

Abstract
Purpose
Uveal melanoma (UM) is a rare disease with a high mortality, and new therapeutic options are being investigated. Preferentially Expressed Antigen in Melanoma (PRAME) is a cancer testis antigen, expressed in the testis, but also in cancers, including uveal melanoma. PRAME is considered a target for immune therapy in several cancers, and PRAME-specific T cell clones have been shown to kill UM cells.
Methods
We studied the literature on PRAME expression in hematological and solid malignancies, including UM, and its role as a target for immunotherapy. The distribution of tumor features was compared between PRAME-high and PRAME-low UM in a 64-patient cohort from the Leiden University Medical Center (LUMC) and in the Cancer Genome Atlas (TCGA) cohort of 80 cases and differential gene expression analysis was performed in the LUMC cohort.
Results
PRAME is expressed in many malignancies, it is frequently associated with a negative prognosis, and can be the target of T cell receptor (TCR)-transduced T cells, a promising treatment option with high avidity and safety. In UM, PRAME is expressed in 26% to 45% of cases and is correlated with a worse prognosis. In the LUMC and the TCGA cohorts, high PRAME expression was associated with larger diameter, higher Tumor-Node-Metastasis (TNM) stage, more frequent gain of chromosome 8q, and an inflammatory phenotype.
Conclusions
We confirm that PRAME is associated with poor prognosis in UM and has a strong connection with extra copies of 8q. We show that PRAME-specific immunotherapy in an adjuvant setting is promising in treatment of malignancies, including UM.
Keywords: uveal melanoma, preferentially expressed antigen in melanoma (PRAME), immunotherapy, prognosis
Uveal melanoma (UM) is a rare disease, with a high rate of metastases.1–3 Metastases usually involve the liver, and, once they are clinically evident, prognosis is grim, with a median survival time of 4 to 15 months.4–6 Based on clinical, histopathological, and genetic markers, one can stratify patients into risk groups and identify those who have a high risk of developing metastases. This would be useful especially when adjuvant therapies become available. However, at this moment, there is no adjuvant therapy available for the treatment of the micro-metastases that have spread before irradiation or surgical removal of the eye.1,7,8 UM-specific survival has not improved over the last 5 decades and finding new therapies is therefore a great unmet need.1,2,5,9 Recently, the US Food and Drug Administration (FDA) and the European Medical Agency (EMA) have approved tebentafusp for use in metastatic UM. Tebentafusp is an immune-mobilizing monoclonal T-cell receptors against cancer (ImmTAC) that consists of a T cell receptor (TCR), which allows binding to tumor cells, fused with an anti-CD3 single-chain variable fragment, which allows binding to T cells. The TCR is specific for tumor antigen gp100/PMEL (Premelanosome Protein), which is expressed in melanosomes, that are melanin-producing organelles in melanocytes and melanoma cells, and its action is restricted to antigen presentation by HLA-A*2:01. Patients treated with tebentafusp had better overall survival compared to controls, but only limited benefit in terms of progression-free survival and objective tumor response, although a decrease in circulating tumor DNA levels was shown in two thirds of patients.10–14 Hence, developing targeted immune therapies against tumor-specific antigens in UM is possible but more targets are needed. This paper will describe patient and tumor characteristics that help to identify high-risk patients with UM, and will present an extensive review of the literature on the expression and role of the cancer-testis antigen (CTA) Preferentially Expressed Antigen in Melanoma (PRAME) in oncology and the possibilities of using it as both a prognostic tool and a potential target for immunotherapy. We will also study the expression of PRAME in two cohorts of patients with UM, a 64-patient cohort from Leiden (The Netherlands) and the 80-patient The Cancer Genome Atlas (TCGA) cohort.
Uveal Melanoma Survival and Prognostic Factors
Clinical, histopathological, and molecular factors can be used to provide information on a patient's chance to develop metastases. The Tumor-Node-Metastases (TNM)/American Joint Committee on Cancer (AJCC) staging combines tumor size (determined by echography or measured in a pathologic specimen), involvement of the ciliary body, and extraocular growth to define specific categories.15 Other histopathological predictors of a poor prognosis include epithelioid cell type, high mitotic count, and extravascular matrix loops, as reviewed by Dogrusöz et al., and Dogrusöz and Jager.16,17
Molecular Prognostic Factors
Molecularly, UM can be subdivided on the basis of chromosomal aberrations, mutations in specific genes and their mRNA gene expression profile (GEP), which are largely overlapping parameters with a predictive value for the clinical outcome. According to the literature, loss of heterozygosity of chromosome 3 (through monosomy 3 [M3] or isodisomy 3), gain of the long arm of chromosome 8, and loss of the short arm of chromosome 1 are associated with a higher risk of metastasis and a poor prognosis,18–21 whereas gain of the short arm of chromosome 6 is correlated with a good prognosis.20,22,23 The TCGA study identified 4 genetic subtypes of UM, 2 of which are associated with disomy of chromosome 3 (D3), and 2 with M3.24 Both the D3 group and the M3 group can be divided into 2 subgroups, based on their 8q status or their mRNA expression pattern. The presence of inflammatory markers (presence of T cell infiltrate, expression of genes for interferon signaling, cytotoxicity, and immunosuppression) also separates the M3 group in two.24,25 The paper by Jager et al. proposed to call the four groups A to D, to prevent confusion with terminology used to identify two types of UM based on mRNA gene expression, that is, class 1 and class 2.25,26
At the gene level, almost every UM carries one of a few specific mutations, which bear prognostic significance and are, in general, mutually exclusive. Mutations in eukaryotic translation initiation factor 1A (EIF1AX) and splicing factor 3B subunit 1 (SF3B1) are associated with D3, whereas an inactivating mutation in the BRCA1-associated protein 1 (BAP1) gene, which is located on the short arm of chromosome 3, is strongly correlated with M3 and with a high risk of metastases.24,27–30 It is important to mention that BAP1 status and chromosome 3 status do not always coincide: some rare M3 tumors do not harbor an inactivating BAP1 mutation, and, even more rarely, some D3 tumors have a BAP1 mutation.24,27,31,32
Another already-mentioned method that allows for prognostication of patients with UM is gene expression profiling (GEP), which identifies two classes of tumors based on their mRNA expression pattern.26,33–36 Using a select group of 15 genes (detailed list in Supplementary Table S1), which have been used to create a commercial prognostication tool, tumors can be classified into low risk GEP class 1 and high risk GEP class 2. Tumors with a GEP class 2 frequently have M3 as well as a BAP1 mutation and are characterized by several negative prognostic features (older age at diagnosis, epithelioid cell type, extravascular loops, and a higher proliferation rate).24,37,38 GEP class 1 tumors can be subclassified, based on the differential expression of CHD1 and RAB31, into class 1A and class 1B.39,40 Class 2 tumors can be further subclassified into class 2A and class 2B on the basis of inflammatory markers and chromosome 8p copy number.17,41,42 The different molecular prognostication methods and their overlap are summarized in Tables 1A and 1B.
Table 1a.
TCGA Prognostication
| Disomy 3 | Group A | Normal 8q | EIF1AX |
| Group B | 8q gain | SF3B1 | |
| Monosomy 3 | Group C | 8q gain | BAP1 |
| Group D | 8q amplification | BAP1 |
Table 1b.
GEP-Based Prognostication
| Class 1 | Class 1A | Low CHD1, low RAB31 |
| Class 1B | High CHD1, high RAB31 | |
| Class 2 | Class 2A | Normal 8p |
| Class 2B | 8p loss |
Combining information on the mutation, chromosome, and/or GEP status with information on tumor size provides excellent insight in the prognosis of the patient. Studies by Bagger and Dogrusöz showed that adding chromosome 3 and chromosome 8q status to AJCC staging provides more accurate patient stratification compared to AJCC stage alone. Moreover, addition of the AJCC stage to the chromosome 3- and chromosome 8q-based stratification improved the accuracy of prognostication.43 Subsequently, Negretti et al. reported that the group of patients with the highest mortality had an advanced AJCC stage and both M3 and 8q gain.44 To help with prognostication of an individual patient, the group of Damato and Coupland has developed Liverpool Uveal Melanoma Prognosticator Online (LUMPO), an online prognostication system that joins histopathological features and chromosome status.45,46
In order to illustrate the relevance of different parameters in the Leiden cohort, we analyzed the predictive value of several individual prognostic factors in our Leiden University Medical Centre data set of 1222 patients (mean follow-up = 9 years, range = 0–46 years) and calculated overall survival and disease-specific mortality as determined for each of these factors. For the whole cohort, we observed a 5-year UM-related survival of 70% and a 10-year UM related-survival of 60%, which is comparable to other centers.7 Our analysis confirms previous studies: age over 60 years, the presence of an epithelioid or mixed cell type, a high largest basal diameter (LBD), a high histopathological AJCC class, a high mitotic count, and loss of chromosome 3 are all associated with worse UM-related survival (Figs. 1A–H). Although it is not included in the main prognostic classifications, tumor pigmentation has been linked to prognosis, and we have recently shown that darker tumors have a shorter survival than lighter ones, in this same cohort of patients.47 We recently also showed in a cohort of 979 patients with UM treated at the Wills Eye Hospital (Philadelphia, PA, USA) that combining chromosome information and AJCC stage yields a higher prognostic power than either parameter alone and is especially informative in the moderate-risk groups.48 We can conclude that in patients undergoing enucleation, proper selection criteria are available to identify those at high risk of developing metastases.
Figure 1.

Impact of risk factors on melanoma-specific mortality, as calculated in the LUMC dataset of 1222 patients with UM.
Preferentially Expressed Antigen in Melanoma
As one can quite reliably predict which patient is most likely to develop metastases, finding adjuvant therapies to prevent the outgrowth of metastases would be of great importance for these patients.
Immunotherapy is increasingly being applied to treat cancers, among which is cutaneous melanoma. The most widespread immunotherapy modality is immune checkpoint inhibitors, which is achieved by targeting the programmed death 1/programmed death ligand 1 (PD-1/PD-L1) axis or CTLA-4 and is currently used in several cancers such as cutaneous melanoma and non-small cell lung cancer (NSCLC).49–52 The response to immune checkpoint therapy and its clinical benefit have been shown to be correlated to the tumor mutational burden, which is high in cutaneous melanoma but low in UM.53,54 Moreover, PD-L1 expression in UM has been reported to occur only in 10% of the primary tumors and 5% of UM metastases.55,56 Several clinical trials have been carried out using the PD-1/PD-L1 blockade and CTLA4 blockade but the results have not shown promise in UM.8,57–60 The only exception seems to be the small subset of patients with a high mutational burden and a germline mutation in methyl-CpG-binding domain protein 4 (MBD4): 2 isolated case reports described 2 patients with these features who responded well to immune checkpoint therapy and a more recent retrospective cohort study confirmed a higher response rate in 5 out of 131 UM with an MBD4 mutation.61–63 Because this is a very rare occurrence, it is reasonable to look for alternative targets and strategies for immune therapy. Targeting gp100/PMEL with tebentafusp, on the other hand, has shown a moderate improvement in overall survival in patients with UM metastases, but only a limited benefit in terms of progression-free survival and tumor response.13
Cancer/testis antigens are attractive targets for immunotherapy because of their peculiar expression patterns: they are highly expressed in various tumors, which makes such tumors potentially susceptible to CTA-specific immunotherapy, as outlined below. Although these antigens are expressed in the testes, these are immune-privileged sites and do not express HLA molecules and are therefore protected from an immune reaction. Some CTAs have been studied in the context of UM but they did not show evidence of high expression in UM. MAGE-1, MAGE-2, and MAGE-3 expression has been reported in cells derived both from the primary tumor and the metastasis of one single patient with UM in 1 study and in 1 out of 5 primary UM cell lines and 1 of 3 metastasis-derived UM cell lines in another study.64,65 However, studies carried out on a larger number of primary tumors found only a low expression of both the MAGE antigens and NY-ESO1.66,67 PRAME is being used to differentiate between low and high-risk UM: PRAME has been found to be expressed in 26.5% to 45% of primary UMs,68–70 and in UM liver metastases.70 Moreover, its expression has been correlated with an increased risk of developing metastases. PRAME has also been identified as a potential target for T cell mediated immunotherapies. Indeed, PRAME was originally identified as a potential target for immune responses, hence its name.71 We will describe the expression of PRAME in normal and malignant tissues, its function, its role as prognostic indicator, and its distribution in UM.
Expression of PRAME
PRAME was first described when T cells from a patient with a cutaneous melanoma recognized a specific antigen on melanoma cells: when peripheral-blood derived T cells were stimulated with autologous tumor cells derived from a metastasis, the T cells were able to lyse the autologous tumor cells. The antigen they recognized was identified and called PRAME.71 PRAME belongs to the CTAs, which are highly expressed in the testis and in several types of cancer, and it has a low expression in the endometrium, in mature dendritic cells and in kidney epithelial cells.71,72 The CTAs so far include 70 antigen families with a total of 140 members, more than half of which are coded by genes clustered on chromosome X (X-CTAs), such as the MAGE-A and MAGE-C genes, and NY-ESO-1. The remaining CTAs are coded by genes on somatic chromosomes. The PRAME gene is located on chromosome 22q11.22. In most normal tissues, expression of CTAs is often inhibited by promoter methylation, whereas during gametogenesis, the promoter is demethylated and the CTAs can be expressed.73,74 This is also the case for the PRAME gene, having its highest expression in spermatocytes. Its exact function is partly understood (see below).
Expression in Other Tumors and Relation to Prognosis
Like other CTAs, PRAME is highly expressed in several solid tumors and hematological malignancies. Supplementary Figure S1 shows the distribution of PRAME in the different types of malignancies examined by the TCGA.
Supplementary Table S2 shows the expression and prognostic significance of PRAME in several types of solid cancer. As outlined in the table, different methods were used in different studies to evaluate PRAME expression and its influence on prognosis. Cutaneous melanoma, synovial sarcoma, myxoid and round cell liposarcoma, osteosarcoma, and neuroblastoma are among the tumors with the highest proportion of PRAME positive tumors (>90%), whereas urothelial carcinoma and hepatocellular carcinoma show PRAME expression in a low percentage of cases (<30%). Studies on NSCLC and breast cancer report discordant percentages of PRAME expression, possibly because of differences in the techniques used or in the patient cohorts.
In several of the tumor types analyzed in the studies reported in Supplementary Table S2, PRAME was correlated with a worse prognosis. These data are in agreement with a meta-analysis75 that included 14 studies (2421 patients) and found PRAME to be associated with higher tumor stage, positive lymph node metastasis, and shorter survival (disease free, progression free, metastasis free, and overall survival) in many malignancies. Exceptions are NSCLC, epithelial ovarian cancer, and renal cell carcinoma, where the prognostic significance of PRAME is as yet unclear.
As shown in Supplementary Table S3, the expression of PRAME in hematological malignancies varies greatly and it has a different prognostic significance in different entities. In acute myelogenous leukemia,76,77 PRAME expression is associated with a better prognosis and with some specific chromosomal translocations typical of low-risk cases (see Supplementary Table S3). However, overexpression of PRAME is associated with a poorer prognosis in some other disease entities, such as Hodgkin’s disease and non-Hodgkin’s lymphoma and myelodysplastic syndromes.78–80 In one type of non-Hodgkin’s lymphoma, diffuse large B cell lymphoma, however, different studies report different results: Mitsuhashi et al. showed that patients with high PRAME expression had worse prognosis, whereas Takata et al. recently reported that cases with low PRAME expression and with PRAME deletion had worse survival and worse treatment outcome.78,81 Because the expression level of PRAME in relapses rises to levels comparable to the disease onset and usually precedes the clinical and cytological signs of relapse, it has been proposed as a marker for minimal residual disease monitoring in hematologic malignancies.82
Function in Oncology
PRAME has been found to be expressed in the nucleus and in the cytoplasm of several cancer cell lines, such as the cervical cancer HeLa cell line, osteosarcoma U2OS cells, melanoma A375 cell line, and leukemic cell lines.71,83,84 PRAME is involved in the regulation of the cell cycle and cell differentiation, probably in the phase of tumor induction, and was found to influence the role of retinoic acid (RA). RA is known to induce cell cycle arrest and stimulate cell differentiation. PRAME is a dominant repressor of RA signaling, mainly through its interaction with the retinoic acid receptor (RAR): it binds to the RAR in the presence of RA and then prevents normal RAR activation and gene transcription through recruitment of polycomb proteins. The polycomb proteins enhancer of zeste homolog 2 (EZH2) and embryonic ectoderm development domain (EED) are involved in this process.85 The involvement of PRAME in RA receptor function in UM is supported by the fact that 16% of the genes downregulated in PRAME-positive UMs had a retinoic acid response element (RARE) close to the transcription start or gene end.68
However, because the repression of RA signaling does not seem to have a role in all types of cancers with high PRAME expression,86 several studies have focused on other potential roles of this cancer-testis antigen. There is evidence that PRAME interacts with members of the family of Cullin-based ubiquitin ligases and the nuclear factor Y (NFY) promoter. PRAME binds Cullin2 (Cul2) and Elongin BC (EloBC): the Cul2/EloBC/PRAME complex is located in the nucleus and binds to a subset of transcriptionally active promoter and enhancer regions that can also bind NFY.87 The same group also showed that PRAME can interact with members of the kinase, endopeptidase and other proteins of small size/endopeptidase-like and kinase associated to transcribed chromatin (KEOPS/EKC) complex, which is an evolutionarily conserved multiprotein complex involved in telomere maintenance and transcriptional regulation.88 In the same study, they report a PRAME-dependent association between members of the EKC complex and Cul2 ubiquitin ligases. NFY is a transcription factor that is important for early embryonic development, for maintenance of the high proliferation rate of embryonic stem cells, and for inhibition of differentiation. Because PRAME cannot directly bind DNA, NFY has been postulated to mediate the interaction of the Cul2/PRAME/EKC complex with chromatin.88 These theories are supported by the fact that PRAME expressing UM overexpress genes that contain an NFY-binding site in their promoter when compared to PRAME-negative UM.68
In triple-negative breast cancer cell lines, overexpression of PRAME promotes the epithelial-to-mesenchymal transition (EMT), which stimulates the motility and the invasive potential of these cancer cells in vitro. This PRAME-driven transition is accompanied by overexpression of vimentin and redistribution of E-cadherin from the cell surface to the cytosol. Overexpression of PRAME led to the expression of SOX10, SNAI1, and TWIST1, which are related to a change from EMT behaviour.89
Recently, Harbour et al. studied the effect of PRAME in UM cells and in normal uveal melanocytes through overexpression in PRAME-low cell lines and downregulation in PRAME-high cell lines: they showed that PRAME upregulation caused an increase in genomic instability, telomere loss and aneuploidy.90 The authors proposed that this effect may be mediated by ubiquitination of Structural Maintenance of Chromosomes 1A (SMC1A), which would then alter the interaction between SMC1A and other proteins associated with centromere cohesion.90
In leukemic cell lines, cytoplasmic PRAME has been shown to have structural similarities with human Toll-like receptors (TLRs) and to be upregulated by bacterial pathogen-associated molecular patterns (PAMPs) and interferon γ (IFN-γ). This finding could be related to the known role of chronic inflammation in the origin and progression of many hematological malignancies and to the fact that a pro-inflammatory microenvironment may promote growth of some solid tumors. Once stimulated by PAMPs and IFN-γ, PRAME accumulates in Golgi-like structures and it co-localizes with Elongin C, which is a component of the E3 ubiquitin ligase complex. The authors hypothesize that PRAME may be involved in targeting substrates, like PAMPs, to the Golgi for ubiquitination in response to pro-inflammatory stimuli.83 Interestingly, transient expression of PRAME has been found to induce caspase-independent cell death in CHO-K1 cells in vitro, and to reduce the transcription of proteins involved in resistance to chemotherapy in KG-1 leukemic cells (S100A4, heat shock protein 27, and p21). Furthermore, transient downmodulation of PRAME with si-RNA in K562 leukemic cells led to increased tumorgenicity in nude mice.84
PRAME and Prognosis in UM
Several studies examined PRAME expression in UM. The first study regarding PRAME expression and survival was set up to identify a marker that could differentiate between class 1 tumors that gave rise to metastases and class 1 tumors that did not.68 In a series of 64 patients with GEP class 1 UM, it was shown that the PRAME-positive cases were larger than PRAME-negative cases. This study reported a 5-year actuarial probability of metastasis of 38% among the 39 patients with PRAME-positive class 1 tumors, and of 0% in the 25 patients with a PRAME-negative class 1 tumor. The median follow-up of this study was 8.2 months (mean = 31.5 months, interquartile range = 3.3–57.1 months).68 Field et al. carried out a larger study involving 678 patients with UM and reported PRAME expression in 18% of class 1A, 29% of class 1B, and 37% of class 2 tumors. In this cohort, PRAME-positive tumors had a higher mean LBD and thickness and showed a significantly shorter time to metastasis and shorter time to UM-related death than PRAME-negative tumors, both when considering class 1 and class 2 patients together and class 2 tumors alone. The median follow-up of this study was 19 months (range = 0–125 months).69 It is important to note that a short follow-up may bias survival analysis, especially in the low-risk group (disomy 3 or GEP class 1), because the time to metastasis and the time to UM-related death is longer than in high-risk cases.29 Schefler et al. carried out a retrospective multicenter chart review on 148 cases diagnosed over a period of 17 months and that underwent GEP and PRAME mRNA expression measurement with a commercially available test.91 Schefler et al. reported that PRAME expression was significantly associated with a greater LBD and a larger tumor volume.91 No survival analysis was reported. In a study from our laboratory on 64 patients with primary Ums, PRAME was expressed in 45% of primary UM cases and its expression correlated with a larger diameter, ciliary body involvement, 8q amplification, and a shorter disease-specific survival.70 The 25 D3 tumors in the Leiden UM cohort had previously been reported in the Field et al.’s original paper.68 A recent study by Kumar et al. analyzed 66 UM cases and reported nuclear PRAME-positivity in 24% of cases by IHC and PRAME mRNA upregulation compared to normal choroid in 36% by quantitative real-time PCR (qRT-PCR).92 In their subset of 11 patients with metastatic cancer, 7 showed upregulation of PRAME. A high PRAME expression (immunohistochemical [IHC] or mRNA) was associated with the presence of negative rgani-pathological prognostic factors, such as epithelioid cell type, high mitotic activity and ciliary body involvement, and with a shorter overall survival.92 As further proof of the general interest in this antigen in the field of UM, PRAME will be included the Collaborative Ocular Oncology Group 2 (COOG2) study, along with GEP class and BAP1, SF3B1, and EIF1AX mutations, with the aim of providing a more accurate genetic classification system.93
Materials and Methods
We carried out survival analysis in a data set of 1222 patients with UM who underwent enucleation at Leiden University Medical Center (LUMC), Leiden, The Netherlands, between 1972 and 2019, studying age at enucleation, cell type, LBD, ciliary body involvement, mitotic count, AJCC stage, and chromosome 3 status. For the sake of statistical survival analysis, continuous variables were classified in groups as follows: age at enucleation lower or higher than 60 years, LBD lower than 10 mm, between 10 and 15 mm, and greater than 15 mm, and mitotic count lower or higher than 5 mitoses per 40 high-power fields.
LUMC Cohort
A retrospective study was performed at the LUMC on a cohort of 64 patients with UM who underwent enucleation between September 21, 1999, and October 6, 2008. The database that was used for data collection was updated in 2019 and contained the results of histopathological analyses and DNA and mRNA studies carried out on formalin-fixed, paraffin-embedded, or fresh frozen tissue from UM. Survival and clinical data were obtained from patient medical records and the Integral Cancer Center West. The patient database study focused on the following features: sex, age at enucleation, largest basal diameter, thickness, mitotic count, cell type, ciliary body involvement, extrascleral extension, TNM stage, pigmentation, chromosome 3, 6, and 8 status, BAP1 immunohistochemistry status, and the development of metastases. We do not have access to information on BAP1 mutation or on EIF1AX and SF3B1 mutation. However, previous studies showed very high concordance between BAP1 mutation and BAP1 immunohistochemical staining.94,95 Therefore, we think the use of BAP1 IHC in the LUMC cohort is a good proxy for BAP1 mutation. Chromosome status was determined through single-nucleotide polymorphism with the Affymetrix 250K_NSP-chip and Affymetrix Cytoscan HD chip Chromosome 8 copy number was obtained by droplet digital polymerase chain reaction.96 Follow-up time was calculated as the time from enucleation to death or last follow-up. Tumor pigmentation was scored macroscopically after enucleation on a 4-point scale: 1 – unpigmented (white), 2 – lightly pigmented (grey), 3 – moderately pigmented (brown), and 4 – heavily pigmented (dark brown – black). For the sake of the analysis, groups 1 and 2 were classified as light, and groups 3 and 4 were classified as dark. The TNM stages were classified into two groups: I to IIB and IIIA to IIIC, whereas chromosome 8q status was classified as follows: normal 1.9 to 2.1 copies, gain 2.2 to 3.1 copies, and amplification >3.1 copies.96 The mRNA expression of PRAME, EMT, and inflammation genes was measured with the Illumina HT-12v4 chip (Illumina). PRAME expression was classified as positive or negative according to a cutoff value set at the inflection point of the PRAME expression curve obtained using probe ILMN_1700031.70 For the expression of CD3, CD4, CD8, and CD68, we used the probes that were validated by immunohistochemistry in a previous study by Gezgin.97 For HLA-A and HLA-B, we selected the probes that were validated in a study by Van Essen.98
The study was approved by the Scientific Committee of the Ophthalmology Department of the Leiden University Medical Center (project number 29.1). Tumor material was made available for research according to the Dutch FEDERA regulations of leftover material of pathological specimens. The research adhered to Dutch law and the tenets of the Declaration of Helsinki (World Medical Association of Declaration 2013; ethical principles for medical research involving human subjects).
TCGA Cohort
A retrospective study was carried out on the TCGA database, which contains clinicopathological data from 80 primary UM cases collected in 6 centers (http://cancergenome.nih.gov/). Information on tumor dimension (LBD and thickness) was determined clinically, whereas ciliary body involvement and extrascleral extension were determined through histopathological analysis. Cytological examination was used to identify the cell types in the samples and chromosome status was determined through single nucleotide polymorphism analysis with the ABSOLUTE algorithm. Tumor pigmentation was scored microscopically into three groups: light, mixed, and heavy. More detailed information can be found in Robertson et al.’s paper.24 Differently from the LUMC analyses, in the TCGA study, gene expression (hence PRAME expression) was measured through RNAseq (Illumina HiSeq 2000). Because PRAME expression had a regular distribution in the TCGA database and there was no change of slope, we split the sample into 2 halves: 40 patients with lower PRAME expression and 40 patients with higher PRAME expression. For BAP1, we classified a case as mutated if a mutation in either RNA seq or DNA seq was reported. For EIF1AX and SF3B1, we classified any alteration as a mutation. In cases with overlapping mutations, we considered the more prognostically severe one of the two.
Statistical Analysis
Statistical analysis was performed with SPSS, versions 23 and 25 (IBM Corp). In the first phase of the study, clinical, histopathological, and genetic features were compared between the PRAME-positive and PRAME-negative groups in the TCGA cohort. In the second phase of the study, the monosomy 3 and disomy 3 groups were analyzed separately in both the LUMC and the TCGA cohort, and the variables were compared between the PRAME-positive and PRAME-negative cases. Pearson's χ2 test was used to compare categorical variables, and whenever the assumptions of the χ2 test were violated, the Fisher's exact test or the likelihood ratio were used. Continuous variables were compared using the Mann-Whitney U test. Disease-specific survival was analyzed with the Kaplan-Meier curve and log-rank test, and patients who died of another or unknown cause were censored. A P value ≤ 0.05 was considered significant. The differential expression analysis and gene set enrichment analysis were carried out in the statistical software R. The probe with the highest mean expression was selected for each gene in the microarray. The package limma was used for differential expression analysis and the significance threshold for the volcano plot included an adjusted P value < 0.05 and log fold change (FC) >0.6 or <−0.6. The top 20 differentially expressed genes were selected after filtering for genes with P value < 0.05 and log FC >0.6 or <−0.6. The gene set enrichment analysis (GSEA) was performed with the R package fgsea and significance was established as a P value ≤ 0.01. The data used to plot Supplementary Figure S1 contains mRNA expression across several cancer types (Batch normalized from Illumina HiSeq_RNASeqV2) and was downloaded from www.cbioportal.org. The graphs present in figures were plotted with R, version 4.05, and GraphPad Prism, version 9.3.1.
Results
LUMC Cohort
We decided to study further the relation between PRAME expression and survival in our cohort of 64 patients with UM, by looking at longer follow-up of the patients reported by Gezgin et al.70 Patients in the LUMC database had a mean age at enucleation of 60.6 years (range = 13–88 years) and a mean follow-up of 86.6 months (range = 2–252 months); 33 patients were male (52%) and 31 were female (31%), and the mean LBD was 13.5 mm (range = 8–30 mm). Because we now have longer follow-up, we repeated the survival analyses. As already reported by Gezgin et al., the total cohort included 64 patients with UM, of which 45% (n = 29) were PRAME-positive and 55% (n = 35) were PRAME-negative.70
Gezgin et al.’s paper showed that a positive PRAME expression had a statistically significant correlation with increased LBD (P = 0.005), ciliary body involvement (P = 0.008), and the presence of metastases (P = 0.03). Moreover, tumors with a positive PRAME expression more frequently showed 8q gain/amplification than PRAME-negative tumors (P = 0.002), but there was no correlation with loss of chromosome 3 (P = 0.21).70 In addition, we found a positive correlation between PRAME expression and dark tumor pigmentation (P = 0.025; see Fig. 2). When testing survival in all 64 cases with longer follow-up, we confirmed patients with PRAME-positive tumors showed a worse UM-specific survival than PRAME-negative ones (P = 0.028; Fig. 3A).
Figure 2.
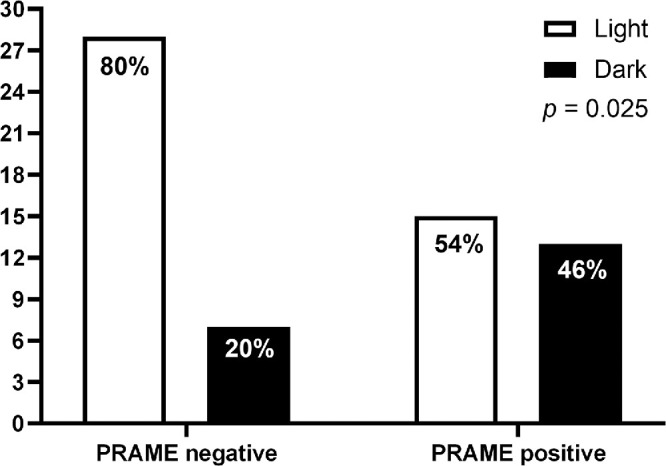
Association between tumor pigmentation and PRAME status in 64 patients with UM (LUMC cohort). PRAME negative: n = 35, PRAME positive: n = 29. The P value calculated with Pearson's chi square test.
Figure 3.

Relation between PRAME expression and Melanoma-related survival in 64 patients with UM (A), in 24 patients with D3 UM (B), and in 40 patients with M3 UM (C).
We subsequently analyzed the D3 and M3 groups separately.
The D3 group included 24 patients, of whom 33% (n = 8) were PRAME-positive and 67% (n = 16) were PRAME-negative. Within the D3 subset, a statistically significant correlation was found between PRAME expression and LBD (15.1 vs. 11.3, P = 0.001, as published by Field et al.68), dark tumor pigmentation (P = 0.028), with chromosome 8q gain/amplification (P = 0.047), and with disease-specific death (P = 0.015; Supplementary Table S4, Fig. 3B).
The M3 group included 40 patients: 52.5% (n = 21) were PRAME-positive and 47.5% (n = 19) tumors were PRAME-negative. When considering the M3 subgroup separately, PRAME-positive tumors more frequently showed ciliary body involvement (14/21 vs. 6/19, P = 0.03) and chromosome 8q amplification (P = 0.01; Supplementary Table S5). However, within the M3 group, disease-specific survival was not influenced by PRAME expression (P = 0.46; Fig. 3C).
The Cancer Genome Atlas Cohort
Next, we examined the TCGA cohort to analyze the correlation between PRAME expression and several prognostic factors. Patients in the TCGA database had a mean age at diagnosis of 61.7 years (range = 22–80 years) and a mean follow-up time of 27.3 months (range = 0–87 months); 45 of the patients were male (56%) and 35 were female (47%) and the mean LBD was 16.7 mm (range = 8–25 mm). Because there was no clear inflection point in the distribution of PRAME mRNA expression across the TCGA cohort, we separated the group into the 50% with the lowest PRAME mRNA expression and the 50% with the highest expression and carried out the tests in the total cohort and in the D3 and M3 subgroups separately. The TCGA cohort included 80 patients: 38 patients with D3 tumors (47.5%) and 42 patients with M3 tumors (52.5%).
As shown in Table 2, in the total cohort, tumors with a high PRAME expression had a higher LBD (P = 0.03), more frequently showed extrascleral extension (P = 0.01), an epithelioid/mixed cell type (P = 0.04), darker tumor pigmentation (P = 0.001) and chromosome 8q gain or amplification (P < 0.001). In the total TCGA cohort, the level of PRAME expression, however, did not show a statistically significant association with UM-related survival (P = 0.40; Fig. 4A).
Table 2.
Distribution of Clinical, Histopathological, and Genetic Features in Patients With UM with Low PRAME and High PRAME (total 80) from the TCGA Study
| Patients, no. (%)§,|| | |||
|---|---|---|---|
| Feature | Low PRAME n = 40 (50%) | High PRAME n = 40 (50%) | P Value |
| Sex | |||
| Male | 22 (55%) | 23 (57%) | 0.82* |
| Female | 18 (45%) | 17 (42%) | |
| Age at enucleation | 63.68 (39–86) | 59.63 (22–86) | 0.24‡ |
| Largest basal diameter | 15.65 (8–25) | 17.52 (11–25) | 0.03‡ |
| Thickness | 10.67 (6–16) | 10.91 (4–16) | 0.54‡ |
| Mitotic count | |||
| 0–5 | 32 (80%) | 31 (77%) | 0.47† |
| >5–10 | 4 (10%) | 7 (17%) | |
| >11 | 4 (10%) | 2 ( 5%) | |
| Cell type | |||
| Spindle cell | 26 (65%) | 17 (42%) | 0.04* |
| Epithelioid-mixed cell | 14 (35%) | 23 (57%) | |
| Ciliary body involvement | |||
| No | 28 (70%) | 26 (65%) | 0.63* |
| Yes | 12 (30%) | 14 (35%) | |
| Extrascleral extension | |||
| None | 40 (100%) | 34 (85%) | 0.01† |
| < 5 mm | 0 (0%) | 3 (7.5%) | |
| >= 5 mm | 0 (0%) | 3 (7.5%) | |
| TNM stage | |||
| I-IIB | 21 (52%) | 15 (37%) | 0.18* |
| IIIA-IV | 19 (19%) | 25 (44%) | |
| Tumor pigmentation | |||
| Light | 28 (70%) | 11 (28%) | 0.001* |
| Mixed | 7 (18%) | 20 (50%) | |
| Heavy | 5 (13%) | 5 (23%) | |
| Chromosome 3 status | |||
| Disomy | 21 (52%) | 17 (42%) | 0.37* |
| Monosomy | 19 (47%) | 23 (57%) | |
| 8q status | |||
| Normal | 19 (47%) | 2 ( 5%) | <0.001* |
| Gain | 11 (27%) | 16 (40%) | |
| Amplification | 10 (25%) | 22 (55%) | |
| 6p status | |||
| Normal | 21 (52%) | 14 (35%) | 0.11* |
| Gain | 19 (47%) | 26 (65%) | |
| BAP1 mutation | |||
| BAP1 wild type | 25 (63%) | 20 (50%) | 0.26* |
| BAP1 mutated | 15 (38%) | 20 (50%) | |
Pearson's χ2 test.
Likelihood ratio.
Mann Whitney U test.
Percentages are rounded and may not total 100.
Percentages were calculated excluding missing data.
Figure 4.

Relation between PRAME expression and survival in 80 patients with UM in the 80 TCGA database (A), 38 patients with D3 (B), and 42 patients with M3 (C).
When looking at only the TCGA D3 subgroup (38 patients), PRAME expression did not show a statistically significant association with UM-related survival (P = 0.38; Fig. 4B). However, in patients with D3 tumors, a high PRAME expression had a lower age at enucleation (P = 0.05), and their tumors showed a higher mitotic count (P = 0.045), and more frequently chromosome 8q gain/amplification (P < 0.001) and chromosome 6p gain (P = 0.05; Supplementary Table S6). As evident from the values presented in the table, most of the cases in the D3 cohort had <5 mitoses, and the association of PRAME expression with age at enucleation and chromosome 6p gain was only borderline significant. As patients with D3 tumors tend not to develop metastases or do so later than patients with M3 tumors, the total follow-up time of this sub-cohort (a mean of 32.5 months) may not have been long enough to properly evaluate a relation between PRAME and the development of metastases.
In the M3 subgroup (42 patients), a higher PRAME expression was associated with a darker tumor pigmentation (P = 0.007), with a more extensive extrascleral extension (P = 0.04) and with chromosome 8q gain or amplification (P = 0.007; Supplementary Table S7). However, in this group, UM-related survival did not show a significant association with PRAME status (P = 0.20; Fig. 4C).
Because BAP1 is not the only prognostically relevant mutation in UM, we analyzed the correlation of PRAME with the presence of SF3B1 and EIF1AX mutations as well. Two cases had both a BAP1 and an SF3B1 mutation and they were classified as BAP1 mutated; 1 case had both a SF3B1 and an EIF1AX mutation and it was classified as SF3B1 mutated. Twenty cases did not harbor any of these mutations and were excluded from this statistical analysis. When considering the full TCGA cohort, PRAME-low and PRAME-high cases showed a significant difference in mutations, even though the frequency of BAP1 mutation was similar (P = 0.002; Supplementary Table S8). We then split the cohort in disomy 3 and monosomy 3. The D3 subgroup did not include any case with BAP1 mutation: patients with SF3B1 mutation had significantly higher PRAME expression than EIF1AX-mutated cases (P < 0.001; see Supplementary Table S8). The monosomy 3 sub-cohort did not contain any EIF1AX mutated case and PRAME expression did not differ between those with an SF3B1 (n = 2) or a BAP1 mutation (n = 35, P = 0.20; see Supplementary Table S8).
Role for PRAME in Tumor Progression and Immunologic Response
We wondered why PRAME is an indicator of bad prognosis and considered the possibility that PRAME positivity indicates a change from EMT transition, as this had previously been observed in breast cancer.89 We therefore analyzed the expression of the EMT markers that were available from mRNA microarray data of the LUMC cohort and compared it between 35 PRAME-negative and 29 PRAME-positive UM. As shown in Supplementary Table S9, we only observed a statistically significant difference with ZEB2, which is a transcription factor that drives EMT. We found no difference in other well-known EMT markers, such as STAT3, SOX10, and SNAI2. However, the GSEA reported in Figure 5 shows EPITHELIAL_TO_MESENCHYMAL_TRANSITION among the Hallmark pathways significantly enriched in the PRAME-positive UM (see Fig. 5).
Figure 5.

Hallmark pathways GSEA enrichment in 29 patients with PRAME-positive UM versus 35 patients with PRAME-negative UM in the LUMC cohort.
Our current analyses and previous papers by our and other groups,68–70,91 show that, in general, PRAME expression in UM is associated with a worse prognosis, and it is known that Ums with a grim prognosis have an inflammatory phenotype.25,99,100 We compared the distribution of inflammatory markers in PRAME-positive and PRAME-negative tumors. Even though there is some overlap in expression levels, we found significantly higher levels of infiltrate markers in PRAME-positive tumors than in PRAME-negative ones (T cell markers CD3, CD4, and CD8A, macrophage marker CD68 and HLA-A and HLA-B; Table 3, Fig. 6). In further support of this connection, the GSEA analysis in Figure 5 shows that the most significantly enriched pathways in PRAME-positive UM are related to inflammation (INTERFERON_GAMMA_RESPONSE, INTERFERON_ALPHA_RESPONSE, INFLAMMATORY_RESPONSE, and ALLOGRAFT_REJECTION, COMPLEMENT; see Fig. 5). Moreover, the volcano plot in Figure 7 shows CD68 among the most upregulated genes in PRAME-positive UM (see Fig. 7).
Table 3.
Distribution of Infiltrate Markers in PRAME Positive and PRAME Negative Tumors in a Cohort of 64 Patients With UM, Mann Whitney U test
| PRAME Negative | PRAME Positive | P Value | |
|---|---|---|---|
| CD3 | 6.88 (±1.1) | 7.43 (±1.1) | 0.002 |
| CD4 | 6.57 (±0.3) | 6.76 (±0.3) | 0.005 |
| CD8A | 6.97 (±1.3) | 7.61 (±1.2) | 0.003 |
| CD68 | 10.46 (±0.9) | 11.29 (±0.8) | 0.001 |
| HLA-A pr 1 | 11.08 (±0.8) | 11.77 (0.8) | 0.001 |
| HLA-A pr 2 | 13.63 (±0.8) | 14.10 (±0.8) | 0.004 |
| HLA-B | 10.97 (±1.6) | 11.77 (±1.7) | 0.03 |
Figure 6.

Comparison of mRNA expression of CD68 and HLA expression in PRAME positive and PRAME negative tumors in a cohort of 64 patients with UM.
Figure 7.

Volcano plot showing the differentially expressed genes in 29 patients with PRAME-positive UM versus 35 patients with PRAME-negative UM. The right side of the plot shows genes upregulated in patients with PRAME-positive UM, the left side of the plot shows genes upregulated in patients with PRAME-negative UM.
As shown in Supplementary Table S10, the difference in CD68 expression was significant in both the D3 and M3 sub-cohorts, whereas CD4 showed a significant difference only in the D3 group and the T cell markers CD3 and CD8A, HLA-A and HLA-B only in the M3 group.
Co-expression of PRAME and inflammation may provide an advantage for immunotherapeutic approaches, as inflammatory markers include the expression of HLA class I antigens, that are needed for proper antigen presentation. Because our expression data comes from microarray analysis of mRNA expression and immune cell markers are high in a good proportion of tumors (see Table 3, Fig. 6), one may suggest that the HLA expression values in our data set may come from expression in infiltrating immune cells, such as macrophages. However, previous IHC work has shown that HLA class I antigens were expressed in uveal melanoma cells, in variable percentages, with HLA-A expression being usually higher than HLA-B.99,101–104 These studies also showed that a higher expression correlated with the presence of epithelioid cells, monosomy of chromosome 3, and more frequent metastases.99,102,103 In the study by Gezgin et al., samples from metastatic UM were examined by mRNA fluorescence in situ hybridization (FISH) to determine PRAME expression, which was present on 11 of 16 metastases.70 These metastases were also tested by immunofluorescence staining for HLA expression, which was observed in 10 of 16 metastases. A total of 8 of 16 UM co-expressed PRAME and HLA class I, which is needed for presentation of PRAME to the immune system. As an effective T cell response needs expression of PRAME with the appropriate HLA class I molecule, these data suggest the possibility to use PRAME-directed immune therapy in UM metastases.
Differential Expression Analysis and Gene Set Enrichment Analysis
We did not only analyze differences in inflammation but carried out a general differential expression analysis and gene set enrichment analysis in order to better understand the differences between PRAME-positive and PRAME-negative tumors (Table S11, see Figs. 5, 7).
Regarding the differential expression analysis, Supplementary Table S11a shows that most of the upregulated genes in PRAME-positive UM in the LUMC cohort are located on chromosome 8q24.3. This finding strengthens the correlation between PRAME expression and chromosome 8q gain, and points to a specific region on chromosome 8q that might be relevant for prognosis. PRAME itself is located on chromosome 22. Among the upregulated genes, we did not find any that have been reported to become upregulated after PRAME overexpression. DGAT1 expression has been correlated to poor survival in patients with gastric cancer and confers protection from oxidative damage to cancer cells in vitro.105–107 Even if it is upregulated in PRAME-negative UM in our data, NAPRT1 is usually downregulated in several cancer types due to promoter hypermethylation, which makes cancer cells more susceptible to treatment with NAPMT inhibitors with nicotinic acid.108–110 Solute Carrier family 52 member 2 (SLC52A2) has been reported to be upregulated in several types of cancer, and its expression has been linked to a worse survival, an increase in T cell exhaustion markers, and in M2 macrophages in several cancers among which is UM.111 Lymphocyte antigen 6 family member E (LY6E) is part of the LY6 gene family, which is involved in immune cell proliferation and differentiation and regulation of tumor progression in mice. Studies on human cancers have reported LY6E to be overexpressed and associated with a poor prognosis in a variety of cancers, as reviewed in ref. 112 to favor tumor growth through TGF-β signaling and promote immune escape,113 and to increase HIF-1α transcription through the PTEN-PI3K/AKT pathway.114 Poly(ADP-Ribose) (PARP) Polymerase family member 10 (PARP10) catalyzes the transfer of ADP-ribose molecules and it has been shown to promote cell proliferation in vitro and tumor formation in vivo from HeLa cells.115 Koning et al. also showed that PARP10 was increased in UM samples and that a PARP inhibitor could increase the efficacy of dacarbazin treatment on UM PDX and cell lines.116 Protein tyrosine phosphatase 4A3 (PTP4A3) has been reported to be increased in UM cases with early metastases compared to cases without or with late metastases, and its overexpression in UM cell lines increased cell migration in vitro and invasiveness in vivo.117 The same group later showed that PTP4A3 interacts with MMP-14, which is involved in cell migration,118 and with CRPM2, which is responsible for actin fiber organisation.119
Of the upregulated genes located on chromosome 8q24.3, we could not find relevant information about binding splicing factor 60 (PUF60) and 5-oxoprolinase, ATP-hydrolyzing (OPLAH).
Several other upregulated genes are located on other chromosomes, such as CCAAT enhancer binding protein beta (CEBPB), epoxide hydrolase 1 (EPHX1), deoxyribonuclease 1 like 3 (DNASE1L3), alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase (MGAT1), apolipoprotein C1 (APOC1), and the already-mentioned CD68. CEBPB is located on 20q13.13 and has been implicated in the aggressiveness of breast cancer, even though the exact regulatory network in not fully understood (reviewed in ref. 120). EPHX1 is located on 1q42.12 and has been associated with an increased recurrence rate and a worse prognosis in acute myeloblastic leukemia (AML).121 DNASE1L3 expression has been reported as downregulated in several types of cancer, but has also been correlated with immune infiltration.122 Increased expression of MGAT1 has been shown to increase progression and invasiveness of cervical cancer, prostate cancer, and hepatocellular carcinoma cell lines.123,124 Last, APOC1 is highly expressed in many types of cancers, among which are colorectal cancer, gastric cancer, and renal cell carcinoma, and is associated with a worse prognosis.125–127
Potential Role as a Therapeutic Target
Ever since its discovery, the PRAME antigen has raised an interest not only as a diagnostic and prognostic marker, but also as a therapeutic target. An antigen can be considered a good target for cancer immunotherapy if it is highly expressed in the tumor and not expressed in healthy tissues, and if it is able to induce an immune response. As mentioned above, PRAME is expressed in several types of tumors, and, among healthy tissues, it is highly expressed in the testes.71 However, because PRAME is an intracellular antigen, lysis of cells mediated by PRAME recognition is an HLA-restricted process. The fact that the testes are an immune-privileged site and that spermatocytes are devoid of HLA molecules (as reviewed in refs. 128 and 129) may help to protect them from such immune responses, although they express PRAME.71 In the first paper that described PRAME, PRAME was expressed on a metastatic cutaneous melanoma cell line and was recognized by HLA-A24 restricted T cells through the recognition of the nonapeptide LYVDSLFFL.71
Another study identified four novel T cell epitopes in the PRAME protein which are presented in HLA-A*02:01: PRA100-108 (VLDGLDVLL), PRA142-151 (SLYSFPEPEA), PRA300-309 (ALYVDSLFFL), and PRA425-433 (SLLQHLIGL). T cells specific for these epitopes demonstrated HLA-A*02:01 restricted lysis of cutaneous melanoma, renal cell carcinoma, lung carcinoma, and mammary carcinoma cell lines.130 The SLLQHLIGL epitope was later shown to mediate recognition of PRAME-positive cells by PRAME-specific T cells derived from a patient with AML who had a severe graft-versus host disease (GVHD) immune response following an HLA-A2 mismatched stem cell transplant (SCT).72 Three further PRAME epitopes able to elicit a T cell response were identified in a study on ovarian cancer: QLLALLPSL (HLA-A*02:01-restricted), LYVDSLFFL (HLA-A*24:02-restricted), and SPSVSQLSVL (HLA-B*07:02-restricted).131
Therapeutic options that are under investigation include cancer vaccine and adoptive T cell therapy.
Cancer Vaccines
Several studies have investigated anti-PRAME vaccines for their feasibility, safety profile, and their efficacy in eliciting a tumor-specific cytotoxic immune response. After a preliminary study in mice demonstrated PRAME's ability to induce humoral immunity, a CD4+ T cell response and long-lasting immunity,132 there were 2 clinical studies tested a vaccine consisting of recombinant PRAME protein together with the immunostimulant AS15 on 60 patients with NSCLC and 66 patients with cutaneous melanoma, respectively.133,134 The authors of both clinical studies reported a good safety profile and showed the development of a humoral immune response in all patients, and a CD4+ T cell response in most patients (more frequently when the highest dose was administered). No CD8+ T cell responses were observed.133,134 Two further trials with AS15 on NSCLC (NCT01159964 and NCT01853878) were stopped by the company because of lack of efficacy from two phase III studies with another CTA (recMAGE-A3 + AS15).
As an alternative strategy, a phase I study investigated a vaccine co-targeting PRAME and Prostate-Specific Membrane Antigen (PSMA) injected into the lymph nodes in HLA-A*02:01+ patients that expressed PSMA and PRAME with progressing metastatic solid tumors (prostate cancer, renal cell carcinoma, and cutaneous melanoma).135 The injections were well tolerated and achieved a T-cell immune response in 15 of 24 patients. Interestingly, a proportion of patients had detectable PRAME-specific and PSMA-specific T cells in their blood at baseline, but these patients were less likely to have T cell expansion than patients without pre-existing antigen-specific T cells, possibly because of anergy or inactivation of naturally occurring PRAME-specific and PSMA-specific T cells. This vaccine was not pursued further by the company.
A different type of vaccination that is effective in sensitizing T cells to a specific antigen is dendritic cell vaccination. Autologous dendritic cells (DCs) derived from AML blasts harvested from patients with AML were confirmed to express PRAME by real-time PCR and administered to five patients with AML; this treatment was tolerated well. After DC vaccination, a significant increase in PRAME-specific granzyme B-releasing CD8+ T cells was observed in the patients who completed the course of vaccination. Moreover, the patients showed an increase in Th1 cytokines and in IFN-γ production by CD4+ T cells.136 Two autologous DC vaccines expressing WT-1 and PRAME antigens (NCT02405338 and NCT01734304, which also targeted CMVpp65) have been tested for post-remission therapy in AML in a phase I/II trial, that has been completed, with positive safety and feasibility results reported.137 Trial NCT01734304 gave rise to an increase in PRAME-specific CD8+ T cells in 4/10 cases by ELISpot but this finding could not be confirmed by multimer staining.137 Trial NCT02405338 has been completed and an interim analysis performed at 12 months showed the presence of PRAME and/or WT-1 specific IFN-γ T cell responses in 2 of 4 cases with early relapse, 4 of 4 cases with late relapse, and 3 of 12 cases with no relapse, but higher percentage of CD3+HLA-DR+ T cells in the bone marrow and peripheral blood of patients in remission compared to relapsing patients.138 A further phase I trial with an autologous DC vaccine targeting several tumor-associated antigens, among which PRAME, in high-grade serous ovarian cancer is being carried out (NCT04739527).
The main issue with using vaccines to target a self-antigen, like PRAME, is self-tolerance: high-avidity self-reactive T cells are eliminated through negative selection in the thymus. This mechanism limits the potential benefits of PRAME-targeting vaccines.
Adoptive T Cell Therapy
Adoptive T cell therapy can be performed with two different approaches: isolation of pre-existing PRAME-specific T cells, with ex vivo expansion and selection, followed by re-infusion, or genetic engineering of high affinity T cells.
Pre-Existing Prame-Specific T Cells
Griffioen et al. tested by IFN-g ELISPOT assays and tetramer staining the four HLA-A*02:01-restricted PRAME peptides identified by Kessler130 and reported that the epitope that was most frequently associated with specific T cells in vitro and ex vivo was PRA100-108 (VLDGLDVLL).139 Stimulation of T cells with this epitope led to T cells which were able to recognize and lyse HLA-A*02:01+/PRAME+ cutaneous melanoma cells but not acute lymphoblastic leukemia (ALL) cells. According to the author, this difference could be partially explained by a difference in the expression level of PRAME.139
In another study, antigen-presenting cells (APCs) were loaded with a peptide library spanning the entire PRAME protein and used to generate polyclonal, high-avidity PRAME-specific T cell lines from autologous peripheral blood mononuclear cells (PBMCs) in vitro. These T cells proved to be able to kill HLA-A*02:01+/PRAME+ leukemic blasts and leukemic progenitors but not PRAME-negative normal hematopoietic cells. Moreover, this study led to the discovery of a novel HLA-A*02:01 restricted peptide, NLTHVLYPV.140
Several clinical trials involving infusion of ex vivo expanded T cells targeting multiple tumor-associated antigens have been and are being performed in hematological diseases and solid tumors. The trials involving patients with leukemia used allogeneic T cells derived from the stem cell donors, whereas trials involving solid tumors used autologous patient-derived T cells. All the antigen combinations showed good safety profiles. Preliminary results from a trial targeting WT1, PRAME, and Cyclin A1 (NCT04284228) in AML showed detection of the adoptive CD8+ T cells in circulation and a reduction in myeloblasts in bone marrow and peripheral blood.141 T cells targeting PRAME, NY-ESO1, MAGEA4, Survivin, and SSX were studied in lymphoma (NCT01333046) and resulted in complete remission in all patients when used as an adjuvant therapy and in a portion of patients with active disease. Responders and non-responders had similar expansion of T cells directed against the target antigens, but responders showed higher expansion of T cells directed against non-targeted antigens.142 In patients with pancreatic cancer (NCT03192462), they are treated with this same product had had a partial response in 3 of 13 and a complete response in 1 of 13 cases.143 When tested in 10 patients with breast cancer (NCT03093350), only one had disease stabilization for more than 3 months. This patient had elevated T cells against both targeted and non-targeted antigens.144 Lulla et al. reported the results of a trial with T cells targeting WT1, NY-ESO-1, PRAME, and Survivin in AML and myelodysplastic syndrome, either as an adjuvant therapy or in active disease (NCT02494167) and showed absence of relapse in 11 of 17 cases in the adjuvant arm and objective response in 2 of 8 cases with active disease.145 T cells targeting WT1, PRAME, and Survivin were administered to patients with AML and acute lymphoblastic leukemia and resulted in complete remission in 9 of 11 patients with relapsed/refractory disease and persistent remission in 9 of 12 patients at risk of relapse after SCT.146 In a study on patients with central nervous system tumors, these same T cells showed specificity for 1 to 3 tumor antigens in 8 of 9 cases and the presence of infusion-related immune cytokine response.147 The advantages of stimulating T cells with multiple tumor antigens are the lack of HLA-restriction and the higher chance of the tumor being sensitive to at least one of the targets. However, these studies do not show if the mechanisms of action of these T cells is related to the specific antigens or to immune stimulation in general. Moreover, in solid tumors, autologous T cells are used: because they target self-antigens, high-avidity clones are likely to be eliminated through negative selection.
Studies on chondrosarcoma and melanoma cell lines have shown that PRAME expression can be enhanced by treatment with the demethylating agent 5-aza-2′ deoxycytidine (DAC)148,149 and a study on UM showed that PRAME expression correlated with hypomethylation at 12 CpG sites near the PRAME promoter.69 Moreover, in cutaneous melanoma cells lines, HLA-A*02:01 restricted T cells targeting the PRA100-108 (VLDGLDVLL) epitope had weak specific killing and the addition of 5-aza-2′ deoxycytidine (DAC) to target cell lines increased not only PRAME expression but also HLA class I expression. Addition of DAC to T cells increased cytotoxic killing; the best results were obtained by pre-treating both the target and the T cells with DAC.149 This supports epigenetic regulation of PRAME expression and offers a way of sensitizing tumor cells to PRAME-targeted therapy. However, one should balance the benefit of increasing sensitivity to PRAME-targeted therapy vs the possible side effects of DAC treatment, such as myelosuppression.150
Engineering Prame-Specific T Cell Therapies
An approach to create high-avidity PRAME-specific T cells, involves the transfer of high affinity T cell receptors (TCRs) specific for PRAME. Previously, PRAME specific T cells were described that were derived from a patient with AML who had a severe GVH immune response following an HLA-A2 mismatched SCT. Two high-avidity HLA-A2-restricted T cell clones specific for the epitope SLLQHLIGL of PRAME were identified.72 Transfer of the PRAME-TCR resulted in PRAME-specific TCR engineered T cells recognizing cutaneous melanoma, AML, colon carcinoma, cervix carcinoma, lung cancer, and breast cancer cells. These T cells also exerted low reactivity against mature dendritic cells and kidney epithelial cells, which was shown to be related to low PRAME expression.72 These PRAME-specific TCR-engineered T cells were also able to kill cultured and patient-derived primary HLA-A*02:01+ medulloblastoma cells, independent of pretreatment with IFN-γ. Moreover, these TCR T cells allowed control of tumor growth in an orthotopic mouse model. The co-expression of the PRAME-TCR with an inducible caspase-9 suicide switch prompted elimination of the genetically modified T cells, thereby providing a safety switch.151
Interestingly, these PRAME-specific T cells72 recognized 4 out of 7 tested UM cell lines, and recognition strictly correlated with PRAME and HLA-A*02:01 expression.70 Recently, even more high affinity PRAME specific TCRs restricted to HLA-A*24:01, HLA-B*07:02, and HLA-A*02:01 were identified.131
To improve persistence of TCR engineered T cells the PRAME-specific TCR was combined with the chimeric PD1-41BB co-stimulatory receptor. The addition of PD1-41BB to CD8+ T cells expressing the transgenic PRAME-TCR enhanced IFN-γ secretion, improved cytotoxic capacity, and prevented exhaustion upon repetitive re-challenge with tumor cells in vitro without altering the in vitro safety profile.152 This combination also allowed tumor clearance in mice, even in PD-L1 positive tumors with low PRAME expression that were not affected by TCR T cells without PD1-41BB.152
At the moment, in vivo clinical studies with PRAME-specific TCR engineered T cells are ongoing. One trial involving patients with AML, myelodysplastic syndrome, and multiple myeloma targeted an HLA-A*02:01 restricted PRAME epitope and showed these PRAME-specific TCR T cells (MDG1011) to be well tolerated and to induce clinical response in 2 of 9 patients. Moreover, TCR-T cells were detected in peripheral blood of 6 of 8 patients, and PRAME was decreased in bone marrow of 4 patients (NCT03503968).153 Further development of MDG1011 has not been planned yet. A second study involving an HLA-A2 restricted PRAME-directed TCR and an inducible safety switch is being carried out on relapsed AML and previously treated myelodysplastic syndrome and metastatic UM (NCT02743611). At the time of writing, no information on the results have been published. Two further trials are being performed in patients with recurrent or relapsing advanced or metastatic solid tumors, among which is UM (NCT03686124 and NCT04262466). An interim report of trial NCT03686124 was published on the company's website and reported manageable tolerability, an objective response rate of 64% (7/11) at week 6 and of 67% (6/9) at month 3, and the presence of TCR T cells in all evaluable tumors.154 Results of the phase I dose escalation phase of the NCT04262466 trial presented at the European Society for Medical Oncology (ESMO) congress in 2022 showed good tolerability and evidence of durable partial responses in some of the cancer types analyzed, especially in UM (Tebentafusp-naïve), cutaneous melanoma, and serous ovarian cancer.155
Because PRAME cannot be directly targeted by antibodies or chimeric antigen receptor (CAR) T cells because PRAME is an intracellular antigen, Chang et al. developed the antibody Pr20, which is a TCR-mimic antibody, able to recognize a complex consisting of the PRAME peptide ALY and HLA-A2.156 Recently, Kirkey et al. developed CAR T cells composed of the Pr20 and tested them against AML cells.157 Although specificity controls were missing, they showed that these CAR T cells were able to kill PRAME+/HLA-A2+ in patients with AML blasts and engineered T cells that decreased tumor burden and prolonged survival in mouse models with PRAME+ tumors.
Discussion
The cancer/testis antigen PRAME has been reported to have variable expression in solid and hematological malignancies. Cutaneous melanoma, synovial sarcoma, myxoid and round cell sarcoma, and neuroblastoma show the highest rates of PRAME expression, whereas in urothelial and hepatocellular carcinoma, chronic lymphoblastic leukemia, and Hodgkin’s lymphoma, the rate of PRAME expression is low. Moreover, PRAME has been shown to have prognostic value in several types of cancer, including UM, as reported in previous studies by Field, Gezgin, and Schefler.68,70,91
We revisited the role of PRAME in a cohort of 64 patients with UM that had been part of prior publications, to which we now added a longer follow-up, and added the analysis of the TCGA cohort of 80 cases. We additionally studied the literature about the use of PRAME as prognostic marker, and we reviewed the current evidence supporting the use of PRAME as a target for immunotherapy. We confirmed the correlation of PRAME expression with negative prognostic factors, as has previously been reported70,91: in the total LUMC cohort, the LUMC disomy 3 cohort, and in the TCGA total cohort, PRAME expression was correlated with a higher LBD (see Table 2,70 Supplementary Table S4), as also reported by Schefler and Field.69,91 To add to PRAME's negative prognostic profile, in both the LUMC and the TCGA cohort, PRAME expression was higher in darker tumors (see Table 2, Fig. 2). We did not find a significant correlation of PRAME with chromosome 3 status or BAP1 expression in either cohort, and we saw a borderline correlation with 6p gain in the TCGA disomy 3 subcohort and a matched near-significant correlation in the LUMC disomy 3 subcohort. Field et al. had previously reported an association between high PRAME expression and the presence of 6p gain, among other chromosomal alterations, such as 6q loss, 8q gain and 16q loss, in both class 1 and class 2 UM.69 At the moment, however, because we only found this association in one of the subcohorts, it is hard to say if 6p gain itself has a role or if it is part of the genomic instability postulated by Harbour et al.90 More consistent correlations were found with the presence of an inflammatory phenotype, with enrichment in inflammation-related pathways (see Table 3, Figs. 5, 6) and with gain of chromosome 8q (Table 2,70 Supplementary Tables S4–S7, S11). Our differential expression analysis highlighted 8q24.3 as strictly related to PRAME overexpression, because most of the upregulated genes are located in this region. Interestingly, this specific region has been reported as relevant in the progression of several types of cancers, and in UM in particular.96 Speicher et al. studied 11 patients with UM with comparative genomic hybridization (CGH) and reported gain of chromosome 8 as the most frequent copy number alteration and the segment 8q24→qter as the smallest represented segment.158 Similarly, Anbunathan et al. analyzed data from 182 patients with UM and reported gain of 8q and loss of heterozygosity in chromosome 3 to be present in >50% of cases and identified 8q24.3 as one of the focal copy number regions associated with metastasis and worse survival.159 A literature search into the region 8q24 highlighted some interesting genes that have been related to UM progression and prognosis. C-MYC (MYC Proto-Oncogene, BHLH Transcription Factor), a known proto-oncogene located on 8q24.1, has been studied in UM and its expression has been correlated with a high proliferation, the presence of monosomy 3 or BAP1 mutation, but its association with survival is not unanimous.160–165 DDEF1 (ArfGAP with SH3 domain, ankyrin repeat and PH domain 1), located on 8q24.21, was identified as the gene, the expression of which correlated best with the number of 8q copies in a study involving 25 patients with primary UM, was significantly higher in class 2 UM compared to class 1 UM and its overexpression increased cell motility in the Mel202 UM cell line.166 The current study, however, found no significant increase in C-MYC in UM with 8q gain. Protein tyrosine kinase 2 (PTK2) on 8q24.3 has been shown to be amplified in breast cancer167 and hepatocellular carcinoma,168 and to be among the upregulated genes that exhibited hypomethylation in a study involving 182 UM cases.159 Protein tyrosine phosphatase 4A3 (PTP4A3), located on 8q.24.3, as mentioned previously, showed a significant association with early metastases and caused an increase in cell migration and invasiveness in UM cell lines.117 Moreover, PTP4A3 was among the genes that showed upregulation and hypomethylation in the study by Anbunathan et al.159 Considering this information from the literature and our data, we think a good candidate that is worth studying further, both in the context of PRAME and of UM with 8q gain in general, might be PTP4A3. In our cohort, only PTP4A3 was among the most differentially expressed genes in PRAME-positive UM (adjusted P value = 0.026, logFC = 0.819; see Fig. 7, Supplementary Table S11), whereas MYC had an adjusted P value > 0.05 and DDEF1 and PTK2 had an adjusted P value of 0.049 and 0.041, respectively, and a lower logFC (0.643 and 0.461, respectively).
Another interesting result of the differential expression analysis and GSEA is the link between PRAME and inflammation. As previously mentioned, inflammation-related pathways showed an enrichment in PRAME-positive tumors, and among the most upregulated genes we identified macrophage marker CD68 and SLC52A2, which has been linked to T cell exhaustion and the presence of M2 macrophages in several cancers, among which is UM. Moreover, the presence of 8q gain has been connected specifically to an increased influx of macrophages in UM, independently of BAP1.97 We confirmed the prognostic value of PRAME on UM-related survival, as reported by Field and Gezgin when we analyzed a longer follow-up68,70 and we identified the D3 population as the cohort where PRAME has the highest correlation with metastases formation (see Fig. 3). We could not confirm this finding in the TCGA cohort of 80 patients, but we think it may partly be due to the shorter follow-up that is available (see Fig. 4) or to the threshold of PRAME expression used to define PRAME-positive and PRAME-negative groups. A further source of variation between the two cohorts may be slight differences in tumor features between the two study populations: centers that provided the tumor to include in the TCGA database may have different criteria for enucleation/eye sparing treatment. Others reported that PRAME has prognostic value in both class 1 GEP as well as class 2 GEP tumors.68,69
Because a study on breast cancer suggests that PRAME drives EMT and that PRAME expression correlates with EMT markers,89 we tested this hypothesis in the LUMC database. Although only one marker (ZEB2) showed a significant difference between PRAME-negative and PRAME-positive tumors (see Supplementary Table S9), our GSEA analysis identified EMT as one of the pathways enriched in PRAME-positive UM (see Fig. 5).
As survival in UM has not improved during the last 5 decades, there is an urgent need to develop effective therapies to treat metastases, or to prevent them from becoming clinically significant. Because immune checkpoint therapy has given unsatisfactory results so far,8,57–59 alternative targets for immunotherapy were considered. CTAs are attractive targets because of their expression pattern (low or not expressed in normal tissues, and high in tumors), and, among the most common CTAs, PRAME has the highest expression in primary UM and its metastases.68–70,91
Cancer vaccines are a feasible option and have given promising results, especially in studies involving the use of dendritic cells or the co-targeting of PRAME and PSMA.135,136 However, PSMA is not expressed in UM. Moreover, vaccines are used to sensitize pre-existing T cells to a new antigen but, because PRAME is a self-antigen, high-avidity T cells may be eliminated by self-tolerance.
Adoptive T cell therapy turned out to be an interesting strategy, both in studies carried out using pre-existing PRAME-specific T cells and with TCR-transduced T cells. The main issue with adoptive T cell therapy with pre-existing T cells is the low avidity of these CTL, which has been tackled in several ways. PRAME specific CTLs were obtained in vitro either through stimulation of APCs with a peptide library spanning the entire PRAME protein140 or with 5-aza-2′deoxycitidine.149 However, negative selection of high-avidity T cells against self-antigens remain an issue. T cell therapy with PRAME TCR engineered T cells is currently in the clinic and showed 67% confirmed objective response rate (ORR) in an interim clinical update on heavily pretreated patients in the phase I dose expansion cohort. Objective responses were observed across multiple tumor types, including checkpoint-refractory cutaneous melanoma, platinum-resistant ovarian cancer, uveal melanoma, head and neck cancer, and synovial sarcoma.72
Previously, UM metastases were demonstrated to be positive for PRAME and HLA class I, and co-expression was documented in 50% of the samples, opening up the possibility of using TCR-mediated PRAME-specific immunotherapy in the treatment of UM.70 In addition, it was shown that PRAME-specific T cells targeting the SLLQHLIGL epitope demonstrated specific killing of PRAME positive UM cell lines. This observation identified the SLLQHLIGL epitope as a suitable target epitope and the use of high-avidity allo-HLA-restricted TCR-transduced T cells as feasible potential strategy for immunotherapy in UM.70 Two previous studies from our group analyzed cohorts of 23 and 45 patients with enucleated UM by IHC and HLA typing. De Waard-Siebinga et al. reported positive IHC staining for HLA-A2 in 12 of 22 and 16 of 23 cases (with 2 different monoclonal antibodies),101 whereas Van Essen et al. showed that HLA-A2 had a frequency of 53% in 45 patients with HLA-typed UM.169 These percentages show that a large proportion of patients would be eligible for an HLA-A2 targeted therapy, but that other HLA alleles should be targeted as well. The study by van Amerongen et al., that identified three further high affinity PRAME specific TCRs with different HLA restriction (HLA-A*24:01, HLA-B*07:02, and HLA-A*02:01), is a promising step in this direction.
One limitation of this study was the small sample size in the LUMC cohort, and especially the low number of disomy 3 PRAME + patients, which may lead to over- or under-estimation of the correlation between PRAME expression and prognostic factors. The TCGA cohort has a larger sample size (80 patients) but it has a shorter follow-up, which may make the survival analyses less reliable. Several American studies included large numbers of patients and show that PRAME is a valid prognostic marker. It would be interesting to see studies on the relation between PRAME and tumor characteristics, such as angiogenesis, in order to determine whether it is PRAME itself that is important or whether it is a correlation with other biological factors. We, for example, showed a relation between expression of PRAME and an inflammatory phenotype.
In conclusion, PRAME is expressed in many different solid and hematological malignancies. In UM, it is expressed in a subset of primary UM and in the majority of metastases. PRAME is a useful prognostic marker in class 1/disomy 3 tumors, PRAME expression significantly decreased survival and was correlated with a greater LBD and 8q amplification, whereas in class 2/monosomy 3 tumors, it was correlated with 8q amplification and ciliary body involvement. About 70% of UM metastases express PRAME, and patients with UM should be the target population for a PRAME-specific immunotherapy. Among the immunotherapy options, TCR-transduced T cells targeting PRAME are promising, and the first clinical data show encouraging results.
Supplementary Material
Acknowledgments
M.C. Gelmi was funded by the Bontius Foundation, Oogfonds, the Sam Fund, the LUF, P.A. Jager-van Gelder Fund, the Blinden-Penning foundation, and ASROO (Associazione Scientifica Retinoblastoma ed Oncologia Oculare).
Disclosure: M.C. Gelmi, None; G. Gezgin, None; P.A. van der Velden, None; G.P.M. Luyten, None; S.J. Luk, None; M.H.M. Heemskerk, None; M.J. Jager, None
References
- 1. Singh AD, Turell ME, Topham AK.. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011; 118: 1881–1885. [DOI] [PubMed] [Google Scholar]
- 2. Singh AD, Topham A.. Survival rates with uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003; 110: 962–965. [DOI] [PubMed] [Google Scholar]
- 3. Hawkins BS, Collaborative Ocular Melanoma Study Group. The Collaborative Ocular Melanoma Study (COMS) randomized trial of pre-enucleation radiation of large choroidal melanoma: IV. Ten-year mortality findings and prognostic factors. COMS report number 24. Am J Ophthalmol. 2004; 138: 936–951. [DOI] [PubMed] [Google Scholar]
- 4. Augsburger JJ, Correa ZM, Shaikh AH.. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009; 148: 119–127. [DOI] [PubMed] [Google Scholar]
- 5. Rantala ES, Hernberg M, Kivela TT.. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2019; 29: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diener-West M, Reynolds SM, Agugliaro DJ, et al.. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: collaborative Ocular Melanoma Study Group Report No. 26. Arch Opthalmol. 2005; 123: 1639–1643. [DOI] [PubMed] [Google Scholar]
- 7. Virgili G, Gatta G, Ciccolallo L, et al.. Survival in patients with uveal melanoma in Europe. Arch Opthalmol. 2008; 126: 1413–1418. [DOI] [PubMed] [Google Scholar]
- 8. Jager MJ, Shields CL, Cebulla CM, et al.. Uveal melanoma. Nat Rev Dis Primers. 2020; 6: 24. [DOI] [PubMed] [Google Scholar]
- 9. Roelofsen CDM, Wierenga APA, van Duinen S, et al.. Five decades of enucleations for uveal melanoma in one center: more tumors with high risk factors, no improvement in survival over time. Ocul Oncol Pathol. 2021; 7: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Middleton MR, Steven NM, Evans TJ, et al.. Safety, pharmacokinetics and efficacy of IMCgp100, a first-in-class soluble TCR-antiCD3 bispecific t cell redirector with solid tumour activity: Results from the FIH study in melanoma. J Clinic Oncol. 2016; 34: 3016. [Google Scholar]
- 11. Sacco JJ, Carvajal R, Butler MO, et al.. 64MO A phase (ph) II, multi-center study of the safety and efficacy of tebentafusp (tebe) (IMCgp100) in patients (pts) with metastatic uveal melanoma (mUM). Ann Oncol. 2020; 31: S1442–S1443. [Google Scholar]
- 12. Shoushtari AN, Collins L, Espinosa E, et al.. 1757O Early reduction in ctDNA, regardless of best RECIST response, is associated with overall survival (OS) on tebentafusp in previously treated metastatic uveal melanoma (mUM) patients. Ann Oncol. 2021; 32. [Google Scholar]
- 13. Nathan P, Hassel JC, Rutkowski P, et al.. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021; 385: 1196–1206. [DOI] [PubMed] [Google Scholar]
- 14. Carvajal RD, Butler MO, Shoushtari AN, et al.. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: a phase 2 trial. Nat Med. 2022; 28: 2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amin MB, Greene FL, Edge SB, et al.. The Eighth Edition AJCC Cancer Staging Manual: continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA Cancer J Clin. 2017; 67: 93–99. [DOI] [PubMed] [Google Scholar]
- 16. Dogrusöz M, Jager MJ, Damato B.. Uveal melanoma treatment and prognostication. Asia Pac J Ophthalmol. 2017; 6: 186–196. [DOI] [PubMed] [Google Scholar]
- 17. Dogrusöz M, Jager MJ.. Genetic prognostication in uveal melanoma. Acta Ophthalmol. 2018; 96: 331–347. [DOI] [PubMed] [Google Scholar]
- 18. Horsman DE, Sroka H, Rootman J, White VA.. Monosomy 3 and isochromosome 8q in a uveal melanoma. Cancer Genet Cytogenet. 1990; 45: 249–253. [DOI] [PubMed] [Google Scholar]
- 19. Kilic E, van Gils W, Lodder E, et al.. Clinical and cytogenetic analyses in uveal melanoma. Invest Opthalmol Vis Sci. 2006; 47: 3703–3707. [DOI] [PubMed] [Google Scholar]
- 20. Damato B, Dopierala JA, Coupland SE.. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010; 16: 6083–6092. [DOI] [PubMed] [Google Scholar]
- 21. Ewens KG, Kanetsky PA, Richards-Yutz J, et al.. Genomic profile of 320 uveal melanoma cases: chromosome 8p-loss and metastatic outcome. Invest Ophthalmol Vis Sci. 2013; 54: 5721–5729. [DOI] [PubMed] [Google Scholar]
- 22. White VA, Chambers JD, Courtright PD, Chang EY, Horsman DE.. Correlation of cytogenetic abnormalities with the outcome of patients with uveal melanoma. Cancer. 1998; 83: 354–359. [PubMed] [Google Scholar]
- 23. Damato B, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland SE.. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest Ophthalmol Vis Sci. 2009; 50: 3048–3055. [DOI] [PubMed] [Google Scholar]
- 24. Robertson AG, Shih J, Yau C, et al.. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell. 2017; 32: 204–220.e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jager MJ, Brouwer NJ, Esmaeli B.. The Cancer Genome Atlas Project: an integrated molecular view of uveal melanoma. Ophthalmology. 2018; 125: 1139–1142. [DOI] [PubMed] [Google Scholar]
- 26. Onken MD, Worley LA, Ehlers JP, Harbour JW.. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004; 64: 7205–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harbour JW, Onken MD, Roberson EDO, et al.. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010; 330: 1410–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin M, Masshofer L, Temming P, et al.. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013; 45: 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yavuzyigitoglu S, Koopmans AE, Verdijk RM, et al.. Uveal melanomas with SF3B1 mutations: a distinct subclass associated with late-onset metastases. Ophthalmology. 2016; 123: 1118–1128. [DOI] [PubMed] [Google Scholar]
- 30. Furney SJ, Pedersen M, Gentien D, et al.. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013; 3: 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ewens KG, Kanetsky PA, Richards-Yutz J, et al.. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Invest Ophthalmol Vis Sci. 2014; 55: 5160–5167. [DOI] [PubMed] [Google Scholar]
- 32. Ewens KG, Lalonde E, Richards-Yutz J, Shields CL, Ganguly A.. Comparison of germline versus somatic BAP1 mutations for risk of metastasis in uveal melanoma. BMC Cancer. 2018; 18: 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zuidervaart W, van der Velden PA, Hurks MH, et al.. Gene expression profiling identifies tumour markers potentially playing a role in uveal melanoma development. Br J Cancer. 2003; 89: 1914–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tschentscher F, Huesing J, Hoelter T, et al.. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003; 63: 2578–2584. [PubMed] [Google Scholar]
- 35. van Gils W, Lodder EM, Mensink HW, et al.. Gene expression profiling in uveal melanoma: two regions on 3p related to prognosis. Invest Ophthalmol Vis Sci. 2008; 49: 4254–4262. [DOI] [PubMed] [Google Scholar]
- 36. Worley LA, Onken MD, Person E, et al.. Transcriptomic versus chromosomal prognostic markers and clinical outcome in uveal melanoma. Clin Cancer Res. 2007; 13: 1466–1471. [DOI] [PubMed] [Google Scholar]
- 37. Onken MD, Worley LA, Tuscan MD, Harbour JW.. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010; 12: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harbour JW. A prognostic test to predict the risk of metastasis in uveal melanoma based on a 15-gene expression profile. Methods Mol Biol. 2014; 1102: 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Field MG, Harbour JW.. Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol. 2014; 25: 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plasseraud KM, Wilkinson JK, Oelschlager KM, et al.. Gene expression profiling in uveal melanoma: technical reliability and correlation of molecular class with pathologic characteristics. Diagn Pathol. 2017; 12: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Lange MJ, van Pelt SI, Versluis M, et al.. Heterogeneity revealed by integrated genomic analysis uncovers a molecular switch in malignant uveal melanoma. Oncotarget. 2015; 6: 37824–37835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Onken MD, Worley LA, Harbour JW.. A metastasis modifier locus on human chromosome 8p in uveal melanoma identified by integrative genomic analysis. Clin Cancer Res. 2008; 14: 3737–3745. [DOI] [PubMed] [Google Scholar]
- 43. Dogrusöz M, Bagger M, van Duinen SG, et al.. The prognostic value of AJCC staging in uveal melanoma is enhanced by adding chromosome 3 and 8q status. Invest Ophthalmol Vis Sci. 2017; 58: 833–842. [DOI] [PubMed] [Google Scholar]
- 44. Negretti GS, Gurudas S, Gallo B, et al.. Survival analysis following enucleation for uveal melanoma. Eye (Lond). 2021; 36: 1669–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eleuteri A, Damato B, Coupland SE, Taktak AFG.. Enhancing survival prognostication in patients with choroidal melanoma by integrating pathologic, clinical and genetic predictors of metastasis. Int J Biomed Eng Technol. 2012; 8: 18–35. [Google Scholar]
- 46. Eleuteri A, Taktak AFG, Coupland SE, Heimann H, Kalirai H, Damato B.. Prognostication of metastatic death in uveal melanoma patients: a Markov multi-state model. Comput Biol Med. 2018; 102: 151–156. [DOI] [PubMed] [Google Scholar]
- 47. Gelmi MC, Wierenga APA, Kroes WGM, et al.. Increased histological tumor pigmentation in uveal melanoma is related to eye color and loss of chromosome 3/BAP1. Ophthalmol Sci. 2023; 3: 100297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gelmi MC, Bas Z, Malkani K, Ganguly A, Shields CL, Jager MJ.. Adding the cancer genome atlas chromosome classes to American Joint Committee on cancer system offers more precise prognostication in uveal melanoma. Ophthalmology. 2022; 129: 431–437. [DOI] [PubMed] [Google Scholar]
- 49. Hodi FS, O'Day SJ, McDermott DF, et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garon EB, Rizvi NA, Hui R, et al.. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372: 2018–2028. [DOI] [PubMed] [Google Scholar]
- 51. Robert C, Long GV, Brady B, et al.. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015; 372: 320–330. [DOI] [PubMed] [Google Scholar]
- 52. Borghaei H, Paz-Ares L, Horn L, et al.. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015; 373: 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yarchoan M, Hopkins A, Jaffee EM.. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017; 377: 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goodman AM, Kato S, Bazhenova L, et al.. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017; 16: 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaunitz GJ, Cottrell TR, Lilo M, et al.. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab Invest. 2017; 97: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Javed A, Arguello D, Johnston C, et al.. PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy. 2017; 9: 1323–1330. [DOI] [PubMed] [Google Scholar]
- 57. Wessely A, Steeb T, Erdmann M, et al.. The role of immune checkpoint blockade in uveal melanoma. Int J Mol Sci. 2020; 21: 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wierenga APA, Cao J, Luyten GPM, Jager MJ.. Immune checkpoint inhibitors in uveal and conjunctival melanoma. Int Ophthalmol Clin. 2019; 59: 53–63. [DOI] [PubMed] [Google Scholar]
- 59. van der Kooij MK, Joosse A, Speetjens FM, et al.. Anti-PD1 treatment in metastatic uveal melanoma in the Netherlands. Acta Oncol. 2017; 56: 101–103. [DOI] [PubMed] [Google Scholar]
- 60. Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD.. Metastatic disease from uveal melanoma: treatment options and future prospects. Br J Ophthalmol. 2017; 101: 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rodrigues M, Mobuchon L, Houy A, et al.. Outlier response to anti-PD1 in uveal melanoma reveals germline MBD4 mutations in hypermutated tumors. Nat Commun. 2018; 9: 1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johansson PA, Stark A, Palmer JM, et al.. Prolonged stable disease in a uveal melanoma patient with germline MBD4 nonsense mutation treated with pembrolizumab and ipilimumab. Immunogenetics. 2019; 71: 433–436. [DOI] [PubMed] [Google Scholar]
- 63. Saint-Ghislain M, Derrien AC, Geoffrois L, et al.. MBD4 deficiency is predictive of response to immune checkpoint inhibitors in metastatic uveal melanoma patients. Eur J Cancer. 2022; 173: 105–112. [DOI] [PubMed] [Google Scholar]
- 64. Luyten GPM, van der Spek CW, Brand I, et al.. Expression of MAGE, gp100 and tyrosinase genes in uveal melanoma cell lines. Melanoma Res. 1998; 8: 11–16. [DOI] [PubMed] [Google Scholar]
- 65. Chen PW, Murray TG, Uno T, Salgaller ML, Reddy R, Ksander BR.. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin Exp Metastasis. 1996; 15: 509–518. [DOI] [PubMed] [Google Scholar]
- 66. Mulcahy KA, Rimolde D, Brasseur F, et al.. Infrequent expression of the MAGE gene family in uveal melanomas. Int J Cancer. 1996; 66: 738–742. [DOI] [PubMed] [Google Scholar]
- 67. Errington JA, Conway RM, Walsh-Conway N, et al.. Expression of cancer-testis antigens (MAGE-A1, MAGE-A3/6, MAGE-A4, MAGE-C1 and NY-ESO-1) in primary human uveal and conjunctival melanoma. Br J Ophthalmol. 2012; 96: 451–458. [DOI] [PubMed] [Google Scholar]
- 68. Field MG, Decatur CL, Kurtenbach S, et al.. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin Cancer Res. 2016; 22: 1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Field MG, Durante MA, Decatur CL, et al.. Epigenetic reprogramming and aberrant expression of PRAME are associated with increased metastatic risk in class 1 and class 2 uveal melanomas. Oncotarget. 2016; 7: 59209–59219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gezgin G, Luk SJ, Cao J, et al.. PRAME as a potential target for immunotherapy in metastatic uveal melanoma. JAMA Ophthalmol. 2017; 135: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ikeda H, Lethé B, Lehmann F, et al.. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997; 6: 199–208. [DOI] [PubMed] [Google Scholar]
- 72. Amir AL, van der Steen DM, van Loenen MM, et al.. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin Cancer Res. 2011; 17: 5615–5625. [DOI] [PubMed] [Google Scholar]
- 73. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ.. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005; 5: 615–625. [DOI] [PubMed] [Google Scholar]
- 74. Fratta E, Coral S, Covre A, et al.. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. 2011; 5: 164–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li J, Yin J, Zhong J, Yang Z, Tang A, Li S.. Clinicopathological and prognostic significance of PRAME overexpression in human cancer: a meta-analysis. Biomed Res Int. 2020; 2020: 8828579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Van Baren N, Chambost H, Ferrant A, et al.. PRAME, a gene encoding an antigen recognized on a human melanoma by cytolytic T cells, is expressed in acute leukaemia cells. Br J Haematol. 1998; 102: 1376–1379. [DOI] [PubMed] [Google Scholar]
- 77. Steinbach D, Hermann J, Viehmann S, Zintl F, Gruhn B.. Clinical implications of PRAME gene expression in childhood acute myeloid leukemia. Cancer Genet Cytogenet. 2002; 133: 118–123. [DOI] [PubMed] [Google Scholar]
- 78. Mitsuhashi K, Masuda A, Wang YH, Shiseki M, Motoji T.. Prognostic significance of PRAME expression based on immunohistochemistry for diffuse large B-cell lymphoma patients treated with R-CHOP therapy. Int J Hematol. 2014; 100: 88–95. [DOI] [PubMed] [Google Scholar]
- 79. Ercolak V, Paydas S, Bagir E, et al.. PRAME expression and its clinical relevance in Hodgkin's lymphoma. Acta Haematol. 2015; 134: 199–207. [DOI] [PubMed] [Google Scholar]
- 80. Huang QS, Wang JZ, Qin YZ, et al.. Overexpression of WT1 and PRAME predicts poor outcomes of patients with myelodysplastic syndromes with thrombocytopenia. Blood Adv. 2019; 3: 3406–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Takata K, Chong LC, Ennishi D, et al.. Tumor-associated antigen PRAME exhibits dualistic functions that are targetable in diffuse large B cell lymphoma. J Clin Invest. 2022; 132: e145343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsushita M, Ikeda H, Kizaki M, et al.. Quantitative monitoring of the PRAME gene for the detection of minimal residual disease in leukaemia. Br J Haematol. 2001; 112: 916–926. [DOI] [PubMed] [Google Scholar]
- 83. Wadelin FR, Fulton J, Collins HM, et al.. PRAME is a golgi-targeted protein that associates with the Elongin BC complex and is upregulated by interferon-gamma and bacterial PAMPs. PLoS One. 2013; 8: e58052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tajeddine N, Gala JL, Louis M, Van Schoor M, Tombal B, Gailly P.. Tumor-associated antigen preferentially expressed antigen of melanoma (PRAME) induces caspase-independent cell death in vitro and reduces tumorigenicity in vivo. Cancer Res. 2005; 65: 7348–7355. [DOI] [PubMed] [Google Scholar]
- 85. Epping MT, Wang L, Edel MJ, Carlee L, Hernandez M, Bernards R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell. 2005; 122: 835–847. [DOI] [PubMed] [Google Scholar]
- 86. Steinbach D, Pfaffendorf N, Wittig S, Gruhn B.. PRAME expression is not associated with down-regulation of retinoic acid signaling in primary acute myeloid leukemia. Cancer Genet Cytogenet. 2007; 177: 51–54. [DOI] [PubMed] [Google Scholar]
- 87. Costessi A, Mahrour N, Tijchon E, et al.. The tumour antigen PRAME is a subunit of a Cul2 ubiquitin ligase and associates with active NFY promoters. EMBO J. 2011; 30: 3786–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Costessi A, Mahrour N, Sharma V, et al.. The human EKC/KEOPS complex is recruited to Cullin2 ubiquitin ligases by the human tumour antigen PRAME. PLoS One. 2012; 7: e42822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Al-Khadairi G, Naik A, Thomas R, Al-Sulaiti B, Rizly S, Decock J.. PRAME promotes epithelial-to-mesenchymal transition in triple negative breast cancer. J Transl Med. 2019; 17: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kurtenbach S, Sanchez MI, Kuznetsoff J, et al. PRAME induces genomic instability in uveal melanoma. Oncogene. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schefler AC, Koca E, Bernicker EH, Correa ZM.. Relationship between clinical features, GEP class, and PRAME expression in uveal melanoma. Graefes Arch Clin Exp Ophthalmol. 2019; 257: 1541–1545. [DOI] [PubMed] [Google Scholar]
- 92. Kumar N, Singh MK, Singh L, et al.. Diagnostic utility of immunohistochemistry in concordance with mRNA analysis of PRAME in the stratification of high-risk uveal melanoma patients. Hum Cell. 2023; 36: 342–352. [DOI] [PubMed] [Google Scholar]
- 93. Schefler AC, Mruthyunjaya P, Decatur C, et al.. Design, methods, and rationale for the Collaborative Ocular Oncology Group 2 (COOG2) study. Invest Ophthalmol Vis Sci. 2021; 62: 2870. [Google Scholar]
- 94. van de Nes JA, Nelles J, Kreis S, et al.. Comparing the prognostic value of BAP1 mutation pattern, chromosome 3 status, and BAP1 immunohistochemistry in uveal melanoma. Am J Surg Pathol. 2016; 40: 796–805. [DOI] [PubMed] [Google Scholar]
- 95. Koopmans AE, Verdijk RM, Brouwer RW, et al.. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod Pathol. 2014; 27: 1321–1330. [DOI] [PubMed] [Google Scholar]
- 96. Versluis M, de Lange MJ, van Pelt SI, et al.. Digital PCR validates 8q dosage as prognostic tool in uveal melanoma. PLoS One. 2015; 10: e0116371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gezgin G, Dogrusoz M, van Essen TH, et al.. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol Immunother. 2017; 66: 903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. van Essen TH, van Pelt SI, Bronkhorst IH, et al.. Upregulation of HLA expression in primary uveal melanoma by infiltrating leukocytes. PLoS One. 2016; 11: e0164292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Maat W, Ly LV, Jordanova ES, de Wolff-Rouendaal D, Schalij-Delfos NE, Jager MJ.. Monosomy of chromosome 3 and an inflammatory phenotype occur together in uveal melanoma. Invest Ophthalmol Vis Sci. 2008; 49: 505–510. [DOI] [PubMed] [Google Scholar]
- 100. Bronkhorst IH, Jager MJ.. Uveal melanoma: the inflammatory microenvironment. J Innate Immun. 2012; 4: 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. de Waard-Siebinga I, Houbiers JG, Hilders CG, de Wolff-Rouendaal D, Jager MJ.. Differential expression of HLA-A and B-alleles on uveal melanoma as determined by immuno-histology. Ocul Immunol Inflamm. 1996; 4: 1–14. [DOI] [PubMed] [Google Scholar]
- 102. Blom DJ, Luyten GP, Mooy C, Kerkvliet S, Zwinderman AK, Jager MJ.. Human leukocyte antigen class I expression. Marker of poor prognosis in uveal melanoma. Invest Ophthalmol Vis Sci. 1997; 38: 1865–1872. [PubMed] [Google Scholar]
- 103. Ericsson C, Seregard S, Bartolazzi A, et al.. Association of HLA class I and class II antigen expression and mortality in uveal melanoma. Invest Ophthalmol Vis Sci. 2001; 42: 2153–2156. [PubMed] [Google Scholar]
- 104. Jager MJ, Hurks MH, Levitskaya J, Kiessling R.. HLA expression in uveal melanoma: there is no rule without some exception. Human Immunol. 2002; 63: 444–451. [DOI] [PubMed] [Google Scholar]
- 105. Cheng X, Geng F, Pan M, et al.. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 2020; 32: 229–242.e228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. He P, Cheng S, Hu F, Ma Z, Xia Y.. Up-regulation of DGAT1 in cancer tissues and tumor-infiltrating macrophages influenced survival of patients with gastric cancer. BMC Cancer. 2021; 21: 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wilcock DJ, Badrock AP, Wong CW, et al.. Oxidative stress from DGAT1 oncoprotein inhibition in melanoma suppresses tumor growth when ROS defenses are also breached. Cell Rep. 2022; 39: 110995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Watson M, Roulston A, Belec L, et al.. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol Cell Biol. 2009; 29: 5872–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cerna D, Li H, Flaherty S, Takebe N, Coleman CN, Yoo SS.. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J Biol Chem. 2012; 287: 22408–22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Shames DS, Elkins K, Walter K, et al.. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin Cancer Res. 2013; 19: 6912–6923. [DOI] [PubMed] [Google Scholar]
- 111. Zhang J, Shen Y, Ma D, Li Z, Zhang Z, Jin W. SLCO4A1-AS1 mediates pancreatic cancer development via miR-4673/KIF21B axis. Open Med (Wars). 2022; 17: 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112. Upadhyay G. Emerging role of lymphocyte antigen-6 family of genes in cancer and immune cells. Front Immunol. 2019; 10: 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. AlHossiny M, Luo L, Frazier WR, et al.. Ly6E/K signaling to TGFbeta promotes breast cancer progression, immune escape, and drug resistance. Cancer Res. 2016; 76: 3376–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yeom CJ, Zeng L, Goto Y, et al.. LY6E: a conductor of malignant tumor growth through modulation of the PTEN/PI3K/Akt/HIF-1 axis. Oncotarget. 2016; 7: 65837–65848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Schleicher EM, Galvan AM, Imamura-Kawasawa Y, Moldovan GL, Nicolae CM.. PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 2018; 46: 8908–8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. de Koning L, Decaudin D, El Botty R, et al.. PARP inhibition increases the response to chemotherapy in uveal melanoma. Cancers (Basel). 2019; 11: 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Laurent C, Valet F, Planque N, et al.. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011; 71: 666–674. [DOI] [PubMed] [Google Scholar]
- 118. Maacha S, Anezo O, Foy M, et al.. Protein tyrosine phosphatase 4A3 (PTP4A3) promotes human uveal melanoma aggressiveness through membrane accumulation of matrix metalloproteinase 14 (MMP14). Invest Ophthalmol Vis Sci. 2016; 57: 1982–1990. [DOI] [PubMed] [Google Scholar]
- 119. Duciel L, Anezo O, Mandal K, et al.. Protein tyrosine phosphatase 4A3 (PTP4A3/PRL-3) promotes the aggressiveness of human uveal melanoma through dephosphorylation of CRMP2. Sci Rep. 2019; 9: 2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Matherne MG, Phillips ES, Embrey SJ, Burke CM, Machado HL.. Emerging functions of C/EBPbeta in breast cancer. Front Oncol. 2023; 13: 1111522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cheng H, Huang C, Tang G, et al.. Emerging role of EPHX1 in chemoresistance of acute myeloid leukemia by regulating drug-metabolizing enzymes and apoptotic signaling. Mol Carcinog. 2019; 58: 808–819. [DOI] [PubMed] [Google Scholar]
- 122. Deng Z, Xiao M, Du D, et al.. DNASE1L3 as a prognostic biomarker associated with immune cell infiltration in cancer. Onco Targets Ther. 2021; 14: 2003–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Beheshti Zavareh R, Sukhai MA, Hurren R, et al.. Suppression of cancer progression by MGAT1 shRNA knockdown. PLoS One. 2012; 7: e43721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Akiva I, Birgul Iyison N.. MGAT1 is a novel transcriptional target of Wnt/beta-catenin signaling pathway. BMC Cancer. 2018; 18: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ren H, Chen Z, Yang L, et al.. Apolipoprotein C1 (APOC1) promotes tumor progression via MAPK signaling pathways in colorectal cancer. Cancer Manag Res. 2019; 11: 4917–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yi J, Ren L, Wu J, et al.. Apolipoprotein C1 (APOC1) as a novel diagnostic and prognostic biomarker for gastric cancer. Ann Transl Med. 2019; 7: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cui Y, Miao C, Hou C, Wang Z, Liu B.. Apolipoprotein C1 (APOC1): a novel diagnostic and prognostic biomarker for clear cell renal cell carcinoma. Front Oncol. 2020; 10: 1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Fiszer D, Kurpisz M.. Major histocompatibility complex expression on human, malegerm cells: a review. Am J Reprod Immunol. 1998; 40: 172–176. [DOI] [PubMed] [Google Scholar]
- 129. Zhao S, Zhu W, Xue S, Han D.. Testicular defense systems: immune privilege and innate immunity. Cell Mol Immunol. 2014; 11: 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kessler JH, Beekman NJ, Bres-Vloemans SA, et al.. Efficient identification of novel HLA-A * 0201–presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J Exp Med. 2001; 193: 73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. van Amerongen RA, Tuit S, Wouters AK, et al.. PRAME and CTCFL-reactive TCRs for the treatment of ovarian cancer. Front Immunol. 2023; 14: 1121973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Gérard C, Baudson N, Ory T, Segal L, Louahed J.. A comprehensive preclinical model evaluating the recombinant PRAME antigen combined with the AS15 immunostimulant to fight against PRAME-expressing tumors. J Immunother. 2015; 38: 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Gutzmer R, Rivoltini L, Levchenko E, et al.. Safety and immunogenicity of the PRAME cancer immunotherapeutic in metastatic melanoma: results of a phase I dose escalation study. ESMO Open. 2016; 1: e000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pujol JL, De Pas T, Rittmeyer A, et al.. Safety and immunogenicity of the PRAME cancer immunotherapeutic in patients with resected non-small cell lung cancer: a phase I dose escalation study. J Thorac Oncol. 2016; 11: 2208–2217. [DOI] [PubMed] [Google Scholar]
- 135. Weber JS, Vogelzang NJ, Enstoff MS, et al.. A phase 1 study of a vaccine targeting preferentially expressed antigen in melanoma and prostate-specific membrane antigen in patients with advanced solid tumors. J Immunother. 2011; 34: 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Li L, Giannopoulos K, Reinhardt P, et al.. Immunotherapy for patients with acute myeloid leukemia using autologous dendritic cells generated from leukemic blasts. Int J Oncol. 2006; 28: 855–861. [PubMed] [Google Scholar]
- 137. Lichtenegger FS, Schnorfeil FM, Rothe M, et al.. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: results of a phase I trial. Clin Transl Immunol. 2020; 9: e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Eckl J, Raffegerst S, Schnorfeil F, et al.. DC vaccination induces antigen specific immune responses in AML patients: a 1-year interim assessment. Available at: https://medigene.com/fileadmin/download/publications/191204_Poster_Ash_2019_final_JE.pdf; 2019.
- 139. Griffioen M, Kessler JH, Borghi M, et al.. Detection and functional analysis of CD8+ T cells specific for PRAME: a target for T-cell therapy. Clin Cancer Res. 2006; 12: 3130–3136. [DOI] [PubMed] [Google Scholar]
- 140. Quintarelli C, Dotti G, Hasan ST, et al.. High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood. 2011; 117: 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Al Malki M, Vasu S, Modi D, et al.. Preliminary analysis of a phase 1/2 study of NEXI-001 donor-derived multi-antigen-specific CD8+ T-cells for the treatment of relapsed acute myeloid leukemia (AML) after allogeneic hematopoietic cell transplantation (HCT). J Clinic Oncol. 2021; 39: 2538. [Google Scholar]
- 142. Vasileiou S, Lulla PD, Tzannou I, et al.. T-Cell therapy for lymphoma using nonengineered multiantigen-targeted T cells is safe and produces durable clinical effects. J Clin Oncol. 2021; 39: 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Smaglo BG, Musher BL, Vasileiou S, et al.. A phase I trial targeting advanced or metastatic pancreatic cancer using a combination of standard chemotherapy and adoptively transferred nonengineered, multiantigen specific T cells in the first-line setting (TACTOPS). J Clinic Oncol. 2020; 38: 4622–4622. [Google Scholar]
- 144. Hoyos V, Vasileiou S, Kuvalekar M, et al.. Multi-antigen-targeted T-cell therapy to treat patients with relapsed/refractory breast cancer. Ther Adv Med Oncol. 2022; 14: 17588359221107113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Lulla PD, Naik S, Vasileiou S, et al.. Clinical effects of administering leukemia-specific donor T cells to patients with AML/MDS after allogeneic transplant. Blood. 2021; 137: 2585–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kinoshita H, Cooke KR, Grant M, et al.. Outcome of donor-derived TAA-T cell therapy in patients with high-risk or relapsed acute leukemia post allogeneic BMT. Blood Adv. 2022; 6: 2520–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Grant M, Fortiz MF, Wang L, et al.. EPCT-15. The remind trial: multi-antigen targeted t cells for pediatric CNS tumors. Neuro Oncol. 2020; 22: iii306. [Google Scholar]
- 148. Pollack SM, Li Y, Blaisdell MJ, et al.. NYESO-1/LAGE-1s and PRAME are targets for antigen specific T cells in chondrosarcoma following treatment with 5-Aza-2-deoxycitabine. PLoS One. 2012; 7: e32165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Yan M, Himoudi N, Basu BP, et al.. Increased PRAME antigen-specific killing of malignant cell lines by low avidity CTL clones, following treatment with 5-Aza-2'-Deoxycytidine. Cancer Immunol Immunother. 2011; 60: 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Karahoca M, Momparler RL.. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin Epigenetics. 2013; 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Orlando D, Miele E, De Angelis B, et al.. Adoptive immunotherapy using PRAME-specific T cells in medulloblastoma. Cancer Res. 2018; 78: 3337–3349. [DOI] [PubMed] [Google Scholar]
- 152. Sailer N, Fetzer I, Salvermoser M, et al.. T-cells expressing a highly potent PRAME-specific T-cell receptor in combination with a chimeric PD1-41BB co-stimulatory receptor show a favorable preclinical safety profile and strong anti-tumor reactivity. Cancers (Basel). 2022; 14: 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Medigene. Medigene presents final phase I data of TCR-T cell therapy MDG1011 in patients with high-risk blood cancers. Available at: https://medigene.com/medigene-presents-final-phase-i-data-of-tcr-t-cell-therapy-mdg1011-in-patients-with-high-risk-blood-cancers/; 2023.
- 154. Immatics. Immatics reports interim clinical data from ongoing phase 1b cohort a monotherapy with ACTengine IMA203 TCR-T targeting PRAME. Available at: https://investors.immatics.com/news-releases/news-release-details/immatics-reports-interim-clinical-data-ongoing-phase-1b-cohort/; 2023.
- 155. Immunocore. Phase 1 dose escalation of IMC-F106C, the first PRAME × CD3 ImmTAC bispecific protein in solid tumors. Available at: https://ir.immunocore.com/news-releases/news-release-details/immunocore-presents-promising-initial-phase-1-data-first-shelf; 2022.
- 156. Chang AY, Dao T, Gejman RS, et al.. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest. 2017; 127: 2705–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kirkey DC, Loeb AM, Castro S, et al.. Therapeutic targeting of PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. 2023; 7: 1178–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Speicher MR, Prescher G, du Manoir S, et al.. Chromosomal gains and losses in uveal melanomas detected by comparative genomic hybridization. Cancer Res. 1994; 54: 3817–3823. [PubMed] [Google Scholar]
- 159. Anbunathan H, Verstraten R, Singh AD, Harbour JW, Bowcock AM.. Integrative copy number analysis of uveal melanoma reveals novel candidate genes involved in tumorigenesis including a tumor suppressor role for PHF10/BAF45a. Clin Cancer Res. 2019; 25: 5156–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Royds JA, Sharrard RM, Parsons MA, et al.. C-myc oncogene expression in ocular melanomas. Graefes Arch Clin Exp Ophthalmol. 1992; 230: 366–371. [DOI] [PubMed] [Google Scholar]
- 161. Mooy CM, Luyten GP, de Jong PT, et al.. Immunohistochemical and prognostic analysis of apoptosis and proliferation in uveal melanoma. Am J Pathol. 1995; 147: 1097–1104. [PMC free article] [PubMed] [Google Scholar]
- 162. Chana JS, Cree IA, Foss AJ, Hungerford JL, Wilson GD.. The prognostic significance of c-myc oncogene expression in uveal melanoma. Melanoma Res. 1998; 8: 139–144. [DOI] [PubMed] [Google Scholar]
- 163. Chana JS, Wilson GD, Cree IA, et al.. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. Br J Ophthalmol. 1999; 83: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Parrella P, Caballero OL, Sidransky D, Merbs SL.. Detection of c-myc amplification in uveal melanoma by fluorescent in situ hybridization. Invest Ophthalmol Vis Sci. 2001; 42: 1679–1684. [PubMed] [Google Scholar]
- 165. Kim YJ, Park SJ, Maeng KJ, Lee SC, Lee CS.. Multi-platform omics analysis for identification of molecular characteristics and therapeutic targets of uveal melanoma. Sci Rep. 2019; 9: 19235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ehlers JP, Worley L, Onken MD, Harbour JW.. DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clin Cancer Res. 2005; 11: 3609–3613. [DOI] [PubMed] [Google Scholar]
- 167. Naylor TL, Greshock J, Wang Y, et al.. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005; 7: R1186–R1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Okamoto H, Yasui K, Zhao C, Arii S, Inazawa J.. PTK2 and EIF3S3 genes may be amplification targets at 8q23-q24 and are associated with large hepatocellular carcinomas. Hepatology. 2003; 38: 1242–1249. [DOI] [PubMed] [Google Scholar]
- 169. van Essen TH, Bronkhorst IH, Maat W, et al.. A comparison of HLA genotype with inflammation in uveal melanoma. Invest Ophthalmol Vis Sci. 2012; 53: 2640–2646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.