorlatinib is a third-generation macrocyclic ALK TKI, designed to be highly potent, selective and CNS penetrant53. Preclinical studies demonstrated robust activity of lorlatinib against wild-type ALK and most known ALK mutations refractory to first- and second-generation TKIs, including the predominant ALKG1202R solvent-front mutation46. Lorlatinib initially received accelerated FDA approval in the second- or third-line setting for advanced ALK+ disease based on efficacy in patients with exposure to at least one prior ALK TKI in the phase 1–2 study, with an objective response rate (ORR) of 47% in this patient population54. The global randomized phase 3 CROWN trial9 evaluated lorlatinib in the front-line setting in comparison to crizotinib and demonstrated significantly longer PFS (HR for disease progression or death of 0.28) and dramatic reduction in the risk for CNS progression (HR for CNS progression or death of 0.07), resulting in its FDA approval as a first-line agent in 2021 (refs. 9,55).
Despite the success of iterative generations of ALK TKIs, the durability of response remains limited by drug resistance. Approximately half of patients treated with first-line alectinib for ALK+ NSCLC will experience disease progression within approximately 2 years50, an unfortunate but expected consequence of tumor evolution and emergence of resistance.
Mechanisms of ALK-dependent resistance
Resistance to ALK-targeted therapies can be broadly classified as ALK dependent and ALK independent. ALK-dependent, or ‘on-target’, resistance is largely defined by the emergence of single or compound mutations in the ALK gene, rendering tumor cells persistently dependent on ALK activity (Fig. 2). ALK-independent, or ‘off-target’, resistance is defined by lineage changes or activation of ALK-independent signaling pathways that obviate ALK dependency in ALK+ tumor cells.
Fig. 2 |. Resistance in ALK+ lung cancer and therapeutic interventions.
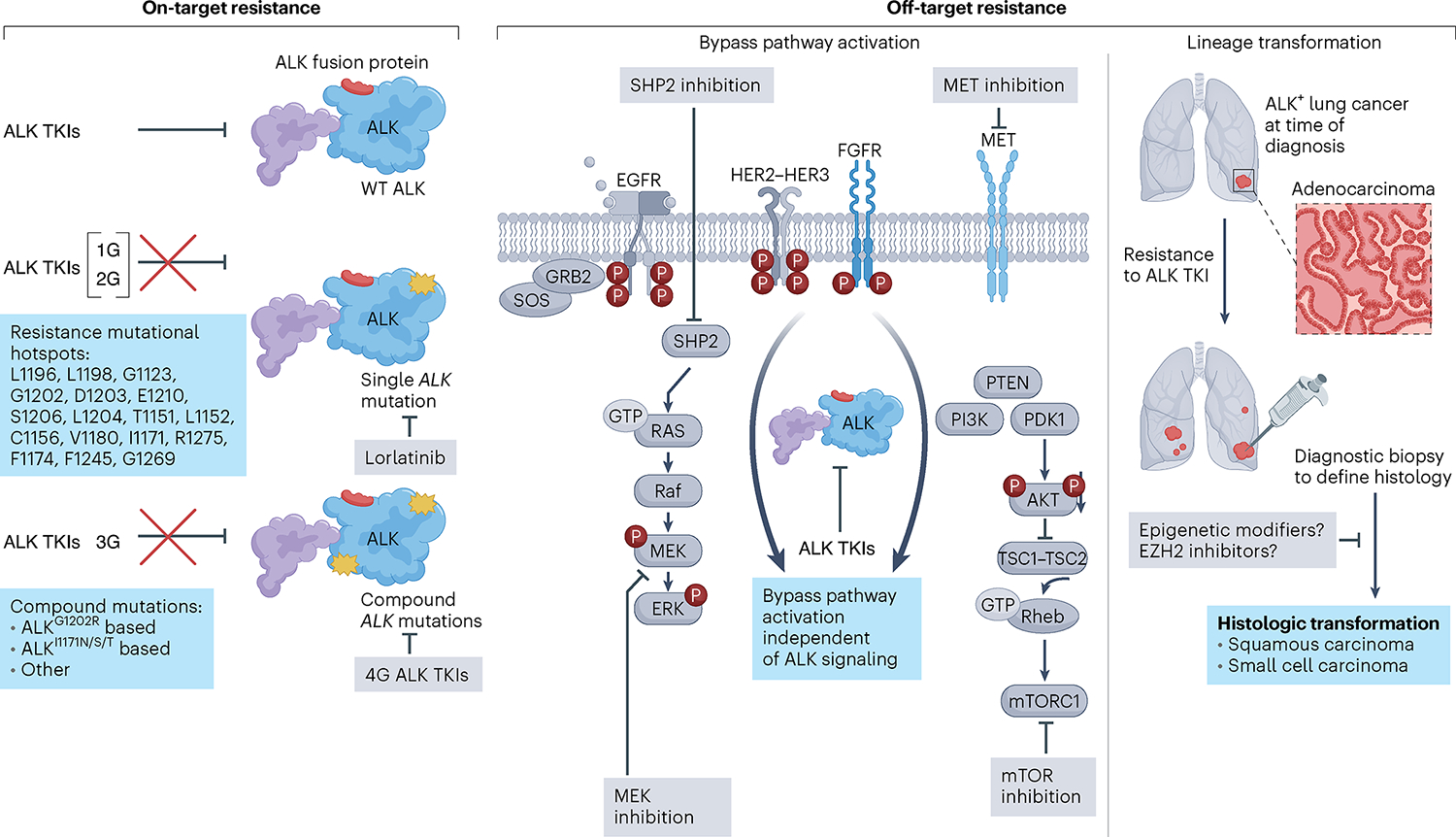
Resistance to ALK TKIs occurs through three main mechanisms. Left, on-target resistance is mediated by mutations in the ALK tyrosine kinase domain, which disrupt TKI binding to ALK, rendering tumor cells insensitive to ALK inhibition. ALK residues involved in ALK TKI resistance are listed. Single ALK mutations are most common after first- or second-generation ALK TKIs, while compound mutations are most common after sequential use of early-generation inhibitors culminating with a third-generation inhibitor, lorlatinib. This stepwise accumulation of ALK mutations confers resistance to ALK TKIs, with fourth-generation (4G) ALK TKIs designed to target compound mutations that are refractory to current FDA-approved ALK inhibitors. Middle, off-target resistance is mediated by bypass signaling activation or lineage transformation. Bypass pathway activation can occur through genetic mechanisms (amplifications, activation mutations, structural alterations) and non-genetic mechanisms (receptor hyperactivation), resulting in activation of signaling pathways that bypass ALK dependency. Rational combinations of ALK plus bypass pathway inhibition are being evaluated and are depicted in gray boxes. Right, lineage transformation is another off-target resistance mechanism that can lead to ALK TKI insensitivity. Diagnostic biopsies to define histology are necessary to select histology-specific chemotherapy regimens in squamous cell- or small cell-transformed tumors. Studies are underway to determine whether histologic changes are reversible and whether epigenetic modifiers may resensitize tumor cells to ALK inhibition. GRB2, growth factor receptor-bound protein 2; PTEN, phosphatase and tensin homolog; Rheb, Ras homolog enriched in brain; SOS, son of sevenless; SHP2, SH2 containing protein tyrosine phosphatase-2; TSC, tuberous sclerosis proteins 1 and 2; WT, wild type.
ALK mutations in acquired resistance
Sequencing of paired treatment-naive and ALK TKI-resistant tumor specimens has been key to discovery of ALK resistance mutations46 that confer resistance by re-inducing kinase activation despite the presence of a TKI. Functional validation of putative resistance mechanisms in PDCs and xenograft models helped distinguish drivers of resistance from bystander mutations56. As tissue biopsies are not always feasible at the time of clinical relapse, analysis of circulating tumor DNA (ctDNA) from liquid biopsies has offered a complementary tool to track the evolution of resistance57.
In 50–60% of patients treated with a second-generation ALK inhibitor, resistance arises through acquisition of a secondary ALK mutation46. These mutations occur universally in the kinase domain and confer resistance by direct steric hindrance of TKI binding, alteration in protein kinase conformation and/or changes in ATP binding46,58,59. In contrast to EGFR-mutant lung cancers in which T790M was the predominant EGFR alteration refractory to early-generation inhibitors60, an impressively broad spectrum of ALK mutations confer resistance in ALK+ NSCLC46. The first ALK resistance mutation identified was the L1196M gatekeeper mutation61,62. ALKG1269A similarly affects the ATP-binding pocket, impairing crizotinib docking. Another class of alterations known as solvent-front mutations, including G1202R, G1202del, D1203N, S1206Y and S1206C, disrupt the solvent-facing surface of ALK and impair drug binding through steric hindrance63. ALKG1202R confers resistance to both first- and second-generation TKIs at clinically achievable doses and accounts for approximately half of on-target resistance across all second-generation ALK TKIs46. Each ALK TKI, even those within the same generation, is associated with a distinct spectrum of secondary ALK resistance mutations. For example, although ALKG1202R is the most common ALK mutation identified across specimens after treatment with ceritinib, alectinib and brigatinib, ALKI1171N, ALKI1171T and ALKI1171S mutations are seen in 10–15% of alectinib-resistant samples but in no ceritinib- or brigatinib-resistant samples46.
Lorlatinib was specifically designed to overcome ALK mutations refractory to first- and second-generation ALK TKIs64,65 and has shown efficacy against most single ALK mutations including G1202R and I1171X54. However, resistance to lorlatinib also emerges, with a distinct profile of ALK mutations arising with sequential TKI use culminating with lorlatinib37,66,67. Sequencing of biopsies from patients relapsing on lorlatinib revealed that on-target resistance accounts for approximately a third of cases, composed of diverse compound (that is, two or more ALK mutations occurring on the same allele or in cis) and notably not single ALK mutations37. For instance, the ALKG1202R + L1196M compound mutation has been identified in patients at the time of relapse and was independently identified in an untransformed cell line mutagenesis screen yielding lorlatinib-refractory clones37. Analyses of serial clinical biopsies demonstrated that sequential ALK TKI therapy culminating in lorlatinib induces compound ALK mutations, with ALKG1202R- or ALKI1171N-based compound mutations being the most common37,68. The multitude of single ALK mutations existing after treatment with prior ALK TKIs likely serves as the substrate for compound ALK mutations to develop on lorlatinib, supporting the notion of stepwise accumulation of resistance mutations.
A substantial fraction of lorlatinib-resistant compound ALK mutations such as G1202R + L1196M and G1202R + F1174C/L are refractory to all approved ALK TKIs68,69, highlighting the need for fourth-generation ALK inhibitors. However, a limited range of double mutants are sensitive to currently available agents. An in vitro study showed that select ALKI1171N-based compound mutations are sensitizing to brigatinib and ceritinib69. Furthermore, in an illustrative case report, a patient with ALK+ lung cancer who had received sequential TKIs (crizotinib, ceritinib, lorlatinib) and ultimately acquired a lorlatinib-resistant ALKL1198F + C1156Y mutation had a durable response to retreatment with crizotinib, as the acquired ALKL1198F mutation re-sensitized tumor cells to crizotinib59. Although these examples are rare, they underscore the importance of serial biopsies in guiding next-line therapeutic strategies and the continued need to catalog and functionally test emerging ALK resistance mutations.
Mechanisms of ALK-independent resistance
In approximately half of the patients with ALK+ NSCLC who progress on a second-generation ALK TKI, ALK mutations are not identified at the time of clinical relapse, suggesting ALK-independent resistance and only modest benefit by subsequent-generation ALK inhibitors for this subset of patients68,70. Diverse off-target mechanisms that confer ALK TKI resistance can occur across patients, making ALK-independent resistance challenging to overcome.
Bypass pathway activation
One important category of ALK-independent resistance is activation of bypass signaling, which arises from genetic alterations, changes in protein expression and/or activation or dysregulation of autocrine feedback signaling. Multiple bypass tracks have been described in ALK TKI-resistant tumors including activation of RTKs MET71, EGFR72, SRC56, IGF-1R73, HER2 and HER3 (ref. 74) and KIT72 and alterations in downstream signaling factors MAP2K1 (refs. 23,56), DUSP6 (ref. 23), STAT3 (ref. 75) and NF2 (ref. 66). These co-occurring genetic alterations mediating resistance are not present at the time of diagnosis in treatment-naive patients76.
The first bypass mechanisms were described in the context of crizotinib resistance. Comparison of crizotinib-sensitive and crizotinib-resistant cells and biopsies before and after treatment revealed increased EGFR tyrosine phosphorylation72,77. EGFR autophosphorylation can occur in the absence of acquired EGFR mutations or amplification, indicating non-genetic mechanisms such as altered EGFR dynamics and expression, enhanced EGFR ligand binding or dysregulation of feedback loops. Hyperactivation of other RTKs has been reported, including HER2 and HER3 (ref. 74) and activation of protein kinase C (PKC) signaling through P2Y purinergic receptor family G-protein-coupled receptors78. Characterization of the bypass resistance landscape after next-generation ALK inhibitors revealed similar findings, including increased activation of RTKs IGF-1R and HER3 and overexpression of the HER3 ligand neuregulin 1 (NRG1)73,74,78,79.
MET alteration is a well-established driver of RTK-mediated resistance in ALK+ and other NSCLC subsets80, with amplification detected in ~15% of tumor biopsies from patients relapsing on next-generation ALK TKIs71. Patients receiving second-generation ALK TKIs in the first-line setting are more likely to develop MET amplification than those on next-generation ALK inhibitors following treatment with crizotinib, which inhibits MET71. Combined ALK–MET inhibition using crizotinib alone or lorlatinib plus a MET-selective TKI can effectively suppress proliferation of ALK+, MET-amplified tumors71, supporting the clinical testing of combined ALK and MET inhibitors (NCT04292119).
Intracellular signaling mediators have also been implicated in acquired resistance. Oncogenic ALK signaling requires activation of the MAPK pathway. ALK-independent MAPK pathway reactivation can occur through multiple mechanisms including KRAS copy number gain, mitogen-activated protein kinase kinase 1 (MAP2K1)-activating mutations56 or loss of DUSP6, a negative regulator of MAPK23. Upfront co-inhibition of mitogen-activated protein kinase kinase (MEK) enhanced the therapeutic efficacy of ALK inhibition through diminished residual MAPK signaling23, providing the rationale for clinical testing of dual ALK and MEK blockade (NCT03202940) to enable more durable responses by limiting tumor cell persistence and clonal expansion.
Functional inhibition of a protein with pleiotropic effects may offer a superior strategy for overcoming bypass pathways. For example, a short hairpin RNA dropout screen of multiple ALK TKI-resistant PDCs identified SH2-containing protein tyrosine phosphatase 2 (SHP2) as a potential target42. SHP2 mediates GTP loading of RAS downstream of multiple RTKs including EGFR, FGFR and MET for modulation of JAK–STAT, PI3K–AKT and MAPK pathways. Pharmacological inhibition of SHP2 attenuates ceritinib-induced ERK kinase reactivation, and combined inhibition of ALK and SHP2 restores sensitivity and overcomes resistance in drug-tolerant cell lines42. Dual ALK and SHP2 inhibition is being evaluated in early-phase trials (NCT04292119 and NCT04800822).
That bypass track engagement renders cells fully independent of ALK may be an oversimplified notion. ALK TKI-resistant PDCs often retain partial dependency on ALK, and maximal cytotoxicity is achieved with dual ALK and bypass inhibition77. In the clinic, disease flares can occur upon discontinuation of ALK TKIs, even in the context of known ALK-independent resistance mechanisms81. These observations may reflect intratumoral or intertumoral heterogeneity in which subsets of cells remain addicted to ALK. More studies are needed on the dynamics of sustained ALK dependency during acquired resistance. The field has been limited by tissue availability, as obtaining matched pretreatment and post-resistance biopsy specimens for functional analyses can be challenging. Comparative analysis of specimens before and after TKI treatment paired with functional validation of putative pathways will be integral to fully elucidate genetic and non-genetic mechanisms of resistance and the breadth of off-target mechanisms.
Histologic transformation
Transformation of a tumor to a different histologic subtype is associated with loss of reliance on the oncogenic driver, leading to drug resistance82. Although virtually all cases of newly diagnosed ALK+ NSCLC are adenocarcinoma, small cell lung cancer transformation has been identified in patients with ALK+ lung cancer after treatment with all generations of ALK TKIs, albeit at low frequency (<3% according to retrospective analysis)83–86. In small cell-transformed EGFR-mutant and ROS1 fusion-positive lung cancers, even though the original driver gene alteration was retained, its expression was lost upon transformation87,88. Transformation to squamous cell carcinoma has also been reported following treatment with alectinib89 and lorlatinib68.
Identifying histological transformation in the clinic is critical for the selection of subsequent histology-matched therapy. Given the rarity of transformed ALK+ lung cancers, randomized prospective trials to inform treatment strategies following phenotypic changes are not feasible. Ongoing research centers on elucidating the molecular changes that rewire cellular phenotypes during ALK-targeted therapy and understanding their reversibility, with the goal of restoring adenocarcinoma histology and re-sensitizing cells to ALK inhibition.
Challenges of polyclonal resistance
Therapeutic targeting of ALK TKI-resistant tumors is complicated by the heterogeneity of resistance mechanisms (Fig. 3). Divergent pathways may evolve in distinct metastatic foci within one patient or in clusters of tumor cells within one disease site, resulting in polyclonal resistance. For instance, concomitant ALK mutations with ALK or KIT amplifications have been identified in biopsies after crizotinib treatment72,90. A report of a patient with ALK+ NSCLC progressing on ceritinib followed by alectinib noted the concomitant detection of sequence encoding ALKG1202R in ctDNA and small cell transformation in tumor91. In instances in which ALK resistance mutations and off-target resistance mechanisms co-occur, addressing ALK dependency alone is not sufficient.
Fig. 3 |. Forward-looking treatment paradigms in advanced ALK+ lung cancer.
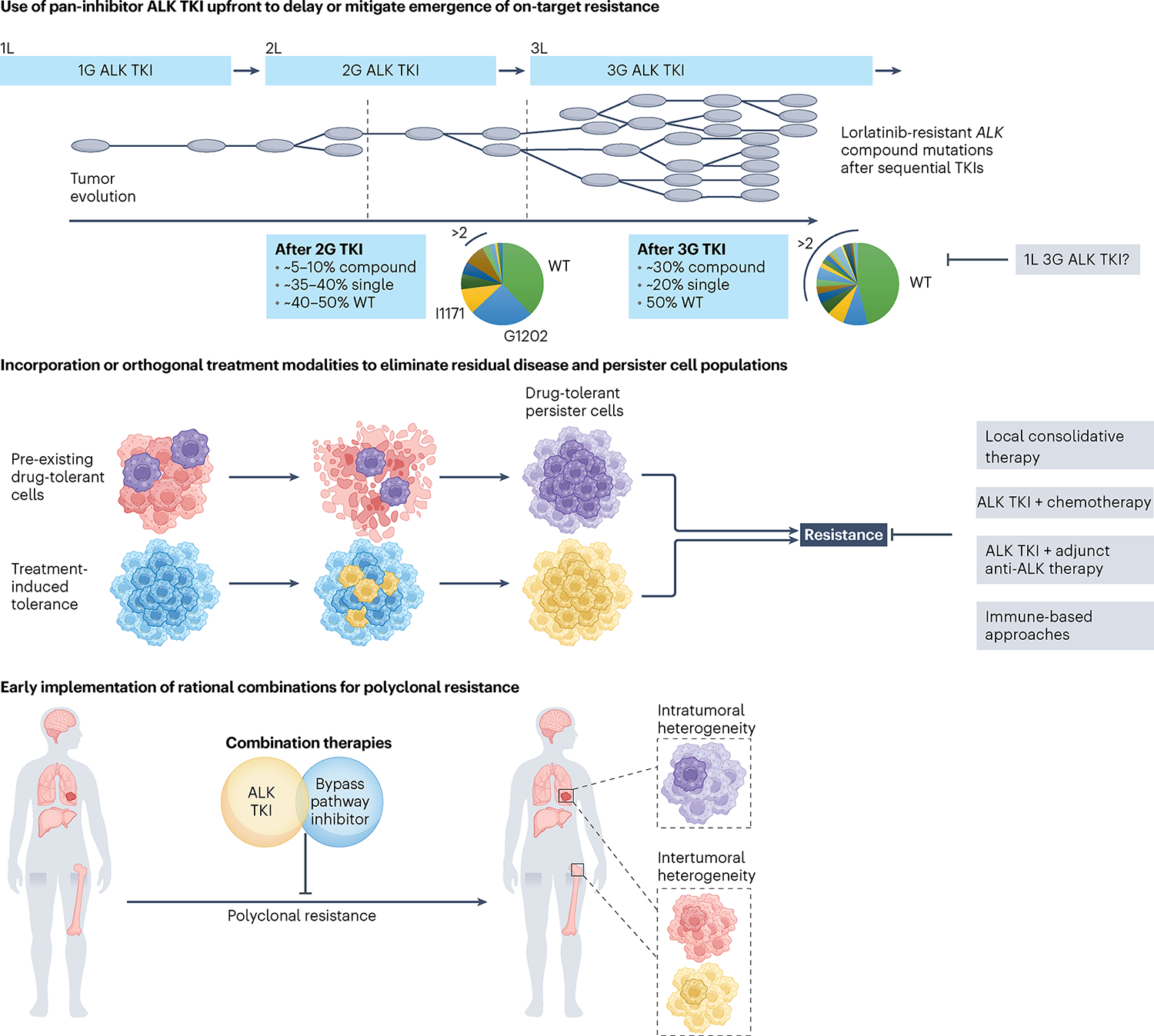
Top, sequential ALK TKI therapy culminating in lorlatinib induces compound ALK mutations, with ALKG1202R- or ALKI1171N-based compound mutations being the most common. The schematic depicts tumor clonal evolution with the multitude of single ALK mutations serving as a substrate for compound ALK mutations, highlighting the notion of stepwise accumulation of resistance mutations. Treatment with a highly potent pan-inhibitory third-generation ALK TKI in the first-line (1L) setting may allow for maximal cytoreduction and depth of response, limiting tumor heterogeneity that can emerge with less potent ALK TKIs. Middle, drug-tolerant cells that are present at the time of treatment may undergo expansion under therapeutic selective pressure, leading to treatment failure and clinical relapse. In parallel, persister cells that survive initial treatment may acquire de novo resistance alterations, serving as a nidus for the development of polyclonal resistance. Depicted in gray are potential adjunctive therapeutic strategies aimed at eliminating persister cells. Bottom, intratumoral heterogeneity can occur across different regions of the primary tumor and/or metastatic sites, with spatial heterogeneity represented by the presence of subclones with different genetic features. Intertumoral heterogeneity can also occur across different metastatic sites, which can be missed using single-site tissue biopsies. Studies are underway to evaluate the utility of early rational combinations and to stem polyclonal resistance. In parallel, efforts are ongoing to develop ultrasensitive diagnostic tools to track tumor response and detect microscopic disease. 2L, second line; 3L, third line.
Tissue biopsies are not always feasible at the time of disease progression. ctDNA analysis from liquid biopsies offers a complementary tool for monitoring the emergence and temporal evolution of acquired ALK mutations and capturing mechanisms of resistance across metastatic sites57. Comparative analysis of plasma and tumor specimens after alectinib treatment in a study with ~90% sensitivity of plasma genotyping to detect ALK resistance mutations in relapsing patients revealed that plasma was more likely to harbor at least two ALK mutations, indicating polyclonal resistance that was not captured with single-site tissue biopsies57. Various other gene alterations implicated in off-target resistance can be detected by plasma biopsies, including BRAF, MAP2K1 and PIK3CA mutations92, although certain copy number changes or structural gene alterations, such as MET amplification, may be harder to detect.
The increasing ability to identify disparate mechanisms of resistance in an individual patient may pose clinical dilemmas about which, if any, warrants therapeutic targeting, especially in cases in which multiple FDA-approved or investigational drugs are available.
Therapeutic strategies in ALK+ NSCLC
Below, we discuss new therapeutic strategies that focus on new ways to maximally inhibit ALK and to overcome or prevent ALK TKI resistance.
Sequencing of ALK TKIs
With several FDA-approved ALK TKIs as first-line treatment for advanced ALK+ NSCLC, the optimal front-line next-generation ALK TKI (in particular, alectinib, brigatinib or lorlatinib) remains controversial93,94. Second-generation ALK TKIs such as alectinib are more commonly used as the initial therapy (with lorlatinib reserved as salvage therapy93) on the basis of efficacy that can be achieved with upfront second-generation TKIs and a favorable toxicity profile with alectinib in particular. Whether this achieves the optimal clinical outcome is unclear. Starting with a less potent agent has the increased probability of selecting for refractory compound mutations, many of which are recalcitrant to all currently FDA-approved ALK inhibitors as discussed above37,95.
Moving the most potent ALK inhibitor to first line may suppress or delay the emergence of on-target resistance and prolong the duration of response. In the phase 3 CROWN trial of first-line lorlatinib versus crizotinib in patients with advanced ALK+ NSCLC9, lorlatinib resulted in significantly longer PFS with an impressive HR for disease progression or death of 0.27 based on an updated analysis9,55, as compared to the HR for progression or death of close to 0.50 seen for the second-generation TKIs alectinib, brigatinib and ensartinib when compared to crizotinib7,8,49. Furthermore, lorlatinib has the highest level of CNS penetration and provides robust CNS protection55,96, which is noteworthy given the CNS tropism of ALK+ disease97. Although there is no head-to-head comparison of lorlatinib versus a second-generation ALK TKI and the resistance landscape for first-line lorlatinib remains to be determined, these data support starting with a pan-inhibitory, highly potent and CNS-penetrant ALK TKI to ensure maximal cytoreduction and depth of response, limiting the tumor heterogeneity that can emerge with less potent second-generation ALK TKIs and delaying on-target resistance and CNS recurrence.
As efforts to reach consensus on ALK TKI sequencing continue, patients whose tumors harbor an ALK resistance mutation can be treated with an ALK TKI targeting that particular mutation, if available. Patients whose tumors lack ALK mutations can be considered for ALK-based combinatorial strategies or other investigational approaches to tackle ALK-independent resistance.
Overcoming on-target resistance with new ALK TKIs
A subset of ALK-driven tumors remains addicted to ALK even after lorlatinib treatment due to compound ALK mutations. Functional screening of a panel of lorlatinib analogs in vitro and in vivo indicated that distinct molecules have differential selectivity for ALKG1202R- versus ALKI1171-based compound mutations68. For example, two lorlatinib analogs, LA7 and LA9, demonstrated selectivity against ALKI1171N and ALKG1202R single and compound mutants, respectively. These data suggest that distinct ALK TKIs may be required against different classes of compound ALK mutations, with one new ALK TKI being unlikely to overcome all on-target lorlatinib resistance.
TPX-0131 (ref. 98) and NVL-655 (ref. 99) are fourth-generation ALK TKIs designed to target compound ALK mutations, with preclinical activity against single and some compound ALK mutations (for example, ALKG1202R + L1196M, ALKG1202R + G1269A, ALKG1202R + L1198F)100. Both agents are currently in phase 1 testing (NCT04849273 and NCT05384626, respectively). Consistent with lorlatinib analogs, TPX-0131 lacks activity against ALKLI1171 mutations but has high potency against ALKG1202R and ALKG1202R-based double and triple mutations98. By contrast, gilteritinib, an agent used in FLT3-mutated acute myelogenous leukemia101, acts against ALKI1171N-based but not ALKG1202R-based compound mutations102.
The challenge of overcoming the diverse array of compound ALK mutations aside, should early-phase trials of fourth-generation ALK TKIs demonstrate favorable safety profiles, studies will be needed on how to integrate them into the already crowded landscape of ALK-targeted therapy. Each TKI will likely have a distinct potency spectrum against ALK mutants, and thus may be most useful after next-generation TKIs with known ALK-dependent resistance. Mutations conferring resistance to these new agents are anticipated, highlighting the challenge of perpetually chasing resistance in ALK+ NSCLC. One potential advantage of starting with a pan-single mutant-inhibitory ALK TKI such as lorlatinib is the prevention of refractory compound ALK mutations and obviation of the need for higher-generation TKIs.
Alternative ALK-centric approaches
Use of sequential ALK TKIs has exposed the problem of increasingly complex on-target resistance mutations. Thus, alternative approaches of targeting ALK outside of small-molecule TKIs merit attention. Targeting ALK through allosteric or covalent inhibition or protein degradation may serve as a complementary therapeutic approach to circumvent the increasingly complex on-target resistance mutations exposed by using sequential ALK TKIs. One strategy involves covalent ALK inhibitors targeting cysteine residues located outside of the active site103. Proteolysis targeting chimeric (PROTAC) technology is another approach, used to direct endogenous protein degradation by linking a protein of interest and an E3 ubiquitin ligase104. A flurry of PROTACs have been developed against oncoproteins including estrogen or androgen receptors, BTK and BCR–ABL1, several of which are in early-phase clinical trials105. Aside from those using an ALK TKI106 as the PROTAC’s ALK ligand, for example, using allosteric ALK inhibitors, ALK-directed PROTACs could be agnostic to ALK kinase domain mutations, thus overcoming TKI resistance. Preclinical testing of ALK-directed PROTACs reported potent decreases in total and phosphorylated ALK levels in a concentration- and time-dependent fashion106,107. In vivo evaluation is needed to clarify the biologically relevant intracellular ALK concentrations, as even residual amounts may be sufficient to activate proliferative pathways.
Another ALK-centric approach involves the disruption of protein–protein interactions. EML4–ALK homodimerizes through a coiled-coil domain within EML4 (ref. 21) that requires a conserved pattern of hydrophobic residues and salt bridges108. Disruption of this interaction abrogates the transforming ability of the ALK fusion21, and introduction of competitive coiled-coil-mimetic compounds abrogates tumor formation109.
Whether these agents will be successfully developed clinically and whether they will be integrated into practice together with or in lieu of ALK TKIs remains unknown. However, despite maximal ALK inhibition, a substantial proportion of patients will develop ALK-independent resistance requiring alternative treatment strategies such as targeting bypass pathways or tumor microenvironmental factors.
Combinatorial strategies against ALK-independent resistance
The functional characterization of bypass pathways mediating ALK TKI resistance has led to development of rational combinatorial approaches for patients who relapse on ALK TKIs. MET amplification is a prototypical actionable bypass pathway in ALK+ NSCLC71 and has similarly been established as a bona fide bypass track in EGFR-mutant110 and RET fusion-positive111 NSCLC, in which co-targeting of the oncogenic driver and MET can overcome MET-driven resistance112,113. Dual ALK– MET inhibition re-sensitizes ALK+ PDC models with MET-driven resistance71. Case reports have documented disease responses to crizotinib monotherapy or lorlatinib-based combinations with a MET inhibitor in patients who are ALK+ with acquired MET amplification71,114. A phase 1–2 study is evaluating the combination of lorlatinib plus crizotinib to address MET-driven resistance (NCT04292119). Several additional ALK TKI-based combination trials are underway (Table 2). Most trials (outside of ALK–MET inhibitor combinations) do not require biomarkers to guide stratification. Further investigation is needed to identify biomarkers associated with responses to combinations.
Table 2 |.
Summary of ongoing trials with combination therapy in ALK+ NSCLC
| Clinical trial identifier | Drugs | Bypass pathway targeted | Phase | Biomarker required outside of ALK | |
|---|---|---|---|---|---|
|
| |||||
| ALK TKI | Other agent | ||||
|
| |||||
| NCT02321501 | Ceritinib | Everolimus | mTOR | 1 | No |
| NCT03202940 | Alectinib | Cobimetinib | MAPK (MEK) | 1–2 | No |
| NCT04005144 | Brigatinib | Binimetinib | MAPK (MEK) | 1 | No |
| NCT04227028 | Brigatinib | Bevacizumab | VEGF | 1 | No |
| NCT04292119 | Lorlatinib | Binimetinib | MAPK (MEK) | 1–2 | No |
| Crizotinib | MET | Yes (MET amplification) | |||
| TNO155 | SHP2 | No | |||
| NCT04800822 | Lorlatinib | PF-07284892 | SHP2 | 1 | No |
mTOR, mammalian target of rapamycin; VEGF, vascular endothelial growth factor.
These rational combination strategies have thus far yielded underwhelming results without notable efficacy. One challenge is the augmentation in toxicities that can limit dosing of each drug. Another potential pitfall is the timing of treatment, as combinations may exert a greater impact in forestalling the emergence of resistance, rather than overcoming resistance once established23. In light of these challenges, whether the evolutionary trajectory of an ALK+ lung cancer could be discerned at initial diagnosis and tumors preemptively pressured toward a particular path to exploit targetable vulnerabilities remains in question.
Targeting persister cell populations
Despite marked responses typically seen upon ALK TKI initiation, residual disease often remains and can lead to relapse, even after several years of stability on therapy. Growing evidence indicates that drug-tolerant persister cells are responsible for residual disease (Fig. 3). The persister state is thought to be reversible, with most cells remaining in cell cycle arrest in the presence of drug but with a small subset having the capacity to re-enter the cell cycle. Persisters may subvert TKI inhibition through adaptive mechanisms, including epigenetic modifications, bypass activation, metabolic reprogramming and altered interactions with the tumor microenvironment115.
Non-genetic mechanisms have emerged as important drivers of persister cell survival. For instance, cycling and non-cycling drug-resistant persister cells from EGFR-mutant lung cancer were shown to arise from distinct cell lineages, with discrete transcriptional and metabolic programs116. A shift to fatty acid oxidation was associated with persister proliferative capacity in EGFR-mutant NSCLC cells and across multiple cancer subtypes, underscoring a proliferative response that may enable transition to oncogene independence following treatment. As an example, the transcriptional regulator YAP1 was shown to be activated following treatment of ALK+ cells with alectinib in vitro and in vivo, an effect attenuated by combinatorial inhibition of ALK and YAP1 (ref. 117). Genetic and pharmacologic blockade of YAP1 suppressed tumor growth in drug-resistant cells, xenograft models and EML4-ALK-transgenic mice118. High expression of YAP1 in treatment-naive samples was found to be a negative prognostic sign for response to ALK TKIs118. Selective inhibitors of YAP, its isoform TAZ and its binding partner TEAD are in early-phase clinical trials119 (NCT04665206, NCT04857372, NCT05228015 and NCT04659096). Illuminating the full spectrum of non-mutational mechanisms that bolster persister fitness will be critical in targeting pro-survival escape pathways promoting clinical relapse.
In the clinic, eradication of persister cells is one rationale for using local consolidative approaches (for example, radiation) to ablate sites of residual disease after TKI initiation. More data are needed to ascertain clinical benefit of ablating persister populations after targeted therapies before relapse120. One prospective study is evaluating the impact of integrating stereotactic body radiation therapy after induction with TKIs in stage IV oncogene-driven NSCLC (NCT02314364). Adding chemotherapy or epigenetic modulators to an ALK TKI after initial cytoreduction could serve as alternative strategies to eliminate persisters, although not yet clinically evaluated. One caveat with existing strategies is the reliance on radiographic findings to identify residual disease at the macroscopic rather than microscopic level. Ultrasensitive, blood-based techniques may prove beneficial for monitoring occult persistent and/or recurrent disease.
Immune-based therapies for ALK+ lung cancer
Although programmed cell death protein 1 (PD-1)–programmed cell death ligand 1 (PD-L1) checkpoint inhibitors have revolutionized the management of NSCLC, their efficacy has been minimal in patients with ALK+ lung cancer121, even among patients with high PD-L1 expression121,122. Retrospective and real-world studies show lower response rates and shorter PFS for immune checkpoint monotherapy among patients with ALK+ or EGFR-mutant NSCLC than those with ALK–EGFR-wild-type disease121,123,124. Although most prospective or randomized phase 3 studies have excluded patients with EGFR mutations or who are ALK+ (ref. 125), those that allowed patients with ALK+ NSCLC have been largely underpowered to draw conclusions126,127. ALK+ tumors have low tumor mutational burden, effectively limiting the neoantigen landscape, and low colocalization of PD-L1 expression with CD8+ tumor-infiltrating lymphocytes, which likely underlies the limited anti-tumor immune responses regardless of oncogene-mediated upregulation of PD-L1 (refs. 121,122,128). Despite data supporting refractoriness of ALK+ NSCLC to immune checkpoint inhibitor monotherapy, the impact of combining them with other treatments (such as chemotherapy) remains to be clarified129,130. Combinations with ALK TKIs have been explored, largely demonstrating lack of synergistic efficacy and, in some cases, heightened toxicities131–133. Alternative immune-based strategies may be a more promising avenue in the context of ALK-driven disease.
For instance, ALK represents an attractive target for vaccines as it is recognized as a tumor antigen and autoantibodies against it are detected in NSCLC and ALCL, suggesting that some patients may be able to generate a spontaneous anti-ALK immune response134,135. Mapping of specific epitope sequences of anti-ALK autoantibodies in a small cohort of patients with NSCLC demonstrated clustering outside the kinase domain134. High levels of these spontaneous anti-ALK antibodies were detected in 17% of patients with ALK+ NSCLC, underscoring the fact that ALK can be spontaneously immunogenic in a small subset of patients. Hypothetically, an ALK vaccine could potentiate an anti-tumor response in patients who already have autoantibodies and may induce an immune response for those without autoantibodies. The first ALK vaccine developed in 2015 consisted of a DNA plasmid coding for the intracytoplasmic domain of ALK and produced remodeling of the immune microenvironment and a CD8+-mediated cytotoxic response in mouse models of orthotopic ALK+ lung tumors136. The first-in-human trial of an ALK peptide vaccine is anticipated to open in 2023. The optimal place for an ALK vaccine in the therapeutic landscape remains to be determined.
Because the ALK fusion protein is localized intracellularly in NSCLC, certain therapeutic modalities such as chimeric antigen receptor T cells and antibody–drug conjugates that require the presence of a tumor antigen on the cell surface are not feasible. However, ALK-targeted antibody–drug conjugates137 and chimeric antigen receptor T cells138 are being developed in ALK-mutated neuroblastoma and other pediatric tumors where ALK is expressed on the plasma membrane. Identification of intracellular ALK antigen fragments presented by major histocompatibility complex (MHC) molecules may serve as a foundation for engineering modified T cell receptor (TCR) T cells. In advanced uveal melanoma, tebentafusp, a bispecific protein consisting of an affinity-enhanced TCR specific for an HLA-A*02:01-presented glycoprotein 100 (gp100) epitope fused to an anti-CD3 effector, had a survival benefit compared to the control group of pembrolizumab, ipilimumab or dacarbazine139. This result serves as a proof of concept that tumors with low tumor mutational burden that are refractory to immune checkpoint inhibitors can be responsive to immune strategies.
A deeper understanding of the immunobiology of ALK+ NSCLC will be critical for devising immune-based therapies. ENIGMA+ (‘Elucidating novel immune and genomic markers for ALK+’; NCT04881916) is a research platform aiming to enable remote consent and participation of patients with ALK+ lung cancer nationwide to illuminate vulnerabilities for immune-based therapeutics in ALK+ NSCLC.
Addressing lineage plasticity
The molecular mediators that govern lineage plasticity in ALK+ lung cancer and how this leads to TKI resistance remain to be determined. More exploratory studies are needed to elucidate the role of repressor RB1 and tumor protein p53 (TP53) in ALK+ SCLC transformation and associated dependencies in tumors that have undergone lineage changes140,141. In patients with EGFR-mutant lung cancers, in which small cell transformation accounts for 10–15% of acquired resistance to EGFR TKIs142,143, patients with concurrent TP53 and RB1 loss at diagnosis are at significantly higher (43×) risk of small cell transformation than those without144,145. One study implicated Aurora kinases as a unique dependency in RB1-deficient SCLC146, raising the possibility that Aurora kinase inhibitors may be used to target subpopulations of EGFR-mutant and ALK+ cancers that acquire RB1 loss in the context of lineage change.
Modulation of the epigenetic landscape is also being studied as an avenue for reversing lineage plasticity and re-sensitizing cells to ALK inhibitors. EZH2 is an epigenetic modulator that is upregulated in SCLC147 and inhibition of which suppresses lineage plasticity in prostate cancer148. EZH2 inhibitors are being evaluated as single agents or in combination in relapsed small cell lung cancer (NCT03460977). Whether transformed ALK+ SCLC exhibits upregulation of EZH2 or other epigenetic modulators is unknown. Moreover, questions remain about appropriate timing of epigenetic therapy. If applied early in ‘at risk’ patients, it may block lineage changes and prevent transformation. If phenotypic changes are reversible, epigenetic therapies may be used at the time of transformation aimed at restoring TKI sensitivity.
Future directions
The treatment of ALK+ lung cancer represents a paradigm of precision oncology, offering lessons on targeted therapies applicable across cancer types. Over a short timeframe since the discovery of the EML4-ALK fusion in NSCLC, tremendous strides have been made in the development of ALK-directed therapies, resulting in dramatic improvements in patient survival50. The field has exemplified how integration of bench and bedside investigations can uncover important molecular insights that translate into real-time clinical benefit for patients.
Yet metastatic ALK+ lung cancer remains incurable, and many challenges remain in the treatment of this disease. Although genotyping of clinical samples at the time of relapse has informed resistance biology and led to development of later-generation ALK TKIs, it has also revealed increasingly TKI-refractory ALK resistance mutations and heterogeneous off-target ALK escape mechanisms. Elucidating the molecular drivers of resistance and continuing to develop innovative therapies to overcome resistance is critical. For instance, tumor microenvironment factors that modulate ALK TKI sensitivity remain to be elucidated. More work is needed to illuminate how manipulation of the microenvironment, including local immune cells and fibroblasts, could be exploited for therapeutic purposes.
Fundamental questions remain regarding how to stem the emergence of polyclonal resistance, eradicate persister cells and ultimately take the transformative step toward curing patients with metastatic disease. A singular ALK TKI or even a combination approach is unlikely to achieve this goal. An optimal treatment approach will require encompassing the following aspects: (1) the use of a pan-inhibitory ALK TKI upfront to block on-target resistance; (2) routine incorporation of orthogonal treatment modalities such as radiation, chemotherapy, epigenetic or immune-based strategies to eliminate residual disease; (3) development of highly sensitive diagnostics to track tumor response and detect microscopic residual or recurrent disease; and (4) early implementation of rational combinations before the clinical evidence of relapse. Plasma monitoring to define the disease status remains investigational, and further studies are needed to clarify whether adaptive therapeutic escalation or de-escalation based on serial plasma monitoring may improve outcomes.
Furthermore, questions remain regarding the potential use of ALK TKIs in the adjuvant and neoadjuvant settings for early-stage ALK+ lung cancer. The seminal phase 3 trial ADAURA demonstrated significantly longer disease-free survival in patients with surgically resected EGFR-mutant NSCLC who received the EGFR TKI osimertinib149. A phase 3 study of adjuvant alectinib versus chemotherapy in patients with resected ALK+ NSCLC is ongoing (NCT03456076). In addition, the NAUTIKA1 phase 2 trial is evaluating various targeted therapies in the neoadjuvant setting for patients with early-stage resectable NSCLC harboring appropriate biomarkers, including alectinib for ALK+ disease (NCT04302025). Results from these adjuvant and neoadjuvant studies will inform the optimal management of early-stage ALK+ lung cancer.
A deeper understanding of the unique biology of ALK+ lung cancer and continued therapeutic advances will ultimately catalyze waves of translational research efforts dedicated to the overarching goal of prolonging lives and inducing cures in patients with ALK+ lung cancer.
Acknowledgements
We thank patients and their families for their heroic efforts in contributing to and advocating for ALK+ lung cancer research. We apologize to the numerous colleagues whose important contributions may not have been cited in this Review due to space constraints. J.L.S. is supported by A Breath of Hope Lung Foundation and the Lung Cancer Research Foundation. J.J.L. is supported in part by NIH/NCI R01CA164273.
Footnotes
Competing interests
J.L.S. has received honoraria from the Academy of Continued Healthcare Learning, Springer Healthcare and Targeted Oncology. J.J.L. has served as a compensated consultant for Genentech, Regeneron, C4 Therapeutics, Blueprint Medicines, Nuvalent, Bayer, Elevation Oncology, Novartis, Mirati Therapeutics and Turning Point Therapeutics; received honorarium and travel support from Pfizer; received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Bayer, Elevation Oncology, Roche, Linnaeus Therapeutics, Nuvalent and Novartis; and received CME funding from OncLive, MedStar Health and Northwell Health. A.T.S. is currently employed by Novartis.
Peer review information Nature Cancer thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Howlader N et al. The effect of advances in lung-cancer treatment on population mortality. N. Engl. J. Med. 383, 640–649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thai AA, Solomon BJ, Sequist LV, Gainor JF & Heist RS Lung cancer. Lancet 398, 535–554 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Kris MG et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311, 1998–2006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 368, 2385–2394 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Solomon BJ et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 371, 2167–2177 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Soria JC et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 389, 917–929 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Peters S et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 377, 829–838 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Camidge DR et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 379, 2027–2039 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Shaw AT et al. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N. Engl. J. Med. 383, 2018–2029 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Lin JJ, Riely GJ & Shaw AT Targeting ALK: precision medicine takes on drug resistance. Cancer Discov. 7, 137–155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris SW et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263, 1281–1284 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Wellstein A ALK receptor activation, ligands and therapeutic targeting in glioblastoma and in other cancers. Front. Oncol. 2, 192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao S et al. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PLoS ONE 8, e63757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du Z & Lovly CM Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao R et al. Transcriptome regulation by oncogenic ALK pathway in mammalian cortical development revealed by single-cell RNA sequencing. Cereb. Cortex 31, 3911–3924 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fadeev A et al. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl Acad. Sci. USA 115, E630–E638 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallberg B & Palmer RH Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 13, 685–700 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Ducray SP, Natarajan K, Garland GD, Turner SD & Egger G The transcriptional roles of ALK fusion proteins in tumorigenesis. Cancers 11, 1074 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu TL et al. NPM–ALK fusion kinase of anaplastic large-cell lymphoma regulates survival and proliferative signaling through modulation of FOXO3a. Blood 103, 4622–4629 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Butrynski JE et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 363, 1727–1733 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soda M et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Ou SI, Zhu VW & Nagasaka M Catalog of 5′ fusion partners in ALK-positive NSCLC circa 2020. JTO Clin. Res. Rep. 1, 100015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hrustanovic G et al. RAS–MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 21, 1038–1047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L et al. Blocking the PI3K pathway enhances the efficacy of ALK-targeted therapy in EML4-ALK-positive nonsmall-cell lung cancer. Tumour Biol. 35, 9759–9767 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Li Y et al. EML4–ALK-mediated activation of the JAK2–STAT pathway is critical for non-small cell lung cancer transformation. BMC Pulm. Med. 21, 190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heuckmann JM et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin. Cancer Res. 18, 4682–4690 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Lin JJ et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J. Clin. Oncol. 36, 1199–1206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Childress MA et al. ALK fusion partners impact response to ALK inhibition: differential effects on sensitivity, cellular phenotypes, and biochemical properties. Mol. Cancer Res. 16, 1724–1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y et al. Detection of nonreciprocal/reciprocal ALK translocation as poor predictive marker in patients with first-line crizotinib-treated ALK-rearranged NSCLC. J. Thorac. Oncol. 15, 1027–1036 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Touriol C et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 95, 3204–3207 (2000). [PubMed] [Google Scholar]
- 31.Tulpule A et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell 184, 2649–2664 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sampson J, Richards MW, Choi J, Fry AM & Bayliss R Phase-separated foci of EML4–ALK facilitate signalling and depend upon an active kinase conformation. EMBO Rep. 22, e53693 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin Z et al. Phase separation of EML4–ALK in firing downstream signaling and promoting lung tumorigenesis. Cell Discov. 7, 33 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song Z et al. Deep RNA sequencing revealed fusion junctional heterogeneity may predict crizotinib treatment efficacy in ALK-rearranged NSCLC. J. Thorac. Oncol. 17, 264–276 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Schneider JL et al. The aging lung: physiology, disease, and immunity. Cell 184, 1990–2019 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sacher AG et al. Association between younger age and targetable genomic alterations and prognosis in non-small-cell lung cancer. JAMA Oncol. 2, 313–320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoda S et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 8, 714–729 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyanaga A et al. EML4–ALK induces cellular senescence in mortal normal human cells and promotes anchorage-independent growth in hTERT-transduced normal human cells. BMC Cancer 21, 310 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soda M et al. A mouse model for EML4–ALK-positive lung cancer. Proc. Natl Acad. Sci. USA 105, 19893–19897 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pyo KH et al. Establishment of a conditional transgenic mouse model recapitulating EML4–ALK-positive human non-small cell lung cancer. J. Thorac. Oncol. 12, 491–500 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Maddalo D et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dardaei L et al. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 24, 512–517 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SY et al. Patient-derived cells to guide targeted therapy for advanced lung adenocarcinoma. Sci. Rep. 9, 19909 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ou SH Crizotinib: a novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des. Devel. Ther. 5, 471–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwak EL et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gainor JF et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 6, 1118–1133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camidge DR et al. Brigatinib versus crizotinib in advanced ALK inhibitor-naive ALK-positive non-small cell lung cancer: second interim analysis of the phase III ALTA-1L trial. J. Clin. Oncol. 38, 3592–3603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camidge DR et al. Brigatinib versus crizotinib in ALK inhibitor-naive advanced ALK-positive NSCLC: final results of phase 3 ALTA-1L trial. J. Thorac. Oncol. 16, 2091–2108 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Horn L et al. Ensartinib vs crizotinib for patients with anaplastic lymphoma kinase-positive non-small cell lung cancer: a randomized clinical trial. JAMA Oncol. 7, 1617–1625 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mok T et al. Updated overall survival and final progression-free survival data for patients with treatment-naive advanced ALK-positive non-small-cell lung cancer in the ALEX study. Ann. Oncol. 31, 1056–1064 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Gadgeel S et al. Alectinib versus crizotinib in treatment-naive anaplastic lymphoma kinase-positive (ALK+) non-small-cell lung cancer: CNS efficacy results from the ALEX study. Ann. Oncol. 29, 2214–2222 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camidge DR et al. Updated efficacy and safety data and impact of the EML4-ALK fusion variant on the efficacy of alectinib in untreated ALK-positive advanced non-small cell lung cancer in the global phase III ALEX study. J. Thorac. Oncol. 14, 1233–1243 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Shaw AT et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 18, 1590–1599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solomon BJ et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 19, 1654–1667 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Solomon BJ et al. Efficacy and safety of first-line lorlatinib versus crizotinib in patients with advanced, ALK-positive non-small-cell lung cancer: updated analysis of data from the phase 3, randomised, open-label CROWN study. Lancet Respir. Med. 10.1016/S2213-2600(22)00437-4 (2022). [DOI] [PubMed] [Google Scholar]
- 56.Crystal AS et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dagogo-Jack I et al. Treatment with next-generation ALK inhibitors fuels plasma ALK mutation diversity. Clin. Cancer Res. 25, 6662–6670 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Toyokawa G et al. Secondary mutations at I1171 in the ALK gene confer resistance to both crizotinib and alectinib. J. Thorac. Oncol. 9, e86–e87 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Shaw AT et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N. Engl. J. Med. 374, 54–61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kobayashi S et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Choi YL et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 363, 1734–1739 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Katayama R et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl Acad. Sci. USA 108, 7535–7540 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Awad MM & Shaw AT ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin. Adv. Hematol. Oncol. 12, 429–439 (2014). [PMC free article] [PubMed] [Google Scholar]
- 64.Akamine T, Toyokawa G, Tagawa T & Seto T Spotlight on lorlatinib and its potential in the treatment of NSCLC: the evidence to date. Onco Targets Ther. 11, 5093–5101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou HY et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl Acad. Sci. USA 112, 3493–3498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Recondo G et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin. Cancer Res. 26, 242–255 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koopman B et al. Actionability of on-target ALK resistance mutations in patients with non-small cell lung cancer: local experience and review of the literature. Clin. Lung Cancer 23, e104–e115 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Shiba-Ishii A et al. Analysis of lorlatinib analogs reveals a roadmap for targeting diverse compound resistance mutations in ALK-positive lung cancer. Nat. Cancer 3, 710–722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okada K et al. Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine 41, 105–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shaw AT et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J. Clin. Oncol. 37, 1370–1379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dagogo-Jack I et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin. Cancer Res. 26, 2535–2545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katayama R et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 4, 120ra117 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lovly CM et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 20, 1027–1034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tanizaki J et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin. Cancer Res. 18, 6219–6226 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Lee HJ et al. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 26, 207–221 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Gainor JF et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin. Cancer Res. 19, 4273–4281 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sasaki T et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 71, 6051–6060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilson FH et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 27, 397–408 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Isozaki H et al. Non-small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res. 76, 1506–1516 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Coleman N et al. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open 6, 100319 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuriyama Y et al. Disease flare after discontinuation of crizotinib in anaplastic lymphoma kinase-positive lung cancer. Case Rep. Oncol. 6, 430–433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quintanal-Villalonga A et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 17, 360–371 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fujita S, Masago K, Katakami N & Yatabe Y Transformation to SCLC after treatment with the ALK inhibitor alectinib. J. Thorac. Oncol. 11, e67–e72 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Takegawa N et al. Transformation of ALK rearrangement-positive adenocarcinoma to small-cell lung cancer in association with acquired resistance to alectinib. Ann. Oncol. 27, 953–955 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Cha YJ, Cho BC, Kim HR, Lee HJ & Shim HS A case of ALK-rearranged adenocarcinoma with small cell carcinoma-like transformation and resistance to crizotinib. J. Thorac. Oncol. 11, e55–e58 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Levacq D, D’Haene N, de Wind R, Remmelink M & Berghmans T Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer: a new mechanism of resistance to ALK inhibitors. Lung Cancer 102, 38–41 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Lin JJ et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ Precis. Oncol. 4, 21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oser MG, Niederst MJ, Sequist LV & Engelman JA Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–e172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaiho T, Nakajima T, Iwasawa S, Yonemori Y & Yoshino I ALK rearrangement adenocarcinoma with histological transformation to squamous cell carcinoma resistant to alectinib and ceritinib. Onco Targets Ther. 13, 1557–1560 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Doebele RC et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 18, 1472–1482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ou SI et al. Dual occurrence of ALKG1202R solvent front mutation and small cell lung cancer transformation as resistance mechanisms to second generation ALK inhibitors without prior exposure to crizotinib. Pitfall of solely relying on liquid re-biopsy? Lung Cancer 106, 110–114 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Horn L et al. Monitoring therapeutic response and resistance: analysis of circulating tumor DNA in patients with ALK+ lung cancer. J. Thorac. Oncol. 14, 1901–1911 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagasaka M & Ou SI Lorlatinib should be considered as the preferred first-line option in patients with advanced ALK-rearranged NSCLC. J. Thorac. Oncol. 16, 532–536 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Camidge DR Lorlatinib should not be considered as the preferred first-line option in patients with advanced ALK rearranged NSCLC. J. Thorac. Oncol. 16, 528–531 (2021). [DOI] [PubMed] [Google Scholar]
- 95.Zhu VW et al. A novel sequentially evolved EML4-ALK variant 3 G1202R/S1206Y double mutation in cis confers resistance to lorlatinib: a brief report and literature review. JTO Clin. Res. Rep. 2, 100116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bauer TM et al. Brain penetration of lorlatinib: cumulative incidences of CNS and non-CNS progression with lorlatinib in patients with previously treated ALK-positive non-small-cell lung cancer. Target Oncol. 15, 55–65 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gainor JF et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis. Oncol. 2017, PO.17.00063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Murray BW et al. TPX-0131, a potent CNS-penetrant, next-generation inhibitor of wild-type ALK and ALK-resistant mutations. Mol. Cancer Ther. 20, 1499–1507 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pelish HE et al. Abstract 1465: NUV-520 (NVL-520) is a brain-penetrant and highly selective ROS1 inhibitor with antitumor activity against the G2032R solvent front mutation. Cancer Res. 81 (13_Supplement), 1465 (2021). [Google Scholar]
- 100.Ou SI, Nagasaka M, Brazel D, Hou Y & Zhu VW Will the clinical development of 4th-generation ‘double mutant active’ ALK TKIs (TPX-0131 and NVL-655) change the future treatment paradigm of ALK+ NSCLC? Transl. Oncol. 14, 101191 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perl AE et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 381, 1728–1740 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Mizuta H et al. Gilteritinib overcomes lorlatinib resistance in ALK-rearranged cancer. Nat. Commun. 12, 1261 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yan G et al. Targeting cysteine located outside the active site: an effective strategy for covalent ALKi design. J. Med. Chem. 64, 1558–1569 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Schneider M et al. The PROTACtable genome. Nat. Rev. Drug Discov. 20, 789–797 (2021). [DOI] [PubMed] [Google Scholar]
- 105.Mullard A Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 20, 247–250 (2021). [DOI] [PubMed] [Google Scholar]
- 106.Yan G et al. Discovery of a PROTAC targeting ALK with in vivo activity. Eur. J. Med. Chem. 212, 113150 (2021). [DOI] [PubMed] [Google Scholar]
- 107.Zhang C et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem. 151, 304–314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Richards MW et al. Microtubule association of EML proteins and the EML4-ALK variant 3 oncoprotein require an N-terminal trimerization domain. Biochem. J. 467, 529–536 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Hirai N et al. Monomerization of ALK fusion proteins as a therapeutic strategy in ALK-rearranged non-small cell lung cancers. Front. Oncol. 10, 419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Piotrowska Z et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 8, 1529–1539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhu VW et al. Acquired tertiary MET resistance (METD1228N and a novel LSM8–MET fusion) to selpercatinib and capmatinib in a patient with KIF5B-RET-positive NSCLC with secondary MET amplification as initial resistance to selpercatinib. J. Thorac. Oncol. 16, e51–e54 (2021). [DOI] [PubMed] [Google Scholar]
- 112.Wu YL et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 36, 3101–3109 (2018). [DOI] [PubMed] [Google Scholar]
- 113.Rosen EY et al. Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clin. Cancer Res. 27, 34–42 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sakakibara-Konishi J et al. Response to crizotinib re-administration after progression on lorlatinib in a patient with ALK-rearranged non-small-cell lung cancer. Clin. Lung Cancer 20, e555–e559 (2019). [DOI] [PubMed] [Google Scholar]
- 115.Cabanos HF & Hata AN Emerging insights into targeted therapy-tolerant persister cells in cancer. Cancers 13, 2666 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oren Y et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tsuji T et al. YAP1 mediates survival of ALK-rearranged lung cancer cells treated with alectinib via pro-apoptotic protein regulation. Nat. Commun. 11, 74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yun MR et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK-rearranged lung cancer. EMBO Mol. Med. 11, e10581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tang TT et al. Small molecule inhibitors of TEAD auto-palmitoylation selectively inhibit proliferation and tumor growth of NF2-deficient mesothelioma. Mol. Cancer Ther. 20, 986–998 (2021). [DOI] [PubMed] [Google Scholar]
- 120.Gan GN et al. Stereotactic radiation therapy can safely and durably control sites of extra-central nervous system oligoprogressive disease in anaplastic lymphoma kinase-positive lung cancer patients receiving crizotinib. Int. J. Radiat. Oncol. Biol. Phys. 88, 892–898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gainor JF et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin. Cancer Res. 22, 4585–4593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Govindan R et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150, 1121–1134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mazieres J et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann. Oncol. 30, 1321–1328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jahanzeb M et al. Immunotherapy treatment patterns and outcomes among ALK-positive patients with non-small-cell lung cancer. Clin. Lung Cancer 22, 49–57 (2021). [DOI] [PubMed] [Google Scholar]
- 125.Bylicki O et al. Targeting the PD-1/PD-L1 immune checkpoint in EGFR-mutated or ALK-translocated non-small-cell lung cancer. Target Oncol. 12, 563–569 (2017). [DOI] [PubMed] [Google Scholar]
- 126.Garon EB et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 127.Garassino MC et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. Lancet Oncol. 19, 521–536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marzec M et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl Acad. Sci. USA 105, 20852–20857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nogami N et al. IMpower150 final exploratory analyses for atezolizumab plus bevacizumab and chemotherapy in key NSCLC patient subgroups with EGFR mutations or metastases in the liver or brain. J. Thorac. Oncol. 17, 309–323 (2022). [DOI] [PubMed] [Google Scholar]
- 130.Socinski MA et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 378, 2288–2301 (2018). [DOI] [PubMed] [Google Scholar]
- 131.Patel M, Jabbour SK & Malhotra J ALK inhibitors and checkpoint blockade: a cautionary tale of mixing oil with water? J. Thorac Dis. 10, S2198–S2201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Spigel DR et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation—positive advanced non-small cell lung cancer (CheckMate 370). J. Thorac. Oncol. 13, 682–688 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Felip E et al. Ceritinib plus nivolumab in patients with advanced ALK-rearranged non-small cell lung cancer: results of an open-label, multicenter, phase 1B study. J. Thorac. Oncol. 15, 392–403 (2020). [DOI] [PubMed] [Google Scholar]
- 134.Awad MM et al. Epitope mapping of spontaneous autoantibodies to anaplastic lymphoma kinase (ALK) in non-small cell lung cancer. Oncotarget 8, 92265–92274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Passoni L et al. ALK as a novel lymphoma-associated tumor antigen: identification of 2 HLA-A2.1-restricted CD8+ T-cell epitopes. Blood 99, 2100–2106 (2002). [DOI] [PubMed] [Google Scholar]
- 136.Voena C et al. Efficacy of a cancer vaccine against ALK-rearranged lung tumors. Cancer Immunol. Res. 3, 1333–1343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sano R et al. An antibody–drug conjugate directed to the ALK receptor demonstrates efficacy in preclinical models of neuroblastoma. Sci. Transl. Med. 11, eaau9732 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Walker AJ et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther. 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nathan P et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N. Engl. J. Med. 385, 1196–1206 (2021). [DOI] [PubMed] [Google Scholar]
- 140.Tanimoto A et al. Proteasome inhibition overcomes ALK-TKI resistance in ALK-rearranged/TP53-mutant NSCLC via Noxa expression. Clin. Cancer Res. 27, 1410–1420 (2021). [DOI] [PubMed] [Google Scholar]
- 141.Qin K, Hou H, Liang Y & Zhang X Prognostic value of TP53 concurrent mutations for EGFR-TKIs and ALK-TKIs based targeted therapy in advanced non-small cell lung cancer: a meta-analysis. BMC Cancer 20, 328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sequist LV et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 3, 75ra26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shaurova T, Zhang L, Goodrich DW & Hershberger PA Understanding lineage plasticity as a path to targeted therapy failure in EGFR-mutant non-small cell lung cancer. Front. Genet. 11, 281 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Niederst MJ et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 6, 6377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Offin M et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J. Thorac. Oncol. 14, 1784–1793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oser MG et al. Cells lacking the RB1 tumor suppressor gene are hyperdependent on Aurora B kinase for survival. Cancer Discov. 9, 230–247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Byers LA et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2, 798–811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ku SY et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wu YL et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 383, 1711–1723 (2020). [DOI] [PubMed] [Google Scholar]