

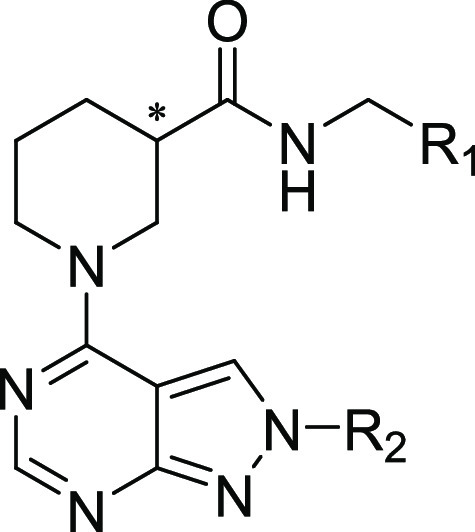
Abstract
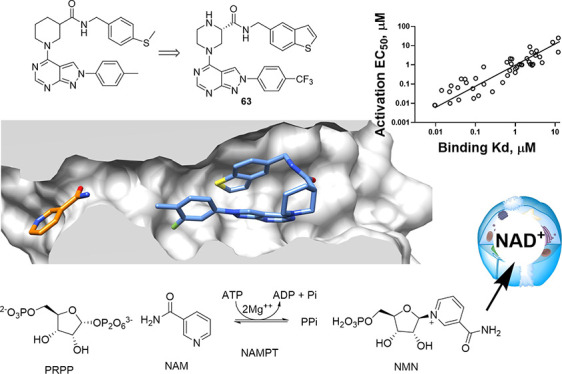
Depletion of nicotinamide adenine dinucleotide (NAD+) is associated with aging and disease, spurring the study of dietary supplements to replenish NAD+. The catabolism of NAD+ to nicotinamide (NAM) requires the salvage of NAM to replenish cellular NAD+, which relies on the rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT). Pharmacological activation of NAMPT provides an alternative to dietary supplements. Screening for activators of NAMPT identified small molecule NAMPT positive allosteric modulators (N-PAMs). N-PAMs bind to the rear channel of NAMPT increasing enzyme activity and alleviating feedback inhibition by NAM and NAD+. Synthesis of over 70 N-PAMs provided an excellent correlation between rear channel binding affinity and potency for enzyme activation, confirming the mechanism of allosteric activation via binding to the rear channel. The mechanism accounts for higher binding affinity leading to loss of efficacy. Enzyme activation translated directly to elevation of NAD+ measured in cells. Optimization led to an orally bioavailable N-PAM.
Introduction
Cellular electron transfer relies on nicotinamide adenine dinucleotide cofactors, specifically NAD+/NADH and NADP+/NADPH. In cellular metabolism and metabolic homeostasis, these coenzymes facilitate electron transfer reactions. For instance, glyceraldehyde-3-phosphate dehydrogenase is crucial for glycolysis, while NADH:ubiquinone oxidoreductase is necessary for ATP synthesis. Additionally, lipid and fatty acid biosynthesis and processing rely on enzymes that are dependent on NAD+/NADP+. Beyond the role of NAD+ as a cofactor for multiple dehydrogenases, oxidoreductases, and other enzymes, several important classes of proteins use NAD+ as a substrate. The NADase enzymic activity of these proteins leads to catabolism of NAD+ to nicotinamide (NAM). When levels of cellular NAD+ plummet, cell death can be triggered via apoptosis, oncosis, necrosis, and Wallerian neurodegeneration.1,2
The level of cellular NAD+ is regulated by the balance between biosynthesis of NAD+ and catabolism of NAD+ to NAM (Figure 1). Poly-ADP-ribose polymerases (PARPs) are enzymes that use NAD+ to add ADP-ribose to proteins. In the case of poly ADP-ribosylation, this process is called PARylation and it is a significant posttranslational modification for cell signaling. Sirtuins (SIRTs) consume NAD+ to drive protein deacylation, regulating posttranslational modification that is important in cell signaling and epigenetic regulation. Taken together, the cellular activity of PARPs and SIRTs accounts for roughly two-thirds of cellular NAD+ consumption.3 Along with these enzyme families, the cyclic-ADP-ribose hydrolase activity of the glycoprotein CD38 also consumes NAD+. In the tissues of CD38 knockout mice, NAD+ levels are increased leading to protection from obesity and metabolic syndrome. This observation highlights both the importance of CD38 in catabolism of NAD+ and the effects of modulating cellular NAD+ levels on metabolic disorders.4−6
Figure 1.
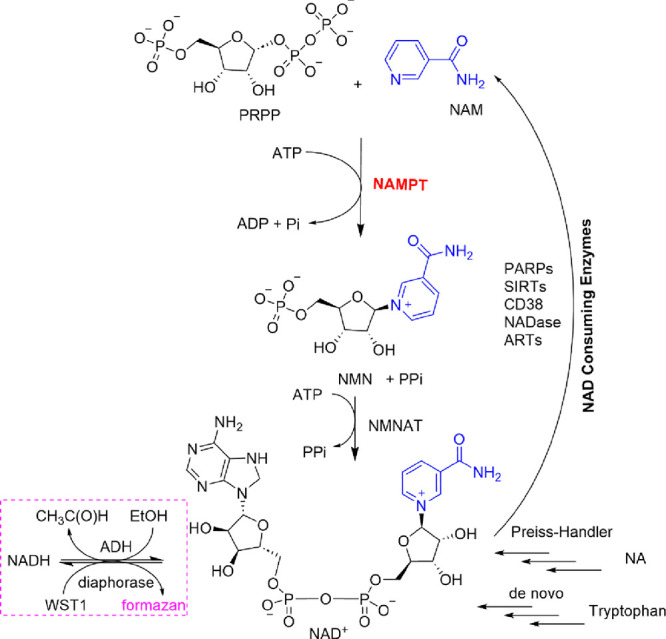
NAD+ biosynthesis, consumption, and assay. Salvage of NAM by NAMPT leads to NMNAT-mediated NAD+ biosynthesis. The Preiss-Handler pathway from nicotinic acid (NA) and de novo synthesis provide alternate biosynthetic pathways. Enzymes that catabolize NAD+ to NAM include poly-ADP-ribose polymerase (PARPs), sirtuins (SIRTs), CD38, and other proteins with NAD glycohydrolase (NADase) and ADP ribosyl transferase (ART) activity. Coupling of ADH/diaphorase to NAMPT/NMNAT1 was used to measure NAMPT activity for HTS (pink box).
PARPs, SIRTs, and CD38 all play a significant role in the physiology and pathophysiology of aging and disease, and there is a significant interplay between their signaling pathways in cells. NAD+ depletion leads to reduced SIRT1-mediated deacetylation of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), the master regulator of mitochondrial biogenesis. Reduced SIRT activity leads to defective mitochondria, release of reactive oxygen species, and DNA damage.5 PARPs play a significant role in repairing DNA damage by engaging in PARylation of target proteins. This process leads to the further consumption of NAD+.7 Chronic inflammation associated with aging and cellular senescence is linked to the observed depletion of NAD+. Inflammation is increased by CD38 expression and increased SIRT3 activity.4,8 Although upregulation of SIRTs, in response to cellular stress, can provide cellular protection, SIRTs themselves catabolize NAD+. Other enzymes with ADP-ribosyltransferases (ART) activity include sterile alpha and TIR motif containing 1 (SARM1) that, when activated by low NAD+ levels, mediates a precipitous depletion of NAD+ in neurons, causing axonal degeneration.9
Having an adequate supply of cellular NAD+ is essential for the proper functioning of PARPs, SIRTs, and CD38. It also helps inhibit the NADase activity of SARM1 and axonal degeneration. The cellular production of NAD+ is regulated by three pathways: de novo biosynthesis, the Preiss-Handler pathway that uses nicotinic acid, and the salvage pathway (Figure 1). In mammals, the salvage of NAM by NAMPT is the dominant pathway outside of the liver. The rate-determining step in the salvage pathway is mediated by the enzyme nicotinamide phosphoribosyltransferase (NAMPT) that catalyzes the ATP-dependent production of nicotinamide mononucleotide (NMN) from the reaction of NAM with α--5-phosphoribosyl-1-pyrophosphate (PRPP).
In cancer therapy, extensive efforts have been made to block synthesis of NAD+ by inhibiting NAMPT.10 Many NAMPT inhibitors were discovered in screening for small molecules that induce cell death. A more targeted rationale for NAMPT inhibitors in cancer therapy is based upon observations that the increased metabolic demand for NAD+ in cancer can lead to cellular addiction to the salvage pathway.11 To date, NAMPT inhibitors, such as FK866, have not progressed through clinical trials. The interest in design and development of NAMPT inhibitors has led to publication of multiple NAMPT structures, including cocrystal structures with the prototypical NAMPT inhibitor, FK866.10,12 These will be important in understanding small molecules that activate NAMPT.
The depletion of NAD+ in metabolic disorders and diseases of aging requires a diametric pharmacological approach. In this context, it is the replenishment or supplementation of NAD+ and its precursors that is potentially of therapeutic benefit.13 Given the evidence for disruption of cellular NAD+ supply and demand in both normal and accelerated aging, many NAD+ supplementation efforts have been targeted at enhanced lifespan and healthspan.7,14,15 These antiaging strategies have included clinical trials on dietary supplements, which have garnered media attention.16,17 Treatments with the NAD+ precursors, NAM,18 nicotinamide riboside (NR),19 or NMN,13 were shown to improve aspects of healthspan or to increase lifespan in mice.
Under pathophysiological conditions, both increased NAD+ catabolism and reduced NAMPT enzyme activity may contribute to NAD+ depletion.20 This consequently lowers levels of cellular NAD+. An alternative to dietary supplements in replenishing cellular NAD+ is to increase the activity of NAMPT, the rate-determining enzyme in the salvage pathway, to increase turnover of NAM to NMN. In drug discovery, designing small molecules that activate enzymes is inherently more challenging than designing enzyme inhibitors. FK866 and all reported potent NAMPT inhibitors simply block the binding of NAM at the nucleobase pocket of the NAMPT active site, by competing for this site. In the absence of a clear design blueprint, enzyme activators are often discovered by a medium or high-throughput screening (HTS).
The carbazole, P7C3-A20, was discovered using a phenotypic screen of a small focused library for neuroprotective agents and was claimed to be a NAMPT activator.21 In biochemical assays with recombinant NAMPT, P7C3-A20 does not cause increased enzyme activity. This has been demonstrated both in recent reports describing another class of NAMPT activators, typified by SBI-797812, and in our own mechanistic studies of the compounds described herein.22,23 Under most conditions, SBI-797812 increases the enzyme activity of recombinant NAMPT. SBI-797812 and related compounds have been described as NAMPT “boosters”.24 Both P7C3-A20 and SBI-797812 have shown activity in animal models. Most importantly, the effects of SBI-797812 have been quantified, measuring levels of NAD+ (and its metabolites and precursors) in cell cultures22 and measuring increased NAD+ levels in cell cultures and mouse liver tissues.25 These and other synthetic activators are compared in more detail below.
Theoretically, an N-PAM or other NAMPT activator will have therapeutic utility in disease states where NAD+ is depleted and in which NMN and NR supplementation have shown efficacy. The list of disease states is long because human cellular physiology is ubiquitously dependent on NAD+ being at the right concentration, in the right place, at the right time.26 Many relevant diseases have a foundation of metabolic dysfunction, including neurodegeneration,5,27 blindness,28 liver disease,20,27,29 and Type-2 diabetes (T2D) and its complications.20,27,30 NMN and NR are metabolized to NAM in the liver3 suggesting that their effects in peripheral tissues are dominated by salvage of NAM. Translation of preclinical studies on NMN and NR to the clinic have been underwhelming.29 Pharmacological enhancement of NAMPT activity represents an untapped approach to unlocking the therapeutic benefits of increased cellular NAD+ flux.
In cell-free systems, increased enzyme activity can result from increased kcat or decreased substrate Km, leading to an increase in kcat/Km. Alternatively, relief of enzyme inhibition may lead to increased substrate turnover either dependent or independent of kcat/Km. Metabolic enzymes are often subject to inhibition by substrates and products. The enzymes that mediate NAD+ biosynthetic and catabolic pathways have extensive feedback inhibition mechanisms, presumably to exert tight control over cellular NAD+ levels. NAMPT enzyme activity is dependent on the concentrations of substrates (ATP, PRPP, NAM) and products. NAMPT is inhibited by NAM, NAD+, NADH, and at higher concentrations by ATP. Put simply, allosteric relief of this inhibition can lead to increased NAM turnover to NMN. We recently proposed a mechanism of allosteric activation of NAMPT by novel NAMPT positive allosteric modulators (N-PAM).23 Herein, we report the optimization of N-PAMs that increase the enzyme activity of NAMPT up to 5-fold. Structure–activity relationships were obtained from over 70 compounds extending our mechanistic understanding of the allosteric activation mechanism. This mechanism is also supported by novel cocrystal structures. Furthermore, N-PAM biochemical activity translates into elevation of NAD+ in cell cultures.
Results and Discussion
Screening for Small Molecules That Increase NAMPT Activity
We adapted a NAMPT enzyme activity assay for HTS, by coupling the production of NMN, catalyzed by NAMPT, with the conversion of NMN to NAD+, catalyzed by nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT) (Figure 1). The coupled enzyme assay cycles the NAD+ product allowing the continuous assay of NAMPT activity. The assay was optimized and validated for HTS in 384-well plates. A pilot screen demonstrated low variability of signal across the 384-well plate with an acceptable Z-factor (Z′ = 0.82). Several chemical libraries were screened, including 20,000 compounds from two ChemDiv libraries (the SMART Library and PPI Library) chosen to balance the diversity of chemical scaffolds with synthetic feasibility and to exclude all PAINs compounds. In addition, 2000 compounds from the Microsource Spectrum library were screened.
To counter-screen against potential NMNAT1 inhibitors, active compounds were tested in the coupled assay without NAM, but with added NMN. Actives were further triaged to obtain validated hits by retesting repurchased or resynthesized compounds in triplicate at 8 concentrations and by use of a biophysical assay described below. To develop an orthogonal enzyme activity assay, we miniaturized and validated an assay in low-volume 384-well plates based on Zhang et al.31 This assay uses the acetophenone trapping of NMN in basic solution followed by acidification to yield a fluorescent adduct (380 nm excitation, 450 nm emission). In contrast to the coupled NAMPT assay used for HTS, in which NMN is captured by NMNAT1, NMN accumulates in the trapping assay. NAMPT is subject to feedback inhibition by substrates and products; therefore, this is a useful orthogonal assay to corroborate findings from the primary, coupled enzyme assay and was used to further examine selected N-PAMs.
Owing to inhibition of NAMPT by ATP, NAM, and NAD+, the assay conditions can strongly influence measured activity. In the study of NAMPT-boosters, such as SBI-797812, NAMPT activity was assayed in the presence of ATP (120 μM), MgCl2 (5 mM), NAM (5 μM), PRPP (6.25 μM), and with added pyrophosphatase to remove the PPi product.22,24,32 Cellular ATP concentrations are high, varying from 2 to 7 mM depending on the cell type.33 Measurements of NAM in tissues vary from double digit micromolar to at least 500 μM.34−37 Our HTS assay conditions (ATP 2.5 mM, NAM 30 μM, PRPP 40 μM, MgCl2 5 mM) reflect cellular concentrations.
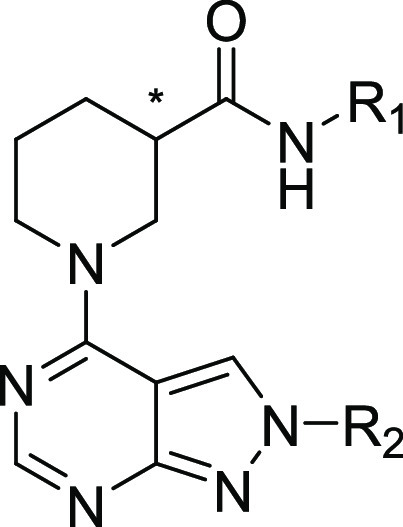
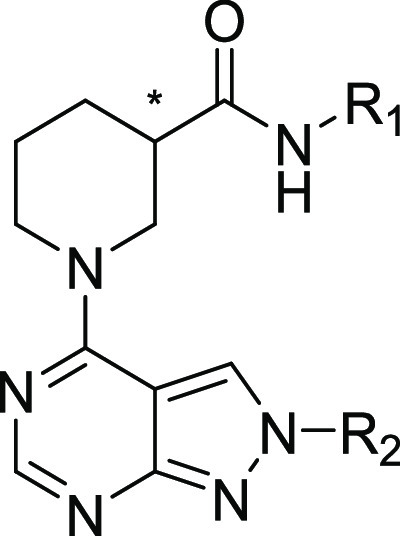
A hit chemical series, containing a 4-piperidyl-substituted pyrazolopyrimidine ring system, was identified and validated. We refer to this series as the NAMPT positive allosteric modulator (N-PAM) A1 (NP-A1) series. NAMPT activity induced by several NP-A1 series hits was measured as 1.6- to 2.6-fold greater than in the absence of the N-PAM, with potency varying from 1 to 16 μM (Table 1). Two resolved isomers (8 and 9) of the initial NP-A1 hit were synthesized, revealing a stereochemical requirement indicative of a well-defined binding site (Table 1). The R-isomer was significantly more potent, and the S-isomer showed significantly higher efficacy (Figure 2A).
Table 1. Biochemical Efficacy, Potency, and Affinity of Compounds 1–9.


Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from the FP displacement assay performed in triplicate.
Figure 2.

NAMPT enzyme activation and binding. (A) Efficacy and potency diverge for activation of NAMPT by NP-A1 isomers 8 (open circles; NP-A1R) and 9 (solid circles; NP-A1S). (B) FP-probe displacement curves showing binding affinity for N-PAMs and other putative NAMPT activators or inhibitors. Data show mean and SD from at least triplicate measurements.
Assay of Binding Affinity Informed by Cocrystal Structures
We were able to obtain NAMPT cocrystal structures relatively early in the process of hit-to-lead optimization (PDB: 8DSC, 8DSD, 8DTJ).23 There are more than 70 crystal structures of NAMPT available in the PDB database, with most of them containing inhibitors bound to human NAMPT. NAMPT inhibitors, such as the prototypical FK866, occupy both the rear channel and nucleobase pocket. The majority of inhibitors contain a nitrogenous base as a “warhead” that reaches into the nucleobase pocket to compete with the binding of substrate NAM.12 Our cocrystal structures showed that N-PAMs bind to the rear channel without occupying the nucleobase pocket.23 The knowledge that N-PAMs bind to the same site occupied by inhibitors allowed us to design and synthesize a fluorescence polarization (FP) probe (ZN-2-102), based on the structure of FK866. Displacement of ZN-2-102 in the FP assay was benchmarked with FK866, giving Kd = 10 nM (Figure 2B). The FP assay allowed measurement of the affinity of N-PAM binding to the rear channel of NAMPT.
Early Optimization of the NP-A1 Series
Initial chemical modifications of the NP-A1 pyrazolopyrimidines retained the 4-methylsulfanyl substituent of compound 9, since this compound displayed the best efficacy. It was found that some variations to the N-phenyl ring were acceptable. Nevertheless, attempts to enhance potency were hampered by a decrease in efficacy (Table 2). The enantiomeric pairs, 15/16, 17/18, and 22/23, demonstrate this phenomenon. For each pair, the R-isomer was significantly more potent, as was observed for the initial enantiomeric pair, 8/9 (Figure 2A). The superior potency of the R-isomer, in each case, was accompanied by a reduction in efficacy (Amax). Although the R-isomer achieved low nanomolar potency, the improved EC50 was accompanied by a decrease in maximal efficacy to 120% of control.
Table 2. Biochemical Efficacy, Potency, and Affinity of Compounds 10–24.

Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from the FP displacement assay performed in triplicate.
Adding a CH3 to the benzyl position was accommodated without a major loss of activity. The S-isomers, 13 and 15, compared to the parent NP-A1 pyrazolopyrimidine, 8, lost binding affinity but gained efficacy. The S-isomers, 14 and 16, also lost affinity compared to the parent NP-A1, 9, but did not gain efficacy (Tables 1 and 2). In this series, the highest efficacy was seen for compound 13, although potency was low. Substitution of the pyrazolyl-phenyl group (R2) with 4-CF3 led to efficacy of 220 and 250% (for the R- and S-isomers, respectively), and submicromolar potency was maintained.
To enhance metabolic stability and develop SAR, structural modifications were made, taking into account the potential hotspots for phase 1 metabolism in the benzyl, tolyl, and sulfanyl positions. It was expected that the sulfanyl group would undergo metabolic oxidation to sulfinyl and sulfonyl and this was clearly undesirable as the resulting derivatives (38 and 36) showed very little activity in the NAMPT enzyme assay (Table 3).
Table 3. Biochemical Efficacy, Potency, and Affinity of Compounds 25–41.

Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from the FP displacement assay performed in triplicate.
By replacing the methyl sulfanyl group with bromide, this metabolic liability can be eliminated. When comparing the isomeric bromides 25/27 with the methyl sulfanyl congeners 8/9, it was found that the efficacy and potency of the bromides were only slightly inferior. It was feasible to modify the benzyl ring with alternative substituents while maintaining NAMPT activation, except for the sulfinyl and sulfonyl derivatives, which are both bulky and polar and therefore essentially inactive (Table 3).
Structure-Based Design
The novel crystal structure of N-PAM 43 bound to NAMPT illustrates the binding mode of N-PAMs in the rear channel (Figure 3). NAMPT is active as a homodimer with two symmetrical active sites formed at the interface. The active site consists of a catalytic cavity containing the essential His-247 and other polar residues and the nucleobase pocket that is formed by a Phe-193/Tyr-18 pi-clamp that binds NAM. The third structural component is the rear channel that abuts the nucleobase pocket (Figure 3A,B).
Figure 3.

N-PAM 43 bound to NAMPT. (A) Cutaway revealing the active site that is formed by the catalytic cavity (containing bound Pi) and the nucleobase pocket (containing NAM, gold). N-PAM 43 (blue) is bound in the rear channel in a hairpin conformation. (B) View from the entrance to the rear channel showing in the foreground 43 (blue) and background NAM (gold) in the nucleobase pocket. H-Bonds between K189 and 43 are shown as a dotted yellow line, and the two NAMPT monomers are colored gray or peach. (C) Superposition of 43 (blue with residues in gray and NAM in gold) (PDB 8F7L) with FK866 (yellow with residues in cyan) bound to NAMPT (PDB 2GVJ). The 3-pyridyl group of FK866 (yellow) superposes on NAM (gold), and the cap group of FK866 superposes with 43 (blue).
The pi-clamp binding interaction in the nucleobase pocket, which is responsible for binding NAM, contributes to the high affinity of FK866 and other NAMPT inhibitors (Figure 3C). The nucleobase pocket cannot be accessed by N-PAMs, which generally results in the weaker binding affinity of N-PAMs relative to NAMPT inhibitors. While this was generally true, N-PAMs 11, 17, and 42 exhibited a binding affinity equivalent to FK866 in the FP displacement assay (Tables 1–4).
Table 4. Biochemical Efficacy, Potency, and Affinity of Compounds 42–55.

Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from the FP displacement assay performed in triplicate.
The rear channel and nucleobase pocket are primarily formed by residues of the first monomer. Tyr-18 and hydrophilic residues in the active site are formed by the second monomer. The cocrystal structure of 43 (Figure 3A) and the superposition of the structures of 43 and FK866 (Figure 3C) emphasize that the N-PAM does not interact with the nucleobase pocket (unlike FK866) and does not interact with NAM bound to the nucleobase pocket. Interestingly, the N-PAM adopts a hairpin conformation in the rear channel (Figure 3).
In human NAMPT, the β14 and β15 strands located at the dimer interface create the rear channel, a unique characteristic not found in the closely related nicotinic acid phosphoribosyltransferase (NAPRT).38 N-PAM 43 binds in the rear channel with a single H-bonding interaction with Lys-189 (Figures 3B and 4A,B). When comparing the 43:NAMPT crystal structure with other NAMPT structures, with and without inhibitors bound, there appears very little perturbation of the side chains lining the rear channel on N-PAM binding. The lysine side chain that forms an H-bond with 43 and other NP-A1 series N-PAMs is superimposable in all liganded and unliganded crystal structures (PDB: 8F7L, 8DSC, 8DSD, 8DSE, 8DSI, 8DSH, 8DTJ), despite the potential flexibility of the lysine side chain. N-PAM binding in the rear channel cavity is largely hydrophobic (Figure 4A). Intermolecular pi-stacking with Tyr-188 and intramolecular pi-stacking fill the cavity with the exclusion of water (Figure 4B). At the doorsill of the rear channel and nucleobase pocket lies an H-bonding network formed by three water molecules interacting with NAM, Ser-241, Ser-275, and Asp-219. The tolyl methyl group of 43 points toward this H-bonding network without disturbing it (Figures 3C and 4B).
Figure 4.

NAMPT residues interacting with N-PAM 43. (A) LigPlot representation of key amino acid interactions with 43 in the rear channel. (B) Key amino acid residues labeled with the nucleobase pi-clamp (raspberry), NAM (green), 43 (blue), and three water molecules at the doorsill of the rear channel (yellow dotted lines show H-bonding (PDB: 8F7L). The H-bond with K189 is also apparent.
In the cocrystal structure of 43, the C–O distance (4-Me---–OH2) is 2.6 Å; however, it is unlikely that a Me group can form stabilizing interactions with the doorsill H-bonding network. To benefit from the polar interactions with the doorsill H-bonding network, several modifications of the Me group were attempted. The phenol, 19, can theoretically form an H-bond with the doorsill water molecules; however, only weak potency and affinity were observed (Table 2). Compound 40 docks with the benzyl alcohol OH replacing the doorsill water. Although this N-PAM exhibited high potency among the tested S-isomers, the increased potency did not result in increased efficacy (Table 3).
We turned our attention to modification/replacement of the carboxamide benzyl group (Table 4). “Cyclization” of the methylsulfanyl substituent in a benzothiophene ring led to the enantiomeric pairs 42/43 and 44/45 with promising double digit nanomolar potency for the R-isomers, as seen previously. The S-isomer potency was now significantly improved over the early lead 9 (Table 2). Based upon compounds 43 and 44, a series of mostly bicyclic replacements of the benzothiophene ring were made to further expand the SAR in this region of the NP-A1 structure (Table 4). Compounds 47 and 54 gave a promising combination of efficacy and potency.
When bound to NAMPT, the N-PAM 43 adopts an unusual hairpin conformation with intramolecular pi-stacking between the benzothiophene and pyrazolopyrimidine ring planes (interplane distance ≈4.0 Å). Even though it may seem strained, molecular orbital calculations in the gas phase indicate that this hairpin structure accommodates a chair conformation for the piperidine ring and is situated in a local energy minimum (Table S2). The hairpin conformation fills the rear channel and excludes water molecules. The volume of the rear channel is calculated as approximately 490–540 Å3 (dependent on where the plane is drawn for the mouth of the channel), which is compatible with the calculated volume of 43 (525 Å3). Pyrrolidinyl analogues of the piperidyl lead compounds were synthesized and tested yielding active compounds without any overall improvement in potency, affinity, or efficacy (Table 5). Removing the conformational lock (65) and even increasing the ring size (66) led to good potency and affinity, without improvement in efficacy, and moreover, these and the pyrrolidinyl compounds did not result in increases in cellular NAD+ that were quantitatively superior to earlier compounds tested (Table S1). Finally, several modifications and substitutions of the pyrrolo moiety of the pyrrolopyrimidine group were attempted (67–77), although most such derivatives were only tested in cells, the thienopyrimidines, 70, 71, gave an interesting balance of properties (Table 5 and Table S1).
Table 5. Biochemical Efficacy, Potency, and Affinity of Compounds 56–79.


Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from fitting concentration-response data from the coupled enzyme assay performed in triplicate.
Means derived from the FP displacement assay performed in triplicate.
Regulation of Cellular NAD+
To assess the effect of N-PAMs on modulating NAD+ in cells, an NADglo assay was adapted and miniaturized to measure NAD+, using FK866 and NMN as negative and positive controls, respectively. We studied a number of cell lines to identify a highly reproducible model system with an excellent dynamic range. The most robust cell system for measuring NAD+ levels was found to be THP-1 human leukemic monocyte cell cultures, incubating test compounds for 24 h. The assay was validated in both 96- and 384-well low-volume plates. All data were normalized to DMSO (100%) with FK866 (3 nM) and NMN (2 mM) used as negative and positive in-plate controls. In initial experiments, cells treated with compound 9 (10 μM) responded with an increase in cellular NAD+ of over 2-fold (Figure 5A), whereas the R-isomer 8 (10 μM) only increased cellular NAD+ 1.2-fold. These observations align with the efficacy observed in the enzyme activity assay (Figure 2A).
Figure 5.

Affinity, efficacy, and potency for NP-A1 series N-PAMs. (A) Concentration response for N-PAMs in THP-1 cells at 24 h. Data show mean and SD from at least triplicate incubations relative to vehicle control (100%). (B–D) Correlations of efficacy, affinity, and potency (open black circles are racemates, green dots S-isomers, and red dots R-isomers). (B) Cellular increase in NAD+ as % of DMSO control at 3.3 μM N-PAM vs biochemical maximal activity (fold over vehicle) with the best-fit line. (C) Biochemical maximal efficacy vs binding affinity. (D) Biochemical maximal efficacy vs biochemical potency for NAMPT activation. Data for correlations are taken from Tables 1–5 and Table S1.
A reasonable correlation of cellular efficacy (compounds at 3.3 μM in THP-1 cell cultures) with biochemical efficacy was observed across the synthesized NP-A1 series compounds (Figure 5B). Due to the positive correlation of biochemical/cellular assay data, as optimization progressed, some compounds were tested either in the enzyme assay (Tables 1–4) or the cellular assay (Table S1).
Correlations of Efficacy, Potency, and Affinity
The goal of concurrent optimization of potency and efficacy for NAMPT activation was elusive. Analogues prepared during optimization, such as 54, 60, 63, and 70, demonstrated positive attributes and the ability to elevate cellular NAD+ significantly (Figure 5A). However, further improvements in NAMPT binding affinity ultimately led to lower efficacy for enzyme activation. From the initial SAR development, a trend was observed with R-isomers possessing superior affinity and their enantiomeric pairs demonstrating weaker affinity accompanied by superior maximal efficacy (Tables 1 and 2). Retrospectively, with all data in hand, R-isomers did not all cluster with low efficacy, nor did all S-isomers possess higher potency for NAMPT activation or affinity (Figure 5C). A similar interpretation can be made of the response to N-PAMs in cells (Figure 5B). Correlations of maximal efficacy with either affinity (Figure 5C) or potency (Figure 5D) showed that improving affinity and potency did not translate to increased efficacy for NAMPT activation. Indeed, increased binding affinity generally led to decreased maximal efficacy with no high affinity N-PAMs delivering high efficacy (Figure 5C,D).
In contrast to correlations with efficacy (Figure 5B–D), the correlation of binding affinity to the rear channel of NAMPT with potency for NAMPT enzyme activation was excellent (Figure 6). When considered together with our cocrystal structures showing rear channel binding, this correlation definitively demonstrates that rear channel binding causes enzyme activation for NP-A1 series N-PAMs.
Figure 6.

Affinity correlates with potency for NP-A1 series N-PAMs (open black circles are racemates, green dots S-isomers, and red dots R-isomers). Data are taken from Tables 1–5.
Improvement of Physicochemical Properties and Bioavailability
The combined bioassay and structural data support a model in which N-PAMs occupy the rear channel of NAMPT to increase enzyme activity. The hydrophobic cavity of the rear channel accommodates N-PAMs with a hydrophobic surface, fitting snuggly within the NAMPT rear channel. Unfortunately, the hydrophobic surface of the NP-A1 series N-PAMs is not compatible with good aqueous solubility. Furthermore, snapshot PK experiments on compounds 9, 43, 45, and 60 using oral administration to mice were not encouraging (Table 6). To improve solubility and potentially bioavailability, we examined the hydrazides, 78, 79. We also prepared piperazine derivatives (60–64), for which salt formation was possible. Piperazines 60 and 63 yielded a promising balance of biochemical and cellular properties (Table 5 and Table S1).
Table 6. PK Data for Oral Administration in Mice (30 mg/kg)a.
| plasma
concentration ng/mL |
|||||
|---|---|---|---|---|---|
| compound | 30 min | 60 min | 120 min | 180 min | 300 min |
| 9b | 112 | 13 | |||
| 43 | 164 | 59 | 42 | 21 | |
| 45 | 99 | 92 | 57 | 47 | |
| 60 | 34 | 19 | |||
| 63·HCl | 1595 | 608 | 111 | 90 | 60 |
N = 3 per time point.
10 mg/kg p.o.
Although 9 contains potential phase 1 metabolic hot spots, potent CYP inhibition was not observed (CYP2D6 >25 μM; CYP3A4 = 7.75 μM). Furthermore, the stability of compound 63 was further improved (CYP2D6 >25 μM; CYP3A4 = 13.6 μM). Perplexingly, stabilities in human and mouse liver microsomes were equally low for both 9 (MLM t1/2 = 3.2 min; HLM t1/2 = 1.1 min) and 63 (MLM t1/2 = 2.3 min; HLM t1/2 = 4.2 min). The primary identified metabolite from 9 added two oxygens, with fragmentation indicating oxidation of both the sulfur and carboxamide benzyl carbon (M + 1 = 505 → 292), whereas for 63, the benzyl carbon was likely oxidized (M + 1 = 554 → 147, 292, 348). Removal of the alternative tolyl metabolic liability to create compound 26 did not increase stability (MLM t1/2 = 9.0 min; HLM t1/2 = 7.1 min), providing evidence that the carboxamide benzyl carbon is the site of metabolic oxidation. The hydrazides 78 and 79 were designed to overcome this metabolic liability by replacing the benzyl carbon; however, this modification led to reduced potency (Table 5).
We proceeded to measure the plasma PK of 63 administered to mice by gavage. The HCl salt of 63 gave a maximal plasma concentration of 3.2 μM and 3 h after administration (30 mg/kg p.o.); a plasma concentration of 180 nM was measured. The in vitro EC50 for 63 in cell-free (58 nM) and cell-based assays (85 nM) is supportive of using 63 as an in vivo chemical probe for NAMPT activation.
Allosteric Modulation via the Rear Channel
The N-PAMs, described in this paper, enhance the turnover of NAM to NMN catalyzed by NAMPT. With respect to NAMPT, these compounds can be classified as enzyme activators. The crystallographic data show that N-PAMs occupy the NAMPT rear channel some 20 Å distant from the catalytically important His-247. Furthermore, the structural data show that N-PAM binding does not directly displace NAM from the nucleobase pocket, nor perturb the binding of NAM in the pocket. Therefore, N-PAMs meet the definition of positive allosteric modulators.
N-PAMs bind in the rear channel displacing an FP-probe that is based on the structure of FK866 (Figure 2B). Therefore, we predict an N-PAM to displace a NAMPT inhibitor such as FK866 from the rear channel. When coadministered to cells, we anticipate a right-shift in the concentration–response curve for FK866 as the N-PAM competes for binding to NAMPT. We observed that treatment of THP-1 cells with N-PAMs (9, 43, and 63) significantly shifts the response to the NAMPT inhibitor FK866 to the right (Figure 7A).
Figure 7.

NAMPT activity and NAM dependence. (A) The inhibition of NAD+ formation in cells by NAMPT inhibitor FK866 (purple) is right-shifted by N-PAMs (3 μM; 43 (red), 63 (sky), 9 (orange)), which compete for binding to the rear channel of NAMPT. (B) Calculations of KM (250 nM) and KI (55 μM) for NAMPT in the absence of N-PAMs measured using the NAMPT orthogonal enzyme assay. (C) Comparison of NAMPT activity using the orthogonal enzyme assay for N-PAMs and putative literature NAMPT activators (all 10 μM). SBI-797812 (blue), NAT1 (green), 9 (orange), 43 (red), 63 (sky), P7C3-A20 (white), relative to vehicle (1.0). Data show mean and SD from at least triplicate readings.
Binding affinity to the NAMPT rear channel was measured for all N-PAMs using FP-probe displacement, revealing an excellent correlation with the potency for enzyme activation by these N-PAMs (Figure 6). Taken together with the structural data, the conclusion is that the activation of NAMPT by N-PAMs results from binding to the rear channel.
The highest maximal activation of NAMPT is achieved by N-PAMs with the modest potency and affinity. The observation that neither binding affinity, nor activation potency, correlated with efficacy for NAMPT activation requires explanation. As demonstrated by Schramm and co-workers, a key aspect of the mechanism of NAMPT catalysis is the lowering of the NAM KM to a submicromolar value, which increases catalytic efficiency (kcat/KM). This is achieved by sequential ATP-mediated phosphorylation of His-247 and binding of PRPP at the active site. However, at higher concentrations, NAM inhibits NAMPT, as graphically demonstrated by the U-shaped dependence of activity on NAM concentration (Figure 7B).39,40 Logically, this low affinity binding mode occurs when PRPP is not bound. In the absence of PRPP, NAM binding cannot lead to product, and therefore, low affinity binding is nonproductive. There is evidence that NAM binding in the absence of PRPP causes breakdown of the phospho-enzyme and enhanced ATP turnover.23 Therefore, low affinity NAM binding diverts reaction flux from the productive pathway to the nonproductive pathway.
If we hypothesize that binding of NAM occurs via the rear channel, the competitive binding of an N-PAM to the rear channel will inhibit nonproductive NAM binding. However, as N-PAM binding affinity increases, the N-PAM will compete with high affinity NAM binding. The ability of an N-PAM to block the productive binding of NAM, via the rear channel, leads to inhibition of the NAMPT-catalyzed turnover of NAM to NMN. This is the simplest explanation for the observation that increasing the affinity of an N-PAM does not increase turnover of NAM to NMN and, in fact, leads to a generally lower maximal activity. We recently provided further support for this mechanism.23
Comparison with Other Small Molecules That Increase NAMPT Activity
We have compared the enzyme kinetics of N-PAMs 8 and 9 with the NAMPT-booster, SBI-797812.23 In the same paper, we also communicated crystal structures of the biogenic phenol, quercitrin, bound to the NAMPT rear channel and reported the activation of NAMPT by quercitrin and a variety of biogenic/bioactive phenols. The synthetic phenol NAT1 was recently reported as a NAMPT activator.41 NAT1 was studied as a neuroprotective agent in a mouse model. The first compound claimed to be a NAMPT activator, P7C3-A20, was studied in a similar mouse model. In contrast to P7C3-A20, the NAT1 phenol does increase the enzyme activity of recombinant NAMPT (Figure 7C),23 and unlike SBI-797812 and NAT1, P7C3-A20 does not bind to the rear channel of NAMPT (Figure 2B).
In the report describing the discovery of NAT1, it was claimed that the NAMPT-booster, SBI-797812, only activated NAMPT at high NAM concentrations (>15 μM) and it was claimed that SBI-797812 was a NAMPT inhibitor at lower concentrations of NAM.42 We also observed enzyme inhibition using samples of SBI-797812 from commercial sources (Figure S1). However, the NAMPT-booster, SBI-797812, synthesized and rigorously purified in-house, is a genuine NAMPT activator (Figure 7C and Figure S1). SBI-797812 is less effective at lower NAM concentration, as were all compounds that increase NAMPT activity (Figure 7C). The mechanism we have proposed for NAMPT activation by N-PAMs is intrinsically linked to the concentration of NAM. The comparison of NAMPT activation induced by N-PAMs (9, 43, and 63), NAT1, and SBI-797812 shows activation dependent on NAM concentration (Figure 7C).22,23,41
In the original report on SBI-797812, various NAMPT activation mechanisms were proposed but without the support of a cocrystal structure defining the binding mode. In contrast, for NAT1, a cocrystal structure (at 2.2 Å resolution) was reported but with no mechanism of action provided for NAMPT activation. In our FP-probe assay, both SBI-797812 and NAT1 are shown to bind to the rear channel of NAMPT with similar affinity (Figure 2B). The phenol NAT-5r, reported as an optimized derivative of NAT1, binds to NAMPT with higher affinity (Figure 2B; Kd = 570 nM).41 The binding affinity of NAT-5r is comparable to the higher efficacy NP-A1 derivatives and suggests that the mechanism of NAMPT activation may be similar.
Paradoxically, the NAMPT-booster, SBI-797812, resembles a NAMPT inhibitor with a 4-pyridyl in place of the 3-pyridyl warhead of NAMPT inhibitors. As we previously reported, the activation mechanism of SBI-797812 is different from N-PAMs and from phenolic activators such as quercitrin and NAT1.23 The most notable difference is the dependence on ATP, where SBI-797812 loses effectiveness when ATP concentration exceeds one millimolar. In similarity with NAMPT-boosters, N-PAMs attenuate NAMPT inhibition by NAD+, which may be an important contributor to cellular regulation of NAD+.23
It has been reported that SBI-797812 (20 mg/kg i.p.), delivered to healthy, young mice, effectively increased tissue NAD+ levels in the liver, although a significant effect was not observed in the heart nor muscle tissues.22 As far as we are aware, this is the only evidence for in vivo NAD+ modulation by a small molecule that increases the activity of recombinant NAMPT. A recent study by Tumpa et al. measured NAD+, NMN, NAM, NADH, and related metabolites in cells and in healthy humans, using stable isotopologue LCMS tracers (measurements on SBI-797812 were in cell cultures).25 In cells, the NAMPT-booster increased both NAD+ production and consumption. This work also proposed that NAD+ regulation and NAMPT activation were cell compartment specific, in accord with other studies.26
Synthesis of N-PAMs and Analogs
The synthesis of N-PAMs 8, 9, 10–18, 21–55, 78, and 79 was carried out using a three-step reaction sequence (Scheme 1). The condensation of 4,6-dichloropyrimidine-5-carbaldehyde with substituted phenylhydrazines in the presence of triethylamine as a base afforded the key intermediates S1–S6,43 which were reacted with commercially available chiral nipecotic acid to give S7–S12. Coupling with benzylamines or phenylhydrazines through HATU condensation reactions led to the desired products.
Scheme 1. Reagents and Conditions: (I) Phenylhydrazines, TEA, THF, 70°C; (II) DIPEA, EtOH, 70°C; (III) HATU, DMAP, DMF.

Compounds 19 and 20 could be obtained easily by bromine hydrolysis or elimination, respectively, from compound 21 (Scheme 2). Similarly, the ester reduction of 39 with LiAlH4 afforded 40.
Scheme 2. Reagents and Conditions: (I) KOH, Pd2(dba)3, tBuXPhos, Dioxane/H2O (1:1), 95°C; (II) n-BuLi, THF −78 °C then H2O; (III) LiAlH4, THF, 0°C to rt.

For the synthesis of analogs 56–59 and 62–66, intermediates S4 and S6 were substituted by different types of amines under basic conditions to afford S13–S18 and following amide coupling reaction and/or Boc deprotection gave the desired, final products (Scheme 3). N-PAMs 60 and 61 were generated from 62 and 63, respectively, through reductive amination with formaldehyde.
Scheme 3. Reagents and Conditions: (I) EtOH, DIPEA, 70°C; (II) HATU, DMAP, DMF; (III) HCl (4M in dioxane), DCM; (IV) HCHO, NaBH3CN, MeOH.
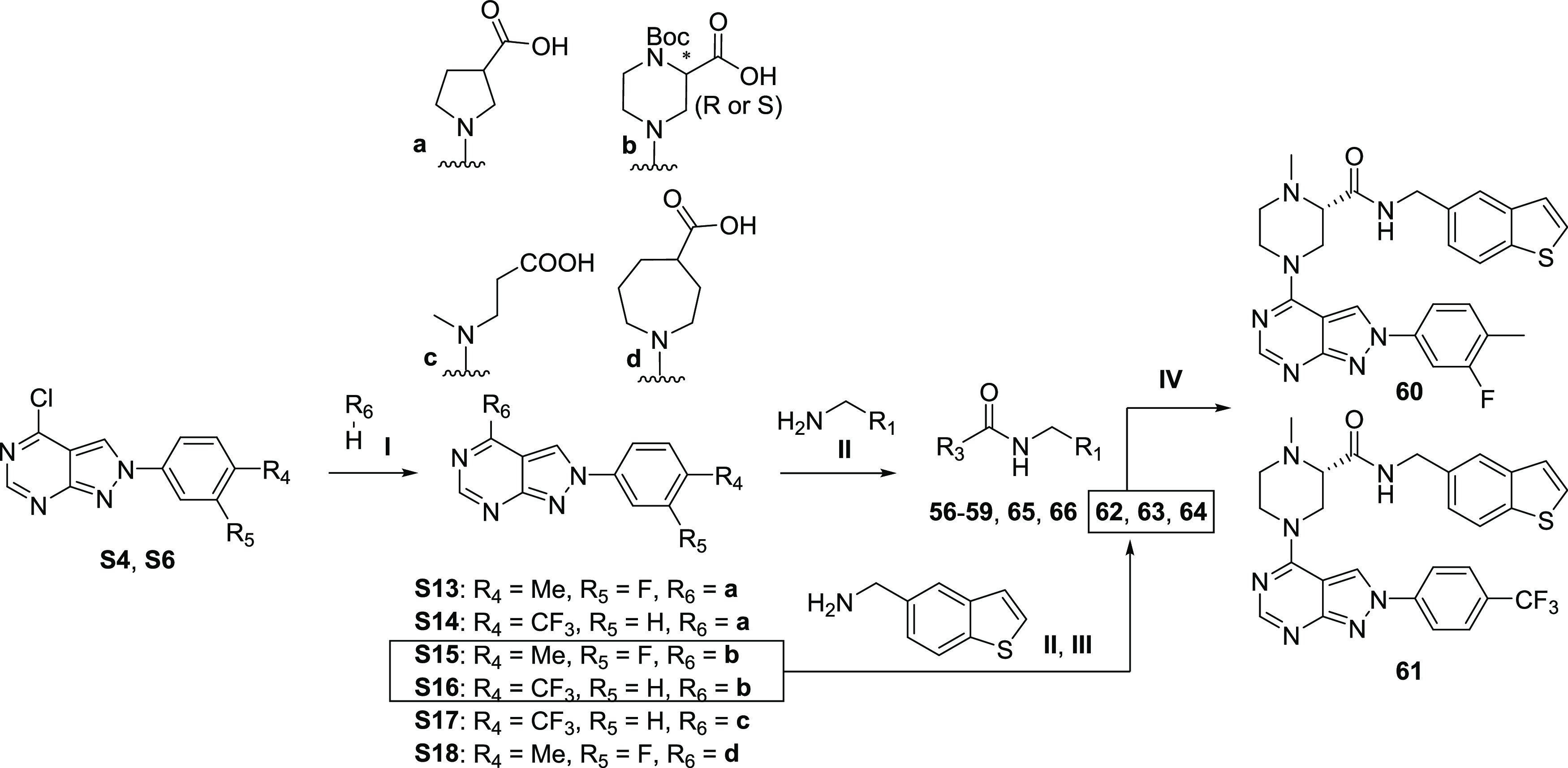
The synthesis of 70 and 71 started with the amine substitution reaction between 6-bromo-4-chlorothieno[2,3-d]pyrimidine and (S)-nipecotic acid to give S19 (Scheme 4). After condensation with (1-benzothien-5-ylmethyl)amine to afford synthon S20, Suzuki–Miyaura cross-coupling reactions with boronic pinacol ester were used to obtain the desired products 70 and 71.
Scheme 4. Reagents and Conditions: (I) EtOH, DIPEA, 70°C; (II) HATU, DMAP, DMF; (III) Pd(dppf)Cl2, Na2CO3, Dioxane/H2O, 90°C.

Conclusions
Modulation of NAMPT activity and control of cellular NMN and NAD+ levels in health, aging, and disease is a subject of intensive current interest in medicine and therapeutic development.20,27 A recent report highlighted the benefits of increasing NAMPT activity in heart tissues, in a model of diabetic cardiomyopathy.30 In prediabetic postmenopausal women, administration of NMN for 10 weeks was shown to restore muscle insulin sensitivity and signaling.44 These reports encourage the pursuit of NAMPT positive allosteric modulators (N-PAMs) as pharmacological probes and therapeutics.
We identified and developed the novel NP-A1 series of small molecule N-PAMs. These N-PAMs increase NAMPT enzyme activity, resulting in increased rates of NMN and NAD+ production and higher levels of NAD+ in cells. Structure-based optimization used crystal structures and extensive SAR correlations for >70 compounds based on data from both cell-free and cell-based assays. This work supports a definitive mechanism for allosteric modulation of NAMPT and a proof-of-concept for the translation of N-PAM activity from biochemical and cellular models. Lead optimization led to an N-PAM that significantly increased NAD+ levels in cells and is orally bioavailable in mice. Collectively, N-PAMs and small molecule NAMPT activators represent a new therapeutic approach to enhance cellular NAD+ production with multiple potential therapeutic applications. Furthermore, given the differences observed between the mechanisms of N-PAMs and NAMPT-boosters, it is likely that these different classes will yield different phenotypes in vivo. Further work is in progress to characterize the in vivo modulation of NAD+ by N-PAMs including the NP-A1 series as a prelude to testing in models of T2D.
Experimental Section
Cell Lines and Reagents
The human monocytic leukemia cell line (THP-1) was obtained from the American Type Culture Collection, and cells were maintained in the RPMI 1640 medium (ATCC) supplemented with 100 units of penicillin, 100 μg/mL streptomycin, and 10% heat inactive fetal bovine serum. All cells were grown at 37 °C, under 5% CO2 in a humidified incubator. Low passage THP-1 cells (37,500 cells/well) were seeded in 96-well plates and incubated at 37 °C and 5% CO2 for 1 1/2 h prior to a 24 h treatment.
Cellular NAD+ Assay
Using FK866 and NMN as negative and positive controls, respectively, we explored a number of cell lines to select a highly reproducible model system with good dynamic range. We optimized a commercial NADglo assay in the THP-1 human leukemic monocyte cell line in 96-well plates, measuring NAD+ after incubation with test compounds for 24 h. All compounds were dissolved in DMSO, and final DMSO concentrations never exceeded 1%. The NAD+ levels in the cells are measured using the NAD/NADH-Glo assay (Promega). The assays were performed in triplicate for each concentration, and the IC50 values were determined from nonlinear regression analysis of the dose–response curve generated in GraphPad Prism 9.
Biochemicals and Reagents
pET21a vector with human wild-type NAMPT in frame with a C-terminal His6-tag was obtained from Genscript. NAMPT protein expression was induced using an autoinduction medium described.45 Cells were grown for 24h at 20 °C, harvested by centrifugation, and lysed by sonication in Buffer A (20 mM Tris-HCl, pH 8, 0.5 M NaCl, 8 mM imidazole) containing Roche EDTA-free protease inhibitor tablets, 10 ug/mL DNaseI, and 10 ug/mL lysozyme. The lysate was clarified by centrifugation and loaded onto a 5 mL Talon cobalt column equilibrated with Buffer A. The NAMPT protein was eluted with Buffer B (20 mM Tris-HCl, pH 8.0, 0.5 M NaCl, 50 mM imidazole). Fractions containing protein were pooled, concentrated, and exchanged into 20 mM Tris-HCl, pH 8, 150 mM NaCl, 5 mM DTT and used directly for crystallization or flash frozen and stored at −80 °C.
NAMPT Coupled Enzyme Activity Assay
The NAMPT primary enzyme assay is based on conditions from Burgos and Schramm39 and adapted to include a cycling reaction to quantitate NAD+ production colorimetrically. The assay follows the NAMPT-catalyzed production of NMN from substrates NAM and PRPP by coupling NMN formation to the NMNAT1 reaction, which produces NAD+ from NMN and ATP. The NAD+ is then cycled by alcohol dehydrogenase (ADH) and diaphorase to continuously produce WST-1 formazan, which can be detected at 450 nm. The assay was optimized and validated for HTS in 384-well plates, monitoring reaction progress and normalizing the slopes to baseline (1.0-fold or 100%: NAMPT in the absence of activator in 32 control wells/plate) and NAMPT inhibitor control (zero: NAMPT with 20 nM FK866 in 32 control wells/plate). Pilot screen validation demonstrated low variability of signal across the 384-well plate with an acceptable Z-factor (Z′ = 0.82). Assays are performed at 25 °C in clear 384-well plates, with a final assay volume of 30 uL, and contain the following: 50 mM HEPES, pH 7.5, 5 mM MgCl2, 50 mM NaCl, 0.01% Triton-X 100, 2.5 mM ATP, 40 μM PRPP, 30 μM NAM, 1.5 uL of WST-1 (Roche Cell Proliferation Reagent), 1U/mL ADH, 0.083 U/mL diaphorase, 1.5% ethanol, 1% DMSO, 30 nM NAMPT, and 7.4 nM purified human NMNAT1. All assay reagents were acquired from Sigma-Aldrich unless otherwise specified. N-Terminal His6-NMNAT1 was overexpressed and purified as detailed for NAMPT from an expression vector obtained from Genscript. Following assay assembly, well signals were measured continuously at 450 nm on a Tecan Infinite M200 plate reader for 1 h with intermittent shaking. Slopes of the linear portions of the reaction progress curves were recorded and corrected for background by subtracting the average slope of control wells containing NAMPT inhibitor FK866.
NAMPT Cycling Assay for HTS
The continuous primary assay described above was used for high-throughput screening of compound libraries. Pilot experiments demonstrated low variability of the signal across the 384-well plate with an excellent Z-factor (0.82). Using this HTS assay, we screened: (1) a 10,000 compound subset of the ChemDiv SMART Library and a 10,000 compound subset of the ChemDiv PPI Library, both chosen to balance the diversity of chemical scaffolds with favorable characteristics for future derivatization and to remove PAINs (pan-assay interference compounds; including oxidants, electrophiles, and undesirable functional groups), in addition to (2) 2000 compounds from the Microsource Spectrum library. Inhibition of NMNAT1 by active compounds was ruled out by testing in the same assay with NAM replaced by NMN. Actives were further triaged to obtain validated hits by retesting repurchased or resynthesized compounds in triplicate at 8 concentrations and by use of orthogonal assays described below.
Orthogonal NAMPT Assay: NMN Trapping
The method of Zhang et al.31 was modified and miniaturized to establish an orthogonal assay for NAMPT activity. The assay uses the trapping of NMN by acetophenone in basic solution followed by acidification to form a fluorescent adduct (380 nm excitation, 450 nm emission). Reactions were performed at RT for 60 min in 50 mM Hepes (pH 7.5), 5 mM MgCl2, 50 mM NaCl, 30 nM NAMPT, 2.5 mM ATP, 30 μM NAM, 40 μM PRPP, and test compound in 1% DMSO/buffer. After 60 min, to the reaction were added 6.7 μL of 20% acetophenone in DMSO and 6.7 μL of 2 M KOH and then incubated on ice for 10 min. The reaction is quenched with 29 μL of 88% formic acid, incubated for 15 min at 37 °C, and read immediately. Fluorescence was measured with a Clariostar Reader (excitation wavelength, 380 nm; emission wavelength, 450 nm). The NAMPT inhibitor FK866 was used to validate the assay. This is an end point assay in which NMN accumulates, in contrast to the primary NAMPT assay in which NMN is immediately captured by NMNAT. Since NAMPT is subject to feedback inhibition by substrates and products (including NMN and NAD+), the orthogonal assay is not expected to yield identical data to the primary assay.
NAMPT Fluorescence Polarization (FP) Displacement Assay
All FP measurements were performed at room temperature in PBS buffer containing 0.01% Triton-X 100 and 1% DMSO in black 384-well plates. Measurements were performed on a Tecan F200 Pro plate reader fitted with polarized 485(20) nm emission filters and 535(25) nm emission filters. Initial titration of 20 nM FP-probe with varying concentrations of NAMPT protein was performed to estimate a Kd value for the probe (∼750 nM), which defined the NAMPT concentration for all subsequent experiments. Anisotropy data from equilibrium competition binding experiments were fit to a four-parameter dose response equation in GraphPad prism. All FP experiments included the following control sets: unbound probe (probe alone), bound probe (probe + NAMPT), and background signal (NAMPT alone).
Protein Crystallography, Data Collection, and Structure Refinement
Crystals of NAMPT complexes were grown by hanging drop vapor diffusion at 16 °C. Prior to crystallization, 11 mg/mL NAMPT protein was incubated with excess molar concentrations of ligand for 30 min on ice. Crystals of the complexes were grown by mixing 1–2 uL of NAMPT complex with 2 uL of reservoir solution containing 0.1 M Tris-HCl, pH 8, 0.1 M KCl, and 24–28% PEG 2000 MME or 0.1 M Tris-HCl, pH 8.5, 0.2 M NaCl, 20% glycerol, and 13–18% PEG 3350. Crystals grew overnight from the PEG 2000 MME conditions and were used to seed drops of NAMPT complex equilibrating against the PEG 3350 conditions. Further details are provided in the Supporting Information.
Animals
All experiments follow the Institutional Animal Care and Use Committee protocols of the University of Arizona. Pharmacokinetic analysis was performed by Pharmaron US Inc. Waltham, MA. Male C57BL/6 mice (20–40 g, Charles River Laboratories) were randomly divided into groups (n = 3) to receive the test compounds by oral gavage (p.o.). The vehicle for 63 was 40% PEG400 and 60% of 20% HP-β-CD in water (v/v).
Chemistry Methods
Unless otherwise specified, reactions were performed under an inert atmosphere of argon and monitored by thin-layer chromatography (TLC) and/or LCMS. All reagents were purchased from commercial suppliers and used as provided. Synthetic intermediates were purified using CombiFlash chromatography system on 230–400 mesh silica gel. 1H and 13C NMR spectra were obtained using a Bruker DPX-400 or AVANCE-400 spectrometer at 400 and 100 MHz, respectively. NMR chemical shifts were described in δ (ppm) using residual solvent peaks as a standard (chloroform-d, 7.26 ppm (1H), 77.16 ppm (13C); methanol-d4, 3.31 ppm (1H), 49.00 ppm (13C); DMSO-d6, 2.50 ppm (1H), 39.52 ppm (13C); acetone-d6, 2.05 ppm (1H), 29.84 ppm (13C)). Data were reported in a format as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, br = broad, m = multiplet, abq = ab quartet), number of protons, and coupling constants. High-resolution mass spectral data were measured in-house using a Shimadzu IT-TOF LC/MS for all final compounds. All compounds submitted for biological testing were confirmed to be ≥95% pure by analytical HPLC. Synthetic methods, spectral data, and HRMS for novel compounds are described in detail below.
4-Chloro-2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidine (S1)
To a round-bottom flask were added 4,6-dichloropyrimidine-5-carboxaldehyde (500 mg, 2.83 mmol), p-tolylhydrazine hydrochloride (449 mg, 2.83 mmol), TEA (1.2 mL, 8.49 mmol), and THF (20 mL). The reaction was allowed to stir, under a nitrogen atmosphere, at 70 °C for 1 h. After removal of solvent, the residue was purified by silica gel column chromatography (hexene/EtOAc, 2:1) to provide the 4-chloro-2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidine (291 mg, 42%) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.86 (s, 1H), 8.33 (s, 1H), 8.02 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 2.44 (s, 3H); LRMS (ESI) calcd for C12H10ClN4 [M + H]+ 245.1, found 245.0.
4-Chloro-2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidine (S4)
Using a similar procedure to the preparation of S1. Light yellow solid (yield 32%): 1H NMR (400 MHz, chloroform-d) δ 8.90 (s, 1H), 8.59 (s, 1H), 7.71 (dd, J = 10.0, 2.3 Hz, 1H), 7.65 (dd, J = 8.2, 2.4 Hz, 1H), 7.43–7.36 (m, 1H), 2.38 (d, J = 2.0 Hz, 3H); LRMS (ESI) calcd for C12H9ClFN4 [M + H]+ 263.1, found 263.0.
2-(4-Bromophenyl)-4-chloro-2H-pyrazolo[3,4-d]pyrimidine (S5)
Using a similar procedure to the preparation of S1. Light brown solid (yield 27%): 1H NMR (400 MHz, chloroform-d) δ 8.91 (s, 1H), 8.62 (s, 1H), 7.90–7.84 (m, 2H), 7.77–7.71 (m, 2H); LRMS (ESI) calcd for C11H7BrClN4 [M + H]+ 309.0, found 308.9.
4-Chloro-2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidine (S6)
Using a similar procedure to the preparation of S1. Light brown solid (yield 23%): 1H NMR (400 MHz, chloroform-d) δ 8.93 (s, 1H), 8.70 (s, 1H), 8.14 (d, J = 8.5 Hz, 2H), 7.87 (d, J = 8.5 Hz, 2H); LRMS (ESI) calcd for C12H7ClF3N4 [M + H]+ 299.0, found 299.1.
1-(2-(p-Tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxylic Acid (S7)
To a solution of S1 (50 mg, 0.20 mmol) and piperidine-3-carboxylic acid (40 mg, 0.31 mmol) in EtOH (2 mL), DIPEA (200 μL) was added. After stirring at 70 °C for 1 h and cooldown, the solvent was removed under vacuum. The residue was purified by Prep-HPLC to provide the desired compound S7 (59 mg, yield 87%) as a white solid: 1H NMR (500 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.27 (s, 1H), 7.98 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 5.01–4.26 (m, 2H), 3.53 (s, 2H), 2.56–2.52 (m, 1H), 2.38 (s, 3H), 2.07–1.97 (m, 1H), 1.87–1.71 (m, 2H), 1.62–1.52 (m, 1H); LRMS (ESI) calcd for C18H20N5O2 [M + H]+ 338.2, found 338.1.
1-(2-(4-Fluorophenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxylic Acid (S8)
Using a similar procedure to the preparation of S7. Yield 93%: 1H NMR (500 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.28 (s, 1H), 8.14 (dd, J = 9.1, 4.6 Hz, 2H), 7.46 (t, J = 8.8 Hz, 2H), 5.00–4.89 (m, 1H), 4.31 (d, J = 12.8 Hz, 1H), 3.64–3.44 (m, 2H), 2.60–2.55 (m, 1H), 2.06–1.98 (m, 1H), 1.87–1.73 (m, 2H), 1.64–1.53 (m, 1H); LRMS (ESI) calcd for C17H17FN5O2 [M + H]+ 342.1, found 342.2.
1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxylic Acid (S10)
Using a similar procedure to the preparation of S7. Yield 90%: 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.28 (s, 1H), 7.96 (dd, J = 11.0, 2.2 Hz, 1H), 7.88 (dd, J = 8.4, 2.2 Hz, 1H), 7.50 (t, J = 8.3 Hz, 1H), 4.30 (d, J = 13.3 Hz, 2H), 3.66–3.28 (m, 2H), 2.54 (s, 1H), 2.30 (d, J = 1.8 Hz, 3H), 2.10–1.97 (m, 1H), 1.90–1.71 (m, 2H), 1.65–1.52 (m, 1H); LRMS (ESI) calcd for C18H19FN5O2 [M + H]+ 356.2, found 356.2.
1-(2-(4-Bromophenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxylic Acid (S11)
Using a similar procedure to the preparation of S7. Yield (92%): 1H NMR (400 MHz, methanol-d4) δ 9.17 (s, 1H), 8.30 (s, 1H), 8.03–7.93 (m, 2H), 7.80–7.68 (m, 2H), 4.60–4.34 (m, 2H), 3.82–3.62 (m, 2H), 2.77–2.63 (m, 1H), 2.21–2.10 (m, 1H), 2.00–1.86 (m, 2H), 1.77–1.66 (m, 1H); LRMS (ESI) calcd for C17H17BrN5O2 [M + H]+ 402.1, found 402.1.
1-(2-(4-(Trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxylic Acid (S12)
Using a similar procedure to the preparation of S7. Yield (93%): 1H NMR (400 MHz, methanol-d4) δ 9.20 (s, 1H), 8.28 (s, 1H), 8.19 (t, J = 7.5 Hz, 2H), 7.84 (d, J = 8.4 Hz, 2H), 4.51–4.14 (m, 2H), 3.84–3.58 (m, 2H), 2.79–2.63 (m, 1H), 2.23–2.11 (m, 1H), 2.00–1.86 (m, 2H), 1.78–1.64 (m, 1H); LRMS (ESI) calcd for C18H19FN5O2 [M + H]+ 392.1, found 392.1.
1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidine-3-carboxylic Acid (S13)
Using a similar procedure to the preparation of S7. Yield (93%): 1H NMR (400 MHz, methanol-d4) δ 9.04 (d, J = 23.5 Hz, 1H), 8.28 (s, 1H), 7.83–7.74 (m, 2H), 7.44 (t, J = 8.2 Hz, 1H), 4.18 (d, J = 7.1 Hz, 1H), 4.11–3.99 (m, 2H), 3.96–3.81 (m, 1H), 3.51–3.40 (m, 1H), 2.54–2.45 (m, 1H), 2.41–2.29 (m, 4H); LRMS (ESI) calcd for C17H17FN5O2 [M + H]+ 342.14, found 342.17.
1-(2-(4-(Trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidine-3-carboxylic Acid (S14)
Using a similar procedure to the preparation of S7. Yield (86%): 1H NMR (400 MHz, methanol-d4) δ 9.22–9.12 (m, 1H), 8.33–8.24 (m, 3H), 7.90 (d, J = 8.5 Hz, 2H), 4.25–4.17 (m, 1H), 4.12–4.02 (m, 2H), 3.97–3.81 (m, 1H), 2.55–2.45 (m, 1H), 2.41–2.27 (m, 2H); LRMS (ESI) calcd for C17H15F3N5O2 [M + H]+ 378.1, found 378.1.
(S)-1-(tert-Butoxycarbonyl)-4-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxylic Acid (S15)
Using a similar procedure to the preparation of S7. Yield (88%): 1H NMR (400 MHz, methanol-d4) δ 9.17 (s, 1H), 8.33 (s, 1H), 7.82–7.72 (m, 2H), 7.47–7.40 (m, 1H), 5.26–5.03 (m, 1H), 4.08–3.45 (m, 6H), 2.34 (s, 3H), 1.59–1.40 (m, 9H); LRMS (ESI) calcd for C22H26FN6O4 [M + H]+ 457.2, found 457.1.
(S)-1-(tert-Butoxycarbonyl)-4-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxylic Acid (S16)
Using a similar procedure to the preparation of S7. Yield (84%): 1H NMR (400 MHz, methanol-d4) δ 9.38–9.28 (m, 1H), 8.35 (s, 1H), 8.27 (d, J = 8.5 Hz, 2H), 7.91 (d, J = 8.5 Hz, 2H), 5.30–5.08 (m, 1H), 4.08–3.81 (m, 2H), 3.76–3.40 (m, 4H), 1.54–1.43 (m, 9H); LRMS (ESI) calcd for C22H24F3N6O4 [M + H]+ 493.2, found 493.1.
3-((2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)(methyl)amino)propanoic Acid (S17)
Using a similar procedure to the preparation of S7. Yield (87%): 1H NMR (400 MHz, methanol-d4) δ 9.05 (s, 1H), 8.30 (s, 1H), 7.81–7.75 (m, 2H), 7.47–7.41 (m, 1H), 4.15 (t, J = 7.1 Hz, 2H), 3.35 (s, 3H), 2.76 (t, J = 7.0 Hz, 2H), 2.35 (d, J = 2.0 Hz, 3H); LRMS (ESI) calcd for C16H17FN5O2 [M + H]+ 330.1, found 330.2.
1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)azepane-4-carboxylic Acid (S18)
Yield (81%): 1H NMR (400 MHz, methanol-d4) δ 8.99 (s, 1H), 8.29 (s, 1H), 7.84–7.75 (m, 2H), 7.44 (t, J = 8.2 Hz, 1H), 4.33–4.06 (m, 2H), 4.01–3.88 (m, 2H), 2.63–2.50 (m, 1H), 2.35 (d, J = 1.9 Hz, 3H), 2.29–1.95 (m, 4H), 1.90–1.67 (m, 2H); LRMS (ESI) calcd for C19H21FN5O2 [M + H]+ 370.2, found 370.2.
(R)-N-(4-(Methylthio)benzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (8)
(R)-1-(2-(p-Tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxylic acid (34 mg, 0.10 mmol), (4-(methylthio)phenyl)methanamine (30 mg, 0.2 mmol), HATU (57 mg, 0.15 mmol), and DMAP (37 mg, 0.3 mmol) were dissolved in dry DMF (1 mL) and stirred at room temperature overnight. The mixture was diluted with ethyl acetate and was then washed with saturated aq. NaHCO3, water, and brine, respectively. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by Prep-HPLC to provide the 8 (41 mg, yield 87%) as a white solid: [α]D25 = −87.8 (c 0.7, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.46 (t, J = 6.0 Hz, 1H), 8.27 (s, 1H), 7.97 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.19 (s, 4H), 5.03–4.47 (m, 2H), 4.33–4.16 (m, 2H), 3.43–3.25 (m, 2H), 2.47–2.46 (m, 1H), 2.44 (s, 3H), 2.39 (s, 3H), 2.00–1.75 (m, 3H), 1.61–1.46 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 160.9, 157.4, 155.8, 137.7, 137.1, 136.3, 136.2, 130.0, 127.9, 126.1, 123.3, 120.1, 102.3, 46.7, 45.3, 42.1, 41.5, 27.6, 24.4, 20.6, 14.9; HRMS (ESI) calcd for C26H29N6OS [M + H]+ 473.2118, found 473.2125.
(S)-N-(4-(Methylthio)benzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (9)
Using a similar procedure to the preparation of 8. White solid (yield 87%): [α]D25 = +94.5 (c 0.5, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.45 (t, J = 5.7 Hz, 1H), 8.27 (s, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.26–7.04 (m, 4H), 5.02–4.43 (m, 2H), 4.31–4.17 (m, 2H), 3.56–3.14 (m, 2H), 2.47–2.46 (m, 1H), 2.44 (s, 3H), 2.39 (s, 3H), 1.98–1.77 (m, 3H), 1.61–1.46 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 172.9, 161.3, 157.9, 156.3, 138.2, 137.6, 136.8, 136.7, 130.4, 128.4, 126.6, 123.8, 120.5, 102.7, 47.2, 46.6, 42.6, 42.0, 28.1, 24.8, 21.0, 15.41; HRMS (ESI) calcd for C26H29N6OS [M + H]+ 473.2118, found 473.2127.
(R)-N-((S)-1-(4-(Methylthio)phenyl)ethyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (13)
Using a similar procedure to the preparation of 8. White solid (yield 87%): [α]D25 = −91.2 (c 0.3, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.39 (d, J = 8.0 Hz, 1H), 8.28 (s, 1H), 8.07–7.96 (m, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.26–7.16 (m, 4H), 4.88 (p, J = 7.1 Hz, 1H), 4.62–4.41 (m, 2H), 3.27–3.02 (m, 2H), 2.47 (s, 1H), 2.44 (s, 3H), 2.39 (s, 3H), 1.93–1.67 (m, 3H), 1.58–1.45 (m, 1H), 1.32 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 171.6, 160.9, 157.4, 155.9, 141.7, 137.7, 137.1, 136.0, 130.0, 126.5, 126.1, 123.4, 120.1, 102.2, 47.2, 46.9, 46.4, 42.0, 27.7, 24.3, 22.4, 20.6, 15.0; HRMS (ESI) calcd for C27H31N6OS [M + H]+ 487.2275, found 487.2277.
(S)-N-((S)-1-(4-(Methylthio)phenyl)ethyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (14)
Using a similar procedure to the preparation of 8. White solid (yield 91%): [α]D25 = +29.1 (c 0.3, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.38 (d, J = 8.0 Hz, 1H), 8.27 (s, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.28–7.10 (m, 4H), 4.98–4.83 (m, 1H), 4.79–4.33 (m, 2H), 3.30–3.12 (m, 2H), 2.48–2.46 (m, 1H), 2.44 (s, 3H), 2.40 (s, 3H), 1.96–1.77 (m, 3H), 1.61–1.46 (m, 1H), 1.31 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 160.9, 157.5, 155.9, 141.3, 137.7, 137.1, 136.1, 130.0, 126.7, 126.1, 123.4, 120.1, 102.3, 47.1, 46.5, 45.9, 42.0, 27.6, 24.2, 22.2, 20.6, 14.9; HRMS (ESI) calcd for C27H31N6OS [M + H]+ 487.2275, found 487.2280.
(R)-N-((R)-1-(4-(Methylthio)phenyl)ethyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (15)
Using a similar procedure to the preparation of 8. White solid (yield 84%): [α]D25 = −32.0 (c 0.4, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.27 (s, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.28–7.21 (m, 2H), 7.20–7.11 (m, 2H), 4.89 (p, J = 7.1 Hz, 1H), 4.70–4.31 (m, 2H), 3.46–3.33 (m, 2H), 2.48–2.47 (m, 1H), 2.44 (s, 3H), 2.40 (s, 3H), 1.88 (d, J = 25.1 Hz, 3H), 1.53 (d, J = 12.9 Hz, 1H), 1.31 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 171.6, 160.9, 157.4, 155.9, 142.4, 137.7, 137.1, 136.1, 130.0, 126.7, 126.0, 123.4, 120.1, 102.3, 47.1, 46.9, 46.3, 42.1, 27.5, 24.6, 22.2, 20.5, 14.9; HRMS (ESI) calcd for C27H31N6OS [M + H]+ 487.2275, found 487.2280.
(S)-N-((R)-1-(4-(Methylthio)phenyl)ethyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (16)
Using a similar procedure to the preparation of 8. White solid (yield 88%): [α]D25 = +87.7 (c 0.4, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.38 (d, J = 8.0 Hz, 1H), 8.28 (s, 1H), 8.04–7.97 (m, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.29–7.16 (m, 4H), 4.88 (p, J = 7.1 Hz, 1H), 4.72–4.37 (m, 2H), 3.52–3.16 (m, 2H), 2.48–2.47 (m, 1H), 2.45 (s, 3H), 2.39 (s, 3H), 1.96–1.68 (m, 3H), 1.60–1.44 (m, 1H), 1.32 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 171.6, 160.9, 157.4, 155.9, 141.7, 137.7, 137.1, 136.0, 130.0, 126.5, 126.1, 123.4, 120.1, 102.2, 47.2, 46.6, 46.0, 42.0, 27.7, 24.3, 22.4, 20.5, 14.9; HRMS (ESI) calcd for C27H31N6OS [M + H]+ 487.2275, found 487.2280.
(R)-1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(4-(methylthio)benzyl)piperidine-3-carboxamide (17)
Using a similar procedure to the preparation of 8. Light brown solid (yield 84%): [α]D25 = −94.2 (c 1.2, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.47–8.40 (m, 1H), 8.28 (s, 1H), 7.98 (dd, J = 11.0, 2.3 Hz, 1H), 7.88 (dd, J = 8.3, 2.2 Hz, 1H), 7.51 (t, J = 8.4 Hz, 1H), 7.24–7.16 (m, 4H), 5.08–4.39 (m, 2H), 4.31–4.18 (m, 2H), 3.44–3.24 (m, 2H), 2.49–2.47 (m, 1H), 2.44 (s, 3H), 2.31 (d, J = 1.8 Hz, 3H), 1.99–1.77 (m, 3H), 1.61–1.48 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 160.9, 160.7 (d, J = 243.3 Hz), 157.4, 156.2, 138.5 (d, J = 10.4 Hz), 136.3, 136.2, 132.4 (d, J = 6.1 Hz), 127.9, 126.1, 124.1 (d, J = 17.3 Hz), 123.9, 115.7 (d, J = 3.3 Hz), 107.1 (d, J = 27.8 Hz), 102.5, 46.9, 46.4, 42.2, 41.5, 27.6, 24.2, 14.9, 13.9; HRMS (ESI) calcd for C26H28FN6OS [M + H]+ 491.2024, found 491.2039.
(S)-1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(4-(methylthio)benzyl)piperidine-3-carboxamide (18)
Using a similar procedure to the preparation of 8. Light brown solid (yield 87%): [α]D25 = +84.2 (c 0.7, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.49–8.39 (m, 1H), 8.28 (s, 1H), 7.98 (dd, J = 11.0, 2.3 Hz, 1H), 7.88 (dd, J = 8.2, 2.2 Hz, 1H), 7.50 (t, J = 8.4 Hz, 1H), 7.23–7.15 (m, 4H), 5.07–4.43 (m, 2H), 4.30–4.17 (m, 2H), 3.37 (s, 2H), 2.48–2.46 (m, 1H), 2.44 (s, 3H), 2.31 (d, J = 1.8 Hz, 3H), 2.02–1.76 (m, 3H), 1.62–1.47 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 160.9, 160.7 (d, J = 243.5 Hz), 157.4, 156.1, 138.5 (d, J = 10.2 Hz), 136.3, 136.2, 132.4 (d, J = 5.9 Hz), 127.9, 126.1, 124.1 (d, J = 17.5 Hz), 123.8, 115.7 (d, J = 2.6 Hz), 107.1 (d, J = 27.7 Hz), 102.5, 46.6, 45.9, 42.2, 41.5, 27.6, 24.3, 14.9, 13.8; HRMS (ESI) calcd for C26H28FN6OS [M + H]+ 491.2024, found 491.2032.
1-(2-(4-Bromophenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(4-(methylthio)benzyl)piperidine-3-carboxamide (21)
Using a similar procedure to the preparation of 8. White solid (yield 87%): 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 8.48–8.41 (m, 1H), 8.28 (s, 1H), 8.08 (d, J = 8.7 Hz, 2H), 7.88–7.73 (m, 2H), 7.25–7.11 (m, 4H), 5.08–4.44 (m, 2H), 4.29–4.16 (m, 2H), 3.59–3.17 (m, 2H), 2.54–2.52 (m, 1H), 2.44 (s, 3H), 2.00–1.74 (m, 3H), 1.59–1.47 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 161.0, 157.4, 156.2, 138.5, 136.3, 136.2, 132.5, 127.9, 126.1, 124.0, 122.1, 120.8, 102.6, 47.1, 46.3, 42.2, 41.5, 27.6, 24.3, 14.9; HRMS (ESI) calcd for C25H26BrN6OS [M + H]+ 537.1067, found 537.1070.
N-(4-(Methylthio)benzyl)-1-(2-phenyl-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (20)
Compound 21 (10 mg, 0.019 mmol) was dissolved in THF (1 mL) and stirred in a −78 °C dry ice acetone bath. n-BuLi (2.5 M in hexenes, 76 μL) was added slowly into the reaction system. After 30 min, the reaction was quenched with water (50 μL, 1.9 mmol). The mixture was purified by Prep-HPLC to provide the desired product 20 (4 mg, yield 46%) as a white solid: 1H NMR (400 MHz, chloroform-d) δ 8.47 (s, 1H), 8.31 (s, 1H), 7.90 (d, J = 8.0 Hz, 2H), 7.58–7.50 (m, 2H), 7.46–7.41 (m, 1H), 7.20–7.12 (m, 4H), 4.38 (qd, J = 14.6, 5.6 Hz, 2H), 4.26–4.16 (m, 3H), 3.75–3.62 (m, 1H), 2.68–2.58 (m, 1H), 2.45 (s, 3H), 2.28–2.20 (m, 1H), 2.05–1.95 (m, 1H), 1.84–1.67 (m, 2H); HRMS (ESI) calcd for C25H27N6OS [M + H]+ 459.1962, found 459.1967.
(R)-N-(4-(Methylthio)benzyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (22)
Using a similar procedure to the preparation of 8. Light brown solid (yield 84%): [α]D25 = −94.4 (c 0.7, MeOH); 1H NMR (400 MHz, chloroform-d) δ 8.57 (s, 1H), 8.32 (s, 1H), 8.06 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.5 Hz, 2H), 7.19–7.13 (m, 4H), 4.46–4.34 (m, 2H), 4.28–4.09 (m, 2H), 3.66 (d, J = 3.8 Hz, 2H), 2.67–2.59 (m, 1H), 2.45 (s, 3H), 2.28–2.15 (m, 1H), 2.08–1.98 (m, 1H), 1.91–1.67 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 161.1, 157.5, 156.5, 142.2, 136.3, 136.2, 127.9 (q, J = 49.3 Hz), 127.9, 126.9, 126.1, 124.6, 124.0 (q, J = 273.2 Hz), 120.6, 102.9, 47.0, 46.4, 42.2, 41.5, 27.6, 24.5, 14.9; HRMS (ESI) calcd for C26H26F3N6OS [M + H]+ 527.1835, found 527.1841.
(S)-N-(4-(Methylthio)benzyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (23)
Using a similar procedure to the preparation of 8. Light brown solid (yield 84%): [α]D25 = +81.7 (c 0.3, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.49 (s, 1H), 8.48–8.42 (m, 1H), 8.36 (d, J = 8.4 Hz, 2H), 8.30 (s, 1H), 7.99 (d, J = 8.5 Hz, 2H), 7.29–7.15 (m, 4H), 5.03–4.46 (m, 2H), 4.32–4.16 (m, 2H), 3.22–3.02 (m, 2H), 2.47–2.40 (m, 4H), 2.00–1.78 (m, 3H), 1.62–1.49 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 161.1, 157.5, 156.5, 142.2, 136.3, 136.2, 127.9 (q, J = 49.2 Hz), 127.9, 126.9, 126.1, 124.6, 124.0 (q, J = 272.1 Hz), 120.6, 102.9, 47.0, 46.0, 42.2, 41.5, 27.6, 24.5, 14.9; HRMS (ESI) calcd for C26H26F3N6OS [M + H]+ 527.1835, found 527.1844.
1-(2-(4-Fluorophenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(4-(methylthio)benzyl)piperidine-3-carboxamide (24)
Using a similar procedure to the preparation of 8. Light brown solid (yield 73%): 1H NMR (500 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.45 (s, 1H), 8.28 (s, 1H), 8.14 (dd, J = 9.0, 4.7 Hz, 2H), 7.46 (t, J = 8.8 Hz, 2H), 7.19 (s, 4H), 5.03–4.48 (m, 2H), 4.29–4.19 (m, 2H), 3.70–3.48 (m, 2H), 2.48–2.41 (m, 4H), 1.99–1.78 (m, 3H), 1.59–1.49 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.4, 162.4, 161.0, 160.4, 157.5, 156.0, 136.3, 135.9 (d, J = 2.7 Hz), 127.9, 126.1, 123.9, 122.4 (d, J = 8.7 Hz), 116.4 (d, J = 23.2 Hz), 102.4, 46.7, 46.4, 42.0, 41.5, 27.6, 24.3, 14.9; HRMS (ESI) calcd for C25H26FN6OS [M + H]+ 477.1867, found 477.1862.
(R)-N-(4-Bromobenzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (25)
Using a similar procedure to the preparation of 8. White solid (yield 82%): [α]D25 = −98.4 (c 0.5, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.55–8.47 (m, 1H), 8.27 (s, 1H), 8.14 (s, 1H), 8.01–7.92 (m, 2H), 7.55–7.45 (m, 1H), 7.44–7.36 (m, 2H), 7.27–7.10 (m, 2H), 5.06–4.45 (m, 2H), 4.31–4.19 (m, 2H), 3.23–3.02 (m, 2H), 2.47 (s, 1H), 2.39 (s, 3H), 1.97–1.76 (m, 3H), 1.71–1.48 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.6, 163.1, 160.9, 157.5, 155.8, 139.0, 137.7, 137.1, 131.2, 130.0, 129.4, 129.3, 123.4, 120.1, 119.8, 102.3, 47.7, 46.8, 42.2, 41.3, 27.6, 24.4, 20.6; HRMS (ESI) calcd for C25H26BrN6O [M + H]+ 505.1346, found 505.1353.
N-(4-Bromobenzyl)-1-(2-(4-bromophenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (26)
Using a similar procedure to the preparation of 8. Light brown solid (yield 83%): 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 8.54–8.49 (m, 1H), 8.28 (s, 1H), 8.08 (d, J = 8.5 Hz, 2H), 7.88–7.74 (m, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.26–7.16 (m, 2H), 5.07–4.43 (m, 2H), 4.25 (d, J = 5.9 Hz, 2H), 3.61–3.36 (m, 2H), 2.44 (s, 1H), 2.05–1.75 (m, 3H), 1.61–1.44 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.5, 161.0, 157.4, 156.2, 139.0, 138.5, 132.5, 131.1, 129.4, 124.0, 122.1, 120.8, 119.7, 102.6, 46.8, 46.4, 42.1, 41.3, 27.6, 24.3; HRMS (ESI) calcd for C24H23Br2N6O [M + H]+ 569.0295, found 569.0288.
(S)-N-(4-Bromobenzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (27)
Using a similar procedure to the preparation of 8. White solid (yield 78%): [α]D25 = +90.4 (c 0.7, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.50 (t, J = 6.0 Hz, 1H), 8.27 (s, 1H), 8.14 (s, 1H), 8.03–7.92 (m, 2H), 7.55–7.44 (m, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 5.03–4.50 (m, 2H), 4.29–4.19 (m, 2H), 3.21–3.01 (m, 2H), 2.48–2.45 (m, 1H), 2.39 (s, 3H), 1.98–1.78 (m, 3H), 1.62–1.48 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.6, 163.1, 160.9, 157.5, 155.8, 138.8, 137.7, 137.2, 131.2, 130.0, 129.4, 129.3, 123., 120.1, 119.8, 102.3, 47.8, 46.6, 42.2, 41.3, 27.6, 24.4, 20.6; HRMS (ESI) calcd for C25H26BrN6O [M + H]+ 505.1346, found 505.1353.
N-(4-Bromobenzyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (28)
Using a similar procedure to the preparation of 8. Light brown solid (yield 78%): 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.56–8.45 (m, 1H), 8.29 (s, 1H), 7.97 (dd, J = 11.0, 2.2 Hz, 1H), 7.87 (dd, J = 8.3, 2.3 Hz, 1H), 7.56–7.42 (m, 3H), 7.27–7.17 (m, 2H), 5.12–4.40 (m, 2H), 4.34–4.18 (m, 2H), 3.30–3.05 (m, 2H), 2.48 (s, 1H), 2.32 (s, 3H), 2.02–1.78 (m, 3H), 1.63–1.48 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.6, 160.7, 160.7 (d, J = 243.3 Hz), 156.7 (d, J = 135.9 Hz), 139.0, 138.5 (d, J = 10.2 Hz), 132.4 (d, J = 5.7 Hz), 131.1, 129.4, 124.1 (d, J = 17.3 Hz), 123.9, 119.8, 115.7, 107.1 (d, J = 27.8 Hz), 102.5, 47.0, 46.2, 42.1, 41.4, 27.6, 24.3, 13.9; HRMS (ESI) calcd for C25H25BrFN6O [M + H]+ 523.1252, found 523.1258.
(S)-N-(4-Bromo-2-fluorobenzyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (30)
Using a similar procedure to the preparation of 8. Light brown solid (yield 74%): [α]D25 = +86.7 (c 0.5, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.50 (t, J = 5.8 Hz, 1H), 8.27 (s, 1H), 7.96 (dd, J = 11.0, 2.2 Hz, 1H), 7.87 (dd, J = 8.3, 2.2 Hz, 1H), 7.54–7.46 (m, 2H), 7.39–7.22 (m, 2H), 5.06–4.39 (m, 2H), 4.34–4.20 (m, 2H), 3.52 (s, 2H), 2.55 (s, 1H), 2.31 (s, 3H), 1.98–1.76 (m, 3H), 1.61–1.48 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.6, 160.8, 160.7 (d, J = 243.4 Hz), 159.9 (d, J = 249.6 Hz), 157.4, 156.1, 138.5 (d, J = 10.4 Hz), 132.4 (d, J = 6.1 Hz), 131.1 (d, J = 5.0 Hz), 127.4, 125.7 (d, J = 14.7 Hz), 124.1 (d, J = 17.1 Hz), 123.8, 120.1 (d, J = 9.8 Hz), 118.4 (d, J = 25.0 Hz), 115.7, 107.1 (d, J = 28.0 Hz), 102.4, 46.6, 45.9, 42.0, 35.5, 27.6, 24.1, 13.8; HRMS (ESI) calcd for C25H24BrF2N6O [M + H]+ 541.1158, found 541.1166.
(S)-N-(4-Bromo-2,6-difluorobenzyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (31)
Using a similar procedure to the preparation of 8. Light brown solid (yield 83%): [α]D25 = +84.5 (c 0.7, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.39 (t, J = 5.3 Hz, 1H), 8.25 (s, 1H), 7.94 (d, J = 11.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.53–7.47 (m, 1H), 7.44–7.38 (m, 2H), 4.96–4.45 (m, 2H), 4.33–4.23 (m, 2H), 3.41–3.33 (m, 2H), 2.46–2.40 (m, 1H), 2.31 (s, 3H), 1.91–1.73 (m, 3H), 1.57–1.45 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.2, 163.4, 161.0 (d, J = 251.6 Hz), 160.9 (d, J = 251.8 Hz), 160.82, 160.66 (d, J = 243.3 Hz), 157.4, 156.1, 138.5 (d, J = 10.2 Hz), 132.4 (d, J = 6.0 Hz), 124.1 (d, J = 17.5 Hz), 123.7, 120.6, 115.7, 115.3 (d, J = 28.2 Hz), 114.1, 107.1 (d, J = 27.5 Hz), 102.4, 47.5, 46.4, 41.8, 30.5, 27.5, 24.3, 13.8; HRMS (ESI) calcd for C25H23BrF3N6O [M + H]+ 559.1063, found 559.1070.
N-(4-Methoxybenzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (34)
Using a similar procedure to the preparation of 8. White solid (yield 91%): 1H NMR (500 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.40 (t, J = 5.9 Hz, 1H), 8.27 (s, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.3 Hz, 2H), 7.24–7.11 (m, 2H), 6.85 (d, J = 7.8 Hz, 2H), 5.04–4.50 (m, 2H), 4.27–4.16 (m, 2H), 3.71 (s, 3H), 3.44–3.13 (m, 2H), 2.49–2.44 (m, 1H), 2.39 (s, 3H), 1.96–1.78 (m, 3H), 1.57–1.48 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.3, 160.9, 158.2, 157.4, 155.8, 137.7, 137.1, 131.4, 130.0, 128.5, 123.3, 120.1, 113.7, 102.3, 55.0, 46.6, 46.2, 42.2, 41.4, 27.6, 24.6, 20.6; HRMS (ESI) calcd for C26H29N6O2 [M + H]+ 457.2347, found 457.2341.
N-(4-Iodobenzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (35)
Using a similar procedure to the preparation of 8. Light yellow solid (yield 87%): 1H NMR (500 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.52–8.44 (m, 1H), 8.27 (s, 1H), 7.97 (d, J = 8.0 Hz, 2H), 7.69–7.60 (m, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 8.1 Hz, 2H), 5.04–4.47 (m, 2H), 4.24 (qd, J = 15.3, 5.7 Hz, 2H), 3.69–3.15 (m, 2H), 2.46 (s, 1H), 2.40 (s, 3H), 2.00–1.76 (m, 3H), 1.53 (d, J = 12.6 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 163.9, 161.4, 157.9, 156.3, 139.9, 138.2, 137.6, 137.5, 130.5, 130.1, 123.8, 120.6, 102.7, 92.9, 47.1, 46.8, 42.6, 41.9, 28.1, 24.9, 21.1; HRMS (ESI) calcd for C25H26IN6O [M + H]+ 553.1207, found 553.1201.
N-(4-(Methylsulfonyl)benzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (36)
Using a similar procedure to the preparation of 8. Light yellow solid (yield 81%): 1H NMR (500 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.61 (t, J = 6.0 Hz, 1H), 8.28 (s, 1H), 7.98 (d, J = 8.2 Hz, 2H), 7.86 (d, J = 7.9 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.2 Hz, 2H), 5.01–4.52 (m, 2H), 4.45–4.34 (m, 2H), 3.64–3.34 (m, 2H), 3.19 (s, 3H), 2.57–2.52 (m, 1H), 2.39 (s, 3H), 2.00–1.95 (m, 1H), 1.90–1.80 (m, 2H), 1.60–1.50 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.7, 163.3, 160.9, 157.5, 155.8, 139.3, 137.7, 137.1, 130.0, 127.9, 127.1, 123.4, 120.1, 102.3, 46.5, 45.9, 43.6, 42.1, 41.6, 27.6, 24.3, 20.6; HRMS (ESI) calcd for C26H29N6O3S [M + H]+ 505.2016, found 505.2010.
N-(4-Acetylbenzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (37)
Using a similar procedure to the preparation of 8. Light yellow solid (yield 67%): 1H NMR (500 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.60–8.56 (m, 1H), 8.28 (s, 1H), 7.98 (d, J = 8.1 Hz, 2H), 7.90–7.86 (m, 2H), 7.39 (dd, J = 8.4, 3.8 Hz, 4H), 5.03–4.51 (m, 2H), 4.45–4.28 (m, 2H), 3.73–3.52 (m, 1H), 3.29–3.07 (m, 1H), 2.57–2.51 (m, 4H), 2.39 (s, 3H), 1.99–1.93 (m, 1H), 1.91–1.81 (m, 2H), 1.59–1.50 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 197.5, 163.6, 160.9, 157.5, 155.9, 137.7, 137.1, 135.5, 130.0, 128.3, 128.3, 127.2, 123.4, 120.1, 102.3, 46.1, 45.3, 42.2, 41.7, 27.6, 26.7, 24.4, 20.6; HRMS (ESI) calcd for C27H29N6O2 [M + H]+ 469.2347, found 469.2345.
N-(4-(Methylsulfinyl)benzyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (38)
Using a similar procedure to the preparation of 8. Light yellow solid (yield 87%): 1H NMR (500 MHz, DMSO-d6) δ 9.29 (s, 1H), 8.56 (t, J = 6.1 Hz, 1H), 8.27 (s, 1H), 7.98 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.45 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 8.3 Hz, 2H), 5.04–4.53 (m, 2H), 4.42–4.29 (m, 2H), 3.10 (s, 2H), 2.70 (d, J = 2.3 Hz, 3H), 2.54 (s, 1H), 2.39 (s, 3H), 2.01–1.79 (m, 3H), 1.55 (d, J = 12.8 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 163.2, 160.9, 157.5, 155.9, 144.7, 142.5, 137.7, 137.1, 130.0, 127.9, 123.7, 123.4, 120.1, 102.3, 46.7, 46.1, 43.3, 42.2, 41.6, 27.6, 24.3, 20.6; HRMS (ESI) calcd for C26H29N6O2S [M + H]+ 489.2067, found 489.2062.
Methyl (S)-4-(4-(3-((4-(Methylthio)benzyl)carbamoyl)piperidin-1-yl)-2H-pyrazolo[3,4-d]pyrimidin-2-yl)benzoate (39)
Using a similar procedure to the preparation of 8. Light gray solid (yield 83%): 1H NMR (500 MHz, DMSO-d6) δ 9.45 (s, 1H), 8.46 (s, 1H), 8.32–8.24 (m, 3H), 8.14 (d, J = 8.5 Hz, 2H), 7.19 (s, 4H), 5.06–4.46 (m, 2H), 4.31–4.18 (m, 2H), 3.90 (s, 3H), 3.65–3.28 (m, 2H), 2.48–2.40 (m, 4H), 2.02–1.78 (m, 3H), 1.55 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.4, 165.5, 161.1, 157.5, 156.5, 142.6, 136.3, 130.7, 128.7, 127.9, 126.1, 124.5, 120.0, 102.9, 52.3, 47.0, 46.5, 42.0, 41.5, 27.6, 24.6, 14.9; HRMS (ESI) calcd for C27H29N6O3S [M + H]+ 517.2016, found 517.2013.
(S)-1-(2-(4-(Hydroxymethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(4-(methylthio)benzyl)piperidine-3-carboxamide (40)
Compound 39 (20 mg, 0.04 mmol) was dissolved in THF (1 mL) and stirred in the ice water bath. LiAlH4 (16 mg, 0.40 mmol) was added slowly into the reaction system. After 30 min, the water bath was removed and kept stirring at room temperature until the reaction finish. The reaction was quenched with water (100 μL, 5.5 mmol). The mixture was purified by Prep-HPLC to provide the desired product 40 (9 mg, yield 46%) as a white solid: 1H NMR (500 MHz, DMSO-d6) 1H NMR (500 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.58 (s, 1H), 8.51 (s, 1H), 8.04 (d, J = 7.8 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H), 7.25–7.15 (m, 4H), 4.97 (s, 1H), 4.60 (s, 2H), 4.47 (d, J = 12.9 Hz, 1H), 4.23 (d, J = 5.8 Hz, 2H), 3.82–3.44 (m, 2H), 2.69–2.54 (m, 1H), 2.44 (s, 3H), 2.06–1.84 (m, 3H), 1.66 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.0, 158.0, 157.8, 156.9, 151.6, 143.6, 137.2, 136.4, 127.9, 127.5, 126.1, 120.2, 101.6, 62.2, 48.0, 41.7, 41.6, 27.2, 24.3, 14.9; HRMS (ESI) calcd for C26H29N6O2S [M + H]+ 489.2067, found 489.2063.
(R)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (42)
Using a similar procedure to the preparation of 8. Light brown solid (yield 84%): [α]D25 = −90.4 (c 1.0, MeOH); 1H NMR (400 MHz, chloroform-d) δ 8.35 (s, 1H), 8.22 (s, 1H), 7.77 (d, J = 8.3 Hz, 1H), 7.67 (d, J = 1.7 Hz, 1H), 7.55 (dd, J = 10.2, 2.1 Hz, 1H), 7.49–7.42 (m, 1H), 7.42 (d, J = 5.4 Hz, 1H), 7.28–7.18 (m, 4H), 4.55 (d, J = 5.5 Hz, 2H), 4.33–4.02 (m, 3H), 3.66–3.47 (m, 1H), 2.73–2.63 (m, 1H), 2.32 (d, J = 1.9 Hz, 3H), 2.26–2.14 (m, 1H), 2.07–1.98 (m, 1H), 1.87–1.77 (m, 1H), 1.75–1.62 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.5, 160.8, 160.7 (d, J = 242.3 Hz), 157.5, 156.2, 139.6, 138.5 (d, J = 10.2 Hz), 137.7, 135.7, 132.5 (d, J = 5.8 Hz), 127.8, 124.3, 124.1, 124.0, 123.8, 122.5, 122.1, 115.8 (d, J = 2.8 Hz), 107.2 (d, J = 27.6 Hz), 102.5, 46.9, 46.2, 42.3, 42.1, 27.7, 24.3, 13.9; HRMS (ESI) calcd for C27H26FN6OS [M + H]+ 501.1867, found 501.1868.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (43)
Using a similar procedure to the preparation of 8. Light brown solid (yield 87%): [α]D25 = +82.7 (c 2.1, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.58–8.47 (m, 1H), 8.28 (s, 1H), 8.00–7.82 (m, 3H), 7.77–7.72 (m, 2H), 7.49 (t, J = 8.4 Hz, 1H), 7.43–7.37 (m, 1H), 7.27 (dd, J = 8.4, 1.6 Hz, 1H), 5.07–4.46 (m, 2H), 4.40 (d, J = 5.8 Hz, 2H), 3.32–3.09 (m, 2H), 2.55–2.52 (m, 1H), 2.31 (d, J = 1.8 Hz, 3H), 2.03–1.79 (m, 3H), 1.63–1.46 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.5, 160.8, 160.7 (d, J = 243.4 Hz), 157.5, 156.2, 139.6, 138.5 (d, J = 10.3 Hz), 137.7, 135.7, 132.5 (d, J = 5.8 Hz), 127.8, 124.3, 124.1, 124.0, 123.8, 122.5, 122.1, 115.8 (d, J = 2.8 Hz), 107.2 (d, J = 27.6 Hz), 102.5, 46.9, 46.2, 42.3, 42.1, 27.7, 24.3, 13.9; HRMS (ESI) calcd for C27H26FN6OS [M + H]+ 501.1867, found 501.1876.
(R)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (44)
Using a similar procedure to the preparation of 8. Light brown solid (yield 87%): [α]D25 = −80.4 (c 0.8, MeOH); 1H NMR (400 MHz, chloroform-d) δ 8.55 (s, 1H), 8.31 (s, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.83–7.75 (m, 3H), 7.68 (s, 1H), 7.44 (d, J = 5.5 Hz, 1H), 7.26–7.17 (m, 2H), 4.61–4.50 (m, 2H), 4.33–4.11 (m, 3H), 3.71–3.59 (m, 1H), 2.69–2.61 (m, 1H), 2.22 (s, 1H), 2.09–2.00 (m, 1H), 1.90–1.81 (m, 1H), 1.78–1.67 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 161.1, 157.5, 156.5, 142.1, 139.5, 137.6, 135.7, δ 128.1 (q, J = 33.1 Hz), 127.8, 126.9, 124.6, 124.1, 124.0 (q, J = 271.4 Hz), 123.8, 122.4, 122.1, 120.6, 102.9, 47.0, 46.4, 42.2, 42.0, 27.6, 24.7; HRMS (ESI) calcd for C27H24F3N6OS [M + H]+ 537.1679, found 537.1685.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (45)
Using a similar procedure to the preparation of 8. Light brown solid (yield 87%): [α]D25 = +83.4 (c 0.5, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.54 (t, J = 5.5 Hz, 1H), 8.34 (d, J = 8.3 Hz, 2H), 8.30 (s, 1H), 8.01–7.88 (m, 3H), 7.79–7.70 (m, 2H), 7.48–7.37 (m, 1H), 7.28 (d, J = 8.3 Hz, 1H), 5.13–4.52 (m, 2H), 4.41 (d, J = 5.7 Hz, 2H), 3.48–3.37 (m, 2H), 2.64–2.54 (m, 1H), 2.08–1.77 (m, 3H), 1.56 (d, J = 12.1 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 161.1, 157.5, 156.5, 142.1, 139.5, 137.6, 135.7, δ 128.0 (q, J = 32.5 Hz), 127.75, 126.9, 124.6, 124.1, 124.0 (q, J = 271.5 Hz), 123.8, 122.4, 122.1, 120.6, 102.9, 47.0, 46.4, 42.2, 42.0, 27.6, 24.7; HRMS (ESI) calcd for C27H24F3N6OS [M + H]+ 537.1679, found 537.1686.
(S)-N-(Benzo[b]thiophen-3-ylmethyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (46)
Using a similar procedure to the preparation of 8. Light brown solid (yield 78%): [α]D25 = +90.1 (c 0.8, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.48 (t, J = 5.7 Hz, 1H), 8.26 (s, 1H), 8.01–7.94 (m, 2H), 7.90–7.82 (m, 2H), 7.55 (s, 1H), 7.53–7.47 (m, 1H), 7.41–7.34 (m, 2H), 5.09–4.35 (m, 4H), 3.93–3.55 (m, 2H), 2.58–2.53 (m, 1H), 2.31 (s, 3H), 1.99–1.79 (m, 3H), 1.60–1.45 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.5, 160.8, 160.7 (d, J = 243.3 Hz), 157.4, 156.1, 139.9, 138.5 (d, J = 10.4 Hz), 137.7, 133.8, 132.4 (d, J = 6.0 Hz), 124.4, 124.1, 124.1, 124.0, 123.8, 122.9, 121.8, 115.7, 107.1 (d, J = 27.6 Hz), 102.4, 47.0, 46.0, 42.1, 36.4, 27.7, 24.5, 13.8; HRMS (ESI) calcd for C27H26FN6OS [M + H]+ 501.1867, found 501.1873.
(S)-1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-((5-fluorobenzo[b]thiophen-2-yl)methyl)piperidine-3-carboxamide (47)
Using a similar procedure to the preparation of 8. [α]D25 = +84.4 (c 0.4, MeOH); Light brown solid (yield 87%): 1H NMR (400 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.74 (t, J = 5.9 Hz, 1H), 8.28 (s, 1H), 8.00–7.82 (m, 3H), 7.56 (d, J = 9.9 Hz, 1H), 7.51–7.42 (m, 1H), 7.29–7.24 (m, 1H), 7.21–7.13 (m, 1H), 5.10–4.40 (m, 4H), 3.55–3.35 (m, 2H), 2.58–2.54 (m, 1H), 2.29 (d, J = 1.8 Hz, 3H), 2.01–1.80 (m, 3H), 1.62–1.50 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 172.6, 160.8, 160.7 (d, J = 243.5 Hz), 160.2 (d, J = 238.6 Hz), 157.4, 156.1, 146.9, 140.4 (d, J = 9.8 Hz), 138.4 (d, J = 10.2 Hz), 134.6, 132.4 (d, J = 5.9 Hz), 124.1, 124.0, 123.8, 121.2 (d, J = 4.2 Hz), 115.7, 112.4 (d, J = 25.0 Hz), 108.6 (d, J = 23.2 Hz), 107.1 (d, J = 27.7 Hz), 102.5, 47.0, 46.3, 42.0, 37.9, 27.5, 24.4, 13.8; HRMS (ESI) calcd for C27H25F2N6OS [M + H]+ 519.1773, found 519.1778.
(S)-N-((1H-Indol-5-yl)methyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (48)
Using a similar procedure to the preparation of 8. Light brown solid (yield 87%): [α]D25 = +91.1 (c 0.5, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 9.34 (s, 1H), 8.45–8.36 (m, 1H), 8.27 (s, 1H), 7.98 (dd, J = 11.0, 2.2 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.51–7.44 (m, 1H), 7.43–7.40 (m, 1H), 7.35–7.29 (m, 2H), 7.00 (dd, J = 8.3, 1.7 Hz, 1H), 6.38–6.33 (m, 1H), 5.15–4.27 (m, 4H), 3.23–3.05 (m, 2H), 2.48–2.42 (m, 1H), 2.31 (d, J = 1.8 Hz, 3H), 1.98–1.79 (m, 3H), 1.61–1.47 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.2, 160.9, 160.7 (d, J = 243.0 Hz), 157.4, 156.2, 138.5 (d, J = 10.0 Hz), 135.0, 132.4 (d, J = 6.2 Hz), 129.5, 127.6, 125.6, 124.1 (d, J = 17.0 Hz), 123.8, 121.0, 118.8, 115.7 (d, J = 2.6 Hz), 111.2, 107.1 (d, J = 27.8 Hz), 102.5, 100.8, 46.4, 45.9, 42.6, 42.2, 27.7, 24.7, 13.9; HRMS (ESI) calcd for C27H27FN7O [M + H]+ 484.2256, found 484.2261.
(S)-1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(quinolin-6-ylmethyl)piperidine-3-carboxamide (49)
Using a similar procedure to the preparation of 8. Light brown solid (yield 86%): 1H NMR (500 MHz, DMSO-d6) 9.37 (s, 1H), 8.86 (d, J = 2.7 Hz, 1H), 8.63 (s, 1H), 8.32–8.25 (m, 2H), 8.01–7.94 (m, 2H), 7.90–7.85 (m, 1H), 7.80 (s, 1H), 7.68–7.64 (m, 1H), 7.53–7.44 (m, 2H), 5.11–4.40 (m, 4H), 3.29–3.09 (m, 2H), 2.57–2.53 (m, 1H), 2.31 (s, 3H), 2.05–1.81 (m, 3H), 1.61–1.52 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.6, 160.9, 160.7 (d, J = 243.6 Hz), 157.5, 156.2, 150.2, 147.0, 138.5 (d, J = 10.1 Hz), 137.7, 135.7, 132.4 (d, J = 6.0 Hz), 129.3, 129.0, 127.6, 125.5, 124.1 (d, J = 17.5 Hz), 123.9, 115.7 (d, J = 3.2 Hz), 107.1 (d, J = 27.6 Hz), 102.5, 47.2, 46.4, 42.5, 41.9, 27.7, 24.5, 13.9; HRMS (ESI) calcd for C28H27FN7O [M + H]+ 496.2256, found 496.2251.
(S)-N-(Benzo[d]thiazol-5-ylmethyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (50)
Using a similar procedure to the preparation of 8. Light brown solid (yield 90%): 1H NMR (500 MHz, DMSO-d6) δ 9.41–9.31 (m, 2H), 8.60 (s, 1H), 8.28 (s, 1H), 8.11–8.05 (m, 1H), 7.98–7.94 (m, 2H), 7.89–7.85 (m, 1H), 7.48 (t, J = 8.2 Hz, 1H), 7.40 (d, J = 8.1 Hz, 1H), 5.07–4.40 (m, 4H), 3.48–3.35 (m, 2H), 2.58–2.53 (m, 1H), 2.30 (s, 3H), 2.03–1.81 (m, 3H), 1.62–1.51 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.6, 160.9, 160.7 (d, J = 243.1 Hz), 157.4, 156.5, 156.2, 138.5 (d, J = 10.1 Hz), 138.0, 132.4 (d, J = 6.0 Hz), 131.9, 125.1, 124.1 (d, J = 17.2 Hz), 123.8, 122.2, 121.3, 115.7 (d, J = 3.2 Hz), 107.1 (d, J = 27.9 Hz), 102.5, 47.1, 46.3, 42.1, 41.8, 27.7, 24.5, 13.9 (d, J = 2.9 Hz); HRMS (ESI) calcd for C26H25FN7OS [M + H]+ 502.1820, found 502.1813.
(S)-N-(Benzo[d]thiazol-5-ylmethyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (51)
Using a similar procedure to the preparation of 8. Light brown solid (yield 90%): 1H NMR (500 MHz, DMSO-d6) δ 9.49 (s, 1H), 9.38 (s, 1H), 8.60 (s, 1H), 8.37–8.33 (m, 2H), 8.30 (s, 1H), 8.12–8.07 (m, 1H), 7.99–7.95 (m, 3H), 7.40 (d, J = 8.2 Hz, 1H), 5.11–4.38 (m, 4H), 3.47–3.19 (m, 2H), 2.59–2.53 (m, 1H), 2.02–1.83 (m, 3H), 1.62–1.49 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.6, 161.1, 157.5, 156.5 (d, J = 7.3 Hz), 142.1, 138.0, 131.9, 128.0 (q, J = 32.3 Hz), 126.9 (d, J = 4.0 Hz), 125.1, 124.6, 124.0 (q, J = 272.1 Hz), 122.3, 121.4, 120.6, 102.9, 46.5, 46.0, 42.1, 41.8, 27.7, 24.4; HRMS (ESI) calcd for C26H23F3N7OS [M + H]+ 538.1631, found 538.1626.
(S)-N-((1H-Indol-3-yl)methyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (52)
Using a similar procedure to the preparation of 8. Light brown solid (yield 83%): [α]D25 = +88.2 (c 0.4, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.31 (s, 1H), 8.27–8.20 (m, 2H), 7.96 (dd, J = 11.0, 2.2 Hz, 1H), 7.88–7.81 (m, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.52–7.46 (m, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.25 (d, J = 2.2 Hz, 1H), 7.10–7.03 (m, 1H), 6.99–6.93 (m, 1H), 5.00–4.34 (m, 4H), 3.26–3.10 (m, 2H), 2.48–2.40 (m, 1H), 2.30 (s, 3H), 1.93–1.78 (m, 3H), 1.57–1.44 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.1, 160.8, 160.7 (d, J = 243.3 Hz), 157.4, 156.1, 138.5 (d, J = 10.4 Hz), 136.3, 132.4 (d, J = 5.9 Hz), 126.5, 124.0 (d, J = 17.2 Hz), 123.8, 123.7, 121.1, 118.7, 118.5, 115.7, 111.4, 107.1 (d, J = 27.7 Hz), 102.4, 47.2, 46.2, 42.2, 34.0, 27.7, 24.6, 13.8; HRMS (ESI) calcd for C27H27FN7O [M + H]+ 484.2256, found 484.2261.
(S)-N-((1H-Indol-2-yl)methyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (53)
Using a similar procedure to the preparation of 8. Light brown solid (yield 84%): [α]D25 = +79.2 (c 0.4, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 10.95 (d, J = 2.1 Hz, 1H), 9.35 (s, 1H), 8.49 (t, J = 5.5 Hz, 1H), 8.28 (s, 1H), 7.98 (dd, J = 11.0, 2.2 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.50–7.40 (m, 2H), 7.34–7.28 (m, 1H), 7.05–6.92 (m, 2H), 6.26 (dd, J = 2.1, 0.9 Hz, 1H), 5.17–4.33 (m, 4H), 3.79–3.46 (m, 2H), 2.58–2.52 (m, 1H), 2.31 (d, J = 1.8 Hz, 3H), 2.01–1.79 (m, 3H), 1.60–1.47 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 172.5, 160.9, 160.7 (d, J = 243.5 Hz), 157.4, 156.2, 138.5 (d, J = 10.2 Hz), 137.0, 136.1, 132.4 (d, J = 6.1 Hz), 127.9, 124.1 (d, J = 17.4 Hz), 123.8, 120.6, 119.5, 118.8, 115.7, 111.0, 107.1 (d, J = 27.7 Hz), 102.5, 99.1, 47.1, 46.5, 42.2, 36.1, 27.7, 24.4, 13.9; HRMS (ESI) calcd for C27H27FN7O [M + H]+ 484.2256, found 484.2261.
(S)-N-((5-Chloro-1H-indol-2-yl)methyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (54)
Using a similar procedure to the preparation of 8. Light brown solid (yield 74%): [α]D25 = +89.3 (c 0.7, MeOH); 1H NMR (400 MHz, DMSO-d6) δ 11.19 (s, 1H), 9.34 (s, 1H), 8.52 (t, J = 5.6 Hz, 1H), 8.28 (s, 1H), 7.97 (dd, J = 11.0, 2.2 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.51–7.42 (m, 2H), 7.35–7.29 (m, 1H), 7.05–7.00 (m, 1H), 6.26 (d, J = 1.9 Hz, 1H), 5.13–4.29 (m, 4H), 3.81–3.49 (m, 2H), 2.57–2.52 (m, 1H), 2.30 (s, 3H), 2.04–1.80 (m, 3H), 1.62–1.47 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 172.6, 160.9, 160.7 (d, J = 243.4 Hz), 157.4, 156.1, 139.0, 138.5 (d, J = 10.5 Hz), 134.6, 132.4 (d, J = 6.2 Hz), 129.1, 124.1 (d, J = 17.1 Hz), 123.8, 123.4, 120.5, 118.6, 115.7 (d, J = 3.5 Hz), 112.5, 107.1 (d, J = 27.6 Hz), 102.5, 98.9, 46.8, 46.2, 42.2, 36.0, 27.6, 24.6, 13.9; HRMS (ESI) calcd for C27H26ClFN7O [M + H]+ 518.1866, found 518.1870.
N-(Pyridin-3-ylmethyl)-1-(2-(p-tolyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carboxamide (55)
Using a similar procedure to the preparation of 8. Light yellow solid (yield 87%): 1H NMR (500 MHz, DMSO-d6) δ 9.29 (d, J = 4.0 Hz, 1H), 8.54 (t, J = 5.9 Hz, 1H), 8.50–8.49 (m, 1H), 8.46 (dd, J = 4.8, 1.6 Hz, 1H), 8.27 (s, 1H), 7.98 (d, J = 8.1 Hz, 2H), 7.65 (dt, J = 7.9, 2.0 Hz, 1H), 7.41–7.38 (m, 2H), 7.33 (dd, J = 7.8, 4.8 Hz, 1H), 4.98–4.51 (m, 2H), 4.32 (d, J = 5.8 Hz, 2H), 3.63–3.11 (m, 2H), 2.48 (s, 1H), 2.42–2.36 (m, 3H), 2.04–1.77 (m, 3H), 1.62–1.49 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.7, 163.2, 160.9, 157.5, 155.8, 148.7, 148.1, 137.7, 137.1, 135.0, 130.0, 123.5, 123.4, 120.1, 102.3, 46.2, 42.1, 27.6, 24.3, 20.6; HRMS (ESI) calcd for C24H26N7O [M + H]+ 428.2193, found 428.2190.
N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidine-3-carboxamide (56)
Using a similar procedure to the preparation of 8. Light brown solid (yield 71%): 1H NMR (400 MHz, DMSO-d6) δ 9.22 (d, J = 14.7 Hz, 1H), 8.79–8.64 (m, 1H), 8.25 (s, 1H), 8.04–7.92 (m, 2H), 7.94–7.85 (m, 1H), 7.82–7.73 (m, 2H), 7.54–7.44 (m, 1H), 7.44 (dd, J = 7.0, 5.4 Hz, 1H), 7.30 (ddd, J = 10.1, 8.3, 1.7 Hz, 1H), 4.55–4.38 (m, 2H), 4.15–3.63 (m, 4H), 3.26–3.12 (m, 1H), 2.44–2.05 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 171.9, 160.7 (d, J = 243.4 Hz), 160.2, 156.6, 155.9, 139.6, 138.6 (d, J = 10.2 Hz), 137.7, 135.6 (d, J = 15.3 Hz), 132.4, 127.8, 124.1, 124.0, 123.9, 123.8, 122.5, 122.1 (d, J = 11.5 Hz), 115.6, 106.9 (d, J = 27.7 Hz), 103.5, 50.4, 48.0, 43.7, 42.0, 29.1, 13.8; HRMS (ESI) calcd for C26H24FN6OS [M + H]+ 487.1711, found 487.1720.
N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidine-3-carboxamide (57)
Using a similar procedure to the preparation of 8. Light brown solid (yield 77%): 1H NMR (400 MHz, DMSO-d6) δ 9.36 (d, J = 13.7 Hz, 1H), 8.80–8.65 (m, 1H), 8.38 (dd, J = 8.7, 3.5 Hz, 2H), 8.27 (s, 1H), 8.02–7.92 (m, 3H), 7.82–7.73 (m, 2H), 7.48–7.40 (m, 1H), 7.35–7.25 (m, 1H), 4.51–4.39 (m, 2H), 4.17–3.66 (m, 4H), 3.24–3.19 (m, 1H), 2.42–2.07 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.4, 160.9, 157.4, 156.4, 142.7, 140.1, 138.2, 136.2, 128.32 (q, J = 32.6 Hz), 128.31, 127.4, 125.4, 124.7, 124.5 (q, J = 272.2 Hz), 122.5, 120.9, 104.4, 50.9, 47.8, 44.2, 42.4, 29.6; HRMS (ESI) calcd for C26H22F3N6OS [M + H]+ 523.1522, found 523.1527.
1-(2-(3-Fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-N-(4-(methylthio)benzyl)pyrrolidine-3-carboxamide (58)
Using a similar procedure to the preparation of 8. Light brown solid (yield 80%): 1H NMR (400 MHz, DMSO-d6) δ 9.21 (d, J = 13.1 Hz, 1H), 8.70–8.58 (m, 1H), 8.24 (s, 1H), 8.03–7.95 (m, 1H), 7.92–7.87 (m, 1H), 7.54–7.46 (m, 1H), 7.27–7.19 (m, 4H), 4.36–4.22 (m, 2H), 4.11–3.62 (m, 4H), 3.24–3.12 (m, 1H), 2.45 (s, 3H), 2.37–2.06 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 171.9, 160.7 (d, J = 243.0 Hz), 160.2, 156.6, 155.9, 138.6 (d, J = 10.3 Hz), 136.3, 136.2, 132.4, 127.9, 126.1, 124.1, 123.9 (d, J = 17.4 Hz), 115.5 (d, J = 4.7 Hz), 106.9 (d, J = 27.7 Hz), 103.5, 50.3, 48.0, 43.7, 41.9, 29.1, 14.9, 13.8; HRMS (ESI) calcd for C25H26FN6OS [M + H]+ 477.1867, found 477.1876.
N-(4-(Methylthio)benzyl)-1-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)pyrrolidine-3-carboxamide (59)
Using a similar procedure to the preparation of 8. Light brown solid (yield 74%): 1H NMR (400 MHz, DMSO-d6) δ 9.35 (d, J = 12.5 Hz, 1H), 8.73–8.57 (m, 1H), 8.41–8.35 (m, 2H), 8.27 (s, 1H), 7.97 (dd, J = 8.7, 4.5 Hz, 2H), 7.29–7.20 (m, 4H), 4.36–4.22 (m, 2H), 4.13–3.65 (m, 4H), 3.22–3.15 (m, 1H), 2.45 (s, 3H), 2.41–2.03 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.3, 160.9, 157.4, 156.4, 142.7, 136.8, 136.5, 128.4, 128.2, 127.3, 126.6, 125.4, 124.5 (q, J = 271.9 Hz), 120.9, 104.4, 50.9, 48.5, 44.1, 42.4, 29.6, 15.4; HRMS (ESI) calcd for C25H24F3N6OS [M + H]+ 513.1679, found 513.1683.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-4-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxamide (62)
(S)-1-(tert-Butoxycarbonyl)-4-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxylic acid (46 mg, 0.10 mmol), benzo[b]thiophen-5-ylmethanamine (33 mg, 0.20 mmol), HATU (57 mg, 0.15 mmol), and DMAP (37 mg, 0.3 mmol) were dissolved in dry DMF (1 mL) and stirred at room temperature overnight. After purification by Prep-HPLC, the product was dissolved in DCM (2 mL) and treated with HCl (4 M in dioxane, 200 μL). The mixture was stirred at room temperature for 2 h and was dried under vacuum. The residue was purified by Prep-HPLC to provide the product 62 (38 mg, yield 76% over 2 steps) as a white solid: [α]D25 = +78.8 (c 1.2, MeOH); 1H NMR (400 MHz, methanol-d4) δ 8.99 (s, 1H), 8.25 (s, 1H), 7.78–7.59 (m, 4H), 7.51 (d, J = 5.4 Hz, 1H), 7.37–7.30 (m, 1H), 7.26–7.18 (m, 2H), 4.74–4.47 (m, 3H), 4.30–4.18 (m, 1H), 4.03–3.79 (m, 3H), 3.40–3.32 (m, 1H), 3.22–3.10 (m, 1H), 2.31 (d, J = 2.0 Hz, 3H); 13C NMR (100 MHz, methanol-d4) δ 171.2, 162.6 (d, J = 245.0 Hz), 161.1, 159.5, 156.8, 141.3, 140.1, 139.8 (d, J = 9.5 Hz), 135.8, 133.5 (d, J = 5.7 Hz), 128.2, 126.5 (d, J = 17.5 Hz), 125.1, 125.0, 124.6, 123.5, 123.5, 116.8 (d, J = 3.6 Hz), 108.9 (d, J = 28.0 Hz), 103.8, 58.1, 46.4, 46.2, 44.3, 43.5, 14.2 (d, J = 3.2 Hz); HRMS (ESI) calcd for C26H25FN7OS [M + H]+ 502.1820, found 502.1829.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-4-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxamide (63)
Using a similar procedure to the preparation of 62. Light brown solid (yield 84%): [α]D25 = +84.5 (c 0.4, MeOH); 1H NMR (400 MHz, methanol-d4) δ 9.12 (s, 1H), 8.26 (s, 1H), 8.12 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.4 Hz, 2H), 7.74–7.63 (m, 2H), 7.49 (d, J = 5.4 Hz, 1H), 7.22–7.12 (m, 2H), 4.76–4.45 (m, 3H), 4.34–4.19 (m, 1H), 4.05–3.81 (m, 3H), 3.40–3.33 (m, 1H), 3.24–3.14 (m, 1H); 13C NMR (100 MHz, methanol-d4) δ 170.7, 161.4, 159.6, 157.2, 143.3, 141.3, 140.1, 135.7, 131.2 (q, J = 32.9 Hz), 128.2, 127.95, 127.91, 125.5, 125.2 (q, J = 271.4 Hz), 125.1, 124.6, 123.5, 123.4, 121.9, 104.2, 58.0, 47.3, 46.1, 44.3, 43.4; HRMS (ESI) calcd for C26H23F3N7OS [M + H]+ 538.1631, found 538.1634.
(R)-N-(Benzo[b]thiophen-5-ylmethyl)-4-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxamide (64)
Using a similar procedure to the preparation of 62. Light brown solid (yield 84%):1H NMR (500 MHz, DMSO-d6) δ 9.47 (s, 1H), 8.62 (t, J = 6.0 Hz, 1H), 8.32 (d, J = 13.9 Hz, 3H), 7.97 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.3 Hz, 1H), 7.72 (d, J = 5.8 Hz, 2H), 7.36 (d, J = 5.4 Hz, 1H), 7.25 (d, J = 8.3 Hz, 1H), 4.98–4.83 (m, 1H), 4.48–4.37 (m, 2H), 4.29–4.05 (m, 2H), 3.71–3.28 (m, 2H), 3.20–3.03 (m, 1H), 2.93–2.82 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 170.6, 163.3, 161.1, 157.8, 156.5, 142.1, 139.5, 137.6, 135.6, 128.0 (q, J = 32.3 Hz), 127.7, 126.9 (q, J = 4.0 Hz), 124.7, 124.1, 124.0 (q, J = 271.6 Hz), 123.8, 122.4, 120.6, 57.4, 42.1; HRMS (ESI) calcd for C26H23F3N7OS [M + H]+ 538.1631, found 538.1627.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-4-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)-1-methylpiperazine-2-carboxamide (61)
To a solution of 62 (20 mg, 0.04 mmol) and formaldehyde (37 wt % in H2O, 100 μL) in MeOH, HOAc (100 μL) was added. After stirring at room temperature for 20 min, NaBH3CN (8 mg, 0.12 mmol) was added. The reaction was continued at room temperature overnight and then concentrated under vacuum. The residue was purified by Prep-HPLC to provide the 61 (17 mg, yield 83%) as a light brown solid: [α]D25 = +87.2 (c 0.6, MeOH); 1H NMR (400 MHz, methanol-d4) δ 8.97 (s, 1H), 8.27 (s, 1H), 8.15 (s, 1H), 7.75–7.61 (m, 4H), 7.50 (d, J = 5.4 Hz, 1H), 7.41–7.31 (m, 1H), 7.26–7.14 (m, 2H), 4.70–4.27 (m, 4H), 3.93–3.82 (m, 1H), 3.75–3.64 (m, 1H), 3.17–3.07 (m, 1H), 3.08–2.98 (m, 1H), 2.68–2.56 (m, 1H), 2.39 (s, 3H), 2.32 (s, 3H); 13C NMR (101 MHz, methanol-d4) δ 173.0, 162.6 (d, J = 245.1 Hz), 160.8, 159.4, 156.8, 141.3, 140.1, 139.9 (d, J = 9.6 Hz), 136.0, 133.7–133.0 (m), 128.1, 126.5 (d, J = 17.2 Hz), 125.2, 124.7, 123.6, 123.4, 116.9, 108.8 (d, J = 28.3 Hz), 103.9, 68.1, 52.8, 47.4, 46.3, 44.1, 43.9, 14.2; HRMS (ESI) calcd for C27H27FN6OS [M + H]+ 516.1976, found 516.1984.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-1-methyl-4-(2-(4-(trifluoromethyl)phenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-2-carboxamide (60)
Using a similar procedure to the preparation of 61. Light brown solid (yield 84%): [α]D25 = +82.1 (c 0.6, MeOH); 1H NMR (400 MHz, methanol-d4) δ 9.07 (s, 1H), 8.29 (s, 1H), 8.17–8.10 (m, 3H), 7.82 (d, J = 8.4 Hz, 2H), 7.71–7.67 (m, 2H), 7.48 (d, J = 5.5 Hz, 1H), 7.23–7.17 (m, 2H), 4.69–4.29 (m, 4H), 3.98–3.86 (m, 1H), 3.77–3.65 (m, 1H), 3.18–3.10 (m, 1H), 3.09–3.02 (m, 1H), 2.68–2.59 (m, 1H), 2.40 (s, 3H); 13C NMR (100 MHz, methanol-d4) δ 173.0, 161.2, 159.5, 157.3, 143.4, 141.3, 140.1, 136.1, 131.1 (q, J = 33.1 Hz), 128.2, 127.94, 127.90, 125.6, 125.3 (q, J = 271.1 Hz), 125.2, 124.6, 123.6, 123.4, 121.9, 104.3, 68.0, 52.7, 47.0, 46.4, 44.1, 43.9; HRMS (ESI) calcd for C27H25F3N7OS [M + H]+ 552.1788, found 552.1789.
N-(Benzo[b]thiophen-5-ylmethyl)-3-((2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)(methyl)amino)propenamide (65)
Using a similar procedure to the preparation of 8. White solid (yield 88%): 1H NMR (400 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.56–8.50 (m, 1H), 8.27 (s, 1H), 8.02–7.81 (m, 3H), 7.76–7.68 (m, 2H), 7.54–7.46 (m, 1H), 7.39–7.34 (m, 1H), 7.23 (dd, J = 8.3, 1.7 Hz, 1H), 4.39 (d, J = 5.8 Hz, 2H), 4.15–4.00 (m, 2H), 3.45 (s, 3H), 2.70–2.55 (m, 2H), 2.30 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.2, 160.7 (d, J = 243.6 Hz), 160.6, 158.0, 156.2, 139.5, 138.5 (d, J = 10.3 Hz), 137.6, 135.7, 132.4 (d, J = 6.1 Hz), 127.8, 124.7, 124.1, 123.7, 122.4, 122.0, 115.6, 107.0 (d, J = 27.5 Hz), 103.3, 47.8, 46.9, 42.2, 33.3, 13.9; HRMS (ESI) calcd for C25H24FN6OS [M + H]+ 475.1711, found 475.1715.
N-(Benzo[b]thiophen-5-ylmethyl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)azepane-4-carboxamide (66)
Using a similar procedure to the preparation of 8. Light brown solid (yield 88%): 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.33 (t, J = 6.0 Hz, 1H), 8.26 (s, 1H), 7.98 (d, J = 11.1 Hz, 1H), 7.94–7.87 (m, 2H), 7.75–7.69 (m, 2H), 7.53–7.48 (m, 1H), 7.41 (dd, J = 11.4, 5.4 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 4.39–4.32 (m, 2H), 4.29–4.15 (m, 1H), 4.13–4.03 (m, 1H), 4.00–3.64 (m, 2H), 2.40–2.28 (m, 4H), 2.23–1.52 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δ 175.1, 160.7 (d, J = 243.2 Hz), 157.7, 156.3, 139.5, 138.5 (d, J = 10.5 Hz), 137.5, 136.0 (d, J = 3.8 Hz), 132.4 (d, J = 5.9 Hz), 127.7, 124.3 (d, J = 9.1 Hz), 124.1, 124.0, 123.8, 122.4, 121.9, 115.7 (d, J = 3.5 Hz), 107.1 (d, J = 27.8 Hz), 102.8, 46.5, 44.7, 41.9, 30.4, 30.2, 25.8, 13.8 (d, J = 2.9 Hz); HRMS (ESI) calcd for C28H28FN6OS [M + H]+ 515.2024, found 515.2031.
(S)-1-(6-Bromothieno[2,3-d]pyrimidin-4-yl)piperidine-3-carboxylic acid (S19)
To a solution of 6-bromo-4-chlorothieno[2,3-d]pyrimidine (250 mg, 1.0 mmol) and (S)-piperidine-3-carboxylic acid (194 mg, 1.5 mmol) in EtOH (5 mL), DIPEA (1 mL) was added. After stirring at 70 °C for 1 h and cooldown, the solvent was removed under vacuum. The residue was purified by Prep-HPLC to provide S19 (311 mg, yield 91%) as a white solid: 1H NMR (500 MHz, methanol-d4) δ 8.32 (s, 1H), 7.68 (s, 1H), 4.51 (dd, J = 13.6, 3.7 Hz, 1H), 4.26 (dd, J = 13.5, 4.4 Hz, 1H), 3.53 (dd, J = 13.4, 9.6 Hz, 1H), 3.43 (ddd, J = 13.5, 10.7, 3.2 Hz, 1H), 2.63–2.57 (m, 1H), 2.16–2.09 (m, 1H), 1.91–1.82 (m, 2H), 1.70–1.61 (m, 1H); LRMS (ESI) calcd for C12H13BrN3O2S [M + H]+ 341.99, found 341.82.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(6-bromothieno[2,3-d]pyrimidin-4-yl)piperidine-3-carboxamide (S20)
S19 (200 mg, 0.58 mmol), benzo[b]thiophen-5-ylmethanamine (142 mg, 0.87 mmol), HATU (331 mg, 0.87 mmol), and DMAP (213 mg, 1.74 mmol) were dissolved in dry DMF (4 mL) and stirred at room temperature overnight. The mixture was purified by Prep-HPLC to provide S20 (237 mg, yield 84%) as a white solid: 1H NMR (500 MHz, methanol-d4) δ 8.28 (s, 1H), 7.84 (d, J = 8.3 Hz, 1H), 7.75 (s, 1H), 7.61–7.53 (m, 2H), 7.30 (dd, J = 24.5, 6.9 Hz, 2H), 4.51 (dt, J = 27.6, 14.3 Hz, 4H), 3.56–3.48 (m, 1H), 3.38 (t, J = 12.7 Hz, 1H), 2.66–2.59 (m, 1H), 2.11–1.84 (m, 3H), 1.72–1.61 (m, 1H); LRMS (ESI) calcd for C21H20BrN4OS2 [M + H]+ 487.03, found 487.10.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(6-(p-tolyl)thieno[2,3-d]pyrimidin-4-yl)piperidine-3-carboxamide (70)
A flask fitted with a rubber septum was charged with S20 (49 mg, 0.10 mmol), 4,4,5,5-tetramethyl-2-(p-tolyl)-1,3,2-dioxaborolane (33 mg, 0.15 mmol), Pd(dppf)Cl2 (8 mg, 0.01 mmol), Na2CO3 (32 mg, 0.3 mmol), dixoane/H2O (4 mL/1 mL) and then purged with argon. The mixture was stirred at 90 °C overnight. The reaction mixture was then cooled to room temperature, diluted with ethyl acetate (20 mL), filtered through Celite, and concentrated in vacuo. The purification by Prep-HPLC afforded product 70 (34 mg, yield 68%) as a light yellow solid: 1H NMR (500 MHz, DMSO-d6) δ 8.55 (s, 1H), 8.38 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.83 (s, 1H), 7.74 (d, J = 5.3 Hz, 2H), 7.66 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 5.3 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.23 (d, J = 8.1 Hz, 2H), 4.67–4.48 (m, 2H), 4.47–4.33 (m, 2H), 3.40–3.23 (m, 2H), 2.62–2.54 (m, 1H), 2.34 (s, 3H), 1.99 (s, 1H), 1.89–1.79 (m, 2H), 1.64–1.52 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.6, 167.8, 157.5, 152.6, 139.5, 138.3, 137.6, 135.8, 130.2, 129.7, 127.8, 126.0, 124.0, 123.8, 122.5, 122.0, 117.3, 116.3, 49.4, 47.2, 42.2, 42.0, 27.5, 24.2, 20.8; HRMS (ESI) calcd for C28H27N4OS2 [M + H]+ 499.1621, found 499.1615.
(S)-N-(Benzo[b]thiophen-5-ylmethyl)-1-(6-(3-fluoro-4-methylphenyl)thieno[2,3-d]pyrimidin-4-yl)piperidine-3-carboxamide (71)
Using a similar procedure to the preparation of 70. Light yellow solid (yield 76%): 1H NMR (500 MHz, DMSO-d6) δ 8.53 (t, J = 5.9 Hz, 1H), 8.39 (s, 1H), 7.95 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.75–7.69 (m, 3H), 7.50–7.45 (m, 1H), 7.39–7.30 (m, 2H), 7.26 (d, J = 8.2 Hz, 1H), 4.67–4.47 (m, 2H), 4.40 (qd, J = 14.9, 5.8 Hz, 2H), 3.40–3.26 (m, 2H), 2.60–2.54 (m, 1H), 2.27 (s, 3H), 1.97–1.94 (m, 1H), 1.88–1.79 (m, 2H), 1.64–1.53 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 172.6, 168.0, 161.0 (d, J = 243.6 Hz), 157.6, 152.8, 139.5, 137.6, 136.0, 135.8, 132.7 (d, J = 8.3 Hz), 132.3, 127.7, 124.0, 123.8, 122.4, 122.0, 117.7, 117.1, 112.4 (d, J = 23.9 Hz), 49.3, 47.3, 42.2, 42.0, 27.5, 24.2, 14.0; HRMS (ESI) calcd for C28H26FN4OS2 [M + H]+ 517.1527, found 517.1523.
(S)-N′-(Benzo[b]thiophen-5-yl)-1-(2-(3-fluoro-4-methylphenyl)-2H-pyrazolo[3,4-d]pyrimidin-4-yl)piperidine-3-carbohydrazide (79)
Using a similar procedure to the preparation of 8. Light brown solid (yield 87%): 1H NMR (500 MHz, DMSO-d6) δ 9.94 (s, 1H), 9.64 (s, 1H), 8.62 (s, 1H), 8.02–7.95 (m, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.74–7.51 (m, 4H), 7.29–7.20 (m, 1H), 7.11 (d, J = 2.3 Hz, 1H), 6.88 (dd, J = 8.7, 2.2 Hz, 1H), 5.06–4.29 (m, 2H), 3.93–3.53 (m, 2H), 2.86–2.66 (m, 1H), 2.32 (s, 3H), 2.17–1.70 (m, 4H); 13C NMR (125 MHz, DMSO-d6) δ 172.1, 160.7 (d, J = 244.0 Hz), 158.2, 157.0, 151.8, 146.9, 140.3, 137.6 (d, J = 10.3 Hz), 132.7 (d, J = 6.2 Hz), 129.9, 127.6, 127.1, 125.2 (d, J = 17.2 Hz), 123.5, 122.5, 116.0 (d, J = 3.2 Hz), 112.4, 107.4 (d, J = 27.8 Hz), 105.0, 101.8, 48.2, 47.0, 27.1, 24.3, 13.9; HRMS (ESI) calcd for C26H25FN7OS [M + H]+ 502.1820, found 502.1814.
Acknowledgments
This study is supported by NIH grant RF1AG067771 and in part by P30CA023074. J.M.G.-B. was supported, in part, by NIH T32AG57468 and M.S.L. and J.M.C. by NIH T32 GM008804. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Dr Bhargava Karumudi is thanked for contributions to compound synthesis. Dr Steven Gardell of Advent Health is thanked for helpful discussions.
Glossary
Abbreviations Used
- ADH
alcohol dehydrogenase
- ADRD
Alzheimer’s disease and related dementia
- aq.
aqueous solution
- Ar
argon
- ARTs
ADP-ribosyltransferases
- Boc
tert-butyloxycarbonyl
- DCM
dichloromethane
- DMAP
4,4-dimethylaminopyridine
- DMF
dimethylformamide
- DIPEA
N,N-diisopropylethylamine
- EtOAc or EA
ethyl acetate
- equiv. or eq
equivalent
- Et
ethyl
- ESI
electron spray ionization
- FP
fluorescence polarization
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uranium
- HOAc
acetic acid
- HRMS
high-resolution mass spectrometry
- LRMS
low-resolution mass spectrometry
- m/z
ratio of mass to charge
- Me
methyl
- MeCN
acetonitrile
- N-PAMs
NAMPT positive allosteric modulators
- NAD+
nicotinamide adenine dinucleotide
- NADP+
2-phosphate derivative
- NAM
nicotinamide
- NAMPT
nicotinamide phosphoribosyltransferase
- NAPRT
nicotinic acid phosphoribosyltransferase
- NMN
nicotinamide mononucleotide
- NMNAT1
nicotinamide/nicotinic acid mononucleotide adenylyltransferase
- NMR
nuclear magnetic resonance
- PARP
poly-ADP-ribose polymerase
- PDB
protein data bank
- PRPP
α--5-phosphoribosyl-1-pyrophosphate
- rt
room temperature
- SARM1
sterile alpha and TIR motif containing 1
- SIRTs
sirtuins
- TEA
triethanolamine
- TLR4
toll-like receptor 4
- T2D
type-2 diabetes
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01406.
Author Contributions
G.R.J.T. conceived the project. K.R., Z.S., and G.R.J.T. contributed to all data interpretation. Z.S. directed and performed the synthetic design and isolation of chemical probes with synthetic assistance from G.R.V., J.M.G.-B., R.X., and J.F. with modeling assistance from N.C., M.S.L., and I.K. K.R., M.A.-B., and C.P. optimized biochemical and biological assays with contributions from S.R.M. and J.M.C. N.M. and S.R.M. performed microsomal stability studies. K.R. performed all structural biology experimental and data analysis tasks. G.R.J.T. wrote the manuscript. All authors contributed to editing the manuscript.
The authors declare the following competing financial interest(s): G.T. is an inventor on patents owned by the University of Illinois.
Special Issue
Published as part of Journal of Medicinal Chemistryvirtual special issue “Celebrating the 60th Anniversary of the MIKIW Meeting-in-Miniature”.
Supplementary Material
References
- Del Nagro C.; Xiao Y.; Rangell L.; Reichelt M.; O’Brien T. Depletion of the central metabolite NAD leads to oncosis-mediated cell death. J. Biol. Chem. 2014, 289, 35182–35192. 10.1074/jbc.M114.580159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsefield S.; Burdett H.; Zhang X.; Manik M. K.; Shi Y.; Chen J.; Qi T.; Gilley J.; Lai J. S.; Rank M. X.; Casey L. W.; Gu W.; Ericsson D. J.; Foley G.; Hughes R. O.; Bosanac T.; von Itzstein M.; Rathjen J. P.; Nanson J. D.; Boden M.; Dry I. B.; Williams S. J.; Staskawicz B. J.; Coleman M. P.; Ve T.; Dodds P. N.; Kobe B. NAD(+) cleavage activity by animal and plant TIR domains in cell death pathways. Science 2019, 365, 793–799. 10.1126/science.aax1911. [DOI] [PubMed] [Google Scholar]
- Liu L.; Su X.; Quinn W. J.; Hui S.; Krukenberg K.; Frederick D. W.; Redpath P.; Zhan L.; Chellappa K.; White E.; Migaud M.; Mitchison T. J.; Baur J. A.; Rabinowitz J. D. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27 (1067), e1065 10.1016/j.cmet.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Pereira J.; Tarrago M. G.; Chini C. C. S.; Nin V.; Escande C.; Warner G. M.; Puranik A. S.; Schoon R. A.; Reid J. M.; Galina A.; Chini E. N. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab 2016, 23, 1127–1139. 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S.; Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J.; Baur J. A.; Imai S. I. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab 2018, 27, 513–528. 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang E. F.; Scheibye-Knudsen M.; Brace L. E.; Kassahun H.; SenGupta T.; Nilsen H.; Mitchell J. R.; Croteau D. L.; Bohr V. A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini C. C. S.; Peclat T. R.; Warner G. M.; Kashyap S.; Espindola-Netto J. M.; de Oliveira G. C.; Gomez L. S.; Hogan K. A.; Tarrago M. G.; Puranik A. S.; Agorrody G.; Thompson K. L.; Dang K.; Clarke S.; Childs B. G.; Kanamori K. S.; Witte M. A.; Vidal P.; Kirkland A. L.; De Cecco M.; Chellappa K.; McReynolds M. R.; Jankowski C.; Tchkonia T.; Kirkland J. L.; Sedivy J. M.; van Deursen J. M.; Baker D. J.; van Schooten W.; Rabinowitz J. D.; Baur J. A.; Chini E. N. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab 2020, 2, 1284–1304. 10.1038/s42255-020-00298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K.; Summers D. W.; Sasaki Y.; Mao X.; DiAntonio A.; Milbrandt J. The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron 2017, 93 (1334), e1335 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli U.; Travelli C.; Massarotti A.; Fakhfouri G.; Rahimian R.; Tron G. C.; Genazzani A. A. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 2013, 56, 6279–6296. 10.1021/jm4001049. [DOI] [PubMed] [Google Scholar]
- Chowdhry S.; Zanca C.; Rajkumar U.; Koga T.; Diao Y.; Raviram R.; Liu F.; Turner K.; Yang H.; Brunk E.; Bi J.; Furnari F.; Bafna V.; Ren B.; Mischel P. S. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. 10.1038/s41586-019-1150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan J. A.; Tao X.; Tong L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct Mol. Biol. 2006, 13, 582–588. 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- Uddin G. M.; Youngson N. A.; Doyle B. M.; Sinclair D. A.; Morris M. J. Nicotinamide mononucleotide (NMN) supplementation ameliorates the impact of maternal obesity in mice: comparison with exercise. Sci. Rep. 2017, 7, 15063. 10.1038/s41598-017-14866-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel W.; Rabeh W. M.; Bogan K. L.; Belenky P.; Wojcik M.; Seidle H. F.; Nedyalkova L.; Yang T.; Sauve A. A.; Park H. W.; Brenner C. Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS Biol. 2007, 5, e263 10.1371/journal.pbio.0050263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L.; Houtkooper R. H.; Moullan N.; Katsyuba E.; Ryu D.; Canto C.; Mottis A.; Jo Y. S.; Viswanathan M.; Schoonjans K.; Guarente L.; Auwerx J. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens C. R.; Denman B. A.; Mazzo M. R.; Armstrong M. L.; Reisdorph N.; McQueen M. B.; Chonchol M.; Seals D. R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. 10.1038/s41467-018-03421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellinger R. W.; Santos S. R.; Morris M.; Evans M.; Alminana D.; Guarente L.; Marcotulli E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. npj Aging Mech. Dis. 2017, 3, 17. 10.1038/s41514-017-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S. J.; Bernier M.; Aon M. A.; Cortassa S.; Kim E. Y.; Fang E. F.; Palacios H. H.; Ali A.; Navas-Enamorado I.; Di Francesco A.; Kaiser T. A.; Waltz T. B.; Zhang N.; Ellis J. L.; Elliott P. J.; Frederick D. W.; Bohr V. A.; Schmidt M. S.; Brenner C.; Sinclair D. A.; Sauve A. A.; Baur J. A.; de Cabo R. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metab. 2018, 27 (667), e664 10.1016/j.cmet.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani M.; Sorrentino V.; Oh C. M.; Li H.; de Lima T. I.; Zhang H.; Shong M.; Auwerx J. NAD(+) boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep 2021, 34, 108660 10.1016/j.celrep.2020.108660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A.; Schuster S.; Penke M.; Gorski T.; de Giorgis T.; Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat. Rev. Endocrinol 2015, 11, 535–546. 10.1038/nrendo.2015.117. [DOI] [PubMed] [Google Scholar]
- Pieper A. A.; McKnight S. L. Benefits of Enhancing Nicotinamide Adenine Dinucleotide Levels in Damaged or Diseased Nerve Cells. Cold Spring Harb Symp. Quant Biol. 2018, 83, 207–217. 10.1101/sqb.2018.83.037622. [DOI] [PubMed] [Google Scholar]
- Gardell S. J.; Hopf M.; Khan A.; Dispagna M.; Hampton Sessions E.; Falter R.; Kapoor N.; Brooks J.; Culver J.; Petucci C.; Ma C. T.; Cohen S. E.; Tanaka J.; Burgos E. S.; Hirschi J. S.; Smith S. R.; Sergienko E.; Pinkerton A. B. Boosting NAD(+) with a small molecule that activates NAMPT. Nat. Commun. 2019, 10, 3241. 10.1038/s41467-019-11078-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K. M.; Shen Z.; Gordon-Blake J.; Lee H.; Laham M. S.; Krider I. S.; Christie N.; Ackerman-Berrier M.; Penton C.; Knowles N. G.; Musku S. R.; Fu J.; Velma G. R.; Xiong R.; Thatcher G. R. J. Mechanism of Allosteric Modulation of Nicotinamide Phosphoribosyltransferase to Elevate Cellular NAD(). Biochemistry 2023, 62, 923–933. 10.1021/acs.biochem.2c00655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkerton A. B.; Sessions E. H.; Hershberger P.; Maloney P. R.; Peddibhotla S.; Hopf M.; Sergienko E.; Ma C. T.; Smith L. H.; Jackson M. R.; Tanaka J.; Tsuji T.; Akiu M.; Cohen S. E.; Nakamura T.; Gardell S. J. Optimization of a urea-containing series of nicotinamide phosphoribosyltransferase (NAMPT) activators. Bioorg. Med. Chem. Lett. 2021, 41, 128007 10.1016/j.bmcl.2021.128007. [DOI] [PubMed] [Google Scholar]
- Dutta T.; Kapoor N.; Mathew M.; Chakraborty S. S.; Ward N. P.; Prieto-Farigua N.; Falzone A.; DeLany J. P.; Smith S. R.; Coen P. M.; DeNicola G. M.; Gardell S. J. Source of nicotinamide governs its metabolic fate in cultured cells, mice, and humans. Cell Rep 2023, 42, 112218 10.1016/j.celrep.2023.112218. [DOI] [PubMed] [Google Scholar]
- Cambronne X. A.; Kraus W. L. Location, Location, Location: Compartmentalization of NAD(+) Synthesis and Functions in Mammalian Cells. Trends Biochem. Sci. 2020, 45, 858–873. 10.1016/j.tibs.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N.; Zhang L.; Gao W.; Huang C.; Huber P. E.; Zhou X.; Li C.; Shen G.; Zou B. NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Sig. Transduct Target Ther. 2020, 5, 227. 10.1038/s41392-020-00311-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. B.; Kubota S.; Ban N.; Yoshida M.; Santeford A.; Sene A.; Nakamura R.; Zapata N.; Kubota M.; Tsubota K.; Yoshino J.; Imai S. I.; Apte R. S. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep 2016, 17, 69–85. 10.1016/j.celrep.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall M.; Hassing A. S.; Treebak J. T. NAD(+) and NAFLD - caution, causality and careful optimizm. J. Physiol 2022, 600, 1135–1154. 10.1113/JP280908. [DOI] [PubMed] [Google Scholar]
- Oka S. I.; Byun J.; Huang C. Y.; Imai N.; Ralda G.; Zhai P.; Xu X.; Kashyap S.; Warren J. S.; Alan Maschek J.; Tippetts T. S.; Tong M.; Venkatesh S.; Ikeda Y.; Mizushima W.; Kashihara T.; Sadoshima J. Nampt Potentiates Antioxidant Defense in Diabetic Cardiomyopathy. Circ. Res. 2021, 129, 114–130. 10.1161/CIRCRESAHA.120.317943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R. Y.; Qin Y.; Lv X. Q.; Wang P.; Xu T. Y.; Zhang L.; Miao C. Y. A fluorometric assay for high-throughput screening targeting nicotinamide phosphoribosyltransferase. Anal. Biochem. 2011, 412, 18–25. 10.1016/j.ab.2010.12.035. [DOI] [PubMed] [Google Scholar]
- Akiu M.; Tsuji T.; Sogawa Y.; Terayama K.; Yokoyama M.; Tanaka J.; Asano D.; Sakurai K.; Sergienko E.; Sessions E. H.; Gardell S. J.; Pinkerton A. B.; Nakamura T. Discovery of 1-[2-(1-methyl-1H-pyrazol-5-yl)-[1,2,4]triazolo[1,5-a]pyridin-6-yl]-3-(pyridin-4- ylmethyl)urea as a potent NAMPT (nicotinamide phosphoribosyltransferase) activator with attenuated CYP inhibition. Bioorg. Med. Chem. Lett. 2021, 43, 128048 10.1016/j.bmcl.2021.128048. [DOI] [PubMed] [Google Scholar]
- Greiner J. V.; Glonek T. Intracellular ATP Concentration and Implication for Cellular Evolution. Biology (Basel) 2021, 10, 1166. 10.3390/biology10111166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W.; Yang T.; Ho L.; Zhao Z.; Wang J.; Chen L.; Zhao W.; Thiyagarajan M.; MacGrogan D.; Rodgers J. T.; Puigserver P.; Sadoshima J.; Deng H.; Pedrini S.; Gandy S.; Sauve A. A.; Pasinetti G. M. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Smythe G. A.; Braga O.; Brew B. J.; Grant R. S.; Guillemin G. J.; Kerr S. J.; Walker D. W. Concurrent quantification of quinolinic, picolinic, and nicotinic acids using electron-capture negative-ion gas chromatography-mass spectrometry. Anal. Biochem. 2002, 301, 21–26. 10.1006/abio.2001.5490. [DOI] [PubMed] [Google Scholar]
- Hwang E.; Song S. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules 2020, 687. 10.3390/biom10050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino J.; Schluter U.; Kroger H. Nicotinamide methylation and its relation to NAD synthesis in rat liver tissue culture. Biochemical basis for the physiological activities of 1-methylnicotinamide. Biochim. Biophys. Acta 1984, 801, 250–258. 10.1016/0304-4165(84)90074-6. [DOI] [PubMed] [Google Scholar]
- Marletta A. S.; Massarotti A.; Orsomando G.; Magni G.; Rizzi M.; Garavaglia S. Crystal structure of human nicotinic acid phosphoribosyltransferase. FEBS Open Bio 2015, 5, 419–428. 10.1016/j.fob.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E. S.; Schramm V. L. Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 2008, 47, 11086–11096. 10.1021/bi801198m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E. S.; Vetticatt M. J.; Schramm V. L. Recycling nicotinamide. The transition-state structure of human nicotinamide phosphoribosyltransferase. J. Am. Chem. Soc. 2013, 135, 3485–3493. 10.1021/ja310180c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Liu M.; Zu Y.; Yao H.; Wu C.; Zhang R.; Ma W.; Lu H.; Xi S.; Liu Y.; Hua L.; Wang G.; Tang Y. Optimization of NAMPT activators to achieve in vivo neuroprotective efficacy. Eur. J. Med. Chem. 2022, 236, 114260 10.1016/j.ejmech.2022.114260. [DOI] [PubMed] [Google Scholar]
- Yao H.; Liu M.; Wang L.; Zu Y.; Wu C.; Li C.; Zhang R.; Lu H.; Li F.; Xi S.; Chen S.; Gu X.; Liu T.; Cai J.; Wang S.; Yang M.; Xing G. G.; Xiong W.; Hua L.; Tang Y.; Wang G. Discovery of small-molecule activators of nicotinamide phosphoribosyltransferase (NAMPT) and their preclinical neuroprotective activity. Cell Res. 2022, 32, 570–584. 10.1038/s41422-022-00651-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu S.; Morrill C.; Almstead N. G.; Moon Y.-C. Selective Synthesis of 1-Substituted 4-Chloropyrazolo[3,4-d]pyrimidines. Org. Lett. 2013, 15, 1882–1885. 10.1021/ol4005382. [DOI] [PubMed] [Google Scholar]
- Yoshino M.; Yoshino J.; Kayser B. D.; Patti G. J.; Franczyk M. P.; Mills K. F.; Sindelar M.; Pietka T.; Patterson B. W.; Imai S. I.; Klein S. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 2021, 372, 1224–1229. 10.1126/science.abe9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 2005, 41, 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.